
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of bis(2,2,2-trifluoroethyl) carbonate flame retarding co-solvent on interfacial chemistry in carbonate ester lithium-ion battery electrolytes†
Mohammad Baghban
Shemirani
 ,
Florian
Gebert
and
Andrew J.
Naylor
*
,
Florian
Gebert
and
Andrew J.
Naylor
*
Department of Chemistry – Ångström Laboratory, Uppsala University, Uppsala, SE-75121, Sweden. E-mail: andy.naylor@kemi.uu.se
First published on 19th February 2025
Abstract
The development of flame-retarding battery electrolytes may be achieved by the inclusion of non-flammable solvents in existing conventional electrolyte formulations. Here the use of one such promising solvent, bis(2,2,2-trifluoroethyl) carbonate (TFEC), mixed with conventional lithium-ion battery solvents ethylene carbonate and ethyl methyl carbonate, achieves comparable or superior electrochemical performance to a state-of-the-art benchmark (up to 90% capacity retention between 5th and 200th cycle, compared with 76% for the benchmark). Further electrochemical analysis indicates comparable cell resistance and rate capability, though a TFEC content beyond 90 vol% leads to increased resistance and rapid capacity fading. This was found to be caused by lithium trapping in the graphite electrodes and formation of a thinner solid electrolyte interphase with a distinct chemistry, as determined by X-ray photoelectron spectroscopy. TFEC's low Li+-solvating ability likely significantly influences these electrolytes' physico-chemical and electrochemical behaviour.
Introduction
There is a growing attention to the safety of lithium-ion batteries (LIBs) used in portable electronic devices and electric vehicles. Improved safety can be considered to also contribute to greater sustainability through increased reliability and adoption by end users. Though there are many components of a battery that can be addressed when improving safety, one of the primary aspects is the thermal instability of the non-aqueous electrolyte.1 Many electrolyte components, including the electrolyte salt, are subject to breakdown during a thermal event of the battery, while many organic solvents employed are highly flammable.2,3One strategy to make the electrolyte less flammable is employing high salt concentrations (“electrolyte in salt”), decreasing the amount of flammable liquids in the cell and potentially reducing the risk of dendrite formation. However, this improvement in the battery's overall stability comes at the cost of higher internal resistances.4 Other approaches involve using ceramic, polymeric or composite solid-state electrolytes, which eliminate the need for flammable liquid electrolytes and offer increased thermal and chemical stability but poorer ionic conductivity.5–7 Another solution being explored is adding flame retarding components to the liquid electrolyte, often as phosphorous-containing or fluorine-containing additives or co-solvents, thereby maintaining many of the beneficial properties of the liquid electrolyte.3,8 The non-flammability of some fluorinated solvents is attributed to their ability to scavenge oxygen radicals during combustion, through the generation of fluorine radicals at elevated temperatures.9,10 This quenching action disrupts the chain reactions of combustion, leading to greater thermal stability within the battery.11 One fluorinated solvent which has shown promising non-flammable properties for LIB electrolytes is bis(2,2,2-trifluoroethyl) carbonate (TFEC). First used by Smart et al. in 2003,12 it has been demonstrated as a co-solvent in propylene carbonate-based electrolytes.13–16 Its excellent flame retarding properties have also been well-established in many such studies. Further benefits of TFEC include its ability to suppress nickel ion dissolution in nickel-rich cathode materials and to form reliable surface layers on silicon–graphite composite anodes.17,18 Unusually for carbonate esters, TFEC has a very low affinity for Li+-ions, which severely limits its ability to dissolve lithium salts.14,19 However, it also makes it an effective diluent for highly concentrated electrolytes, reducing their viscosity without disrupting the Li-solvation shell that is key to their stability and non-flammability.3,19,20
TFEC has been tested in full cells in proportions up to 30%12,21 and as a co-solvent with several unconventional main solvents (e.g. propylene carbonate, fluoroethylene carbonate).13–16,22 However, it has not thus far been investigated in detail as a co-solvent in higher proportions with conventional carbonate ester electrolyte solvents. The interfaces formed by such electrolyte systems have also not yet been studied. Here, we investigate the use of TFEC as a co-solvent in the LP57 (1 M LiPF6 in ethylene carbonate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl methyl carbonate – EC:EMC 3:7 v/v) electrolyte system, a commonly employed commercial electrolyte, in LiNi0.6Mn0.2Co0.2O2 (NMC622)|graphite full cells. Through varying the proportion of TFEC in LP57 at a constant LiPF6 concentration, the stability and performance of such electrolyte systems are studied in cells with industrially-relevant electrode materials using various electrochemical techniques. A key consideration affecting the performance of cells is the solid electrolyte interphase (SEI), a passivation layer that forms from electrolyte decomposition on the surface of the graphite electrode during the first charge–discharge cycles of a cell. An SEI should ideally be ionically conductive and electrically insulating, to allow Li-ions to move through it while preventing further electrolyte decomposition. The composition and effectiveness of the SEI are highly dependent on the electrolyte makeup. For this reason, the SEI formed from the TFEC-based electrolytes is studied in detail using surface-sensitive X-ray photoelectron spectroscopy (XPS) at multiple probing depths. XPS is selected as the main characterisation technique due to its excellent surface sensitivity matching the nanometre-thickness of the SEI, its ability to distinguish different oxidation states or bonding environments, and the large analysis area that negates the effects of inhomogeneity.23–25 This is in contrast to other possible techniques such as electron microscopy where the SEI is typically not easily observed and chemical information is limited to the elemental composition on a small sample size.
:ethyl methyl carbonate – EC:EMC 3:7 v/v) electrolyte system, a commonly employed commercial electrolyte, in LiNi0.6Mn0.2Co0.2O2 (NMC622)|graphite full cells. Through varying the proportion of TFEC in LP57 at a constant LiPF6 concentration, the stability and performance of such electrolyte systems are studied in cells with industrially-relevant electrode materials using various electrochemical techniques. A key consideration affecting the performance of cells is the solid electrolyte interphase (SEI), a passivation layer that forms from electrolyte decomposition on the surface of the graphite electrode during the first charge–discharge cycles of a cell. An SEI should ideally be ionically conductive and electrically insulating, to allow Li-ions to move through it while preventing further electrolyte decomposition. The composition and effectiveness of the SEI are highly dependent on the electrolyte makeup. For this reason, the SEI formed from the TFEC-based electrolytes is studied in detail using surface-sensitive X-ray photoelectron spectroscopy (XPS) at multiple probing depths. XPS is selected as the main characterisation technique due to its excellent surface sensitivity matching the nanometre-thickness of the SEI, its ability to distinguish different oxidation states or bonding environments, and the large analysis area that negates the effects of inhomogeneity.23–25 This is in contrast to other possible techniques such as electron microscopy where the SEI is typically not easily observed and chemical information is limited to the elemental composition on a small sample size.
Experimental methods
The following chemicals and materials were employed to formulate the electrolytes: bis(2,2,2-trifluoroethyl) carbonate (TCI America, 98.0%), tris(2,2,2-trifluoroethyl) phosphate (TCI America, 94.0%), LiPF6 (Solvionic, 99.99%), ethylene carbonate (Solvionic, 99.99%), ethyl methyl carbonate (Solvionic, 99.99%), and LP57 [1 M LiPF6 in EC:EMC, 3:7 v/v] (Solvionic, 99.9%). Electrolytes were formulated from the individual components apart from LP57, which was used as received. All electrolytes had a LiPF6 concentration of 1 M, except for that containing 90% TFEC, in which LiPF6 was not soluble at 1 M. The excess salt was removed from the 90% TFEC electrolyte by filtration; it consequently had a LiPF6 concentration of <1 M. No purification processes were carried out on the chemicals. Sample preparation was conducted in a glove box with an O2 content of less than 1.0 ppm and an H2O content of less than 1.5 ppm. Electrolytes were prepared by mixing all solvent components and drying them over molecular sieves for at least 3 days. The liquid was then filtered, and 1 mol L−1 LiPF6 was added. Before use, LiPF6 was dried at 70 °C under vacuum for 48 hours. All materials were dried at 60 °C for at least 3 hours prior to being stored in an argon-filled environment.
Electrolytes (30% TFEC, 50% TFEC, LP57) were tested for their flammability by placing 300 μL of the electrolyte into a stainless steel casing (from a CR2025 coin cell) inside a fume hood and setting alight with a lighter. The self-extinguishing time (SET) was calculated using the density of the electrolyte and the time taken for complete loss of the flame. Measurements were performed three times for each electrolyte.
For fabrication of CR2025 coin cells, commercial graphite (2.46 mA h cm−2) and NMC622 (LiNi0.6Mn0.2Co0.2O2) (2.0 mA h cm−2, 165 mA h g−1) electrodes obtained from Custom Cells GmbH were employed. The electrodes were dried at 120 °C under vacuum for 12 hours. Celgard 2400 served as the separator material in these cells. Cells were assembled inside an argon glovebox. Overall, each cell was made with 70 μl of electrolyte and they were rested for 12 hours prior to any electrochemical analysis.
Cathode disks with a diameter of 13 mm contained 16.8 mg of active material, providing a nominal capacity of 2.65 mA h. Anode disks with a diameter of 13 mm contained 19.9 mg of active material, providing a nominal capacity of 3.18 mA h. The electrode capacities were balanced to achieve an anode-to-cathode (N/P) ratio of 1.3.
Conductivity measurements were performed using a portable Mettler Toledo Seven2Go conductivity meter under inert conditions as mentioned above. Electrolytes for these measurements contained 98% purity LiPF6 (Solvionic). Calibration was performed using solutions with conductivities of 1413 μS cm−1 and 12.88 mS cm−1. Electrolytes were poured into 1.5 mL vials. Measurements were taken at approximately room temperature. Each measurement was performed twice and an average result taken, with the probe re-inserted into the vial each time. To prevent electrolyte evaporation, a lid was placed on the vial between measurements.
Neware battery testing systems were employed for electrochemical testing (cycling, rate capability, and intermittent current interruption – ICI – experiments) including constant current – constant voltage (CCCV) cycling: each cell was charged at constant current (CC) to 4.3 V, after which the voltage was held at 4.3 V until the current reached 1/10th of the current used during the preceding CC step. The cell was then discharged at constant current to 3 V. For long-term cycling, each cell underwent 2 cycles at C/10 (0.265 mA) and an additional 2 cycles at C/5 (0.53 mA) to allow SEI formation. Subsequent cycles were conducted at C/2 (1.325 mA). C-rates are based on a theoretical capacity of 165 mA h g−1. For rate capability experiments, no additional formation cycles beyond the initial five cycles at C/10 were performed. All tests were carried out at room temperature (∼20 °C). ICI experiments were conducted following the method described by Lacey et al.26,27 In short, this method involves pausing the battery cell for very short periods at regular intervals and calculating the cell resistance from the voltage response. In this study, 1 s pauses at 30 minute intervals were used, excluding the constant voltage steps, which had no interruptions. The resistance, Rcell at each pause was calculated using:
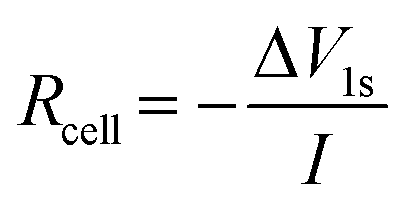
To allow SEI formation, each cell was first subjected to 2 cycles at C/10 and 2 cycles at C/5 under ICI conditions. The data were then analyzed using an in-house Python script. Cyclic voltammetry (CV) measurements were carried out using a BioLogic MPG-2 potentiostat. These measurements were conducted utilizing two-electrode Li|graphite cells. The scan rate was 0.1 mV s−1, and the potential swept initially down to 0.01 V vs. Li+/Li and up to 3.0 V vs. Li+/Li.
X-ray photoelectron spectroscopy (XPS) measurements were performed on graphite electrode samples cycled for 2 cycles at C/10 and 2 cycles at C/5 in the various electrolytes containing between 30 and 90% TFEC. Prior to measurement, the electrodes were removed from cells inside an argon glovebox, washed by dripping 1 ml of EMC over the surface of each electrode with a pipette, and dried in the glovebox atmosphere for 30 minutes. Pieces of the electrodes were cut and mounted on a sample holder in a floating configuration, using a piece of insulating tape between holder and sample. The sample holder was sealed inside the glovebox and the samples transferred under inert atmosphere to the instrument before being exposed to vacuum. A Kratos AXIS Supra+ spectrometer was used for measurements with photon energies of 1487 eV (Al Kα) and 2984 eV (Ag Lα), produced by a monochromatized dual anode X-ray source. Charge neutralisation was used during measurements. The analysis spot size was approximately 700 × 300 μm. Data analysis was performed using CasaXPS, employing the Gaussian-Lorentzian GL(30) peak shape and the Kratos relative sensitivity factors library for quantification from survey spectra. Energy calibration was achieved by setting the hydrocarbon (C–C) peak to 285.0 eV and the C–O peak (the most intense for C 1s) at close to 285.7 eV; spectral features for other core levels were considered to be at reasonable binding energies for the species that should be expected in the samples. Probing depths (3× IMFP) were calculated using the TPP-M2 equation as detailed in the NIST database, using parameters for polyethylene, as a low-density material representative of the surface layer studied here.28,29 Approximate depths are estimated as 14 and 24 nm for the excitation energies of 1487 and 2984 eV, respectively. These depths should be considered as upper limits, since electron kinetic energies are used in calculations, while it should also be noted that smaller probing depths would be expected for denser inorganic materials, including those making up the bulk electrode.
A graphite electrode sample cycled in LP57 electrolyte was analysed by XPS using the I09 beamline at the Diamond Light Source (UK) synchrotron facility. The sample was prepared in a similar way as that previously described, mounted using carbon tape on a sample plate, and transferred to the beamline end-station under vacuum via an Ar-filled glovebox without being exposed to air. The sample was measured using a photon energy of 2.35 keV, monochromatized by a Si (111) double-crystal monochromator. A hemispherical VG Scienta EW4000 analyser set to a pass energy of 200 eV. No charge neutralizer was used during the measurements.
Results and discussion
The electrolyte systems studied, containing various ratios of bis(2,2,2-trifluoroethyl) carbonate (TFEC) and ethylene carbonate (EC):ethyl methyl carbonate (EMC) (3:7 v/v) with 1 M LiPF6, are summarised in Table 1.
| EC:EMC (3:7) v/v% |
TFEC v/v% | [LiPF6]/M | Notation |
|---|---|---|---|
| 100 | 0 | 1 | LP57 |
| 70 | 30 | 1 | 30% TFEC |
| 50 | 50 | 1 | 50% TFEC |
| 30 | 70 | 1 | 70% TFEC |
| 10 | 90 | Saturated (<1) | 90% TFEC |
90 vol% represents an upper limit of TFEC content: cells containing electrolytes with 95% and 100% TFEC solvent, saturated with LiPF6 (<1 M), display no charge capacities (Fig. S1†), suggesting very high cell resistances. This is probably related to the very low Li+-solvating ability of TFEC (discussed in detail later on). Electrolytes containing at least 30% TFEC were found to be flame retarding, with at least 50% TFEC considered non-flammable (Fig. S2†). Galvanostatic cycling data for NMC622|graphite cells over 200 cycles for the electrolytes up to 90% TFEC is presented in Fig. 1(a). With 90% TFEC, the capacity fades rapidly within the initial 10 cycles. 30, 50, and 70% TFEC display more stable cycling, although cycling stability is better with lower amounts of TFEC, delivering capacities of 157, 142 and 136 mA h g−1, respectively, after 200 cycles. All three formulations offer higher capacities than for the LP57 benchmark and higher capacity retention: 90, 87, and 85%, respectively, from 5th to 200th cycle, compared with 76% for LP57.
 | ||
| Fig. 1 Electrochemical performance of NMC622|graphite cells with electrolytes containing up to 90% TFEC. (a) Galvanostatic cycling (discharge) over 200 cycles. (b) Coulombic efficiency over 20 cycles. The shaded areas represent the standard deviation of duplicate cells. The erratic coulombic efficiencies during the first five cycles are an artefact of increases in the C-rate. | ||
All the electrolytes offer similar first-cycle coulombic efficiencies (∼80–85%), reaching close to 100% by the 6th cycle (Fig. 1(b)). However, while most cells continue cycling with close to 100% coulombic efficiency for 200 cycles (Fig. S3†), the cells with 90% TFEC exhibit an initial drop to ∼85–90% efficiency from the 5th cycle, when the C-rate is increased to C/2. This is concurrent to the rapid capacity decline of these cells. The rate capability of cells containing these electrolytes is discussed in more detail later on. The higher capacities and irregular coulombic efficiencies in the first four cycles result from changes in the C-rate during the formation cycling protocol imposed for the cells (see the Experimental section). Overall, electrolytes containing TFEC, with the exception of 90%, show similar efficiencies compared to the LP57 benchmark.
Rate capability tests of these electrolytes show an overall similar performance to LP57 up to 2C, apart from 90% TFEC, as can be seen in Fig. 2(a). All electrolytes up to 70% TFEC display excellent rate capability for C-rates up to 1C, recovering the capacity well at C/10 after 30 cycles. However, 2C appears to be a significant limitation for TFEC-containing electrolytes, while LP57 continues to perform well at 2C but much more poorly at 5C. With 90% TFEC, the performance falls dramatically with each increasing C-rate. Although the capacity falls in a similar fashion at a constant current (Fig. 1(a)), the capacities achieved at a given C-rate for this electrolyte are lower, suggesting high resistivities. This is probably in part due to its lower ionic conductivity (Table S1†).30 Based on TFEC's poor Li-solvating ability, it is likely that the EC and EMC co-solvents play an important role, even in low concentrations, in the conduction of Li+ ions and in the performance of these electrolytes. In the base electrolyte LP57, EC is responsible for the bulk of Li-ion solvation and conduction.31 At 90% TFEC, the estimated combined molar ratio of EC and EMC to Li+ is less than 1.1 (see Table S2†), well below the combined Li+ coordination number of 4–6 for these solvents.32,33 The low availability of effective Li+-conducting species is likely responsible for the poor rate capability and cycling stability of the 90% TFEC. Further to the apparent ionic conductivity limitation for TFEC-containing cells suggested in the rate capability tests, intermittent current interruption (ICI) measurements (Fig. 2(b)) show the evolution of the cell resistance during galvanostatic cycling. Many parallels can be drawn between ICI and electrochemical impedance spectroscopy (EIS); however, ICI offers the benefits of a more facile, less time-consuming experiment with simpler interpretation. Changes in cell resistance can result from formation and evolution of the electrode–electrolyte interfaces, which are known to contribute heavily to cell ageing.34 Cells with electrolytes of higher TFEC content generally exhibit higher resistance. In particular, for 70% and 90% TFEC, the 5th cycle (when formation cycling is complete) resistance is ∼40 and ∼80 Ω, respectively, versus ∼25 Ω for the 30% and 50% TFEC. The resistance of the cells with 30 and 50% TFEC increases only slightly over long-term cycling, and for 70% drops from its initial value to stabilise at a similar value of ∼20–25 Ω. In contrast, the resistance of the cells with 90% TFEC increases markedly to ∼120 Ω within the first 50 cycles. These changes in the cell resistance are probably linked to the evolution of the electrode–electrolytes interfaces, such as changes in SEI thickness or composition. It is clear, again, that the EC:EMC co-solvent mixture plays a key role in defining the performance of TFEC-containing electrolytes, influencing electrolyte ionic conductivity and likely also electrode–electrolyte interface properties, which in turn manifest in the rate capability and internal cell resistance. Cyclic voltammetry (CV) supports this: in all electrolytes, even those with high TFEC content, the major reduction peak at ca. 0.5–0.6 V vs. Li+/Li likely corresponds to the reduction of EC (as it is the only peak visible in the TFEC-free electrolyte LP57),35 while TFEC reduction (likely the peak at 0.7–0.8 V) plays a comparatively minor role (Fig. S4†).
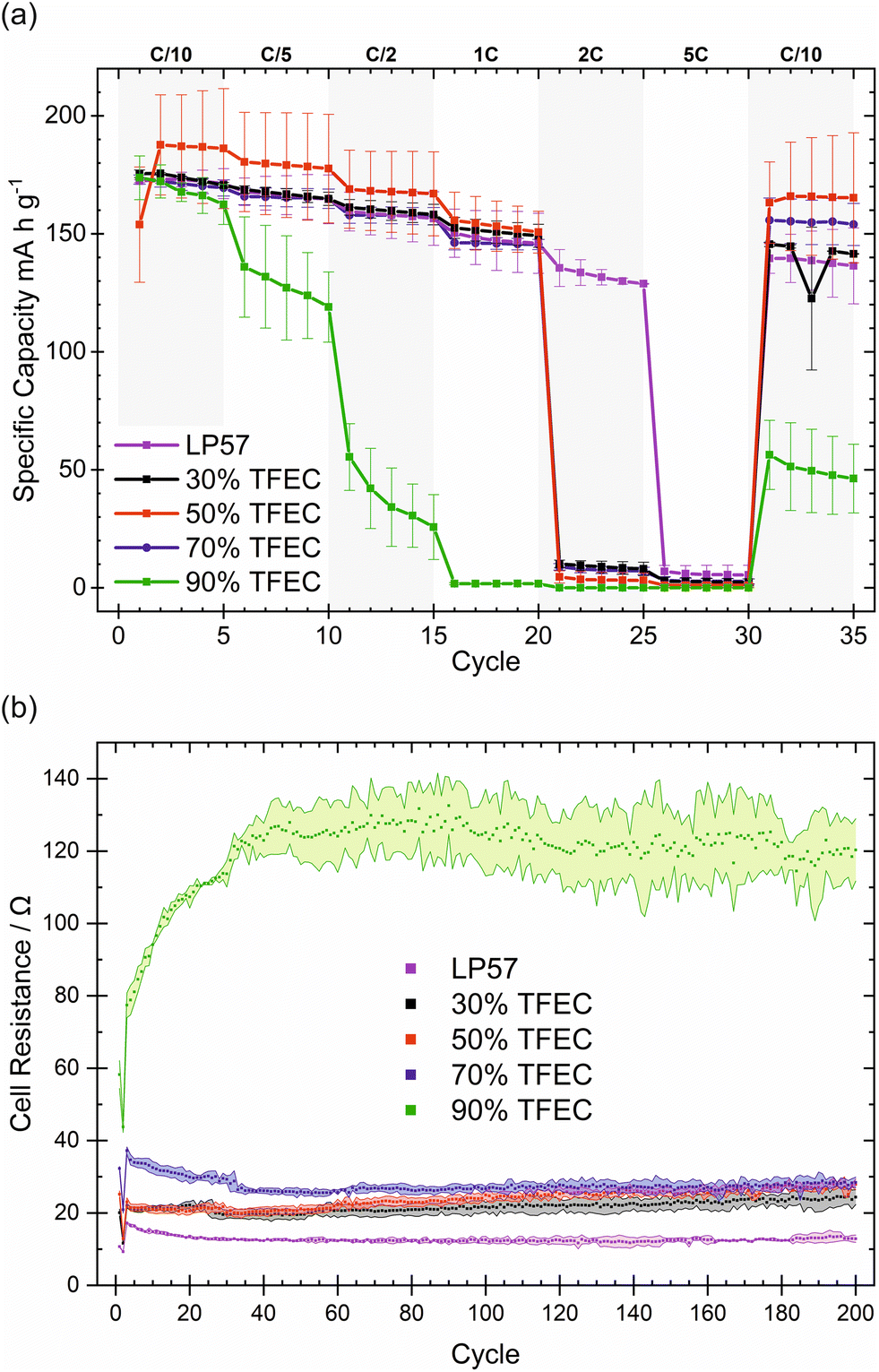 | ||
| Fig. 2 (a) Rate capability (discharge capacity) of TFEC containing electrolytes compared to LP57. (b) Internal resistance of the cell derived from the ICI measurement recorded at C/2. The resistance values are the averaged charge step for each cycle. The shaded area represents the standard deviation for two cells. | ||
In addition to studies with TFEC, the phosphate-analogue tris(2,2,2-trifluoroethyl) phosphate was tested in the same way, and is demonstrated to have a poorer performance than TFEC (Fig. S5†). The general trend of fluorinated hydrocarbons exhibiting greater performance over phosphate and phosphonate-based solvents has been reported previously.16 This further underscores the key role of the solvent mixture in determining cell performance, most often as a result of electrode–electrolyte interfacial properties.
XPS was employed to investigate the surface chemistry of graphite electrodes after cycling in electrolytes with between 30 and 90% TFEC. Fig. 3 presents the C 1s spectra measured at two different excitation energies, 1487 and 2984 eV, corresponding to approximately 14 and 24 nm depth, respectively. Spectra for the graphite electrode cycled with LP57 are presented in Fig. S6.† C 1s spectra are fitted with peaks commonly reported in the literature for aged graphite electrodes,36–50 with components representing bonding environments in the active material, binder and SEI, as tabulated in Table S3.† The main peak (∼286 eV) in each case is attributed to C–O, a typical component of the SEI found to form from carbonate ester electrolytes on graphite.36–40 At ∼285 eV and ∼284 eV peaks are assigned for C–C and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, typically associated with organic SEI species and the graphite electrode, respectively.36,41–47 Other species, CO at ∼287.5 eV and OC–O at ∼288.5 eV, are also frequently observed for the SEI formed from carbonate ester electrolytes.38,44,46 Some differences are observed for the spectra between the different electrolytes. For 30% and 50% TFEC, peaks for carbonates at ∼290–291 eV46,48–50 have a greater relative intensity, compared with the electrolytes containing higher TFEC fractions. This is also observed in O 1s spectra (∼533 eV) in Fig. S7.† While at the lower excitation energy for 30% TFEC no signal for the bulk graphite could be detected, such a signal is present with the higher excitation energy. This indicates an SEI thickness of between 14 and 24 nm. For all other electrolytes with TFEC contents over 30%, the bulk graphite signal is observed at the lower excitation energy, indicating SEI layers thinner than 14 nm. Particularly at TFEC contents of 70% and above, a second bulk peak is also observed at a binding energy (∼283 eV) lower than that for graphite. This is consistent with a lithiated form of graphite, and the observation suggests trapping of lithium in the graphite structure upon cycling with the higher TFEC fraction electrolytes.51 A greater peak intensity is also observed below 56 eV in Li 1s spectra (Fig. S8†) for such electrolytes, indicative of intercalated lithium, rather than SEI components such as LiF or Li2CO3.49,52 For C 1s, the lithiated graphite is observed almost exclusively (without the unlithiated graphite) at the lower excitation energy for both 70 and 90% TFEC, while at the higher excitation energy both phases are observed. This suggests that a layered bulk structure exists beneath the SEI, with the lithiated phase closer to the particle surfaces.
C, typically associated with organic SEI species and the graphite electrode, respectively.36,41–47 Other species, CO at ∼287.5 eV and OC–O at ∼288.5 eV, are also frequently observed for the SEI formed from carbonate ester electrolytes.38,44,46 Some differences are observed for the spectra between the different electrolytes. For 30% and 50% TFEC, peaks for carbonates at ∼290–291 eV46,48–50 have a greater relative intensity, compared with the electrolytes containing higher TFEC fractions. This is also observed in O 1s spectra (∼533 eV) in Fig. S7.† While at the lower excitation energy for 30% TFEC no signal for the bulk graphite could be detected, such a signal is present with the higher excitation energy. This indicates an SEI thickness of between 14 and 24 nm. For all other electrolytes with TFEC contents over 30%, the bulk graphite signal is observed at the lower excitation energy, indicating SEI layers thinner than 14 nm. Particularly at TFEC contents of 70% and above, a second bulk peak is also observed at a binding energy (∼283 eV) lower than that for graphite. This is consistent with a lithiated form of graphite, and the observation suggests trapping of lithium in the graphite structure upon cycling with the higher TFEC fraction electrolytes.51 A greater peak intensity is also observed below 56 eV in Li 1s spectra (Fig. S8†) for such electrolytes, indicative of intercalated lithium, rather than SEI components such as LiF or Li2CO3.49,52 For C 1s, the lithiated graphite is observed almost exclusively (without the unlithiated graphite) at the lower excitation energy for both 70 and 90% TFEC, while at the higher excitation energy both phases are observed. This suggests that a layered bulk structure exists beneath the SEI, with the lithiated phase closer to the particle surfaces.
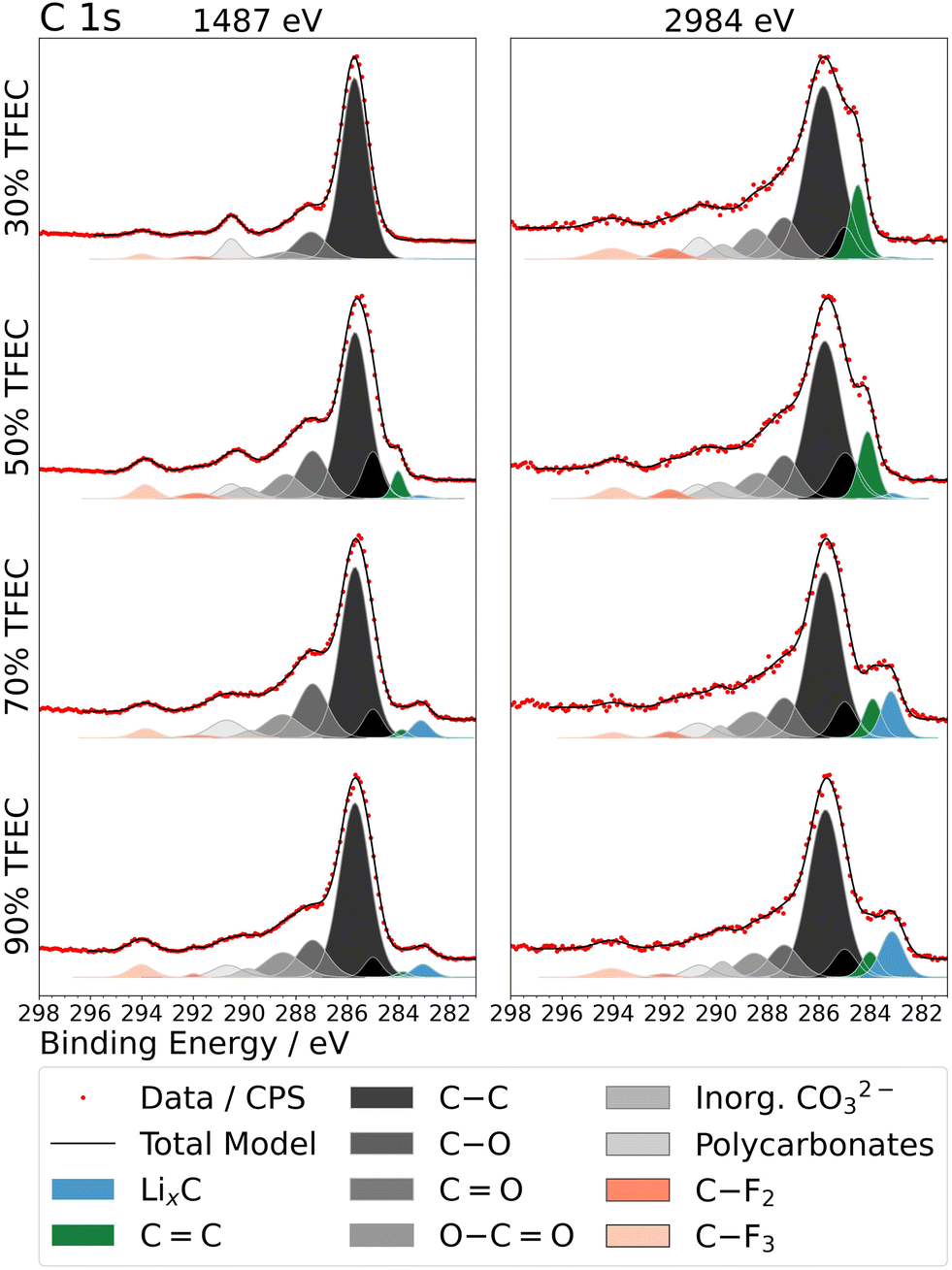 | ||
| Fig. 3 C 1s X-ray photoelectron spectra, measured at two excitation energies, for graphite electrodes cycled to 4 cycles in electrolytes with up to 90% TFEC. | ||
A more pronounced peak for polycarbonate species (∼291 eV) is observed for 30% TFEC, while the –CF3 peak is seen to be slightly more intense for those electrolytes with greater than 30% TFEC. This suggests a greater participation by EC or EMC in SEI formation at 30% TFEC than for the other electrolytes and a larger extent of decomposition or inclusion of TFEC at the higher TFEC-content electrolytes. A similar trend is also observed in the O 1s spectra (Fig. S7†), where the PO peak has greater relative intensity for the electrolytes with higher TFEC fractions. In the same spectra, the appearance of a peak attributed to Li2O also occurs,52 particularly for 90% TFEC, but also somewhat for 70% TFEC. The Li2O peak is observed to have a greater relative intensity at the higher probing depth, corresponding to it being localised close to the electrode particle surfaces.
The organic fluorine (C–F) peak observed in the F 1s spectra (Fig. 4) is quite consistent with that observed for C 1s. In general, the spectra obtained with the higher excitation energy are very similar, independent of the electrolyte. The LiF peak makes up the majority of the signal in each case, tending to be stronger in the deeper measurement, suggesting that LiF resides at a higher concentration nearer to the graphite electrode.53,54 C–F and P–F species are observed closer to the surface of the SEI layer. The samples from the 30% and 50% TFEC electrolytes show significantly higher P–F in the lower-excitation energy measurement. Such a trend is also noticed in P 2p spectra (Fig. S9†). This suggests that the LiPF6 has a greater role in SEI formation for the lower TFEC concentrations.
 | ||
| Fig. 4 F 1s X-ray photoelectron spectra, measured at two excitation energies, for graphite electrodes cycled to 4 cycles in electrolytes with up to 90% TFEC. | ||
Overall, the XPS data shows broad similarities for the SEI/electrode composition and structure between the 30 and 50% TFEC electrolytes as well as between 70 and 90% TFEC. Lithium trapping in graphite is already evident for 70% TFEC, while it appears to occur to a larger extent for 90% TFEC. The SEI at lower TFEC content (30 and 50%) appears to contain less PO species but more C–O/P–O species, and is more LiF-deficient particularly at the SEI surface. Despite the many similarities for 70 and 90% TFEC in the XPS analysis, the electrochemical cycling performance is much poorer in the case of 90% TFEC. The cell resistance, however, indicates the likely tendency of the interfacial properties for 70% TFEC towards those of 90%. One major difference between 70 and 90% TFEC in the XPS analysis is the presence of Li2O in O 1s spectra, located close to the electrode surface, which may be determined as a critical characteristic that negatively affects the electrochemical performance in this case. 30, 50 and 70% TFEC electrolytes are shown to perform well electrochemically even though the SEI and electrode properties are evidently varying with TFEC content and in some cases resembling more closely that from 90% TFEC which displayed poor performance.
Conclusions
In summary, electrolytes with 30–100% TFEC solvent have been demonstrated in cells with graphite and NMC622 electrodes, with electrochemical and interfacial characterisation presented. It can be concluded that comparable or greater specific capacities and capacity retentions than the benchmark LP57 can be achieved over 200 cycles with up to 70% TFEC, while also exhibiting comparable coulombic efficiency. Beyond 70%, rapid capacity fading is observed. Rate capabilities are also comparable, although increased cell resistances for all formulations compared with LP57 results in a generally poorer performance at the highest rates tested. Due to the low Li+-solvating ability of TFEC, the EC/EMC co-solvents are expected to play the dominant role in Li+ conduction, electrode–electrolyte interphase formation and thereby be highly influential on overall performance. Characterisation of the SEI layers on the graphite electrodes indicates a greater carbonate content for lower TFEC contents and LiF-rich SEI layers for all formulations. The SEI is found to become thinner with increasing TFEC content, but with decreasing concentrations of PFx species. The higher cell resistances for electrolytes with 70% TFEC and higher are consistent with the lithium trapping in graphite observed in XPS data and with the lower ionic conductivities of these electrolytes, which are likely related to the scarcity of effective Li+-solvating species in them. Overall, the use of high fractions (at least 50%) of TFEC in typical carbonate-based electrolytes presents a promising route to safer – and therefore more sustainable – LIBs, while maintaining or exceeding state-of-the-art performance. Further work could potentially investigate the use of electrolyte additives to improve the rate capability of such electrolytes.Data availability
Data for this article, including electrochemical and XPS data are available at figshare at https://doi.org/10.6084/m9.figshare.26403511.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Swedish Energy Agency (projects P2021-90019 and P2022-00045) and STandUP for Energy for support to the Ångström Advanced Battery Centre. This work was carried out with the support of Diamond Light Source, instrument I09 (proposal SI30357-1).References
- Y. Chen, Y. Kang, Y. Zhao, L. Wang, J. Liu, Y. Li, Z. Liang, X. He, X. Li, N. Tavajohi and B. Li, J. Energy Chem., 2021, 59, 83–99 CrossRef CAS.
- Q. Wang, L. Jiang, Y. Yu and J. Sun, Nano Energy, 2019, 55, 93–114 CrossRef CAS.
- R. Gond, W. van Ekeren, R. Mogensen, A. J. Naylor and R. Younesi, Mater. Horiz., 2021, 8, 2913–2928 RSC.
- L. Suo, Y.-S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1481 CrossRef PubMed.
- M. Grünebaum, M. M. Hiller, S. Jankowsky, S. Jeschke, B. Pohl, T. Schürmann, P. Vettikuzha, A. C. Gentschev, R. Stolina, R. Müller and H. D. Wiemhöfer, Prog. Solid State Chem., 2014, 42, 85–105 Search PubMed.
- N. Boaretto, L. Meabe, S. Lindberg, H. Perez-Furundarena, I. Aldalur, E. Lobato, F. Bonilla, I. Combarro, A. Gutiérrez-Pardo, A. Kvasha, M. Lechartier, R. Vincent, J. Cognard, S. Genies, L. Daniel and M. Martinez-Ibañez, Adv. Funct. Mater., 2024, 2404564 CrossRef CAS.
- S. Kundu, A. Kraytsberg and Y. Ein-Eli, J. Solid State Electrochem., 2022, 26, 1809–1838 CrossRef CAS.
- J. Xie and Y. C. Lu, Adv. Mater., 2024, 2312451 Search PubMed.
- X. Fan, L. Chen, O. Borodin, X. Ji, J. Chen, S. Hou, T. Deng, J. Zheng, C. Yang, S. C. Liou, K. Amine, K. Xu and C. Wang, Nat. Nanotechnol., 2018, 13, 715–722 Search PubMed.
- N. von Aspern, G.-V. Röschenthaler, M. Winter and I. Cekic-Laskovic, Angew. Chem., Int. Ed., 2019, 58, 15978–16000 Search PubMed.
- Q.-K. Zhang, X.-Q. Zhang, H. Yuan and J.-Q. Huang, Small Sci., 2021, 1, 2100058 Search PubMed.
- M. C. Smart, B. V. Ratnakumar, V. S. Ryan-Mowrey, S. Surampudi, G. K. S. Prakash, J. Hu and I. Cheung, J. Power Sources, 2003, 119–121, 359–367 CrossRef CAS.
- H. Q. Pham, E.-H. Hwang, Y.-G. Kwon and S.-W. Song, Chem. Commun., 2019, 55, 1256–1258 RSC.
- T. Doi, R. Matsumoto, T. Endo, Z. Cao, T. Sato, M. Haruta, M. Hashinokuchi, Y. Kimura and M. Inaba, ACS Appl. Energy Mater., 2019, 2, 7728–7732 CrossRef CAS.
- H. Q. Pham, H. Y. Lee, E. H. Hwang, Y. G. Kwon and S. W. Song, J. Power Sources, 2018, 404, 13–19 CrossRef CAS.
- F. Gebert, M. Longhini, F. Conti and A. J. Naylor, J. Power Sources, 2023, 556, 232412 CrossRef CAS.
- L. Zhou, S. Qian, C. Yang, T. Han, Y. Song, Y. Jiang, H. Zhang, J. Zhang, L. Li, J. Liu and T. Qian, ACS Appl. Energy Mater., 2022, 5, 14201–14210 CrossRef CAS.
- F. Demelash, A. Gomez-Martin, B. Heidrich, E. Adhitama, P. Harte, A. Javed, A. Arifiadi, M. M. Bela, P. Yan, P. Harte, D. Diddens, M. Winter and P. Niehoff, Small Struct., 2024, 2400063 CrossRef CAS.
- R. Okada, Y. Aoki, M. Oda, M. Nakazawa, M. Inaba and T. Doi, ACS Appl. Energy Mater., 2023, 6, 546–553 CrossRef CAS.
- X. Cao, P. Gao, X. Ren, L. Zou, M. H. Engelhard, B. E. Matthews, J. Hu, C. Niu, D. Liu, B. W. Arey, C. Wang, J. Xiao, J. Liu, W. Xu and J.-G. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2020357118 CrossRef CAS PubMed.
- H. Nakagawa, Y. Shibata, Y. Fujino, T. Tabuchi, T. Inamasu and T. Murata, Electrochemistry, 2010, 78, 406–408 CrossRef CAS.
- M. He, C.-C. Su, Z. Feng, L. Zeng, T. Wu, M. J. Bedzyk, P. Fenter, Y. Wang, Z. Zhang, M. He, C. Su, P. Fenter, Z. Zhang, Y. Wang, Z. Feng, L. Zeng, M. J. Bedzyk and T. Wu, Adv. Energy Mater., 2017, 7, 1700109 CrossRef.
- I. Källquist, R. Le Ruyet, H. Liu, R. Mogensen, M. T. Lee, K. Edström and A. J. Naylor, J. Mater. Chem. A, 2022, 10, 19466–19505 RSC.
- P. Verma, P. Maire and P. Novák, Electrochim. Acta, 2010, 55, 6332–6341 Search PubMed.
- Z. Huey, Y. Ha, S. Frisco, A. Norman, G. Teeter, C. S. Jiang and S. C. DeCaluwe, J. Power Sources, 2023, 564, 232804 Search PubMed.
- M. J. Lacey, ChemElectroChem, 2017, 4, 1997–2004 CrossRef CAS.
- Z. Geng, T. Thiringer and M. J. Lacey, IEEE Trans. Transp. Electrif., 2022, 8, 2985–2995 Search PubMed.
- C. J. Powell and A. Jablonski, NIST Electron Inelastic-Mean-Free-Path Database, Version 1.2, SRD 71, National Institute of Standards and Technology, Gaithersburg, MD, 2010 Search PubMed.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 1994, 21, 165–176 CrossRef CAS.
- H. Buqa, D. Goers, M. Holzapfel, M. E. Spahr and P. Novák, J. Electrochem. Soc., 2005, 152, A474 CrossRef CAS.
- K. Xu, J. Electrochem. Soc., 2007, 154, A162 CrossRef CAS.
- Q. Li, Z. Cao, W. Wahyudi, G. Liu, G. T. Park, L. Cavallo, T. D. Anthopoulos, L. Wang, Y. K. Sun, H. N. Alshareef and J. Ming, ACS Energy Lett., 2021, 6, 69–78 CrossRef CAS.
- T. Hou, K. D. Fong, J. Wang and K. A. Persson, Chem. Sci., 2021, 12, 14740–14751 RSC.
- C. R. Birkl, M. R. Roberts, E. McTurk, P. G. Bruce and D. A. Howey, J. Power Sources, 2017, 341, 373–386 CrossRef CAS.
- R. Lundström, N. Gogoi, T. Melin and E. J. Berg, J. Phys. Chem. C, 2024, 128, 8147–8153 CrossRef.
- B. Philippe, R. Dedryvère, J. Allouche, F. Lindgren, M. Gorgoi, H. Rensmo, D. Gonbeau and K. Edström, Chem. Mater., 2012, 24, 1107–1115 CrossRef CAS.
- F. Jeschull, F. Lindgren, M. J. Lacey, F. Björefors, K. Edström and D. Brandell, J. Power Sources, 2016, 325, 513–524 CrossRef CAS.
- R. Azmi, F. Lindgren, K. Stokes-Rodriguez, M. Buga, C. Ungureanu, T. Gouveia, I. Christensen, S. Pal, A. Vlad, A. Ladam, K. Edström and M. Hahlin, ACS Appl. Mater. Interfaces, 2024, 16, 34266–34280 CrossRef CAS PubMed.
- V. Eshkenazi, E. Peled, L. Burstein and D. Golodnitsky, Solid State Ionics, 2004, 170, 83–91 CrossRef CAS.
- M. Nie, J. Demeaux, B. T. Young, D. R. Heskett, Y. Chen, A. Bose, J. C. Woicik and B. L. Lucht, J. Electrochem. Soc., 2015, 162, A7008–A7014 CrossRef CAS.
- M. Phaner-Goutorbe, A. Sartre and L. Porte, Microsc., Microanal., Microstruct., 1994, 5, 283–290 Search PubMed.
- P. Guan, L. Liu and X. Lin, J. Electrochem. Soc., 2015, 162, A1798–A1808 CrossRef CAS.
- H. Hori, M. Shikano, H. Kobayashi, S. Koike, H. Sakaebe, Y. Saito, K. Tatsumi, H. Yoshikawa and E. Ikenaga, J. Power Sources, 2013, 242, 844–847 Search PubMed.
- A. Ponrouch, R. Dedryvère, D. Monti, A. E. Demet, J. M. Ateba Mba, L. Croguennec, C. Masquelier, P. Johansson and M. R. Palacín, Energy Environ. Sci., 2013, 6, 2361–2369 RSC.
- Z. Guo, Z. Xu, F. Xie, J. Jiang, K. Zheng, S. Alabidun, M. Crespo-Ribadeneyra, Y. S. Hu, H. Au and M. M. Titirici, Adv. Mater., 2023, 35, 2304091 CrossRef CAS PubMed.
- R. I. R. Blyth, H. Buqa, F. P. Netzer, M. G. Ramsey, J. O. Besenhard, P. Golob and M. Winter, Appl. Surf. Sci., 2000, 167, 99–106 CrossRef CAS.
- B. Philippe, R. Dedryvère, M. Gorgoi, H. Rensmo, D. Gonbeau and K. Edström, J. Am. Chem. Soc., 2013, 135, 9829–9842 CrossRef CAS PubMed.
- V. Shutthanandan, M. Nandasiri, J. Zheng, M. H. Engelhard, W. Xu, S. Thevuthasan and V. Murugesan, J. Electron Spectrosc. Relat. Phenom., 2019, 231, 2–10 CrossRef CAS.
- K. N. Wood and G. Teeter, ACS Appl. Energy Mater., 2018, 1, 4493–4504 CrossRef CAS.
- N. Schulz, R. Hausbrand, L. Dimesso and W. Jaegermann, J. Electrochem. Soc., 2018, 165, A819–A832 Search PubMed.
- B. Philippe, M. Hahlin, K. Edström, T. Gustafsson, H. Siegbahn and H. Rensmo, J. Electrochem. Soc., 2016, 163, A178–A191 CrossRef CAS.
- S. Oswald, F. Thoss, M. Zier, M. Hoffmann, T. Jaumann, M. Herklotz, K. Nikolowski, F. Scheiba, M. Kohl, L. Giebeler, D. Mikhailova and H. Ehrenberg, Batteries, 2018, 4, 36 CrossRef.
- K. Yang, Z. Shen, J. Huang, J. Zhong, Y. Lin, J. Zhu, J. Chen, Y. Wang, T. Xie, J. Li and Z. Shi, Mater. Chem. Front., 2023, 7, 4152–4163 RSC.
- V. Sharova, A. Moretti, T. Diemant, A. Varzi, R. J. Behm and S. Passerini, J. Power Sources, 2018, 375, 43–52 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lf00405a |
| This journal is © The Royal Society of Chemistry 2025 |