
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vanadium pentoxide mesoporous cathodes for Li-ion batteries†
Andrea Palumboa,
Ullrich Steiner a,
Andrea Dodero*ab and
Ilja Gunkela
a,
Andrea Dodero*ab and
Ilja Gunkela
aAdolphe Merkle Institute, Chemin de Verdiers 4, Fribourg, Switzerland. E-mail: andrea.dodero@unifr.ch
bDepartment of Chemistry and Industrial Chemistry, University of Genoa, Via Dodecaneso 31, Genoa, Italy
First published on 20th May 2025
Abstract
The combination of micro- and nanoporosity is advantageous for Li-ion intercalation in battery electrodes. In this work, we synthesize porous 10 μm-sized poly(styrene-vinylpyridine) block copolymer particles via an emulsion-based approach. The vinylpyridine-phase was then subjected to methanol swelling to enable vanadium ions infiltration, followed by calcination to obtain mesoporous vanadium pentoxide particles. These exhibited a hierarchical porosity, and electrodes manufactured from them displayed a very high specific surface area. Two liquid electrolytes were compared to manage solid-electrolyte-interface growth, which can clog nanopores. Notably, the combination of a lithium bis(trifluoromethane)sulfonimide-containing tetraethylene glycol dimethyl ether tetraglyme electrolyte with the hierarchically porous vanadium pentoxide electrodes demonstrated a substantial enhancement in cycling performance, surpassing established industry benchmarks.
1 Introduction
Given the increasing interest in battery technology, there is an ongoing pursuit of breakthrough materials and fabrication processes to address the rapidly expanding demand for high-performance energy storage solutions. Mesoporous materials have garnered significant attention due to their advantageous properties, including an enhanced effective surface area that facilitates ion intercalation.1,2 This, in turn, exerts a direct influence on battery performance, particularly with regard to power and energy densities.3–6 However, the use of entirely nanostructured materials is often impeded by their inadequate ion transport, which can be attributed to the constrained mass diffusion resulting from the substantial thickness of conventional electrodes (i.e., several 100 μm).7,8 To address this challenge, the incorporation of micrometer-sized interconnected pores into mesoporous materials has emerged as a promising solution.9,10 This approach aims to enhance Li-ion diffusion across the hierarchically structured material while preserving the advantages of interconnected pores with a high sur face area.11–13In energy storage devices, mesoporous electrodes offer distinct advantages over conventional materials, including bulk and nanopowder-based formulations.12,13 The integration of high surface area with optimized ion transport pathways has been demonstrated to result in improvements in energy density and charge/discharge kinetics.9,10,14 Furthermore, these structures demonstrate enhanced mechanical stability during the charge and discharge processes, resulting in a longer cycle life and superior electrochemical performance.15–17 Moreover, their organized structure prevents agglomeration, a common issue in purely nanosized materials.14,15,18
A further critical challenge for nanostructured electrodes is the formation of a solid-electrolyte interphase (SEI) layer through electrolyte degradation if the electrochemical potential exceeds the electrolyte's stability. This can impede the flow of ions, leading to the accumulation of matter within the nanopores and consequently the degradation of battery performance over time.3,6,15
This study proposes a new manufacturing approach that extends for the first time the advantages of mesostructured battery electrodes to vanadium pentoxide (V2O5). Vanadium pentoxide is a promising cathode material for Li-ion batteries, as it offers a high theoretical capacity, a wide voltage range, and good conductivity.19–22 However, V2O5 exhibits considerable sensitivity to volume changes during cycling, resulting in capacity degradation over short time frames and limited cyclability.23–26 To address these challenges, we have developed a fabrication procedure to synthesize mesoporous V2O5 microspheres. These microspheres are capable of effectively mitigating issues related to volume change while utilizing the inherent advantages of vanadium pentoxide. In contrast to earlier studies, which commonly employed the co-assembly of block copolymers with precursor sols followed by calcination into metal oxides, the present approach starts with the oil-in-water emulsification of a block copolymer in the presence of a non-solvent.27,28 The resultant mesostructured block copolymer spheres, obtained after solvent evaporation, are then swollen and selectively infiltrated with V4+ ions. The final step involves calcination to obtain mesoporous V2O5 microspheres. The formation of battery electrodes is then accomplished by implementing the slurry technique, which is a prevalent method within the field.29
The structural and electrochemical properties of the resulting mesoporous V2O5 electrodes were investigated to determine their suitability for energy storage applications. Given that the electrochemical operation of these electrodes is sensitively dependent on the electrolyte formulation, which controls the SEI formation, two electrolytes were compared to optimize device performances.30–33 Notably, by combining mesoporosity with a suitable electrolyte, we achieve a synergistic effect that maximizes energy storage performance while simultaneously minimizing SEI-forming chemical reactions.2,34,35
2 Experimental
2.1 Materials
PS-b-P4VP with molar mass of Mw = 195-b-204 kg mol−1 was purchased from Polymer Source, Inc. PVA with a molar mass of Mw = 13–27 kg mol−1 and a degree of hydrolysis of 87–89% was purchased from Sigma-Aldrich. MeOH 99.9% analytical reagent grade, and CHCl3 99.8% analytical reagent grade were purchased from Fisher Scientific. Dioxane anhydrous, 99.8%, was purchased from Sigma-Aldrich. VOSO4 97% and hexadecane reagent grade 99% were purchased from Sigma-Aldrich. The standard electrolyte of 1 M LiPF6 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) EC:DMC was purchased from Sigma-Aldrich. Conductive carbon black (Super C65) was kindly provided by Imerys Graphite & Carbon, Switzerland Ltd. PVDF (Kynar HSV900) was provided by ARKEMA Innovative Chemistry. NMP anhydrous 99.5% was purchased from Sigma-Aldrich. Lithium chips were purchased from Gelon LIB Group, and GF/B glass microfiber was purchased from Healthcare Life Sciences. LiTFSI 99.95% and tetraglyme 99% were purchased from Sigma-Aldrich.
:1 (v/v) EC:DMC was purchased from Sigma-Aldrich. Conductive carbon black (Super C65) was kindly provided by Imerys Graphite & Carbon, Switzerland Ltd. PVDF (Kynar HSV900) was provided by ARKEMA Innovative Chemistry. NMP anhydrous 99.5% was purchased from Sigma-Aldrich. Lithium chips were purchased from Gelon LIB Group, and GF/B glass microfiber was purchased from Healthcare Life Sciences. LiTFSI 99.95% and tetraglyme 99% were purchased from Sigma-Aldrich.
2.2 Synthesis
PS-b-P4VP was solubilized in chloroform (CHCl3) at a concentration of 20 mg mL−1 under stirring, and PVA was solubilized in deionized water at a 5 mg mL−1 concentration at T = 80 °C for several hours under stirring. Additionally, a solution of hexadecane in CHCl3 was prepared at a concentration of 20 mg mL−1. The BCP and hexadecane solutions were mixed in a glass vial at two ratios (i.e., 70:30 and 100:0) and left under stirring for 30 minutes. The PVA solution was then added to the BCP mixture in a volume ratio of 5 to 1. An oil-in-water emulsion (i.e., small droplets of oil dispersed in water) was then formed by agitating the two phases using a vortex mixer at a speed of 2000 rpm for 30 seconds.
The resulting emulsion was transferred to a covered 5 cm wide Petri dish, and 10 mL of deionized water was added to slow the rate of evaporation of chloroform. The emulsion was left undisturbed for approximately 5 days to allow for the gradual drying of the emulsion droplets, resulting in the formation of solid BCP microparticles ranging in size from 5 to 10 μm. These were then collected, transferred to a 50 mL flask, and centrifuged at 8000 rpm for 30 minutes to separate the self-assembled polymers from the remaining supernatant.
A 100 mg mL−1 solution of VOSO4 in MeOH was then added to the dried microparticles and the resulting suspension was stirred overnight. The addition of MeOH to the microparticles causes the P4VP domains to swell, allowing the vanadium ions to uniformly infiltrate the polymeric matrix.36,37
The mixture was finally centrifuged to remove any remaining MeOH and excess precursor. The resulting solid was transferred to a crucible and calcined at 425 °C for two hours in air with a gradual temperature increase at a rate of 5 °C per minute.
2.3 Slurry formulation and cells preparation
Slurries for electrode preparation were formulated by dispersing active materials, conductive carbon black (Super C65) as the conductive additive, poly(vinylidene fluoride) (PVDF, Kynar HSV900) as the binder, and 1-methyl-2-pyrrolidone (NMP) anhydrous as the solvent. The slurries were prepared with a ratio of active material:carbon black:PVDF of 7:2:1.
The slurry components were mixed thoroughly to ensure homogeneity and coated onto the current collectors using a doctor blade method. For this step a sheet film vacuum coater from MTI Corp. (MSK-AFA-II-VC) and 250 mm doctor blade where used, the coater slid the doctor blade, set at an high of 100 μm over a foil of aluminum paper. The coatings were subsequently dried at 60 °C overnight to ensure complete solvent evaporation.
The coin cells manufacturing process began with preparing the discs to insert into the coin cell from the dried slurry. With a specific tool, the hand-held disc cutter (MSK-T-12) from MTI Corp. discs of 15 mm in diameter where cut out. Subsequently, separator discs of 18 mm in diameter of Whatman glass microfiber grade GF/A filter discs with 1 μm pore size where cut the same way. After, both the slutty and the separator disks, and the coin cell assembly materials were put in vacuum oven overnight at 120 °C. The cell assembly materials, all purchased from MTI Corp. composed of CR2032 Coin Cell Cases, 20 mm current collector stainless steel disks and 20 mm stainless spring disks. The items where then transferred into a glove box under Argon atmosphere with oxygen and moisture levels below 1 ppm for the cell assembly. The cell assembly took place inside the glove box, where the bottom lid of the coin cell was layered with a spring, current collector, an 18 mm lithium disk (2 mm thick), a separator soaked with 150 μl of electrolyte solution, an active material disk, another current collector, and a second spring. Once all the components where stacked the top lid of the coin cell was added and sealed under a mechanical manual press, from MTI corp. at a pressure of 1000 kg.
2.4 Instrumentation
A Tescan Mira 3 LMH scanning electron microscope was used to examine the nanoparticles. Measurements were performed with a sample-detector distance between 5 and 10 mm and accelerating voltages between 10 and 20 kV. A 2–4 nm conductive gold coating was sputtered on the samples to prevent surface charging. A FEI Tecnai Spirit TEM equipped with a 120 kV LaB6 emitter was used for transmission electron microscopy. Images were acquired by a Veleta charge-coupled device (CCD) sensor at room temperature. The samples for TEM analysis were prepared inside the glove box, where the cells were first disassembled, and the electrodes were collected in a glass vial. The cathode materials were washed twice with a solution of DMC to remove the electrolyte, and the vial was again filled with DMC. The vial was removed from the glove box and sonicated for 10 min at room temperature to suspend the metal oxides in DMC. Some of this suspension was deposited on a TEM grid, which was left in fume hood for 24 h, for the DMC to evaporate.Physisorption analysis was performed using a high-precision BET analyzer (model 110 from 3P Instruments). The sample was degassed in a liquid nitrogen bath and subjected to gas adsorption and desorption cycles at different pressures ranging from 0.05 to 0.995 atm. The BET model was used to calculate the specific surface area, while the BJH method was used to estimate the pore size distribution.
An Arbin BT 2043 multi-channel system was used for galvanostatic cycling experiments. The selected current rates, called C-rates, were varied to study the effect of different current intensities on the cycling process.
A BioLogic VMP 300 system was used for the cyclic voltammetry experiments. CV measurements involved sweeping the voltage within a material-specific range where reversible electrochemical reactions occur, typically between 1.0 and 5.0 V (versus Li+/Li). To gain insight into the electrochemical behavior of the material under different conditions, the analysis was performed at different scan rates, namely 0.1, 0.2, 0.5, 1.0, and 2.0 mV s−1. By systematically varying the scan rates, it was possible to study the reaction kinetics associated with the electrochemical processes.
A BioLogic VMP 300 system was used for the electronic impedance spectroscopy measurements. EIS data were collected over a wide frequency range, from 1 MHz to 100 MHz, applying a voltage amplitude of 20 mV.
3 Results
The schematic in Fig. 1 summarizes the preparation of mesoporous vanadium pentoxide microspheres. First, an oil(CHCl3)-in-water emulsion is prepared using polyvinyl alcohol (PVA) as a hydrophilic emulsifier. The CHCl3 minority phase contains a polystyrene-block-poly(4-vinylpyridine) (PS-b-P4VP) diblock copolymer (BCP) with a molar mass of Mw = 195-b-204 kg mol−1, and hexadecane. The weight ratio of BCP to hexadecane was varied between 100:0 and 70:30. The resulting emulsion was left at room temperature to allow the evaporation of CHCl3 by diffusion through the water phase. The slow increase in BCP concentration within the emulsion droplets induces their self-assembly into internally nanostructured microparticles. Since hexadecane does not mix with water or the BCP, it remains within the emulsion droplets and phase separates from the macromolecular chains, creating micrometer-sized inclusions within the final particles. Drying the resulting suspensions in a vacuum oven yields a powder of ca. 10 μm BCP spheres containing micrometer-sized pores resulting from the evaporation of the hexadecane.
 | ||
| Fig. 1 Schematic representation of the formation of the mesoporous vanadium pentoxide microspheres. The starting point is an emulsion of a BCP, CHCl3, and hexadecane mixture in water. As CHCl3 diffuses through the water and evaporates, the organic phase shrinks, forming a self-assembled BCP phase with hexadecane inclusions. The spheres were collected and dried, causing the hexadecane to evaporate, forming micrometer-sized holes. The mesoporous spheres were exposed to a vanadium precursor dissolved in methanol, which swells the BCP and allows the precursor to infiltrate the BCP. Centrifugation, drying, and calcination at 425 °C result in mesoporous V2O5 spheres. | ||
The resulting micropowder was then suspended in a methanol-containing (VOSO4·2H2O) vanadium precursor. Methanol is a solvent for P4VP but not for PS, so the P4VP phase swells selectively while the PS-b-P4VP spheres remain intact. The P4VP swelling allows vanadium ions to diffuse into the BCP microspheres and complex with the vinylpyridine groups. The infiltration is particularly aided by the microporosity of the spheres created by the evaporation of hexadecane. P4VP was chosen as one of the BCP blocks because of its strong ability to form complexes with metal ions, such as V4+.38,39
Finally, the infiltrated particles were heated to 425 °C in air, causing complete evaporation of methanol, oxidation of V4+ to V2O5, and combustion of the polymer phase. The microphase-separated BCP matrix confines the growth of V2O5 crystals during the thermal process and preserves the spherical 10 μm shape of the spheres. The resulting V2O5 is porous on three different length scales: (1) at the 10 nm level defined by the BCP microphase morphology, (2) at the micrometer level through the pores left by the hexadecane evaporation, and (3) through the 10 μm porosity resulting from the packing of the emulsion-derived spheres.40 Details of the sample preparation are given in the Experimental section.
SEM analysis was performed to evaluate the formation of microporous spheres and the effect of hexadecane addition. The SEM micrographs in Fig. 2 show the morphological variations in the synthesized V2O5 microspheres. Fig. 2a and c shows V2O5 microspheres in the absence of hexadecane, i.e. a 100:0 BCP to hexadecane ratio. In Fig. 2b and d, hexadecane was added during the emulsification process in a BCP to hexadecane 70:30 ratio. In general, Fig. 2 shows that the presence of micrometer-sized pores in the V2O5 microsphere is clearly due to the added hexadecane. Porosimetry was used to determine the characteristic surface area and average pore size of the samples. Table 1 lists the characteristic parameters for samples synthesized with and without hexadecane addition in comparison to a commercial V2O5 material. The two samples prepared by emulsion-induced self-assembly have specific surface areas and total pore volumes an order of magnitude higher than the commercial material. Since these values are dominated by the 10–20 nm microporosity resulting from V4+ infiltration into the pyridine domains of the block copolymer particles, these high values demonstrate the effectiveness of the infiltration process.
 | ||
| Fig. 2 SEM images of mesoporous V2O5 microspheres. V2O5 microspheres synthesized from emulsions containing different ratios of BCP to hexadecane. a and c) 100:0; b and d) 70:30. While porosity on the 10 nm scale is present in all samples, the 70:30 spheres exhibit micrometer-sized pores resulting from the addition of hexadecane. | ||
:30) and without (100:0) hexadecane added during their manufacture, compared to a commercial V2O5 material
| Samples | Specific surface area (BET) (m2 g−1) | Average pore size (BJH) (nm) | Total pore volume (BJH) (cm3 g−1) |
|---|---|---|---|
| 70:30 |
55.13 | 11.25 | 0.155 |
| 100:0 |
42.56 | 12.47 | 0.133 |
| Commercial | 3.14 | 14.9 | 0.015 |
As a high-voltage cathode, V2O5 is known to form an SEI layer when in contact with organic electrolytes.30,41,42 Since SEI layers often grow during cycling, they can clog the nanopores and reduce battery performance. In a recent study, an electrolyte based on lithium bis(trifluoromethane)sulfonimide (LiTFSI) in tetraethylene glycol dimethyl ether tetraglyme (tetraglyme), 1:1 in mole ratio was compared to one containing lithium hexafluorophosphate (LiPF6) dissolved in an ethylene carbonate–dimethyl carbonate (EC:DMC) 1 M mixture.30 Here we study the effect of SEI layers by comparing these two electrolytes using theV2O5 electrode material with the highest specific surface area in Table 1, i.e. 70:30 sample.
The results of this comparison are shown in Fig. 3. In the cycling test shown in Fig. 3a, the V2O5 70–30 sample with LiTFSI in tetraglyme exhibits exceptional stability up to a C-rate of C/2 (first 15 cycles), maintaining a specific discharge capacity of 220 mA h g−1 in all cycles. On the contrary, the sample with LiPF6 in EC:DMC has a higher initial discharge capacity, but it drops to 178 mA h g−1 when cycling at C/2. Both samples exhibit capacity decrease when the C-rate is increased to 20C. The LiTFSI in tetraglyme maintains higher capacities, reaching 77 mA h g−1 at 20C compared to 27 mA h g−1 for LiPF6 in EC:DMC. This can be attributed to the fact that while LiPF6 in EC:DMC allows for lower initial Rct, its rate performance is hampered by rapid SEI growth that blocks active sites and restricts pore access, as seen in the calendar test in Fig. 3b and from the TEM images in Fig. 4c and d. In contrast, LiTFSI in tetraglyme enables more stable cycling and better high rate performance by forming a thinner, more stable SEI that preserves the mesoporous structure of the electrode (Fig. 4e and f). In addition, the tetraglyme is a flexible solvent and strongly coordinates with Li+, improving solvation and desolvation kinetics at the electrode surface, which enhances Li-ion transport at high rates.30,43–45
 | ||
| Fig. 3 Electrochemical performance of 70:30 V2O5 electrodes using two electrolytes. (a) Cycling performance of half-cells containing LiTFSI in tetraglyme (black) and LiPF6 in EC:DMC (red). (b) Calendar test of half-cells over 250 cycles at a rate of 1C. (c and d) Cyclic voltammetry of V2O5 70:30 with LiPF6 in EC:DMC and LiTFSI in tetraglyme, respectively. (e and f) Electrochemical impedance spectroscopy spectra of V2O5 70:30 with LiPF6 in EC:DMC and LiTFSI in tetraglyme, respectively. | ||
 | ||
| Fig. 4 Transmission electron microscopy images of V2O5 surfaces before and after electrochemical cycling. (a and b) Pristine V2O5 70:30 surface with well-defined crystalline structures. (c and d) Similar V2O5 surface after 75 cycles with LiPF6 in EC:DMC electrolyte, showing non-uniform SEI layer formation. (e and f) When LiTFSI in tetraglyme is used as electrolyte, only a few lumpy SEI aggregates separated by a very thin, uniform SEI layer are observed. | ||
After cycling at high C rates, the rate is reduced to C/10 and C. While the LiTFSI in the tetraglyme sample returns to its initial C/10 capacity, the C/10 capacity of LiPF6 in EC:DMC is reduced to ca. 200 mA h g−1. Fig. 3b shows a calendar test over 250 cycles at a C-rate of 1C. After an initial capacity of 200 mA h g−1 for 10 cycles of the LiTFSI in tetraglyme half-cell, it declines to 178 mA h g−1 over 250 cycles, corresponding to an 11% loss in specific capacity. The LiPF6 in EC:DMC sample declines from an initial capacity of 145 mA h g−1 to 77 mA h g−1, a 47% capacity loss over 250 cycles.
Fig. 3c and d show cyclic voltammetry (CV) curves of LiPF6 in EC:DMC and LiTFSI in tetraglyme electrolytes, respectively. The CV curves show the three expected redox peaks of V2O5 corresponding to lithium intercalation and deintercalation processes. The initial peak shift around 3.0 V (Fig. 3c) suggests SEI formation during the first cycle, a common phenomenon in lithium-ion batteries, but it is much less pronounced in the LiTFSI in tetraglyme sample shown in Fig. 3d. Subsequent cycles show the expected peaks at their characteristic voltages, indicating reproducible cycling behavior. Fig. 3e and f show electrochemical impedance spectroscopy (EIS) spectra for both electrolytes taken immediately after cell assembly (black line), after CV analysis (red line), and after calendar testing (blue line). The EIS comparison of the pristine materials reveals that LiPF6 initially outperforms LiTFSI. This is particularly evident in the first semicircle, which actually represents the contribution of multiple overlapping semicircles. The impedance, observable on the real axis (Re), is approximately 75 ohms lower for the cells having LiPF6 as electrolyte salt compared to those with LiTFSI. This result is expected, as 1 M LiPF6 in EC:DMC is a highly efficient electrolyte, and is widely used in the field due to its excellent performance. However, its combination with mesoporous V2O5 limits its applicability, as it promotes side reactions, such as SEI formation, significantly more than its counterpart, LiTFSI in tetraglyme, as observed from the EIS collected after cycling the cells.
First, the evolution of the charge transfer resistance (Rct) is highlighted. The LiPF6 in EC:DMC has a lower (Rct, Fig. 3e) due to the higher ionic conductivity and better wetting of the electrode surface by the carbonate solvents. However, the (Rct) increases dramatically during cycling, reflecting the increasing impedance caused by the unstable SEI layer. Conversely, while LiTFSI in tetraglyme shows a higher initial (Rct, Fig. 3f), it shows greater stability during cycling, consistent with the formation of a favorable SEI layer.43,46,47
Furthermore, the spectra reveal changes in impedance behavior across the mid-to-low frequency range, with the semicircles expanding more rapidly over cycles for the LiPF6-based sample. This suggests a significant increase in impedance, likely due to a higher occurrence of side reactions, most notably, the growth of the SEI layer.30 These changes are particularly pronounced in the LiPF6-based system (Fig. 3e), suggesting increased electrode and charge transfer resistance after cycling. This finding supports the assumption that the SEI layer progressively grows during cycling. In contrast, in the LiTFSI-based system (Fig. 3f), the high-frequency semicircles remain similar, indicating stable charge transfer processes, likely due to an SEI layer that remains unchanged during prolonged cycling.
The origin of the different cycling behavior of the two electrolytes can be understood by evaluating the transmission electron micrographs (TEM) shown in Fig. 4. The pristine 70:30 V2O5 electrode shown in Fig. 4a and b has well-resolved 10–50 nm-sized V2O5 crystallites prior to contact with the electrolyte. After electrochemical characterization (GC, CV, and EIS, corresponding to 75 cycles) for the same sample, the growth of an SEI layer on the V2O5 surface is visible, as shown in Fig. 4c and d. In particular, the SEI layer covers the entire surface, but is non-uniform, with distinct thickness variations. Fig. 4e and f show the V2O5 surface after similar cycling using the LiTFSI/tetraglyme electrolyte. While extensive SEI formation is found on isolated areas, the V2O5 surface is predominantly covered by a uniform, nanometer-thick SEI layer, in agreement with previous reports.30
Based on these results, the interplay of micro- and nanoporosity of the V2O5 materials reported in Table 1 is studied in detail in terms of rate capability and calendar tests with cells using LiTFSI in tetraglyme as electrolyte.
Fig. 5a shows the rate performance of three V2O5 electrode types after three cycles of conditioning at C/20. At the low cycle rate of C/2, the two synthesized materials exhibit specific capacities of 275 mA h g−1, exceeding the commercial material's 150 mA h g−1 capacity. However, differences between the 70:30 and 100:0 materials appear with increasing cycling rates, with a complete loss of specific capacity at 20C for the 100:0 and commercial reference materials. The 70:30 V2O5 retains a specific capacity of about 70 mA h g−1 at this high cycling rate. Both samples recover to a specific capacity of ca. 250 mA h g−1 when returned to a low cycling rate. This finding indicates that the low performance of the 100:0 V2O5 material at high C-rates is due to Li-ion diffusion limitation across the nanoporosity rather than irreversible SEI formation.
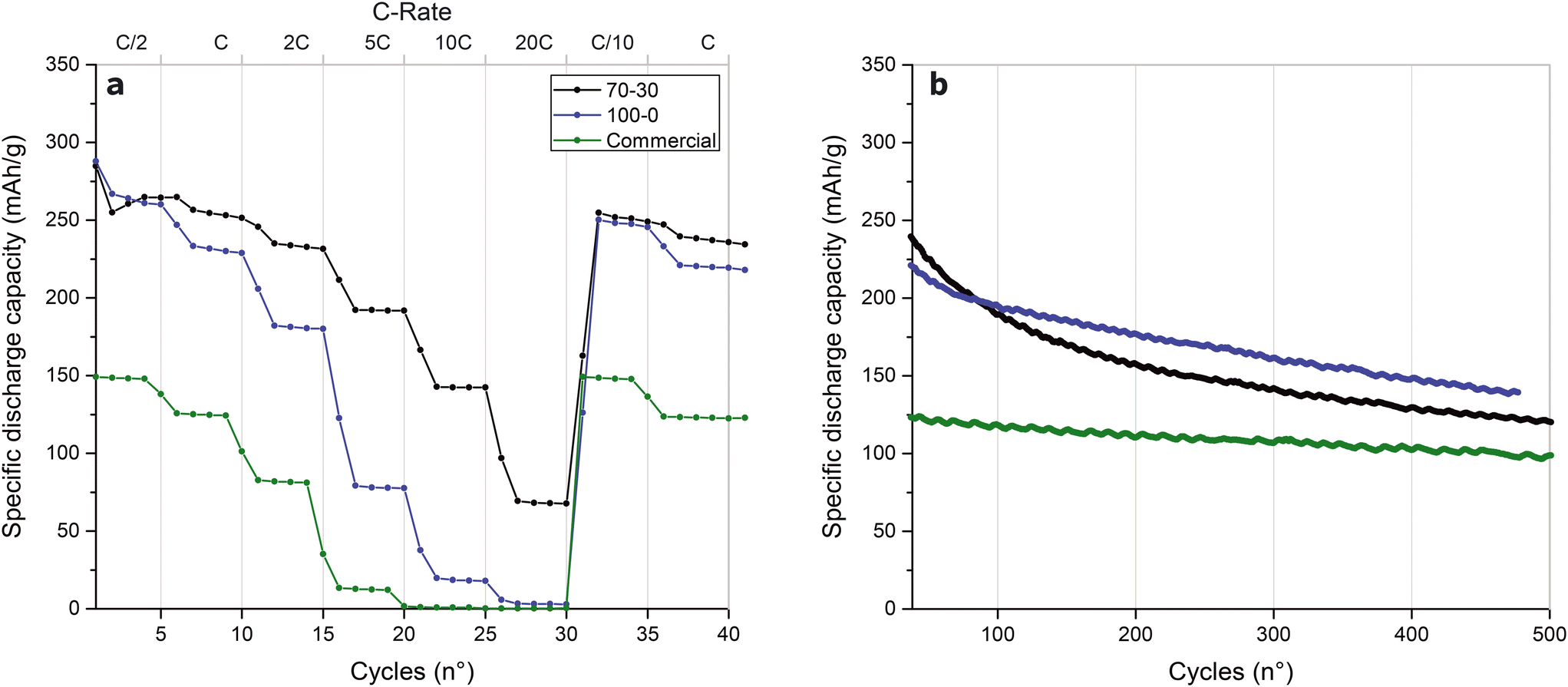 | ||
| Fig. 5 Electrochemical performance of V2O5 electrodes with different microporosities. Rate capability (a) and calendar tests (b) of V2O5 microsphere electrodes prepared from templates made with different BCP:hexadecane ratios (70:30 and 100:0) cycled using LiTFSI in tetraglyme as electrolyte, compared to an unstructured, commercial V2O5. | ||
In particular, the long-term 1C performance in the calendar test of Fig. 5b is superior to that of the commercial material. While the commercial material exhibits a capacity loss of ca. 20% over 500 cycles, the 100:0 and 70:30 materials show capacity losses of 38% and 48%, respectively.
3.1 Discussion
The combination of micro- and nanoporosity is promising for next-generation Li-ion battery electrode materials, as previously shown for cathodes14 and anodes.5,9,10,48 However, these previous studies were limited to stable battery chemistries in which a solid electrolyte interphase remains under control. To address this limitation, the present study focuses on exploring the micro-nanoporosity interplay for the high-voltage V2O5 cathode. In this case, since the growth of an SEI layer during electrochemical cycling is expected to clog the nanopores of the structure, two electrolytes are compared. A comparison of the electrochemical cycling performance of Fig. 3 with the TEM micrographs of Fig. 4 demonstrates the importance of limiting SEI growth during cycling. The stable, thin SEI layer shown in Fig. 4e and f and obtained by using the LiTFSI in tetraglyme enables better rate performance and, in particular, superior rate retention in the calendar test compared to the commonly used carbonate-based electrolyte. The uniform formation of the SEI layer with LiTFSI in tetraglyme can be attributed to the low reactivity of the tetraglyme solvent and the stable nature of the LiTFSI salt, which together suppress uncontrolled decomposition reactions at the electrode-electrolyte interface. TEM images (Fig. 4e and f) clearly show a thin, conformal SEI layer distributed uniformly across the V2O5 surface. Unlike carbonate-based systems, which tend to form thicker and uneven SEI layers due to their higher reduction potential, as shown in Fig. 4c and d, the tetraglyme facilitates slow, surface-limited reactions. This leads to self-limiting SEI growth and maintains consistent ionic pathways during extended cycling.43,44,49 In contrast to previous approaches to synthesize nanoporous microspheres to form mesoporous battery electrodes, the emulsion-based approach developed in this work allows the introduction of an additional hierarchy level into this mesoporosity in the form of micrometer-sized pores. The results reported in Fig. 5 show that this additional hierarchy level is beneficial for the rate performance of the material, further reducing the diffusion-limited Li-ion transport into and out of the mesoporous spheres. The micropores do not significantly affect the overall porosity of the material (cfg. Table 1), nor the long-term performance measured in the calendar test of Fig. 5b. However, the comparison of Fig. 3b, where the cells were cycled using LiPF6 in EC:DMC as electrolyte and Fig. 5b, where the cells were cycled using instead LiTFSI in tetraglyme, shows a considerable batch-to-batch performance. In fact, the two identically prepared sample batches show different capacity retention after 250 cycles. This indicates that further optimization of the material synthesis and electrode preparation steps is needed to produce high performance mesoporous V2O5 cathodes for Li-ion batteries.
Conclusions
In the present work, an emulsion-based method was used to prepare block-copolymer microparticles, which were then used as templates to synthesize V2O5 microparticles with well-defined hierarchical porosity, and finally processed into a Li-ion battery cathode using a standard slurry technique. The final electrode material possessed porosity on three hierarchical levels: (1) the 10 nm level induced by the confined V2O5 crystallization within the BCP microphase morphology, (2) the micrometer length scale caused by the demixing of an added non-solvent (hexadecane) into the fabrication mixture, and (3) the 10 μm length scale arising during the assembly of the microparticles into the final electrode.Two essential conditions were identified to enable better cycling rate performance and calendar lifetime of the thus synthesized mesoporous V2O5 electrodes. First, suppressing the growth of the SEI layer is essential to avoid clogging the nanopores. Second, the introduction of micrometer-sized pores into the microspheres improves the rate capability of the electrode compared to the material without micropores.
While our results show promising electrochemical performance using a scalable emulsion-based technique, the observed batch-to-batch variation suggests that further optimization of this technique is required.
Data availability
Data for this article are available from the Authors upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Adolphe Merkle Foundation for funding.Notes and references
- S.-H. Park, H.-S. Shin, S.-T. Myung, C. S. Yoon, K. Amine and Y.-K. Sun, Chem. Mater., 2005, 17, 6–8 CrossRef CAS.
- C. Sun, S. Rajasekhara, J. B. Goodenough and F. Zhou, J. Am. Chem. Soc., 2011, 133, 2132–2135 CrossRef CAS PubMed.
- F. Wang, S. Xiao, Z. Chang, Y. Yang and Y. Wu, Chem. Commun., 2013, 49, 9209 RSC.
- Z. Zhou, H. Zhang, Y. Zhou, H. Qiao, A. Gurung, R. Naderi, H. Elbohy, A. L. Smirnova, H. Lu, S. Chen and Q. Qiao, Sci. Rep., 2017, 7, 1440 CrossRef PubMed.
- J. Jeong, J. Chun, W.-G. Lim, W. B. Kim, C. Jo and J. Lee, Nanoscale, 2020, 12, 11818–11824 RSC.
- S. Caes, J. C. Arrebola, N. Krins, P. Eloy, E. M. Gaigneaux, C. Henrist, R. Cloots and B. Vertruyen, J. Mater. Chem. A, 2014, 2, 5809–5815 RSC.
- R. Tian, S.-H. Park, P. J. King, G. Cunningham, J. Coelho, V. Nicolosi and J. N. Coleman, Nat. Commun., 2019, 10, 933 CrossRef PubMed.
- K. Fu, X. Li, K. Sun, Z. Zhang, H. Yang, L. Gong, G. Qin, D. Hu, T. Li and P. Tan, Adv. Funct. Mater., 2024, 34, 2409623 CrossRef CAS.
- M. G. Fischer, X. Hua, B. D. Wilts, I. Gunkel, T. M. Bennett and U. Steiner, ACS Appl. Mater. Interfaces, 2017, 9, 22388–22397 CrossRef CAS PubMed.
- M. T. Nguyen, P. Sutton, A. Palumbo, M. G. Fischer, X. Hua, I. Gunkel and U. Steiner, Mater. Adv., 2022, 3, 362–372 RSC.
- Q. Yue, H. Jiang, Y. Hu, G. Jia and C. Li, Chem. Commun., 2014, 50, 13362–13365 RSC.
- J. Shao, X. Li, Z. Wan, L. Zhang, Y. Ding, L. Zhang, Q. Qu and H. Zheng, ACS Appl. Mater. Interfaces, 2013, 5, 7671–7675 CrossRef CAS PubMed.
- Q. Wu, W. s. Miao, Y. du Zhang, H. j. Gao and D. Hui, Nanotechnol. Rev., 2020, 9, 259–273 CrossRef CAS.
- M. G. Fischer, X. Hua, B. D. Wilts, E. Castillo-Martínez and U. Steiner, ACS Appl. Mater. Interfaces, 2018, 10, 1646–1653 CrossRef CAS PubMed.
- Z. Chen, J. Wang, D. Chao, T. Baikie, L. Bai, S. Chen, Y. Zhao, T. C. Sum, J. Lin and Z. Shen, Sci. Rep., 2016, 6, 25771 CrossRef CAS PubMed.
- P. Parant, S. Picart, P. Lhuissier and C. L. Martin, J. Eur. Ceram. Soc., 2020, 40, 1613–1619 CrossRef CAS.
- H. Wang, R. Liu, Y. Li, X. Lü, Q. Wang, S. Zhao, K. Yuan, Z. Cui, X. Li, S. Xin, R. Zhang, M. Lei and Z. Lin, Joule, 2018, 2, 337–348 CrossRef CAS.
- N. Yabuuchi, Y. Koyama, N. Nakayama and T. Ohzuku, J. Electrochem. Soc., 2005, 152, 1434 CrossRef.
- A. Jain, S. R. Manippady, R. Tang, H. Nishihara, K. Sobczak, V. Matejka and M. Michalska, Sci. Rep., 2022, 12, 21024 CrossRef CAS PubMed.
- J. Ren, Y. Li, Z. Wang, J. Sun, Q. Yue, X. Fan and T. Zhao, Int. J. Heat Mass Transfer, 2023, 203, 123818 CrossRef CAS.
- M. S. Whittingham, Chem. Rev., 2014, 114, 11414–11443 CrossRef CAS PubMed.
- P. Hu, T. Zhu, J. Ma, C. Cai, G. Hu, X. Wang, Z. Liu, L. Zhou and L. Mai, Chem. Commun., 2019, 55, 8486–8489 RSC.
- Y. Zhang, Y. Luo, C. Fincher, S. Banerjee and M. Pharr, J. Mater. Chem. A, 2019, 7, 23922–23930 RSC.
- Y. Li, Z. Huang, P. K. Kalambate, Y. Zhong, Z. Huang, M. Xie, Y. Shen and Y. Huang, Nano Energy, 2019, 60, 752–759 CrossRef CAS.
- Y. Rong, Y. Cao, N. Guo, Y. Li, W. Jia and D. Jia, Electrochim. Acta, 2016, 222, 1691–1699 CrossRef CAS.
- P. Hu, P. Hu, T. D. Vu, M. Li, S. Wang, Y. Ke, X. Zeng, L. Mai and Y. Long, Chem. Rev., 2023, 123, 4353–4415 CrossRef CAS PubMed.
- A. Dodero, K. Djeghdi, V. Bauernfeind, M. Airoldi, B. D. Wilts, C. Weder, U. Steiner and I. Gunkel, Small, 2023, 19, 2205438 CrossRef CAS PubMed.
- A. Escher, G. Bravetti, S. Bertucci, D. Comoretto, C. Weder, U. Steiner, P. Lova and A. Dodero, ACS Macro Lett., 2024, 13, 1338–1344 CrossRef CAS PubMed.
- V. Singh, S. Kuthe and N. V. Skorodumova, Batteries, 2023, 9, 3390 Search PubMed.
- X. Liu, M. Zarrabeitia, B. Qin, G. A. Elia and S. Passerini, ACS Appl. Mater. Interfaces, 2020, 12, 54782–54790 CrossRef CAS PubMed.
- E. Strelcov, J. Cothren, D. Leonard, A. Y. Borisevich and A. Kolmakov, Nanoscale, 2015, 7, 3022–3027 RSC.
- Z. Zhang, J. Yang, W. Huang, H. Wang, W. Zhou, Y. Li, Y. Li, J. Xu, W. Huang, W. Chiu and Y. Cui, Matter, 2021, 4, 302–312 CrossRef CAS.
- V. S. K. Sungjemmenla, C. B. Soni, V. Kumar and Z. W. Seh, Energy Technol., 2022, 10, 2200421 CrossRef CAS.
- H. Alptekin, H. Au, E. Olsson, J. Cottom, A. C. Jensen, T. F. Headen, Q. Cai, A. J. Drew, M. Crespo Ribadeneyra and M.-M. Titirici, Adv. Mater. Interfaces, 2022, 9, 2101267 CrossRef CAS.
- V. Velez, G. Ramos-Sánchez, B. Lopez, L. Lartundo-Rojas, I. González and L. Sierra, Carbon, 2019, 147, 214–226 CrossRef CAS.
- J. G. Kennemur, Macromolecules, 2019, 52, 1354–1370 CrossRef CAS.
- D. Kawaguchi and M. Satoh, Macromolecules, 1999, 32, 7828–7835 CrossRef CAS.
- L. A. Belfiore, A. T. N. Pires, Y. Wang, H. Graham and E. Ueda, Macromolecules, 1992, 25, 1411–1419 CrossRef CAS.
- Y. Chen, W. Zhao and J. Zhang, RSC Adv., 2017, 7, 4226–4236 RSC.
- J. Xu, J. Li, Y. Yang, K. Wang, N. Xu, J. Li, R. Liang, L. Shen, X. Xie, J. Tao and J. Zhu, Angew. Chem., Int. Ed., 2016, 55, 14633–14637 CrossRef CAS PubMed.
- E. Strelcov, J. Cothren, D. Leonard, A. Y. Borisevich and A. Kolmakov, Nanoscale, 2015, 7, 3022–3027 RSC.
- B. Yan, X. Li, X. Fu, L. Zhang, Z. Bai and X. Yang, Nano Energy, 2020, 78, 105233 CrossRef CAS.
- S. Cora, B. Key, J. Vaughey and N. Sa, J. Electrochem. Soc., 2023, 170, 020507 CrossRef CAS.
- X. Liu, M. Zarrabeitia, B. Qin, G. A. Elia and S. Passerini, ACS Appl. Mater. Interfaces, 2020, 12, 54782–54790 CrossRef CAS PubMed.
- M. Tang, J.-C. Chang, S. R. Kumar and S. J. Lue, Energy, 2019, 187, 115926 CrossRef CAS.
- M. Dahbi, F. Ghamouss, F. Tran-Van, D. Lemordant and M. Anouti, J. Power Sources, 2011, 196, 9743–9750 CrossRef CAS.
- C. L. Berhaut, D. Lemordant, P. Porion, L. Timperman, G. Schmidt and M. Anouti, RSC Adv., 2019, 9, 4599–4608 RSC.
- Y. Wang, X.-B. Cheng, Y.-X. Yao and R. Xu, Adv. Energy Mater., 2021, 11, 2003156 Search PubMed.
- B. N. Olana, S.-H. Pan, B.-J. Hwang, H. Althues, J.-C. Jiang and S. D. Lin, J. Mater. Chem. A, 2024, 12, 3659–3670 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5lf00033e |
| This journal is © The Royal Society of Chemistry 2025 |