
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterojunction photocatalysts: where are they headed?
Hanggara
Sudrajat
 *abc and
Maya
Nobatova
*d
*abc and
Maya
Nobatova
*d
aQuantum Materials Group, Research Center for Quantum Physics, National Research and Innovation Agency (BRIN), South Tangerang 15314, Indonesia. E-mail: hanggara.sudrajat@brin.go.id
bQuantum Materials and Devices, Indonesian Quantum Initiative (IQI), South Tangerang 15314, Indonesia
cCollaboration Research Center for Advanced Energy Materials, BRIN – Institut Teknologi Bandung, Bandung 40132, Indonesia
dDipartimento di Ingegneria, Università degli Studi di Palermo, Viale delle Scienze Edificio 6, 90128 Palermo, Italy. E-mail: maya.nobatova@unipa.it
First published on 2nd April 2025
Abstract
Heterojunction photocatalysts have gained attention for their potential to enhance performance in light-driven, bias-free redox reactions. By integrating two or more semiconducting materials, these photocatalysts exhibit improved light absorption, more efficient charge separation, and enhanced charge transfer. They also enable better alignment of band edge potentials with the redox potentials of reactants, which can promote the selective formation of desired products with higher yields. Recent advancements in characterization techniques and theoretical calculations have provided deeper insights into optimizing charge transfer processes and effectively managing photoexcited charges on the surface. However, key questions remain: how far can heterojunction photocatalysts progress? And what steps are needed to move beyond lab-scale demonstrations? In this perspective, we discuss the status and challenges of heterojunction photocatalysis, aiming to encourage further discussion within the catalysis community to drive this research area toward meaningful progress rather than mere academic hype.
1. Enhanced charge separation: the key feature of heterojunction photocatalysts
Light-driven catalysis that operates without the need for external potential bias—referred to as photocatalysis—presents alternative routes for energy production1 and environmental remediation.2,3 Developing a highly efficient photocatalyst requires addressing three crucial aspects: (i) expanding light absorption into the long-wavelength region, (ii) enhancing the separation and transfer of photoexcited charges to the surface and subsequently to the adsorbed reactants, and (iii) optimizing band edge positions so they exceed the redox potential of the reactants.4,5 A single semiconducting material, however, often fails to meet all these requirements. Therefore, integrating two or more semiconducting materials to form a heterostructure photocatalyst, characterized by a junction, can potentially overcome these limitations.4,6Creating a heterojunction photocatalyst can enhance both the quality and quantity of charge carriers. Quality refers to the redox potentials of the charge carriers, which determine the material's thermodynamic capability to drive chemical reactions, as well as their mobility, which affects their reactivity. Meanwhile, quantity represents the population of these carriers. It is difficult to optimize both aspects at the same time, since enhancing one aspect often comes at the expense of the other. Thus, designing a heterojunction requires a careful balance between redox potential, mobility, and the number of charge carriers to achieve the desired catalytic behavior. Mobility is strongly related to the number of charge carriers, as highly mobile electrons have a lower probability of recombination—a photophysical event that decreases the number of charge carriers.
The redox potential of charge carriers typically changes upon the formation of a heterojunction, either increasing or decreasing relative to the individual semiconductors that form the heterojunction. This variation depends on the migration pathway and the specific locations where the carriers accumulate. In some cases, a lower redox potential is advantageous; for example, partial (selective) oxidation requires moderate redox potentials, in contrast to the high redox potentials needed for complete oxidation (mineralization). By adjusting these redox potentials, the photocatalyst can be optimized for specific reactions. As charge carriers transfer within a heterojunction, they become spatially separated, which retards recombination and increases their availability for surface reactions.
One critical factor associated with the quantitative aspect of charge carriers, which determines overall photocatalytic performance, is charge separation.7–9 In the case of particulate photocatalysts, however, separating electrons from holes is challenging because the cathodic and anodic sites, or the reduction and oxidation sites, are distributed over very short distances—on the nanometer scale.10 This differs from photoelectrochemical cells (PECs), for instance, where these sites are distributed on the millimeter scale. In a PEC, a membrane is also installed to separate the redox sites, and electrical contacts are introduced to collect charge carriers, preventing their recombination. Based on driving forces, transport physics, and transfer kinetics, charge separation can be categorized into three modes: (i) asymmetric energetics (AE), (ii) asymmetric kinetics (AK), and (iii) a hybrid of both.10 The creation of heterojunction photocatalysts falls under the category of AE.
As illustrated in Fig. 1, AE and AK differ fundamentally in their underlying driving forces, charge transport mechanisms, and design strategies. The primary distinction between AE and AK lies in the driving forces responsible for charge separation. AE relies on an internal electric field within the photocatalyst, which directs electrons and holes to different reaction sites. This electric field is typically created due to spatial variations in electrochemical potential, band bending, built-in potentials, or space-charge regions within the material. The formation of AE occurs naturally in semiconductor-based photocatalysts, where a difference in energy levels across different sites induces charge migration. Conversely, AK does not rely on an internal electric field but instead depends on the differential charge-transfer rates at various reaction sites. In this mechanism, one type of charge carrier is preferentially transferred at a much faster rate than the other. This kinetic asymmetry prevents recombination by ensuring that one charge carrier is rapidly migrated before it has a chance to recombine. AK is commonly observed in molecular-scale or nanostructured systems where quantum confinement prevents the formation of internal electric fields.
 | ||
| Fig. 1 (A) Schematic of charge separation driven by AE: band energetics in the dashed box region under dark equilibrium; strategies for creating AE, with solid arrows representing internal electric fields. (B) Schematic of charge separation driven by AK: charge-transfer energy diagram illustrating photogenerated electron flow and competing processes; spatial arrangements for charge separation in different systems. Reprinted with permission from ref. 10. Copyright 2021, American Chemical Society. | ||
The way charge carriers move in a photocatalyst also varies significantly between AE and AK. In AE-driven systems, charge transport is primarily governed by drift motion induced by the built-in electric field. When a semiconductor absorbs light, electron–hole pairs are generated, and the internal electric field forces electrons toward reductive sites and holes toward oxidative sites. This mechanism is analogous to the charge separation process in PECs, where spatial separation of reaction sites is crucial to preventing recombination. In contrast, AK-driven systems rely on diffusion rather than drift for charge transport. Since no significant internal electric field is present, charge carriers move based on concentration gradients. The faster charge transfer at one site creates a depletion of one type of charge carrier, generating a concentration difference that facilitates diffusion-driven charge separation. However, this pathway is more susceptible to recombination losses, as it lacks an intrinsic field to sustain spatial separation.
AE mechanisms are predominantly found in semiconductor photocatalysts with continuous energy bands. These materials, such as metal oxides and perovskite-structured oxides, exhibit band bending at their interfaces, leading to an internal electric field. The degree of band bending depends on factors such as dopant concentration, defect density, and interfacial properties. To enhance AE-driven charge separation, strategies such as doping, heterojunction formation, and facet engineering are commonly employed. On the other hand, AK mechanisms are characteristic of molecular, quantum-confined, or nanostructured photocatalysts. Examples include quantum dots, dye-sensitized systems, and metal–organic frameworks. These materials lack continuous energy bands and built-in electric fields, necessitating the use of high-turnover co-catalysts or redox mediators to facilitate rapid charge transfer. Enhancing charge-transfer rates through ligand engineering, metal coordination, and molecular tethering is a key strategy in AK-driven systems.
AE-driven photocatalysts face limitations related to charge recombination at defect sites and slow surface reaction kinetics. Although the built-in electric field aids charge separation, if the surface reaction is not fast enough, accumulated charges can recombine before participating in redox reactions. To mitigate this issue, strategies such as passivation of surface states and optimized band alignment can be employed to ensure efficient electron and hole extraction by reactant molecules. For AK-driven photocatalysts, the primary challenge is overcoming charge recombination due to the absence of an internal electric field. The effectiveness of AK relies heavily on maintaining a large disparity between electron and hole transfer rates. However, if the kinetic asymmetry is insufficient, recombination dominates, leading to low quantum yields. Optimizing AK systems involves using highly active co-catalysts, modifying surface chemistry to promote selective charge transfer, and integrating redox mediators to shuttle electrons or holes efficiently.
The choice between AE and AK mechanisms depends on the specific application and material constraints. AE-driven photocatalysts are well-suited for large-scale solar fuel production, such as water splitting, where long-range charge transport and built-in electric fields provide stability. These systems benefit from bulk material optimization, which allows for efficient charge migration over extended distances. AK-driven photocatalysts, in contrast, are advantageous in applications requiring localized charge transfer, such as organic synthesis. The high surface-to-volume ratio and rapid charge transfer rates in AK systems make them effective for reactions that occur at molecular interfaces. AK mechanisms are often employed in hybrid photocatalytic–electrocatalytic systems, where charge carriers are extracted via external electrodes to enhance efficiency.
While AE and AK are distinct mechanisms, many advanced photocatalysts aim to integrate both approaches to maximize efficiency. Hybrid charge-separation strategies utilize both internal electric fields and asymmetric charge transfer kinetics. For example, semiconductor heterojunctions incorporating molecular co-catalysts such as dye molecules and quantum dots or forming plasmonic photocatalysts upon incorporating metallic nanoparticle can provide both a built-in electric field for drift-based separation and fast charge-transfer kinetics to minimize recombination. Such hybrid systems have shown promising results in achieving near-unity quantum yields by combining the strengths of both AE and AK mechanisms. Thus, future research should integrate AE and AK strategies into hybrid photocatalysts to achieve optimal charge separation. In this context, heterojunction photocatalysts are promising platforms for realizing efficient hybrid charge-separation pathways.
In heterojunction photocatalysts, charge separation is influenced by material properties such as band edge potential, work function, Fermi levels, and differences in electronegativity within the framework of AE.9 These factors drive electron migration across interfaces, generating internal electric fields that promote charge separation. Heterojunction photocatalysts use various junction configurations to optimize the efficiency of charge separation and transfer. As illustrated in Fig. 2, the two main types, type-II and S-scheme heterojunctions, have distinct characteristics in managing charge transfer.4,11
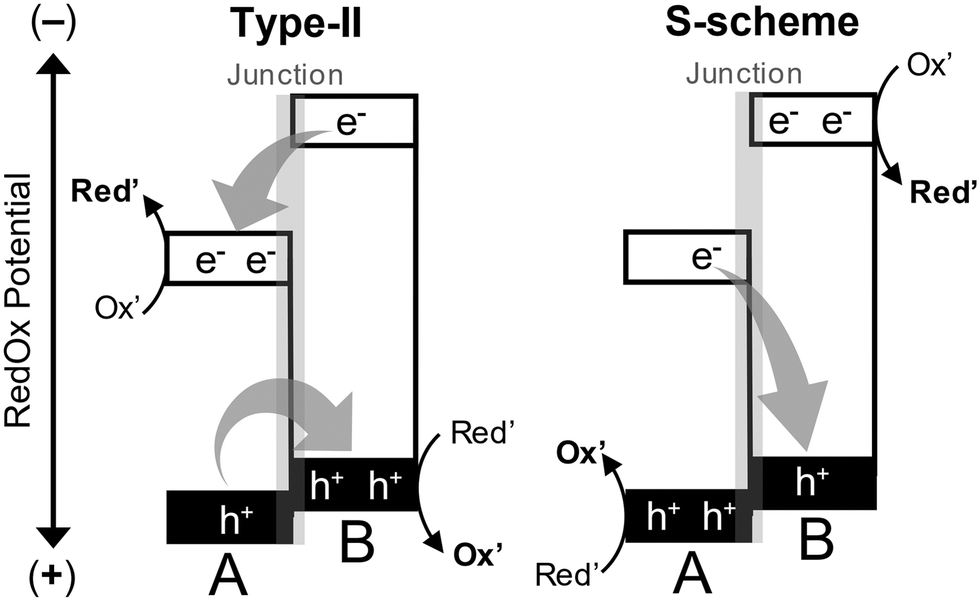 | ||
| Fig. 2 Schematics illustration of charge migration pathways in the type-II heterojunction and S-scheme. The S-scheme heterojunction achieves charge separation while maintaining favorable redox potentials, unlike the type-II heterojunction. However, higher redox potential is not necessarily advantageous. For partial oxidation reactions, moderate oxidation potentials are often preferred to selectively convert reactants into desired products and prevent the complete oxidation of reactants into their inorganic entities. | ||
(1) Type-II (staggered) heterojunctions. Type-II heterojunctions facilitate charge separation by directing two opposite charges across two semiconducting materials with staggered band alignments. In these configurations, the conduction band minimum (CBM) and valence band maximum (VBM) of one semiconductor are aligned with the CBM and VBM of the other, enabling charge carriers to flow toward less negative (for electrons) and less positive (for holes) states. While this spatial separation reduces recombination rates, it also diminishes the redox power, potentially limiting the applicability of heterojunction in reactions that demand high redox potentials.
(2) S-scheme (step-scheme) heterojunctions. S-scheme heterojunctions align their energy bands in a manner similar to type-II heterojunctions but offer enhanced redox capabilities compared to the individual semiconducting materials that form the heterojunction. Unlike type-II structures, S-scheme heterojunctions retain electrons and holes in favorable redox potential states. Thus, thermodynamically, compared to type-II heterojunctions, S-scheme heterojunctions can drive a broader range of reactions. Despite this feature, however, good product selectivity cannot be fully guaranteed, as it also depends on the unique properties and affinity of the photocatalyst surface toward particular reactants, in addition to charge carrier properties.
In AE charge-separation mode, a built-in electric field between reductive and oxidative sites drives charge separation in heterojunctions with type II band alignment, promoting charge accumulation at surface sites.10 This field is created by band edge offsets, built-in potentials, or interfacial dipoles, favoring charge transfer from high to low energy sides. The S-scheme, on the other hand, involves direct or metallic contact for charge transport, which is the opposite of type II heterojunction. Electrons and holes recombine at the interface, while remaining charges move to respective reductive or oxidative sites. In type II heterojunction, charge separation occurs without recombination, dissociating excitons but not increasing photovoltage. Both systems rely on the Fermi-level difference to set the built-in potential, but with opposite directions. Therefore, proper design of band offsets and built-in potentials is essential for functionality. The AE mechanism underpins the functionality of type II and S-scheme heterojunctions in practical applications. For example, Yamakata and co-workers studied electron and hole transfer kinetics in CoOx/BiVO4/SnO2 heterojunctions for the oxidation of HCOOH and CH3OH using transient absorption from visible to mid-IR.12 After exciting BiVO4, electrons transferred to SnO2 in ∼3 ps, while holes stayed in BiVO4 and transferred to CoOx in a few ps. This suppressed recombination, leading to 2.4 and 3.6 times more surviving carriers at 5 μs for BiVO4/SnO2 and CoOx/BiVO4/SnO2, respectively, compared to bare BiVO4. This rapid charge separation indicates that both intra-layer and inter-layer mechanisms work together, with band-bending aiding charge transfer across the heterojunction. The enhanced catalytic activity of CoOx/BiVO4/SnO2 is well explained by the synergistic charge separation driven by the AE mechanism, which involves band-bending and the surface co-catalyst (CoOx) as the oxidative site.
Due to their combined advantages of strong redox power and efficient charge separation, S-scheme heterojunctions are increasingly favored. To illustrate this concept, consider two semiconductors, A and B, forming an S-scheme heterojunction (Fig. 2). Semiconductor A has lower reduction power (a less negative CBM potential) and higher oxidation power (a more positive VBM potential), whereas semiconductor B has higher reduction power (a more negative CBM potential) and lower oxidation power (a less positive VBM potential). When A and B are structurally aligned to form a heterojunction following the S-scheme mechanism, electrons excited in the CBM of A recombine with holes in the VBM of B at the junction. This recombination occurs because the VBM of B is less positive than that of A, making it energetically closer to the CBM of A than to B's own CBM. This migration pathway enhances charge separation while maintaining redox potential. In contrast, when A and B are aligned according to the type-II mechanism, photoexcited electrons in the CBM of B move toward a lower-energy state with a less negative potential, namely the CBM of A. Simultaneously, holes in the VBM of A migrate toward a lower-energy state with a less positive potential, specifically the VBM of B. As a result, electrons accumulate in A, while holes concentrate in B, leading to spatial charge separation. This separation minimizes the recombination probability, as electrons and holes are less likely to come into contact; however, it also decreases the redox power.
2. What type of heterojunction do we get?
Confirming the formation of the heterojunction, the type of heterojunction, and its distribution in the examined sample is a challenging task. This is particularly true for particulate catalyst systems, where observing the junction interface is more difficult than in thin films. Techniques such as transmission electron microscopy (TEM), X-ray diffraction (XRD), and scanning electron microscopy (SEM) can be used to examine the structural and compositional characteristics of the heterojunction. These methods help determine whether the desired interactions between the constituent materials have been achieved. However, when the quantity of one constituent in a heterojunction is low, or when its qualitative properties—such as size—are too small and its crystallinity is not very high, the junction interface often becomes difficult to distinguish under a conventional electron microscope. This occurs because the minor phase is so dispersed that it does not form a continuous or well-defined boundary with the primary material.13–15 Instead, the constituent may exist as isolated nanoscale clusters or be incorporated into the dominant phase in a way that does not produce significant contrast in electron microscopy imaging.15 The resolution limitations of the electron microscope and the potential for atomic-scale mixing at the interface further obscure the visibility of the junction.16 This effect is especially pronounced when light elements, such as carbon and nitrogen, are involved in the heterojunction. As a result, identifying and characterizing the heterojunction in such cases often requires more advanced techniques, such as aberration-corrected scanning TEM (STEM).13,14,16,17 The correction of spherical aberrations enhances the contrast and sharpness of atomic columns, making it possible to detect subtle structural and compositional variations. Aberration-corrected STEM images with sub-nanometer or even Ångström resolution can be captured around the junction interface to probe detailed atomic arrangements. As shown in Fig. 3A, this technique can identify the exact location of the heterojunction even when no dislocation is observed. Heterojunctions often exhibit dislocations at the boundary between two materials, making the junction interface easily identifiable. However, some heterojunctions show no distinct boundary in the junction interface. Combining aberration-corrected STEM with techniques such as electron energy-loss spectroscopy (EELS) enables precise elemental mapping, further enhancing the identification of heterojunction interfaces (Fig. 3A).18 STEM imaging and EELS can also be combined with numerical simulations such as density functional theory (DFT) to understand the atomic structure of junction interfaces (Fig. 3B). Atomic electron tomography (AET) is another advanced imaging technique used to map three-dimensional atomic positions and identify crystal imperfections at the heterointerface with picometer accuracy. This technique enables the detection of various structural defects, such as vacancies, substitutional impurities, bond distortions, and atomic-scale ripples. These observations allow for a quantitative analysis of 3D atomic displacements and the complete strain tensor across the heterointerface (Fig. 3C).19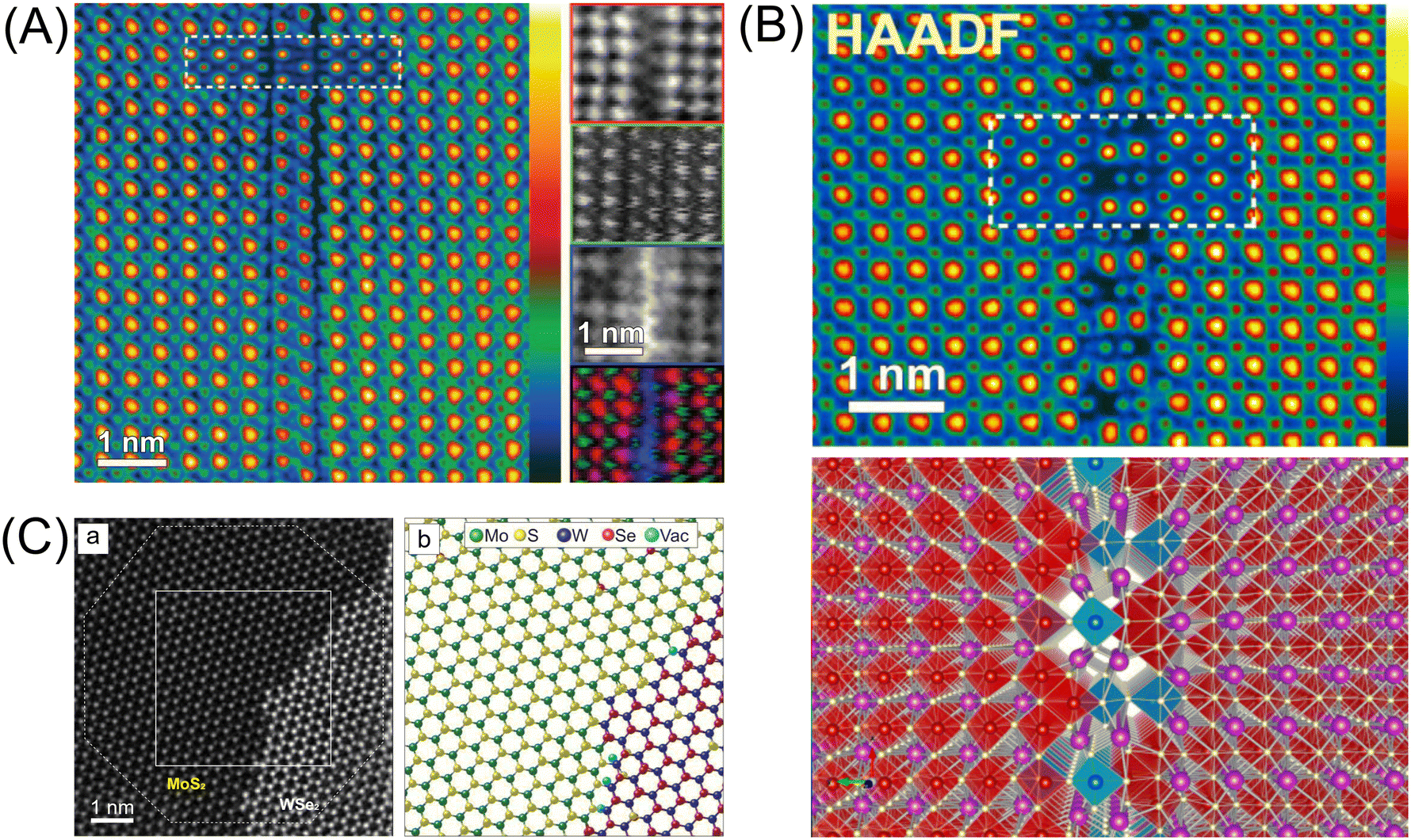 | ||
| Fig. 3 (A) High-angle annular dark-field (HAADF) STEM image and EELS maps of an antiphase boundary forming in a Ti/Nd–BiFeO3 heterojunction along the [010] projection. A simulated image is inset and overlaid on the HAADF image using the same contrast scale, while the EELS maps for individual elements display the full contrast range. In the red–green–blue (RGB) overlay of Fe (R), HAADF (G), and Ti (B) signals, background intensity has been removed to enhance visibility of the primary atomic columns. (B) A 3D model of an antiphase boundary in the Ti/Nd–BiFeO3 heterojunction, reconstructed using HAADF and bright-field (BF) images alongside atomic-resolution chemical maps from two orthogonal projections. The inset, which is outlined by a dotted white box, presents a simulated image of the model, generated with the multi-slice algorithm using the QSTEM software suite. The figures shown in (A) and (B) are reprinted with permission from ref. 18. Copyright 2013, American Institute of Physics. (C) 3D atomic coordinates of a monolayer MoS2–WSe2 heterostructure: (a) a top view of the 3D AET reconstruction of the interface region, where atomic coordinates within the octagonal region were determined with picometer precision, and (b) an experimentally derived 3D atomic model corresponding to the square region in (a). Reprinted with permission from ref. 19. Copyright 2021, American Association for the Advancement of Science. | ||
The formation of a heterojunction can also be identified through changes in its electronic structure. To examine these changes, photoelectrochemical characterizations such as Mott–Schottky analysis, photocurrent measurement, and impedance spectroscopy can be used. For example, the Mott–Schottky analysis helps determine the conductive type and flat band potential.20 To characterize electronic structure spectroscopically, ultraviolet photoemission spectroscopy (UPS) can be an option. UPS measures the energy levels of the VB and the work function, and by analyzing photoemitted electrons, it provides information about the electronic states at the surface and the energy band alignment.21
Once a heterojunction photocatalyst is created and its electronic structure—such as bandgap energy and edge potential levels—is fully characterized, we need to analyze and quantify its charge transfer dynamics to confirm whether the junction effectively facilitates charge transfer processes. Various techniques have been used to verify charge transfer pathways, assess charge separation efficiency, and identify recombination sites.4,9 These include X-ray photoelectron spectroscopy (XPS), which detects binding energy shifts related to electron transfer and provides information on into charge distribution; Kelvin probe force microscopy (KPFM), which maps the contact potential difference to visualize charge flow and density; and photoinduced redox probe reactions, which use high-valent cations to confirm electron transfer pathways by examining redox product deposition on catalyst surfaces. Meanwhile, electron paramagnetic resonance (EPR) can measure charge separation efficiency and reactive oxygen species generation, while femtosecond transient absorption spectroscopy (fs-TAS) captures ultrafast charge transfer events to analyze electron transfer dynamics. Femtosecond time-resolved diffuse reflection spectroscopy (fs-TDR) probes charge carrier lifetimes, and DFT calculations estimate recombination probabilities, complementing experimental data to enhance the understanding of the electronic properties and dynamics affecting photocatalytic performance.
Surface photovoltage (SPV) is another important technique to examine charge carrier dynamics in heterojunction photocatalysts. The absence of an SPV signal indicates rapid recombination. For instance, Xu and co-workers used SPV to examine the ability of CuxO/ZnO heterojunctions to switch the dominant surface carrier type from holes to electrons for the development of visible-light-active photocatalysts that can selectively drive diverse reactions, depending on the nature of the reactions—reduction or oxidation (Fig. 4A).22Fig. 4B and C show the SPV responses of CuxO/ZnO heterojunctions under UV-visible light, with the lower panels illustrating phase values as a function of wavelength. As seen in Fig. 4B, CuxO/ZnO exhibits photovoltage phases in the fourth quadrant under UV light, generating strong positive signals that indicate hole accumulation on the irradiation side. Under visible light, the phases shift to the second quadrant, forming a weak negative photovoltage signal due to CuxO, indicating minimal electron accumulation at the surface. In contrast, the re-engineered CuxO/ZnO(r) sample shows a strong negative SPV response under visible light, with an integral area surpassing ZnO's positive UV-light response (Fig. 4C). This implies predominant p-type conductivity, where the surface is electron-dominated under UV-visible light, potentially enhancing reduction activity. For CuxO/ZnO samples with higher donor impurity concentrations, stronger n-type surface conductivity and high electric field neutralization limit electron migration to the surface under visible light. Conversely, CuxO/ZnO(r) samples with lower donor defect concentrations exhibit stronger p-type conductivity and reduced electric field offset, allowing efficient electron migration to the surface. Based on the bandgaps of ZnO, CuO, and Cu2O, as well as their energy levels relative to NHE, a schematic of the plausible electron flow under visible light (λ > 400 nm) is depicted in Fig. 4D.
 | ||
| Fig. 4 (A) Switching the dominant surface carrier type potentially enhances performance and selectivity in photocatalytic applications. SPV responses of (B) CuxO/ZnO and (C) CuxO/ZnO(r), highlighting distinct photoresponse characteristics due to differences in surface conductivity. (D) Schematic illustrating electron flow on heterojunction surfaces under visible light excitation. Reprinted with permission from ref. 22. Copyright 2025, Cell Press. | ||
3. Key issues
Given the extensive research on heterojunction photocatalysts, it is time to address two critical questions: (1) is the heterojunction effect truly the primary reason for the enhanced photocatalytic activity? (2) What steps are necessary to facilitate industrial adoption?3.1. Fundamental issues
One fundamental issue is verifying whether the heterojunction effect is genuinely responsible for the enhanced performance observed when a heterostructure outperforms its individual components. Schemes such as the S-scheme, p–n, Schottky, type-II, and surface heterojunctions have been proposed to explain enhanced photocatalytic performance, each featuring unique charge separation pathways that suppress recombination.23 To approximate charge flow in a heterojunction for charge separation, the band edge energies of the isolated materials can be examined first.24 In the typical energy scale representation (vacuum at the top), electrons tend to occupy the lowest CB, while holes occupy the highest VB to minimize energy.Recall that photocatalytic reactions occur in the excited state. Therefore, any technique used to propose charge transfer pathways—and consequently the heterojunction type—should not examine the photocatalyst sample in the ground state. XPS, for example, is often used to investigate charge migration pathways in photocatalysts, but it is typically operated in the dark. When XPS is used to elucidate charge transfer pathways, it must sample the material in the excited state, i.e., under illumination. For instance, Yu and co-workers used irradiated XPS to examine the direction of electron transfer in a ZnO/CuInS2 (ZC3) heterojunction photocatalyst.25 After heterostructuring CuInS2 with ZnO, the binding energies of Zn 2p and O 1s in the heterostructure became more negative compared to those in ZnO (Fig. 5). Meanwhile, the binding energies of Cu 2p, In 3d, and S 2p in the heterostructure shifted toward higher values compared to those in CuInS2, confirming that electrons migrate from CuInS2 to ZnO during heterostructure formation. Despite this elegant work, however, we should still be aware that photocatalytic reactions occur in aqueous solution under ambient pressure, whereas light-induced XPS operates in vacuum or near-ambient conditions without a solvent. Thus, we cannot fully ensure that the charge carrier pathway observed in XPS experiments accurately represents what occurs in real photocatalytic reactions.
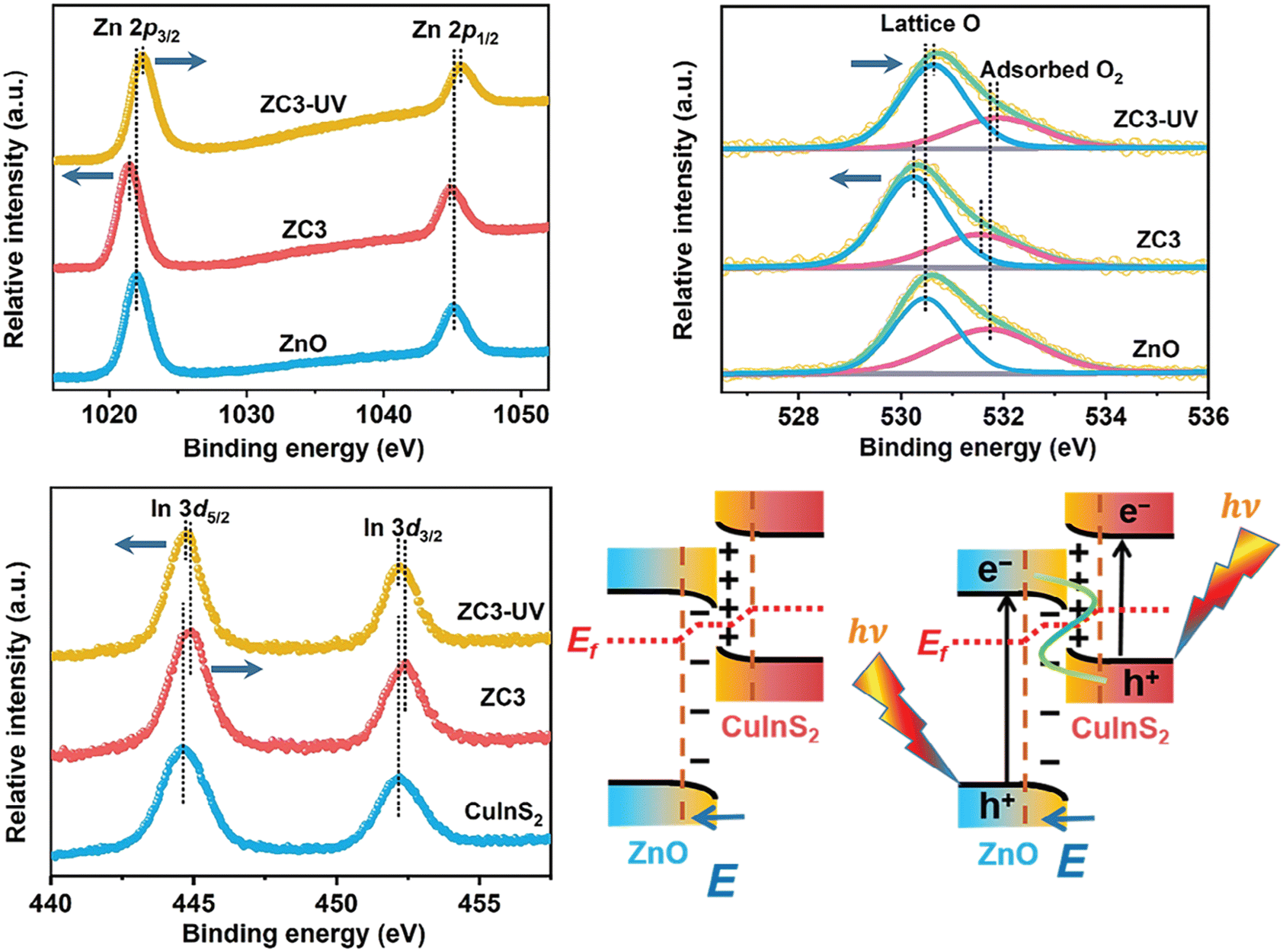 | ||
| Fig. 5 XPS spectra recorded in the dark and under UV light from a 365 nm LED. The proposed interfacial electron transfer is also schematically illustrated. Reprinted with permission from ref. 25. Copyright 2024, Wiley-VCH GmbH. | ||
It is generally believed that controlling the structure and surface states of the semiconducting components can amplify the synergistic effect.7,23 Nevertheless, the heterojunction effect remains more of an interpretative concept rather than a precise regulatory mechanism for improving photocatalytic performance. Therefore, a fundamental question persists: why and how do heterostructure photocatalysts often exhibit better performance compared to single-component systems? While the heterojunction effect is well-established in semiconductor physics, it may not entirely account for the observed catalytic enhancements in heterogeneous (slurry) systems. Since heterogeneous photocatalysis is a surface phenomenon,26,27 surface properties—such as the type and quantity of functional groups, specific surface area, morphology, porosity, and surface defects—may also play a role. Surface defects, for instance, often form unintentionally in heterojunction photocatalysts and can significantly influence their performance. The second law of thermodynamics suggests that defects are inherently present in crystalline materials and thus unavoidable during catalyst synthesis.28 These defects affect band structure, built-in electric fields, charge transfer kinetics, and hence redox reactions.28,29 By modifying charge transfer pathways and introducing energy states within the bandgap, they selectively trap carriers, reducing recombination, and extending carrier lifetimes.30 In TiO2/Ce2S3 heterojunction, for example, photogenerated electrons in TiO2's CB decay into impurity energy levels (defect states), where they are temporarily trapped.31 These electrons are then gradually released to recombine with holes in Ce2S3's VB, effectively extending charge carrier lifetimes. Defects can also induce built-in electric fields that drive charge carriers toward active sites, improving separation and reactivity—especially in systems where interfacial charge transfer is a limiting factor.30 They modulate charge density around active sites, affecting redox power.28,32 As trapping centers for photogenerated carriers, defects can enhance surface charge transport and adsorption of reactants, accelerating charge transfer and boosting reaction rates.32 They also improve surface hydrophilicity, facilitating better mass transport of reactants and products, further optimizing photocatalytic efficiency.33 Equally important, reactive species often form at the interfaces of nanostructured semiconductors or between semiconductors and metallic particles loaded onto their surface, which may have a greater impact on photocatalytic properties than the heterojunction effect itself.4,11 This indicates the need for a more in-depth investigation into the chemical structure and composition of these interfacial reactive species.
Another issue is establishing a quantitative correlation between photocatalytic performance (conversion, selectivity, and productivity) and the characteristic properties of heterojunctions, which remains a challenge. Current explanation of the heterojunction effect are predominantly qualitative,4,34 with limited robust quantitative research available. Despite efforts to calculate built-in electric fields and photogenerated carrier lifetimes, for instance, a direct link between these photophysical properties and photocatalytic performance has yet to be established.34 We need to elucidate how the intensity of built-in electric fields influences the migration of photogenerated charges and how this, in turn, affects reaction rates and product selectivity. The migration of photoexcited charge carriers from the bulk to catalytically active surface sites is characterized by a photophysical parameter known as photoconductivity. This property can be measured using contact-less techniques such as time-resolved terahertz spectroscopy (TRTS) or time-resolved microwave conductivity (TRMC).
3.2. Practical issues
One major bottleneck in charge-carrier-based catalysis for future industrial applications is the limited generation of charge carriers, which restricts product yields to the micro- to millimole scale in solution-based reactions or the gram- to kilogram scale in solid-state synthesis. In contrast, industrial processes require yields on a ton scale with high reaction productivity (yield per unit time), revealing a huge gap. This raises questions about the quantity of charge carriers required for large-scale production and the durability of photocatalysts in sustaining long-term productivity. To meet industrial demands, reaction productivity must be maximized, ensuring that the desired products are generated not only in sufficient quantities but also at a high rate. Overcoming these challenges will necessitate strategies to dramatically improve both the generation and utilization efficiency of charge carriers while maintaining high selectivity.Scaling-up or numbering-up (scaling-down and then scaling-out) the production of lab-engineered heterojunction photocatalysts is a challenging task. Here, scale-up refers to increasing the size of a single reactor while maintaining similar operating conditions, while numbering-up (or scale-out) refers to running multiple microreactors in parallel to achieve higher production capacity while maintaining the advantages of small-scale processing. To meet industrial demands, scalable synthesis methods are required.35–38 We can learn best practices from the more mature field of heterojunction solar cells39–41 and adapt them to suit the context of heterojunction photocatalysts. Hydrothermal and solvothermal techniques can be optimized in terms of temperature, pressure, and precursor concentrations to produce high-quality heterojunctions in large volumes. Flame spray pyrolysis offers a continuous method for producing nanoparticles with controlled morphology and composition. Chemical vapor deposition provides a way to create thin-film heterojunctions with high uniformity, though further cost reduction is needed. Meanwhile, electrospinning allows for the mass production of nanofibrous photocatalysts with large surface areas.
The use of low-cost precursors along with alternative synthesis routes is also necessary. Instead of relying on expensive noble metals like Pt, earth-abundant alternatives need to be explored such as Cu, Fe, or Ni-based co-catalysts. On the other hand, one-step synthesis approaches help eliminate multi-step, energy-intensive fabrication methods, making production more cost-effective. For this purpose, solid-state and mechanochemical approaches offer alternatives for heterojunction synthesis. Ball milling and mechanochemical synthesis eliminate the need for solvents and high temperatures. Molten salt synthesis allows for rapid diffusion and crystallization at lower temperatures, while 3D printing and advanced manufacturing techniques provide additional opportunities for large-scale photocatalyst production. Additive manufacturing may also be used to deposit heterojunction layers with high precision, reducing material waste and improving reproducibility. Equally important, integrating synthesis with industrial processes can be attempted for scaling-up or numbering-up heterojunction production. Continuous flow reactors can be designed to enable in situ synthesis and deposition of photocatalysts onto various substrates, streamlining production while reducing costs. One example is the use of reactive extrusion techniques, which hold potential by allowing direct heterojunction formation during polymer or ceramic processing.
The industrial-scale production of heterojunction photocatalysts requires a balance between efficiency, cost-effectiveness, stability, and reproducibility. While many studies focus on improving photocatalytic performance at the laboratory scale, translating these advancements into large-scale applications presents huge challenges. Among these challenges, ensuring the stability and reproducibility of heterojunction photocatalysts during large-scale production remains a critical concern.
One of the primary issues in large-scale production is maintaining the structural and electronic integrity of heterojunctions. At the lab scale, synthesis methods such as hydrothermal, solvothermal, and sol–gel are carefully optimized to produce high-quality heterojunctions with well-defined interfaces. However, when these methods are scaled up, variations in reaction parameters—such as temperature gradients, precursor concentrations, and mixing dynamics—can lead to inconsistencies in material properties. These inconsistencies may lead to variations in particle size, crystallinity, and surface composition, ultimately impacting photocatalytic performance. To address these issues, scalable synthesis techniques used in semiconductor heterojunction fabrication can be explored. In the industry, semiconductor heterojunctions are typically produced using highly precise and scalable methods. Molecular beam epitaxy (MBE) offers atomic-level control and is widely used for high-performance devices. Chemical vapor deposition (CVD) and its variants, such as metal–organic chemical vapor deposition (MOCVD) and plasma-enhanced chemical vapor deposition (PECVD), are commonly employed in large-scale semiconductor manufacturing, particularly for electronics and photovoltaics.42 Meanwhile, sputtering and physical vapor deposition (PVD) are widely used for thin-film heterojunctions in photovoltaic applications.43 While these techniques have not yet been applied specifically to photocatalysts with heterojunctions, they are known to offer better control over material properties by ensuring uniform precursor distribution and minimizing batch-to-batch variations.
Reproducibility is another issue in scaling up heterojunction photocatalysts. In laboratory settings, small-scale synthesis allows for precise control over reaction conditions, but industrial processes require continuous and high-throughput production methods. This introduces variability in the microstructure and composition of the material, affecting performance consistency. One approach to improving reproducibility is employing automated synthesis and real-time monitoring techniques. Advanced spectroscopic and microscopic methods operated in situ, can provide real-time information on material properties.
The anatase–rutile mixture in commercial P90 and P25 TiO2 from Evonik (Germany) serves as an example of a heterojunction. P25, which consists of approximately 80% anatase and 20% rutile, forms a natural heterojunction where the two phases work synergistically to improve charge separation. The energy band alignment between anatase and rutile facilitates electron transfer from the CB of anatase to that of rutile, reducing charge recombination and enhancing overall performance. This mechanism makes P25 one of the most widely used photocatalysts in both academic research and industrial applications. P90, another TiO2-based photocatalyst from Evonik, has a similar anatase–rutile heterojunction structure but is engineered to have different surface properties. In addition to TiO2-based heterojunctions, several commercially available heterojunction photocatalysts, which are not naturally formed like the anatase–rutile junction, have also been implemented in industrial applications. One example is WO3–TiO2 heterojunction photocatalysts used in air and water purification systems. WO3 has a narrow bandgap,44 enabling it to absorb visible light, while TiO2 provides strong oxidative capabilities.45 When coupled together, WO3–TiO2 heterojunctions extend the light absorption range, enhance charge separation, and provide strong oxidation power, making them effective for degrading organic pollutants. These heterojunctions have been integrated into commercial air purifiers and water treatment systems, such as those developed by companies like Panasonic and Toto (Japan).
Another industrially relevant heterojunction photocatalyst is ZnO–TiO2. ZnO has a similar bandgap to TiO2 but exhibits different charge carrier dynamics.46,47 When combined, the ZnO–TiO2 heterojunction enhances photocatalytic efficiency by improving charge transfer and reducing recombination losses. This heterojunction has been used in self-cleaning coatings for building materials, anti-fogging glass, and antibacterial surfaces. Companies producing such coatings include Pilkington (UK-Japan), which has developed self-cleaning glass.
Taken together, a few photocatalysts with naturally occurring heterojunctions have been adopted for industrial and commercial applications, with the anatase–rutile junction in P25 and P90 being among the most widely recognized. Other photocatalysts with engineered heterojunctions, such as WO3–TiO2 and ZnO–TiO2, have been integrated into commercial products for environmental remediation and self-cleaning surfaces. Future advancements in scalable synthesis and stability improvements will further promote the industrial adoption of heterojunction photocatalysts and broaden their applications, including those in renewable energy.
Regarding the potential applications of heterojunction photocatalysts in real settings, as mentioned before, self-cleaning surfaces—pioneered by Toto (Japan)—have already reached the industrial level in environmental applications.48,49 Air treatment follows with a technology readiness level (TRL) of 5, while water treatment falls within TRL 2–5.49 In chemical synthesis applications, the technology is at TRL 2–4. Energy applications remain the most challenging for photocatalytic technology, still at the university research stage (TRL 1–4).
One may ask: which established and emerging photocatalytic technologies are the most viable for simultaneous industrial-scale adoption in both energy and environmental applications? In our opinion, photocatalytic biomass reforming is the most practical option. While solar-driven water splitting for H2 production is the most mature technology for solar-to-fuel conversion, it primarily addresses energy challenges. In contrast, biomass offers a sustainable feedstock for both energy production and the generation of value-added products potentially from waste. Photoreforming enables the recycling of biomass-derived wastes, such as crude glycerol from biodiesel, into clean H2 fuel. This technology potentially operates under ambient conditions, because well-engineered photocatalysts can be active in the visible light portion of solar irradiation. From a fundamental perspective, photoreforming biomass to produce H2 is thermodynamically less challenging since it does not require oxidizing water into O2. Technically, therefore, it eliminates the need for expensive membrane separation to handle explosive H2–O2 mixtures. With the push toward net-zero emissions, this approach becomes even more attractive due to its ability to harness renewable energy, such as sunlight, while utilizing diverse biomass resources.49 The H2 produced can serve as a feedstock for fuels such as methanol or ammonia or integrate with mild catalytic processes such as non-thermal plasma CO2 hydrogenation and reverse water-gas shift. If H2 production and consumption rates align, the renewable H2 can be immediately used for electricity generation or the synthesis of key chemicals like CH4 and CO.49 Efficient irradiation—primarily from solar energy, supplemented by renewable-powered light-emitting diodes (LEDs)—maximizes photon absorption. On the other hand, immobilizing particulate photocatalysts simplify downstream processing, although they face mass transfer limitations, particularly due to the low solubility of biomass feedstocks.
For efficient biomass conversion, the catalyst should achieve an H2 yield in the millimole range with a photonic efficiency of >50%.49 A solar-to-hydrogen efficiency of 5–10% should be achieved for economic viability. Developing modular photocatalytic units, similar to electrolyzers and fuel cells, is important for scalable deployment, with pilot-scale capacities ranging from 1 to 5 m3. Irradiation research should move beyond lab-standard Xe arc lamps and adopt a dual approach: optimizing solar irradiation by engineering systems for efficient photon distribution and developing LED systems powered by low-carbon renewable energy, such as wind. Even with LED lamps, economic feasibility remains a major challenge. For example, Ortiz and co-workers recently conducted a life cycle assessment of H2 production through waste photoreforming, demonstrating its potential for efficiently recovering H2 from biomass-derived wastes.50 Their findings suggest that achieving H2 production rates above 1 × 10−3 kg h−1 could ensure the sustainability of biomass photoreforming. However, electricity consumption was found to be a key concern, particularly for LED-driven photoreforming. In contrast, sunlight-based photoreforming has been identified as the most sustainable approach. Indeed, photocatalytic reforming faces inherent challenges, particularly the relatively modest H2 production rates compared to the substantial energy consumption of the lamps.
Regarding the choice of photocatalyst materials, we suggest TiO2, such as P25 and P90, as a viable option. They can be modified with Cu-based species, forming Cu–TiO2 heterostructures. Based on our experiences, such heterostructures can stably produce H2 in aqueous alcohol solutions, even after being stored for over five years. This stability is due to the well-preserved chemical composition, oxidation states, and atomic-scale structure of the Cu modifiers. The Cu modifier can be in the form of metallic nanoparticles to form a plasmonic system, partially oxidized Cu as amorphous nanoclusters, or single atoms. These heterostructured photocatalysts meet key criteria for large-scale industrial applications—they are cost-effective, easy to prepare at scale, and highly stable.
One bottleneck is the long-term operational effectiveness of photocatalytic biomass reforming in real industrial environments. The operational effectiveness depends on several factors, including catalyst stability, reactor design, and feedstock variability.49,51,52 While this technology offers a sustainable pathway for H2 production, maintaining consistent efficiency presents significant challenges. Catalyst deactivation, often caused by biofouling, accumulation of carbonaceous by-products, and metal leaching, can severely hinder long-term performance.49,53 Additionally, the recalcitrant nature of lignocellulosic biomass limits the interaction between reactants and the catalyst, necessitating costly pre-treatment steps.52 Light penetration is another challenge, as biomass particles can scatter and absorb photons, reducing the efficiency of photocatalytic reactions.51 Reactor engineering solutions, such as improved mass transfer and immobilized catalyst supports, are important for scalability.51,53 Despite these hurdles, advancements in visible-light-active photocatalysts and hybrid renewable energy-powered photoreactors could enhance long-term viability.49 Indeed, economic feasibility remains a key concern, requiring further research to optimize system efficiency and cost-effectiveness.
All things considered, we identify two primary issues that future research should focus on. First, from a fundamental perspective, it is essential to systematically investigate the migration pathways of photoexcited charge carriers. While it has been demonstrated that these charge carriers can be spatially separated, direct evidence of their migration pathways at the heterojunction interface is still lacking. In situ techniques with operando mode may be useful for monitoring the time-dependent formation of heterostructures and observing how reactants transform into intermediates prior to product formation. Secondly, we need to develop efficient and cost-effective methods for producing heterojunction photocatalysts that can achieve the kilogram- or even ton-scale yields required by industry. Otherwise, we should focus on specialty chemicals, which are very valuable and are produced in much smaller quantities. In fact, the solar-to-fuel conversion efficiency in photocatalytic systems should be significantly improved, from the current ∼1% to over 10%, to enable scalability and have a practical impact.10 These numbers refer to H2 production through water splitting.
The growing number of articles in the literature should not be overlooked when identifying promising photocatalytic materials for prioritization in research. For this purpose, standardized metrics are needed to benchmark performances across different photocatalysts and photocatalytic reaction systems. If the focus of biomass photoreforming is on H2 as an energy carrier, an energy balance approach is required for determining system efficiency.49 In simple terms, if more energy is input than produced by H2, the process is unlikely to be viable in future energy systems. For larger-scale photocatalytic systems, at least three factors must be considered: life cycle primary energy balance, energy return on energy invested, and energy payback time.54
Also, for a more efficient catalyst design, the role of artificial intelligence (AI)55 and machine learning (ML)56 should not be overlooked. These approaches are revolutionizing the discovery and optimization of efficient catalysts by analyzing large datasets to identify patterns and correlations that enhance material properties and performance. AI and ML enable data-driven material design, allowing for the prediction of optimal material combinations, the optimization of synthesis parameters, and the modeling of catalytic performance prior to experimental validation. As well, AI-driven automation facilitates high-throughput screening,57 while in situ and operando characterizations help to elucidate mechanisms58 and enable informed adjustments.
4. Applications for various redox reactions
There are various semiconducting materials that can be coupled together to form heterojunction photocatalysts with different configurations. Our perspective article here cannot cover all of these heterostructured photocatalysts; therefore, we focus on the photocatalytic material that, in our view, is the most promising for photocatalytic redox reactions—carbon nitride (CNx). CNx is considered a leading candidate for practical applications, alongside TiO2 for environmental applications and SrTiO3 for energy applications.24 In addition to being composed of earth-abundant elements, the CNx precursors (e.g., urea, thiourea, melamine, cyanamide, dicyandiamide, and guanidine compounds) are inexpensive, and CNx exhibits good chemical- and photo-stability. Currently, CNx remains mostly at the academic level, but recently, some companies—such as American Elements (USA) and Tokyo Chemical Industry (Japan)—have commercialized it. Its modification with single-atom catalysts, such as Fe and Co, has even been commercialized by MSE Supplies (USA).In general, CNx can be coupled with either a non-light-absorbing co-catalyst or a light-absorbing semiconductor to enhance charge carrier dynamics within the system (Fig. 6A).24,59 The efficiency of photocatalytic junctions is primarily determined by charge transfer across interfaces. An energy offset or band bending at the interface can facilitate charge separation and improve charge yield. By confining electrons and holes in distinct components of a single particulate photocatalyst, the rate of distance-dependent charge recombination can be reduced, thereby extending charge carrier lifetimes.59 If the redox potential of these long-lived charge carriers is sufficiently greater than that of the target reactions, the desired products can be generated efficiently, leading to high productivity. Otherwise, charge carriers with extended lifetimes remain ineffective. Incorporating an additional material to enhance light absorption or catalytic activity typically necessitates charge transfer across the interface to establish a connection between the two materials.
 | ||
| Fig. 6 (A) Schematic of representative heterojunctions with interfacial charge transfer shown by arrows: (a) CNx, (b) CNx coupled with a non-light-absorbing cocatalyst, (c) CNx paired with a semiconductor that absorbs at longer wavelengths, where charge transfer depends on band alignment and interfacial electric fields, and (d) electrons and holes from the two materials recombine near the interface, forming a Z-scheme. Reprinted with permission from ref. 24. Copyright 2021, Royal Society of Chemistry. (B) Band edge potential levels of well-known photocatalysts suitable for reduction and/or oxidation reactions. Reprinted with permission from ref. 60. Copyright 2020, Cell Press. | ||
One important thermodynamic feature of CNx is its suitability for both reduction and oxidation reactions. The band edge potentials of one variant of CNx, specifically C3N4, are suited for many important reactions (Fig. 6B), including the thermodynamically challenging overall water splitting, which produces H2–O2 bubbles. It has a highly negative CB potential (greater than −1 eV vs. NHE at pH 7) and a highly positive VB potential (greater than 1 eV vs. NHE). When aiming to enhance oxidation capability, CNx can be coupled with oxidation photocatalysts such as WO3 through an S-scheme configuration to preserve the reduction potential of its excited electrons.
Table 1 summarizes the performance of CNx-based materials in various photocatalytic applications. Comparing performance is challenging, as even for the same target reaction, experimental conditions—such as reactor configuration and the presence of additives—can vary considerably. To enable performance comparison and benchmarking, apparent quantum yield (AQY) or apparent quantum efficiency (AQE) may be used.61,62 These quantitative metrics are useful to evaluate the efficiency of H2 generation. Although the terms are often used interchangeably, they may have distinctions depending on the context. AQY represents the fraction of incident photons that contribute to H2 yield. It is typically expressed as a percentage and calculated as the ratio of evolved H2 molecules to incident photons at a specific wavelength. This metric helps assess the light utilization efficiency of a photocatalyst under given experimental conditions. However, since AQY considers only incident photons rather than absorbed ones, it does not fully account for the material's intrinsic light-harvesting capability.
| Photocatalyst | Heterojunction type | Bandgap, eV | Photocatalyst dose | Synthesis method | Light source | Target reaction | Rate constant |
|---|---|---|---|---|---|---|---|
| a NA denotes not available. b TEOA refers to triethanolamine, which is typically used as an electron donor to consume holes in the H2 evolution reaction. | |||||||
| NixP/Mn3O4/C3N4/RP | Type I | 1.96 | 5 mg | Photochemical deposition | 300 W Xe lamp, AM 1.5G filter | H2 production in 10 mL of 20 vol% TEOAb | 5851.3 mmol g−1 h−1 |
| P-doped C3N4/O-doped C3N4 | Type I | NAa | 50 mg | Solid state | 300 W Xe lamp, 420 nm filter | H2O2 production in EtOH (5 mL) + water (45 mL) | 179 μM h−1 |
| NiS/ZnIn2S4/C3N4 | Type I | NA | 100 mg | Solid state, solvothermal | 300 W Xe lamp | H2 generation in 100 mL (80 mL H2O and 20 mL TEOA) | 5.02 mmol g−1 h−1 |
| BP quantum dots/S doped C3N4 | Type I | NA | 100 mg | Solid state with ultrasound | 300 W Xe lamp, 420 nm filter | H2 generation in 100 mL with 10 vol% TEOA | 102 μmol h−1 |
| C3N4 quantum dots@SnS2 | Type I | 2.14 | 50 mg | Solid state, hydrothermal | 500 W Xe lamp | Bisphenol A degradation in 50 mL solution (10−5 M) | 0.472 h−1 |
| O-doped C3N4/red P | Type I | NA | 10 mg | Solid-state, mechanical grinding | 300 W Xe lamp | Malachite green in 50 mL solution (20 mg L−1) | 0.116 min−1 |
| CdIn2S4/C3N4 | Type I | 2.4 | NA | Solid-state, hydrothermal, wet impregnation | 300 W Xe lamp, 420 nm cutoff filter | Reactive blue 19 degradation in 250 mL (20 mg L−1) | 0.07357 min−1 |
| (2D/3D) metal-free heterostructures based on C3N3 linkers | Type I | 1.91 | 10 mg | Copper plate-based wet chemical method | 300 W Xe lamp, 395 nm cutoff filter | H2 production in 18 mL of water–acetonitrile mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with 2 mL TEOA :1) with 2 mL TEOA |
34 μmol h−1 g−1 |
| C3N4/Bi2MoO6 | Type II | NA | 50 mg | Ball-milling and corona poling post-treatment | 300 W xenon lamp, λ > 420 nm | Tetracycline degradation in 50 mL (20 mg L−1) | 0.0045 min−1 |
| Nb2O5/C3N4 | Type II | 2.64 | 30 mg | Multistep synthesis | 300 W Xe lamp | H2 generation in 80 mL of 10 vol% TEOA + H2PtCl6 | 2.07 mmol g−1 h−1 |
| h-BN/flower-ring C3N4 | Type II | NA | 20 mg | Thermal polymerization | 300 W Xe lamp, 420 nm cut-off filter | Tetracycline degradation in 100 mL with a concentration of 20 mg L−1 | 0.0703 min−1 |
| Bi@H-TiO2/B-C3N4 | Type II | NA | 20 mg | Solvothermal, thermal polymerization | 300 W Xe lamp, λ > 400 nm | H2 generation in 30 mL with 20 vol% TEOA | 18.84 μmol g−1 h−1 |
| C3N4/MoO3−x | Z-scheme | NA | 0.05 g | Hydrothermal, two-step calcination | 300 W Xe lamp | H2 generation in 100 mL of 10 vol% TEOA | 209.2 μmol h−1 |
| O-doped C3N4/Bi2O3 | Z-scheme | NA | 0.05 g | Hydrothermal, solid state | 300 W Xe lamp, UV cut-off filter | Tetracycline degradation in 100 mL solution (10 mg L−1) | 0.07 min−1 |
| Carbon dot decorated C3N4/TiO2 | Z-scheme | 2.70 | 50 mg | Burning, hydrothermal | Four LED lamps (3 W, λ 420 nm) | H2 generation in 80 mL of 10 vol% TEOA + H2PtCl6 | 580 μmol h−1 g−1 |
| Ag/WO3/C3N4 | Z-scheme | NA | 40 mg L−1 | Multistep synthesis | 500 W Xe lamp, 420–800 nm | Oxytetracycline degradation (10 mg L−1) | 0.1164 min−1 |
| TiO2@C/C3N4 | Z-scheme | 2.69 | 0.2 g | One step calcination | 500 W Xe lamp, 420 nm filter | NO removal on 100 ppm, 8% N2/air balance, 200 mL min−1 | NA |
| V-H3PMo12O40/C3N4 | Z-scheme | NA | 10 mg | Self-assembly | 300 W Xe lamp, 420 nm cut-off filter | Upcycling of various plastic wastes into formic acid in 10 mL acetonitrile + 20 mg polyethylene | 24.66 μmol g−1 h−1 |
| SnS2/RGO/C3N4 | S-scheme | NA | 10 mg | Photoassisted self-assembly | 300 W Xe lamp, filter 420–780 nm | Rhodamine B degradation in 50 mL (10 mg L−1) | 0.55 min−1 |
| O-doped C3N4/N-doped Nb2O5 | S-scheme | 2.81 | NA | One-step polymerization, hydrothermal, aggregation | 300 W Xe lamp | CO2 reduction with 0.12 g of NaHCO3, 0.35 M HCl | CO: 253.34 μmol g−1 h−1 CH4: 68.11 μmol g−1 h−1 |
A more comprehensive and standardized metric for evaluating the overall performance of a photocatalytic system for water splitting is solar-to-hydrogen (STH) efficiency.61 STH efficiency represents the percentage of incident solar energy converted into chemical energy stored in H2. Unlike AQY, which is measured under monochromatic illumination, STH efficiency accounts for the full solar spectrum and is typically calculated under simulated sunlight (e.g., AM 1.5G conditions). The calculation of STH efficiency is based on the measured H2 generation rate, the enthalpy of water splitting (1.23 V per electron transfer), and the total incident solar power. Since STH directly correlates with practical applications, it serves as a benchmark for comparing different photocatalysts. These standardized performance metrics—AQY/AQE and STH—represent the effectiveness of photocatalysts under both controlled laboratory conditions and real-setting solar energy conversion scenarios.
For other redox reactions, such as selective chemical transformations, reaction productivity and product selectivity serve as key performance metrics.64,65 Reaction productivity refers to the efficiency of a chemical reaction in generating the desired product over a given period and under specific conditions. It is often expressed in terms of the amount of product formed per unit time (e.g., moles per hour) or per unit of catalyst (e.g., turnover number or turnover frequency). High productivity indicates that the reaction system efficiently converts reactants into products, making it an essential metric for assessing the practical applicability of a catalytic process. Factors influencing reaction productivity include reaction kinetics, catalyst activity, reactant concentration, temperature, and reaction conditions. In photocatalytic reactions, productivity can also depend on light intensity and photon absorption efficiency.65 Product selectivity, on the other hand, measures how effectively a reaction pathway favors the formation of a specific desired product over undesired side products.64 It is expressed as a percentage, calculated as the ratio of the desired product formed to the total products generated. High selectivity is crucial in chemical transformations, especially when multiple reaction pathways exist, as it minimizes waste, reduces purification costs, and improves process efficiency. Selectivity is particularly important in photocatalytic and electrocatalytic reactions, where controlling electron transfer and reaction intermediates significantly impacts the final product distribution. Both reaction productivity and product selectivity are important to evaluate the efficiency of photocatalytic processes. While productivity determines the overall output and feasibility of a reaction system, selectivity ensures that the process is economically and environmentally viable by maximizing the yield of valuable products while minimizing unwanted byproducts.
For determining their feasibility in real-setting applications such as H2 production, CO2 reduction, and wastewater treatment, we need to assess the environmental impact of heterojunction photocatalysts. While photocatalysis is often regarded as a sustainable technology, its overall environmental benefit depends on factors such as material sourcing, synthesis methods, energy consumption, and waste generation. A photocatalyst with excellent lab-scale performance may still have a significant environmental footprint if its production involves high energy input, toxic chemicals, or scarce materials. Evaluating these impacts ensures that the development of new photocatalysts aligns with sustainability goals, minimizing unintended environmental consequences.
For large-scale deployment, a photocatalyst must not only be efficient but also environmentally responsible throughout its life cycle. Life cycle assessment (LCA) helps quantify emissions, energy use, and resource depletion at different stages, from raw material extraction to disposal or recycling.66 For example, photocatalysts that require high-temperature calcination or rare metals may have a higher environmental burden compared to those made from earth-abundant and recyclable materials. By integrating LCA into research and development, we can identify greener alternatives, optimize low-energy synthesis methods, and improve the recyclability of photocatalysts, ensuring their long-term viability for industrial applications.
A comprehensive environmental evaluation also strengthens the economic and regulatory acceptance of heterojunction photocatalysts in real settings. Industries and policymakers are increasingly prioritizing technologies that align with carbon neutrality goals and circular economy principles. Photocatalysts that demonstrate low environmental impact, high durability, and minimal waste generation are more likely to receive funding, regulatory approval, and commercial adoption. Therefore, assessing the environmental impact is not just a scientific necessity but a critical step toward scaling up sustainable photocatalytic technologies.
For example, Kumar and co-workers developed a cradle-to-grave LCA framework to assess the greenhouse gas (GHG) footprints of four heterojunction photocatalysts, such as C3N4/BiOI.67 Unlike most studies focused on lab-scale fabrication, this study includes utility-scale production, assembly, operation, and end-of-life for a more precise environmental assessment. Results show C3N4/BiOI has the lowest GHG footprint (0.38 kg CO2 eq./kg H2), while CNF: TNR/TiO2 (F-doped C3N4 quantum dots embedded with TiO2) achieves the shortest energy payback time (0.4 years). Across all pathways, material extraction accounts for 83–89% of total GHG emissions. Sensitivity and uncertainty analyses confirm photocatalytic water splitting as a promising mainstream H2 production pathway.
5. Outlook
Among the various heterojunction alignment schemes, the type-II heterojunction is the most widely validated.68,69 In this scheme, the CBM of one semiconductor is positioned at a lower energy level than that of the other, while the VBM of the same semiconductor is at a higher energy level. This configuration facilitates spatial charge separation, as electrons migrate to the less-negative CBM, while holes move to the less-positive VBM, reducing charge recombination and enhancing photocatalytic efficiency. The validity of type-II heterojunctions is supported by numerous experimental techniques, including TAS, photoluminescence (PL) quenching, and KPFM, which directly confirm charge transfer at the heterojunction interface. Theoretical studies using DFT also consistently validate this mechanism, making it a well-established concept. Despite its effectiveness, the major limitation of type-II heterojunctions is the reduction in redox potential, which may lower catalytic activity for certain reactions.To address this issue, alternative configurations such as Z-scheme and S-scheme heterojunctions have been proposed.60,70–72 These designs aim to retain strong redox capabilities while still benefiting from effective charge separation. The Z-scheme mimics natural photosynthesis by facilitating the recombination of specific charge carriers, thereby maintaining high redox potential.71,73 The electron-transfer mechanisms in type-II heterojunctions, liquid-phase Z-scheme systems, and all-solid-state Z-scheme heterojunctions were later found inappropriate to explain the photocatalytic behavior of certain heterojunctions,73 leading to the development of the novel S-scheme heterojunction concept. While S-scheme and traditional type-II heterojunctions share similar structures, where the two photocatalysts are in close contact, their electron-transfer mechanisms are vastly different.
As it is still in its infancy, the S-scheme heterojunction remains one of the most debated alignment schemes in photocatalysis. Unlike the type-II heterojunction, which facilitates bidirectional charge transfer, the S-scheme proposes a selective recombination mechanism where low-energy electrons and holes annihilate each other, while high-energy charge carriers are preserved for redox reactions.60,74 While this concept theoretically improves both charge separation and redox potential, there is a lack of direct experimental validation for the charge transfer mechanism. Most studies rely on indirect measurements, such as band structure calculations and electrochemical characterizations, to infer S-scheme behavior. Also, the distinction between S-scheme and Z-scheme heterojunctions remains ambiguous, as both involve recombination at the heterojunction interface,73,74 leading to conflicting interpretations. We argue that many reported S-scheme heterojunctions may, in fact, function similarly to Z-scheme systems, further complicating their classification. While the S-scheme shows promise in enhancing photocatalytic performance,70 further studies are required to fully validate its mechanism.
Since the type-II heterojunction has been experimentally validated as the most convincing scheme,68 it is reasonable to assume that, while the redox potentials of the CBM and VBM are important, they are not the key factors determining photocatalytic behavior in type-II heterojunction photocatalysts. This is because, when enhanced photocatalytic activity is observed, the redox potential in this type of photocatalyst decreases. Thus, the redox power after heterojunction formation is inversely proportional to photocatalytic activity. We believe that the primary factor contributing to the enhanced photocatalytic activity of heterojunction photocatalysts, especially those with the type-II scheme, is the efficient separation of electrons and holes. This separation prolongs their lifetime, making them more readily available for redox reactions on the surface. When the thermodynamic requirements of the target reaction have been fulfilled (qualitative aspect), the progress of the reaction will primarily depend on the population of charge carriers at the surface (quantitative aspect). A photocatalyst may generate excited charge carriers with strong redox power; however, if the quantity of these charge carriers is insufficient, there will not be enough driving force to initiate the reaction.
On the whole, theoretical studies and experimental validations generally support the idea that constructing heterojunctions by coupling two or more semiconductors is an efficient method for enhancing photocatalytic activity by facilitating electron–hole pair separation. The built-in electric field and potential barriers at the heterojunction interface play important roles in directing carrier movement, thereby preventing recombination losses. Carrier transfer behavior is influenced by several factors, including the type of semiconductors (n-type or p-type), their work functions (or Fermi levels), and their band edge positions. Different types of heterojunctions exist which determine which charge transfer mechanism the system follows. Unfortunately, we often overlook the formation of interface charge regions and built-in electric fields in our discussions of photocatalytic mechanisms, leading to an incomplete understanding of carrier transfer processes. Experimental techniques such as STEM, EELS, UPS, DRS, AET, Mott–Schottky analysis, and transient spectroscopies are crucial for determining semiconductor band structures and charge transport properties. Equally important, factors such as work function differences, defect states, and potential barriers must be considered to evaluate how efficiently a heterojunction can separate charge carriers. These insights help determine whether a particular heterojunction design will enhance photocatalytic activity by promoting charge separation while maintaining strong redox potential. To design more efficient photocatalysts, optimizing band structure alignment, carrier mobility, and interfacial charge transfer efficiency is crucial. S-scheme heterojunctions retain high redox potential by allowing selective recombination of less reactive charge carriers. Meanwhile, type-II heterojunctions improve charge separation but may suffer from reduced redox power, requiring additional modifications such as surface engineering, doping, or co-catalyst loading to enhance performance. Defect engineering can also introduce mid-gap states that extend light absorption and improve carrier lifetime. By systematically tailoring these properties, heterojunction photocatalysts can achieve higher efficiency.
Equally important, a heterojunction photocatalyst can even be prepared from waste materials. For instance, Xie and co-workers developed a feasible method by using recycled graphite target materials to fabricate a heterostructured photocatalyst with ZnO.75 Graphite target waste was successfully recycled into 2D few-layer graphite, serving as raw material for the subsequent synthesis of heterojunction photocatalysts. The resulting photocatalyst exhibits a 19% enhancement in photocatalytic efficiency and good photostability in sunlight-driven degradation of organic pollutants.
5.1. What is the most promising heterojunction?
Up to this point, one might ask: what is the most promising heterojunction, and which heterojunction photocatalyst should be the focus of further research? Answering the question of the best heterojunction configuration is not straightforward, as it depends on the constituents forming the heterojunction. Each target reaction also requires specific photocatalyst properties, depending on the thermodynamic and kinetic nature of the reaction. Factors such as redox potential, bandgap, and other specific characteristics of the photocatalyst play a crucial role in determining its suitability. We tend to suggest the S-scheme heterojunction as the optimal choice when considering charge transfer mechanisms.To understand the reasoning behind this preference, let us first summarize and compare the different heterojunction types based on their charge transfer mechanisms.63 Type I heterojunctions enhance charge separation, with charge density being higher in the component possessing a narrower band gap. However, this comes at the cost of reduced redox potential. Type II heterojunctions also improve charge separation and increase light absorption, leading to a more balanced charge distribution between both components, though this similarly compromises redox potential. In a Z-scheme heterojunction, charge separation efficiency is improved alongside enhanced light absorption while maintaining strong redox potential. Nevertheless, charge recombination between the higher CB and the lower VB is not entirely eliminated. S-scheme heterojunctions promote efficient charge separation and improved light absorption while retaining the strong redox potential of electrons and holes. In addition, they introduce an internal electric field at the interface, facilitating recombination of charges with lower redox capability. There are also less commonly known heterojunction configurations, such as type III and type B heterojunctions.63 Type III heterojunctions often suffer from inefficient or nonexistent charge separation due to their broken-gap band alignment. This drawback can be effectively mitigated by introducing a conductive component, which facilitates hot charge transfer and consequently converts the system into a type B heterojunction.
Overall, from our perspective, the S-scheme heterojunction is the best heterojunction configuration, particularly in terms of thermodynamic properties, due to its enhanced redox capability. Unlike type-II heterojunctions, where charge transfer is often inefficient due to repulsion between like charges, the S-scheme mechanism ensures that only the charge carriers with strong redox potential are retained while those with weaker energy are recombined and eliminated. This selective charge separation enhances photocatalytic efficiency by maintaining a strong driving force for redox reactions, overcoming the limitations of type-II heterojunctions, which often suffer from reduced redox power despite improved charge separation.
Kinetically, however, type II alignment is advantageous due to its strong driving force for charge separation.24 Indeed, determining whether type II or S-scheme is truly superior remains challenging. A comprehensive understanding of the heterointerface must account for factors like interfacial reconstruction and chemical interactions. Charge carrier dynamics can be examined through optical or electrical techniques in both steady-state and time-resolved manners.
In comparison to traditional Z-scheme heterojunctions, which rely on redox couples that introduce instability and undesired side reactions,60 the S-scheme avoids such drawbacks by directly facilitating electron transfer between semiconductors with staggered band structures. All-solid-state Z-scheme heterojunctions attempt to bypass redox couples by using conductive bridges, yet they still suffer from inefficient charge transfer due to unintended electron migration pathways. The S-scheme mechanism effectively mitigates these challenges by promoting a stepwise charge transfer, ensuring high efficiency and stability while preventing charge recombination.
Equally important, the S-scheme heterojunction offers practical advantages in photocatalysis, particularly in pollutant degradation and solar energy conversion. By utilizing an internal electric field and band bending, it promotes spatial charge separation, thereby boosting photocatalytic activity. The recombination of weaker charge carriers further minimizes energy losses, allowing for improved performance in applications such as water splitting and CO2 reduction.60 The combination of extended light absorption, enhanced charge separation, and strong redox potential makes the S-scheme heterojunction a more efficient and reliable option compared to other heterojunction configurations.
5.2. Feasible research directions for future studies
The role of heterojunction effects in photocatalytic activity remains a rapidly evolving area of research with potential for enhancing photocatalytic efficiency in energy conversion and environmental applications. Despite considerable advancements, several challenges persist, including carrier recombination, limited redox potential, material stability, and scalability.34 To address these issues, future research should focus on optimizing charge transfer mechanisms, developing novel heterojunction architectures, improving synthesis methods, utilizing computational modeling, expanding functional applications, and enhancing material stability.One of the most crucial aspects in improving heterojunction-based photocatalysts is optimizing charge transfer and separation pathways. A fundamental limitation in photocatalysis is the rapid recombination of photogenerated electron–hole pairs,34 which substantially reduces efficiency. Future research should employ advanced spectroscopic techniques, such as ultrafast transient absorption microscopy and time-resolved photoluminescence, to directly observe electron migration pathways at heterojunction interfaces. Investigating the built-in electric fields and potential barriers at heterojunction junctions could lead to more efficient charge separation.7 A deeper understanding of band structure engineering can help in designing heterojunctions with favorable energy alignments, ensuring efficient charge carrier dynamics.
Another feasible direction for further research is the development of new heterojunction architecture. Traditional heterojunctions, such as type II, have inherent trade-offs between charge separation efficiency and redox potential.34 S-scheme (or direct Z-scheme) heterojunctions have emerged as efficient alternatives, as they preserve strong redox potential while facilitating directional charge transfer and minimizing recombination losses.60 Note that the S-scheme and direct Z-scheme are essentially similar, except for the mode of Fermi level alteration after junction formation.76 In a direct Z-scheme heterojunction, the Fermi levels of the two semiconductors equilibrate upon contact, allowing photogenerated electrons from the semiconductor with a higher Fermi level to recombine with holes from the semiconductor with a lower Fermi level, thereby preserving strong redox potential. An S-scheme heterojunction also undergoes Fermi level equilibration but exhibits band bending at the interface, which facilitates selective charge transfer and enhances charge separation. Research should focus on designing novel semiconductor combinations with optimized band structures to enable more effective charge separation. van der Waals heterojunctions using 2D materials, such as MXenes and graphene, offer the potential for enhanced interlayer electron transfer.63 Exploring new material combinations and heterojunction morphologies will be crucial in achieving higher efficiencies.
For real-setting applications, the synthesis of heterojunction photocatalysts must be cost-effective and scalable. Many current synthesis methods, such as hydrothermal and solvothermal processes, face challenges in large-scale production. Future research should prioritize the development of simple, environmentally friendly, and economically viable synthesis techniques. Template-assisted fabrication, atomic layer deposition, and solution-processed methods may provide feasible alternatives for scalable production. Engineering hierarchical structures with well-controlled crystallization and morphology further enhances the performance by increasing surface area and optimizing light absorption properties. By refining synthesis techniques, one can bridge the gap between laboratory-scale developments and industrial applications.
Computational modeling and theoretical studies will play a critical role in guiding the design of heterojunction photocatalysts. DFT calculations and molecular dynamics simulations can provide insight into charge carrier behavior, band alignment, and interface interactions. Future research should focus on developing more accurate theoretical models to predict electron–hole dynamics and optimize material selection. Meanwhile, ML and AI techniques can be employed to analyze large datasets, identify promising material combinations, and accelerate the discovery of new heterojunction configurations.
Expanding the functional applications of heterojunction photocatalysts is another important research avenue. While heterojunction photocatalysts have been widely studied for pollutant degradation, H2 evolution, and CO2 reduction, their potential in other fields remains underexplored. Research should investigate their applications in organic transformations. Tailoring heterojunction properties to meet specific application requirements—such as improved selectivity, durability, and efficiency under visible light—will broaden their practical use. Also, the development of multi-functional photocatalysts that can perform multiple reactions, such as water splitting with CO2 reduction, or H2O2 production coupled with the selective oxidation, would represent a significant advancement.
Equally important, enhancing the stability and photostability of heterojunction photocatalysts is also essential for their long-term practical applications. Many heterojunction materials suffer from photocorrosion, structural degradation, or instability under continuous operation. Thus, future studies should focus on strategies to mitigate these issues. Moreover, the exploration of self-healing materials and protective coatings may improve the longevity of heterojunction-based systems. By developing stable and durable photocatalysts, one can ensure that these materials remain viable for practical applications over extended periods.
5.3. Economic feasibility and scalability for industrial applications
The large-scale implementation of heterojunction photocatalysts in industrial applications hinges on their economic feasibility and scalability, yet these aspects have not been comprehensively addressed in the literature. While significant research has been devoted to optimizing charge separation and transfer processes, the transition from laboratory-scale studies to industrial deployment remains a major challenge. One of the fundamental concerns is the cost of raw materials and synthesis methods. Although some heterojunction photocatalysts rely on abundant semiconductors such as TiO2, C3N4, and ZnO, others incorporate expensive noble metals or rare-earth elements, driving up production costs. Replacing costly materials with earth-abundant alternatives, such as transition metal sulfides, phosphides, and carbides, which can provide comparable catalytic performance at a lower cost, is necessary.67,77,78 Furthermore, the development of low-cost synthesis methods, including mechanochemical synthesis and molten salt techniques, has shown promise in reducing the energy and material costs associated with heterojunction fabrication.Scalability, indeed, remains a bottleneck in the commercialization of heterojunction photocatalysts.79,80 Conventional synthesis methods, such as hydrothermal and sol–gel processes, are difficult to adapt to large-scale production due to batch-processing limitations, prolonged reaction times, and high energy input. Continuous flow synthesis and flame spray pyrolysis seem promising alternatives for large-scale production, offering improved control over particle morphology and crystallinity. However, the challenge of maintaining catalyst uniformity during scale-up persists. Variations in temperature gradients, reactant diffusion, and precursor concentrations can lead to inconsistencies in catalyst properties, impacting photocatalytic performance. Advanced characterization techniques, such as in situ spectroscopy81,82 and artificial intelligence-driven process optimization,57,83 are being explored to optimize large-scale catalyst synthesis. Ensuring reproducibility across different production batches is essential for industrial adoption, necessitating the development of standardized synthesis protocols.
From an economic perspective, the cost-effectiveness of heterojunction photocatalysts is contingent on their long-term operational stability and efficiency. Industrial photocatalysis applications, such as wastewater treatment and H2 production, require catalysts that can sustain high performance over extended periods without significant degradation. However, photocatalyst deactivation due to surface fouling, photochemical corrosion, and structural instability remains a major concern. Incorporation of protective surface coatings, such as SiO2 and Al2O3, can potentially enhance the durability of photocatalysts by preventing oxidation and leaching.84 Despite these advances, systematic long-term stability studies under real-setting operating conditions are still lacking.
In assessing the feasibility of heterojunction photocatalysts, techno-economic analysis plays a crucial role in evaluating capital investment, operational costs, and return on investment.85 For example, when comparing photocatalytic water splitting with other H2 production methods, such as electrolysis and thermochemical processes, current photocatalysts likely still fall short in terms of STH efficiency. For practical deployment, an STH efficiency of at least 10% is required to compete with conventional H2 production technologies,86,87 yet most heterojunction photocatalysts achieve efficiencies below this threshold. Moreover, artificial illumination using conventional Xe lamp systems, while effective in controlled environments, increases operational costs. Hybrid reactor designs integrating solar-driven photocatalysis with renewable-powered LED systems could be a potential solution.
Reactor design and process integration are equally important for scaling up heterojunction photocatalysts. Photocatalytic reactors must maximize light absorption while ensuring efficient mass transfer. Common reactor configurations include slurry reactors, fixed-bed reactors, and immobilized photocatalyst systems.88,89 While slurry reactors offer high reaction rates, they present challenges in catalyst recovery and reusability.89 Immobilized systems provide better stability but suffer from lower active surface area. Hierarchical nanostructured photocatalysts, combined with improved reactor designs, potentially enhance both performance and durability. The integration of heterojunction photocatalysts into existing industrial processes, such as CO2 conversion and wastewater treatment, also requires compatibility with existing infrastructure. Hybrid systems,90,91 such as the photocatalytic materials–microbial hybrid system,90 which combines the strengths of photocatalytic materials and microorganisms, have demonstrated improved efficiencies in experimental setups. However, their large-scale implementation remains questionable.
The environmental impact of heterojunction photocatalysts is another critical factor in their industrial viability. While photocatalysis is often promoted as a sustainable technology, the overall environmental benefit depends on factors such as material sourcing, synthesis energy consumption, and end-of-life disposal. LCA studies have revealed that some photocatalysts, particularly those containing toxic heavy metals such as Cd and Pb, pose environmental risks.67,92,93 Research is increasingly focused on developing non-toxic, recyclable photocatalysts using biodegradable or earth-abundant materials.94 As well, regulatory frameworks must be established to ensure the safe disposal and recycling of spent photocatalysts, particularly in large-scale applications. Reducing the carbon footprint of photocatalyst production by optimizing low-energy synthesis methods and minimizing hazardous waste generation will be key to achieving true sustainability.
Future advancements in the commercialization of heterojunction photocatalysts will require interdisciplinary collaboration between material scientists, chemical engineers, and industry stakeholders. The application of AI and ML in catalyst design and optimization has shown great potential in accelerating material discovery and performance prediction. High-throughput screening methods, coupled with computational modeling, can significantly reduce the time and cost associated with developing new photocatalyst formulations. In addition, pilot-scale demonstration projects are essential to validate laboratory findings and assess real-setting feasibility. To bridge the gap between research and commercialization, industries must invest in scaling up production, optimizing reactor designs, and integrating photocatalysis into existing industrial processes. By addressing these economic and scalability challenges, heterojunction photocatalysts have the potential to become a transformative technology in sustainable energy and environmental applications.
Finally, we should address the central question raised in this perspective: where are heterojunction photocatalysts headed? They are advancing toward enhanced efficiency through band structure engineering, novel material combinations, and improved charge separation strategies to maximize photon energy utilization. However, these efforts remain largely confined to lab-scale experiments. Future research is expected to emphasize scalable synthesis methods, stability improvements, and practical implementations, particularly in energy conversion and the production of value-added chemicals, which often require kilogram-scale production within an economically viable timescale.
6. Summary
Heterojunction photocatalysts are designed to extend light absorption, enhance charge separation, and optimize redox properties. However, issues such as lattice mismatch at the interface, suboptimal band alignment, and the complexity of charge transfer mechanisms can hinder performance. The stability of these heterojunctions under operational conditions is also a critical concern. Ongoing research is focused on optimizing interface properties, exploring novel material combinations, and employing advanced characterization techniques to better understand and improve the efficiency and durability of heterojunction photocatalysts. Effective heterojunction design requires a comprehensive understanding of charge migration pathways and the factors influencing charge transfer. Research should focus on establishing quantitative relationships with photocatalytic performance, accurately identifying heterojunction types and their modes of charge transfer, and refining design and synthesis methods tailored to specific photocatalytic reactions. Transient spectroscopies and synchrotron-based methods, especially in operando mode, will be particularly useful. On the other hand, the integration of AI and ML offers new opportunities to combine potential materials, predict heterojunction properties, and optimize their designs more efficiently. Strategies should be developed to achieve future large-scale production while maintaining the high selectivity and reaction productivity currently achieved in laboratory settings.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Research Organization for Nanotechnology and Materials of the National Research and Innovation Agency under Grant No. 3/III.10.4/HK/2023.References
- Q. Wang and K. Domen, Chem. Rev., 2019, 120, 919–985 Search PubMed.
- H. Wang, X. Li, X. Zhao, C. Li, X. Song, P. Zhang and P. Huo, Chin. J. Catal., 2022, 43, 178–214 CAS.
- H. Sudrajat and S. Hartuti, Adv. Powder Technol., 2019, 30, 983–991 CAS.
- M. Lin, H. Chen, Z. Zhang and X. Wang, Phys. Chem. Chem. Phys., 2023, 25, 4388–4407 CAS.
- H. Sudrajat, Mater. Res. Express, 2018, 5, 095501 Search PubMed.
- H. Sudrajat, Y. Zhou, T. Sasaki, N. Ichikuni and H. Onishi, Phys. Chem. Chem. Phys., 2019, 21, 5148–5157 CAS.
- H. Yang, Mater. Res. Bull., 2021, 142, 111406 CAS.
- J. Xue and J. Bao, Surf. Interfaces, 2021, 25, 101265 CAS.
- J. Zhang, Y. Lin and L. Liu, Phys. Chem. Chem. Phys., 2023, 25, 7106–7119 CAS.
- R. Yanagi, T. Zhao, D. Solanki, Z. Pan and S. Hu, ACS Energy Lett., 2021, 7, 432–452 CrossRef.
- B. Zhu, J. Sun, Y. Zhao, L. Zhang and J. Yu, Adv. Mater., 2024, 36, 2310600 CAS.
- A. Yamakata, C. S. K. Ranasinghe, N. Hayashi, K. Kato and J. J. M. Vequizo, ACS Appl. Energy Mater., 2019, 3, 1207–1214 Search PubMed.
- S. J. Pennycook, M. F. Chisholm, A. R. Lupini, M. Varela, A. Y. Borisevich, M. P. Oxley, W. Luo, K. van Benthem, S.-H. Oh and D. Sales, Philos. Trans. R. Soc., A, 2009, 367, 3709–3733 CAS.
- I. MacLaren and Q. M. Ramasse, Int. Mater. Rev., 2014, 59, 115–131 CAS.
- Z. Yuan, J. Ruan, L. Xie, X. Pan, D. Wu and P. Wang, Appl. Phys. Lett., 2017, 110, 171602 Search PubMed.
- D. J. Smith, H. Wu, S. Lu, T. Aoki, P. Ponath, K. Fredrickson, M. D. McDaniel, E. Lin, A. B. Posadas and A. A. Demkov, J. Mater. Res., 2017, 32, 912–920 CAS.
- K. W. Urban, Science, 2008, 321, 506–510 CrossRef CAS.
- I. MacLaren, L. Wang, O. Morris, A. J. Craven, R. L. Stamps, B. Schaffer, Q. M. Ramasse, S. Miao, K. Kalantari and I. Sterianou, APL Mater., 2013, 1, 021102 CrossRef.
- X. Tian, X. Yan, G. Varnavides, Y. Yuan, D. S. Kim, C. J. Ciccarino, P. Anikeeva, M.-Y. Li, L.-J. Li and P. Narang, Sci. Adv., 2021, 7, eabi6699 CrossRef CAS PubMed.
- D. Xu, S.-N. Zhang, J.-S. Chen and X.-H. Li, Chem. Rev., 2022, 123, 1–30 Search PubMed.
- J. E. Whitten, Appl. Surf. Sci. Adv., 2023, 13, 100384 Search PubMed.
- H. Li, Y. Ding, K. Luo, Q. Zhang, H. Yuan, S. Xu and M. Xu, iScience, 2025, 28, 111750 CAS.
- Z. Wang, Z. Lin, S. Shen, W. Zhong and S. Cao, Chin. J. Catal., 2021, 42, 710–730 CAS.
- E. Mitchell, A. Law and R. Godin, Chem. Commun., 2021, 57, 1550–1567 CAS.
- K. Meng, J. Zhang, B. Cheng, X. Ren, Z. Xia, F. Xu, L. Zhang and J. Yu, Adv. Mater., 2024, 2406460 CAS.
- S. Mohd and A. M. Khan, Sustainable Green Catalytic Processes, 2024, pp. 141–163 Search PubMed.
- A. S. Morshedy, E. M. El-Fawal, T. Zaki, A. A. El-Zahhar, M. M. Alghamdi and A. M. El Naggar, Inorg. Chem. Commun., 2024, 112307 CAS.
- D. Zu, H. Wei, Z. Lin, X. Bai, M. N. A. S. Ivan, Y. H. Tsang and H. Huang, Adv. Funct. Mater., 2024, 34, 2408213 CAS.
- Z. Zafar, S. Yi, J. Li, C. Li, Y. Zhu, A. Zada, W. Yao, Z. Liu and X. Yue, Energy Environ. Mater., 2022, 5, 68–114 CAS.
- E. Pastor, M. Sachs, S. Selim, J. R. Durrant, A. A. Bakulin and A. Walsh, Nat. Rev. Mater., 2022, 7, 503–521 CrossRef CAS.
- F. Xu, K. Meng, S. Cao, C. Jiang, T. Chen, J. Xu and J. Yu, ACS Catal., 2021, 12, 164–172 CrossRef.
- D. Maarisetty and S. S. Baral, J. Mater. Chem. A, 2020, 8, 18560–18604 RSC.
- X. Yu, S. F. Ng, L. K. Putri, L. L. Tan, A. R. Mohamed and W. J. Ong, Small, 2021, 17, 2006851 CAS.
- J. Low, J. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Adv. Mater., 2017, 29, 1601694 Search PubMed.
- J. Chen, T. Tang, W. Feng, X. Liu, Z. Yin, X. Zhang, J. Chen and S. Cao, ACS Appl. Nano Mater., 2021, 5, 1296–1307 Search PubMed.
- Z. Lin, A. McCreary, N. Briggs, S. Subramanian, K. Zhang, Y. Sun, X. Li, N. J. Borys, H. Yuan and S. K. Fullerton-Shirey, 2D Mater., 2016, 3, 042001 Search PubMed.
- W. Ahmad, A. K. Tareen, K. Khan, M. Khan, Q. Khan, Z. Wang and M. Maqbool, Appl. Mater. Today, 2023, 30, 101717 Search PubMed.
- J. Li, X. Yang, Z. Zhang, W. Yang, X. Duan and X. Duan, Nat. Mater., 2024, 23, 1326–1338 CAS.
- Y. Liu, Y. Li, Y. Wu, G. Yang, L. Mazzarella, P. Procel-Moya, A. C. Tamboli, K. Weber, M. Boccard and O. Isabella, Mater. Sci. Eng., R, 2020, 142, 100579 Search PubMed.
- B. K. Ghosh, C. N. Weoi, A. Islam and S. K. Ghosh, Renewable Sustainable Energy Rev., 2018, 82, 1990–2004 CAS.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS.
- Z. Cai, B. Liu, X. Zou and H.-M. Cheng, Chem. Rev., 2018, 118, 6091–6133 CrossRef CAS.
- J. Gutpa, H. Shaik, K. N. Kumar and S. A. Sattar, Mater. Sci. Semicond. Process., 2022, 143, 106534 CrossRef CAS.
- H. Sudrajat and S. Babel, Sol. Energy Mater. Sol. Cells, 2016, 149, 294–303 CrossRef CAS.
- S. Babel, P. A. Sekartaji and H. Sudrajat, J. Water Supply: Res. Technol.--AQUA, 2017, 66, 25–35 CrossRef.
- H. Sudrajat, J. Cleaner Prod., 2018, 172, 1722–1729 CrossRef CAS.
- S. Babel, P. A. Sekartaji and H. Sudrajat, Environ. Sci. Pollut. Res., 2021, 28, 31163–31173 CrossRef CAS PubMed.
- N. Skillen, C. Rice, X. Pang, P. K. Robertson, W. McCormick and D. McCrudden, in Nanostructured Photocatalysts, 2021, ch. 4, pp. 85–118 Search PubMed.
- N. Skillen, H. Daly, L. Lan, M. Aljohani, C. W. Murnaghan, X. Fan, C. Hardacre, G. N. Sheldrake and P. K. Robertson, Top. Curr. Chem., 2022, 380, 33 CAS.
- M. Rumayor, J. Corredor, M. Rivero and I. Ortiz, J. Cleaner Prod., 2022, 336, 130430 CAS.
- X. Xu, L. Shi, S. Zhang, Z. Ao, J. Zhang, S. Wang and H. Sun, Chem. Eng. J., 2023, 469, 143972 CAS.
- S. Xu, X. Huang and H. Lu, Fuel Process. Technol., 2024, 255, 108057 CAS.
- C. Shi, F. Kang, Y. Zhu, M. Teng, J. Shi, H. Qi, Z. Huang, C. Si, F. Jiang and J. Hu, Chem. Eng. J., 2023, 452, 138980 CrossRef CAS.
- R. Sathre, C. D. Scown, W. R. Morrow, J. C. Stevens, I. D. Sharp, J. W. Ager, K. Walczak, F. A. Houle and J. B. Greenblatt, Energy Environ. Sci., 2014, 7, 3264–3278 RSC.
- S. Wang, P. Mo, D. Li and A. Syed, Catalysts, 2024, 14, 217 CrossRef CAS.
- L. Ge, Y. Ke and X. Li, Chem. Commun., 2023, 59, 5795–5806 RSC.
- J. Benavides-Hernández and F. Dumeignil, ACS Catal., 2024, 14, 11749–11779 CrossRef.
- F. Zaera, J. Catal., 2021, 404, 900–910 CAS.
- E. Mitchell, A. Law and R. Godin, J. Photochem. Photobiol., C, 2021, 49, 100453 CAS.
- Q. Xu, L. Zhang, B. Cheng, J. Fan and J. Yu, Chem, 2020, 6, 1543–1559 CAS.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 1–17 Search PubMed.
- L. Lin, T. Hisatomi, S. Chen, T. Takata and K. Domen, Trends Chem., 2020, 2, 813–824 Search PubMed.
- A. Balapure, J. R. Dutta and R. Ganesan, RSC Appl. Interfaces, 2024, 1, 43–69 CAS.
- X. Zhang, X. Li, D. Zhang, N. Q. Su, W. Yang, H. O. Everitt and J. Liu, Nat. Commun., 2017, 8, 14542 Search PubMed.
- S. Fukuzumi and K. Ohkubo, Chem. Sci., 2013, 4, 561–574 Search PubMed.
- D. Kralisch, D. Ott and D. Gericke, Green Chem., 2015, 17, 123–145 CAS.
- J. Maurya, E. Gemechu and A. Kumar, Int. J. Hydrogen Energy, 2023, 48, 20077–20095 CAS.
- D. Salazar-Marín, G. Oza, J. D. Real, A. Cervantes-Uribe, H. Pérez-Vidal, M. Kesarla, J. T. Torres and S. Godavarthi, Appl. Surf. Sci. Adv., 2024, 19, 100536 Search PubMed.
- Y. Wang, Q. Wang, X. Zhan, F. Wang, M. Safdar and J. He, Nanoscale, 2013, 5, 8326–8339 CAS.
- F. Li, G. Zhu, J. Jiang, L. Yang, F. Deng and X. Li, J. Mater. Sci. Technol., 2024, 177, 142–180 CAS.
- Y. Yuan, R.-t. Guo, L.-f. Hong, X.-y. Ji, Z.-d. Lin, Z.-s. Li and W.-g. Pan, Mater. Today Energy, 2021, 21, 100829 CAS.
- H. Sudrajat, Mater. Res. Express, 2018, 5, 065519 Search PubMed.
- L. Wang, C. Bie and J. Yu, Trends Chem., 2022, 4, 973–983 CrossRef CAS.
- C. Wang, Y. Zhao, C. Cheng, Q. Li, C. Guo and Y. Hu, Coord. Chem. Rev., 2024, 521, 216177 CrossRef CAS.
- M. Bai, R. Chen, X. Liu, H. Li, J. Li, H. Huang, M. Song, Q. Zhang, Y. Su, H. Wang, M. Xu and G. Xie, J. Environ. Chem. Eng., 2024, 12, 113872 CrossRef CAS.
- A. Shabbir, S. Sardar and A. Mumtaz, J. Alloys Compd., 2024, 175683 CrossRef CAS.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- S. Kahng, H. Yoo and J. H. Kim, Adv. Powder Technol., 2020, 31, 11–28 CrossRef CAS.
- M. Muscetta, P. Ganguly and L. Clarizia, J. Environ. Chem. Eng., 2024, 113073 CAS.
- Y. Li, Y. Ma, K. Li, S. Chen and D. Yue, Catalysts, 2022, 12, 724 CAS.
- A. Chakrabarti, M. E. Ford, D. Gregory, R. Hu, C. J. Keturakis, S. Lwin, Y. Tang, Z. Yang, M. Zhu and M. A. Banares, Catal. Today, 2017, 283, 27–53 CAS.
- U. Caudillo-Flores, M. J. Muñoz-Batista, A. Kubacka and M. Fernández-García, ChemPhotoChem, 2018, 2, 777–785 CAS.
- N. S. Lai, Y. S. Tew, X. Zhong, J. Yin, J. Li, B. Yan and X. Wang, Ind. Eng. Chem. Res., 2023, 62, 17835–17848 Search PubMed.
- Q. Cheng, M. K. Benipal, Q. Liu, X. Wang, P. A. Crozier, C. K. Chan and R. J. Nemanich, ACS Appl. Mater. Interfaces, 2017, 9, 16138–16147 CAS.
- J. Maurya, E. Gemechu and A. Kumar, Energy Convers. Manage., 2023, 279, 116750 CAS.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama and T. Watanabe, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- T. Hisatomi and K. Domen, Nat. Catal., 2019, 2, 387–399 CrossRef CAS.
- R. Binjhade, R. Mondal and S. Mondal, J. Environ. Chem. Eng., 2022, 10, 107746 CrossRef CAS.
- K. P. Sundar and S. Kanmani, Chem. Eng. Res. Des., 2020, 154, 135–150 Search PubMed.
- J. Song, H. Lin, G. Zhao and X. Huang, Micromachines, 2022, 13, 861 Search PubMed.
- I. Muñoz, J. Peral, J. A. Ayllón, S. Malato, P. Passarinho and X. Domènech, Water Res., 2006, 40, 3533–3540 Search PubMed.
- V. B.-Y. Oh, S.-F. Ng and W.-J. Ong, J. Cleaner Prod., 2022, 379, 134673 Search PubMed.
- S. Fernandes, J. C. Esteves da Silva and L. Pinto da Silva, Materials, 2020, 13, 1487 CAS.
- J.-Q. Wang, X.-J. Liu, J.-J. Ma, S. Zhang, H.-R. Liu, Y.-L. Dong and Q.-Y. Yang, Green Chem. Eng., 2024, 5, 383–389 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |