
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of Ni substitution in manganite perovskite Li–O2 battery†
Sandra Sajeev a,
Mewin Vincenta,
Piotr Garbacza,
Marcin Strawskia,
Chunyu Zhub,
Yoshitaka Aokic and
Damian Kowalski*a
a,
Mewin Vincenta,
Piotr Garbacza,
Marcin Strawskia,
Chunyu Zhub,
Yoshitaka Aokic and
Damian Kowalski*a
aFaculty of Chemistry and Biological and Chemical Research Centre, University of Warsaw, Zwirki i Wigury 101, Warsaw 02-089, Poland. E-mail: damian.kowalski@chem.uw.edu.pl
bSchool of Low-carbon Energy and Power Engineering, China University of Mining and Technology, Xuzhou 221116, China
cFaculty of Engineering, Hokkaido University, Kita-Ku Kita 13, Jo Nishi 8, Sapporo 060-8628, Hokkaido, Japan
First published on 23rd April 2025
Abstract
A fundamental understanding of the electrochemical processes in Li–O2 batteries is critical for the further development and commercialization of Li–O2 and air-breathing battery technology. This study explores the electrochemistry of nickel-substituted manganite perovskites, La0.7Sr0.3Mn1−xNixO3 (x = 0, 0.1, 0.3, 0.5), which were subsequently used as catalysts in Li–O2 battery operating in 1 mol dm−3 bis trifluoromethane sulfonimide lithium salt (LiTFSi) in tetra ethylene glycol dimethyl ether (TEGDME) electrolyte. In situ Raman spectroscopy fingerprints on the discharge products correlated with charge–discharge profiles revealed that the electrochemical reaction pathway involves the formation of superoxide (LiO2) followed by reduction to lithium peroxide (Li2O2) during the battery discharge and corresponding two-step oxidation process in the charge phase. The superoxide (LiO2) was exceptionally stable for more than 2 h, which is in contrast to previous studies and expectations for short-lifetime intermediate formations. Electrochemical analysis revealed a significant improvement in the Li–O2 battery performance for oxygen electrodes substituted with 10% of nickel, reaching a specific capacity of 3554 mAh g−1. Substitution of Mn with Ni in La0.7Sr0.3Mn0.9Ni0.1O3 led to enhanced charge transfer kinetics due to a high surface population of the low valence state of B-site ions (Mn3+/Mn4+ ratio) accommodating the presence of eg1 electrons in line with Jahn–Teller disordered metal–oxygen octahedra effect. The current finding offers new insights for designing of aprotic LiO2 batteries.
Introduction
Lithium–air (Li–air) and lithium–oxygen (Li–O2) batteries have a high potential to become an efficient energy storage solution because of their relatively high theoretical energy density estimated to be 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 W h kg−1, comparable to that of gasoline.1,2 One of the benefits over conventional lithium-ion batteries is the advantage of utilizing oxygen from air-breathing electrode or pure oxygen, significantly reducing the battery's weight, which is beneficial for commercial applications in electric vehicles and aviation.1–4 The electrochemical process in Li–O2 battery is based on the reduction of molecular oxygen, O2, on the cathode side to form lithium peroxide (Li2O2) upon discharge process and recovery of O2 from the discharge product during the charging process. It is generally accepted that the main discharge product is lithium peroxide (Li2O2) whereas lithium superoxide (LiO2) and lithium oxide (Li2O) formation, stability, and their impact on battery operation is under debate. The reversibility of discharge products on the cathode side is the main bottleneck of Li–O2 technology, hampering its commercialization. A key enabling component in the battery system is a chemically stable electrocatalyst with inartistic catalytic active sites necessary for efficient oxygen reduction and oxygen evolution reactions, which further decide the battery's power density and energy efficiency. The application of noble metals and platinum group metals, palladium (Pd),5 silver (Ag),6 ruthenium (Ru),7 gold (Au),8 and platinum (Pt)9 catalysts has been extensively studied and is rather limited to fundamental studies due to relatively high costs.10 Among the cost-effective materials, transition metal nitrides/oxides,11–13 carbon-promoted transition materials,14 conductive polymers,15 and functional carbon metals16 have been examined.
400 W h kg−1, comparable to that of gasoline.1,2 One of the benefits over conventional lithium-ion batteries is the advantage of utilizing oxygen from air-breathing electrode or pure oxygen, significantly reducing the battery's weight, which is beneficial for commercial applications in electric vehicles and aviation.1–4 The electrochemical process in Li–O2 battery is based on the reduction of molecular oxygen, O2, on the cathode side to form lithium peroxide (Li2O2) upon discharge process and recovery of O2 from the discharge product during the charging process. It is generally accepted that the main discharge product is lithium peroxide (Li2O2) whereas lithium superoxide (LiO2) and lithium oxide (Li2O) formation, stability, and their impact on battery operation is under debate. The reversibility of discharge products on the cathode side is the main bottleneck of Li–O2 technology, hampering its commercialization. A key enabling component in the battery system is a chemically stable electrocatalyst with inartistic catalytic active sites necessary for efficient oxygen reduction and oxygen evolution reactions, which further decide the battery's power density and energy efficiency. The application of noble metals and platinum group metals, palladium (Pd),5 silver (Ag),6 ruthenium (Ru),7 gold (Au),8 and platinum (Pt)9 catalysts has been extensively studied and is rather limited to fundamental studies due to relatively high costs.10 Among the cost-effective materials, transition metal nitrides/oxides,11–13 carbon-promoted transition materials,14 conductive polymers,15 and functional carbon metals16 have been examined.
Perovskite-type compounds have attained recent attention because of their chemical and structural flexibility, improved oxygen mobility, high catalytic activity and reduced cost, making them promising candidates as bifunctional catalysts in metal–air batteries.17,18 The catalytic activity of perovskite-type compounds may be effectively controlled by the substitution of B-site ion on the corner of the perovskite lattice.19–21 Recent reports indicate that Ni-substitution of double perovskite, La1−xSrxMn1−yNiyO3 creates structure distortion and oxygen vacancies thus enhancing the catalytic activity of cathode in metal-air battery.22
The substitution of B-site ion in the lattice changes the population of oxygen vacancies in the lattice, as well as the configuration of B–O bonds and corresponding surface state, typically boosting oxygen evolution and oxygen reduction reaction performance. By fine-tuning the ratio of nickel to manganese, it is possible to enhance the electronic and structural properties of the material, leading to improved electrocatalytic performance.19,23 La1−xSrxMnO3 has been previously studied as a prominent perovskite-type catalyst for oxygen reduction reaction (ORR).24 The low activity towards oxygen evolution reaction (OER) is possibly associated with low covalency of the Mn–O bond and deficiency of highly active transition metal ions, thereby hindering the rechargeability and degrading the overall cell performance in the battery systems. Recent studies demonstrated that the nickel substitution played an important role in enhancing the OER catalytic activity in various catalytic systems.19,22 In the current study, we investigate the performance of La0.7Sr0.3Mn1−xNixO3 depending on the substitution level of nickel, supported by in situ observations on Li2O2, LiO2 having fundamental aspects on Li–O2 battery operation (Fig. 1).
 | ||
| Fig. 1 Scheme of Li–O2 battery with La0.7Sr0.3Mn1−xNixO3 double perovskite catalyst (LSMN) in the cathode. Red, green and yellow spheres correspond to lithium ions, oxygen, and superoxide/peroxide compounds, respectively. | ||
Results and discussion
La0.7Sr0.3Mn1Ni0O3 perovskite crystalizes in the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c space group (no. 167) with lattice constants a = 5.50300 Å, c = 13.4241 Å and cell volume of 349.916086 Å3 (PDF 96-152-1157).25,26 The double perovskite has a general formula of AA′BB′O3 having BO6 units constructing a three-dimensional structure of corner-sharing MnO6 octahedra with A cations located in the interstitial spaces. The ions La3+/Sr4+ occupying the A-site are nine-fold coordinated, while the Mn3+/Mn4+ ions that occupy the octahedral B-site are six-fold coordinated, according to Rc symmetry (Fig. 2).XRD patterns of La0.7Sr0.3Mn1−xNixO3, where x = 0, 0.1, 0.3, 0.5, are depicted in Fig. 3c. The crystallite size calculated by Scherrer equation is 16.3, 15.8, 16.9, and 17.8 nm, for La0.7Sr0.3Mn1O3, La0.7Sr0.3Mn0.9Ni0.1O3, La0.7Sr0.3Mn0.7Ni0.3O3, and La0.7Sr0.3Mn0.5Ni0.5O3, respectively. The splitting of characteristic peaks of La0.7Sr0.3Mn0.5Ni0.5O3 shows the rhombohedral distortion of the perovskite structure.27 The substantial peak splitting for La0.7Sr0.3Mn0.5Ni0.5O3 is typically observed for all peaks except the (100) reflections. Variations from the ideal cubic form cause a small elongation or compression along the body diagonal of the unit cell. The preservation of the unsplit (100) peaks and the splitting of other reflections gives strong evidence that the distortion is rhombohedral. This structural shift is most noticeable at the highest nickel content (x = 0.5), implying a relationship between nickel doping and the degree of rhombohedral distortion.
c space group (no. 167) with lattice constants a = 5.50300 Å, c = 13.4241 Å and cell volume of 349.916086 Å3 (PDF 96-152-1157).25,26 The double perovskite has a general formula of AA′BB′O3 having BO6 units constructing a three-dimensional structure of corner-sharing MnO6 octahedra with A cations located in the interstitial spaces. The ions La3+/Sr4+ occupying the A-site are nine-fold coordinated, while the Mn3+/Mn4+ ions that occupy the octahedral B-site are six-fold coordinated, according to Rc symmetry (Fig. 2).XRD patterns of La0.7Sr0.3Mn1−xNixO3, where x = 0, 0.1, 0.3, 0.5, are depicted in Fig. 3c. The crystallite size calculated by Scherrer equation is 16.3, 15.8, 16.9, and 17.8 nm, for La0.7Sr0.3Mn1O3, La0.7Sr0.3Mn0.9Ni0.1O3, La0.7Sr0.3Mn0.7Ni0.3O3, and La0.7Sr0.3Mn0.5Ni0.5O3, respectively. The splitting of characteristic peaks of La0.7Sr0.3Mn0.5Ni0.5O3 shows the rhombohedral distortion of the perovskite structure.27 The substantial peak splitting for La0.7Sr0.3Mn0.5Ni0.5O3 is typically observed for all peaks except the (100) reflections. Variations from the ideal cubic form cause a small elongation or compression along the body diagonal of the unit cell. The preservation of the unsplit (100) peaks and the splitting of other reflections gives strong evidence that the distortion is rhombohedral. This structural shift is most noticeable at the highest nickel content (x = 0.5), implying a relationship between nickel doping and the degree of rhombohedral distortion.
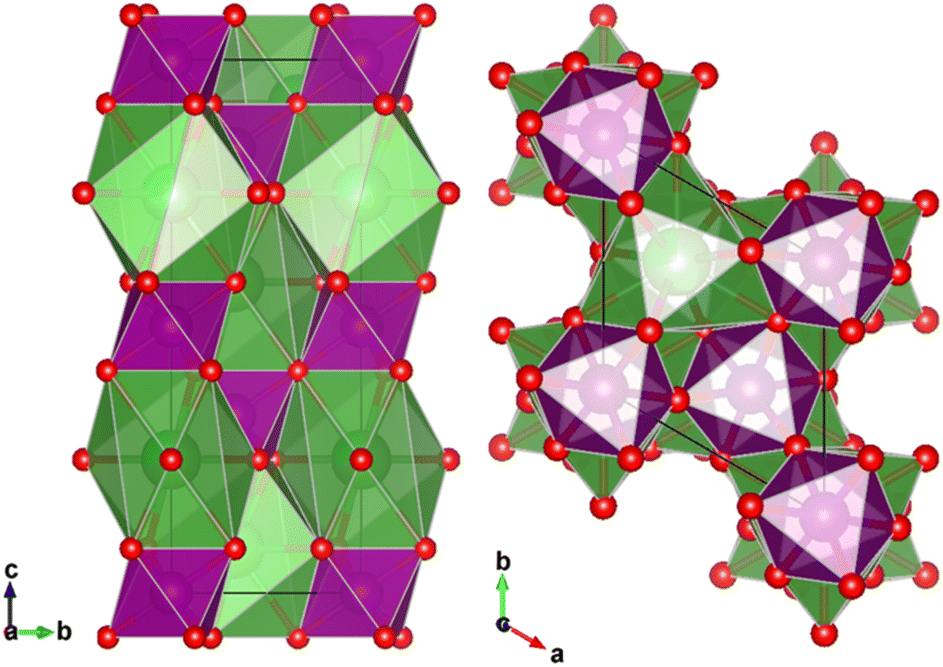 | ||
| Fig. 2 Crystallographic structure of the AA′BB′O3 double perovskite of La0.7Sr0.3Mn1Ni0O3 compound having a 167 Rc symmetry. The unit cell dimensions are a = 5.50300 Å, b = 5.50300 Å, c = 13.4241 Å and cell volume of 349.916086 Å3. | ||
 | ||
| Fig. 3 TEM and corresponding EDS elemental mapping for La, Sr, Mn Ni, O and superimposed MnNi and NiLa (a and b), (c) ex situ XRD patterns and (d) ex situ Raman spectra of La0.7Sr0.3Mn1−xNixO3 (x = 0, 0.1, 0.3, 0.5) pristine catalyst. | ||
The ex situ Raman spectroscopy results for the as-formed catalysts (Fig. 3d), the peaks that have been detected convey significant details about the structural properties of double perovskites. A characteristic of perovskite-type manganites, the Mn–O stretching vibration in the MnO6 octahedra is responsible for the high-intensity peak observed at ca. 660 cm−1.28 The Mn–O–Mn bending mode is most likely represented by the lower intensity peak, which is located at c.a. 531 cm−1. The blue shift of these peaks, which is noticed with increased nickel content, indicates that the metal-oxygen interactions are strengthening.
The Raman peaks move towards higher wavenumbers as a result of the replacement of Mn3+ ions by Ni2+ ions, which also causes a contraction of the lattice and greater metal–oxygen interactions. The observed alterations in peak locations and intensities in response to different nickel contents suggest that nickel incorporation alters the local structure and bonding environment inside the perovskite lattice, which could have an impact on the materials catalytic characteristics. TEM (HAADF) images with corresponding high-resolution EDS elemental maps in Fig. 3a and b show the homogeneous distribution of La, Sr, Mn and O in La0.7Sr0.3Mn0.5Ni0.5O3 structure.
The superimposed EDS elemental maps for MnNi and LaNi reveal nanoregions with enriched nickel content. Those slight compositional alternations most likely contribute to the rhombohedral distortion in the double perovskite. The discharge and charge voltage profiles of cells with La0.7Sr0.3Mn0.5Ni0.5O3 cathode having different ratios of active material, carbon, and binder are demonstrated in Fig. S1.† The 5:4:1 ratio most likely gives a more ideal ionic conductivity and balanced environment for ionic transport, hence increasing initial discharge capacity. Conversely, imbalances were observed in the 2:7:1 and 7:2:1 ratios resulting in higher resistance and less effective ion transport.
Hence for all the following electrochemical studies the electrode ratio of 5:4:1 was used. Fig. 4a depicts the discharge/charge voltage profiles obtained with four distinct perovskites at a discharge current density of 100 mAg−1.
 | ||
| Fig. 4 a) Galvanostatic discharge and charge cycles for La0.7Sr0.3Mn1Ni0O3, La0.7Sr0.3Mn0.9Ni0.1O3, La0.7Sr0.3Mn0.7Ni0.3 O3, and La0.7Sr0.3Mn0.5Ni0.5O3 and corresponding voltage-time curves obtained at first cycle at a current density of 100 mAg−1 b) cyclic voltammograms of first five consecutive cycles of La0.7Sr0.3Mn0.9Ni0.1 at a scan rate of 0.1 mV s−1 in 1 mol dm−3 bis trifluoromethane sulfonimide lithium salt (LiTFSi) in tetra ethylene glycol dimethyl ether (TEGDME). | ||
The cell with La0.7Sr0.3Mn1O3, La0.7Sr0.3Mn0.9Ni0.1O3, La0.7Sr0.3Mn0.7Ni0.3O3, and La0.7Sr0.3 Mn0.5Ni0.5O3 catalysts at the cathode delivered a total discharge capacity of 2500 mAh g−1(electrode), 3554 mAh g−1(electrode), 2507 mAh g−1(electrode) and 2373 mAh g−1(electrode), respectively. Furthermore, the charge voltage plateau of La0.7Sr0.3Mn0.9Ni0.1O3 is approximately 100 mV lower than La0.7Sr0.3Mn1O3. La0.7Sr0.3Mn0.9Ni0.1O3 has better electrochemical performance and a lower discharge–charge voltage gap than other three studied electrocatalysts.
The substitution of 10% manganese with nickel is supposed to create catalytic centers oxygen reduction/oxidation reactions. The cyclic voltammetry (Fig. 4b) results for La0.7Sr0.3Mn0.9Ni0.1O3 show a major decrease in peak current after the first cycle. Despite the initial fading, the CV curves general shape and structural patterns are stable across all the cycles indicating that the fundamental electrochemical process remains constant across the cycles.
A more fundamental understanding of electrochemical processes during discharge/charge was investigated using in situ Raman spectroscopy in Fig. 5.29–32 Raman bands at open circuit voltage originate from the electrode, electrolyte, and optical window. In the first stage of discharge (2.41 V), two peaks appear at 1127 cm−1 and 1517 cm−1. The O–O stretching of lithium superoxide (LiO2) in the discharge products is assigned the Raman shift at 1127 cm−1. The 1517 cm−1 Raman band is generated in conjunction with the 1127 cm−1 band, assigned to the LiO2-C mode, where carbon is used as a conductive additive in the electrode.33
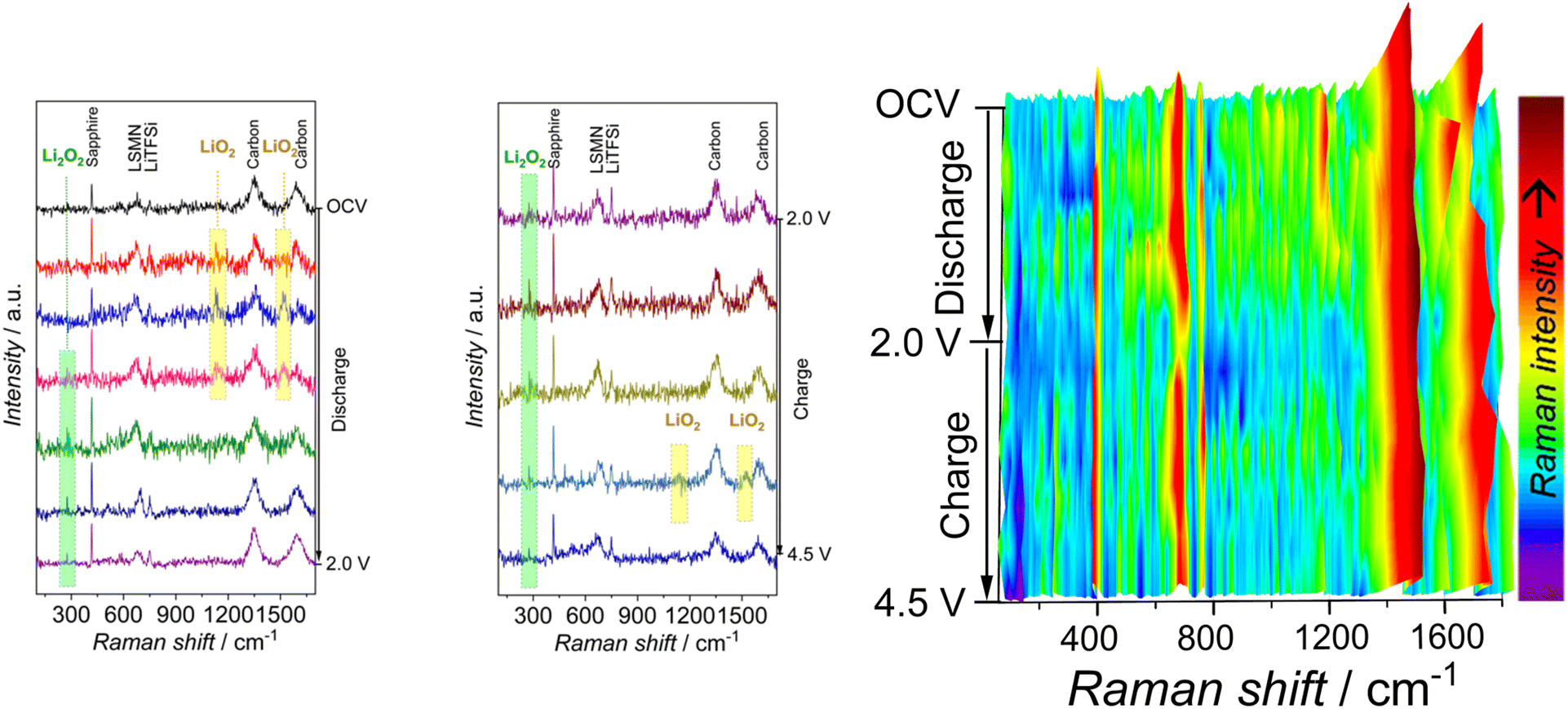 | ||
| Fig. 5 In situ Raman spectroscopy for Li–O2 battery composed La0.7Sr0.3Mn0.9Ni0.1O3 catalyst obtained at the first discharge, charge cycle at a current density of 100 mAg−1 in 1 mol dm−3 bis trifluoromethane sulfonimide lithium salt (LiTFSi) in tetra ethylene glycol dimethyl ether (TEGDME). | ||
The formation and disappearance of LiO2 is displayed in 3D in situ Raman spectrum in Fig. 5. The reversible processes in non-aqueous Li–O2 cell include the development and evolution of mostly lithium peroxide (Li2O2) products, where superoxide (LiO2) plays an essential role. Reference Li2O2 powder Sigma Aldrich gives ex situ Raman spectra with expected O–O stretching frequency of peroxide at around 802 cm−1 and Li–O vibrations located at 256 cm−1.34,35 The Li2O2 peak assigned to Li–O vibrations is detected at 279 cm−1 (Fig. 5), while the O–O stretching at 790 cm−1 is absent in the spectra in the current study.
Most of the studies report difficulties in peroxide detection with low intensity and broad peaks, particularly without surface enhancement.36 The variations in the strength of the signals between the literature data37 and the current study may arise from differences in Raman scattering from peroxide due to surface enhancement issues. Peroxide may be in the form of amorphous, crystalline or a mixture of overlapping amorphous/crystalline layers with complex morphology features. The amorphous Li2O2 is less coordinated than crystalline Li2O2, with a larger population of lithium vacancies and hole polaron defects.38 In addition, the O–O peroxide bond is expected to be shorter for the amorphous phase. Recent studies by others indicate the presence of amorphous Li2O2 as a discharge product, giving unique Raman spectroscopy profiles such as higher O–O vibrational frequencies than crystalline Li2O2.39–41
In situ Raman spectra obtained during the discharge process suggest that the reduction reaction involves one electron transfer to form superoxide (O2−, LiO2) in the first stage of the discharge process:
| O2 + e− → O2− | (1) |
| O2− + Li+ → LiO2 | (2) |
| LiO2 + Li+ + e− → Li2O2 | (3) |
| 2LiO2 → Li2O2 + O2 | (4) |
| Li2O2 → LiO2 + Li+ + e− | (5) |
| LiO2 → Li+ + O2 + e− | (6) |
 | ||
| Fig. 6 Ex situ impedance and ToF-SIMS analysis. Electrochemical impedance spectra (EIS) for Li–O2 battery composed of La0.7Sr0.3Mn1Ni0O3, La0.7Sr0.3Mn0.9Ni0.1O3, La0.7Sr0.3Mn0.7Ni0.3O3, and La0.7Sr0.3Mn0.5Ni0.5O3 catalyst were obtained at open circuit voltage for prisine electrode. EIS obtained after full discharge of Li–O2 battery composed of La0.7Sr0.5Mn0.5Ni0.5O3 and La0.7Sr0.3Mn0.9Ni0.1O3 catalyst are shown for comparition, experimental data were fitted with an equivalent circuit of R1[R2Q2]W. ToF-SIMS spectra were obtained of pristine (green) and fully discharged electrode (red) of La0.7Sr0.3Mn0.9Ni0.1O3. | ||
Electrochemical impedance spectroscopy was utilized to investigate the electrocatalyst's impedance both at open circuit voltage and after discharge process. Fig. 6a and b depicts the Nyquist plots for experimental data as well as simulated data using the equivalent circuit fitting, where R1 denotes the internal resistance of the battery; R2 is the charge transfer resistance, representing the kinetics of an electrochemical process; Q2 is the constant-phase element representing capacitive nature of the process or battery component; W denotes the Warburg impedance.46–49 The simulated data matches well the experimental data across the full range of frequency with χ2/|Z|2 fitting at 0.1. The arc located in the high-frequency zone corresponds to R2, with the diameter depending on type of catalyst; here the reduced diameter of the arc indicates facilitated charge transfer. In the analogous circuit diagram, the straight line located in the low-frequency region represents the diffusion component, i.e., Warburg impedance. The catalyst composed of La0.7Sr0.3Mn0.9Ni0.1O3 possesses lowest charge transfer resistance of 273 Ω compared to 422, 377 and 356 Ω for La0.7Sr0.3Mn1O3, La0.7Sr0.3Mn0.7Ni0.3O3 and La0.7Sr0.3Mn0.5Ni0.5O3, respectively. Hence, the La0.7Sr0.3Mn0.9Ni0.1O3 electrocatalyst facilitates faster electron transfer during the electrochemical reactions in the current battery configuration. The Mn3+/Mn4+ ratio seems to play a critical role in the electrical conductivity of perovskite electrodes. The connection between the Mn3+/Mn4+ ratio and the charge storage capability is discussed further in XPS analysis.
The trend is the same in the spectra after discharge, with an evident increase in the charge transfer resistance by a factor of two. The formation of low conductivity discharge products, i.e. LiO2 and Li2O2 on the electrode surface as indicated by in situ Raman supported by ToF-SIMS analysis is the main reason for a significant increase of charge transfer resistance at completed discharge. The low value of charge transfer resistance of La0.7Sr0.3Mn0.9Ni0.1O3 correlates with the increased discharge–charge performance.
From the electrochemical data it is evident that 10% of nickel substitution has a beneficial impact on the catalytic activity of catalyst. The Mn and Ni valence states in La0.7Sr0.3Mn1−xNixO3 are shown in XPS spectra (Fig. 7 and S2,† respectively). The Ni 3p, does not show a clear peak for x = 0.1 due to relatively low Ni content. Two peaks at 66.0 and 67.5 eV appear for x = 0.3 and x = 0.5 assigned to 3p3/2 and 3p1/2 spin–orbit components of Ni2+ states, respectively.50,51 The 3p3/2 and 3p1/2 peaks of Ni3+ states of reference LaNiO3 appear around 69 and 71 eV,50 which differs from our study indicating that the Ni cations favor remaining in the Ni2+ valence state and the higher valence state Ni3+ is very scarce in all the studied double perovskites. Mn 2p spectra show variable Mn valence states with variation of x. All compositions show a sharp peak around 641 eV and a broad peak around 658 eV, attributed to 2p3/2 and 2p1/2 spin–orbit components, respectively (Fig. 7). The deconvolution of the 2p3/2 peak was performed with binding energy full width at half maximum (FWHM) kept constant. The presence of Mn3+ and Mn4+ valence states in La0.7Sr0.3Mn1−xNixO3 was evident at 641.8 and 643.2 eV, respectively.22,52 The molar ratio of Mn3+/Mn4+ increases with increasing x from 0 to 0.1 and decreases with increasing x above 0.1 (Table S4†) indicating the highest population of Mn3+ oxidation states in La0.7Sr0.3Mn0.9Ni0.1O3, in which more than 90% of the manganese ions occupy the Mn3+ valence state.
 | ||
| Fig. 7 Mn 2p XPS spectra for pristine La0.7Sr0.3Mn1Ni0O3, La0.7Sr0.3Mn0.9Ni0.1O3, La0.7Sr0.3Mn0.7Ni0.3O3, and La0.7Sr0.3Mn0.5Ni0.5O3 catalyst demonstrating population of Mn3+ on the oxide surface depending on the Ni substitution ratio. The molar ratios of Mn3+/Mn4+ determined from fitting analysis are listed in ESI.† | ||
The catalytic centers for oxygen reduction reaction tend to be associated with a high population of lower valence states of manganese ions. One of the most efficient catalysts reported for oxygen reduction reaction are cobalt oxides containing Co3+ ions in the intermediate spin state t2g5eg1.53 The manganese oxides containing Mn3+, which are at the highest population on the surface of La0.7Sr0.3Mn0.9Ni0.1O3 catalyst are in the t2g3eg1 spin state. The similarity between both transition metals is that they have Jahn-Teller disordered metal–oxygen octahedra due to the presence of eg1 electrons.53 Several studies indicated that the presence of eg1 electrons of redox-active transition metal atoms, i.e., B-site cations has a key impact on the catalytic activity of the perovsites.19,53 The presence of a single eg1 electron is expected to improve the electronic communication in the oxide structure54 and, therefore facilitate the charge transfer kinetics as observed for catalysts composed of La0.7Sr0.3Mn1–xNixO3 at the maximum population of Mn3+ states.
Conclusions
Nickel-substituted lanthanum strontium manganite La0.7Sr0.3Mn1−xNixO3 perovskite with x level of 0, 0.1, 0.3, and 0.5 was synthesized using citrate precursor method and studied as an oxygen electrode in an aprotic Li–O2 battery operating in 1 mol dm−3 bis trifluoromethane sulfonimide lithium salt (LiTFSi) in tetra ethylene glycol dimethyl ether (TEGDME) electrolyte. The substitution of manganese with nickel was optimized to achieve high electrocatalytic activity of oxygen electrode; at a current density of 100 mAg−1 in a voltage window of 2.0–4.5 V vs. Li+/Li, the discharge capacity for La0.7Sr0.3Mn0.9 Ni0.1O3 catalyst was 3554 mAh g−1. The in situ Raman spectroscopy revealed lithium superoxide (LiO2) formation during discharge and its further reduction to lithium peroxide (Li2O2). A reversible formation of LiO2 from Li2O2 was observed during the charge stage. The superoxide (LiO2) was electrochemically stable for the first two hours at the discharge, indicating that LiO2 is more stable than expected for short-lifetime intermediates such as superoxides. The formation of stable and reversible LiO2 is in contrast to earlier reports proposed that LiO2 is an intermediate in the formation of Li2O2.Experimental section
θ) was used to calculate the average crystallite size of the catalyst. K is an arbitrary constant having a value of 0.9, the wavelength of the source radiation is λ, the full width at half maximum (FWHM) of the most significant peak in the XRD spectrum is represented by β, and θ is the angle of diffraction. A transmission electron microscope (FEI, Talos F200X) provided with tools for an energy-dispersive X-ray spectroscope (Bruker Super-X EDS system) was used to observe specimens morphology. Raman scattering spectra were collected using a DXR3 Raman Microscope (Thermo Scientific) equipped with a 50× objective having a numerical aperture of 0.25, a spot size of 2.88 μm, and a DPSS laser (532 nm). To ensure the alignment of spectra, the cell was calibrated prior to the experiment. In order to prevent sample deterioration and unintended side reactions, the laser power was kept at 1% maximum intensity.The assembly of ECC-Air electrochemical cells (EL-CELL) was done inside an argon-filled glove box (mBraun Labstar) with the moisture and oxygen levels kept <0.5 ppm. The Li–O2 cells were assembled with a lithium metal anode, a glass fiber separator (GF/B, Whatman), and an electrolyte composed of 1 mol dm−3 LiTFSi (Bis(trifluoromethane) sulfonimide lithium salt, ≥99.0%) in TEGDME (tetra ethylene glycol dimethyl ether), and La0.7Sr0.3Mn1−xNixO3 catalyst cathode.
The electrochemical performance of the batteries was tested using BioLogic SP-300 potentiostat. Before applying electrochemical protocol, rest time was given for all the cells for 8 hours with O2 flow. For the study of the discharge/charge cycles a constant current of 100 mAg−1 was applied in 2.0–4.5 V vs. Li+/Li voltage window. The specific capacities were calculated by normalizing the mass of the catalyst loaded at the cathode. The cyclic voltammetry was performed with a scan rate of 0.1 mV s−1. The electrochemical impedance spectroscopy (EIS) study was performed at open-circuit potential before and after discharge in the frequency range 105 to 10−1 Hz, using a sinusoidal voltage signal amplitude of 5 mV. The impedance spectra were analyzed using an equivalent circuit model R1[R2Q2]W. The experimental data were fitted to the corresponding circuit using BioLogic's EC-Lab software. The distribution of ions over the sample surface was obtained with a time-of-flight secondary ion mass spectrometry (TOF-SIMS). The measurements were performed on a TOF-SIMS.5 spectrometer (ION-TOF GmbH, Germany) operating in Bi3+ mode (at 30 keV energy and 0.48 pA ion current conditions). The LSMN powder was pressed onto copper tape to form a thick layer as a reference sample. The electrode grid after discharge was mounted gently using metal clips. The base pressure in the chamber was below 2 × 10−9 mbar. Analyses were done over 500 μm × 500 μm area. The internal mass calibration was performed using mass a series of ions: Li+, Na+, K+, Mn+, La+. Identification of molecular ions and fragments was performed using SurfaceLab 7.0 software (ION-TOF GmbH, Germany).
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Sandra Sajeev: investigation, methodology, writing – original draft. Mewin Vincent: methodology. Marcin Stawski: methodology, analysis. Piotr Garbacz: resources. Chunyu Zhu: resources. Yoshitaka Aoki: methodology, resources, Damian Kowalski: supervision, manuscript writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The current study was financially supported by National Science Centre (OPUS20) grant number 2020/39/B/ST4/02548. The authors would like to thank K. Sobczak for TEM meas-urements and Szymon Sutula for XRD analysis.Notes and references
- W.-J. Kwak, Rosy, D. Sharon, C. Xia, H. Kim, L. R. Johnson, P. G. Bruce, L. F. Nazar, Y.-K. Sun, A. A. Frimer, M. Noked, S. A. Freunberger and D. Aurbach, Chem. Rev., 2020, 120, 6626–6683 CrossRef CAS PubMed.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- H.-G. Jung, J. Hassoun, J.-B. Park, Y.-K. Sun and B. Scrosati, Nat. Chem., 2012, 4, 579–585 CrossRef CAS.
- A. Kondori, M. Esmaeilirad, A. M. Harzandi, R. Amine, M. T. Saray, L. Yu, T. Liu, J. Wen, N. Shan, H.-H. Wang, A. T. Ngo, P. C. Redfern, C. S. Johnson, K. Amine, R. Shahbazian-Yassar, L. A. Curtiss and M. Asadi, Science, 2023, 379, 499–505 CrossRef CAS.
- Z. Cui, L. Li, A. Manthiram and J. B. Goodenough, J. Am. Chem. Soc., 2015, 137, 7278–7281 CrossRef CAS.
- Q. Hong and H. Lu, Sci. Rep., 2017, 7, 3378 CrossRef.
- B. Sun, P. Munroe and G. Wang, Sci. Rep., 2013, 3, 2247 CrossRef PubMed.
- Y.-C. Lu, D. G. Kwabi, K. P. C. Yao, J. R. Harding, J. Zhou, L. Zuin and Y. Shao-Horn, Energy Environ. Sci., 2011, 4, 2999–3007 RSC.
- G. Zhao, J. Lv, Z. Xu, L. Zhang and K. Sun, J. Power Sources, 2014, 248, 1270–1274 CrossRef CAS.
- J. Zhao, R. Pathak, Z. Zhao, X. Chen, M. B. Saud, H. Li, F. Wu, Q. Qiao, J. W. Elam and X. Wang, Green Chem., 2023, 25, 10182–10208 RSC.
- F. Li, R. Ohnishi, Y. Yamada, J. Kubota, K. Domen, A. Yamada and H. Zhou, Chem. Commun., 2013, 49, 1175–1177 RSC.
- A. Débart, A. J. Paterson, J. Bao and P. G. Bruce, Angew. Chem., Int. Ed., 2008, 47, 4521–4524 CrossRef.
- S. H. Oh, R. Black, E. Pomerantseva, J.-H. Lee and L. F. Nazar, Nat. Chem., 2012, 4, 1004–1010 CrossRef CAS.
- J.-L. Shui, N. K. Karan, M. Balasubramanian, S.-Y. Li and D.-J. Liu, J. Am. Chem. Soc., 2012, 134, 16654–16661 CrossRef CAS PubMed.
- Y. Cui, Z. Wen, X. Liang, Y. Lu, J. Jin, M. Wu and X. Wu, Energy Environ. Sci., 2012, 5, 7893–7897 RSC.
- S. Wang, S. Dong, J. Wang, L. Zhang, P. Han, C. Zhang, X. Wang, K. Zhang, Z. Lan and G. Cui, J. Mater. Chem., 2012, 22, 21051–21056 RSC.
- G. Liu, H. Chen, L. Xia, S. Wang, L.-X. Ding, D. Li, K. Xiao, S. Dai and H. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 22478–22486 CrossRef CAS.
- D. U. Lee, H. W. Park, M. G. Park, V. Ismayilov and Z. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 902–910 CrossRef CAS PubMed.
- Y. Aoki, E. Tsuji, T. Motohashi, D. Kowalski and H. Habazaki, J. Phys. Chem. C, 2018, 122, 22301–22308 CrossRef CAS.
- D. Kowalski, H. Kiuchi, T. Motohashi, Y. Aoki and H. Habazaki, ACS Appl. Mater. Interfaces, 2019, 11, 28823–28829 CrossRef CAS PubMed.
- E. Tsuji, T. Motohashi, H. Noda, D. Kowalski, Y. Aoki, H. Tanida, J. Niikura, Y. Koyama, M. Mori, H. Arai, T. Ioroi, N. Fujiwara, Y. Uchimoto, Z. Ogumi and H. Habazaki, ChemSusChem, 2017, 10, 2864–2868 CrossRef CAS.
- Z. Wang, Y. You, J. Yuan, Y.-X. Yin, Y.-T. Li, S. Xin and D. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 6520–6528 CrossRef CAS.
- Y. Aoki, K. Takase, H. Kiuchi, D. Kowalski, Y. Sato, H. Toriumi, S. Kitano and H. Habazaki, J. Am. Chem. Soc., 2021, 143, 6505–6515 CrossRef CAS.
- W. Wang, W. Liu, M. Kamiko and S. Yagi, New J. Chem., 2022, 46, 13082–13088 RSC.
- M. C. Ramírez Camacho, C. F. Sánchez Valdés, M. Curiel, J. L. Sánchez Llamazares, J. M. Siqueiros and O. Raymond Herrera, Sci. Rep., 2020, 10, 2568 CrossRef.
- H.-S. Lee and H.-H. Park, Adv. Condens. Matter Phys., 2015, 2015, 746475 Search PubMed.
- T. I. M. Vaz, S. M. Gurav and A. V. Salker, Indones. J. Chem., 2021, 21, 1244–1251 CrossRef CAS.
- N. Bajpai, M. Saleem and A. Mishra, J. Mater. Sci.: Mater. Electron., 2021, 32, 12890–12902 CrossRef CAS.
- M. Vincent and D. Kowalski, ACS Appl. Nano Mater., 2023, 6, 6528–6537 CrossRef CAS.
- M. Vincent, S. Sajeev, M. Srivastava, E. Kowalska, S. Srinivasan and D. Kowalski, Electrochim. Acta, 2025, 509, 145309 CrossRef CAS.
- L. Fadillah, D. Kowalski, M. Vincent, C. Zhu, S. Kitano, Y. Aoki and H. Habazaki, ACS Appl. Mater. Interfaces, 2023, 15, 52563–52570 CAS.
- M. Vincent, S. S. Kumar and D. Kowalski, Electrochim. Acta, 2023, 469, 143161 CrossRef CAS.
- A. Halder, H.-H. Wang, K. C. Lau, R. S. Assary, J. Lu, S. Vajda, K. Amine and L. A. Curtiss, ACS Energy Lett., 2018, 3, 1105–1109 CrossRef CAS.
- F. S. Gittleson, W.-H. Ryu and A. D. Taylor, ACS Appl. Mater. Interfaces, 2014, 6, 19017–19025 CrossRef CAS.
- J. Yang, D. Zhai, H.-H. Wang, K. C. Lau, J. A. Schlueter, P. Du, D. J. Myers, Y.-K. Sun, L. A. Curtiss and K. Amine, Phys. Chem. Chem. Phys., 2013, 15, 3764–3771 RSC.
- T. A. Galloway and L. J. Hardwick, J. Phys. Chem. Lett., 2016, 7, 2119–2124 CrossRef CAS PubMed.
- S. Higashi, Y. Kato, K. Takechi, H. Nakamoto, F. Mizuno, H. Nishikoori, H. Iba and T. Asaoka, J. Power Sources, 2013, 240, 14–17 CrossRef CAS.
- F. Tian, M. D. Radin and D. J. Siegel, Chem. Mater., 2014, 26, 2952–2959 CrossRef CAS.
- Y. Wang, S. Pan, H. Li, D. Li, Y. Guo, S. Chi, C. Geng, S. Wu and Q.-H. Yang, EES Catal., 2023, 1, 312–321 RSC.
- Y. Dou, X.-G. Wang, D. Wang, Q. Zhang, C. Wang, G. Chen, Y. Wei and Z. Zhou, Chem. Eng. J., 2021, 409, 128145 CrossRef CAS.
- F. S. Gittleson, K. P. C. Yao, D. G. Kwabi, S. Y. Sayed, W.-H. Ryu, Y. Shao-Horn and A. D. Taylor, ChemElectroChem, 2015, 2, 1446–1457 CrossRef CAS.
- M. Aoki, D. Dilixiati, M. Ushijima, S. Yamada and T. Kondo, J. Phys. Chem. C, 2023, 127, 15051–15061 CrossRef CAS.
- B. M. Gallant, D. G. Kwabi, R. R. Mitchell, J. Zhou, C. V. Thompson and Y. Shao-Horn, Energy Environ. Sci., 2013, 6, 2518–2528 RSC.
- L. Johnson, C. Li, Z. Liu, Y. Chen, S. A. Freunberger, P. C. Ashok, B. B. Praveen, K. Dholakia, J.-M. Tarascon and P. G. Bruce, Nat. Chem., 2014, 6, 1091–1099 CrossRef CAS.
- U. Das, K. C. Lau, P. C. Redfern and L. A. Curtiss, J. Phys. Chem. Lett., 2014, 5, 813–819 CrossRef CAS.
- T. L. Kulova, V. A. Tarnopol'skii and A. M. Skundin, Russ. J. Electrochem., 2009, 45, 38–44 CrossRef CAS.
- M. Gaberšček, Curr. Opin. Electrochem., 2022, 32, 100917 CrossRef.
- M. Gaberšček, Nat. Commun., 2021, 12, 6513 CrossRef.
- B. A. Boukamp, Solid State Ionics, 1986, 20, 31–44 CrossRef CAS.
- J. G. Kim, D. L. Pugmire, D. Battaglia and M. A. Langell, Appl. Surf. Sci., 2000, 165, 70–84 CrossRef CAS.
- M. Saghayezhian, Z. Wang, H. Guo, Y. Zhu, E. W. Plummer and J. Zhang, Phys. Rev. B, 2017, 95, 165434 CrossRef.
- J. Hu, L. Wang, L. Shi and H. Huang, Electrochim. Acta, 2015, 161, 115–123 CrossRef CAS.
- U. Maitra, B. S. Naidu, A. Govindaraj and C. N. R. Rao, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11704–11707 CrossRef CAS.
- H. Yadegari, M. Norouzi Banis, A. Lushington, Q. Sun, R. Li, T.-K. Sham and X. Sun, Energy Environ. Sci., 2017, 10, 286–295 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Charge–discharge curves, ratios of active material, carbon, and binder, analysis of XPS spectra. See DOI: https://doi.org/10.1039/d5lf00050e |
| This journal is © The Royal Society of Chemistry 2025 |