
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vitamin C: friend or foe! A synopsis of ascorbic acid's reduction and oxidation of graphene oxide†
Omar
El-Basha Hassan
 a,
Yves
Chenavier
a,
Vincent
Maurel
a,
Julien
Pérard
b,
Adnane
Bouzina
a,
Lionel
Dubois
a and
Florence
Duclairoir
*a
a,
Yves
Chenavier
a,
Vincent
Maurel
a,
Julien
Pérard
b,
Adnane
Bouzina
a,
Lionel
Dubois
a and
Florence
Duclairoir
*a
aUniv. Grenoble Alpes, CEA, CNRS, Grenoble INP, IRIG, SyMMES, 38000 Grenoble, France. E-mail: florence.duclairoir@cea.fr
bUniv. Grenoble Alpes, CEA, CNRS, Grenoble INP, IRIG, LCBM, 38000 Grenoble, France
First published on 10th December 2024
Abstract
L-Ascorbic acid is being commonly used as a green reducing agent for graphene oxide. However, re-oxidation of reduced Graphene Oxide (rGO) was established when using a moderate to high concentration of L-ascorbic acid and/or long reduction time. This unexpected finding gives important direction about conditions to privilege to limit such re-oxidation.
Ascorbic acid and more specifically its L-enantiomer, commonly known as Vitamin C, is a natural organic anti-oxidant. It has a very important role in the body as it is a vital anti-oxidant, anti-diabetic, anti-tumour, is involved in the metabolism of cholesterol and notably promotes collagen biosynthesis, while also being a non-heme system taking part in iron absorption, transport and storage.1–7L-Ascorbic Acid (L-AA) can mainly be found in fruits and vegetables or taken as a supplement. L-AA is a mild reducing agent reacting with superoxide, hydroxyl and peroxyl radicals. However, under specific conditions, Vitamin C has been shown to act as a prooxidant, for example promoting lipidic peroxidation when in presence of Fe3+ reduced to Fe2+ (Fenton reaction participant).8–10
L-Ascorbic acid has a standard redox potential of E° = −0.39 V vs. SHE11,12 and has been used as a mediator in combination with transition metals to catalyze many different organic chemistry reactions.13,14 This mild-reducing ability of L-AA has also been applied to reduce graphene oxide (GO), providing an efficient and green alternative to hydrazine hydrate reduction.6,15–20 Such a safer reduction option is of high interest as reduced Graphene Oxide (rGO) is a material that is being widely studied, pristine or in composite, for a variety of applications (i.e. sensors, optoelectronic devices, material strengthening applications, batteries, supercapacitors…).21 Over the years it was reported that different L-AA reduction parameters (reaction time, reaction temperature, pH, and concentration of L-AA) can have an impact on the GO's reduction extent and hence lead to samples with different surface chemistries and conductivities. A recent review summarizes the different GO reduction conditions used in a selection of papers (Table S1, ESI† enriched with additional inputs) showing that the concentration of L-AA and the reduction time are different from one paper to another or sometimes not even reported.16
Considering that this reduction method is widely used to obtain rGO, it seemed necessary to rationalize (for a given pH and set temperature) the impact of L-AA concentration and the reduction time on the final material reduction degree (based on the oxygen content). Herein, we report for the first time that when a moderate to high concentration of L-AA is used in combination with a long reaction time, an unexpected rGO re-oxidation occurs. This finding is supported by chemical characterization of a series of GO samples reduced with L-AA. Based on literature about L-AA reactivity, reaction paths possibly explaining such reoxidation have been evoked and discussed. Hence this study stresses that the optimum concentration of L-AA used to reduce graphene oxide should be kept moderate and should not be combined with a long reaction time (<2 h).
Graphene Oxide (GO) was synthesized using a modified Hummer's Method.22 Briefly, graphite was oxidized into graphite oxide using H2SO4/KMnO4 mixture. It was then ultra-sonicated to exfoliate it into GO. The oxidation of GO was confirmed through monitoring the C/O obtained from X-ray Photoelectron Spectroscopy (XPS) (Fig. S2, ESI†) and elemental analysis (EA). The C/O ratio was found to be 2.35 from XPS and 1.06 ± 0.007 from EA (Table 1) which is in agreement with the range of values reported in literature for GO.23 The fitting of the C1s High Resolution (HR) XPS peak reveals various oxygen bearing carbons such as epoxide, hydroxide, carbonyl and carboxylic groups (Fig. S3, ESI†). Thermogravimetric Analysis (TGA), done under Ar, also shows an abrupt weight loss (20%) around 200 °C (Fig. 1) which can be attributed to the loss of loosely bound oxygen functionalities followed by a gradual loss of the strongly bound ones, indicating the abundance of such functionalities post oxidation in line with literature.24 All together, these analyses show that the GO is strongly oxidized. To confirm the success of the exfoliation, X-ray Diffraction (XRD) was carried out (Fig. S4, ESI†). The XRD profile of GO demonstrates the absence of the graphitic 002 peak and the appearance of the GO's 001 peak at larger expanded d-spacing value (0.34 nm for graphite and 0.73 nm for GO).
| Sample | XPS (at%) | EA (wt%) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| C | N | O | C/O | C | N | O | H | C/O | |
| rGO-VitC-24h | 84.99 | 0.84 | 14.17 | 6 | 72.54 ± 0.16 | 0.69 ± 0.01 | 24.86 ± 0.17 | 1.91 ± 0.004 | 2.92 ± 0.026 |
| rGO-VitC-30 min | 88.53 | 1.31 | 10.16 | 8.71 | 77.41 ± 0.11 | 0.67 ± 0 | 20.81 ± 0.09 | 1.11 ± 0.022 | 3.72 ± 0.022 |
| GO | 70.06 | 0.1 | 29.84 | 2.35 | 50.13 ± 0.14 | 0.17 ± 0.01 | 47.17 ± 0.007 | 2.53 ± 0.026 | 1.06 ± 0.007 |
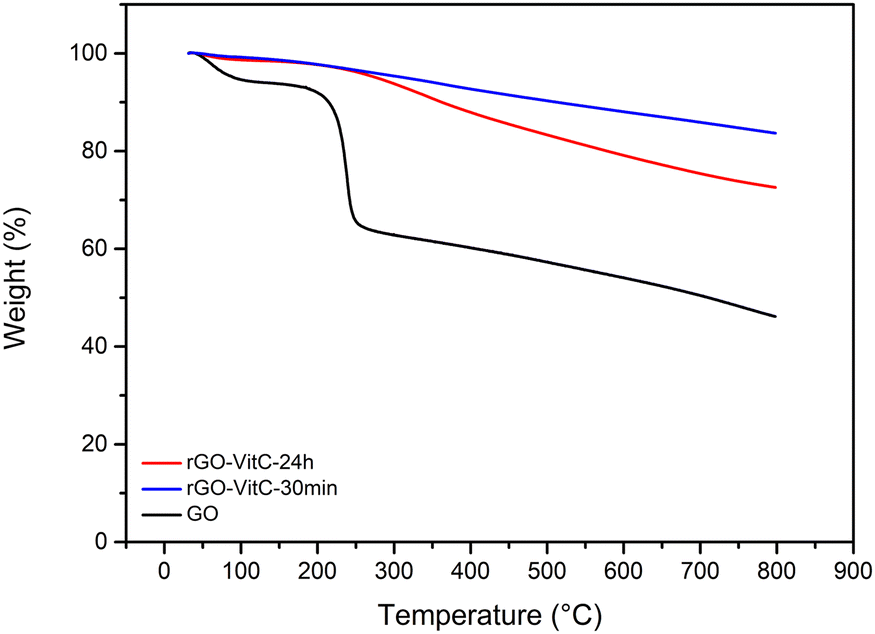 | ||
| Fig. 1 TGA profiles of GO, rGO-VitC-30min and rGO-VitC-24h. | ||
Two rGO samples were synthesized through the reduction of GO using L-Ascorbic Acid (L-AA) as reported in literature.20 A pH of ∼9–10 was reached using NH4OH to promote colloidal stability of the GO solution prior to L-AA addition.20,25 The samples were both reduced at 95 °C using a 60 mM solution of L-AA for a short reaction time (30 min) for the rGO-VitC-30min sample and long reaction time (24 h) for the rGO-VitC-24h sample. Such a concentration of 60 mM is considered a moderate concentration based on the average of the concentrations reported for GO reduction in literature calculated from S1 (65.7 mM). The samples were washed using a Soxhlet setup with water for 48 h prior to being dried in the oven at 80 °C overnight. This washing step was added to ensure the removal of any remaining L-AA and/or any of its oxidized products. XPS and EA were carried out to validate the success of the reduction by comparing the C/O ratio of the reduced samples to that of GO (Fig. S2, ESI†). Table 1 shows that the reduced samples display a higher C/O ratio (8.71 and 6 from XPS and 3.72 ± 0.022 and 2.92 ± 0.026 from EA for rGO-VitC-30min and rGO-VitC-24h, respectively) compared to that of GO mentioned earlier. Such findings, combined with the disappearance of the abrupt weight loss in the TGA profiles of the reduced samples (Fig. 1), corroborate the decrease in oxygen content after the reduction of GO viaL-AA. Such a decrease in oxygen content can also be verified by fitting the C1s HR XPS spectra of the reduced samples (Fig. S5a and b, ESI†). Such fitting highlights a drastic decrease of the signals arising from the various oxygen functionalities present on the GO surface, notably those of hydroxy and epoxide nature (Fig. S3, ESI†). Furthermore, the reduction was confirmed by XRD (Fig. S4, ESI†). The GO peak disappeared upon reduction using L-AA and a broader graphitic peak appeared at higher 2θ values corresponding to d-spacing values of 0.36–0.37 nm. This indicates the partial reconstruction of the π–π stacking between the sheets due to the partial restoration of the sp2 carbon network upon reduction of GO.
Investigating the reduction extent of the reduced samples based on the C/O ratios displayed in Table 1 clearly reveals that the sample subjected to longer reduction time (rGO-VitC-24h) is, counterintuitively, less reduced compared to the one subjected to shorter reduction time (rGO-VitC-30min). rGO-VitC-24h has a C/O ratio lower than that of rGO-VitC-30 min calculated from XPS (6 versus 8.71) and EA (2.92 ± 0.026 versus 3.72 ± 0.022). In addition, it shows a much higher weight loss in its TGA profile (Fig. 1), indicating the presence of more oxygen functionalities in the sample. To the best of our knowledge, such a finding has never been addressed before in any VitC reduced GO papers, which primarily focused on the reduction process and the by-products depicted in Scheme S6 (ESI†). A hypothesis is that this phenomenon might be attributed to the auto-oxidation of L-AA and/or its metal-catalyzed oxidation, leading to probable in situ production of H2O2 and other reactive species such as ˙OH (Scheme S7, ESI†).26–28 The auto-oxidation is usually reported at physiological temperature so it could be expected to be more expressed at higher temperatures and in the abundance of soluble oxygen. In the literature, ascorbic acid has been discussed to be either an anti- or pro- oxidant as a function of its concentration, pH and/or presence of metals.29 When used at pharmacological concentrations (higher than cell concentration), ascorbic acid was shown to serve as a pro-drug, producing H2O2 which would kill selected cancer cells.30,31 The oxidation of L-AA has been described to be potentially metal catalyzed by trace amounts of metal ions such as Fe3+ and Cu2+.27,29 So Inductively Coupled Plasma Optical Emission Spectroscopy (ICP–OES) analysis on the starting GO material and final rGO-VitC-24h was performed and showed the presence of trace amounts (i.e. not detected with XPS) of Mn, Mg, Zn and Fe in GO and rGO-VitC (Table S2, ESI†).
Hydrogen peroxide can decompose, yielding oxygen bearing radicals (such as ˙OH)32 through heating33–35 and/or catalytic decomposition over Fe2+,27,36,37 Cu+,27,36,37 and Mn2+ ions.26,32,38,39 As explained before, Fe and Mn have been shown to be present in the samples. H2O2 is described in literature as an edge-oxidizing agent for graphite33 and rGO sheets.32,34 Therefore, during this reduction of GO, many different side products can be found and among them some very reactive species (Schemes S6 and S7, ESI†), that could react with rGO to functionalize its edges with various possible oxygen functionalities: hydroxyl, carbonyl and/or carboxylic groups. The fittings of the O1s and C1s HR XPS peaks (Fig. S5, ESI†) indicate an increase in oxygen of the C–O type such as hydroxyl groups upon reduction for long time periods (O–C at% represents 7.87 and 11.77 at% of total O calculated from O1s HR and C–O at% represents 18.54 and 21.88 at% of total C calculated from C1s HR for rGO-VitC-30min and rGO-VitC-24h, respectively). These results are in line with the possible oxidation products of rGO when oxidized by H2O2 as postulated earlier.
To verify such hypotheses of auto- and/or metal-catalyzed oxidation of L-AA, Electron Paramagnetic Resonance (EPR) was carried out to look for the ascorbyl radical known to be the first oxidation product in both pathways (Scheme S7, ESI†).27,40 The ascorbyl radicals are meant to be formed due to the transfer of electrons from the ascorbate anions to oxygen molecules in case of the auto-oxidation and to metal ions in case of the metal-catalyzed oxidation. The EPR experiments were done in a flat cell to detect the presence of the ascorbyl radical in a 60 mM L-AA aqueous solution heated at 95 °C. A drop of 28 wt% ammonia solution was added to adjust the pH to 10 to mimic the reduction conditions. GO was excluded from the media to avoid signals from GO and rGO. The presence of the ascorbyl radical can be inferred from the detection of its unique EPR signal: a doublet with a hyperfine coupling constant (aH) of 1.8 G (Fig. 2) in agreement with the literature.41,42 The presence of such radicals confirms the viability of the pathways discussed earlier.
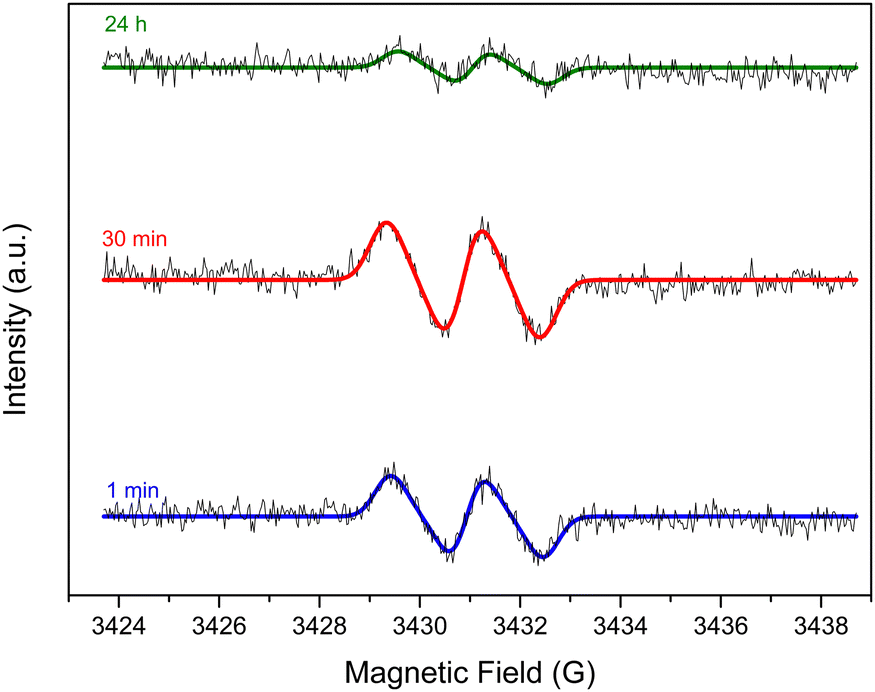 | ||
| Fig. 2 EPR signal of the ascorbyl radical detected in a 60 mM VitC aqueous solution heated at 95 °C with a starting pH of 10 after 1 min, 30 min and 24 h. | ||
To further support the postulated role of the oxidation of L-AA presented so far, two series of rGO samples were obtained through the reduction of GO viaL-AA using different concentrations of L-AA: 26 mM (rGO-VitC-26mM) and 260 mM (rGO-VitC-260mM). Each series of reactions was carried out with three different reduction periods: 30 min, 2 h and 24 h. XPS was carried out on the samples to calculate the C/O ratio of the produced rGO. Fig. 3 displays the evolution of the C/O ratio with time for the two different series of different concentrations. For rGO-VitC-26mM, the C/O ratio decreases after 2 h and after 24 h reaches around 75% of the value obtained after 30 min. For rGO-VitC-260 mM, after 30 min, the C/O decreases with time and after 24 h, it reaches around 50% of that obtained after 30 min. This confirms the fact that longer reaction times may lead to the re-oxidation of the produced rGO sheets. In addition, it can be inferred from Fig. 3 that higher concentrations of L-AA yields a higher C/O ratio after a short time period (C/O ratios were 8.52 and 7.85 after 30 min for rGO-VitC-260mM and rGO-VitC-26 mM, respectively). However, higher concentrations of L-AA lead to higher re-oxidation levels when left for long reduction times (C/O ratios decreased by 50% and 25% after 24 h for rGO-VitC-260mM and rGO-VitC-26mM, respectively). An additional control experiment conducted under Ar in presence of 60 mM of L-AA for 24 h revealed a higher C/O than that observed when the reaction was conducted under aerobic conditions (6.79 from XPS and 3.22 ± 0.034 from EA vs. 6 from XPS and 2.92 ± 0.026 from EA for rGO-VitC-24h) showing that the sample was more reduced when the reduction was carried out under Ar. This observation further stresses that the interaction of O2 with L-AA creates very reactive species that could be responsible for the rGO re-oxidation.
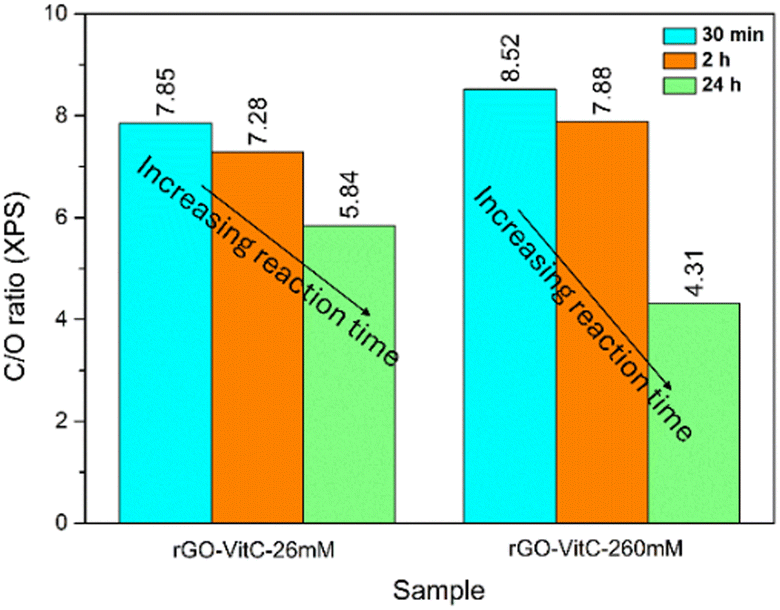 | ||
| Fig. 3 C/O ratios calculated from XPS for rGO-VitC-26mM and rGO-VitC-260mM after 30 min, 2 h and 24 h reduction time periods. | ||
Conclusions
L-AA was shown to be a good reducing agent for GO as reported in literature. However, care must be taken when selecting the reduction conditions of GO using L-AA. Counterintuitively, it was demonstrated herein that higher concentrations and longer reduction time periods do not necessarily lead to higher reduction extent. Therefore, to obtain a high reduction extent, a shorter reduction time (<2 h) is highly recommended for moderate to high concentrations of L-AA used in literature. While a hypothesis about the production of by-products and their reactivity has been proposed, additional in-depth mechanistic studies are required to decipher the exact mechanism driving the oxidation of the rGO sheets when moderate to high concentrations of L-AA are used for long periods of time to reduce GO.Omar El-Basha Hassan was involved in conceptualization, investigation, formal analysis, validation, visualization and writing (original draft and review and editing). Yves Chenavier took part in the investigation/resource (provision of study material at its early stage). Julien Pérard performed formal analysis and edited the paper. Adnane Bouzina was involved in formal analysis. Vincent Maurel performed formal analysis (EPR analysis) and edited the paper. Lionel Dubois co-supervised this study and edited the paper. Florence Duclairoir co-supervised this study and was involved in formal analysis (XPS data recording/interpreting) and writing of the paper.
Data availability
The raw data used to draw the plots included in the paper and ESI† are currently stored on CEA repositories. Hence these data would be made available upon request to the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The access to mutualized equipment of PFNC Nano characterization Minatec platform is acknowledged by the authors. The authors are grateful to ANR for the financial support (SPICS project: grant ANR-19-CE05-0035). This work was also supported by a french government grant from the Agence Nationale de la Recherche under the France 2030 program reference ANR-22-PEBA-0003 (HiPoHyBat project).Notes and references
- R. E. Beyer, J. Bioenerg. Biomembr., 1994, 26, 349–358 CrossRef CAS.
- D. Njus, P. M. Kelley, Y.-J. Tu and H. B. Schlegel, Free Radical Biol. Med., 2020, 159, 37–43 CrossRef CAS.
- S. A. Mason, L. Parker, P. van der Pligt and G. D. Wadley, Free Radical Biol. Med., 2023, 194, 255–283 CrossRef CAS PubMed.
- T. Maekawa, T. Miyake, M. Tani and S. Uemoto, Front. Oncol., 2022, 12, 981547 CrossRef CAS PubMed.
- D. A. Bender and L. Fontana, in Encyclopedia of Human Nutrition, ed. B. Caballero, Academic Press, Oxford, 2023, pp. 504–514 Search PubMed.
- K. K. H. De Silva, H.-H. Huang, R. K. Joshi and M. Yoshimura, Carbon, 2017, 119, 190–199 CrossRef CAS.
- M. Kohlmeier, in Nutrient Metabolism, ed. M. Kohlmeier, Academic Press, London, 2003, pp. 542–551 Search PubMed.
- D. D. Miller, T. Li and R. H. Liu, Antioxidants and Phytochemicals, Reference Module in Biomedical Sciences, Elsevier, 2014 Search PubMed.
- B. Poljšak and J. Ionescu, Handb. Vitam. C Res., 2009, 153–183 Search PubMed.
- I. D. Podmore, H. R. Griffiths, K. E. Herbert, N. Mistry, P. Mistry and J. Lunec, Nature, 1998, 392, 559 CrossRef CAS.
- M. D. Lovander, J. D. Lyon, D. L. Parr, J. Wang, B. Parke and J. Leddy, J. Electrochem. Soc., 2018, 165, G18–G49 CrossRef CAS.
- T. Matsui, Y. Kitagawa, M. Okumura and Y. Shigeta, J. Phys. Chem. A, 2015, 119, 369–376 CrossRef CAS PubMed.
- A. Das, B. Banik and R. Yadav, Encyclopedia. Available online: https://encyclopedia.pub/entry/1722 (accessed May 2024).
- K. Min, H. Gao and K. Matyjaszewski, Macromolecules, 2007, 40, 1789–1791 CrossRef CAS.
- L. G. Guex, B. Sacchi, K. F. Peuvot, R. L. Andersson, A. M. Pourrahimi, V. Ström, S. Farris and R. T. Olsson, Nanoscale, 2017, 9, 9562–9571 RSC.
- M. Palomba, G. Carotenuto and A. Longo, Materials, 2022, 15, 6456 CrossRef CAS.
- J. Gao, F. Liu, Y. Liu, N. Ma, Z. Wang and X. Zhang, Chem. Mater., 2010, 22, 2213–2218 CrossRef CAS.
- S. Eigler, S. Grimm, M. Enzelberger-Heim, P. Müller and A. Hirsch, Chem. Commun., 2013, 49, 7391–7393 RSC.
- J. Zhang, H. Yang, G. Shen, P. Cheng, J. Zhang and S. Guo, Chem. Commun., 2010, 46, 1112–1114 RSC.
- M. J. Fernández-Merino, L. Guardia, J. I. Paredes, S. Villar-Rodil, P. Solís-Fernández, A. Martínez-Alonso and J. M. D. Tascón, J. Phys. Chem. C, 2010, 114, 6426–6432 CrossRef.
- R. Tarcan, O. Todor-Boer, I. Petrovai, C. Leordean, S. Astilean and I. Botiz, J. Mater. Chem. C, 2020, 8, 1198–1224 RSC.
- W. S. HummersJr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- P. P. Brisebois and M. Siaj, J. Mater. Chem. C, 2020, 8, 1517–1547 RSC.
- Y. Wang, Q. He, H. Qu, X. Zhang, J. Guo, J. Zhu, G. Zhao, H. A. Colorado, J. Yu, L. Sun, S. Bhana, M. A. Khan, X. Huang, D. P. Young, H. Wang, X. Wang, S. Wei and Z. Guo, J. Mater. Chem. C, 2014, 2, 9478–9488 RSC.
- K. K. H. De Silva, H.-H. Huang and M. Yoshimura, Appl. Surf. Sci., 2018, 447, 338–346 CrossRef CAS.
- N. Miyake, M. Kim and T. Kurata, Biosci., Biotechnol., Biochem., 1999, 63, 54–57 CrossRef CAS PubMed.
- A. J. Michels and B. Frei, Nutrients, 2013, 5, 5161 CrossRef CAS.
- B. H. J. Bielski and A. O. Allen, J. Phys. Chem., 1977, 81, 1048–1050 CrossRef CAS.
- G. R. Buettner and B. A. Jurkiewicz, Radiat. Res., 1996, 145, 532–541 CrossRef CAS.
- Q. Chen, M. G. Espey, M. C. Krishna, J. B. Mitchell, C. P. Corpe, G. R. Buettner, E. Shacter and M. Levine, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13604–13609 CrossRef CAS.
- Q. Chen, M. G. Espey, A. Y. Sun, C. Pooput, K. L. Kirk, M. C. Krishna, D. B. Khosh, J. Drisko and M. Levine, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11105–11109 CrossRef CAS.
- X. You, S. Yang, J. Li, Y. Deng, L. Dai, X. Peng, H. Huang, J. Sun, G. Wang, P. He, G. Ding and X. Xie, ACS Appl. Mater. Interfaces, 2017, 9, 2856–2866 CrossRef CAS.
- A. Vittore, M. R. Acocella and G. Guerra, Langmuir, 2019, 35, 2244–2250 CrossRef PubMed.
- D. P. Suhas, T. M. Aminabhavi, H. M. Jeong and A. V. Raghu, RSC Adv., 2015, 5, 100984–100995 RSC.
- E. O. Ogunsona, T. Grovu and T. H. Mekonnen, Sustainable Mater. Technol., 2020, 26, e00208 CrossRef CAS.
- J. Shen, P. T. Griffiths, S. J. Campbell, B. Utinger, M. Kalberer and S. E. Paulson, Sci. Rep., 2021, 11, 7417 CrossRef CAS PubMed.
- G. J. Kontoghiorghes, A. Kolnagou, C. N. Kontoghiorghe, L. Mourouzidis, V. A. Timoshnikov and N. E. Polyakov, Medicines, 2020, 7, 45 CrossRef CAS.
- S. Tian, J. Sun, S. Yang, P. He, G. Wang, Z. Di, G. Ding, X. Xie and M. Jiang, Sci. Rep., 2016, 6, 34127 CrossRef CAS.
- M. B. Yim, B. S. Berlett, P. B. Chock and E. R. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 394–398 CrossRef CAS.
- M. Lin and J. Z. Yu, Environ. Sci. Technol., 2020, 54, 1431–1442 CrossRef CAS.
- G. R. Buettner and B. A. Jurkiewicz, Free Radical Biol. Med., 1993, 14, 49–55 CrossRef CAS PubMed.
- O. Augusto, D. R. Truzzi and E. Linares, Redox Biol. Chem., 2023, 5–6, 100009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further experimental and characterization details, summary of the ascorbic acid reduced graphene oxide samples mentioned in literature as well as extra characterization data and illustrative schemes. See DOI: https://doi.org/10.1039/d4ma00798k |
| This journal is © The Royal Society of Chemistry 2025 |