
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of the oxidation level of the graphene oxide substrate on in situ growth of COF-300†
Ying
Quan
ab,
Yizhou
Yang
b,
Qinfu
Liu
*a and
Karl
Börjesson
 *b
*b
aSchool of Geosciences and Surveying Engineering, China University of Mining and Technology (Beijing), 100083 Beijing, China
bDepartment of Chemistry and Molecular Biology, University of Gothenburg, Medicinaregatan 19, 41390 Gothenburg, Sweden. E-mail: karl.borjesson@gu.se
First published on 30th January 2025
Abstract
The synthesis of covalent organic framework (COF) based hybrid materials is highly important for society as it provides materials with a large variety of beneficial properties. However, the COFs in graphene–COF and reduced graphene oxide (rGO)–COF hybrids are mostly two-dimensional (2D) due to the challenge in the design and synthesis of three-dimensional (3D) COFs. rGO-3D COF composites were here synthesized using several different graphene oxide (GO) substrates via a simple ventilation-vial protocol. These composites, as well as the starting materials of GO, were characterized by XRD, Raman spectroscopy, XPS, FT-IR, TG, SEM and EDS. The mechanism of in situ growth of COF-300 on graphene is proposed, where the oxygen-containing functional groups on GO are assumed to play a leading role in anchoring COF-300. Interestingly, a change in the morphology of COF-300 particles on the GO substrate was observed. It is found that GO acts as not only the substrate but also a structure-directing agent for modulating the morphology of COF-300. The high oxidation level and the large interlayer distance of GO are beneficial for growing COF-300 with smaller length, higher loading and more uniform distribution. This finding opens an avenue to control the morphology of COFs just by regulating the GO substrate. This work also covers GO prepared from natural coaly graphite, which promotes the high-value utilization of natural coaly graphite resources.
Introduction
Covalent organic frameworks (COFs),1 consisting of light elements (B, C, N, O, and Si) linked via strong covalent bonds, are an emerging class of new crystalline and porous nanomaterials.2 COFs are classified based on their dimensionality into two- (2D) and three-dimensional (3D) COFs. 2D COFs are planar structures that stack in a similar fashion to graphite. 3D-COFs, on the other hand, have an isotropic structure, with covalent bonds in all three dimensions. 3D COFs are harder to make and characterize compared to their 2D analogues,3–6 but allow for high chemical and mechanical stabilities, tuneable pore structures and large porosity.3,7,8 They have found use in a variety of applications, ranging from gas storage/separation to catalysis and chemical sensing. However, the low electrical conductivity of most COFs limits their use in optoelectronics and electrical energy storage devices.9–14Graphene and reduced graphene oxide (rGO) are 2D materials with high conductivity and specific surface area, and they have been shown to work excellently as a part of composite materials.15,16 Specifically, several composites containing graphene and COFs have been made in order to improve the conductivity of the COFs.17–19 An et al.20 fabricated a rGO-2D COF composite through the electrostatic self-assembly between negatively charged graphene oxide (GO) nanosheets and a positively charged COF. Furthermore, Xiong et al.21 reported 2D COF films that were in situ synthesized on Cu supported graphene, where the two entities were held together through π–π interactions. GO contains functional groups such as carbonyl, carboxyl and epoxy that allow GO to participate in a wide range of coupling reactions, and the covalent coupling between GO and various COFs has been demonstrated.22–25 Such a covalent connection between the two entities in a composite material is beneficial as the strength of the connection is on the same order of magnitude as the strength of the individual materials, thus minimizing weak areas. As mentioned above, researchers have proposed many treatments to synthesise the graphene–COF and rGO–COF composites. Note that COFs in these composites are 2D COFs, not 3D COFs. The construction of rGO-3D COFs is still in its infancy due to the challenge in the design and synthesis of 3D COFs alone.6,26–30 Nonetheless, Feng et al. reported the successful synthesis of GO/3D COF(COF-300)/PPy nanocomposites. By adjusting the ratio of GO/COF-300/PPy, the composites were shown to be effective in the removal of organic contaminants in water.25 However, the influence of GO on the 3D COF growth process and the final morphology of the composites were not explored. Moreover, at present, most research related to tuning the growth of the COFs primarily modifies the monomer used in the COF polymerization reaction, adjusts the reaction concentration or functionalizes the COFs to control the crystallinity, size, and shape of the COFs.18,23,31–33 Thus, to the best of our knowledge, the effect of the GO substrate on the growth of 3D COFs has never been explored.
COF-300 belongs to the group of 3D imine-based COFs, which is synthesized from the condensation of amines and aldehydes, and it can be synthesized conveniently and efficiently.34–36 Here, COF-300 was selected to react and form a composite with GO. The growth mechanism of the COF-300 particles on graphene is proposed, where GO firstly covalently binds to the tetrakis-4-aminophenyl methane (TAM) monomer used in the reaction, thus forming a nucleation seed for further COF-300 growth. This interpretation is supported by two observations: The first one is that no homogeneous composite is formed without the seeding reaction. The other is that the morphology of COF-300 crystals depends on the concentration of GO and thus on the number of seeding sites. Afterwards, three GO sources were used to explore the effect of GO on COF growth. Interestingly, the morphology of COF-300 on rGO varies with the source of the GO, which we ascribe to the oxygen content of the GO, and thus seeding sites. Combined with investigating the structure of GO, we demonstrate that the GO acts as not only the substrate but also as a structure-directing agent for modulating the size, amount and distribution of COF-300. This work reveals a new perspective on the controlled growth of COFs on the GO substrate.
Experimental methods
Materials
One natural coaly graphite sample was collected from the Lutang mining area, in Hunan Province, China, which is labelled as LT. The other natural coaly graphite sample was collected from the Xinhua mining area, in Hunan Province, China, which is labelled as CM powder. Commercial GO powder products were purchased from Leadernano Co. and Sigma-Aldrich Co. Tetrakis-4-aminophenyl methane (TAM, ≥95%) was purchased from TCI Europe. Terephthalaldehyde (TPA, ≥99%), acetic acid (≥99%), anhydrous 1,4-dioxane (≥99.5%), cyclohexane (≥99%), acetone (≥99.5%), ethanol (96%) and 1-methyl-2-pyrrolidinone (NMP, ≥99%) were purchased from Sigma-Aldrich Co. Hydrochloric acid (HCl, 32%) and hydrofluoric acid (HF, 40%) were purchased from Merck Co. All reagents were used without further purification.Preparation of GO dispersions
To investigate the structural effect of the GO on the rGO–COF-300 composites, except the commercial GO powders, another GO sample was synthesized from the LT natural block coaly graphite using the electrochemical exfoliation method as reported in our previous work.37,38 Specifically, GO was obtained in a two-electrode configuration by using the LT natural coaly graphite as an anode and platinum as a cathode, and the distance between electrodes was around 4 cm. The electrolyte solution was 1 M ammonium sulphate ((NH4)2SO4). A direct current bias voltage of +8 V was applied. After the exfoliation process, lasting for 1 h, the dispersion was sonicated for 1 h. Then, it was cleaned several times with DI water by centrifugation, followed by drying in an oven at 60 °C for 24 h. Finally, GO was purified by acid leaching treatment (using HCl and HF).To determine the feasibility of using natural coaly graphite, CM raw graphite was also explored. To be specific, firstly, the CM powder was treated using acid leaching treatment. After removing the inorganic mineral impurities of the natural coaly graphite, they were dispersed in NMP at a concentration of 10 mg mL−1 by sonicating for 800 min. Subsequently, the product was centrifuged to remove the NMP and unexfoliated CM coaly graphite. Finally, the GO powder was obtained by drying at 60 °C for 24 h.
For the preparation of GO dispersions, the GO powder was dispersed into DI water at a certain concentration by sonication for 10 min.
Fabrication of GO–TAM
20 mg of TAM was completely dissolved in 1.6 mL of anhydrous 1,4-dioxane in a 10 mL vial. Then, 0.4 mL of cyclohexane and 1 mL of 3 M acetic acid (aqueous) were added, and the vial was sonicated for 5 min. After that, the solution was dropwise added to a 1 mL GO suspension and stirred for 3 h at 60 °C to obtain a homogeneous dispersion as the intermediary product.Preparation of rGO–COF-300 composites
12 mg of TPA was added to the GO–TAM solution and heated at 65 °C for 72 h. After cooling down to room temperature, the obtained rGO–COF-300 composites were collected by centrifugation, washed with acetone and ethanol, and dried in an oven at 60 °C for 24 h. For comparison, 1 rGO–COF-300, 10 rGO–COF-300, 20 rGO–COF-300 and 30 rGO–COF-300 composites were also prepared via the same procedure using 1, 10, 20 and 30 mg mL−1 GO dispersions, respectively.Moreover, BlankG–COF-300 was synthesized by directly adding the monomers, catalyst and solvents into the GO suspension (20 mg mL−1, 1 mL) simultaneously and then following the same procedure. In addition, pure COF-300 was also synthesized as a comparison under similar experimental conditions without the addition of the GO suspension.
Characterization
X-ray diffraction (XRD) measurements were performed with a Bruker D8 Discover at 40 kV and 40 mA using CuKα radiation (λ = 0.15418 nm). Data were scanned in the continuous mode over the angular range 5–50° (2θ) with a time per step of 0.1 s.Raman spectroscopy was conducted using a WITec alpha 300R equipped with a 532 nm laser. The beam size was 1 μm and the power at the sample was below 10 mW with a typical exposure time of 30 s, in order to avoid laser induced heating. Three spectra were recorded for each sample in the range of 1000 to 2000 cm−1 to ensure that the presented spectra are representative.
Fourier transform infrared spectroscopy (FT-IR) measurements were performed using a Bruker Vertex 70v spectrometer in the range of 400–4000 cm−1 at a resolution of 4 cm−1.
The surface chemical composition of the samples was determined by X-ray photoelectron spectroscopy (XPS) on a Thermo Scientific K-Alpha spectrometer, using an Al Kα X-ray source (hv = 1486.6 eV) with a 900 mm beam diameter. Survey-scan spectra were recorded at a pass energy of 100 eV and an energy step of 1 eV. High-resolution spectra of carbon were recorded at a pass-energy of 50 eV and an energy step of 0.1 eV. A survey scan was required between 1000 and 0 eV. For calibration purposes, the C 1s binding energy of the graphitic peak was referenced as 284.5 eV. The spectra were analysed by peak fitting using Avantage software.
Morphological studies were carried out using a Quanta 200 ESEM scanning electron microscopy (SEM) at the acceleration voltage of 15 kV with an energy dispersive X-ray spectrometer (EDS) measurement at the acceleration voltage of 5 kV. Before the SEM measurements, the samples were placed on a carbon conductive tape and sputter-coated with a thin layer of gold (around 4 nm).
Thermogravimetry (TG) analysis was carried out using a STARe System TGA/DSC 3+ under air conditions from 30 to 950 °C at 10 °C min−1.
Results and discussion
The making of an rGO–COF-300 composite
To make a composite material containing both COF and graphene functionalities, it is important that these two entities contain covalent bonds to each other. The concept used here to achieve a covalent linkage is conceptually outlined in Scheme 1. Graphene oxide (GO) is used which contains a variable amount of carboxyl, epoxy, carbonyl and other oxygen containing functional groups depending on the source of the GO. The GO is first treated with tetrakis-4-aminophenyl methane (TAM), which is one of the monomers used in COF synthesis. The amine groups on TAM react in a Schiff base reaction with the oxygen containing groups on the GO under acidic conditions,39 forming an activated GO species that can anchor to COF particles in the subsequent COF synthesis. It will later be shown that this pretreatment has two important functions. Firstly, it provides steric hindrance to further separate individual GO sheets apart from each other, thus preventing them from aggregating. Secondly, it provides a high concentration of anchoring groups on the GO surface at the start of COF synthesis, in other words, providing a higher concentration of COF nucleation sites on the GO surface.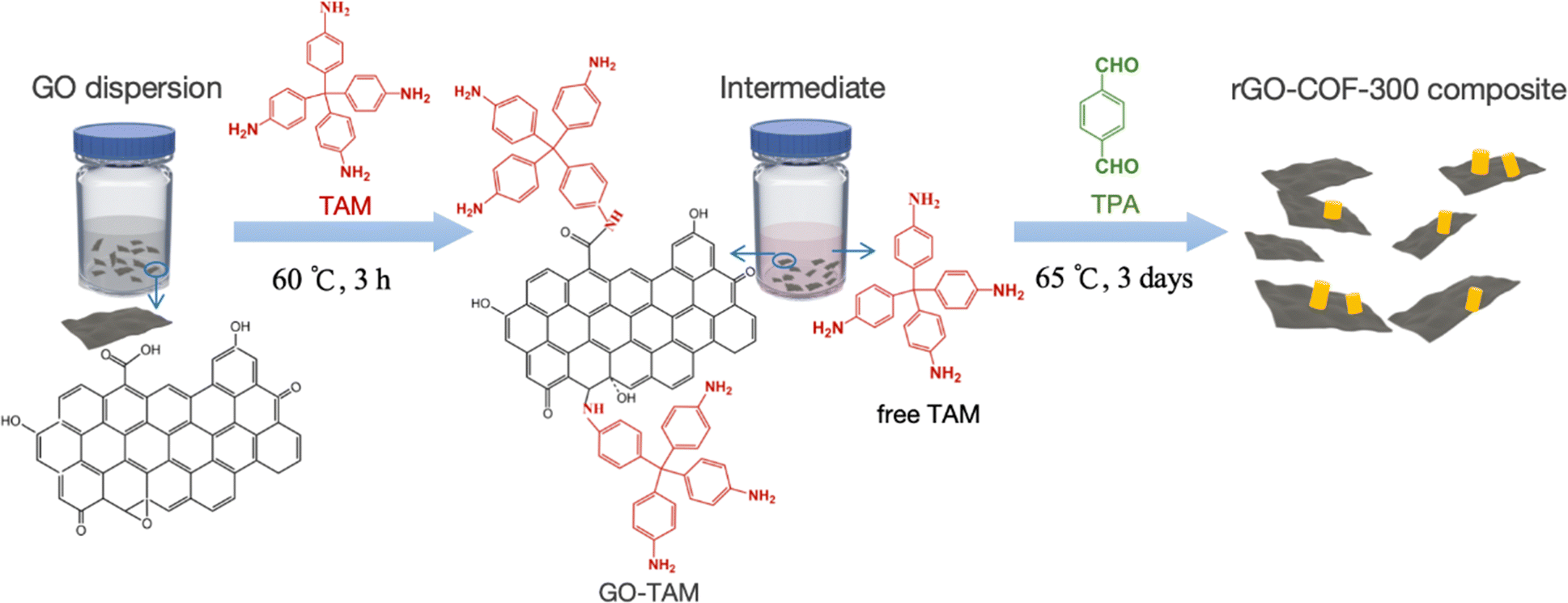 | ||
| Scheme 1 Schematic illustration for in situ growing COF-300 on graphene. The GO first reacts with an excess of TAM forming a high density of anchoring units on GO before terephthalaldehyde (TPA) is added and the COF-300 synthesis starts. | ||
Characterization of GO–TAM and the rGO–COF-300 composites
The binding of the anchoring groups to GO and the subsequent synthesis of the rGO–COF-300 composite was followed by XRD, and Raman and FT-IR spectroscopy. GO shows a typical (001) diffraction plane at a 2θ value of 12.6°, corresponding to an average interlayer spacing of around 7.0 Å, which is attributed to the existence of oxygen-containing functional groups on the surface of the graphene sheets (Fig. 1a). After reacting with TAM, the (001) diffraction plane shifts to a lower angle, at around a 2θ value of 6.5°. This angle corresponds to a larger interlayer spacing (13.65 Å) for the GO–TAM conjugate. Also, the full width at half maximum of the (001) diffraction plane increases. These results indicate that the TAM monomers have intercalated into the GO nanosheets, interfering with the stacking between GO layers, and giving a more disordered carbon structure. The chemical and structural purity of graphene is conveniently assessed by Raman spectroscopy. All graphene containing products exhibit two typical bands related to graphite-based structures (Fig. 1b). The D band, caused by the A1g vibrational mode, represents the presence of structural defects in the graphitic structure. The G band caused by the E2g vibration mode, reflects the in-plane stretching vibration of the sp2 hybrid structure.40 The ratio of the intensity of the D and G bands (ID/IG) represents the degree of defects. After reacting GO with TAM, the G band of the formed GO–TAM redshifts slightly from 1583 cm−1 to 1580 cm−1 accompanied by the decreased ID/IG value from GO (1.01) to GO–TAM (0.97). The Raman signal changes only slightly, but the change could imply partial restoration of some vacancies and defects in the GO carbon lattice. The change in the chemical composition was therefore assessed using FT-IR and XPS. The FT-IR spectrum of GO shows distinct peaks at 1722, 1620, 1400 and 1045 cm−1, which can be attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of carboxyl and carbonyl groups situated at the edges, aromatic CC stretching, O–H deformation and C–O–C stretching vibrations of the epoxy groups, respectively (Fig. 1c).9,41 The FT-IR spectrum of GO–TAM differs from that of GO in several important aspects. Firstly, new characteristic transitions appear at 1185, 1505 and 820 cm−1 in GO–TAM, which can be assigned to C–N stretching, N–H bending and wagging deformation vibrations, respectively,9,31,42 demonstrating the conversion of GO to GO–TAM. Secondly, the transition at around 1610 cm−1 was attributed to amides (inset in Fig. 1c)9,31,42 Although the FT-IR spectrum of GO also has a broad peak in the same region, GO contains almost no N atoms (as demonstrated by EDS in Fig. 3). Combining these observations with that of a decreased intensity of the O–H signal from carboxylic dimers (1400 cm−1),39 the FT-IR data are consistent with amide formation through a addition–elimination reaction between the carboxyl groups (–COOH) on the GO and the amino groups (–NH2) on TAM. Furthermore, the decreased intensity of O–H at around 3390 cm−1 (Fig. S1, ESI†) is either due to the removal of adsorbed water from GO–TAM,25 or due to the consumption of OH when forming amide bonds. Meanwhile, the strong transition at 3150 cm−1, attributed to the C–H stretching vibration, almost disappears in GO–TAM, indicating the simultaneous reduction of GO (Fig. S1, ESI†).40
O stretching vibrations of carboxyl and carbonyl groups situated at the edges, aromatic CC stretching, O–H deformation and C–O–C stretching vibrations of the epoxy groups, respectively (Fig. 1c).9,41 The FT-IR spectrum of GO–TAM differs from that of GO in several important aspects. Firstly, new characteristic transitions appear at 1185, 1505 and 820 cm−1 in GO–TAM, which can be assigned to C–N stretching, N–H bending and wagging deformation vibrations, respectively,9,31,42 demonstrating the conversion of GO to GO–TAM. Secondly, the transition at around 1610 cm−1 was attributed to amides (inset in Fig. 1c)9,31,42 Although the FT-IR spectrum of GO also has a broad peak in the same region, GO contains almost no N atoms (as demonstrated by EDS in Fig. 3). Combining these observations with that of a decreased intensity of the O–H signal from carboxylic dimers (1400 cm−1),39 the FT-IR data are consistent with amide formation through a addition–elimination reaction between the carboxyl groups (–COOH) on the GO and the amino groups (–NH2) on TAM. Furthermore, the decreased intensity of O–H at around 3390 cm−1 (Fig. S1, ESI†) is either due to the removal of adsorbed water from GO–TAM,25 or due to the consumption of OH when forming amide bonds. Meanwhile, the strong transition at 3150 cm−1, attributed to the C–H stretching vibration, almost disappears in GO–TAM, indicating the simultaneous reduction of GO (Fig. S1, ESI†).40
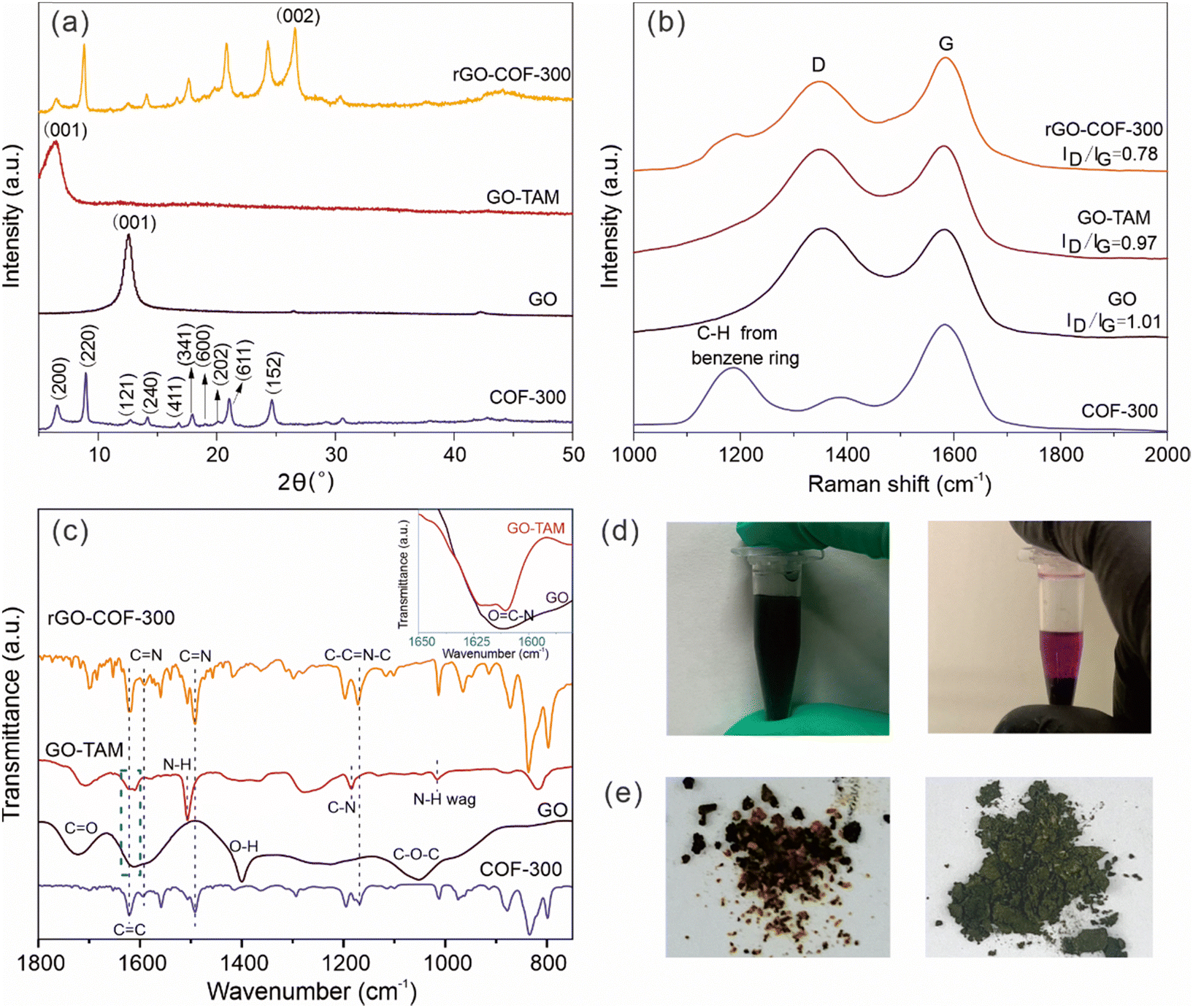 | ||
| Fig. 1 (a) XRD patterns, (b) Raman spectra and (c) FT-IR spectra of GO, GO–TAM, rGO–COF-300 and COF-300. (d) Digital photos of GO (left) and GO–TAM (right) solution (the GO–TAM solution was treated by centrifugation to see the colour change clearly). (e) Digital photos of the reference BlankG–COF-300 (left) and rGO–COF-300 (right). | ||
XPS results are also consistent with coupling between carbonyls and amino functionalities. The wide scan XPS spectra of the samples are shown in Fig. 2a. The O/C atomic ratio is around 44.1% in GO, which was subsequently reduced to 34.0%, after the grafting of TAM. More detailed information about the chemical compositions of GO and GO–TAM is shown in Fig. 2b and c. The C 1s XPS spectrum of GO exhibits five peaks at about 284.8, 287.0, 288.4 and 289.4 corresponding to C–C, C–O (hydroxyl/epoxy), CO (carbonyl/quinone) and O–CO bonds, respectively. It can be seen from Fig. 2c that the ratio of C–O bonds decreases in GO–TAM. Meanwhile, signals at binding energies of 285.1 and 286.9 eV, attributed to the C–N and OC–N bonds, indicate that the oxygen-containing functional groups of GO are bound to the TAM monomers. In addition, there is a C satellite signal at around 291.1 eV due to the energy loss in the measurement. The N 1s XPS spectra also confirm the successful grafting of GO through the appearance of a nitrogen peak in the GO–TAM spectra (Fig. S2, ESI†). Furthermore, SEM-EDX based elemental analysis (Table S1, ESI†) indicates that the GO–TAM sample exhibits a TAM loading of approximately 12.5%.
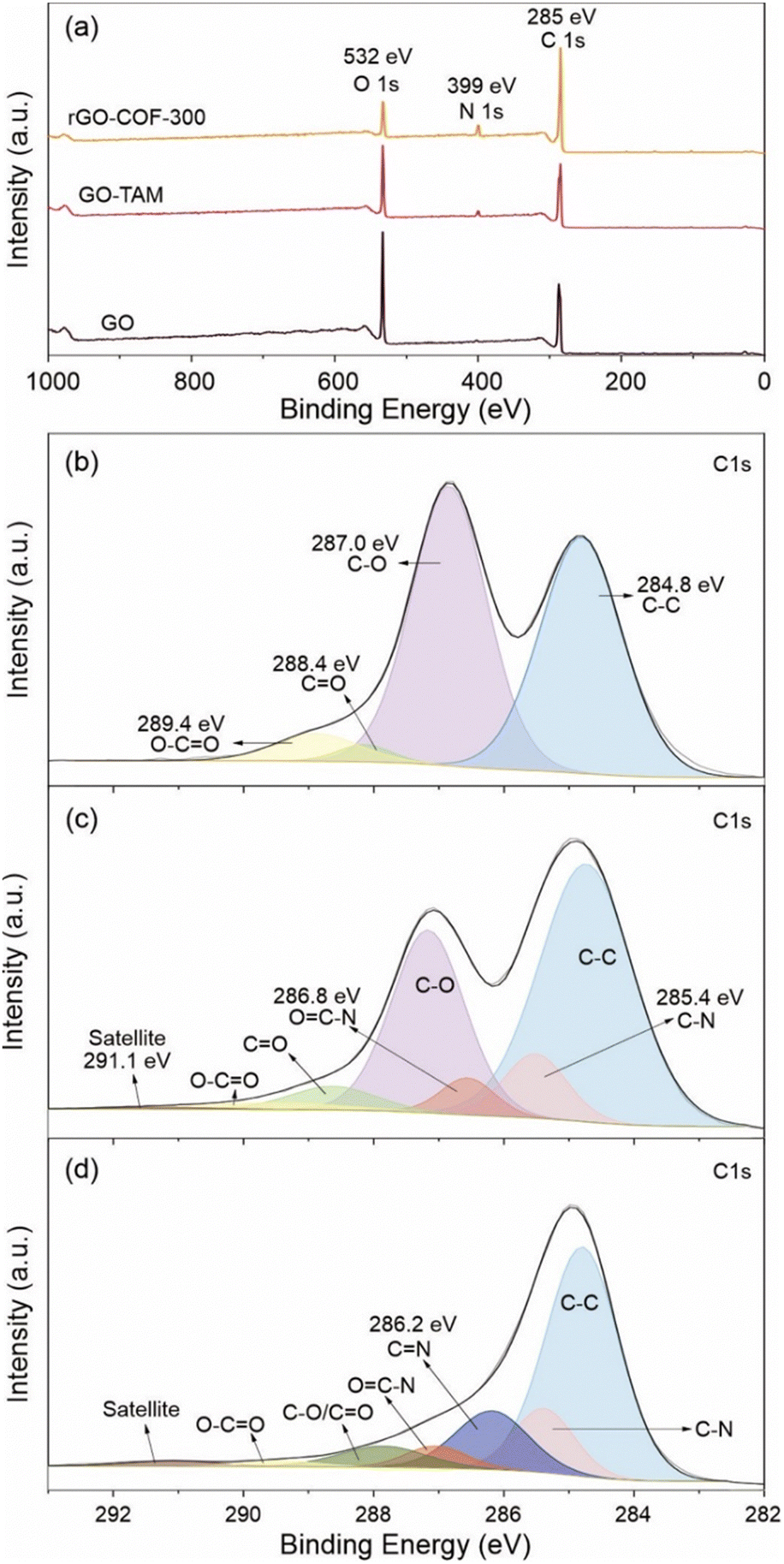 | ||
| Fig. 2 (a) Wide scanning XPS spectra of the GO, GO–TAM and rGO–COF-300. High-resolution C 1s spectra of (b) GO, (c) GO–TAM and (d) rGO–COF-300. The C 1s XPS spectrum of GO exhibits five peaks at about 284.8, 287.0, 288.4, 289.4 and 291.1 eV, which corresponds to C–C, C–O (including hydroxyl, epoxy, etc.), CO, O–CO and C satellite bonds, respectively.43,44 | ||
To summarize, the chemical binding of GO with TAM was confirmed by Raman, FT-IR and XPS, and the consequence of the conjugation of TAM to GO is an increased sheet distance between the graphitic layers. This covalent modification of GO is easily visualized in a clear violet colour of GO–TAM compared to the brownish/black colour of pristine GO (Fig. 1d), while the TAM stirred for 3 h at 60 °C under the same acid conditions but without the GO dispersion was transparent light yellow (Fig. S3, ESI†).
After the reaction of GO–TAM with terephthalaldehyde (TPA) and free TAM, the XRD spectrum of the formed rGO–COF-300 composite resembles that of COF-300. More specifically, the (200), (220), (121), (240), (411), (341), (600), (202), (611) and (152) diffraction planes are clearly visible. The 2θ values of these diffraction planes are consistent with reported COF-300 having a 5-fold interpenetrated dia topology.45,46 Thus, the presence of GO–TAM does not prohibit the formation or radically change the crystal structure of COF-300, and rGO–COF-300 exhibits a COF-300 loading of around 37% (Table S1, ESI†). Furthermore, besides the typical COF-300 peaks, the XRD pattern of rGO–COF-300 also shows a sharp and strong peak at approximately 26.6°, which is assignable to the (002) diffraction plane of rGO.47 It is due to the reduction of GO under hydrothermal conditions. The Raman spectrum of the rGO–COF-300 composite includes the band at 1180 cm−1, corresponding to C-Harom from benzene rings in COFs,24,48 and the typical G and D peaks of graphene, thus confirming the growth of COF-300 on graphene since it preserves the main characteristic bands of both graphene and COF-300.
The FT-IR spectrum of pure COF-300 (Fig. S4, ESI†) shows the appearance of CN imine stretching (1590 cm−1) and C–CN–C (1170 cm−1) vibrations, and the disappearance of the stretching vibration of the amino group on TAM. These changes in the FT-IR spectra are in agreement with previous reports of COF-300.43 Furthermore, for the rGO–COF-300 composite, the existence of bands at 1590 cm−1 and 1170 cm−1 indicates the successful formation of CN and C–CN–C bonds in the in situ grown COF-300 on GO. XPS corroborates the conclusions drawn from the FT-IR analysis. The high-resolution N 1s spectrum of rGO–COF-300 compared to that of GO–TAM shows the buildup of CN double bonds (Fig. S2, ESI†), indicating an increased amount of imine bonds. Further, according to the high-resolution C 1s spectra (Fig. 2), the relative amount of CO bonds reduced for rGO–COF-300, which is in alignment with expectations.
In summary, the grafting of COF-300 on graphene could be followed chemically by Raman, FT-IR and XPS, and XRD, indicating no major change in the crystal structure when COF-300 is grown in the presence of graphene. However, it is difficult from these results to distinguish if COF-300 is covalently attached to GO, or if the two materials are just physically blended. In a first attempt to check the chemical binding between the two materials and if it affects material properties, a reference material (BlankG–COF-300) was made. The reference material was made in the same manner as -COF-300 with the exception that the pretreatment of TAM was removed. The consequence of removing this pretreatment is that no TAM is present on the GO at the start of COF synthesis, reducing the likelihood of nucleation points on the GO and making the GO less well dispersed in the solvent. Fig. 1e shows a picture of rGO–COF-300 and the reference BlankG–COF-300. rGO–COF-300 shows a homogenous greenish colour, while the reference, BlankG–COF-300, contains black and yellow particles physically blended together. As graphene is black and COF-300 is yellow it is possible to interpret these results as the two materials are only physically blended in BlankG–COF-300, and that the pretreatment of TAM is a necessary step to make an rGO–COF-300 composite.
To gain more insight into the as-synthesized materials, the morphologies of GO, GO–TAM and rGO–COF-300 were observed by SEM. Fig. 3a and c show a wrinkled morphology of GO and GO–TAM with a visible layer arrangement. Meanwhile, after the TAM modification, the EDS mapping of GO–TAM shows a relatively uniform introduction of N (Fig. 3b and d), due to the formation of amide bonds and the existence of N in TAM, which could promote the subsequent growth of COF-300. The drop in the oxygen content occurs simultaneously, further demonstrating that some of the oxygen-containing functional groups of GO were reduced during the grafting of TAM. For the composite material, rGO–COF-300, a large number of COF-300 particles are uniformly distributed on the graphene sheets. These particles are rod-shaped, and quite similar to the shape although significantly shorter compared to COF-300 crystals made in the absence of GO (Fig. S5, ESI†).49 Moreover, a further increase in N and a decrease in O is seen in the EDS mapping, which is expected due to the generation of COF-300. To examine if the COF-300 material is mostly present as needles or if it is also present as layers on top of the graphene as previously shown for 2D-COF graphene conjugates,50,51 TEM image and the corresponding EDS mapping are presented in Fig. S6 (ESI†). A high N content is only present on crystallites, indicating that COF-300 does not form a continuous film on the GO but is rather localized to the observed bound crystals.
 | ||
| Fig. 3 SEM images of (a) GO, (c) GO–TAM and (e) rGO–COF-300, and the corresponding EDS mappings of (b) GO, (d) GO–TAM, and (f) rGO–COF-300. | ||
It is interesting to compare the micromorphology of rGO–COF-300 with the reference sample, BlankG–COF-300. For BlankG–COF-300 the COF crystals lay in bundles beside the graphene layers (Fig. S7, ESI†), which is in line with the macroscopic picture (Fig. 1e), showing separate graphene and COF particles. This must be due to the omission of the pretreatment step, where the TAM monomers functionalize GO, thus leading to an insufficient grafting of TAM on the GO substrate and few nucleation sites of COF-300 on the graphene substrate.
To further explore the picture of the functionalized GO acting as nucleation sites for the COF-300 synthesis, a series of rGO–COF-300 samples having a decreasing amount of GO–TAM were prepared. The concentration of the modified graphene relates to the concentration of nucleation sites for the COF on graphene. The graphene/monomer concentration ratio therefore tunes the number of COF crystals vs. the amount of material that can build up the crystals (i.e. their maximal theoretical size). Noticeably, on the basis of the statistical analysis of the size of COF-needles, performed from SEM images (Fig. S8, ESI†), when decreasing the GO content, a gradual increase in the length of COF-300 needles can be observed (Fig. 4). For very low GO concentrations, the size of the needles even approaches that seen in pure COF-300. Overall, the morphology of COF-300 transforms from short rod-like to nanofibrous structures to longer ones with a decrease in GO content in the reaction. These results confirm that the oxygen-containing groups of the GO are taking part in the reaction and play a decisive role in controlling the growth size of COF-300 in the composite material.
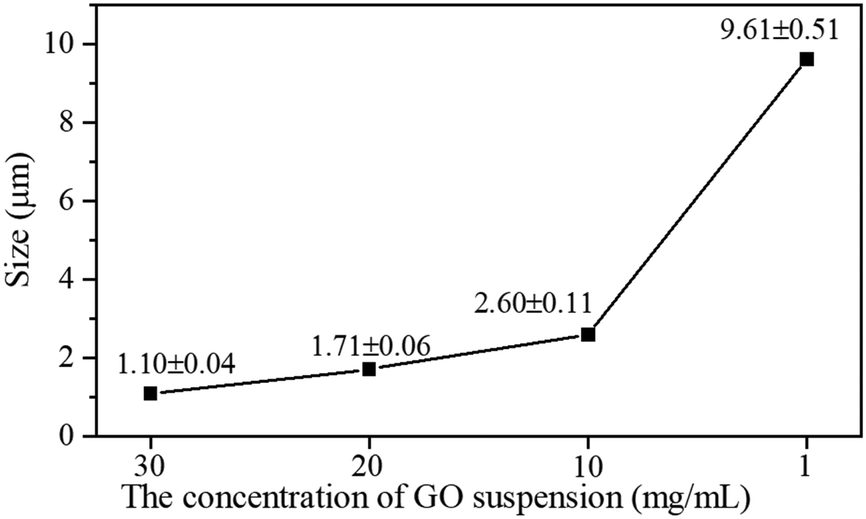 | ||
| Fig. 4 The size of COF-300 crystallites on graphene as a function of the concentration of the GO suspension used in the synthesis. | ||
The effect of GO on the growth of COF-300
With reducing the amount of GO in the synthesis step, the size of COF-300 crystallites approaches the size observed when omitting GO completely in the synthesis step (10 μm; Fig. 5a and Fig. S9 and S10, ESI†). Three different GO, both synthesized in this work and commercial, were therefore selected to examine the effect of the GO source on the morphology of the rGO–COF-300 composites. Commercial GO powders from two different companies are labelled as GO (source 1) and GO (source 3), respectively. GO produced from LT natural coaly graphite by the electrochemical exfoliation method is labelled as GO (source 2). Based on the starting GO (source 1), GO (source 2) and GO (source 3), the corresponding intermediates are labelled as GO (source 1)–TAM, GO (source 2)–TAM and GO (source 3)–TAM under the same reaction conditions (20 mg mL−1 of GO dispersion), and the obtained composites are labelled as rGO (source 1)–COF-300, rGO (source 2)–COF-300 and rGO (source 3)–COF-300, respectively. | ||
| Fig. 5 SEM images of (a) pure COF-300, (b) rGO (source 1)–COF-300, (c) rGO (source 2)–COF-300, and (d) rGO (source 3)–COF-300. | ||
Fig. 5b–d displays the rod-like COF-300 particles grown on all GO substrates. The size of COF-300 crystallites on the substrates of GO (source 1), GO (source 2) and GO (source 3) is distinguished from each other. In the case of the rGO (source 1)–COF-300 composite, the average length of COF-300 was found to be around 1.62 μm, while larger COF-300 particles were observed for rGO (source 2)–COF-300, having an average size of 2.13 μm. The size of COF-300 particles shows a further increase for rGO (source 3)–COF-300, around 2.65 μm. Thus, it can be speculated that the GO substrate may play a crucial role in controlling the morphology and size of COF-300 particles.
To further investigate the effect of the GO on the growth of COF-300, the chemical structure of the three GO sources was studied by XPS. The XPS survey spectra of all GO sources are displayed in Fig. 6. It shows that the GO (source 1) sample has a higher content ratio of O 1s atoms to C 1s atoms (O/C = 44.0%) than GO (source 2) (O/C = 24.8%) and GO (source 3) (O/C = 5.1%). Thus, the study will show the influence of the oxidation level of the GO on COF-300 growth. Fig. 6b–d show the high-resolution C 1s spectra of GO (source 1), GO (source 2) and GO (source 3). Clearly, the strong peaks in the high-resolution C 1s spectrum of GO (source 1) show oxygen-containing functional groups rather than C–C groups, while the C–C groups gradually become the main bonds in GO (source 2) and GO (source 3). For quantitative comparison, the oxidation level of these GO was estimated based on the area ratio of oxygen-containing groups in the high-resolution C1s spectra rather than the atom ratio of O to C. The reason is that the purified GO (source 2) samples, prepared from the natural coaly graphite still may contain some silicate minerals, thus leading to the increase of the O atom content. As a result, the oxidation level of the GO samples varies in the order GO (source 1) (51.8%) > GO (source 2) (36.1%) > GO (source 3) (22.0%). It is worthwhile to point out that the content of C–O bonds in GO samples is much higher than that of OC–O bonds (Fig. 7a), which means the oxidation degree of these GO is dominated by C–O groups. At the same time, the nitrogen-containing functional groups on this intermediate product (including OC–N and C–N) decrease from rGO (source 1)–TAM (12.9%) to rGO (source 2)–TAM (10.8%) and to rGO (source 3)–TAM (8.1%), indicating the decreased content of grafting TAM in GO–TAM samples (Fig. S11, ESI†). As shown in Fig. S12 (ESI†), the N/C ratio of rGO (source 1)–COF-300 is calculated to be 7.0%, which is higher than those of rGO (source 2)–COF-300 (6.7%) and rGO (source 3)–COF-300 (5.2%). It also indicates that the rGO (source 1)–COF-300 sample exhibits the highest content of NC bonds (12.3%), while the value for rGO (source 3)–COF-300 is the lowest, only 3.5%.
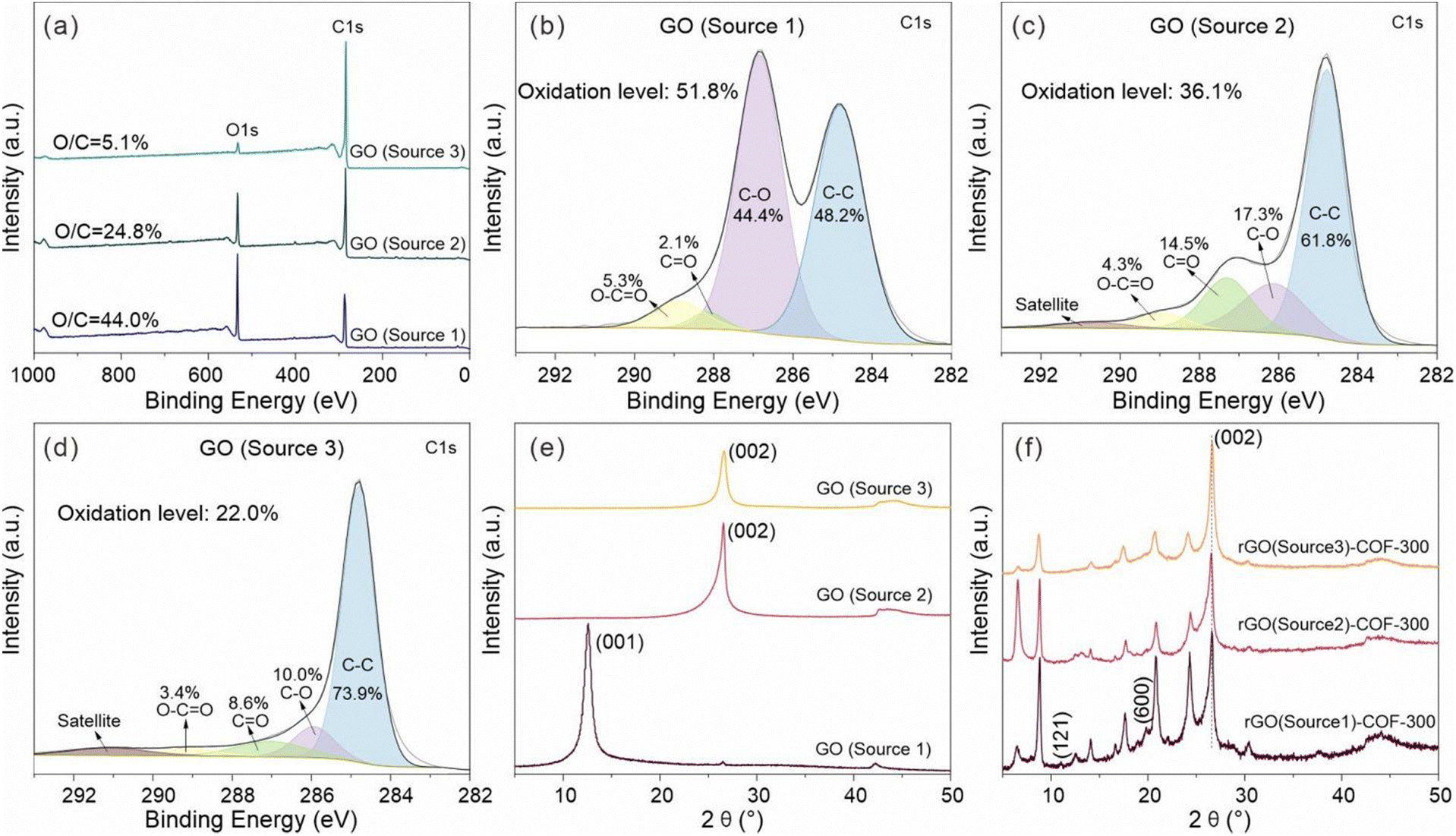 | ||
| Fig. 6 (a) Wide scanning XPS spectra of GO samples, high-resolution C 1s spectra of (b) GO (source 1), (c) GO (source 2) and (d) GO (source 3). The XRD patterns of (e) GO and (f) rGO–COF-300 composites. | ||
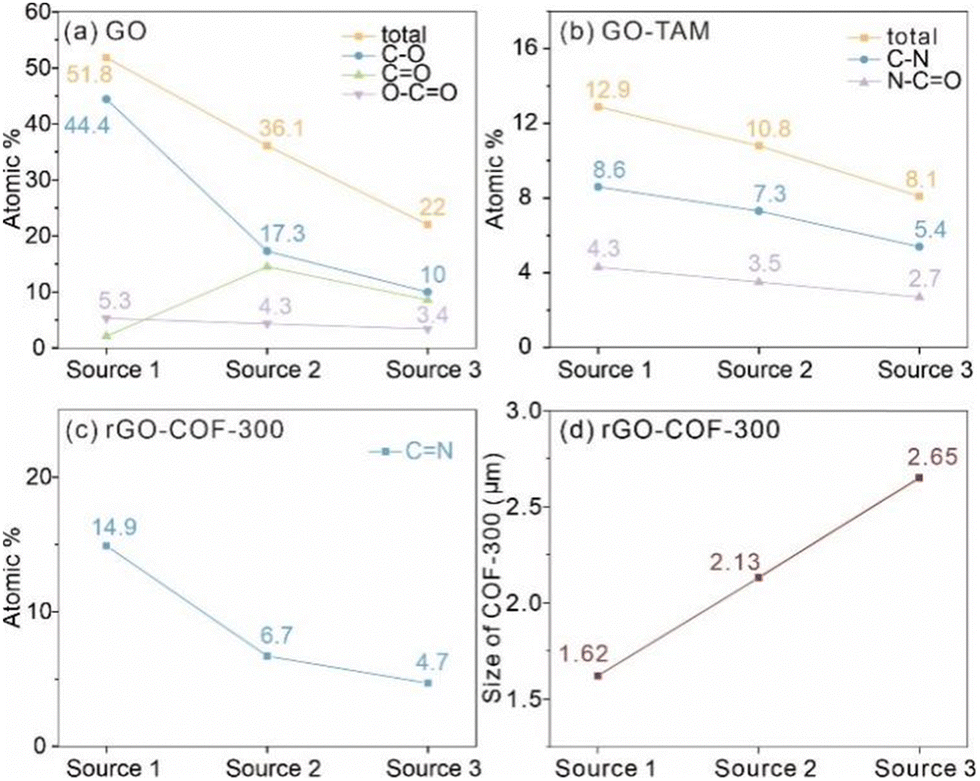 | ||
| Fig. 7 (a) The content of oxygen-containing groups in GO samples, (b) The content of nitrogen-containing groups in GO–TAM samples, (c) The content of CN in rGO–COF-300 samples, and (d) the average size of the COF-300 crystallites on the GO. | ||
These results are in proportion to the oxidation degree of the starting GO sources. The relationship between the oxidation level, TAM grafting, and size of COF crystallites is shown in Fig. 7. The higher the degree of oxidation level of the GO source, the more TAM monomers are bound in the grafting reaction. The linkages connecting GO and TAM are based on amides (as previously suggested by FT-IR) and C–N, which are produced using carboxylic acid and nucleophilic substitution reactions, respectively (Fig. 7b). The functional groups of interest are thus the ones containing carboxylic acids (O–CO) and epoxides (C–O). In other words, with more reactive oxygen-containing groups, more TAM monomers can be bound, thus resulting in more sites to connect to COF-300. Furthermore, it cannot be ignored that the COF-300 particles on graphene vary not only in amount but also in size, as mentioned in the SEM section. It can thus be postulated that the lower number of binding sites on the GO substrate with a low oxidation level results in more available TAM monomers per binding site (if the starting concentration of TAM monomers is the same), thus giving rise to larger COF-300 crystallites on the GO substrate (Fig. 7d). Overall, the size and the amounts of COF-300 particles on the graphene substrate can be controlled simultaneously based on the different oxidation degrees of the GO substrate.
The physical microstructure of GO was also studied to explore the influence of GO on the growth of COF-300. The XRD patterns of samples from the different GO sources are shown in Fig. 6e. It is clear that the diffraction planes at 12.6° disappear in GO (source 2) and GO (source 3) samples when compared with GO (source 1), while typical diffraction planes associated with the graphitic (002) diffraction plane were observed at high diffraction angles at 26.6° corresponding to the d002 value of 3.35 Å. It means that there is a higher ordered carbon structure in GO (source 2) and GO (source 3) samples as a consequence of the small amount of oxidation of the graphene. Additionally, it is observed that the (002) diffraction plane of GO (source 3) is more symmetrical than that of GO (source 2). It implies that GO (source 3) exhibits the most ordered carbon structure with the most tightly stacked layers among the three GO materials, which is consistent with its lowest level of oxidation. GO (source 1), like the typical XRD patterns of GO in previous reports,52,53 shows an enhancement of an interlayer distance from 7.00 Å to 13.65 Å after the addition of TAM into GO. In contrast, the obtained rGO (source 2)–TAM and rGO (source 3)–TAM still maintain a similar (002) diffraction plane position with the starting GO (source 2) and GO (source 3), respectively (Fig. S13, ESI†), which means that the interlayer distance does not change significantly after the treatment. This is because the starting GO (source 1) is highly oxidized, exhibiting the poorest graphitic structural order and largest layer spacing when compared to GO (source 2) and GO (source 3). As the interlayer distance of GO increases, it is easier to further enlarge the interlayer distance of the intermediate graphene–TAM because intercalation of TAM is more feasible. Generally, the increasing interlayer spacing of GO samples comes from its increased oxygen-containing functional groups,52,53 which is also in agreement with our XPS and XRD results. Hence, it also means that the distribution of COF-300 on the graphene substrate can be influenced by the oxidation level of GO. It is feasible to use the parameter of the oxidation level of GO to control the distribution of the growth of the COF-300 particles on the graphene substrate. However, taken together, XPS, XRD and EDS results suggest that the TAM monomers have been attached to all GO substrates.
All rGO–COF-300 samples show the main diffraction planes of COF-300, as shown in Fig. 6f, revealing the successful synthesis of COF-300 composites. However, it could be seen that the (121) and (600) diffractions of COF-300 disappear for rGO (source 3)–COF-300. It indicates that, despite the existence of the crystalline nature of COF-300 in these composites, the crystallinity of COF-300 is poorer in a composite with GO (source 3) compared to GO (sources 1 and 2). Thus, it can be concluded that the GO substrate allows for the tuning of not only the morphology, but also the crystallinity of the COF.
The high oxidation level of GO might be beneficial for the growth of the COF with long-range molecular ordering due to the fixed growth sites, while the low oxidation level of the GO substrate may interfere with the growth of COF-300 with high crystallinity. To test this hypothesis, GO synthesized from the CM natural coaly graphite (source 4) was also investigated by XPS (Fig. S14a and b, ESI†). The result shows that the oxidation level of GO (source 4) is only 19.2%, which is lower than that of GO (source 3) (22%). Fig. S14c (ESI†) shows the XRD pattern of the synthesized rGO (source 4)–COF-300 composite. As expected, some diffraction planes such as (411), (600) and (202) disappear. The result indicates that GO substrates with a low oxidation level are unfavourable for growing COF-300 with high crystallinity. Additionally, as shown in Fig. S14d (ESI†), the average size of the COF-300 particles on the GO (source 4) substrate is approximately 2.70 μm, which is larger than those of rGO (source 3)–COF-300 (2.65 μm), rGO (source 2)–COF-300 (2.13 μm) and rGO (source 1)–COF-300 (1.62 μm), consistent with the above discussion.
Considering the recent trend of rGO–COF composites in optoelectronics,8,51,54,55 the purity of the composite is very important. Hence, GO (source 2) and GO (source 4) in this work were purified via acid leaching of the GO or the natural coaly graphite precursor. The XRD results clearly display the presence of the characteristic diffraction plane of COF-300 and the absence of the diffraction peaks of impurity minerals. Thus, there is no doubt that the purified composite can be fabricated by purifying GO.
However, the traditional process flow to purify GO is complex and has high energy consumption, including crushing, grinding, purifying and oxidizing the natural graphite.56 For example, the purification process needs large amounts of chemical reagents or high temperature treatment (>2700 °C).57 To reduce energy consumption, GO (source 5) was synthesized directly from LT natural block coaly graphite by a one-step electrochemical exfoliation method without purification or independent crushing and grinding. The ash content of the GO was confirmed by TG analysis. Fig. S16a (ESI†) shows the ash content of the GO where acid leaching is only 0.03%, while the ash in the GO without acid leaching is severe, around 8.46%. The ash in GO (source 5) is mainly from impurity minerals, as demonstrated in Fig. S16b (ESI†). It can be seen that apart from the graphitic peak, it also includes the chlorite peak at around 12.5° and the quartz peak at 25.2°. Actually, these impurities in the obtained GO (source 5) were mostly removed when compared to the raw graphite mineral (Fig. S17, ESI†). As our previous studies reported, some impurity minerals can be removed during the electrochemical exfoliation process due to the delamination of the graphite and thus the exposure and separation of impurities.37 The obtained G (source 5)–COF-300 composite was also studied. Interestingly, small amounts of impurities in GO (source 5) did not prohibit the growth of well-crystallized COF-300 (Fig. S16c, ESI†). It should be mentioned that two commercial GO in this study were prepared from the ‘Hummers’ method, which consumes strong oxidants and acids, while GO (source 5) was prepared by employing the neutral salt solution ((NH4)2SO4) and without an acid leaching process. Therefore, natural coaly graphite could be a promising parent material for growing rGO–COF composites using a more environmentally friendly method.
To further understand the mechanism by which the GO substrate regulates the growth of COF-300, Raman spectroscopy was performed (Fig. S15, ESI†). Compared to the GO sources themselves, all ID/IG values of the rGO–COF-300 composites decrease. These changes amply illustrate that all GO samples are reduced during the synthesis of COF-300. The highest ID/IG value is seen for GO (source 1), which means that it exhibits the most active sites in its graphene planes, leading to the highest amount of TAM graft followed by the growth of the most mass proportion of COF-300. As a result, the signal intensity of COF-300 for the rGO (source 1)–COF-300 composite is larger than for the rGO (source 2 and 3)–COF-300 composites. It suggests that more defects on the GO substrate could accommodate more COF-300. In addition, it should be noted that GO (source 2) exhibits a high ID/IG value, which is close to that of GO (source 1). However, the signal intensity of COF-300 in rGO (source 2)–COF-300 is much lower than that of rGO (source 1)–COF-300. This may be due to the tightly stacked graphitic layers in GO (source 2), which is unfavourable for grafting TAM monomers. These results clearly suggest that the defect degree of GO is not the only reason, but also the expanded interlayer distance of GO is highly influential for the size- and content-controlled synthesis of COF-300 on graphene substrates. Moreover, the decrease of the ID/IG value from GO (source 3) to rGO (source 3)–COF-300 is much lower when compared to GO (source 1/2) to rGO (source 1/2)–COF-300. Combined with the morphology results, this could be due to the larger size of COF-300 particles on rGO (source 3)–COF-300, which induced more defects on the graphene plane.
Conclusions
In summary, 3D COF-300 was successfully grown on a variety of GO substrates using a simple ventilation-vial protocol. The carbonyl, carboxyl and epoxy groups of the GO substrate could covalently bind with TAM monomers, which promoted the in situ growth of COF-300. The morphology of COF-300 without the addition of GO is long fiber-like, with a size of 10 μm, whereas the rGO–COF-300 shows short rod-like crystallites with sizes ranging from 1 to 10 μm. The GO substrate serves as a nucleation node, which is capable of tuning the size of the COF crystallites in a controlled manner by tuning the ratio between GO and COF monomers. Furthermore, the oxidation level and interlayer distance of GO play an indispensable role in regulating the growth of COF-300. GO with the highest degree of oxidation, poorest graphitic structural order, and the largest layer spacing resulted in composites with the most uniform growth, smallest particle size, and the highest content of COF-300. The synthesis methodology was also shown to be compatible with GO made from naturally coaly graphite, which was made using an environmentally friendly protocol and therefore contained a relatively large amount of inorganic impurities. In summary, this study provides a methodology to make rGO-3D COF composites and gives insights into controlling the morphology of COFs on the GO substrate. It further highlights the importance of the oxidation level of the GO when making composite materials and broadens the development of natural coaly graphite.Author contributions
Y. Q. and K. B. conceptualized this project. Y. Q. collected data under the supervision of K. B., Y. Y., and Q. L. All authors provided input when writing this article.Data availability
The raw data presented in this article is available upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. B. gratefully acknowledges financial support from the Knut and Alice Wallenberg Foundation (KAW 2017,0192) and the Swedish Research Council (2020-03578). YQ would like to thank the China Scholarship Council, National Natural Science Foundation of China (Grant No. 41672150), for financial support, and the Chalmers Materials Analysis Laboratory for help with the Raman characterization.References
- P. J. Waller, F. Gandara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef CAS PubMed.
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L. H. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Science, 2018, 361, 48–52 CrossRef CAS PubMed.
- X. Guan, F. Chen, Q. Fang and S. Qiu, Chem. Soc. Rev., 2020, 49, 1357–1384 RSC.
- X. Ma and T. F. Scott, Commun. Chem., 2018, 1, 98 CrossRef.
- Y. Yang, Y. Chen, F. Izquierdo-Ruiz, C. Schäfer, M. Rahm and K. Börjesson, Nat. Commun., 2023, 14, 220 CrossRef CAS PubMed.
- Y. Yang, M. Ratsch, A. M. Evans and K. Börjesson, J. Am. Chem. Soc., 2023, 145, 18668–18675 CrossRef CAS PubMed.
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- Y.-H. Yao, J. Li, H. Zhang, H.-L. Tang, L. Fang, G.-D. Niu, X.-J. Sun and F.-M. Zhang, J. Mater. Chem. A, 2020, 8, 8949–8956 RSC.
- F. Vilela, K. Zhang and M. Antonietti, Energy Environ. Sci., 2012, 5, 7819–7832 RSC.
- Y. Yang and K. Börjesson, Trends Chem., 2022, 4, 60–75 CrossRef CAS.
- Y. Yang, C. Schäfer and K. Börjesson, Chem, 2022, 8, 2217–2227 CAS.
- Y. Yang, S. Mallick, F. Izquierdo-Ruiz, C. Schäfer, X. Xing, M. Rahm and K. Börjesson, Small, 2021, 17, 2103152 CrossRef CAS PubMed.
- Y. Yang, A. P. Sandra, A. Idström, C. Schäfer, M. Andersson, L. Evenäs and K. Börjesson, J. Am. Chem. Soc., 2022, 144, 16093–16100 CrossRef CAS PubMed.
- A. Tarhini and A. R. Tehrani-Bagha, Appl. Compos. Mater., 2023, 30, 1737–1762 CrossRef CAS.
- F. Li, L. Long and Y. Weng, Adv. Mater. Sci. Eng., 2020, 2020, 1–16 CrossRef.
- Z. Zha, L. Xu, Z. Wang, X. Li, Q. Pan, P. Hu and S. Lei, ACS Appl. Mater. Interfaces, 2015, 7, 17837–17843 CrossRef CAS PubMed.
- J. Shi, M. Su, H. Li, D. Lai, F. Gao and Q. Lu, ACS Appl. Mater. Interfaces, 2022, 14, 42018–42029 CrossRef CAS PubMed.
- J. Chen, J. Yang, X. Wang, D. Yang, M. Yang, J. Tian, R. Tao, Y. Wang, Q. Wei, R. Wang, Y. Yang and Y. Liu, Int. J. Hydrogen Energy, 2022, 47, 16179–16188 CrossRef CAS.
- N. An, Z. Guo, J. Xin, Y. Y. He, K. F. Xie, D. M. Sun, X. Y. Dong and Z. G. Hu, J. Mater. Chem. A, 2021, 9, 16824–16833 RSC.
- Y. Xiong, Q. Liao, Z. Huang, X. Huang, C. Ke, H. Zhu, C. Dong, H. Wang, K. Xi, P. Zhan, F. Xu and Y. Lu, Adv. Mater., 2020, 32, 1907242 CrossRef CAS PubMed.
- J. Sun, A. Klechikov, C. Moise, M. Prodana, M. Enachescu and A. V. Talyzin, Angew. Chem., 2017, 130, 1046–1050 CrossRef.
- C. Li, S. Cao, J. Lutzki, J. Yang, T. Konegger, F. Kleitz and A. Thomas, J. Am. Chem. Soc., 2022, 144, 3083–3090 CrossRef CAS PubMed.
- C. Zhang, W. Li, C. Liu, C. Zhang, L. Cao, D. Kong, W. Wang and S. Chen, J. Colloid Interface Sci., 2022, 608, 1025–1039 CrossRef CAS PubMed.
- Y. Xiao, Z. Jin, L. He, S. Ma, C. Wang, X. Mu and L. Song, Composites, Part B, 2020, 182, 107616 CrossRef CAS.
- Y. Yang, Y. Chen, F. Izquierdo-Ruiz, C. Schafer, M. Rahm and K. Börjesson, Nat. Commun., 2023, 14, 220 CrossRef CAS PubMed.
- Y. Yang, S. Mallick, F. Izquierdo-Ruiz, C. Schafer, X. Xing, M. Rahm and K. Börjesson, Small, 2021, 17, e2103152 CrossRef PubMed.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Cóté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS PubMed.
- O. Yahiaoui, A. N. Fitch, F. Hoffmann, M. Froba, A. Thomas and J. Roeser, J. Am. Chem. Soc., 2018, 140, 5330–5333 CrossRef CAS PubMed.
- D. N. Bunck and W. R. Dichtel, Angew. Chem., Int. Ed., 2012, 51, 1885–1889 CrossRef CAS PubMed.
- N. A. Khan, J. Yuan, H. Wu, L. Cao, R. Zhang, Y. Liu, L. Li, A. U. Rahman, R. Kasher and Z. Jiang, ACS Appl. Mater. Interfaces, 2019, 11, 28978–28986 CrossRef CAS PubMed.
- R. E. Mow, A. S. Lipton, S. Shulda, E. A. Gaulding, T. Gennett and W. A. Braunecker, J. Mater. Chem. A, 2020, 8, 23455–23462 RSC.
- Y. Bai, Y. Liu, M. Liu, X. Wang, S. Shang, W. Gao, C. Du, Y. Qiao, J. Chen, J. Dong and Y. Liu, Angew. Chem., Int. Ed., 2022, 61, e202113067 Search PubMed.
- Y. Chen, Z. L. Shi, L. Wei, B. Zhou, J. Tan, H. L. Zhou and Y. B. Zhang, J. Am. Chem. Soc., 2019, 141, 3298–3303 CrossRef CAS PubMed.
- J.-B. Feng, Y.-y Li, Y. Zhang, Y.-y Xu and X.-W. Cheng, Chem. Eng. J., 2022, 429, 132499 CrossRef CAS.
- X. L. Wang, Y. Y. Sun, Y. Xiao, X. X. Chen, X. C. Huang and H. L. Zhou, Molecules, 2022, 27, 8002 CrossRef CAS PubMed.
- Y. Quan, Q. F. Liu, K. Li, H. Zhang and L. Yuan, Sep. Purif. Technol., 2022, 281, 119931 CrossRef CAS.
- Y. Quan, Q. F. Liu, K. Li, H. Zhang, Y. J. Yang and J. Zhang, Mater. Today Commun., 2022, 32, 104130 CrossRef CAS.
- L. Lai, L. Chen, D. Zhan, L. Sun, J. Liu, S. H. Lim, C. K. Poh, Z. Shen and J. Lin, Carbon, 2011, 49, 3250–3257 CrossRef CAS.
- T. A. Saleh, M. M. Al-Shalalfeh and A. A. Al-Saadi, Sci. Rep., 2016, 6, 32185 CrossRef CAS PubMed.
- C. Nethravathi and M. Rajamathi, Carbon, 2008, 46, 1994–1998 CrossRef CAS.
- H. Wang, M. Wang, Y. Wang, J. Wang, X. Men, Z. Zhang and V. Singh, Sep. Purif. Technol., 2021, 276, 119268 CrossRef CAS.
- R. Al-Gaashani, A. Najjar, Y. Zakaria, S. Mansour and M. A. Atieh, Ceram. Int., 2019, 45, 14439–14448 CrossRef CAS.
- L. Silipigni, M. Cutroneo, G. Salvato and L. Torrisi, Radiat. Eff. Defects Solids, 2018, 173, 740–750 CrossRef CAS.
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klock, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570–4571 CrossRef CAS PubMed.
- T. Ma, J. Li, J. Niu, L. Zhang, A. S. Etman, C. Lin, D. Shi, P. Chen, L. H. Li, X. Du, J. Sun and W. Wang, J. Am. Chem. Soc., 2018, 140, 6763–6766 CrossRef CAS PubMed.
- E. F. Aboelfetoh, A. H. Gemeay and R. G. El-Sharkawy, Environ. Monit. Assess., 2020, 192, 355 CrossRef CAS PubMed.
- Z. Lei, Q. Yang, Y. Xu, S. Guo, W. Sun, H. Liu, L. P. Lv, Y. Zhang and Y. Wang, Nat. Commun., 2018, 9, 576 CrossRef PubMed.
- Q. Zheng, J. J. Huang, Y. T. He, H. Huang, Y. Ji, Y. F. Zhang and Z. Lin, ACS Appl. Mater. Interface, 2022, 14, 9754–9762 CrossRef CAS PubMed.
- Y. Xiong, Q. Liao, Z. Huang, X. Huang, C. Ke, H. Zhu, C. Dong, H. Wang, K. Xi, P. Zhan, F. Xu and Y. Lu, Adv. Mater., 2020, 32, e1907242 CrossRef PubMed.
- J. W. Colson, J. A. Mann, C. R. DeBlase and W. R. Dichtel, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 378–384 CrossRef CAS.
- K. Krishnamoorthy, M. Veerapandian, K. Yun and S. J. Kim, Carbon, 2013, 53, 38–49 CrossRef CAS.
- J. Shen, Y. Hu, M. Shi, X. Lu, C. Qin, C. Li and M. Ye, Chem. Mater., 2009, 21, 3514–3520 CrossRef CAS.
- J. W. Colson, J. A. Mann, C. R. DeBlase and W. R. Dichtel, J. Polym. Sci., Part A: Polym. Chem., 2014, 53, 378–384 CrossRef.
- S. Kumar and K. Chatterjee, ACS Appl. Mater. Interfaces, 2016, 8, 26431–26457 CrossRef CAS PubMed.
- D. Hou, K. Li, R. Ma and Q. Liu, J. Mater., 2020, 6, 628–641 Search PubMed.
- A. D. Jara, A. Betemariam, G. Woldetinsae and J. Y. Kim, Int. J. Min. Sci. Technol., 2019, 29, 671–689 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma01088d |
| This journal is © The Royal Society of Chemistry 2025 |