
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChitosan–acrylic acid biomaterial with an antimicrobial nature supports biomineralization and is suitable for bone tissue regeneration
Sweta
Agarwal
a,
Abhishek
Singh†
 a,
Satish
Kumar†
b,
Tejas Pravin
Rokade
b,
Chandan
Goswami
b and
Luna
Goswami
*ac
a,
Satish
Kumar†
b,
Tejas Pravin
Rokade
b,
Chandan
Goswami
b and
Luna
Goswami
*ac
aSchool of Biotechnology, KIIT Deemed to be University, Patia, Bhubaneswar, Odisha 751024, India. E-mail: lgoswami@kiitbiotech.ac.in
bSchool of Biological Sciences, National Institute of Science Education and Research, HBNI, Khordha, Jatni, Odisha 752050, India
cSchool of Chemical Engineering, KIIT Deemed to be University, Patia, Bhubaneswar, Odisha 751024, India
First published on 23rd May 2025
Abstract
Rapid increases in different pathophysiologies in mineralized tissues have imposed demands on biomaterials of natural origin to act as a surface for the regeneration of fully functional tissue. It has been reported that chitosan-based biomaterials are biocompatible, biodegradable, nontoxic, and also have antibacterial properties. However, due to the insoluble nature of chitosan, for biomaterial synthesis it needs to be dissolved in an acidic medium followed by pH neutralization. The objective of this study was to synthesize a chitosan-based biomaterial without the use of any acidic medium, followed by modification with acrylic acid in different w/v ratios. The synthesized biomaterial, i.e. chitosan–acrylic acid (chitosan–AA) shows uniform dispersion in water. It was further characterized for various physicochemical properties. The obtained results indicate successful modification of the polymer, exhibiting a porous nature with a high swelling index. It shows biocompatibility against different osteogenic cell lines and supports biomineralization by osteoblasts under osteo-inductive conditions. No hemolytic effect was observed in response to the biomaterial even after prolonged exposure to blood cells. We also show that the synthesized material can be used for controlled drug release. This work demonstrates that the biomaterial can be used as a suitable surface for the adhesion and proliferation of bone cells in vitro.
Introduction
Advances in medical science and reductions in morbidity rates have increased the average lifespan of human beings. However, it is compromised by several lifestyle-related disorders, such as cancer, hormonal problems, changes in food habits, random and unmethodical use of therapeutic agents, and other pathophysiological problems. All of these have led to an increase in bone-related disorders, which have created new challenges for clinicians. Some frequent cases of bone-related disorders involve osteoporosis, rickets, scoliosis, Paget's disease, osteoarthritis, osteomyelitis, bone cancer and tumors.1,2 All of these, slowly or rapidly, lead to an imbalance in skeletal integrity, disruption of vascular structures, and improper flow of nutrients, thus preventing initiation of the repair process. The primary bone repair process involves the production of certain cytokines and growth factors, followed by chondrogenesis and differentiation of bone cells into osteoblasts, osteoclasts, and osteocytes. These cells then carry out mineralization, leading to new bone formation and bone remodeling. This bone healing process is not sufficient for severe bone damage, which demands proper medical assistance and even surgical procedures. Two procedures, namely “autograft” and “allograft”, are widely used, but both come with a few limitations and side effects.3 So better and cheaper alternative options are needed to overcome these limitations.Biomaterials can act as a scaffold for tissue engineering. There are various polymeric compounds, such as gelatin, alginate, collagen, and chitosan,4 which are used for the synthesis of scaffold materials. Due to their diverse chemical properties, naturally occurring polysaccharides show great advantages for tissue engineering purposes. Pure forms of such polysaccharides lack stability and mechanical strength, especially under physiological conditions. To act as a suitable surface for tissue regeneration, these need to be modified with improved physicochemical and mechanical properties.5
Among many polymerization methods, UV-irradiation has various advantages. Due to its shorter wavelength, it holds more energy than visible light, which allows reactions to occur instantaneously with a higher rate of polymerization. Additionally, it is much safer, simpler, pollution-free and cheaper than thermal polymerization because it eliminates the involvement of volatile organic solvents. The method of polymerization by the UV-irradiation method involves the incorporation of the monomer on the polymer chain in the presence of photo-initiators. Photo-initiators are molecules that undergo excitation in response to light for the generation of reactive species, which are responsible for carrying out consecutive reactions. Benzophenones, alpha-hydroxy ketones, phosphine oxides, and thioxanthones are some commonly used photo-initiators. These molecules absorb photons from light to produce reactive species, and free radicals for initiation of the reaction. This method has already been used for the production of optical fibers, composites, microchips, microelectronics, coatings, adhesives, edible films, and 3D-objects.
Chitosan (CS), the second most abundant polymer after cellulose, is a naturally occurring polysaccharide composed of two arbitrarily distributed monomers, N-acetyl-D-glucosamine and β-(1–4)-linked D-glucosamine. It is non-toxic, biocompatible, and biodegradable, and has attracted a lot interest because of its application in the delivery of therapeutic drugs, wound dressing, and tissue engineering. The synthesis of chitosan-based biomaterial demands the initial dissolution of chitosan in acetic acid,6,7 hydrochloric acid8 or other acidic media. Post-synthesis and neutralization of such biomaterials with a base, for biological applications, has also been reported.
Polymers incorporating acrylic acid (as a monomer) exhibit pH sensitivity and can impart high water-absorbing capacity to the biomaterial. This pH sensitivity is imparted by the presence of the ionizable carboxylic acid functional group of the monomer. It can be polymerized with other monomers or polymers of both hydrophobic and hydrophilic nature in the presence of crosslinking agents. This ability of acrylic acid to react easily with free radicals allows the formation of copolymers with desired physicochemical properties.9
So far it has been found that due to lack of solubility in water, chitosan-based biomaterials need to be fabricated by dissolving them in an acidic medium,6,7 requiring an additional step of post-synthesis neutralization for biological applications. Therefore, in this work, a simple approach was designed to synthesize a chitosan-based biomaterial by modifying the chitosan backbone with acrylic acid monomer without the use of any acidic medium to dissolve the polymer and the homogenous dispersion of chitosan–AA in water was observed. The resulting material (chitosan–AA) was further characterized for its physicochemical properties, and possibility for biological applications. To check the extent of the modification, different instrumental techniques, like UV-vis spectroscopy, FTIR and TGA, were performed. Further, to investigate whether this biomaterial is suitable to act as a surface for cell growth, the swelling index and SEM were studied which show its swelling ability and porous structure. Due to this swelling ability, chitosan–AA was utilized for a drug release assay. Additionally, from a well diffusion assay it was determined whether the inherent antimicrobial property of chitosan is retained in chitosan–AA, so that it can also be used for osteomyelitis along with bone tissue regeneration at the site. As chitosan is already known to be beneficial for bone tissue engineering, this biomaterial was used as a surface for both osteoclast and osteoblast and finally for in vitro biomineralization, which was further supported by Ca2+-imaging. Detection of mitochondrial membrane potential and hemolysis experiments suggested no toxic effect of the biomaterial on bone cells or RBCs, respectively.
Materials and methods
Materials
Chitosan (mol wt of chitosan = 3800–20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Daltons, degree of deacetylation ≥75.00%), acrylic acid, sodium chloride, tryptone, yeast extract, tryptic soy broth, penicillin–streptomycin, and trypsin-EDTA (1X) were purchased from Hi-media. Sodium dodecyl sulfate, DMSO, cetyl-pyridinium chloride and MTT were purchased from MP Biomedicals (India). Benzophenone and agar-agar were purchased from Merck India. 4′-Hydroxyacetanilide was procured from TCI Chemicals Co., Ltd (India). Acetone was purchased from Sisco Research Laboratories Pvt. Ltd, (India). Milli-Q water was used for all experiments, where required.
000 Daltons, degree of deacetylation ≥75.00%), acrylic acid, sodium chloride, tryptone, yeast extract, tryptic soy broth, penicillin–streptomycin, and trypsin-EDTA (1X) were purchased from Hi-media. Sodium dodecyl sulfate, DMSO, cetyl-pyridinium chloride and MTT were purchased from MP Biomedicals (India). Benzophenone and agar-agar were purchased from Merck India. 4′-Hydroxyacetanilide was procured from TCI Chemicals Co., Ltd (India). Acetone was purchased from Sisco Research Laboratories Pvt. Ltd, (India). Milli-Q water was used for all experiments, where required.
Synthesis of chitosan–AA
Chitosan–AA 1:0.5, 1:1, 1:1.5 (∼weight ratio) were synthesized where sodium dodecyl sulfate (SDS) and benzophenone (initiator) were added in a 1:1 ratio. Briefly, 250 μL (1:0.5), 500 μL (1:1) and 750 μL (1:1.5) of acrylic acid (AA) were taken in different glass Petri dishes, followed by the addition of SDS and benzophenone. A fixed weight of chitosan, i.e. 500 mg, was added to the respective mixtures. A 1:1 water–ethanol solution was added gradually to obtain a thick uniform paste. The reaction mixture was exposed to UV irradiation of 365 nm for 30 min. This was followed by washing with acetone for the removal of any unreacted monomer, homopolymer, and initiator molecules. The purified sample was kept for 15–20 h in a hot-air oven at 50 °C, until dried. These variables, i.e. 1:0.5, 1:1, and 1:1.5, were used for the ∼weight ratio of chitosan to acrylic acid. The synthesis of the material is shown in a schematic diagram (Fig. 1).
 | ||
| Fig. 1 Schematic diagram for the synthesis of chitosan–AA biomaterial. Schematic representation for the synthesis of the biomaterial chitosan–AA with varying ratios of chitosan to acrylic acid shown. | ||
Solubility of pure chitosan and chitosan–AA in water
To examine the solubility, 1% (w/v) solutions of both pure chitosan and synthesized chitosan–AA were prepared and kept on a magnetic stirrer at 80 °C and 320 rpm for 72 h.Swelling study
This was performed by the tea bag method,10 where a dry sample of a fixed weight was completely submerged in water at room temperature. The biomaterial was weighed at constant intervals, until it attained its maximum swelling weight. The swelling index was then calculated using the following equation:11
The same method was performed by immersing the sample in solutions of different pH, i.e., 6.2, 6.8, 7.4, and in Ringer's solution.
Spectroscopic characterization
UV-visible spectra of pure chitosan and chitosan–AA were measured in the wavelength range 200–500 nm (Agilent, Cary 100 UV-visible spectrophotometer). FTIR analysis was performed within the frequency range 400–4000 cm−1 (FTIR Spectrophotometer–Spectrum Two, PerkinElmer).Thermo-gravimetric analysis (TGA)
A thermo-gravimetric analyzer was utilized to perform thermal stability analysis (TGA 4000, PerkinElmer, USA). It was carried out in the temperature range 40 °C to 700 °C in an inert environment with a constant heating rate of 20 °C min−1.12Microscopic structural analysis
The microscopic structure of the lyophilized sample was studied using SEM (Hitachi S3400 N) at an accelerating voltage of 15.0 kV. Sample preparation was done by freezing the swelled sample at −80 °C overnight and then lyophilizing it for 48 h. Both lateral and cross sections were cut and subjected to gold-sputtering before examination.13 These images were also utilized to measure pore size for frequency distribution analysis.Porosity (%) measurement
A known weight of the lyophilized sample (W1) was immersed in a fixed volume of absolute ethanol at 25 °C. The total weight of ethanol and sample submerged in ethanol (W2) was noted. After 24 h, the swelled sample was removed and the remaining ethanol was weighed (W3). The porosity (%) was calculated by the volumetric displacement method, using the equation:14
Drug release
For drug release analysis, a fixed weight of lyophilized chitosan–AA was immersed in a solution of 4′-hydroxyacetanilide for 24 h at room temperature. The drug-loaded biomaterial was removed, wrapped in a muslin cloth and kept in the rotatory basket of a type II drug dissolution apparatus (Electrolab dissolution tester, TDT-06L).15 2 mL of drug dissolution solution was taken out at regular intervals and replaced by an equal volume of fresh 1X PBS (pH 7.2) solution. 1X PBS was prepared by adding NaCl, KCl, Na2HPO4 and KH2PO4 to 400 mL of distilled water. The pH was adjusted to 7.2 and the final volume was made up to 500 mL. The absorbance of the drug dissolution solution at each time point was measured at a wavelength of 249 nm (λmax of 4′-hydroxyacetanilide) using a UV-visible spectrophotometer (Agilent, Cary 100 UV-visible spectrophotometer) and the drug release pattern was evaluated.Antimicrobial efficacy
The antimicrobial properties were analyzed by a well diffusion assay. Luria–Bertani agar, and tryptic soy agar Petri plates were inoculated with Gram-negative (−ve) bacteria Klebsiella pneumoniae (MTCC 12949) and Gram-positive (+ve) bacteria Staphylococcus aureus (ATCC 6538 and MRSA), respectively. Different concentrations of all three ratios of chitosan–AA were prepared in Milli-Q water i.e., 0.6%, 1.2%, 1.8%, and added to different wells of the agar plate for each microbial strain. The Petri plates were kept at 37 °C overnight in an incubator and checked for any “zone-of-inhibition” by measuring the longest diameter of clear zone around the well.
Cell culture
RAW 264.7 cells and SaOS-2 cells (procured from NCCS Pune) were maintained in DMEM (Gibco) and McCoy's (Gibco) media enriched with 10% FBS (Gibco), and penicillin–streptomycin (Hi-Media), respectively, within an incubator maintained at 37 °C with 5% CO2.Biocompatibility test
To check the biocompatibility, thin films of different concentrations of the biomaterial were coated onto 12–15 mm glass coverslips and air dried. Mouse preosteoclast cells (RAW 264.7) and osteoblast cells (SaOS-2) maintained in DMEM and McCoy's media, respectively, as described earlier, were passaged using trypsin-EDTA (1X), and ∼20000 cells were seeded on the coated and uncoated coverslips and grown for 48 h in the CO2 incubator at 37 °C. Cells were subsequently fixed using paraformaldehyde (PFA).
Nucleus and cytoskeletal staining
Cells were fixed with 4% PFA and treated with Triton-X (0.1%) to permeabilize the cell membrane. The cell cytoskeleton (actin) was subsequently stained with Alexa-488-labeled phalloidin (Invitrogen, 1:200) and Alexa-594-labeled phalloidin (Invitrogen, 1:200). Cells were further labelled with DAPI stain (1:2000, Invitrogen). They were then visualized under a confocal laser-scanning microscope (Olympus FV3000), and images were acquired accordingly.16
Detection of mitochondrial membrane potential by TMRM staining
For measurement of the mitochondrial membrane potential, RAW 264.7 and SaOS-2 cells were seeded on coverslips (25 mm) coated with or without biomaterials and allowed to grow for 48 h within the incubator set at 37 °C and CO2 (5%). For TMRM imaging, the cells were incubated with a TMRM (tetramethylrhodamine methyl ester, perchlorate) probe (Invitrogen, 50 nM) for a duration of 0.5 h at 37 °C. Images of the cells were captured, as described for the detection of cytosolic Ca2+, and TMRM intensity was plotted in GraphPad Prism.Ca2+-imaging
For the measurement of changes in Ca2+-levels of osteogenic cells, SaOS-2 cells were grown for 48 h on both uncoated and biomaterial coated coverslips (25 mm) within the incubator set at 37 °C and CO2 (5%). For Ca2+-imaging, the cells were incubated with a Fluo4-AM probe (Invitrogen, 1 μM) for a duration of 0.5 h at 37 °C. Images of the cells were captured using a confocal laser-scanning microscope (Olympus FV3000). Fluo4-AM intensity and cellular parameters (area and perimeter) were plotted in GraphPad Prism for a clear comparison between the two conditions, i.e. biomaterial uncoated (control) and coated surface (chitosan–AA). Linear regression analysis of Fluo4-AM intensity and cellular area was also performed to determine the correlation between the two (intensity vs. area).17Cell proliferation by MTT assay
To check the cytotoxicity, 48-well plates were coated with chitosan–AA. Approximately, 3 × 104 cells per well (RAW 264.7 and SaOS-2 separately) were seeded on both uncoated and coated surfaces and kept in a CO2 incubator at 37 °C for 48 h. Subsequently, the medium was discarded and 200 μL (0.5 mg mL−1) of MTT solution (prepared in 1X PBS) was added to the wells and kept at 37 °C for the development of formazan crystals.12 After 4 h of incubation, DMSO was added to each well to dissolve the formazan crystals, and the optical density (O.D) was measured at 570 nm in a multimode spectral scanning reader (Thermo-Scientific Varioskan Flash).Hemolytic compatibility
A hemocompatibility assay was performed, as described earlier12 with slight modifications. Goat blood was collected from a slaughter house in vials pre-coated with EDTA to prevent coagulation. The collected blood samples were centrifuged thrice at 1006g for 10 min with washing of the pellet every single time with cold 0.9% NaCl solution. Finally, the biomaterial solution (1 mg mL−1 or 3 mg mL−1) was gradually added to the pellet followed by incubation at 37 °C with gentle mixing for a duration of 1 h. Milli-Q water and PBS were taken as positive (‘+ve’) and negative (‘−ve’) controls, respectively. For collection of the supernatant, the samples were centrifuged at 3000 rpm for 5 min. The optical density (O.D) of the collected supernatant was measured at 540 nm with a Thermo-Scientific NANODROP ONE©, and the release of hemoglobin was determined with the equation:
A (s) = O.D of the sample, A (−) = O.D of the ‘−ve’ control, A (+) = O.D of the ‘+ ve’ control.
Mineralization assay in vitro by SaOS-2 cells
Mineralization takes place as a result of the late differentiation of osteoblastic bone cells and is a crucial phenomenon for the regeneration of bone tissue.18 An osteo-induction medium consisting of L-ascorbic acid (100 mM), β-glycerophosphate (10 mM) and dexamethasone (10 nM) in McCoy's 5A medium was prepared for differentiation of SaOS-2 cells on both biomaterial-coated and uncoated surfaces in a 24-well plate for 10 days. Cells were then fixed using PFA (4%) followed by staining with Alizarin Red S solution (Sigma-Aldrich, India, at pH 4.2, 40 mM). Alizarin Red S selectively binds to calcium salts, developing a red color. Quantitative analysis was also performed by extracting it in 10% cetyl-pyridinium chloride solution and the absorbance (O.D.) was measured at 590 nm. Digital images of each well were also captured for representation.Statistical study
All the graphs were created using OriginPro and GraphPad Prism 8.4.3, while all the statistical analyses were executed in GraphPad Prism 8.4.3. For comparison of two groups, an unpaired t-test was performed, whereas for more than two groups, one-way ANOVA was chosen, and a p-value of less than 0.05 was considered to be statistically significant.Results and discussion
Synthesis of chitosan–AA
Chitin is a natural biopolymer readily available from crustacean waste and its deacetylation results in the production of chitosan. The degree of acetylation plays an immense role in its solubility.19 Chitosan has been commonly modified using various techniques, like etherification, acylation, or alkylation, to improve its solubility.20,21 However, in the last 30 years, acrylic-acid-modified chitosan has been synthesized in the form of microspheres,22 polyelectrolyte complex,23 nano-capsules,24 interpenetrating networks,25 nanofibers,26etc. to enhance the solubility and mechanical properties of this polymer. While most of the routes involved suspension mode synthesis via thermal initiation of free radical polymerization,27–31 only a few reports suggested synthesis via gamma irradiation,32 UV irradiation33 or photochemical initiation.34 To date, these hydrogels/blends have been studied mostly for anticancer/gastric drug delivery applications,35–39 wastewater treatment,40,41 metal ions42–44 and lipoprotein adsorption.45 Whereas for application in bone tissue regeneration,46 nanohydroxyapatite,47 silica48 or calcium carbonate49 have been reported to be additionally incorporated in chitosan–acrylic acid blends. However, in this work chitosan–AA biomaterial was synthesized via UV irradiation, where chitosan was used in the solid state, eliminating the need for any acidic or organic solvent to dissolve chitosan, as required previously (Fig. 1). Hydrolysis of polymeric biomaterials is related to its bioactivity and ability to interact with the surrounding body tissue. The synthesized biomaterial was also found to be dispersed in water and was investigated for application in bone tissue regeneration.Chitosan–AA exhibits improved solubility/dispersibility in water
Unlike pure chitosan, modified chitosan i.e. chitosan–AA, shows dispersion in water on continuous stirring at 80 °C. As acrylic acid is already known to impart water solubility, this change in the solubility/dispersibility pattern of chitosan might indicate the successful incorporation of acrylic acid monomer into the polymeric chitosan backbone.Physicochemical characterization
:0.5). The hydrophilicity further increases from chitosan–AA (1:0.5) to chitosan–AA (1:1) with the swelling index reaching ∼4000%, while it rapidly decreases thereafter for chitosan–AA (1:1.5). This decrease in the swelling index with increased acrylic acid concentration might be due to the incorporation of AA/PAA (polyacrylic acid) onto the polymer backbone.50 Therefore, chitosan–AA (1:1) shows the highest swelling ability and was chosen for further analysis.
 | ||
| Fig. 2 Swelling index and spectroscopic characterization for pure chitosan and chitosan–AA. (a) Swelling indexes of all the ratios of chitosan–AA, where chitosan–AA (1:1) shows the maximum swelling ability. (b) Hydro-swelling behavior of chitosan–AA (1:1) in different pH solutions. Mean values of swelling index (%) from three independent experiments (n = 3). (c)–(f) Representative graphs of all the spectroscopic analysis. UV-vis spectroscopy (c) and FTIR spectra of pure chitosan and chitosan–AA indicating the successful incorporation of acrylic acid in the biomaterial chitosan–AA (d). Thermogram showing the weight retention (%) (e) and comparison of the derivative weight (%) (f) of pure chitosan and chitosan–AA. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1551 (N–H), and 1373 cm−1 (C–N) are due to amides I, II, and III, respectively.56,57 In chitosan–AA, the amide I band undergoes extreme weakening, while the amide II and III bands show an increase in intensity and shift to ∼1556 and ∼1378 cm−1, respectively.52 This shift might correspond to the bending and deformation of methylene (CH2) and methyl (CH3) groups, respectively.58 Sharp peaks at ∼1060, 1024 and 893 cm−1, which correspond to the C–O and C–H bonds, are found to be present in both pure and modified chitosan.55 The extreme broadening and weakening of the characteristic –OH and N–H groups of the chitosan–AA spectrum imply that it took part in the modification reaction.59,60 The presence of such bands might indicate the incorporation of acrylic acid into the chitosan backbone.
O), 1551 (N–H), and 1373 cm−1 (C–N) are due to amides I, II, and III, respectively.56,57 In chitosan–AA, the amide I band undergoes extreme weakening, while the amide II and III bands show an increase in intensity and shift to ∼1556 and ∼1378 cm−1, respectively.52 This shift might correspond to the bending and deformation of methylene (CH2) and methyl (CH3) groups, respectively.58 Sharp peaks at ∼1060, 1024 and 893 cm−1, which correspond to the C–O and C–H bonds, are found to be present in both pure and modified chitosan.55 The extreme broadening and weakening of the characteristic –OH and N–H groups of the chitosan–AA spectrum imply that it took part in the modification reaction.59,60 The presence of such bands might indicate the incorporation of acrylic acid into the chitosan backbone.
 | ||
| Fig. 3 Chitosan–AA has suitable pores. (a) and (b) Microstructure analysis by SEM shows the highly porous structure of chitosan–AA, as depicted in both lateral section (a) and cross section images (b). The inset images show the lyophilized sample view of the respective lateral and cross section. (c and d) Distribution pattern of pore size with pore count of 52 (calculated by SEM images using ImageJ-win64 software) (c) and porosity (%) (d). (e) Release pattern of the drug (4′-hydroxyacetanilide) from the chitosan–AA matrix in controlled pH conditions represented by mean ± SEM values of experiment performed three times. | ||
 | ||
| Fig. 4 Chitosan–AA has antimicrobial activities and is bio-compatible. (a) Enlarged images of the regions of Petri plates of K. pneumoniae (i), S. aureus (MDR) (ii) and S. aureus (iii) with clear “zone-of-inhibition”. (b) Anti-microbial assay indicates that pure chitosan does not show any “zone-of-inhibition” while it increases with an increase in the concentration of chitosan–AA. The graphical representation includes mean ± SEM values of experiments, where n = 3 (i and iii) and n = 2 (ii). One-way ANOVA was performed, where **** denotes p-value < 0.0001. (c) and (d) For biocompatibility assay, mouse macrophage cells (RAW 264.7) (c) and human sarcoma osteogenic cells (SaOS-2) (d) were grown on both uncoated and biomaterial coated surface of various concentrations (i.e. 0.6%, 1.2% and 1.8%). Fixed cells were stained with Alexa Fluor 488-labelled (green) or Alexa Fluor 594 (red)-labelled phalloidin and DAPI (blue). Confocal images were obtained. Scale bar of 30 μm (RAW 264.7) and 50 μm (SaOS-2). | ||
 | ||
| Fig. 5 Chitosan–AA improves mitochondrial membrane potential. (a) and (b) RAW 264.7 (a) and SaOS-2 (b) cells were assessed for any change in the mitochondrial membrane potential in the absence and in the presence of the biomaterial chitosan–AA. Cells were labelled with TMRM, a mitochondrial membrane potential sensor and confocal images were obtained. Scale bar of 30 μm. (c) and (d) Quantification of the images for TMRM intensity reveals a significant increase in the mitochondrial membrane potential of cells grown on the biomaterial coated surface compared to cells grown on only a glass surface for both RAW 264.7 (c) and SaOS-2 (d) cells. Statistical value of unpaired t-test, p-value: ****< 0.0001 and *< 0.05. | ||
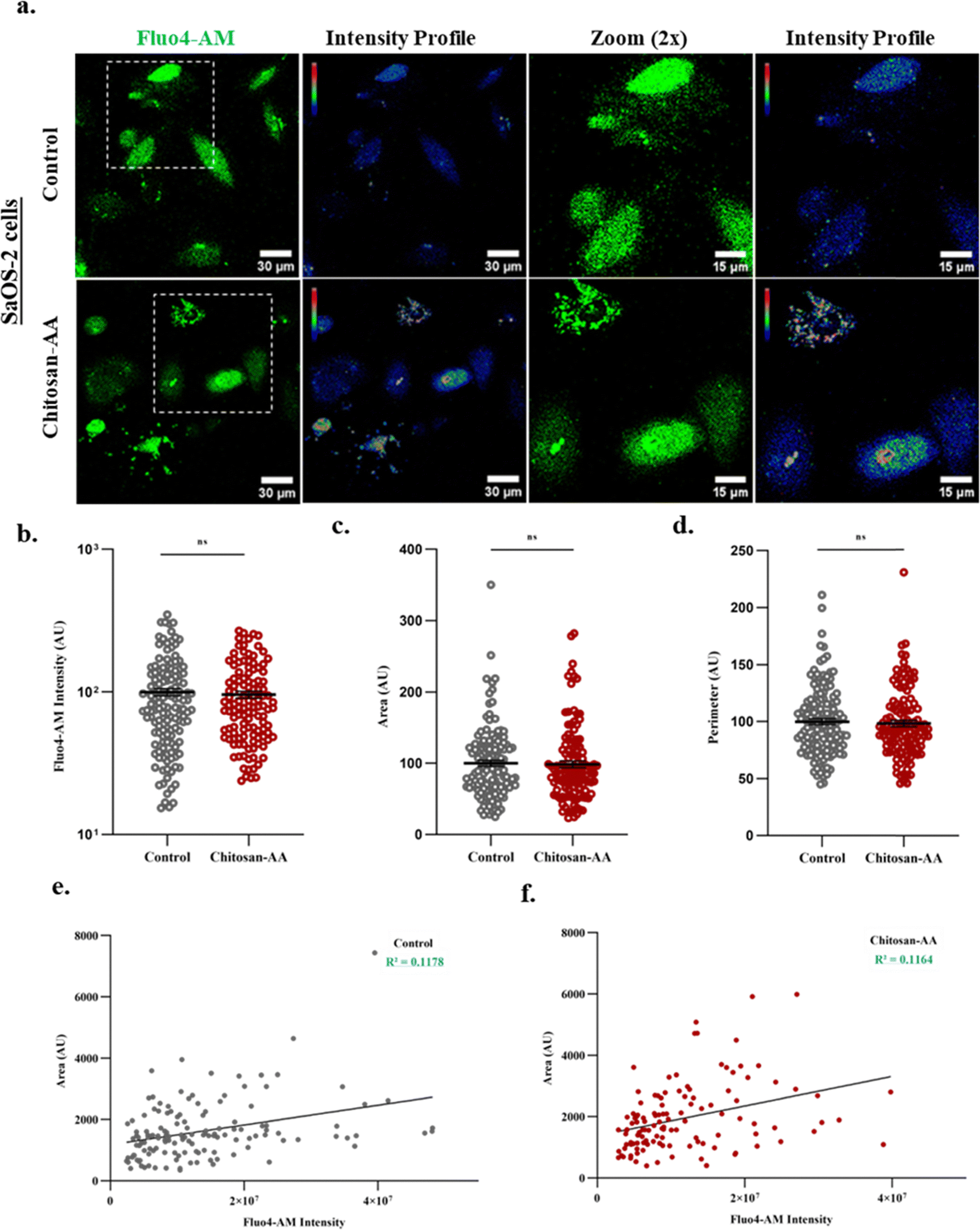 | ||
| Fig. 6 Chitosan–AA does not alter the intracellular basal level Ca2+. (a) Representative images of cytosolic basal-level Ca2+ of SaOS-2 cells grown on glass surface or on chitosan–AA surface (upper panel). Cells were probed with Fluo4-AM (30 min of incubation) followed by acquiring live cell confocal images. Respective RGB intensity profiles for Fluo4 intensity as well as for the enlarged area are shown. (b) Quantification of mean fluorescence intensity of Fluo4-AM, where n > 100 cells per group. (c) and (d) For quantitative analysis, images of Fluo4-AM labelled SaOS-2 cells were processed and all cellular parameters were calculated. Represented graphs depict area (c) and perimeter (d), showing no major effect of cell morphology in response to the chitosan–AA biomaterial. Unpaired t-test was performed by GraphPad Prism. ns (non-significant, p > 0.05). (e) and (f) Correlation graphs of area and Fluo4-AM intensities depict no significant difference in the R2 values between biomaterial uncoated (control) and coated conditions. Linear regression analysis with lower R2 values in both groups are indicative of no considerable correlation between cell morphology and cytosolic Ca2+-levels. | ||
 | ||
| Fig. 7 Chitosan–AA is biocompatible. (a) and (b) Both RAW 264.7 (a) and SaOS-2 (b) cells attain more than 75% cell viability on chitosan–AA surface. (c) The biomaterial shows no sign of hemolysis even after prolonged exposure to blood cells. Double-distilled water is used as a positive control for hemolysis. (d) Quantitative analysis of Alizarin-stained calcium deposits by CPC extraction method. The graphical data is represented in the form of mean ± SEM. Actual images of plates with red-colored deposition of calcium complexes by SaOS-2 cells. Statistical value of unpaired t-test, p-value: **** < 0.0001, *** < 0.001, * < 0.05 and ns = non-significant. | ||
Conclusions
This paper entails a quick and unique approach for the fabrication of a highly porous modified chitosan-based biomaterial, without solubilizing the polymer in any low-pH solutions. Analysis indicates that the biomaterial holds excellent swelling potential, which permits favourable attachment and viability of cells, effective diffusion of nutrients, and mineralization by bone osteoblasts in vitro. Compared to earlier work that used an acidic medium for the dissolution of chitosan prior to biomaterial synthesis, the chitosan–AA surface developed in this work shows similar or better ability for bone cell adherence and mineralization by osteoblasts. This biomaterial can be considered as a suitable framework for bone tissue engineering due to its easy dispersion in water, antimicrobial properties, and lack of noticeable toxicity to blood and bone cells.Author contributions
Conceptualization, investigation and methodology: Sweta Agarwal, Abhishek Singh, Chandan Goswami, Luna Goswami. Data acquisition: Sweta Agarwal, Satish Kumar, Tejas Pravin Rokade. Manuscript drafting: Sweta Agarwal, Luna Goswami. Supervision: Luna Goswami.Data availability
Data will be made available upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Sweta Agarwal was financially supported by DST-INSPIRE Fellowship (IF180735), New Delhi, India. Authors acknowledge infrastructure support from KIIT School of Biotechnology, KIIT, Deemed to be University. CG acknowledge NISER for central instrumental facility and intramural support from NISER/DAE. OUAT, Bhubaneswar is acknowledged for its FTIR and SEM facility. The author also thanks past and present members of the lab for their support.References
- A. Misaghi, A. Goldin, M. Awad and A. A. Kulidjian, SICOT J., 2018, 4, 12 CrossRef PubMed.
- X. Feng and J. M. McDonald, Annu. Rev. Pathol.: Mech. Dis., 2011, 6, 121–145 CrossRef CAS PubMed.
- J. Venkatesan, B. M. Ryu, P. N. Sudha and S. K. Kim, Int. J. Biol. Macromol., 2012, 50, 393–402 CrossRef CAS PubMed.
- Y. H. Lin, K. W. Huang, S. Y. Chen, N. C. Cheng and J. Yu, J. Mater. Chem. B, 2017, 5, 4614–4622 Search PubMed.
- P. Choudhury, S. Chawla, S. Agarwal, A. Singh, A. Nayak, A. Kumar, P. K. Maji, C. Goswami and L. Goswami, Mater. Sci. Eng. C, 2021, 121, 111779 CrossRef CAS PubMed.
- Y. Huang, Y. Cai and Y. Lapitsky, J. Mater. Chem. B, 2015, 3, 5957–5970 Search PubMed.
- M. Liu, C. Wu, Y. Jiao, S. Xiong and C. Zhou, J. Mater. Chem. B, 2013, 1, 2078–2089 Search PubMed.
- L. S. Connell, F. Romer, M. Suárez, E. M. Valliant, Z. Zhang, P. D. Lee, M. E. Smith, J. V. Hanna and J. R. Jones, J. Mater. Chem. B, 2014, 2, 668–680 RSC.
- G. Sennakesavan, M. Mostakhdemin, L. K. Dkhar, A. Seyfoddin and S. J. Fatihhi, Polym. Degrad. Stab., 2020, 180, 109308 CrossRef CAS.
- K. Zhang, W. Feng and C. Jin, MethodsX, 2020, 7, 100779 CrossRef CAS PubMed.
- S. Sanyasi, A. Kumar, C. Goswami, A. Bandyopadhyay and L. Goswami, Carbohydr. Polym., 2014, 101, 1033–1042 CrossRef CAS PubMed.
- P. Choudhury, S. Kumar, A. Singh, A. Kumar, N. Kaur, S. Sanyasi, S. Chawla, C. Goswami and L. Goswami, Carbohydr. Polym., 2018, 189, 87–98 CrossRef CAS PubMed.
- S. Kumar, R. K. Majhi, S. Sanyasi, C. Goswami and L. Goswami, Connect. Tissue Res., 2018, 59, 111–121 CrossRef CAS PubMed.
- A. Singh, S. Kumar, T. K. Acharya, C. Goswami and L. Goswami, Mater. Today Commun., 2022, 31, 103783 CrossRef CAS.
- T. K. Acharya, S. Kumar, N. Tiwari, A. Ghosh, A. Tiwari, S. Pal, R. K. Majhi, A. Kumar, R. Das, A. Singh, P. K. Maji, N. Chattopadhyay, L. Goswami and C. Goswami, Sci. Rep., 2021, 11, 3730 CrossRef CAS PubMed.
- S. Sanyasi, S. Kumar, A. Ghosh, R. K. Majhi, N. Kaur, P. Choudhury, U. P. Singh, C. Goswami and L. Goswami, Macromol. Biosci., 2016, 17, 201600268 Search PubMed.
- R. Chakraborty and C. Goswami, Biochem. Biophys. Res. Commun., 2022, 611, 132–139 CrossRef CAS PubMed.
- A. Singh, S. Kumar, C. Goswami and L. Goswami, Mater. Today Chem., 2024, 38, 102067 CrossRef CAS.
- I. Aranaz, A. R. Alcántara, M. C. Civera, C. Arias, B. Elorza, A. H. Caballero and N. Acosta, Polymers, 2021, 13, 3256 CrossRef CAS PubMed.
- R. Pandey and G. Mathur, Starch - Stärke, 2024, 77(1), 2300248 CrossRef.
- A. El-Araby, W. Janati, R. Ullah, S. Ercisli and F. Errachidi, Front. Chem., 2023, 11, 1327426 CrossRef CAS PubMed.
- C. Peniche, M. Fernández, A. Gallardo, A. López-Bravo and J. San Román, Macromol. Biosci., 2003, 3, 540–545 CrossRef CAS.
- S. Y. Nam and Y. M. Lee, J. Membr. Sci., 1997, 135, 161–171 CrossRef CAS.
- S. Belbekhouche, O. Mansour and B. Carbonnier, J. Colloid Interface Sci., 2018, 522, 183–190 CrossRef CAS PubMed.
- C. Peniche, W. Arguk Elles-Monal, N. Davidenko, R. Sastre, A. Gallardo and J. S. Romah, Biomaterials, 1999, 20, 1869–1878 CrossRef CAS PubMed.
- C. Y. Chen, J. W. Wang and M. H. Hon, Macromol. Mater. Eng., 2006, 291, 123–127 CrossRef CAS.
- Jayanudin, R. S. D. Lestari, D. R. Barleany, A. B. Pitaloka, M. Yulvianti, D. Prasetyo, D. V. Anggoro and A. Ruhiatna, Polymers, 2022, 14, 5175 CrossRef CAS PubMed.
- C. Liu, L. Fu, T. Jiang, Y. Liang and Y. Wei, Polymer, 2021, 230, 124006 CrossRef CAS.
- M. S. P. De Lima, M. S. Freire, J. L. C. Fonseca and M. R. Pereira, Carbohydr. Res., 2009, 344, 1709–1715 CrossRef CAS PubMed.
- H. Ge and S. Wang, Carbohydr. Polym., 2014, 113, 296–303 Search PubMed.
- Y. Hu, X. Jiang, Y. Ding, H. Ge, Y. Yuan and C. Yang, Biomaterials, 2002, 23, 3193–3201 Search PubMed.
- S. Benamer, M. Mahlous, D. Tahtat, A. Nacer-Khodja, M. Arabi, H. Lounici and N. Mameri, Radiat. Phys. Chem., 2011, 80, 1391–1397 CrossRef CAS.
- T. T. Nge, M. Yamaguchi, N. Hori, A. Takemura and H. Ono, J. Appl. Polym. Sci., 2002, 83, 1025–1035 CrossRef CAS.
- N. Davidenko and J. San Roman, Lat. Am. Appl. Res., 2007, 37, 247–253 CAS.
- Y. Hu, Y. Ding, D. Ding, M. Sun, L. Zhang, X. Jiang and C. Yang, Biomacromolecules, 2007, 8, 1069–1076 Search PubMed.
- I. A. Duceac, L. Verestiuc, C. D. Dimitriu, V. Maier and S. Coseri, Polymers, 2020, 12, 1–20 Search PubMed.
- Y. Wang, J. Wang, Z. Yuan, H. Han, T. Li, L. Li and X. Guo, Colloids Surf., B, 2017, 152, 252–259 CrossRef CAS PubMed.
- J.-W. Shim and Y.-C. Nho, J. Appl. Polym. Sci., 2003, 90, 3660–3667 Search PubMed.
- S. Torrado, P. Prada, P. M. De La Torre and S. Torrado, Biomaterials, 2004, 25, 917–923 CrossRef CAS PubMed.
- S. F. Hamza, M. M. El-Sawy, N. A. Alian and N. O. Shaker, Egypt J. Chem., 2021, 64, 6007–6015 Search PubMed.
- A. Shanmugapriya, A. Srividhya, R. Ramya and P. N. Sudha, Int. J. Environ. Sci., 2011, 1, 7 Search PubMed.
- M. Marjub, N. Rahman, N. C. Dafader, F. Sultana Tuhen, S. Sultana and F. Tasneem Ahmed, J. Polym. Eng., 2019, 39(10), 883–891 CrossRef.
- M. M. Rahman, N. N. Lata, S. H. Rimu and A. H. Chisty, Desalin. Water Treat., 2021, 216, 252–262 Search PubMed.
- A. Sepehrianazar, Iran. J. Chem. Chem. Eng., 2023, 42(4), 1099–1110 CAS.
- G. Fu, H. Li, H. Yu, L. Liu, Z. Yuan and B. He, React. Funct. Polym., 2006, 66, 239–246 Search PubMed.
- S. Saber-Samandari and S. Saber-Samandari, Mater. Sci. Eng. C, 2017, 75, 721–732 Search PubMed.
- G. S. Sailaja, P. Ramesh, T. V. Kumary and H. K. Varma, Acta Biomater., 2006, 2, 651–657 Search PubMed.
- Y. J. Lin, F. C. Hsu, C. W. Chou, T. H. Wu and H. R. Lin, J. Mater. Chem. B, 2014, 2, 8329–8337 RSC.
- M. A. E. Cruz and A. P. Ramos, Surf. Coat. Technol., 2016, 294, 145–152 Search PubMed.
- G. Hoti, F. Caldera, C. Cecone, A. R. Pedrazzo, A. Anceschi, S. L. Appleton, Y. K. Monfared and F. Trotta, Materials, 2021, 14, 1–20 Search PubMed.
- R. Komeri, F. G. Thankam and J. Muthu, RSC Adv., 2015, 5, 31439–31449 Search PubMed.
- Z. Bujňáková, E. Dutková, A. Zorkovská, M. Baláž, J. Kováč, M. Kello, J. Mojžiš, J. Briančin and P. Baláž, J. Mater. Sci., 2017, 52, 721–735 CrossRef.
- H. Abdulsahib, H. T. Abdulsahib, A. H. Taobi and S. S. Hashem, Adv. J. Sci. Res., 2016, 1, 1–10 Search PubMed.
- L. Xue and D. J. Kieber, J. Photochem. Photobiol., A, 2024, 449, 115371 CrossRef CAS.
- E. Stodolak-Zych, P. Jeleń, E. Dzierzkowska, M. Krok-Borkowicz, Ł. Zych, M. Boguń, A. Rapacz-Kmita and B. Kolesińska, J. Mol. Struct., 2020, 1211, 128061 CrossRef CAS.
- Y. Liu, X. Shen, H. Zhou, Y. Wang and L. Deng, Appl. Surf. Sci., 2016, 370, 270–278 CrossRef CAS.
- S. Mincke, T. G. Asere, I. Verheye, K. Folens, F. Vanden Bussche, L. Lapeire, K. Verbeken, P. Van Der Voort, D. A. Tessema, F. Fufa, G. Du Laing and C. V. Stevens, Green Chem., 2019, 21, 2295–2306 Search PubMed.
- M. F. Queiroz, K. R. T. Melo, D. A. Sabry, G. L. Sassaki and H. A. O. Rocha, Mar. Drugs, 2015, 13, 141–158 CrossRef PubMed.
- C. Brasselet, G. Pierre, P. Dubessay, M. Dols-Lafargue, J. Coulon, J. Maupeu, A. Vallet-Courbin, H. de Baynast, T. Doco, P. Michaud and C. Delattre, Appl. Sci., 2019, 9, 1321 CrossRef CAS.
- B. Doshi, E. Repo, J. P. Heiskanen, J. A. Sirviö and M. Sillanpää, Carbohydr. Polym., 2017, 167, 326–336 CrossRef CAS PubMed.
- M. Liu, C. Wu, Y. Jiao, S. Xiong and C. Zhou, J. Mater. Chem. B, 2013, 1, 2078–2089 RSC.
- S. Kumar and J. Koh, Int. J. Mol. Sci., 2012, 13, 6102–6116 CrossRef CAS PubMed.
- R. A. Batista, C. G. Otoni and P. J. P. Espitia, in Materials for Biomedical Engineering: Hydrogels and Polymer-based Scaffolds, Elsevier, 2019, pp. 61–81 Search PubMed.
- M. N. Collins, G. Ren, K. Young, S. Pina, R. L. Reis and J. M. Oliveira, Adv. Funct. Mater., 2021, 31, 2010609 Search PubMed.
- S. Kumar, R. K. Majhi, A. Singh, M. Mishra, A. Tiwari, S. Chawla, P. Guha, B. Satpati, H. Mohapatra, L. Goswami and C. Goswami, ACS Appl. Mater. Interfaces, 2019, 11, 42998–43017 CrossRef CAS PubMed.
- N. Kavanagh, E. J. Ryan, A. Widaa, G. Sexton, J. Fennell, S. O’rourke, K. C. Cahill, C. J. Kearney, F. J. O’brien and S. W. Kerrigan, Clin. Microbiol. Rev., 2018, 31, e00084–17 CrossRef CAS PubMed.
- T. Kawamura, D. Ono, A. Shirai, K. Mimura, S. Iida, K. Saita, H. Oka and H. Ohno, IDCases, 2022, 27, e01404 Search PubMed.
- A. Piegat, A. Żywicka, A. Niemczyk and A. Goszczyńska, Polymers, 2021, 13, 1–13 Search PubMed.
- X. Liu, D. P. Rodeheaver, J. C. White, A. M. Wright, L. M. Walker, F. Zhang and S. Shannon, Regul. Toxicol. Pharmacol., 2018, 97, 24–32 Search PubMed.
- A. Singh, S. Kumar, T. K. Acharya, S. Kumar, S. Chawla, C. Goswami and L. Goswami, Int. J. Biol. Macromol., 2024, 264, 130605 CrossRef CAS PubMed.
- A. Singh, C. Muduli, S. P. Senanayak and L. Goswami, Int. J. Biol. Macromol., 2023, 234, 123724 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |