
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual-state emission triphenylamine-coumarin fluorescent polymorphs: halochromic reversible fluorescence switching and π–π stacking facilitated picric acid sensing†
Sasikala
Ravi
a,
Subramanian
Karthikeyan
b,
Mehboobali
Pannipara
c,
Abdullah G.
Al-Sehemi
de,
Dohyun
Moon
 *f and
Savarimuthu Philip
Anthony
*a
*f and
Savarimuthu Philip
Anthony
*a
aSchool of Chemical & Biotechnology, SASTRA Deemed University, Thanjavur- 613401, Tamil Nadu, India. E-mail: philip@biotech.sastra.edu
bPG and Research Department of Chemistry, KhadirMohideen College (affiliated to Bharathidasan University), Adirampattinam, Tamil Nadu, India
cCentral Labs, King Khalid University, AlQuráa, Abha, Saudi Arabia
dResearch center for Advanced Materials Science, King Khalid University, Abha 61413, Saudi Arabia
eDepartment of chemistry, King Khalid University, Abha 61413, Saudi Arabia
fBeamline Department, Pohang Accelerator Laboratory, 80 Jigokro-127 beongil, Nam-gu, Pohang, Gyeongbuk, Korea. E-mail: dmoon@postech.ac.kr
First published on 24th March 2025
Abstract
Organic fluorescence molecules with external stimuli-responsive properties have received significant interest because of their potentiality in various device applications ranging from optoelectronics to biological sciences. Herein, we designed and synthesized dual-state emission (DSE), triphenylamine (TPA)-integrated, coumarin-based donor–acceptor multifunctional fluorophores (DP2C and DP4C) with an acid responsive pyridine unit. DP2C produced fluorescent polymorphs (yellow (DP2C-Y) and orange (DP2C-O)) and showed tunable solid-state fluorescence (λmax = 537 and 560 nm). DP2C-O exhibited relatively strong fluorescence (quantum yield (ϕf) = 20%) compared with DP2C-Y (ϕf = 12%) and DP4C (ϕf = 16%), which could be attributed to the higher molecular twist in the solid state without any close π-stacking. Both DP2C and DP4C showed strong fluorescence in the solution state, especially in non-polar toluene and CHCl3 (ϕf = 0.69) compared with a 0.5 M quinine sulphate standard in H2SO4. Acid responsive pyridine and DSE were utilized to demonstrate reversible fluorescence switching via protonation and deprotonation upon exposure to trifluoroacetic acid (TFA) and triethylamine/ammonia (NEt3)/NH3 in CHCl3 and solids. TFA addition red shifted the fluorescence with a strong reduction in fluorescence intensity, which was reversed to the initial state by adding NEt3. Furthermore, DP2C and DP4C exhibited highly selective fluorescence sensing of picric acid (PA). Limit of detection (LOD) studies for PA sensing indicated sensing values of up to 280.7 and 310 nM corresponding to DP2C and DP4C, respectively. Single crystal analysis revealed π-stacked arrangements between DP2C and PA. Computational studies showed intermolecular charge transfer from DP2C to the PA unit in the lowest unoccupied molecular orbital (LUMO), supporting fluorescence quenching. Fluorescent PMMA and PVA polymer thin films were fabricated and demonstrated reversible fluorescence switching and PA sensing, respectively, in thin films, indicating the practical utility of the developed DSE fluorophores.
Introduction
Functional organic fluorescence molecules are important candidates for many applications, including organic light-emitting diodes (OLEDs), displays, sensors and bioimaging.1–10 Particularly, dual-state emission (solution- and solid-state) molecules with stimuli-responsive properties have received significant attention in recent years since they can be utilized in their molecular and aggregated states for improving device performance and fabrication.11–13 For instance, strong fluorescence in a de-aggregated state allows the fabrication of fluorescent polymer composite thin films for re-usable sensors, data storage and anticounterfeiting applications.14–17 In general, organic π-conjugated planar molecules show strong emission in solution but weak or no emission in the condensed state owing to the aggregation-caused quenching (ACQ) effect.18,19 In contrast, generating fluorophores with a non-planar core unit produces aggregation-induced emission (AIE) and exhibits weak emission in solution due to the molecular flexibility and free rotation of single bonds.20–22 Hence, DSE fluorophores is relatively challenging than designing solution or solid-state emissive molecules and requires a special design strategy.23–25 In general, organic π-conjugated fluorophores with donor–acceptor units connected through an aliphatic linker mostly exhibit strong solid-state fluorescence.26 However, donor–acceptor units interconnected through an aromatic linker in a π-conjugated structure leads to DSE due to the restriction of structural flexibility.27,28 Integrating imidazole and triazole acceptor units results in strongly enhanced fluorescence in the solution and solid states, along with improved stability.29–32 Excited-state intramolecular proton transfer (ESIPT) molecules with a strong intramolecular H-bonding functionality exhibited DSE and stimuli-induced fluorescence switching.33–35 Introducing aliphatic chains or bulky groups into aromatic fluorophores, such as pyrene, perylene diimide and BODIPY, isolates the fluorophores, preventing π-stacking and leading to enhanced fluorescence in the solution and solid states.36–38 Nevertheless, the advantages of DSE fluorophores for demonstrating multi-functional applications are rarely reported. The present work aims to develop DSE molecules with functional units to achieve stimuli-responsive fluorescence switching and sensing in the solution and solid states.Triphenylamine (TPA), a propeller-shaped non-planar unit, has been extensively utilized for synthesising donor–acceptor fluorophores with tunable and switchable emission.39–41 Its non-planar conformation hinders close π-stacking between molecules in the solid state, producing AIEgens with strong fluorescence.42,43 Its structural tailorability allows modulation of π-conjugation and lead to fluorescence tuning from blue to NIR light.44–46 Similarly, heteroaromatic π-conjugated coumarin derivatives have been employed for developing fluorescence chemosensors and room temperature phosphorescent molecules.47–52 The bio-compatibility and structural flexibility of coumarin make it suitable for many biological applications, including bioimaging as well as synthesis of antibiotics and anticoagulants.53–55 Coumarin functionality has also been exploited for synthesizing ESIPT molecules with significantly red-shifted fluorescence.56 Therefore, the integration of planar heteroaromatic coumarin with non-planar TPA is expected to produce a DSE molecule with functional properties. In this study, we synthesized TPA-based pyridine isomers with coumarin units (DP2C and DP4C), which exhibited DSE, polymorphism, switchable fluorescence and picric acid (PA) sensing (Scheme 1). DP2C produced yellow (DP2C-Y) and orange (DP2C-O) emitting fluorescent polymorphs. DP4C displayed yellow fluorescence in the solid state. DP2C-Y exhibited a relatively lower conformational twist between coumarin and pyridine compared with DP2C-O. DP4C showed a nearly coplanar conformation with slipped π-stacking. Both isomers demonstrated strong fluorescence in non-polar solvents, such as toluene, CHCl3 and EtOAc. The fluorescence was significantly enhanced (2.5 times) for both isomers upon integration into a PMMA polymer matrix owing to structural rigidification and a hydrophobic environment. The presence of an acid-responsive pyridine unit was utilized to demonstrate acid/base-dependent fluorescence switching in solution, solid state, and the PMMA polymer matrix. DP2C and DP4C exhibited highly selective fluorescence quenching for PA in solution and the PVA polymer matrix. Single crystal analysis suggested the formation of a supramolecular complex with PA. Computational studies revealed intermolecular charge transfer from DP2C to PA in LUMO, which contributed to fluorescence quenching.
 | ||
| Scheme 1 Molecular structures of DP2C and DP4C, images of fluorescent polymorphs of DP2C, digital fluorescence images, stimuli-responsive fluorescence switching and PA sensing. | ||
Experimental
Diphenylamine, 3-iodoanisole, phosphorus oxychloride, boron tribromide, piperidine, 2-/3-pyridyl acetonitrile, potassium carbonate and solvents were procured from Merck.Synthesis of 4-(diphenylamino)-2-hydroxybenzaldehyde
4-(Diphenylamino)-2-hydroxybenzaldehyde was synthesized according to previous reports.57,58Synthesis of coumarin integrated TPA pyridine isomers (Scheme S1, ESI†)
Aldehyde (0.25 g, 1 mmol), 2/4-pyridylacetonitrile (0.18 g, 1.8 mmol) and piperidine (0.07 g, 1 mmol) were mixed and refluxed for 12 h. After completion of the reaction, the mixture was extracted, concentrated and purified through column chromatography using ethyl acetate: hexane (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85) mixture as the eluent.
:85) mixture as the eluent.
7-(Diphenylamino)-3-(pyridin-2-yl)-2H-chromen-2-one
(DP2C): M.pt: 180 °C; yield: 81%. 1H NMR (600 MHz, CDCl3) δ 8.68 (s, 1H), 8.59 (d, 1H), 8.37–8.36 (d, J = 6 Hz, 1H), 7.74–7.71 (t, J = 9 Hz, 1H), 7.35–7.34 (d, J = 6 Hz, 1H), 7.30–7.27 (m, 4H), 7.22–7.20 (m, 1H), 7.14–7.11 (m, 6H), 6.83–6.81 (dd, J = 9 Hz, 1H), 6.77–6.76 (d, J = 6 Hz, 1H) ppm. 13C NMR (150 MHz, CDCl3) δ 160.6, 155.7, 152.3, 151.5, 148.2, 145.8, 143.1, 129.8, 129. 7, 126.4, 125.5, 124.0, 122.8, 116.6, 112.8, 106.7, 105.5 ppm. HRMS (m/z) C26H18N2O2 calculated: 390.1368; found: 389.9920.7-(Diphenylamino)-3-(pyridin-2-yl)-2H-chromen-2-one
(DP4C): M.pt: 190 °C, yield: 76%. 1H NMR (600 MHz, CDCl3) δ 8.58 (s, 1H), 7.80 (s, 1H), 7.60–7.59 (d, J = 6 Hz, 2H), 7.31–7.25 (m, 6H), 7.14–7.12 (m, 6H), 6.84–6.82 (dd, J = 9 Hz, 1H), 6.77–6.76 (d, J = 6 Hz, 1H) ppm. 13C NMR (150 MHz, CDCl3) δ 160.1, 155.5, 152.2, 149.9, 145.7, 142.9, 141.4, 130.1, 129.8, 128.9, 126.4, 125.5, 122.5, 116.5, 112.4, 105.7 ppm. HRMS (m/z) C26H18N2O2 calculated: 390.1368; found: 390.0590.Characterization
NMR spectra were measured on JEOL 600 MHz. Absorption and fluorescence spectra were recorded using a Jasco V-730ST UV-visible spectrophotometer and a FP-8300 spectrofluorometer. Absolute quantum yields for solids were measured using an integrating sphere attached to the FP-8300 spectrofluorometer. Single crystals were coated with paratone-N oil, and the diffraction data were measured using synchrotron radiation (λ = 0.62998 Å) on an ADSC Quantum-210 detector at 2D SMC with a silicon (111) double crystal monochromator (DCM) at the Pohang Accelerator Laboratory, Korea. CCDC no. 2415627–2415630 contain the supplementary crystallographic data for this study. The HOMO, LUMO and band gap of DP2C/DP4C isomers were studied in the gas phase using the B3PW91/6-31+G(d,p) level of theory (Gaussian 09 package). The molecular structures for the calculations were obtained from the single crystal data and used without structural optimization.Results and discussion
TPA-based coumarin pyridine isomers, DP2C and DP4C, were synthesized via intramolecular cyclisation, as presented in Scheme 2. The 4-(diphenylamino)-2-hydroxybenzaldehyde precursor was synthesized as per the procedure depicted in Scheme S1 (ESI†). The coupling reaction between 4-(diphenylamino)-2-hydroxybenzaldehyde and pyridine acetonitrile in ethanol in the presence of piperidine base produced DP2C and DP4C, respectively, in good yield. The structures of both isomers were confirmed using NMR and mass spectrometry (ESI†). Single crystals of DP2C and DP4C were grown through the slow evaporation method to confirm the structure and analyse the molecular assembly in the solid state (Tables S1–S3, ESI†). DP2C produced yellow and orange-emitting polymorphic crystals (DP2C-Y from CHCl3–MeOH and DP2C-O from MeOH) depending on the solvent of crystallization. DP4C did not exhibit polymorphism and formed yellow fluorescent crystals. PXRD of DP2C-Y and DP2C-O showed different patterns, confirming polymorphism (Fig. S1, ESI†). The polymorphs (DP2C-Y and DP2C-O) and pyridine isomer (DP4C) displayed different molecular twists between the coumarin and pyridine units in the solid state (Fig. 1a). The conformational twist was analysed using the torsional (τ) angle between pyridine and coumarin (Table 1). The comparison of torsion angles suggested higher molecular twist in DP2C-O compared with that in DP2C-Y and DP4C. A coplanar conformation with the lowest molecular twist was observed in DP4C. This variation in the conformational twist resulted in different molecular assemblies and intermolecular interactions in the crystal lattice of DP2C-Y and DP2C-O. The intermolecular H-bonding between the carbonyl oxygen and TPA phenyl hydrogen produced a dimer with an opposite molecular arrangement (Fig. 1b). The slipped π-interactions between pyridine and coumarin carbonyl carbon produced a layered structure with a parallel arrangement (Fig. 1c). Two molecules were present in the asymmetric unit of DP2C-O, and one of the TPA phenyl units showed disorder (Fig. S2, ESI†). The complimentary H-bonding between carbonyl oxygen and phenyl hydrogen connected both the molecules and produced an oppositely arranged dimer (Fig. 1d). Furthermore, each molecule of the dimer formed C–H⋯π and slipped π⋯π interactions and a network structure in the crystal lattice (Fig. 1e). DP4C exhibited a nearly coplanar conformation between coumarin and pyridine (Fig. 1a). The intermolecular H-bonding between coumarin oxygen and phenyl hydrogen produced a sheet structure with a parallel arrangement of molecules in the crystal lattice (Fig. 1f). The sheets were interconnected through π⋯π interactions and H-bonding, resulting in an oppositely arranged structure (Fig. 1g). The molecular conformational twist differences in DP2C polymorphs and DP4C caused distinct molecular packing in the crystal lattice (Fig. S3, ESI†).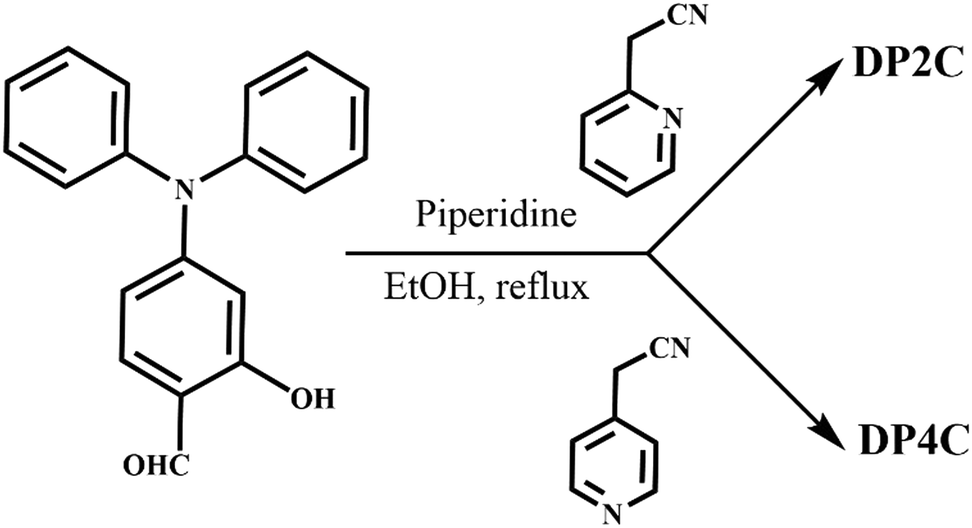 | ||
| Scheme 2 Synthetic scheme for DP2C and DP4C. | ||
 | ||
| Fig. 1 (a) Molecular structure in the crystal lattice of DP2C-Y, DP2C-O and DP4C, and intermolecular interactions in the crystal lattice of (b) and (c) DP2C-Y, (d) and (e) DP2C-O and (f) and (g) DP4C [C (grey), H (white), N (blue) and O (red)]. Dotted lines indicate hydrogen bonding, C–H⋯π and π⋯π interactions in Å. | ||
|
|
||
|---|---|---|
| Torsion angle (τ) | ||
| τ 1 | τ 2 | |
| DP2C-Y | 17.06 | 13.54 |
| DP2C-O | 21.05 (I) | 17.61 (I) |
| 26.16 (II) | 23.46 (II) | |
| DP4C | 6.14 | 4.86 |

The donor–acceptor structure with non-planar molecular confirmation was expected to produce solid-state fluorescence. DP2C-Y showed dual emission in the solid state at 498 and 537 nm, whereas DP2C-O showed strong fluorescence at 560 nm (Fig. 2a). DP4C exhibited yellow fluorescence at 546 nm. Absolute quantum yield measurement revealed the highest efficiency for DP2C-O (20%), which could be attributed to the more twisted molecular conformation (Table 1). DP2C-Y and DP4C showed slightly lower fluorescence efficiencies of 12% and 16%, respectively.
 | ||
| Fig. 2 (a) Solid-state fluorescence, solvent polarity dependent fluorescence tuning of (b) DP2C and (c) DP4C and (d) fluorescence of DP2C/DP4C in CHCl3 and CDCl3. Digital fluorescence images are integrated into the corresponding spectra. λexc = 420 nm (for spectra) and 365 nm (for images). | ||
In solution, the absorption of DP2C and DP4C did not show a significant change with solvent polarity, suggesting a non-polar ground state (Fig. S4, ESI†). Both molecules exhibited an intramolecular charge transfer (ICT) peak between 405 and 410 nm in polar to non-polar solvents. In contrast, DP2C and DP4C showed solvent polarity-dependent tunable fluorescence (Fig. 2b and c, Table 2). DP2C and DP4C displayed dual emission peaks: a locally excited (LE) state (shorter wavelength) and a charge transfer (CT) state emission (longer wavelength), with varied peak intensity depending on solvent polarity. In toluene, LE state shorter emission (λmax = 481 (DP2C) and 491 nm (DP4C)) was more intense compared with CT state emission (λmax = 532 nm). However, CT state emission became stronger in CHCl3, THF and EtOAc.
| DP2C | DP4C | |||
|---|---|---|---|---|
| λ max (nm) | ϕ f | λ max (nm) | ϕ f | |
| a Quantum yields were measured by comparing with standard quinine (0.5 M H2SO4). b Absolute quantum yields. | ||||
| Solventa | ||||
| Toluene | 483, 530 | 0.41 | 491, 532 | 0.37 |
| THF | 495, 536 | 0.45 | 493, 545 | 0.13 |
| EtOAC | 494, 536 | 0.66 | 493, 545 | 0.12 |
| CHCl3 | 494, 536 | 0.69 | 494, 540 | 0.47 |
| DMF | 492, 553 | 0.04 | 488, 564 | — |
| CH3CN | 492, 553 | 0.03 | 490, 569 | — |
| CH3OH | 492, 548 | — | 488, 567 | — |
| Solidsb | ||||
| DP2C-Y | 497, 537 | 12% | ||
| DP2C-O | 560 | 20% | ||
| DP4C | 547 | 16% | ||
| Polymer matrixb | ||||
| DP2C-PMMA | 481, 528 | 55.2% | ||
| DP4C-PMMA | 492, 526 | 63.3% | ||
The fluorescence was very weak in polar protic/aprotic solvents with a further red shift in longer wavelength emission. This insignificant change in absorption and tunable fluorescence with solvent polarity indicated the formation of twisted intramolecular charge transfer (TICT).59 The red shift in emission with efficiency reduction in polar solvents is characteristic of the TICT state.60 Thus, DP2C and DP4C exhibited strong LE state emission in non-polar toluene, whereas strong TICT emission in CHCl3 and THF. Excitation spectra of DP2C/DP4C in toluene revealed similar spectra except for an increase in intensity for LE state emission compared with CT state emission (Fig. S5, ESI†). Excited state lifetime analysis was performed to understand the LE and CT state emission of DP2C and DP4C (Fig. S6 and Table S4, ESI†). Toluene was chosen as a representative solvent. Both DP2C and DP4C revealed biexponential decay components with nanosecond lifetimes (Table S4, ESI†). Blue-shifted emission showed a shorter lifetime compared with red-shifted emission in both compounds (Fig. S6a, ESI†). This shorter and longer lifetime of blue and red-shifted emissions could be attributed to the LE and CT states, respectively. Moreover, biexponential decay with a shorter lifetime for blue-shifted emission was observed in the solid state (Fig. S6b and Table S4, ESI†).
DP2C and DP4C displayed unexpected behavior when dissolved in CDCl3. Both compounds showed a significant red shift in the ICT peak and strong quenching of fluorescence in CDCl3 compared with that in CHCl3 (Fig. 2d and Fig. S7, ESI†). The peaks at 418 and 410 nm were red shifted to 467 and 473 nm for DP2C and DP4C, respectively (Fig. S7, ESI†). Fluorescence also red-shifted for both molecules with strong reduction in intensity (Fig. 2d). This unusual behavior was observed while recording NMR spectra in CDCl3. This unusual absorption and fluorescence modulation in CDCl3 could be attributed to the solute–solvent interaction of CHCl3 present in the solvent in trace amounts. NMR spectra of DP2C/DP4C showed a CHCl3 peak at 7.19 ppm compared with the usual 7.26 ppm, further supporting the solvent–solute interaction with the pyridine nitrogen.61 To gain insight into ICT, density functional theory (DFT) calculations were performed for DP2C/DP4C. Electron density was mostly occupied in the triphenylamine unit in HOMOs but shifted to the coumarin unit in LUMOs in all three structures (Fig. 3). The comparison of the optical band gap suggested a lower band gap for DP2C-O (3.47 eV) compared with that for DP2C-Y (3.58 eV), supporting fluorescence tuning through polymorphism.
 | ||
| Fig. 3 HOMO–LUMO diagram for DP2C-Y, DP2C-O, DP4C and DP2C with PA. | ||
The strong dual-state fluorescence and acid-responsive pyridine functionality of DP2C and DP4C provided an excellent opportunity for achieving halochromic fluorescence switching through protonation and deprotonation. DP2C powder showed strong fluorescence at 550 nm, which was red-shifted to 635 nm with strong quenching upon exposure to TFA (Fig. 4a). The protonation of nitrogen could increase the electron-withdrawing character and red-shift the emission. To confirm the protonation of pyridine, NMR was recorded in the presence of TFA. The addition of 1 eq. TFA to DP2C in CDCl3 showed a downfield shift of NMR signals (Fig. S8, ESI†). Additionally, a new peak appeared at δ 9.5 ppm, corresponding to the Py-NH+ proton, confirming pyridine nitrogen protonation. Fluorescence was completely reversed to the initial state upon NH3 exposure. The strong fluorescence of DP4C powder was red-shifted from 557 to 632 nm upon TFA exposure (Fig. S9, ESI†). Unlike the NH3 exposure in DP2C, that in DP4C did not show complete reversal of fluorescence and reversed only up to 593 nm. Furthermore, protonated DP4C showed relatively higher fluorescence intensity compared with DP2C after TFA exposure. In CHCl3, DP2C showed π–π* and ICT absorption at 340 and 409 nm, respectively (Fig. S10a, ESI†). TFA addition red-shifted ICT absorption to 502 nm, while the intensity of π–π* absorption increased without changing the peak position. The red-shifted absorption was reversed to the initial state upon NEt3 addition (Fig. S10b, ESI†). DP4C in CHCl3 was more sensitive to TFA and NEt3 (Fig. S10c and d, ESI†). The absorption immediately changed from 413 to 495 nm upon the addition of 1 μL of TFA but gradually reversed with the addition of NEt3. DP2C showed dual emission at 494 and 536 nm in CHCl3 (Fig. 4b). The fluorescence was completely quenched upon the addition of TFA and reversed to the initial state upon the addition of NEt3. DP4C in CHCl3 also exhibited similar off–on fluorescence switching upon the addition of TFA and NEt3 (Fig. S11, ESI†).
 | ||
| Fig. 4 Acid (TFA)–base (NEt3/NH3) exposure-dependent fluorescence switching of (a)–(c) DP2C and (d) DP4C. (a) Solid-state, (b) CHCl3 and (c, d) PMMA matrix. Digital fluorescence images are integrated into the corresponding spectra. λexc = 420 nm (for spectra) and 365 nm (for images). | ||
The DP2C and DP4C fluorophores were integrated into a PMMA polymer matrix to demonstrate reversible fluorescence switching in a thin film. DP2C/DP4C (25 μL of 10−3 M) was added to PMMA (1 wt%) toluene solution and stirred at 70 °C temperature for 30 min. Thin films of PMMA-DP2C/DP4C were fabricated using the drop casting method. Interestingly, integrating the fluorophore into the PMMA matrix significantly enhanced its fluorescence efficiency (Table 2). DP2C-PMMA and DP4C-PMMA thin films showed fluorescence efficiencies of 55.2% and 63.3%, respectively (Fig. 4c and d). The enhancement of fluorescence efficiency was attributed to the rigidification of the molecular structure under a hydrophobic environment. In the PMMA matrix, LE state emission (481 nm) became stronger than CT state emission (528 nm, Fig. 4c). FTIR spectra indicated an increase in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O wavenumber after integrating DP2C/DP4C fluorophores with the PMMA matrix (Fig. S12 and S13, ESI†). PMMA carbonyl stretching appeared at 1731 cm−1. DP2C and DP4C carbonyl stretching increased from 1720 and 1699 to 1734 and 1743, respectively. This suggested the interaction between the fluorophore and the PMMA matrix, which might have contributed to the rigidification of the fluorophore. The fluorescence was completely quenched upon TFA exposure and reversed to the initial state upon NH3 exposure. Digital fluorescence images also confirmed reversible fluorescence switching due to TFA/NH3 exposure. The off–on fluorescence switching by TFA/NEt3, could be achieved for several cycles without significant changes in the fluorescence intensity or peak position (Fig. S14, ESI†). The DP4C-PMMA thin film also showed strong LE state emission (492 nm) than CT state emission (526 nm, Fig. 4d). TFA exposure red-shifted the fluorescence (583 nm) with a strong reduction in intensity. The fluorescence intensity was reversed upon NH3 exposure, but CT emission (536 nm) appeared stronger than LE state emission after NH3 exposure. Both thin films showed slight self-recovery of fluorescence in 24 h (Fig. S15a and b, ESI†). Absorption spectra of PMMA-DP2C/DP4C fluorophore thin films showed a strong ICT peak around 410 nm, which was red-shifted upon TFA exposure and reversed to the initial state upon NH3 exposure (Fig. S15c and d, ESI†).
O wavenumber after integrating DP2C/DP4C fluorophores with the PMMA matrix (Fig. S12 and S13, ESI†). PMMA carbonyl stretching appeared at 1731 cm−1. DP2C and DP4C carbonyl stretching increased from 1720 and 1699 to 1734 and 1743, respectively. This suggested the interaction between the fluorophore and the PMMA matrix, which might have contributed to the rigidification of the fluorophore. The fluorescence was completely quenched upon TFA exposure and reversed to the initial state upon NH3 exposure. Digital fluorescence images also confirmed reversible fluorescence switching due to TFA/NH3 exposure. The off–on fluorescence switching by TFA/NEt3, could be achieved for several cycles without significant changes in the fluorescence intensity or peak position (Fig. S14, ESI†). The DP4C-PMMA thin film also showed strong LE state emission (492 nm) than CT state emission (526 nm, Fig. 4d). TFA exposure red-shifted the fluorescence (583 nm) with a strong reduction in intensity. The fluorescence intensity was reversed upon NH3 exposure, but CT emission (536 nm) appeared stronger than LE state emission after NH3 exposure. Both thin films showed slight self-recovery of fluorescence in 24 h (Fig. S15a and b, ESI†). Absorption spectra of PMMA-DP2C/DP4C fluorophore thin films showed a strong ICT peak around 410 nm, which was red-shifted upon TFA exposure and reversed to the initial state upon NH3 exposure (Fig. S15c and d, ESI†).
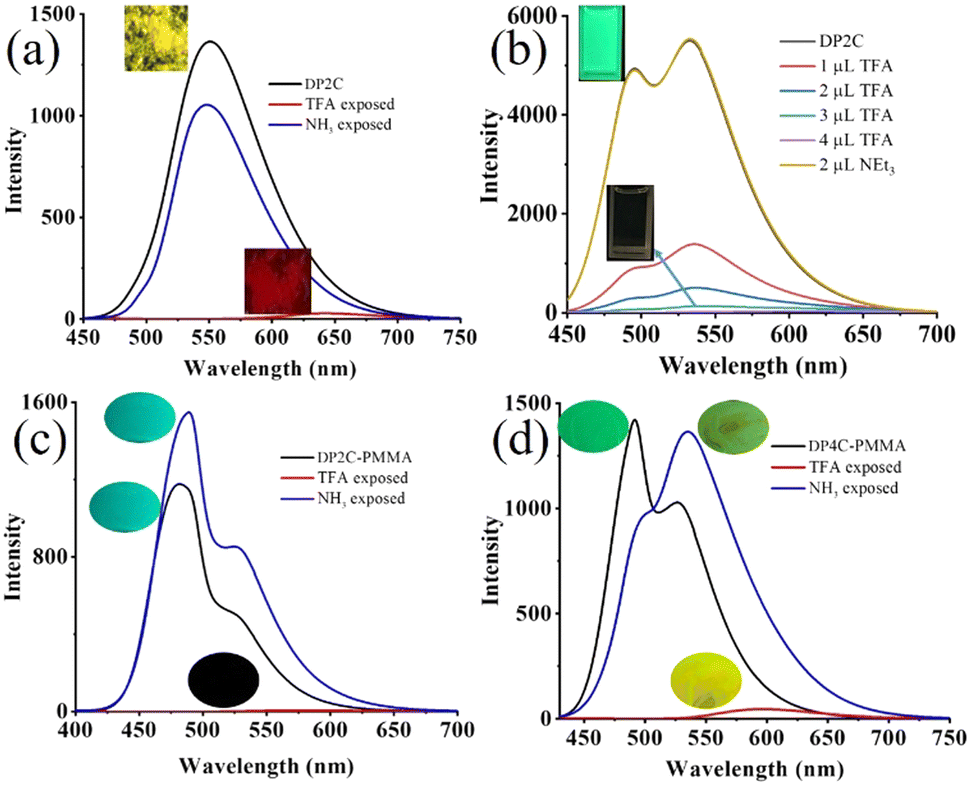
Dual state emission of DP2C and DP4C was further explored for nitroaromatic compound (NACs) sensing because of their intermolecular interacting functionality and planar coumarin π-conjugated ring. NACs were dissolved in CHCl3 (10−3 M) and added to a CHCl3 solution of DP2C/DP4C (10−4 M). The fluorescence of DP2C/DP4C was selectively quenched by PA, while the addition of other NACs did not significantly alter the fluorescence (Fig. 5a and b). Digital fluorescence images also showed the complete disappearance of green fluorescence after the addition of PA. Concentration-dependent studies showed a gradual decrease in fluorescence intensity with increasing PA concentration (Fig. 5c and Fig. S16a, ESI†). The LOD obtained from concentration-dependent studies on DP2C/DP4C with PA indicated a sensing value of up to 280.7/310 nM (Fig. S16b and c, ESI†). Furthermore, absorption spectra of DP2C/DP4C probes with NACs showed a selective response to PA (Fig. S17, ESI†). DP2C with PA showed appearance of a new peak at 340 nm and a reduction of the ICT peak at 409 nm (Fig. S17a, ESI†). 4-Nitroaniline (4-NA) exhibited a change in absorption, whereas other NACs did not show any significant change. DP4C showed a more sensitive response, with a new red-shifted CT peak at 482 nm and a new peak at 340 nm (Fig. S17b, ESI†). PA concentration-dependent absorption studies revealed a gradual increase in peak intensity at 340 nm and an extension of λcut-off beyond 475 nm for DP2C (Fig. S17c, ESI†). DP4C exhibited a gradual disappearance of the ICT peak at 410 nm and the emergence of a new red-shifted peak at 482 nm and 340 nm (Fig. S17d, ESI†). The absorption and fluorescence studies indicated a selective interaction between DP2C/DP4C and PA. DP2C/DP4C could interact with PA via (i) protonation of pyridine nitrogen and (ii) intermolecular H-bonding/π⋯π interactions.
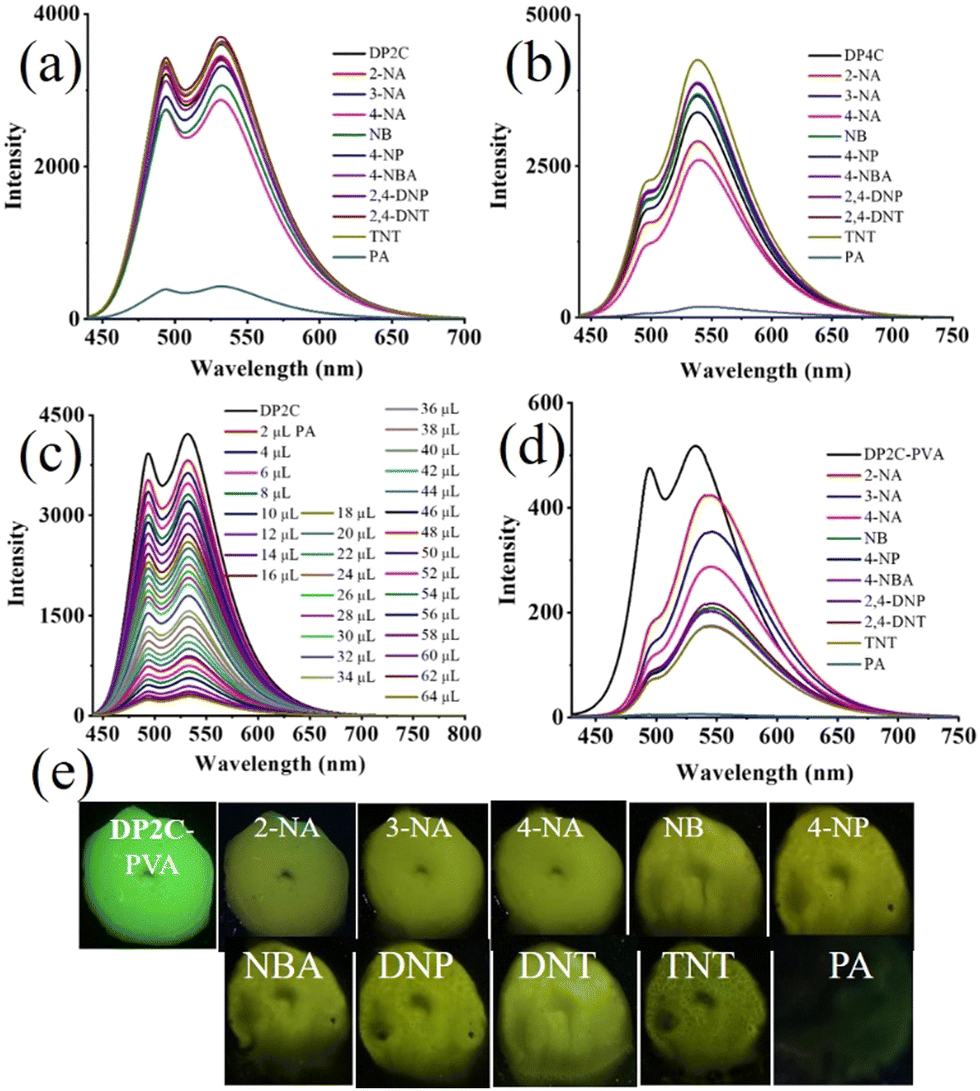 | ||
| Fig. 5 Fluorescence sensing of PA (10−4 M) using (a) DP2C (10−5 M) and (b) DP4C (10−5 M), (c) PA concentration-dependent fluorescence change of DP4C in CHCl3, (d) fluorescence sensing of PA in an aqueous medium using the DP2C-PVA thin film and (e) digital fluorescence images of DP2C-PVA after dipping in aqueous solution of NACs. λexc = 420 nm (for spectra) and 365 nm (for images). | ||
To understand the mechanism of DP2C/DP4C selective fluorescence quenching by PA, single crystals were attempted to be grown along with PA from a CHCl3–methanol mixture via slow evaporation of the solvent. DP2C produced quality dark-red single crystals with PA (Fig. 6a). In contrast, DP2C crystals without PA appeared pale yellow. The crystal lattice of DP2C-PA comprised two DP2C molecules and one PA, along with a solvent methanol molecules (Fig. 6a and Table S5, ESI†). Oxygen in one of the nitro groups showed disorder in the crystal lattice. The PA hydroxy group either underwent deprotonation or participated in intermolecular H-bonding. The methanol solvent formed intermolecular H-bonding with DP2C carbonyl oxygen and the nitro group of PA. Another PA molecule exhibited direct intermolecular interactions with DP2Cvia nitro with carbonyl oxygen and pyridine nitrogen. These intermolecular interactions produced a layered structure in the crystal lattice (Fig. 6a). PA further showed strong π⋯π interactions with coumarin and produced alternative packing of PA and coumarin in the crystal lattice (Fig. 6b). The π⋯π stacking of PA and coumarin might facilitate charge transfer from coumarin to PA, which could result in fluorescence quenching. Computational studies indicated that electron density was primarily located in the TPA-coumarin unit in HOMO but was completely transferred to PA in LUMO (Fig. 4). Thus, HOMO–LUMO calculations supported the fluorescence quenching by intermolecular charge transfer from DP2C/DP4C to PA. DP2C/DP4C showed a red shift of fluorescence with a reduction in intensity upon TFA exposure, further indicating that fluorescence quenching was not due to protonation of pyridine. To improve the practical utility of PA sensing, PVA-DP2C/DP4C polymer thin films were fabricated and PA sensing was performed in an aqueous medium. PVA (1 wt%) was dissolved in water and 25 μL of DP2C/DP4C (10−3 M in CH3CN) was introduced. The resulting solution was stirred at room temperature for 1 h to ensure uniform distribution. Thin films were fabricated using the drop casting method. DP2C-PVA showed green fluorescence with dual peaks at 495 and 532 nm (Fig. 5d and e). Dipping the film into an aqueous solution of PA (10−3 M) resulted in complete quenching of fluorescence. Dipping into other NAC solutions showed yellow-green fluorescence with reduced intensity. LE state emission appeared as a hump, and CT emission was slightly red-shifted to 543 nm, accompanied by a reduction in intensity with some of the NACs. The digital fluorescence image also showed transformation from green to yellow-green (Fig. 5e). It was observed that the as-fabricated thin film of DP2C-PVA showed fluorescence color transformation from green to yellow-green when dipped into an aqueous solution without NACs (Fig. S18, ESI†). The fluorescence color was reversed to green upon drying. Hence, fluorescence changes in DP2C/DP4C-PVA after dipping into aqueous solutions of other NACs might not be due to the influence of NACs but rather due to dipping into water. Only dipping into PA solution caused complete selective quenching of fluorescence. The DP4C-PVA thin film also exhibited similar behavior (Fig. S19, ESI†).
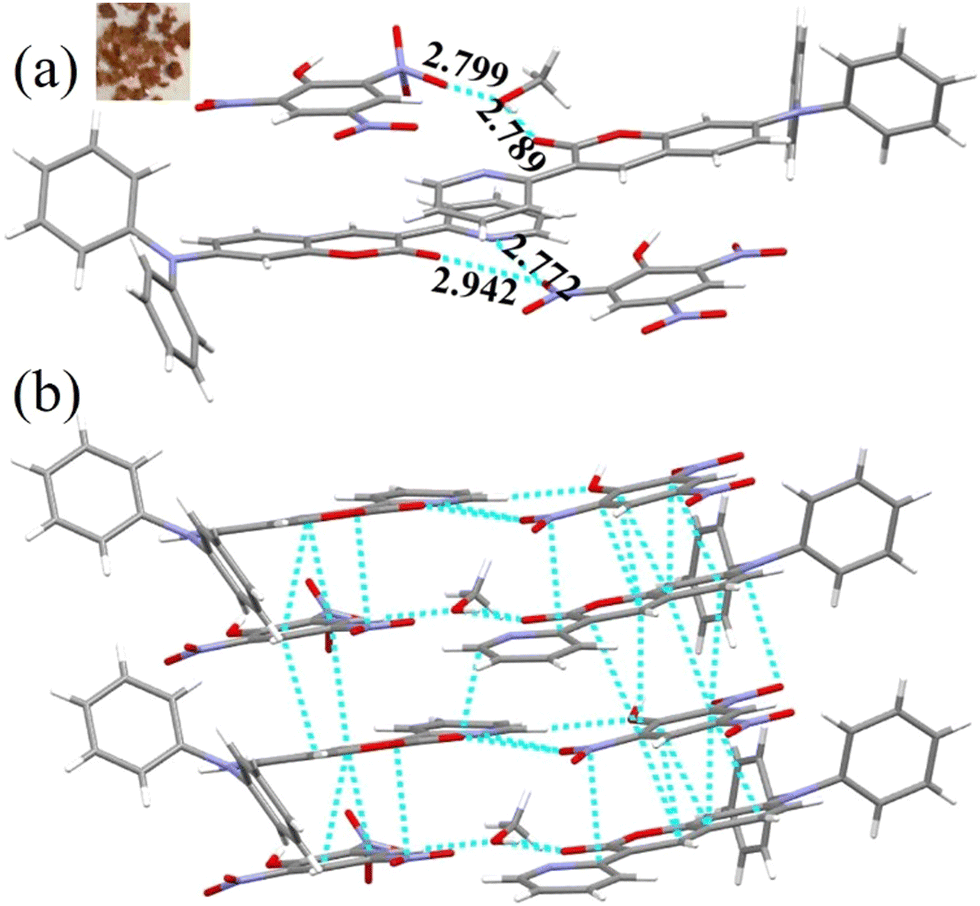 | ||
| Fig. 6 (a) Molecular structure in the crystal lattice of DP2C-PA and (b) π⋯π stacked arrangements of DP2C and PA in the crystal lattice [C (grey), H (white), N (blue) and O (red)]. Dotted lines indicate hydrogen bonding, C–H⋯π and π⋯π interactions in Å. The distances between PA and DP2C range between 3.030 and 3.396 Å. DP2C-PA crystal image is shown in the inset. | ||
Conclusion
In conclusion, dual state emissive TPA-coumarin-based donor–acceptor pyridine isomers (DP2C and DP4C) were synthesized and demonstrated fluorescence tuning, reversible fluorescence switching and highly selective PA sensing. The DP2C and DP4C isomers showed strong fluorescence in the solution and solid states. Integration of fluorophores into the PMMA polymer matrix resulted in highly enhanced fluorescence efficiency (up to 2.5–3 times). DP2C produced fluorescent polymorphs with yellow (DP2C-Y) and orange (DP2C-O) emitting crystals, while DP4C crystals exhibited only yellow emission. DP2C-O displayed relatively higher molecular conformational twist with well-separated packing in the crystal lattice, leading to enhanced solid-state fluorescence efficiency (ϕf = 20%). The slipped π-stacked arrangements in DP2C-Y and DP4C produced slightly lower efficiencies (ϕf = 16% and 12%, respectively). Both isomers exhibited strong fluorescence in non-polar solvents. The presence of the acid-responsive pyridine functionality was utilized to achieve reversible off–on fluorescence switching upon acid–base exposure in solution, solids and polymer-fluorophore thin films. Furthermore, the DP2C and DP4C isomers exhibited highly selective fluorescence sensing of PA in CHCl3, with LODs of up to 280.7 and 310 nM, respectively. Moreover, PVA-fluorophore thin films exhibited highly selective fluorescence quenching upon dipping into an aqueous solution of PA, suggesting potential for PA sensing device fabrication. Single crystal analysis showed the formation of π-stacking arrangements between DP2C and PA via intermolecular interactions, which facilitated energy transfer from the fluorophore to PA and fluorescence quenching.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Science and Engineering Research Board (SERB), CRG/2020/003978, New Delhi, India is acknowledged with gratitude. The Deanship of Research and graduate studies at King Khalid University is greatly appreciated for funding through large research project (RGP2/397/46).References
- H. Liu, Y. Fu, B. Z. Tang and Z. Zhao, Nat. Commun., 2022, 13, 5154 Search PubMed.
- H. Li, N. Xie, J. Wang, Y. Zhao and B. Liang, Org. Electron., 2021, 88, 106004 CrossRef CAS.
- L. Xing, J. Wang, W.-C. Chen, B. Liu, G. Chen, X. Wang, J.-H. Tan, S. S. Chen, J.-X. Chen, S. Ji, Z. Zhao, M.-C. Tang and Y. Huo, Nat. Commun., 2024, 15, 6175 CrossRef CAS PubMed.
- S. R. Nayak, I. Siddiqui Shahnawaz, J.-H. Jou and S. Vaidyanathan, ACS Appl. Opt. Mater., 2023, 1, 94–106 CrossRef CAS.
- S. Zuo, Y. Li, T. Ren and L. Yuan, Sens. Diagn., 2024, 3, 28–39 RSC.
- Y. T. Nguyen, S. Shin, K. Kwon, N. Kim and S. W. Bae, J. Chem. Res., 2023, 47, 17475198231168961 CrossRef CAS.
- M. K. Goshisht, G. K. Patra and N. Tripathi, Mater. Adv., 2022, 3, 2612–2669 RSC.
- H. Liu, G. Jiang, G. Ke, T. Ren and L. Yuan, ChemPhotoChem, 2024, 8, 202300277 Search PubMed.
- R. Gui and H. Jin, Talanta, 2024, 275, 126171 CrossRef CAS.
- Y. Li, Y. Liu, Q. Li, X. Zeng, T. Tian, W. Zhou, Y. Cui, X. Wang, X. Cheng, Q. Ding, X. Wang, J. Wu, H. Deng, Y. Li, X. Meng, Z. Deng, X. Hong and Y. Xiao, Chem. Sci., 2020, 11, 2621–2626 RSC.
- L. A. Rodriguez-Cortes, A. Navarro-Huerta and B. Rodriguez-Molina, Matter, 2021, 4, 2622–2624 CrossRef CAS.
- G. Xia, L. Si and H. Wang, Mater. Today Chem., 2023, 30, 101596 CrossRef CAS.
- N. A. Kukhta and M. R. Bryce, Mater. Horiz., 2021, 8, 33–55 RSC.
- B. Ma, S. Wu and F. Zeng, Sens. Actuators, B, 2010, 145, 451–456 CrossRef CAS.
- R. A. Carrillo-Betancourt, A. D. López-Camero and J. Hernández-Cordero, Polymers, 2023, 15, 505 CrossRef CAS PubMed.
- J. Jiang, P. Zhang, L. Liu, Y. Li, Y. Zhang, T. Wu, H. Xie, C. Zhang, J. Cui and J. Chen, Chem. Eng. J., 2021, 425, 131557 CrossRef CAS.
- J. C.-M. Lee, J.-W. Li, K.-F. Cheng, J.-X. Chen, Y.-S. Ciou, J.-H. Wang, M.-C. Lu, Y.-F. Chen and C.-W. Chiu, ACS Appl. Electron. Mater., 2024, 6, 1617–1627 CrossRef CAS.
- Y. Zhan, Z. Yang, J. Tan, Z. Qiu, Y. Mao, J. He, Q. Yang, S. Ji, N. Cai and Y. Huo, Dyes Pigm., 2020, 173, 107898 Search PubMed.
- P. Das, A. Kumar, A. Chowdhury and P. S. Mukherjee, ACS Omega, 2018, 3, 13757–13771 Search PubMed.
- M. Bonnot, N. Ibrahim, M. Allain and P. Frère, Molecules, 2024, 29, 3135 Search PubMed.
- L. A. Rodríguez-Cortés, F. J. Hernández, M. Rodríguez, R. A. Toscano, A. Jiménez-Sánchez, R. Crespo-Otero and B. Rodríguez-Molina, Matter, 2023, 6, 1140–1159 CrossRef.
- G. Yashwantrao, P. Gosavi, V. Naik, M. Debnath, S. Seth, P. Badani, R. Srivastava and S. Saha, J. Mater. Chem. C, 2025, 13, 3955–3968 CAS.
- J. L. Belmonte-Vázquez, Y. A. Amador-Sánchez, L. A. Rodríguez-Cortés and B. Rodríguez-Molina, Chem. Mater., 2021, 33, 7160–7184 CrossRef.
- T. Stoerkler, T. Pariat, A. D. Laurent, D. Jacquemin, G. Ulrich and J. Massue, Molecules, 2022, 27, 2443 CAS.
- Q. Pei, Y. Yin, F. Tang and A. Ding, J. Lumin., 2023, 263, 120021 CAS.
- W. Jiang, G. Zhao, W. Tian and Y. Sun, Molecules, 2022, 27, 8099 CAS.
- K. Panthi, R. M. Adhikari and T. H. Kinstle, J. Phys. Chem. A, 2010, 114, 4542–4549 CAS.
- P. Gayathri, S. B. Subramaniyan, A. Veerappan, M. Pannipara, A. G. Al-Sehemi, D. Moon and S. P. Anthony, Cryst. Growth Des., 2022, 22, 633–642 CAS.
- S. Ravi, P. R. Nithiasri, S. Karthikeyan, M. Pannipara, A. G. Al-Sehemi, D. Moon and S. P. Anthony, RSC Adv., 2023, 13, 12476–12482 Search PubMed.
- A. Da Lama, J. P. Sestelo, L. Valencia, D. Esteban-Gómez, L. A. Sarandeses and M. M. Martínez, Dyes Pigm., 2022, 205, 110539 CAS.
- M. Rajasekar, C. Narendran, J. Mary, M. Sivakumar and M. Selvam, Results Chem., 2024, 7, 101543 Search PubMed.
- S. K. Pathak, Y. Xiang, M. Huang, T. Huang, X. Cao, H. Liu, G. Xie and C. Yang, RSC Adv., 2020, 10, 15523–15529 CAS.
- P. Gayathri, S. Ravi, S. Karthikeyan, M. Pannipara, A. G. Al-Sehemi, D. Moon and S. P. Anthony, J. Mol. Struct., 2023, 1284, 135429 CAS.
- P. Gayathri, S. Ravi, K. Akshaya, S. Karthikeyan, M. Pannipara, A. G. Al-Sehemi, D. Moon and S. P. Anthony, CrystEngComm, 2022, 24, 8126–8133 RSC.
- S. Petdee, C. Chaiwai, W. Benchaphanthawee, P. Nalaoh, N. Kungwan, S. Namuang, T. Sudyoadsuk and V. Promarak, Dyes Pigm., 2021, 193, 109488 CrossRef CAS.
- A. K. Tripathi, Spectrochim. Acta, Part A, 2020, 228, 117806 CrossRef CAS PubMed.
- M. Zhu, Y. Zhuo, H. Guo, F. Yang and J. Qiu, J. Lumin., 2018, 194, 264–270 CrossRef CAS.
- C. T. Arranja, A. Aguiar, T. Encarnação, S. M. Fonseca, L. L. G. Justino, R. A. E. Castro, A. Benniston, A. Harriman, H. D. Burrows and A. J. F. N. Sobral, J. Mol. Struct., 2017, 1146, 62–69 CrossRef CAS.
- P. Gayathri, S. Ravi, S. Karthikeyan, M. Pannipara, A. G. Al-Sehemi, V. Madhu, D. Moon and S. P. Anthony, J. Lumin., 2023, 264, 120173 CrossRef CAS.
- P. Gayathri, M. Pannipara, A. G. Al-Sehemi and S. P. Anthony, New J. Chem., 2020, 44, 8680–8696 RSC.
- J. Issac, S. Ravi, K. Chidambaranathan, S. Karthikeyan, M. Pannipara, A. G. Al-Sehemi, S. P. Anthony and V. Madhu, Cryst. Growth Des., 2024, 24, 3388–3398 CrossRef CAS.
- Y. Cao, C. Pan and J. Xu, Mater. Adv., 2024, 5, 3281–3288 RSC.
- S. Ravi, S. Karthikeyan, M. Pannipara, A. G. Al-Sehemi, D. Moon and S. P. Anthony, Spectrochim. Acta, Part A, 2024, 315, 124303 CrossRef CAS.
- S. Sambathkumar, S. Priyadharshini, M. Fleisch, D. W. Bahnemann, G. Gnana Kumar, S. Senthilarasu and R. Renganathan, Mater. Lett., 2019, 242, 28–31 CrossRef CAS.
- S.-N. Zou, C.-C. Peng, S.-Y. Yang, Y.-K. Qu, Y.-J. Yu, X. Chen, Z.-Q. Jiang and L.-S. Liao, Org. Lett., 2021, 23, 958–962 CrossRef CAS PubMed.
- J. Zhu, J. Zou, Z. Zhang, J. Zhang, Y. Sun, X. Dong and Q. Zhang, Mater. Chem. Front., 2019, 3, 1523–1531 RSC.
- R. Dondon, V. P. Khilya, A. D. Roshal and S. Fery-Forgues, New J. Chem., 1999, 23, 923–927 RSC.
- S.-Y. Park, M. Ebihara, Y. Kubota, K. Funabiki and M. Matsui, Dyes Pigm., 2009, 82, 258–267 CrossRef CAS.
- I. Cazin, E. Rossegger, G. Guedes de la Cruz, T. Griesser and S. Schlögl, Polymers, 2020, 13, 56 CrossRef PubMed.
- D. Cui, L. Zhang, J. Zhang, W. Li, J. Chen, Z. Guo, C. Sun, Y. Wang, W. Wang, S. Li, W. Huang, C. Zheng and R. Chen, Angew. Chem., 2024, 63, 11588 Search PubMed.
- D. Zúñiga-Núñez, F. Mura, N. Mariño-Ocampo, B. Zúñiga, J. Robinson-Duggon, R. A. Zamora, H. Poblete, A. Aspée and D. Fuentealba, Dyes Pigm., 2024, 229, 112290 Search PubMed.
- D. Cao, Z. Liu, P. Verwilst, S. Koo, P. Jangjili, J. S. Kim and W. Lin, Chem. Rev., 2019, 119, 10403–10519 Search PubMed.
- X.-Y. Liu, K. Xing, Y. Li, C.-K. Tsung and J. Li, J. Am. Chem. Soc., 2019, 141, 14807–14813 CrossRef CAS PubMed.
- Y. Chen, B. Yu, Y. Cui, S. Xu and J. Gong, Chem. Mater., 2019, 31, 1289–1295 CAS.
- M. J. Fasco, E. F. Hildebrandt and J. W. Suttie, J. Biol. Chem., 1982, 257, 11210–11212 CAS.
- M. Mathivanan, J. G. Malecki and B. Murugesapandian, Spectrochim. Acta, Part A, 2025, 326, 125270 CAS.
- A. Kundu, V. V. Kumar and S. P. Anthony, Inorg.Chem. Commun., 2021, 123, 108301 CAS.
- P. Gayathri, S. Ravi, S. Karthikeyan, A. Mohitkar, S. Jayanty, M. Pannipara, A. G. Al-Sehemi, D. Moon and S. P. Anthony, New J. Chem., 2023, 47, 7288–7298 CAS.
- A. M. El-Zohry, E. A. Orabi, M. Karlsson and B. Zietz, J. Phys. Chem. A, 2021, 125, 2885–2894 CAS.
- A. Kundu, S. Karthikeyan, Y. Sagara, D. Moon and S. P. Anthony, ACS Omega, 2019, 4, 5147–5154 CAS.
- A. L. Guzman and T. R. Hoye, J. Org. Chem., 2022, 87, 905–909 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and crystal data and structure, photophysical studies. CCDC 2415627–2415630. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ma00054h |
| This journal is © The Royal Society of Chemistry 2025 |