
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of translucent silicalite-1 bulk ceramics by spark plasma sintering†
Masanori
Takemoto
 a,
Yoshiaki
Ito
a,
Yuka
Yoshihara
a,
Shiori
Odagiri
b,
Yuta
Shuseki
c,
Kenta
Iyoki
a,
Tatsuya
Okubo
a,
Atsunobu
Masuno
bc and
Toru
Wakihara
*ad
a,
Yoshiaki
Ito
a,
Yuka
Yoshihara
a,
Shiori
Odagiri
b,
Yuta
Shuseki
c,
Kenta
Iyoki
a,
Tatsuya
Okubo
a,
Atsunobu
Masuno
bc and
Toru
Wakihara
*ad
aDepartment of Chemical System Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: wakihara@chemsys.t.u-tokyo.ac.jp
bGraduate School of Science and Technology, Hirosaki University, Hirosaki, Aomori 036-8505, Japan
cGraduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto 615-8520, Japan
dInstitute of Engineering Innovation, The University of Tokyo, 2-11-16 Yayoi, Bunkyo-ku, Tokyo 113-8656, Japan
First published on 24th March 2025
Abstract
Fabrication of binderless or binder-free zeolite ceramics is an ideal strategy to achieve outstanding performance. In this study, bulky, translucent ceramics composed of silicalite-1, a pure silica zeolite with MFI topology, is prepared by spark plasma sintering (SPS) without adding binder. The effects of SPS treatment conditions, such as silica source, temperature, pressure, holding time, ramping rate, and sample dose, are systematically investigated. A comparison of silica sources indicated that zeolite nanoparticles (NPs) have better sinterability than a silica source with large particle sizes of several micronmeters. SPS treatment using silicalite-1 NPs under optimised conditions allows the sintering of zeolite compacts while retaining their crystal structure.
Introduction
Zeolites are a family of inorganic materials with ordered microporous structures. Their frameworks are basically composed of SiO4 tetrahedra, and various three-dimensional (3D) pore systems are formed through the common apex of the oxygen bridge, endowing the materials with high specific surface areas. Furthermore, partially substituting the Si atoms with heteroatoms, such as Al atoms, imparts cation exchange ability and acidity. Owing to these features, zeolites are widely used in the fields of catalysis1–4 and separation.5,6 According to the practical application, zeolite powder must be moulded into an appropriate shape, such as granules, pellets, or films. As is generally known, applying a conventional heat process to prepare zeolite ceramics is difficult owing to poor sinterability,7 which is a barrier to the fabrication of binder-free zeolite ceramics. Inorganic and/or organic materials are generally mixed as a binder with zeolite powder for shaping. Even though the mechanical properties of the final products are enhanced by these binders, a decrease in the content of the zeolite moieties is unavoidable. These binders probably block the micropores of the zeolites, leading to a decrease in cation exchange capacity and disturbing the diffusion of molecular reactants. Therefore, binderless or binder-free fabrication methods are of interest for producing zeolite ceramics with outstanding properties.Sintering is a promising method for densifying compacts of powdery zeolites without the addition of binders. To overcome the poor sinterability of zeolites, unusual approaches have been proposed in the last two decades. For example, Nakahira et al. synthesized transparent, bulky zeolite ceramics by hydrothermal hot pressing,8–10 which involves adding tiny amounts of aqueous sodium hydroxide to the zeolite body. The hydrothermal reaction that occurs in the presence of sodium hydroxide induces the dissolution–recrystallization of zeolites, forming a highly condensed zeolite matrix. Several research studies have proposed cold sintering as an alternative technique for preparing highly crystalline bulk zeolites.11,12 Cold sintering is performed on the open system and involves milder conditions than hydrothermal hot pressing.11–14 The metastability of zeolites is well known, and hydrothermal conditions devastatingly induce the conversion of the original zeolites into amorphous materials or other phases with higher density. In particular, inorganic cations originating from the addition of a base could possibly induce the conversion of the original zeolites into zeolites with different topologies through hydrothermal reactions; this process is known as interzeolite conversion.15,16 Thus, base-free processing should prevent the occurrence of this accidental reaction. Spark plasma sintering (SPS), also called “electric current-activated sintering,” has great potential in the base-free ceramic fabrication of zeolites.17–22 Although SPS methods have already been applied to zeolite precursors in the synthesis of zeolite-derived glasses,23–34 the crystal structures of the zeolite precursors were not retained after the SPS treatment. In other words, the fabrication of zeolite ceramics using SPS techniques is yet to be achieved.
Herein, translucent ceramics composed of zeolite nanoparticles (NPs) were prepared using the SPS technique. MFI zeolite was chosen as a target zeolite to prove our concept because it is commercially available and widely used in acid catalysis3 and separation.35 The sinterabilities of various silica sources were compared, and the results indicated that the use of zeolites with small particle sizes were favourable for sintering owing to their high specific surface area. Several parameters in the SPS treatment (e.g., sample dose, temperature, ramping rate, pressure, and holding time) were systematically investigated to yield bulk ceramics composed of MFI zeolite particles. Excessively high temperatures and pressures induced the degradation of silicalite-1, leading to the formation of undesirable phases. SPS treatment under optimised conditions allowed us to homogeneously sinter a translucent zeolite ceramic while retaining the original crystal structure.
Experimental
Materials
Sodium hydroxide solution (50 wt% in water, Wako), ammonium fluoride (NH4F, Wako), colloidal silica (LUDOX® AS-40, Sigma Aldrich), tetraethylammonium hydroxide (TEAOH) solution (35 wt% in water, Sigma-Aldrich), and tetrapropylammonium hydroxide (TPAOH) solution (40 wt% in water, Sigma-Aldrich) were used as received. A commercially available zeolite with MFI topology (890HOA, Si/Al = 750) was purchased from Tosoh company. Silicalite-1, a pure silica zeolite with MFI topology, was prepared in varying particle sizes according to a method reported in the literature.36Synthesis of silicalite-1 NPs
Silicalite-1 was synthesized according to the literature, with minor modifications.36 TPAOH solution (3.7 g), sodium hydroxide solution (1.7 g), and distilled water (2.3 g) were mixed and stirred for 5 min. Colloidal silica (16.5 g) was added to the basic solution, and the mixture was stirred for 30 min. The mixture was heated at 70 °C for 120 h or at 90 °C for 30 h in a preheated oven. After heating, the products were obtained by centrifugation, washed with distilled water, and dried in an oven at 80 °C. The dried samples were calcined at 550 °C for 6 h. Hereafter, silicalite-1 NPs prepared at 70 and 90 °C are denoted as silicalite-1_70 and silicalite-1_90, respectively. Defect-healing of silicalite-1 was also performed as described in a previous study, with minor modifications.37 Healed samples were labelled “healed” after the sample name. Further details on the defect-healing procedure are given in the ESI.†Spark plasma sintering
A graphite die (10 mm in diameter) and tungsten carbide die (10 mm in diameter) were purchased from Fuji Electronic Industrial Co., Ltd. The SPS experiments were conducted using SPS apparatus (Fig. S1, Dr Sinter Lab. Jr, Fujidempa Kogyo Co., Ltd, ESI†) The sample powder was loaded in the die/punch set, which was placed in the chamber under vacuum (<6 Pa). Uniaxial pressure was applied to the sample powder, followed by pulsed electrical current pass under PID control. The sintered samples were weighed on an analytical balance, and then the height and diameter of the sintered samples were measured with a digital caliper. Bulk density of the samples was calculated by dividing the mass by volume of the samples.Characterisation
Powder X-ray diffraction (PXRD) patterns of the samples were collected on a Rigaku SmartLab diffractometer equipped with a Cu Kα radiation source (λ = 0.15406 nm). Diffraction patterns were recorded in the 2θ range of 3–50° with a scanning speed of 10° min−1. The relative crystallinities of the samples were calculated from the integrated peak intensities of the diffraction peaks in the 2θ range of 20–30°. The parent zeolites were used as references for 100% crystallinity. Nitrogen adsorption–desorption isotherms at −196 °C were obtained using volumetric gas-adsorption apparatus (Autosorb-iQ2-MP, Anton Paar). Before the measurements, sample powders were pretreated at 300 °C under vacuum for 4 h. The morphologies of the samples were investigated using a field-emission scanning electron microscope (FE-SEM; JSM-IT800, JEOL).Results and discussion
Characterisation of translucent silicalite-1 ceramic
Fig. 1 shows the characteristics of a bulk silicalite-1 ceramic prepared under optimised SPS conditions, where 100 mg of silicalite-1_70 was pressed at 400 MPa. The temperature was increased to 500 °C at a ramping rate of 32 °C min−1, and the temperature was maintained for 5 min. The product with a thickness of 0.8 mm was translucent, which was further improved by the elimination of surface scattering by adding a drop of water (Fig. 1(a) and Fig. S2, ESI†). As a result of consolidation, the bulk density of the product was 1.7 g cm−1. The cross-sectional SEM image shows that the spherical particles are similar to the original silicalite-1 particles (as described below), and necking between silicalite-1 NPs also occurs (Fig. 1(b)). The PXRD pattern of the translucent ceramic confirmed that the crystal structure originating from an MFI topology was retained after SPS treatment (Fig. 1(c)). The relative crystallinity of the product decreased to 63%, indicating that precursory silicalite-1_70 was partially converted into amorphous materials. Fig. 1(d) shows the N2 adsorption–desorption isotherm of the sample. The isotherm exhibited a type-IV shape with a combination of H1 and H4 hysteresis loops. The steep increase at low relative pressure (P/P0 < 0.1) is due to the presence of micropores (Vmicro = 0.083 cm g−1), which support the retention of the 3D microporous structures originating from the MFI topology after SPS treatment. The H1 and H4 hysteresis loops (0.2 < P/P0 < 0.9, P/P0 > 0.9) are indicative of mesoporosity originating from interparticle voids, which is in good agreement with the SEM images. It can be concluded that optimised SPS treatment yields a translucent bulk ceramic composed of silicalite-1 NPs with a high micropore volume. Several previous studies have also reported SPS treatments for zeolite precursors.23–34 However, as far as we know, this is the first success to prepare translucent zeolite ceramics using the SPS technique while retaining the original crystalline structure. During hydrothermal hot pressing and cold sintering, the addition of NaOH promotes the consolidation of zeolite particles.9–11 However, sintering in the presence of additional cations would probably cause the undesirable conversion of the parent zeolites into zeolites with other topologies,9 inhibiting the application of sintering process to zeolites.38 Although binderless zeolite coatings have been prepared by combining wash coating with dry-gel conversion, this method requires the addition of amines.39 It should be noted that the SPS technique has the advantage of allowing zeolite sintering in the presence of bases and/or binders.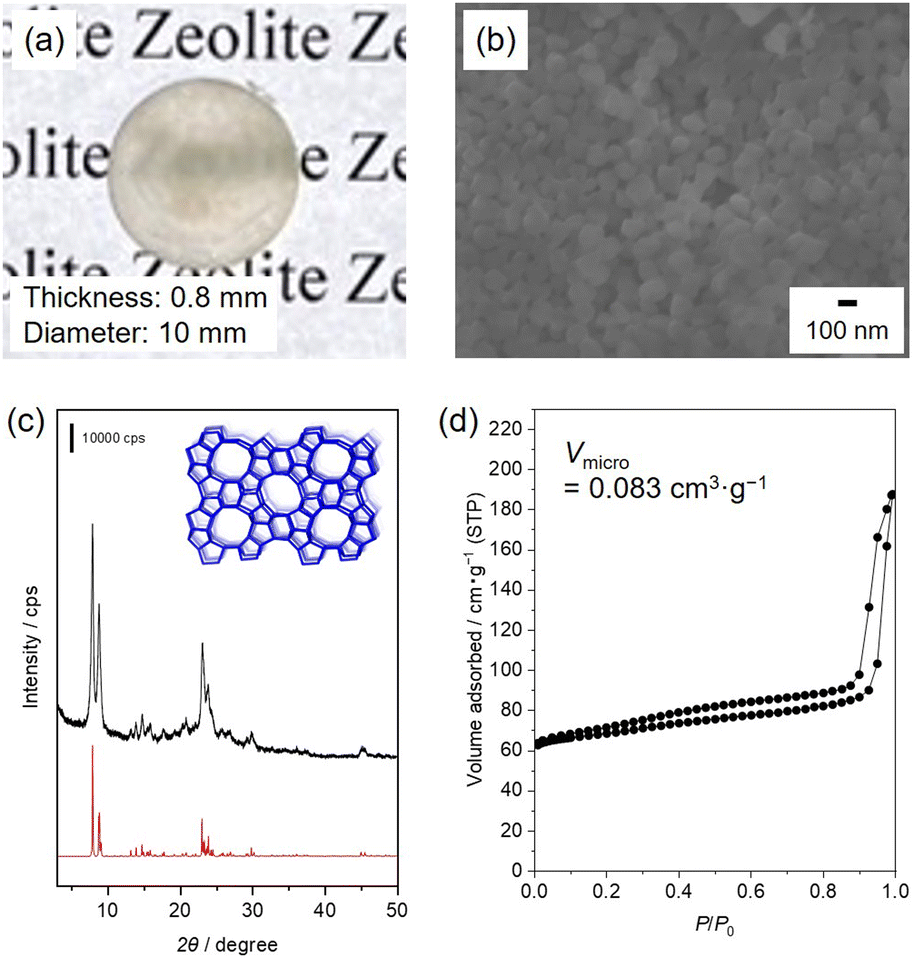 | ||
| Fig. 1 Characterisation of a translucent silicalite-1 ceramic prepared under optimised SPS conditions. (a) Photograph, (b) cross-sectional SEM image, (c) PXRD pattern, and (d) N2 adsorption–desorption isotherm. Silicalite-1_70 (100 mg) was sintered at 400 MPa and 500 °C for 5 min using a tungsten carbide die. The ramping rate was 32 °C min−1. | ||
Optimisation of preparation conditions
Several parameters were systematically varied (e.g., silica source, temperature, pressure, ramping rate, and sample dose) to investigate the sintering behaviour under different SPS conditions. Fig. 2(a) shows the relative crystallinities and bulk densities of the sintered 890HOA samples at 50 MPa for 5 min with varying temperatures. The PXRD patterns of the sintered samples are also shown in Fig. S3 (ESI†). The crystal structure of the MFI zeolite was retained after SPS treatment at 600 °C, and the relative crystallinity decreased with an increase in treatment temperature, but the bulk density of 890HOA did not significantly increase. 890HOA exhibits a prism-like morphology and smooth surface (Fig. 2(b)). In the SEM images of the samples subjected to SPS, spherical particles were observed, in addition to coarse crystals with a similar morphology to the original 890HOA (Fig. 2(c)–(g)). Although part of the original 890HOA was converted into amorphous particles by the SPS treatments, the particle sizes and morphologies did not drastically change after SPS treatment. These results indicate the poor sinterability of 890HOA, which is probably due to its large particle size and non-equiaxed particle morphology (2 × 5 μm by catalogue value).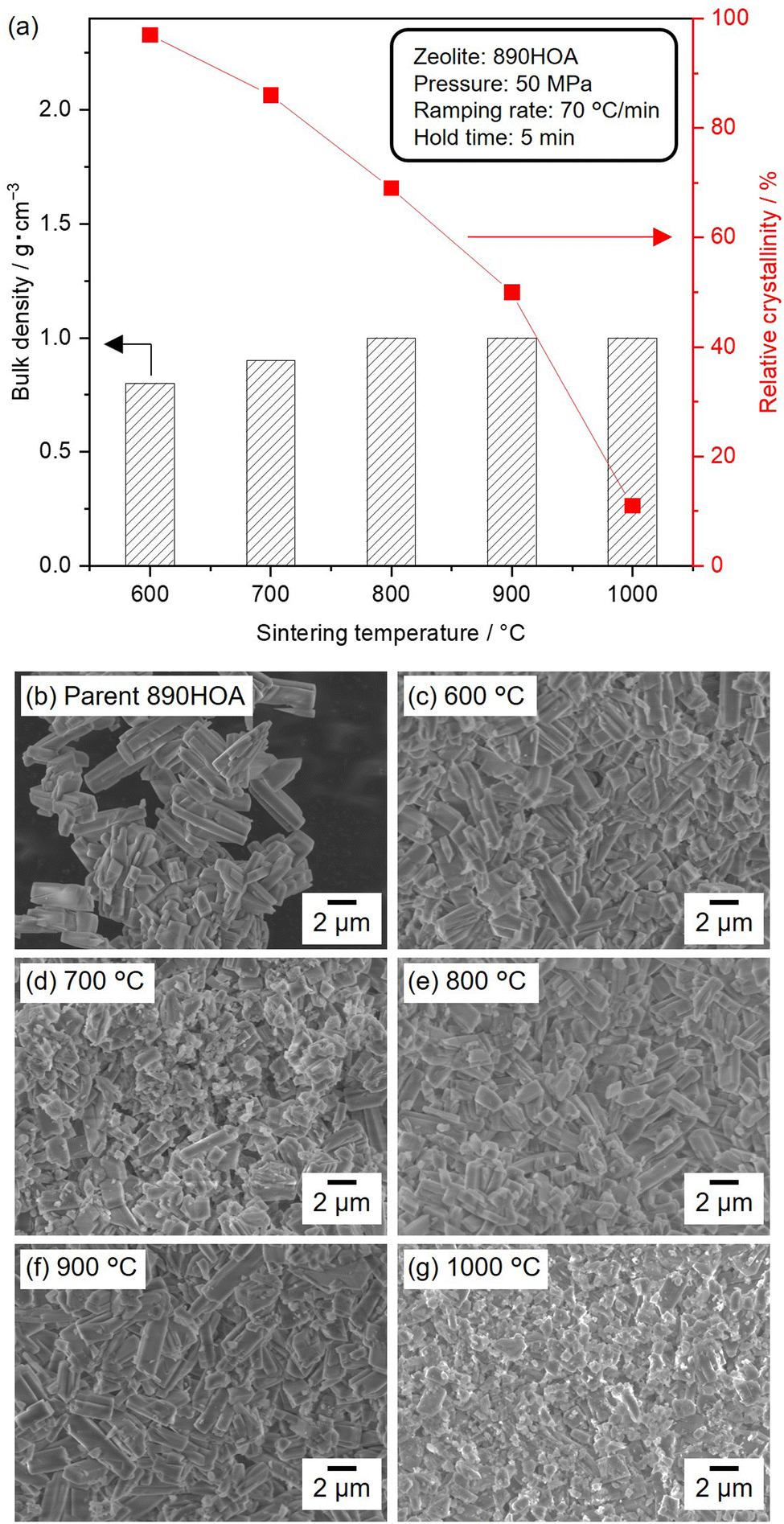 | ||
| Fig. 2 (a) Bulk densities and relative crystallinities of sintered 890HOA samples at varying temperatures. All samples (330 mg) were sintered at 50 MPa for 5 min using the graphite die. The ramping rate was 70 °C min−1. (b)–(g) SEM images of parent 890HOA and cross-sections of sintered samples. | ||
The use of zeolite with smaller particle sizes was considered to improve sinterabilities. Fig. 3(a)–(d) show SEM images of a series of silicalite-1 samples used as silica sources in this study. The SEM image of silicalite-1_70 reveals a uniform particle size distribution of spherical particles with ≈100 nm diameters (Fig. 3(a)). The same features were observed for silicalite-1_90, except for the average particle diameter of ≈250 nm (Fig. 3(b)). The diffraction peaks in the PXRD patterns of 890HOA, silicalite-1_70, and silicalite-1_90 are consistent with an MFI topology (Fig. S4, ESI†). No changes in the particle size and morphology were observed after the defect-healing treatment for silicalite-1_70 and silicalite-1_90 (Fig. 3(c) and (d)).
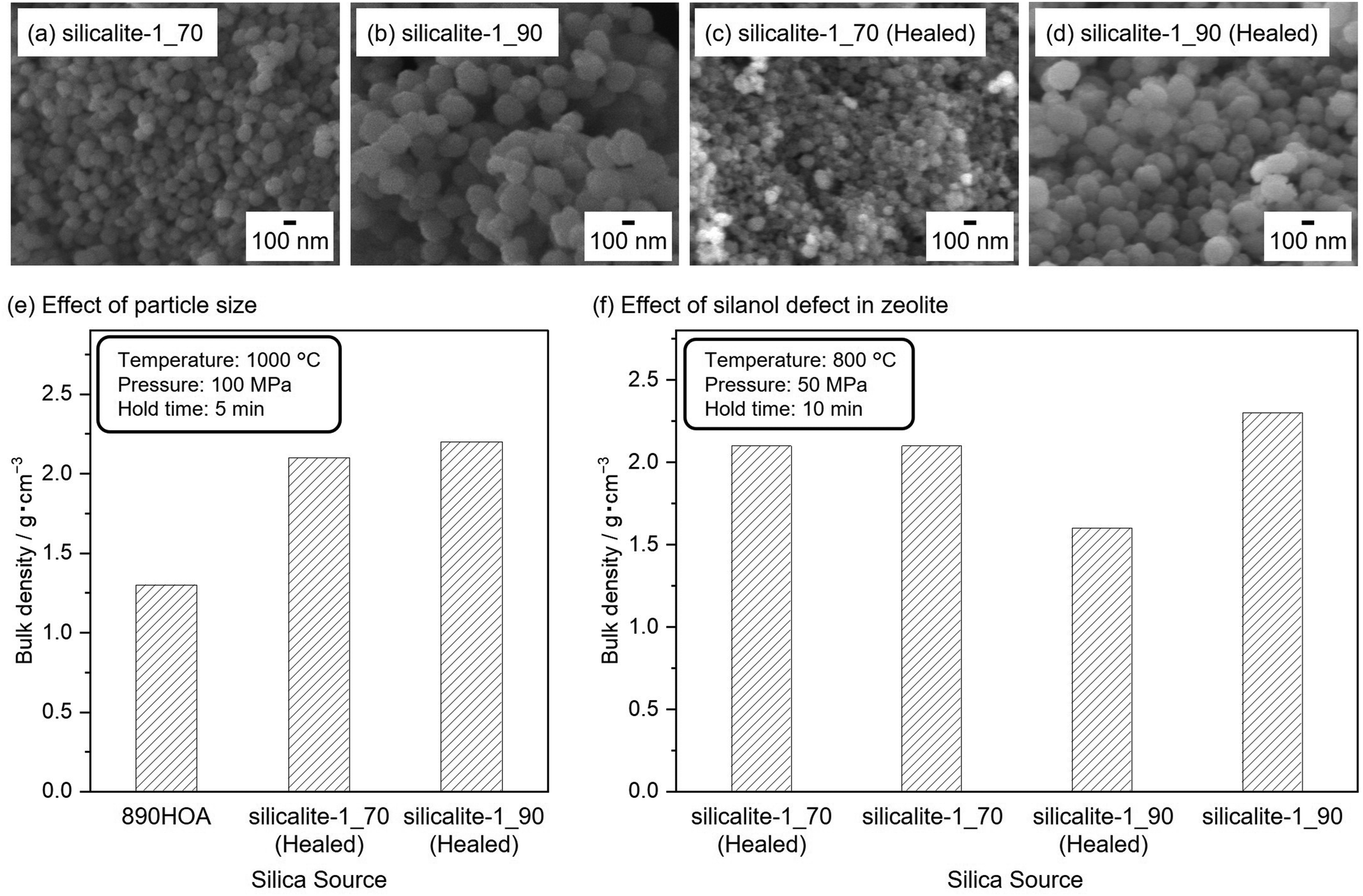 | ||
| Fig. 3 SEM images of (a) silicalite-1_70, (b) silicalite-1_90, (c) silicalite-1_70 (healed), and (d) silicalite-1_90 (Healed). Effects of (e) particle size and (f) defects in silicalite-1 on the sinterability of various silica sources. All samples (330 mg) were sintered using the graphite die. The ramping rate was 70 °C min−1, and the other SPS conditions are given in the inset. | ||
The influence of particle size and morphology on the sinterabilities is compared in Fig. 3(e), where the temperature of the samples increased at a ramping rate of 70 °C min−1. All samples were sintered at 1000 °C for 5 min under 100 MPa. In contrast to the 890HOA samples, the bulk densities of the healed silicalite-1 NP samples increased to >2.0 g cm−1. While 890HOA has large particle sizes of several micrometers and non-equiaxed particle morphology (2 × 5 μm by catalogue value), silicalite-1 samples have smaller particle sizes and spherical morphologies (Fig. 2(b) and Fig. 3(a)–(d)). This indicates that the particle sizes and morphologies of the healed silicalite-1 NP samples would contribute to their superior sinterabilities compared to 890HOA.
The effect of the amount of silanol defect in the zeolites is compared in Fig. 3(f). It can be seen that the bulk density of silicalite-1_70 and silicalite-1_70 (healed) is the same, while silicalite-1_90 (healed) had a lower bulk density than silicalite-1_90. Recently, our group demonstrated that the defect-healing treatment enhanced the mechanical stability of zeolites by reducing the amount of silanol defects.40 The superior stability of healed silicalite-1 can be assumed to account for its poorer sinterability than that of non-healed silicalite-1. Non-healed silicalite-1 NPs should be suitable as a precursor based on their sinterability; however, the PXRD patterns of the sintered samples (not shown) contain almost no diffraction peaks of the MFI zeolite. This indicates amorphization of the MFI zeolite during SPS treatment under such severe conditions. Therefore, the SPS conditions had to be optimised in order to obtain bulk ceramics composed of crystalline moieties.
To avoid complete degradation of the crystalline structure of the MFI zeolite, silicalite-1_70 was subjected to SPS treatments under milder conditions. Fig. 4(a) shows the bulk densities and relative crystallinities of silicalite-1_70 sintered at 400 MPa and varying temperatures. The ramping rate and holding time were set to 32 °C min−1 and 0 min, respectively. With increasing temperature, the bulk densities of the resultant samples gradually increased, and the relative crystallinities decreased while retaining the crystal structure of the MFI zeolite. The effect of pressure on the bulk density and relative crystallinity was investigated using Fig. 4(b). The target temperature was set to 200 °C, with a ramping rate of 32 °C min−1. The SPS treatments at 400 MPa did not change the bulk density and relative crystallinity. SPS treatment at 500 MPa for 5 min increased the bulk density and decreased the relative crystallinity. These results indicate that higher pressure and temperature induced the amorphization of silicalite-1 and densification.
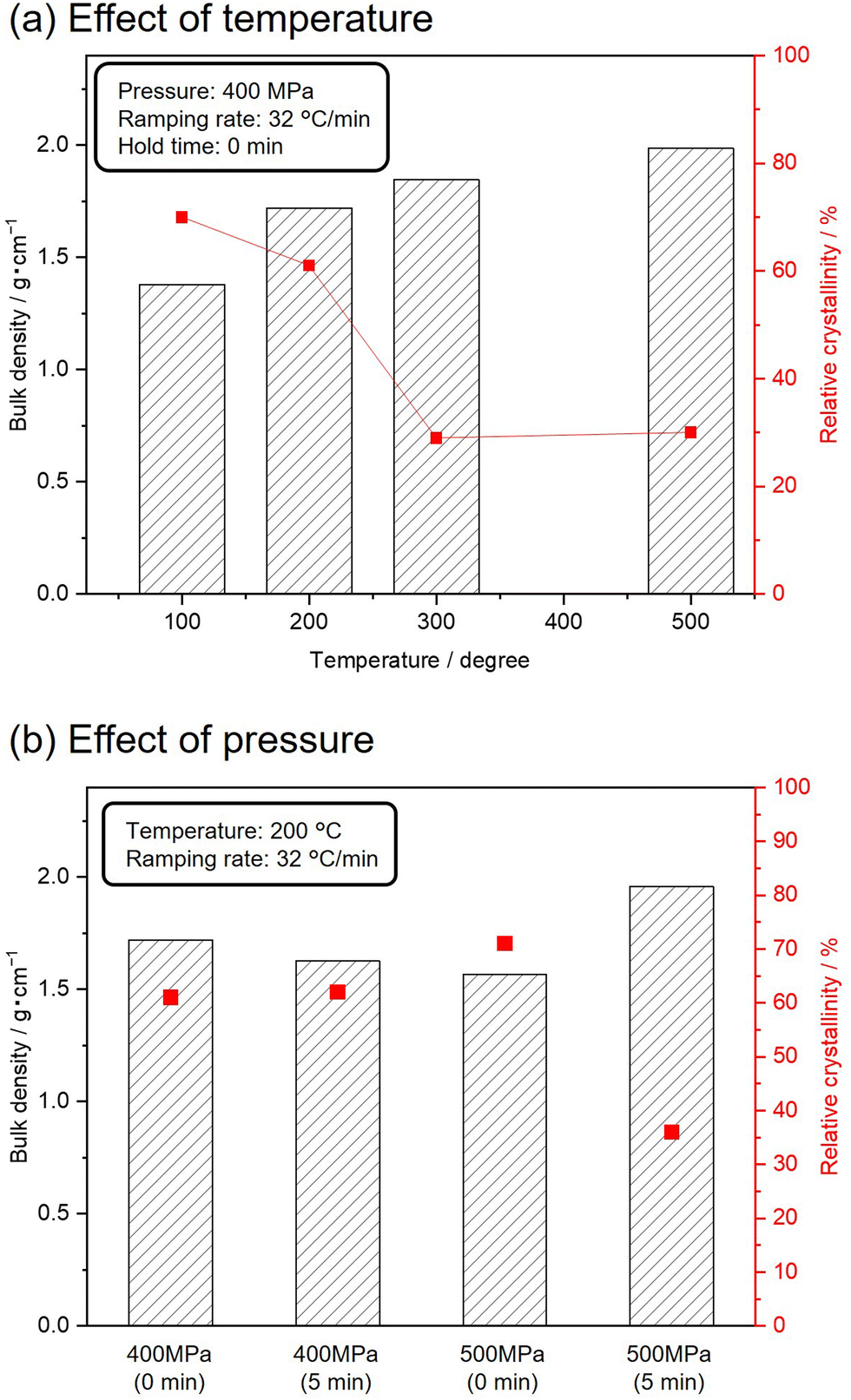 | ||
| Fig. 4 Effect of (a) temperature and (b) pressure for bulk densities and relative crystallinities of sintered silicalite-1_70 samples. All samples (250 mg) were sintered by using the tungsten carbide die. Fixed conditions were described in the inset of each figure. | ||
Fig. 5(a) shows a photograph of bulk ceramic silicalite-1 obtained by SPS treatment at 500 °C with a ramping rate of 32 °C min−1 under 400 MPa for 0 min. The obtained ceramic was non-uniform in appearance. The edge side of the ceramic was translucent, whilst the interior appeared opaque. Longer SPS treatments (5 and 20 min) did not affect the sample appearance (not shown). PXRD patterns of the different sides of the resultant ceramics prepared by SPS of varying durations are shown in Fig. 5(b) and (c). Diffraction peaks originating from the MFI zeolite and broad, halo patterns were observed for the edge side at 0 and 5 min. Longer SPS treatment (20 min) promoted the degradation of the MFI zeolite and the formation of quartz. The crystalline structure of the MFI zeolite was not retained at the centre of the samples during the initial stage of SPS treatment (0 and 5 min), and diffraction peaks with high intensities arose after 20 min of SPS treatment. This implies that the silicalite-1 degrades more quickly at the centre of the samples than at the edge. The SEM images of the edge indicate that spherical silicalite-1 particles were sintered after 5 min of SPS treatment, and a certain amount of quartz crystals in a fibre-like form were formed after 20 min (Fig. 5(d)–(f)). A similar densification phenomenon owing to the sintering of the spherical silicalite-1 particles was also observed at the centre, and angular crystals were formed after SPS treatment for 20 min (Fig. 5(g)–(i)). In a previous study, heat distribution during the SPS process was simulated in non-conducting samples, such as zeolites.41 In particular, the authors claimed that the temperature at the centre of the zeolites became higher than that at the edge. As evidenced in Fig. 4(a), SPS treatment at high temperatures accelerated silicalite-1 degradation. Thus, the inhomogeneous temperature distribution was responsible for the different degradation phenomena of silicalite-1, resulting in the formation of bulk ceramics with non-uniform appearance on the macroscopic scale.
 | ||
| Fig. 5 (a) Photograph of bulk ceramic silicalite-1_70 obtained by SPS treatment at 500 °C with a ramping rate of 32 °C min−1 under 400 MPa for 0 min. (b) and (c) PXRD patterns and (d)–(i) cross-sectional SEM images of the edge and centre of sintered silicalite-1_70 °C ceramics at varying times. | ||
The rapid temperature increase is generally accepted as a major advantage of the SPS technique. However, several studies demonstrated that SPS treatments with low ramping rates yield ceramics with improved transparency.42–44 The effect of the ramping rate is illustrated by comparing runs 1–5 in Table 1. At a high ramping rate of 100 °C min−1, the resultant ceramic was opaque at the centre and translucent at the edge (run 1). SPS treatments at lower heating rates improved the uniformity of the resultant ceramics (runs 2–5). To improve the temperature distribution, the sample dose was also varied (runs 4, and 6–8). SPS treatments of a small amount of silicalite-1_70 resulted in the formation of highly uniform, translucent ceramics (runs 4 and 6), whilst SPS treatments for larger amounts failed to yield ceramics that were homogeneous on the macroscopic scale (runs 7 and 8). As discussed above, optimisation of the ramping rate and sample dose helped to regulate the temperature distribution during SPS treatment, leading to the formation of homogeneous ceramics on the millimetre scale.
| No. | Ramping rate/°C min−1 | Sample dose/mg | Uniformity | Apparent (C; centre, E; edge) | Crystalline phase |
|---|---|---|---|---|---|
| 1 | 100 | 100 | Non-uniform | C: opaque E: translucent | C: amorphous E: MFI |
| 2 | 70 | 100 | Uniform | Opaque | MFI |
| 3 | 50 | 100 | Uniform | Opaque | MFI |
| 4 | 32 | 100 | Uniform | Translucent | MFI |
| 5 | 10 | 100 | Uniform | Translucent | MFI |
| 6 | 32 | 70 | Uniform | Translucent | MFI |
| 7 | 32 | 150 | Non-uniform | C: opaque | C: amorphous |
| E: translucent | E: MFI | ||||
| 8 | 32 | 250 | Non-uniform | C: opaque | C: amorphous |
| E: translucent | E: MFI |
Conclusions
Highly translucent bulk silicalite-1 ceramic with a high micropore volume was prepared using the SPS technique. The use of fine zeolite particles was favourable for sintering owing to their high specific external surface areas. SPS treatments under relatively mild conditions allowed the sintering of silicalite-1 particles while retaining their original crystal structures. To obtain ceramics that appeared homogenous on the macroscopic scale, the temperature distribution was improved by reducing the ramping rate and sample dose. To the best of our knowledge, this is the first demonstration of the fabrication of zeolite ceramics using the SPS technique, which will encourage further research on various binderless/binder-free zeolites and open up new opportunities for zeolites ceramics towards applications utilizing light transparency.Data availability
The data that support the findings of this study are openly available.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by New Energy and Industrial Technology Development Organization (NEDO) under the Moonshot Project, the Japan Society for the Promotion of Science (JSPS), KAKENHI Grant-in-Aid for Transformative Research Areas (A) JP20A206/20H05880, Grant-in-Aid for Scientific Research (S) JP23H05454, and the Materials Processing Science project (“Materealize”) of MEXT, Grant Number JPMXP0219192801.References
- S. N. Khadzhiev, M. V. Magomedova and E. G. Peresypkina, Pet. Chem., 2014, 54, 245–269 CrossRef CAS.
- M. Jablonska, RSC Adv., 2022, 12, 25240–25261 RSC.
- P. Tian, W. Yingxu, Y. Mao and L. Zhongmin, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS.
- P. Li, Y. Xin, H. Zhang, F. Yang, A. Tang, D. Han, J. Jia, J. Wang, Z. Li and Z. Zhang, Front. Chem., 2022, 10, 1033255 CrossRef CAS PubMed.
- S. Kumar, R. Srivastava and J. Koh, J. CO2 Util., 2020, 41, 101251 CrossRef CAS.
- M. M. Zagho, M. K. Hassan, M. Khraisheh, M. A. A. Al-Maadeed and S. Nazarenko, Chem. Eng. J. Adv., 2021, 6, 100091 CrossRef CAS.
- M. Biesuz, L. Spiridigliozzi, A. Marocco, G. Dell'Agli, V. M. Sglavo and M. Pansini, J. Am. Ceram. Soc., 2017, 100, 5433–5443 CrossRef CAS.
- T. Fujii, M. Yoshida, R. Yoshino and M. Matsuda, J. Eur. Ceram. Soc., 2022, 42, 3510–3514 CrossRef CAS.
- A. Nakahira, S. Takezoe, Y. Yamasaki, Y. Sasaki and Y. Ikuhara, J. Am. Ceram. Soc., 2007, 90, 2322–2326 CrossRef CAS.
- A. Nakahira, S. Takezoe and Y. Yamasaki, Chem. Lett., 2004, 33, 1400–1401 CrossRef CAS.
- S. Lee, Y. I. Kim, M. Akmal and H. J. Ryu, ACS Appl. Mater. Interfaces, 2023, 15, 36489–36499 CrossRef CAS PubMed.
- F. W. Zhou, J. Z. Shi, T. L. Sun, X. L. Zhu and X. M. Chen, J. Porous Mater., 2023, 30, 1843–1850 CrossRef CAS.
- J. Gao, K. Wang, W. Luo, X. Cheng, Y. Fan and W. Jiang, J. Adv. Ceram., 2022, 11, 1714–1724 CrossRef CAS.
- S. H. Bang, K. Tsuji, A. Ndayishimiye, S. Dursun, J. H. Seo, S. Otieno and C. A. Randall, J. Am. Ceram. Soc., 2020, 103, 2322–2327 CrossRef CAS.
- T. Sano, M. Itakura and M. Sadakane, J. Jpn Pet. Inst., 2013, 56, 183–197 CrossRef CAS.
- D. V. Bruter, V. S. Pavlov and I. I. Ivanova, Pet. Chem., 2021, 61, 251–275 CrossRef CAS.
- P. Vasiliev, F. Akhtar, J. Grins, J. Mouzon, C. Andersson, J. Hedlund and L. Bergstrom, ACS Appl. Mater. Interfaces, 2010, 2, 732–737 CrossRef CAS PubMed.
- D. Zou and Y. Fan, Ceram. Int., 2021, 47, 14966–14987 CrossRef CAS.
- X. Zhang, X. Yu, B. Zhou, W. Luo, W. Jiang, W. Jiang, Z. Shen, L. Wang and Y. Yuxiang, J. Am. Ceram. Soc., 2015, 98, 1056–1059 CrossRef CAS.
- E. K. Papynov, O. O. Shichalin, A. A. Belov, V. S. Pechnikov, A. V. Ognev, A. L. Shkuratov, I. Y. Buravlev, M. I. Dvornik, P. G. Chigrin, N. M. Vlasova, A. N. Fedorets, S. A. Azon, O. V. Kapustina, A. O. Lembikov, V. A. Nepomnyushchaya, Z. E. Kornakova, E. A. Gridasova, I. G. Tananaev, Y. Shi and A. I. Ivanets, Mater. Chem. Phys., 2023, 302, 127648 CrossRef CAS.
- O. O. Shichalin, E. K. Papynov, V. Y. Maiorov, A. A. Belov, E. B. Modin, I. Y. Buravlev, Y. A. Azarova, A. V. Golub, E. A. Gridasova, A. E. Sukhorada, I. G. Tananaev and V. A. Avramenko, Radiochemistry, 2019, 61, 185–191 CrossRef CAS.
- E. K. Papynov, O. O. Shichalin, V. Y. Mayorov, V. G. Kuryavyi, T. A. Kaidalova, L. V. Teplukhina, A. S. Portnyagin, A. B. Slobodyuk, A. A. Belov, I. G. Tananaev, V. A. Avramenko and V. I. Sergienko, J. Hazard. Mater., 2019, 369, 25–30 CrossRef CAS PubMed.
- B. J. Riley, S. Chong, M. Zhao and J. Lian, Ind. Eng. Chem. Res., 2023, 62, 8779–8792 CrossRef CAS.
- Y. Zhao, S. Sun, X. Cai, Y. Fan, W. Jiang, B. Zhou, S. Gu, N. Shi, W. Luo and L. Wang, J. Am. Ceram. Soc., 2020, 103, 5654–5663 CrossRef CAS.
- C. Gao, H. Lin, D. Zhang, R. Hong, C. Tao and Z. Han, Ceram. Int., 2018, 44, 19547–19553 CrossRef CAS.
- O. O. Shichalin, E. K. Papynov, V. A. Nepomnyushchaya, A. I. Ivanets, A. A. Belov, A. N. Dran’kov, S. B. Yarusova, I. Y. Buravlev, A. E. Tarabanova, A. N. Fedorets, S. A. Azon, Z. E. Kornakova, S. Y. Budnitskiy, I. G. Tananaev, Y. Shi, Y. Xiong and H. Wang, J. Eur. Ceram. Soc., 2022, 42, 3004–3014 CrossRef CAS.
- L. Wang, L. Wang, W. Jiang and H. Lin, J. Solid State Chem., 2014, 212, 128–133 CrossRef CAS.
- Z. Xu, H. Lin, R. Hong, D. Zhang and S. Zhou, J. Lumin., 2022, 244, 118664 CrossRef CAS.
- S. Gu, B. Zhou, W. Luo, L. Wang, W. Jiang, W. Jiang and J. Ballato, J. Am. Ceram. Soc., 2015, 99, 121–127 CrossRef.
- A. E. Panasenko, O. O. Shichalin, S. B. Yarusova, A. I. Ivanets, A. A. Belov, A. N. Dran'kov, S. A. Azon, A. N. Fedorets, I. Y. Buravlev, V. Y. Mayorov, D. K. Shlyk, A. A. Buravleva, E. B. Merkulov, N. V. Zarubina and E. K. Papynov, Nucl. Eng. Technol., 2022, 54, 3250–3259 CrossRef CAS.
- E. K. Papynov, O. O. Shichalin, I. Y. Buravlev, A. A. Belov, A. N. Fedorets, A. I. Ivanets and I. G. Tananaev, Ceram. Int., 2024, 50, 2759–2771 Search PubMed.
- M. Koide, M. Kato, T. Sato and S. Kudo, Electrochemistry, 2015, 83, 459–461 CrossRef CAS.
- L. C. Harnett, L. J. Gardner, S.-K. Sun, C. Mann and N. C. Hyatt, J. Nucl. Sci. Technol., 2019, 56, 891–901 CrossRef CAS.
- R. Geng, B. Zhou, J. Wang, Q. Yuan, Z. Pan, Y. Zhao, L. Wang and W. Jiang, J. Am. Ceram. Soc., 2022, 105, 4709–4718 CrossRef CAS.
- Y. Li, G. Zhu, Y. Wang, Y. Chai and C. Liu, Microporous Mesoporous Mater., 2021, 312, 110790 Search PubMed.
- M. Deguchi, K. Iyoki, C. Anand, Y. Yanaba, T. Yoshikawa, T. Okubo and T. Wakihara, Microporous Mesoporous Mater., 2018, 270, 200–203 CrossRef CAS.
- K. Iyoki, K. Kikumasa, T. Onishi, Y. Yonezawa, A. Chokkalingam, Y. Yanaba, T. Matsumoto, R. Osuga, S. P. Elangovan, J. N. Kondo, A. Endo, T. Okubo and T. Wakihara, J. Am. Chem. Soc., 2020, 142, 3931–3938 CrossRef CAS PubMed.
- L. Van Tendeloo, E. Gobechiya, E. Breynaert, J. A. Martens and C. E. Kirschhock, Chem. Commun., 2013, 49, 11737–11739 RSC.
- Z. You, G. Liu, L. Wang and X. Zhang, Microporous Mesoporous Mater., 2013, 170, 235–242 CrossRef CAS.
- M. Takemoto, Y. Yoshihara, Y. Ito, H. Yamada, K. Iyoki, T. Okubo and T. Wakihara, Microporous Mesoporous Mater., 2024, 372, 113087 CrossRef CAS.
- S. Gu, X. Zhang, L. Wang, X. Gan, Z. Shen and W. Jiang, J. Eur. Ceram. Soc., 2015, 35, 1599–1603 CrossRef CAS.
- K. Morita, B. N. Kim, K. Hiraga and H. Yoshida, Scr. Mater., 2008, 58, 1114–1117 Search PubMed.
- I. B. Oparina, A. G. Kolmakov, M. A. Sevost’yanov and A. S. Lysenkov, Inorganic Mater.: Appl. Res., 2019, 10, 825–835 Search PubMed.
- C. Wang and Z. Zhao, Scr. Mater., 2009, 61, 193–196 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ma00075k |
| This journal is © The Royal Society of Chemistry 2025 |