
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unraveling the structural complexity of niobate units in aluminosilicate glasses and glass–ceramics†
Maria Rita Cicconi *a,
Koji Kimurabcd,
Henrik Bradtmüllere,
Hongyi Denga,
Shinji Koharacd,
Yohei Onoderac,
Tomokatsu Hayakawaf,
Seiya Shimonod,
Koichi Hayashibd and
Dominique de Lignya
*a,
Koji Kimurabcd,
Henrik Bradtmüllere,
Hongyi Denga,
Shinji Koharacd,
Yohei Onoderac,
Tomokatsu Hayakawaf,
Seiya Shimonod,
Koichi Hayashibd and
Dominique de Lignya
aDepartment Werkstoffwissenschaften, Institut für Glas und Keramik, Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Martenstrasse 5, 91058 Erlangen, Germany. E-mail: maria.rita.cicconi@fau.de; Tel: +49 9131 85-27555
bDepartment of Physical Science and Engineering, Nagoya Institute of Technology, Gokiso-cho, Showa-ku, Nagoya 466-8555, Japan
cCenter for Basic Research on Materials, National Institute for Materials Science, 1-2-1 Sengen, Tsukuba, Ibaraki 305-0047, Japan
dJapan Synchrotron Radiation Research Institute, 1-1-1, Kouto, Sayo-cho, Sayo-gun, Hyogo 679-5198, Japan
eDepartment of Materials Engineering, Vitreous Materials Laboratory, Federal University of São Carlos, CP 676, 13565-905, São Carlos, SP, Brazil
fDepartment of Life Science and Applied Chemistry, Nagoya Institute of Technology, Gokiso-cho, Showa-ku, Nagoya 466-8555, Japan
First published on 28th April 2025
Abstract
Niobium-containing glasses and glass–ceramics play an important role in several technological applications, but our understanding of the structure–property relationships of many Nb-containing compositions is still rudimentary. To address the current limitations, the present contribution reports data from synchrotron high-energy X-ray diffraction data to unravel the structural evolution of niobate entities in alkali–aluminosilicate glasses. The data obtained are compared with complementary Raman and solid-state NMR spectroscopy data to provide a better interpretation of the macroscopic properties of the glasses and their crystallization behavior in terms of the glass structure. The data show that the incorporation of niobium into the glass network (from 0.2 mol% to 10 mol%) causes a rearrangement of the units and induces large modifications, particularly in the medium-range order. Nb5+ is present in all glasses predominantly in the form of 6-coordinated [NbO6] units with rather invariant 〈Nb–O〉 bond distances around 2.0 Å. This observation correlates well with the 93Nb NMR data showing similarly small changes in the chemical shift values. A contrasting scenario is presented when looking beyond the first coordination sphere, with a particular focus on the A–Nb (A = alkali) and Nb–Nb correlations. Both are strongly dependent on bulk chemistry, which, in turn, is influenced by the availability and nature of charge-compensating alkali ions. The addition of Nb has a relatively minimal effect on Si and Al units, promoting the association of Nb with other Nb species, thereby initiating the formation of a subnetwork of [NbO6] units in a corner shared environment. Both alkali species and Nb5+ ions in the amorphous state tend to favor a structural arrangement very similar to that of the stable crystalline phase.
1. Introduction
Niobium-containing glasses have applications in many fields and have been studied for their enhanced optical and dielectric properties, radiation shielding capabilities, and energy storage applications.1–5 Nb ions can be found in various coordination environments in glasses and crystalline materials. In glasses, Nb2O5 is considered an intermediate oxide: it can play a network modifying role, inducing a greater number of non-bridging oxygens, or it can help to crosslink the structural units, improving the overall glass connectivity, e.g., ref. 6–8. Based on previous studies, it seems that this dual role depends on the bulk composition but also on the total Nb2O5 content.8–10 Indeed, there is a fair consensus in the literature that, regardless of the network former cation selected, Nb2O5 tends to form 3D corner-shared [NbO6] clusters1,8–11 at high Nb-contents, independently of the present network former. These clusters provide specific glass properties, such as the enhancement of the electro-optical Kerr coefficient.12,13 However, it has been observed that the transition between the different Nb environments/structural roles does not purely depend on a specific niobium content but also strongly on the chemistry of the glass.9Recently, in order to develop niobate perovskite-like crystals in stable glass matrices, the Nb2O5 solubility and the properties of three aluminosilicate glass series have been investigated, starting from very low doping contents (0.2 mol% Nb2O5) up to 15 mol%.9,14 It was observed that the Nb2O5 incorporation limit varied quite a lot depending on the degree of glass polymerization (alkali/Al ratio), being three times lower in more polymerized glasses. Most importantly, it was observed that the properties of the developed glasses showed different trends and even opposite behavior in a few cases.9 Raman spectroscopy was used to identify some fingerprint vibrations that were correlated to the different connectivity between [NbO6] entities and to the availability of charge compensator ions as the driving forces in determining the solubility and properties evolution.9 The differences in the glass topology also give rise to quite dissimilar behavior in terms of optical properties and, in particular, regarding the use of these glasses as self-activated lanthanides for photoluminescence applications.14 Another distinctive feature of Nb-containing glasses concerns the crystallization kinetics and crystallization mechanisms, which are strongly influenced by the bulk chemistry. Indeed, the glass–ceramics obtained from peralkaline compositions (Na2O/Al2O3 molar ratio = 2.3) show preferential surface crystallization with alkali–niobate crystals propagating from the edge towards the center. A polymerized glass (Na2O/Al2O3 molar ratio = 1), on the other hand, shows a volume crystallization of sub-micron crystals, resulting in a glass–ceramic with a high degree of transparency even after prolonged thermal treatments.9,14 Similar crystallization behavior has been observed in a glass in the SiO2–Li2O–Nb2O5 ternary system with a Li2O/Nb2O5 molar ratio of 1.4.15
While Raman9,13 and NMR15–17 spectroscopy have provided some insight into the compositional dependence of the Nb structural units, more specific information on the local environment and connectivity of the Nb units, which is critical for the design of Nb-based multifunctional materials remains elusive. A major challenge, especially when considering cation arrangements beyond the first coordination sphere, is to unravel the “order” in disordered materials. In this work, we have addressed this issue through a combination of synchrotron X-ray scattering, Raman, and NMR data. The collected data documents the structural evolution of the niobate entities in aluminosilicate glasses in more detail, helping to shed some light on the structural behavior of this technologically important element.
2. Experimental methods
Details of the glass synthesis, bulk chemistries, and physical, thermal, and optical properties, along with the study of the glass structure via Raman of the present glasses and glass–ceramics, were previously described in ref. 9, 14 and 16. The glasses belong to the ternary system SiO2–Na2O–Al2O3, and the pristine materials have a constant SiO2 molar content (∼66.6 mol%) and Na2O/Al2O3 molar ratios of 2.3 and 1, respectively corresponding to a peralkaline composition (Na2O > Al2O3, sample label NA66.10), and a polymerized glass at the metaluminous joint (Na2O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Al2O3, label NA66.17). Portions of the base glass were finely ground, doped with commercially available K0.5Na0.5NbO3 ceramic powder (KNN, d50 = 0.95 μm, CerPoTech, Norway) from 0.4 up to 20 mol%, then remelted in air with a dwell time of 1 h at 1500 °C and 1650 °C, respectively for the NA66.10 and the NA66.17 series (the Nb2O5 content is about half that of KNN. The chemical compositions of the glasses obtained by ICP analysis9 are reported in Table S0, ESI†). In the polymerized series NA66.17, the sample containing 10 mol% Nb2O5 crystallized during quenching (label xx17-20); therefore, the maximum amount of Nb2O5 in this glass series is ∼4.8 mol% (label NA66.17-10KNN). Glass–ceramic (GC) samples were prepared from polished glass specimens (containing 10 mol% KNN) through heating at the respective exothermic event with a 10 K min−1 heating rate and a holding time of 2 to 4 hours (see ESI,† S0). The samples are labeled GC10-10 and GC17-10, respectively, for the peralkaline and metaluminous series. Both GCs show the presence of a single crystalline phase KxNa(1−x)NbO3 (XRD patterns and unit cell parameters are reported in ref. 14).
Al2O3, label NA66.17). Portions of the base glass were finely ground, doped with commercially available K0.5Na0.5NbO3 ceramic powder (KNN, d50 = 0.95 μm, CerPoTech, Norway) from 0.4 up to 20 mol%, then remelted in air with a dwell time of 1 h at 1500 °C and 1650 °C, respectively for the NA66.10 and the NA66.17 series (the Nb2O5 content is about half that of KNN. The chemical compositions of the glasses obtained by ICP analysis9 are reported in Table S0, ESI†). In the polymerized series NA66.17, the sample containing 10 mol% Nb2O5 crystallized during quenching (label xx17-20); therefore, the maximum amount of Nb2O5 in this glass series is ∼4.8 mol% (label NA66.17-10KNN). Glass–ceramic (GC) samples were prepared from polished glass specimens (containing 10 mol% KNN) through heating at the respective exothermic event with a 10 K min−1 heating rate and a holding time of 2 to 4 hours (see ESI,† S0). The samples are labeled GC10-10 and GC17-10, respectively, for the peralkaline and metaluminous series. Both GCs show the presence of a single crystalline phase KxNa(1−x)NbO3 (XRD patterns and unit cell parameters are reported in ref. 14).
The variation in the local structures as a function of KNN molar content was investigated by high energy X-ray total scattering. The experiments were carried out at the high-energy X-ray diffraction beamline BL04B2 at SPring-8 (Japan) by using a horizontal two-axis diffractometer.18–21 Bulk glass samples and crushed glass–ceramics samples were measured, with the latter inserted into silica glass capillaries. Additionally, the crystalline KNN material was measured as a reference. Only sample with significant amound of Nb were studied to obtain a good enough signal. Samples were irradiated with a monochromatic X-ray beam with an incident energy of 112.826 keV (λ = 0.1099 Å), and the scattered X-rays were detected by seven-point-type detectors. The highest scattering vector Q (Q = 4π![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ/λ) was 25 Å−1. The raw data were corrected for polarization, absorption, and background, and the contribution of Compton scattering was subtracted using the data analysis software developed by the beamline scientists (see ref. 20). The fully corrected data were normalized to give a Faber–Ziman total structure factor S(Q). The reduced pair distribution function (PDF), G(r), and total correlation function T(r) were obtained by the inverse-Fourier transform of S(Q) with a Lorch function.20,22 The data for the Fourier transform were terminated at Qmax = 23.5 Å−1 beyond which the signal-to-noise ratio of the unsmoothed data was considerably low. The concept of pair-function developed by ref. 23 and 24 to analyze the average distances and coordination numbers of vitreous SiO2 and B2O3 was used here to estimate the interatomic distances and the cation–oxygen coordination numbers of the glasses. The procedure is described in detail in ref. 21. The parameters for Si–O, Na–O, O–O, and K–O correlations were extracted from the RMC-MD model of alkali–silicate glasses reported in ref. 25, while the parameter for Al–O was extracted from MD simulations of a sodium aluminosilicate glass.26
sinθ/λ) was 25 Å−1. The raw data were corrected for polarization, absorption, and background, and the contribution of Compton scattering was subtracted using the data analysis software developed by the beamline scientists (see ref. 20). The fully corrected data were normalized to give a Faber–Ziman total structure factor S(Q). The reduced pair distribution function (PDF), G(r), and total correlation function T(r) were obtained by the inverse-Fourier transform of S(Q) with a Lorch function.20,22 The data for the Fourier transform were terminated at Qmax = 23.5 Å−1 beyond which the signal-to-noise ratio of the unsmoothed data was considerably low. The concept of pair-function developed by ref. 23 and 24 to analyze the average distances and coordination numbers of vitreous SiO2 and B2O3 was used here to estimate the interatomic distances and the cation–oxygen coordination numbers of the glasses. The procedure is described in detail in ref. 21. The parameters for Si–O, Na–O, O–O, and K–O correlations were extracted from the RMC-MD model of alkali–silicate glasses reported in ref. 25, while the parameter for Al–O was extracted from MD simulations of a sodium aluminosilicate glass.26
Multinuclear 23Na, 27Al, 29Si, and 93Nb solid-state NMR experiments were carried out at 5.7, 7.05, and 14.1 T on an Agilent DD2, a Varian INOVA 300 MHz, and a Bruker Avance 600 Neo spectrometer. 29Si MAS NMR experiments were recorded at a Larmor frequency of 59.2 MHz in a 7 mm double resonance probe operating at υMAS = 5 kHz. 64–128 transients of 4096 data points were recorded using 90° pulses of 6 μs duration and recycle delays of 500–1000 s. 29Si chemical shifts are reported w.r.t. tetramethylsilane (TMS – 0 ppm) using kaolinite (−91.2 ppm) as a secondary standard. 23Na central transition (CT) MAS NMR experiments were performed at 14.1 T (Larmor frequency of 158.8 MHz) in a 2.5 mm triple-resonance probe at υMAS = 20 kHz. Short 1 μs pulses corresponding to a 30° tip angle were used for excitation and 256–1024 transients were collected at recycle delays of 0.5–5 s. 23Na chemical shifts are reported w.r.t. a 0.1 M NaCl solution (0 ppm). 27Al CT MAS NMR experiments were performed at 14.1 T (Larmor frequency of 156.4 MHz) in a 2.5 mm triple-resonance probe at υMAS = 20 kHz. Short 0.8 μs pulses corresponding to a 25° tip angle were used for excitation and 256 transients were collected at recycle delays of 0.5–1 s. 27Al chemical shifts are reported w.r.t. a 0.1 M Al(NO3)3 solution (0 ppm) using AlF3 as a secondary standard (−16 ppm). 93Nb CT MAS NMR experiments were conducted at 14.1 T (Larmor frequency of 146.80 MHz) in a 1.3 mm double-resonance probe at υMAS = 60.0 kHz and a nutation frequency of υrf = 150 kHz. For the rotor-synchronous Hahn-Echo spectra, excitation pulse lengths of 0.2 μs and 0.6 μs were used at a recycle delay of 0.1 s. To minimize coil ringing effects, spectra were acquired after 2 rotor cycles. Chemical shifts were referenced against a saturated solution of NbCl5 in acetonitrile (MeCN 0 ppm), using LiNbO3 (δiso = −1004 ppm) as a secondary standard.
3. Results
3.1 High energy X-ray total scattering
Fig. 1 shows the total structure factor S(Q) of the samples under study. The signals show distinct oscillations up to high Q values, indicating the presence of well-defined coordination shells in the glass network. By adding KNN, there is a general damping of the oscillations at high scattering vector values (Q > 10 Å−1), even though there is a large similarity in the oscillation width and position. At low Q vectors, the most prominent features are the first two peaks at 1.64 Å−1 and 2.17 Å−1, with the latter becoming dominant as KNN is added. In pure silica glass, the first peak at ∼1.6–1.7 Å−1, also referred to as first sharp diffraction peak (FSDP), is associated with the medium-range order of amorphous materials.19,27,28 The absolute position of these two peaks does not seem to change as a function of the KNN content, and only the relative intensities vary. On the contrary, the local signal maxima in the higher Q range show rather strong changes with the increase of alkali–niobate species, i.e., the peaks at 5 Å−1 and 9 Å−1 become suppressed, while the peak at ∼4.5 Å−1 shifts toward smaller Q vectors.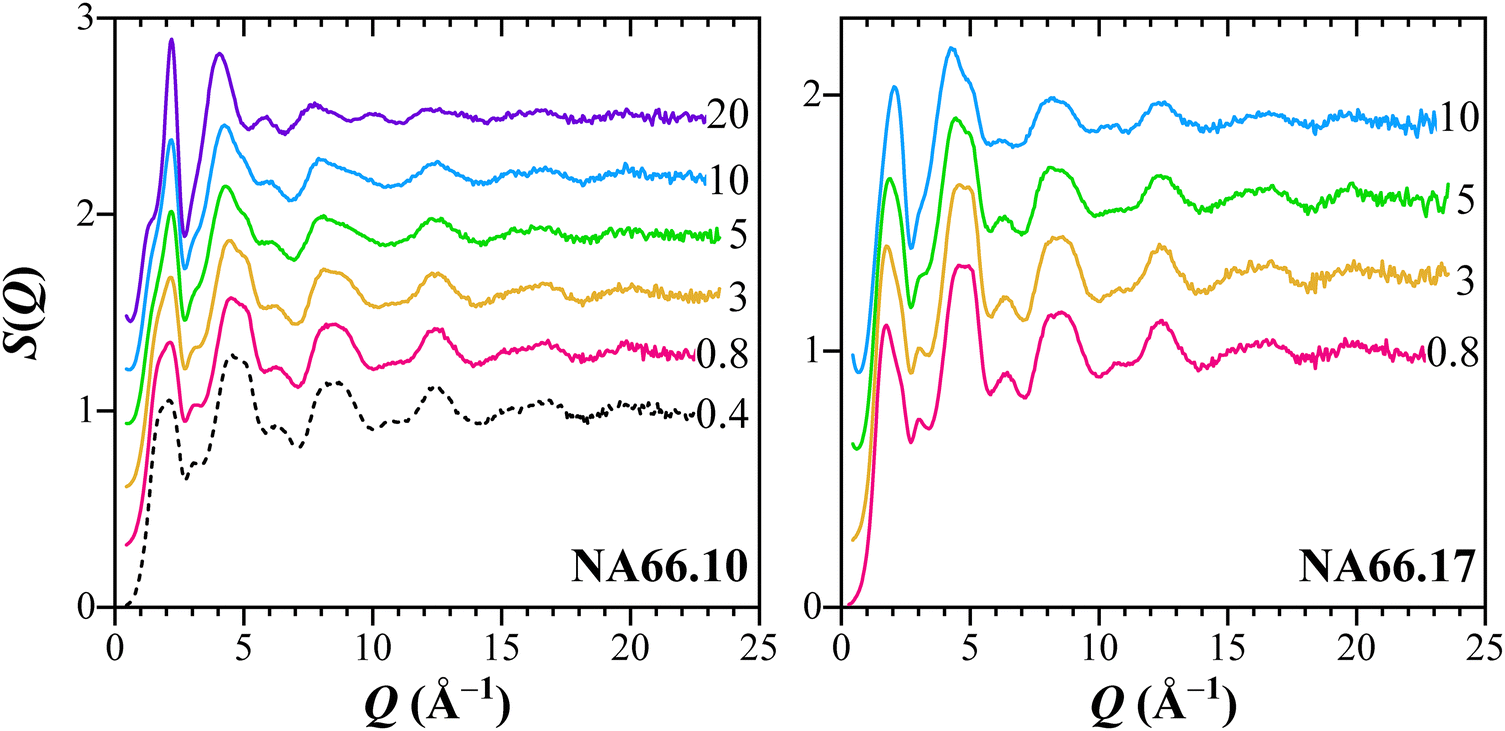 | ||
| Fig. 1 Measured X-ray total structure factors for the two aluminosilicate glass series having different KNN contents (from 0.4 mol% to 20 mol%). | ||
The total correlation function T(r) for an aluminosilicate glass is presented in Fig. 2, along with data of crystalline (K0.5Na0.5)NbO3 (KNN), used as a reference for typical Nb-related distances. The assignment of the different peaks is done by comparing the interatomic distances given by the radial distribution curve with the interatomic distances of crystalline materials and previous reports on aluminosilicate glasses. The T(r) of the glass having only 0.2 mol% Nb2O5 (NA66.10-0.4KNN) shows the expected peaks of aluminosilicate glass, with no strong contributions from Nb. The intense first peak at 1.65 Å, related to nearest neighbor intra tetrahedral bonds (T–O, T = Si, Al) derives from the partial contributions of Si–O (∼1.6 Å) and Al–O pairs (∼1.75 Å).29,30 The two following smaller correlations arise from the alkali cations–oxygen (A–O, ∼2.30 Å) and oxygen–oxygen (O–O, ∼2.63 Å) distances. The strong contribution peaking around 3.10 Å derives mainly from tetrahedra–tetrahedra (T–T) distances.31 The reference alkali–niobate crystalline compound (KNN) shows well-defined, strong peaks. The experimental PDF data analysis of the crystalline material was refined by PDFgui32 and the results are reported in the ESI† (Fig. S1 and Table S1). The refined structure of the KNN crystalline compound results in an average bond length of 2.00 Å for the Nb–O octahedron, and a distortion index of the bond length of 0.06 (Baur's polyhedra distortion index33) compatible with the broad Nb–O correlation observed in Fig. 2 (see Table S1 for the refined correlations, ESI†). Furthermore, mathematical functions (Gaussians) were used to decompose the real-space data both for the KNN crystalline material and the glass NA66.10-0.4(KNN). The latter shows four main contributions that well agree with the expected distances in a sodium aluminosilicate glass. Regarding the KNN crystal compound, the first broad contribution peaking around 2.00 Å is split and can be deconvoluted into two Gaussian functions centered at 1.90 and 2.14 Å (Fig. 2). The position of both functions stands in agreement with the PDF refined structure, and the displacement of the Nb cations observed by Levin et al.,34 reported to be predominantly along the polar c-axis of the orthorhombic unit cell. In a similar fashion, the second broad peak around 2.8 Å derives from two sets of distances associated with the alkali cations–oxygen pairs (A–O). The A–Nb and Nb–Nb correlations are very prominent, and the Gaussian functions peak, respectively, around 3.44 Å and 3.98 Å. These values are consistent with the average distances obtained from the refinement of the crystalline compound (respectively, at 3.40 Å and 4.00 Å; see Table S1.2, ESI†).
 | ||
| Fig. 2 Total correlation function T(r) for the aluminosilicate glass NA66.10-0.4(KNN) and the crystalline (K0.5Na0.5)NbO3 (KNN), used respectively as references for T–O, A–O and T–T distances in the glasses and Nb-related distances. T = tetrahedra (Si, Al); A = alkali ions (Na, K). Grey dotted and dashed curves are the Gaussian functions used to decompose, respectively, the signals of the crystalline and glass materials. | ||
The distances obtained from these two materials will be used as a guideline for the assignment of the correlations of the glass samples under study.
An overview of the real-space transforms of the data for the two glass series is presented in Fig. 3 (and Fig. S2, ESI†). The experimental reduced pair distribution function G(r) obtained from the Fourier transformation of the total scattering structure factor S(Q) (in Fig. S2, ESI†) and the total correlation function T(r) show the presence of well-defined peaks, even at longer distances (i.e., >4–5 Å). In Fig. 3, the correlations related to the aluminosilicate matrix are the most prominent ones, and gradually, their intensities decrease as KNN is added. Only for the richest composition (20KNN) do the Nb-related correlations dominate the functions. Despite the significant changes in the relative intensity, the distances related to the aluminosilicate matrix appear to be rather constant. Indeed, the strong first peak at 1.65 Å, related to T–O bonds, and the T–T contribution peaking at ∼3.10 Å, show just lower relative intensities but no strong changes in their positions. Regarding the Nb-related correlations, it is clear that the addition of Nb2O5 results in a peak around 2 Å, whose intensity increases as the fraction of Nb in the glass increases. Therefore, this peak can be attributed to Nb–O bonds. Based on the previous structural characterization of these glasses,9 we assume those distances to be related to six-fold coordinated Nb5+ units. Still, a more detailed discussion on the coordination will be done in the Discussion section. The other two features that are clearly emerging with increasing KNN content are centered at ∼3.45 Å and ∼3.85 Å. Worth to be noted is also the occurrence of an isosbestic point around 3.21–3.24 Å in the peralkaline NA66.10 glasses. Usually, the occurrence of an invariant point represents the presence of two predominant species whose ratio changes depending on external factors (in our case, the increase in KNN and a matrix relatively depleted in the aluminosilicate fraction).
 | ||
| Fig. 3 The upper panels display the experimental total correlation functions T(r) for the two aluminosilicate glass series having increasing KNN contents (from 0.4 mol% to 20 mol%). The T(r)s of the glass–ceramics (GC) for each respective composition are reported as well. In the lower panels, the correlation functions of the two glass systems (red lines: NA66.10; black lines: NA66.17) and of the two GCs are compared at equal KNN content. To highlight the changes, their second derivative signals are shown. | ||
The T(r) of the glass–ceramics (GC) are compared to the glass series in Fig. 3. The two partially crystallized samples contain (Kx, Na1−x)NbO3 crystals with a perovskite-like structure.14 The GC10-10 signal resembles well the one of the parent glass (NA66.10-10KNN, blue line) with the exception of a stronger correlation centered close to 4 Å. The same considerations can be applied to the GC17-10 signal, which has a very prominent contribution peaking at 3.96 Å.
To better evaluate the evolution of the interatomic distances depending on the niobate content and to highlight how these distances differ depending on the glass polymerization, the T(r) and their second derivatives are shown on the lower panels of Fig. 3. The direct comparison highlights the significant differences in the atom–atom correlations of the two glass series, and especially notable are the more pronounced features at 2.0–2.05 Å and around 3.45–3.48 Å in the NA66.10 series. Therefore, the bulk chemistry influences the local environment of the Nb species not only with respect to the nearest neighbors but predominantly beyond the first coordination shell. Another aspect to highlight is the lower intensity of the Nb–O correlation in the NA66.17 series for the same niobate content: it suggests that the probability of finding Nb in a particular distance is much lower than in the peralkaline bulk composition. This difference could be explained by a larger distribution of distances stemming from a potentially greater distortion of the polyhedra when Nb is incorporated into a peraluminous system.
3.2 Nuclear magnetic resonance spectroscopy – NMR
Fig. 4a shows the 29Si MAS NMR spectra of selected specimens under study. The spectra of the aluminosilicate glasses without KNN are centered around −93 and −95 ppm, respectively, for the NA66.10 and NA66.17 systems, indicating the presence of mostly Q3 and Q4 units (see also Table 1). In the present case, we refrained from fitting the 29Si MAS NMR spectra because of the potentially great number of possible SinmAl and SinmNb species, which cannot be resolved due to the intrinsically limited resolution.35 The addition of KNN produces a subtle positive shift of the center of gravities of both glass systems. Typically, positive shifts are interpreted in terms of depolymerization of the Si network, which can be expected since the introduction of each mole of KNN introduces a mole equivalent of (K/Na2O), increasing the alkali/silica ratio. However, Si species bonded to Al35 and Nb10 also resonate at higher frequencies than their Si(OSi)4 counterparts, all of which are possible outcomes in the present glass systems. It should be noted that the width of the 29Si line decreases slightly as a function of KNN content. Fig. 4b shows the 27Al MAS NMR spectra of selected specimens under study. The spectra of the samples corresponding to the NA66.10 and NA66.17 series show a single line centered at about 58 ppm, independently of KNN content, corresponding to four-coordinated [AlO4] units. All spectra were successfully simulated using a single component according to the Czjzek distribution model of quadrupolar parameters36 (see Fig. S3, ESI†). No significant amounts of higher coordinated Al species can be detected, and crystallization has no apparent effect on the NMR spectra. Thus, it can be established that Al ions do not partition into the B site of the crystallized perovskite in the GC samples. The 23Na NMR spectra in Fig. 4c show more pronounced variations, both in terms of isotropic chemical shift and line width. The 23Na center of gravity of the NA66.17 series is located near −15 ppm compared to −13 ppm for the NA66.10 series, in agreement with previous data on aluminosilicate glasses.37 In particular, the lower 23Na average isotropic chemical shift in the NA66.17 metaluminous glass compared to the peralkaline one is expected because of the lower number of non-bridging oxygens (NBO) in the composition.37 The addition of KNN induces similar variations in the two glass series in terms of chemical shift and line broadening: the spectra of the metaluminous NA66.17 series show a shift toward even lower frequencies with a peak maximum around −16 ppm, while no significant effect is seen for the peralkaline glasses. In the crystallized sample xx17-20, the 23Na signal clearly shows a shoulder corresponding to at least a second contribution, with maxima peaking around −9 ppm and −16 ppm. Interestingly, in the GC sample for the same metaluminous composition (GC17-10), both contributions are visible, albeit the much stronger intensity of the one at higher frequencies (−9 ppm). According to previous studies on silicate glasses and minerals, e.g., ref. 37 and 38, the 23Na average isotropic chemical shift generally decreases for an increase in the ion coordination and/or the average 〈Na–O〉 bond distances. In ANbO3 (A = Na, K) perovskites, the positional disorder of the alkali atoms is mirrored by the broadening of the NMR signals. Nevertheless, two main peaks have been assigned to the Na–O distances in hydrothermally synthesized NaNbO3 and KNN, respectively, at −13 ppm and −21 ppm.39 In aluminosilicate glasses, the 〈Na–O〉 distances reported are between 2.30 and 2.43 Å, whereas, in perovskites, these distances are in the range of 2.4–2.8 Å, in agreement with the low-frequency contribution observed in the GCs. The line width in the peralkaline series is very large and likely reflects the many structural environments of Na in this composition as (i) network modifier, introducing NBOs, (ii) charge compensator for 4-fold coordinated Al3+ ions (Na–BO), and (iii) charge compensator of the [NbO6] units. In the metaluminous composition NA66.17, all Na+ ions are charge-compensators for 4-fold coordinated Al3+ ions. For each mole of KNN added there is one atom of Nb and 0.5 atoms of both alkaline elements (Na/K) inserted into the structure: the newly added alkaline elements can disrupt the aluminosilicate network and create NBO or favour the charge balance of 6-fold coordinated Nb5+ species, enhancing the formation of an alkali compensated-[NbO6] sub-network. Both scenarios are likely; however, the very limited shift observed for the 29Si NMR of the NA66.17 series, tends to favor the charge compensating role as the main one.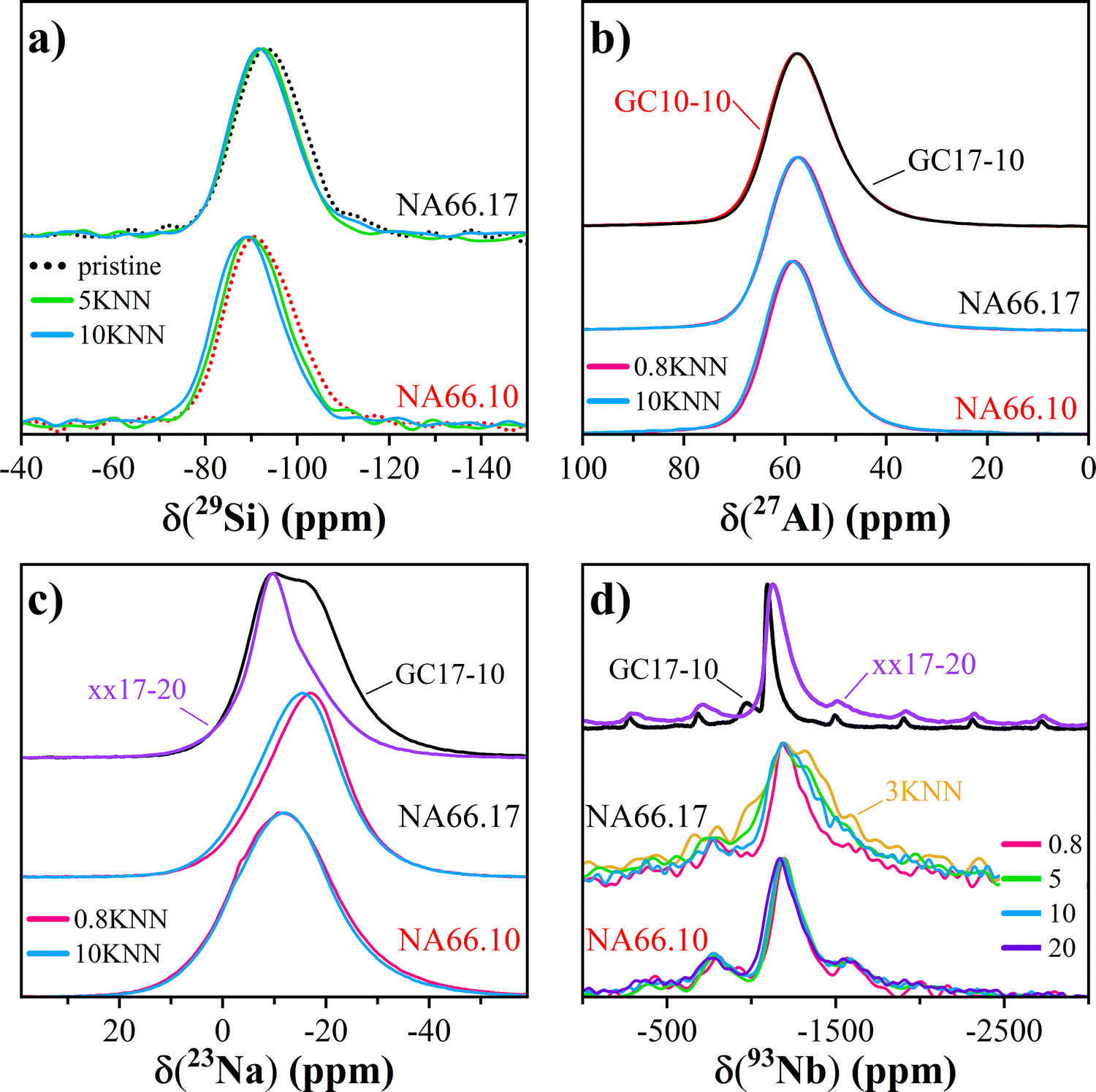 | ||
| Fig. 4 29Si, 27Al, 23Na and 93Nb MAS NMR spectra for selected samples in the two glass series (in panels a to d, respectively). | ||
| Sample | δCSCG/ppm (± 1) | FWHM/ppm (± 1) |
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| 29Si | 23Na | 27Ala | 93Nbb | 29Si | 23Na | 27Alc | 93Nbd | 27Al | |
| a Isotropic chemical shift, δCSiso.b (±5).c FWHM of δCSiso distribution.d (±10). | |||||||||
| 66.10 | −93 | — | — | — | 18 | 23 | — | — | — |
| 66.10-0.8 | — | — | — | −1204 | — | — | — | 210 | — |
| 66.10-1.5 | — | −13 | 62 | −1194 | — | — | 9 | 230 | 4.0 (2.0) |
| 66.10-3 | — | — | — | −1201 | — | — | — | 210 | — |
| 66.10-5 | −91 | — | — | −1208 | 17 | — | — | 240 | — |
| 66.10-10 | −90 | −13 | 63 | −1194 | 17 | 22 | 9 | 240 | 4.1 (2.1) |
| 66.10-20 | — | — | — | −1180 | — | — | — | 250 | — |
| GC10-10 | — | −15 | 62 | — | — | 19 | 9 | — | 4.5 (2.2) |
| 66.17 | −95 | — | — | — | 17 | — | — | — | — |
| 66.17-0.8 | — | −16 | 62 | −1209 | — | 18 | 9 | 260 | 4.5 (2.2) |
| 66.17-3 | — | — | — | −1245 | — | — | — | 578 | — |
| 66.17-5 | −93 | — | — | −1228 | 16 | — | — | 450 | — |
| 66.17-10 | −93 | −15 | 62 | −1242 | 16 | 19 | 9 | 370 | 4.6 (2.4) |
| xx17-20 | — | −12 | 63 | −1135 | — | 12 | 9 | 160 | 4.2 (2.1) |
| GC17-10 | — | −15 | 62 | −1095 | — | 20 | 9 | 60 | 4.5 (2.3) |
| −980 | 340 | ||||||||

The 93Nb MAS NMR spectra in Fig. 4d (and Fig. S3, ESI†) show an interesting compositional effect: while all spectra are rather large and asymmetrically broadened due to the presence of distributions of electric field gradient (EFG) tensor and chemical shift anysotropy (CSA) tensor components, the 93Nb linewidth of the NA66.10 glasses stays rather invariant as a function of KNN content. On the other hand, its center of gravity moves systematically from ca. −1197 ppm to around −1168 ppm when adding KNN. Different is the situation in the NA66.17 series: the lines become significantly larger for KNN contents above 1.5 mol%, and, on average, the center of gravity shifts to about −1220 ppm, indicating the formation of multiple unresolved Nb local environments or significant differences in the Nb second-coordination sphere. The crystallized xx17-20 and the glass ceramic sample GC17-10 (Fig. 4d) exhibit narrower linewidths of about 160 and 60 ppm that peak, respectively, around −1135 ppm and −1095 ppm. These values are very similar to those reported for nanofibers Pbcm NaNbO3 (−1094 ppm) by ref. 39, but smaller than the isotropic chemical shift found in various NaNbO3 (−1078 ppm40) or KNbO3 (−1056 ppm39) polymorphs. According to the authors, the smaller shift in the nanofibers is due to the occurrence of a higher degree of structural disorder, and in particular, a mixture of coexisting orthorhombic and monoclinic phases having different local environments of the alkali ions.
Noteworthy is the difference observed in the 93Nb MAS NMR spectrum of the sample NA66.17-3 (see Table 1), which is the broadest of all the glasses with a FWHM of 578 ± 1 ppm, twice that of the 0.8 mol% KNN sample, and has a lower center of gravity δCSCG −1245 ± 1 ppm. This out-of-trend spectrum indicates a different Nb structural environment that cannot be easily resolved by NMR. Additional considerations will be presented in the Discussion section.
4. Discussion – the Nb structural environment's evolution
To provide an overview of the structural evolution of the Nb species in aluminosilicate glasses, newly acquired structural information by nuclear magnetic resonance (NMR) and high-energy X-ray total scattering can be coupled with the polarization information of the vibrations from Raman spectroscopy and the physical, thermal and optical properties previously reported.9,14The high-energy X-ray total scattering data show well-defined coordination shells in the glass network and the distances associated with the aluminosilicate network agree well with previous reports. The addition of niobium (as KNN) into the glass network results in a rearrangement of the units, leading to significant modifications, particularly extending beyond the first coordination spheres. Prior to examining the atomic ordering in the niobo-aluminosilicate glasses, it is important to recall the interatomic distances of Nb5+ in crystalline materials to facilitate an assessment of the probable local environments of the Nb species.
In crystalline materials, there are many possible arrangements for the Nb5+ cations, since they can be found in different coordination environments, with different connectivity (edge, face, and corner shared) and with polyhedra having different degrees of distortion. For instance, in CeNbO4, Nb5+ is coordinated by four oxygens arranged in a distorted tetrahedron, with an average 〈Nb–O〉 bond length of 1.88 Å.41 Its high-temperature tetragonal polymorph is more regular and has shorter Nb–O distances (1.85 Å).42 [NbO5] trigonal bipyramids have been reported in Na5NbO543,44 with Nb–O bond lengths of 1.99 (×2) Å, 1.90 (×2) Å and 1.88 Å. The six-fold coordination is the most common in Nb-crystalline compounds: for example, KNbSi2O7 (P4bm) has [SiO4] tetrahedral linked to chains of corner-shared [NbO6] units.45 These octahedra are highly distorted with alternating short (1.76/1.78 Å) and long (2.28/2.32 Å) Nb–O bonds and Nb–Nb distances of ∼4.16 Å. The closest Nb–Si correlation is around 3.59 Å. CaNb2O6 exhibits chains of edge-sharing [NbO6] octahedrons linked to each other along their shortest edge and an average bond length of 2.02 Å (quadratic elongation = 1.04). The closest Nb–Nb interatomic distance is 3.66 Å, while the longest one is at 3.83 Å.46 ANbO3 (A = Na, K) compounds have a perovskite-type structure and atom arrangements similar to the KNN reference material reported in this study. All octahedral units present a slightly distorted environment due to the displacement of the central cations, and the 〈Nb–O〉 bond lengths are ∼2 Å, while the Nb–Nb distances range between 3.91 and 4.04 Å.47–49 In NaNbO3 (Pmc21), K0.5Na0.5NbO3 (Amm2), and KNbO3 (Bmm2), the A–Nb distances range between ∼3.30 and 3.55 Å. Higher coordination environments for Nb5+ ions have been reported in NaNb3O850 where there are chains of two Nb5+ sites: a dodecahedral one (〈Nb–O〉 = 2.075 Å) and a distorted pentagonal bipyramid with an average 〈Nb–O〉 bond length of 2.09 Å.
As observed in Fig. 3, in glasses, the interatomic distances associated with KNN incorporation are centered around 2 Å, ∼3.40/3.45 Å and ∼3.85 Å. Additionally, the NA66.17 composition presents a correlation around 3.25–3.30 Å. The crystalline KNN material (Fig. 2 and Fig. S1 and related text in the ESI†) can be used as a reference for the 6-fold coordinated Nb5+-related distances: the 〈Nb–O〉 correlation at 2.00 Å, the 〈A–O〉 at 2.83 Å, the 〈A–Nb〉 at 3.40 Å and the 〈Nb–Nb〉 correlation at 4.00 Å.
The incorporation of KNN in the aluminosilicate glasses results in an enhancement of the correlation associated with Nb–O bonds and a shift toward shorter distances: from 2.07 to 2.0 Å in the peralkaline series and from 2.02 to 1.98 Å in the polymerized glasses. However, given the difficulties in estimating the exact position of the correlation at low Nb2O5 contents, the Nb–O distances should be considered almost unaffected by the KNN content. Nevertheless, the correlations are visibly at a shorter distance in the polymerized samples. The real space functions, the NMR data, and the observed invariance in the 〈Nb–O〉 distance suggest that the octahedral environment is the predominant one. Indeed, if species with lower coordination were present, the Nb–O correlation should be expected at much shorter distances, considering the average 〈Nb–O〉 of 1.88 Å in the 4-fold coordinated CeNbO4, and the 〈Nb–O〉 of 1.93 Å in the 5-fold coordinated Na5NbO5. In turn, the first correlation related to T–O should have a larger width. However, this is not the case. It is, therefore, reasonable to conclude that a prevailing 6-fold coordination environment for Nb is present in all glasses. In order to verify these considerations, the cation–oxygen coordination numbers were estimated on the basis of the pair-function concept developed by ref. 23 for SiO2 glass. The pair-functions obtained are compared to the experimental data in Fig. 5 for the structure factors and the total correlation functions. The average coordination numbers and distances for NA66.10 and NA66.17 glass series obtained by the pair-function method are reported in Table S2 (ESI†), along with the results of the two glass–ceramics. Samples with low Nb2O5 contents (0.4 and 0.8 mol% KNN) could not be accurately analyzed due to the very weak Nb–O correlations, leading to large errors in both coordination numbers and distances. The pair-function method confirms the near invariability of the two network former cations (Al and Si) and, most importantly, confirms that the Nb coordination number is predominantly 6, regardless of the bulk chemistry and Nb content. Another important aspect is obtained for sample GC17-10, where the Nb–O correlation peak shows an asymmetric shape. The Nb coordination number was studied by considering two Nb–O functions located at 1.94 Å and 2.21 Å, as shown in Fig. 5, and an average Nb coordination number of 5.9 (5.2 and 0.7; Table S2, ESI†). This Nb structural environment is very similar to that of the reference KNN material, where the distortion is mainly caused by the displacement of the Nb cations along the polar c-axis.
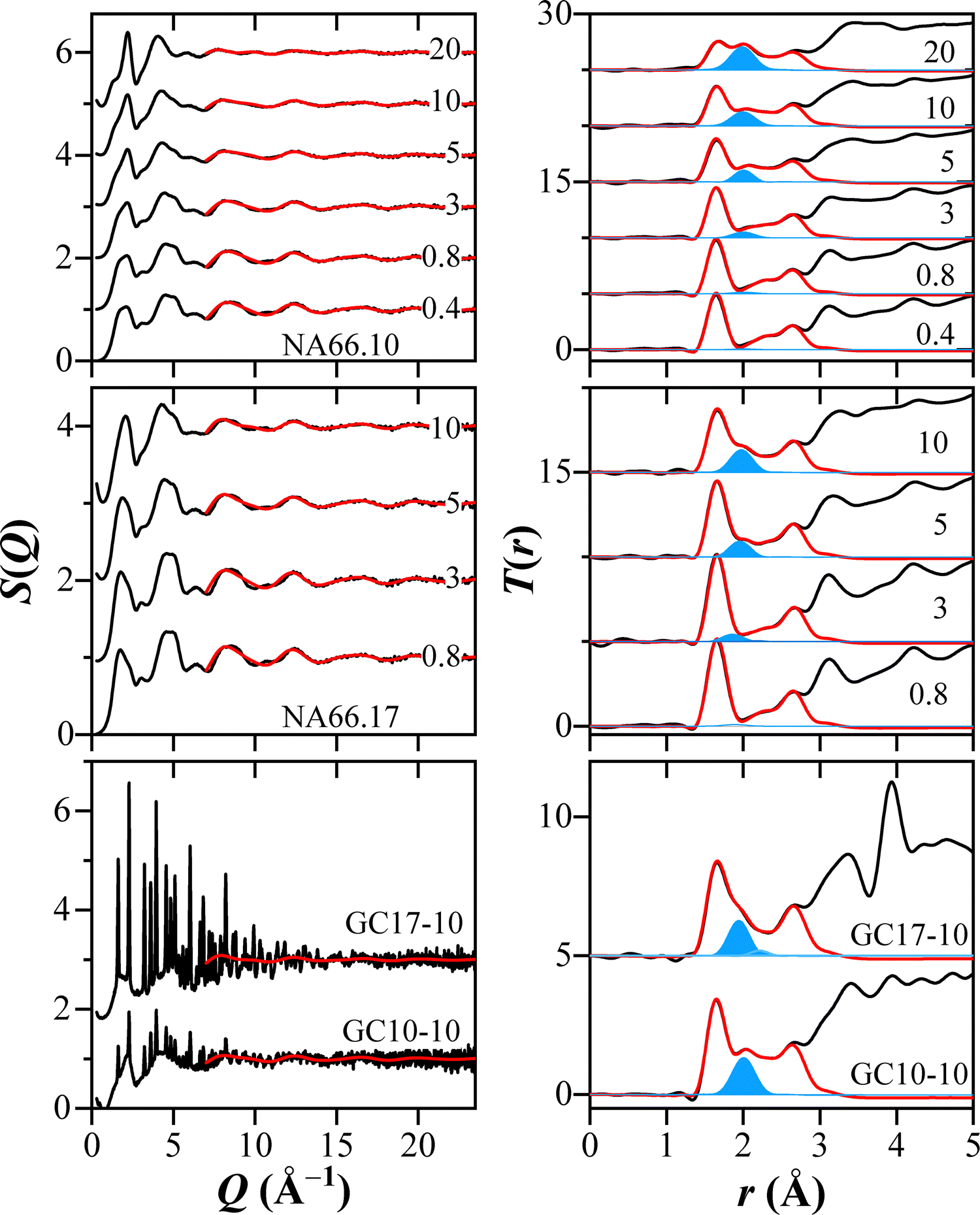 | ||
| Fig. 5 X-ray structure factors and total correlation functions of the two glass series having different KNN contents (from 0.4 mol% to 20 mol%), and GCs samples. The red solid curves represent the calculated structure factors and total correlation functions using the pair function methods for Si–O, Al–O, Nb–O, Na–O, O–O, and K–O correlations, while the blue lines are the estimated total correlation functions for the Nb–O correlation. | ||
It is worth highlighting the results obtained for the sample NA66.17-3KNN (see Fig. 3, Table 1 and Fig. S3, Table S2, ESI†). The first observation concerns the lower intensity of the Nb–O correlation around 2 Å compared to the peralkaline glass, suggesting that the probability of finding Nb at a given distance is much lower in a metaluminosilicate composition (Fig. 3). Furthermore, the Nb–O pair-function gives a lower bond distance and coordination number (respectively, 1.86 Å and 4.1), a unique result among all compositions. However, in the real space function, the contribution around 2 Å is visible, and the pair-function method suggests the presence of a correlation at 2.17 Å, albeit very low (0.4 coordination) (all data in Table S2, ESI†). Therefore, similarly to the glass–ceramic, the polyhedra units in NA66.17 show a distribution of both shorter and longer Nb–O distances. These observations, together with the very broad 93Nb NMR signal observed in Fig. 4, lead to the conclusion that there is a greater distortion of the NbO6 polyhedra in the metaluminous composition than in the peralkaline one. The reason for this distortion could be the preferential use of high-field strength Na+ ions to balance the negative charge of the [AlO4/2]− units compared to the larger K+ ions, leaving the latter to preferentially associate with the [NbO6/2]− units. This hypothesis also explains the preferential partitioning of K+ ions into the perovskite crystal phase observed in the resulting glass–ceramics.14
Although the Nb coordination remains predominantly the same in both glass series, it is evident that the bulk chemistry plays a key role in effecting the distortion, with the most significant differences occurring between 3.2 Å and 4.0 Å: correlations related to Nb – 2nd nearest neighbor distances. As previously observed in the series NA66.10, the contribution at ∼3.15 Å, associated with T–T distances, decreases steadily with the inclusion of KNN, while at the same time, the ∼3.42–3.45 Å distance increases in intensity. The latter distance is consistent with the A–Nb correlation reported in crystalline materials, and the isosbestic point marks the equilibrium point between the two arrangements. Therefore, the invariant point is between distances mainly related to T–T and A–Nb pairs, which in turn suggests a progressive increase of [NbO6] units charge-balanced by alkali species, and a very limited interaction between the aluminosilicate network and the Nb units. Only above 10 mol% of KNN, the peralkaline series shows a clear correlation at higher distances (∼3.85 Å) related to Nb–Nb. This evolution is in agreement with the structural data obtained by 23Na NMR, where the broad Na distribution remains almost invariant in the NA66.10 series, confirming that there is no drastic or sudden change at a specific Nb2O5 content, but rather is a continuous adjustment of the network connectivity. Additionally, the structural evolution observed well agrees with the polarized Raman spectroscopy and the physical and thermal properties of the NA66.10 glass series.9 Indeed, the occurrence of many intermediated vibrational Nb-units was observed in the polarized Raman, with their proportions shifting as Nb2O5 content increased, ultimately leading to [NbO6] cluster formation at the highest KNN contents. Also, the increase of the glass transition temperature of the peralkaline glass series (∼50 °C higher than its pristine glass) explained by the incorporation of Nb species in the modifier channels fits well with the observed A–Nb correlations that further crosslink the network.
The metaluminous NA66.17 series shows a quite different evolution in the 2nd coordination shell, as observed in Fig. 3. Namely, the correlation at ∼3.14 Å becomes broader, and another contribution appears at longer distances (∼3.50 Å). Additionally, the Nb–Nb correlation moves from 3.80 Å to ∼3.94 Å by increasing the Nb2O5 content. The change in the Nb–O–Nb bond angle distribution could explain the shift of the Nb–Nb correlation toward longer distances; in fact, by considering the average 〈Nb–O〉 distance of 2.00 Å, a 180° corner-shared (opposite bond angle) would imply that the niobium–niobium distance is approximatively twice that of niobium–oxygen, at about 4.00 Å. If edge-shared octahedrons were present, the minimum distance would be reduced to ∼2.83 Å, a correlation not seen in any signal. Therefore, the most reasonable explanation is the formation of [NbO6] chains with Nb–O–Nb interatomic angle lower than 180° (estimated as 165°). The charge-compensator K+/Na+ ions would change their average bond distances accordingly, as observed from the 23Na isotropic chemical shift of the NA66.17 series. A comparison of the GC and their respective parent glasses reveals no significant variations in the distances of the aluminosilicate matrix (both T–O and T–T). Therefore, it can be concluded that both alkaline species and Nb5+ ions in the amorphous state already favour a topology very similar to that of the stable crystalline phase.
The different evolution of the glass network depending on the glass chemistry or rather, on the alkali/Al ratio explains the different Nb2O5 solubility observed in the NA66.10 and NA66.17 series and also explains the different macroscopic properties. Also, it might clarify why the solubility of Nb2O5 is much lower in aluminosilicate glasses compared to other systems, such as the borophosphate1 or aluminophosphate glasses.51 Indeed, 31P and 27Al NMR measurements on aluminophosphate compositions with Nb2O5 contents ranging from 0 to 30 mol% have shown a drastic change in both the P and Al coordination environment and the predominant formation of Nb–O–P linkages, while Nb5+ speciation was rather constant and independent of its content.51 In the aluminosilicate glasses studied here, there is a very limited interaction between Nb and the aluminosilicate network, since a linkage between Nb–O–Si(Al) does not provide an efficient charge distribution, as in the case of the phosphate matrix. In the future, it would be interesting to test whether the inclusion of P5+ ions in an aluminosilicate glass would boost Nb solubility due to the Nb–O–P crosslinking.
The good agreement between polarized Raman spectra, NMR, and synchrotron X-ray scattering is remarkable. To highlight this agreement and schematically represent the correlations found in this work, in Fig. 6, the radial distribution function RDF, the perpendicularly polarised (VH) Raman spectra, and the 93Nb MAS NMR spectra are compared. The details and analysis of the Raman spectra have already been reported in ref. 9. Based on these data, we propose the presence of four main fingerprint features in the Raman spectra that can be used on other systems to identify the local environment of the Nb and forecast the glass properties:
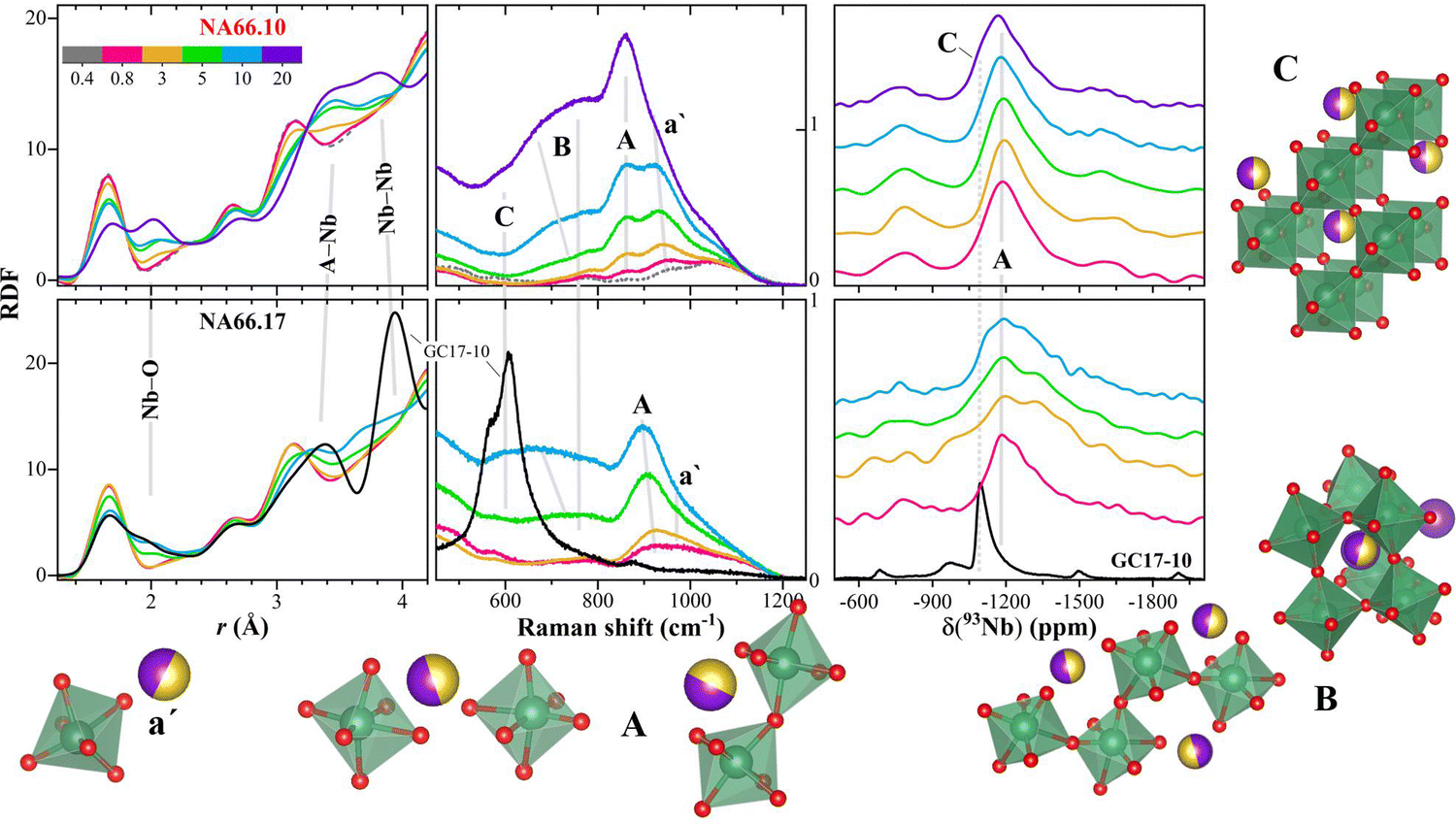 | ||
| Fig. 6 The structural information obtained from synchrotron X-ray scattering, perpendicularly polarised (VH) Raman spectroscopy,9 and NMR are summarized here, together with a schematic representation of the different possible structural units associated with specific fingerprint features in NMR and Raman spectra (labeled a′, A, B, and C; see text for details). Upper panels: data for glasses in the NA66.10 series; lower panels: data for glasses in the NA66.17 series. The vertical axis of the Raman signals represents the raw intensity of the scattered light in counts (×103). | ||
(a′) Alkali compensated-[NbO6] structural units. The large NMR peak width and the frequency of the Raman vibration indicates a highly distorted environment. This feature dominates only in the compositions having low Nb2O5 contents (≤5 mol% KNN) and prevails in compositions with high amounts of charge-compensating ions (i.e., peralkaline). As the Nb content increases, this contribution diminishes in intensity. The higher distortion of the [NbO6] structural units of the NA66.17 series, and in turn, the broader bond distribution around these units, is mirrored by the lower intensity of the Nb–O correlation in the radial distribution function and the Raman vibrational modes, and probably, also from the NMR component at lower shifts (as observed in sample NA66.17-3KNN).
(A) This contribution is related to the center of the NMR band and to the main band associated with Nb-vibrations in the Raman spectra. In the latter, the high polarizability of the bond9 indicates that there is a directional character of the vibrations, while the lower frequency position compared to a′ indicates a decrease in [NbO6] distortion. These considerations are consistent with the appearance of a correlation at longer distances related to Nb–Nb pairs. The formation of Si(Al)–O–Nb bonds would also be compatible with a more polarizable bond (more sensitive to light polarization because of the deformation of the electron cloud). However, Al NMR data do not show strong changes in the Al local environment, and the vibrational modes related to Si–O and O–Si–O are unchanging. Therefore, the formation of bridging bonds Nb–O–Nb is the most probable one.
(B/C) The Raman vibrations between 680 and 800 cm−1 have lower polarizability9 (and constant polarization ratio), compatible with the formation of more ionic clusters of [NbO6] units with no preferential direction, charge compensated by the alkali ions. These clusters do not seem to have edge-shared units. Indeed, the Nb–Nb correlations in the T(r) are consistent with the formation of [NbO6] chains with Nb–O–Nb interatomic angle of about 165°, which will evolve in the more regular 3D arrangement of corner-sharing [NbO6] units, similar to those in the perovskite structure (C). The formation of these 3D arrangement could be recognized also in the 93Nb NMR from the component at higher chemical shift values that overlaps with the NMR band found in perovskite-bearing glass–ceramics.
The fingerprint features identified can be used to predict preferred bonding and network connectivity in many glass systems. This information will allow us to identify promising compositions with tailored functionalities, such as highly distorted niobate units able to enhance the Nb photoluminescence emission,14 or higher fractions of edge-shared units, useful for electro-optical devices.13
5. Conclusions
This work presents, for the first time, high-energy X-ray diffraction data for two series of alkali–aluminosilicate glasses to unravel the evolution of the niobate units. In order to understand the structure–properties relationship, these data are coupled with complementary solid-state NMR and polarised Raman spectroscopy data.The results show that Nb5+ is present in all aluminosilicate glasses predominantly as 6-fold coordinated units, and with a nearly constant 〈Nb–O〉 bond distance of 2.0 Å. The addition of Nb to the aluminosilicate network has a marginal effect on the Si and Al tetrahedral units with very limited crosslinking. The unchanging T–O and T–T correlations, together with the 27Al NMR data confirm that the aluminosilicate matrix preserves its network, while the Nb5+ ions in the amorphous state already favour a topology very similar to that of the stable crystalline phase. Indeed, strong changes can be seen in the A–Nb (A = alkaline) and Nb–Nb correlations. Therefore, the overall glass connectivity is influenced by the availability and nature of charge-compensating alkali ions. The different properties and structural arrangements observed in other glass systems such as the aluminophosphate are rationalised based on favourite connectivity due to efficient charge distribution.
The structural information obtained from NMR and synchrotron X-ray scattering is complemented by that from perpendicularly polarised Raman spectra, and we propose specific fingerprint features in the NMR and Raman spectra that represent the different possible structural units. In particular, we show that we can use polarised Raman spectra to predict preferred bonding and network connectivity, and use this information to identify promising compositions with tailored functionalities.
Author contributions
MRC: conceptualization, resources, project administration, funding acquisition, investigation, formal analysis, writing – original draft, review & editing; KK: conceptualization, investigation, formal analysis, writing – review & editing; HB, HD, SK, YO, TH, SS: investigation, data curation, validation, writing – review & editing; KH, DdL: resources, funding acquisition, validation, writing – review & editing. All authors each made a significant contribution to the research reported and have seen and approved the final version of the manuscript being submitted.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing interests that could influence the work in this paper.Acknowledgements
The authors thank SPring-8 for the provision of synchrotron radiation (Proposal 2023A1202) and the staff of Beamline BL04B2 for assistance. The authors gratefully acknowledge the financial support from the Deutsche Forschungsgemeinschaft under grant numbers CI294/2-1, and GRK2495/2/G and the Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for Transformative Research Areas (A) “Hyper-Ordered Structures Science” (Grant numbers 20H05878 and 20H05881). H. B. is grateful to FAPESP for infrastructural and post-doctoral support received under grant numbers 2013/07793-6 (CEPID program) and 2019/26399-3, respectively. M. R. C., H. B., and D. d. L. acknowledge the support for the Bayern-São Paulo workshops (FAPESP-Baylat grant 2022/01286-4).References
- T. Cardinal, E. Fargin, G. Le Flem and S. Leboiteux, J. Non-Cryst. Solids, 1997, 222, 228–234 Search PubMed.
- L. Petit, T. Cardinal, J. J. Videau, E. Durand, L. Canioni, M. Martines, Y. Guyot and G. Boulon, Opt. Mater., 2006, 28, 172–180 Search PubMed.
- A. Flambard, J. J. J. Videau, L. Delevoye, T. Cardinal, C. Labrugère, C. A. A. Rivero, M. Couzi and L. Montagne, J. Non-Cryst. Solids, 2008, 354, 3540–3547 CrossRef CAS.
- S. Sanghi, S. Rani, A. Agarwal and V. Bhatnagar, Mater. Chem. Phys., 2010, 120, 381–386 Search PubMed.
- S. Liu, B. Shen, H. Hao and J. Zhai, J. Mater. Chem. C, 2019, 7, 15118–15135 Search PubMed.
- M. A. Villegas and J. M. F. Navarro, J. Eur. Ceram. Soc., 2007, 27, 2715–2723 CrossRef CAS.
- R. L. Ciceo, M. Todea, D. Toloman, M. Muresan-Pop and V. Simon, J. Non-Cryst. Solids, 2020, 542, 120102 Search PubMed.
- T. Komatsu, T. Honma, T. Tasheva and V. Dimitrov, J. Non-Cryst. Solids, 2022, 581, 121414 Search PubMed.
- M. R. Cicconi, D. K. Dobesh, B. Schroeder, T. Otsuka, T. Hayakawa and D. de Ligny, Opt. Mater. X, 2023, 18, 100228 Search PubMed.
- H. Bradtmüller, Q. Zheng, A. Gaddam, H. Eckert and E. D. Zanotto, Acta Mater., 2023, 255, 119061 CrossRef.
- D. E. Vernacotola and J. E. Shelby, Phys. Chem. Glasses, 1994, 35, 153–159 CAS.
- A. A. Lipovskii, D. K. Tagantsev, B. V. Tatarintsev and A. A. Vetrov, J. Non-Cryst. Solids, 2003, 318, 268–283 Search PubMed.
- A. A. A. Lipovskii, D. K. K. Tagantsev, A. A. A. Vetrov and O. V. V. Yanush, Opt. Mater., 2003, 21, 749–757 Search PubMed.
- M. R. Cicconi, H. Deng, T. Otsuka, A. Telakula Mahesh, N. H. Khansur, T. Hayakawa and D. de Ligny, Materials, 2024, 17, 2283 Search PubMed.
- H. Bradtmüller, Q. Zheng, H. Eckert and E. D. Zanotto, J. Non-Cryst. Solids, 2024, 641, 123096 CrossRef.
- T. Otsuka, M. R. Cicconi, D. Dobesh, B. Schroeder and T. Hayakawa, Phys. Status Solidi B, 2022, 259, 2200016 CrossRef CAS.
- L. M. Marcondes, H. Bradtmüller, S. N. Carvalho dos Santos, L. K. Nolasco, C. R. Mendonça, S. H. Santagneli, G. Y. Poirier and M. Nalin, Ceram. Int., 2022, 48, 20801–20808 CrossRef CAS.
- S. Kohara, K. Suzuya, Y. Kashihara, N. Matsumoto, N. Umesaki and I. Sakai, Nucl. Instrum. Methods Phys. Res., Sect. A, 2001, 467–468, 1030–1033 CrossRef CAS.
- H. Ohno, S. Kohara, N. Umesaki and K. Suzuya, J. Non-Cryst. Solids, 2001, 293–295, 125–135 CrossRef CAS.
- S. Kohara, M. Itou, K. Suzuya, Y. Inamura, Y. Sakurai, Y. Ohishi and M. Takata, J. Phys.: Condens. Matter, 2007, 19, 15 CrossRef.
- K. Ohara, Y. Onodera, S. Kohara, C. Koyama, A. Masuno, A. Mizuno, J. T. Okada and S. Tahara, Int. J. Microgravity Sci. Appl., 2020, 37, 1–7 Search PubMed.
- S. Kohara and P. S. Salmon, Adv. Phys. X, 2016, 1, 640–660 Search PubMed.
- R. L. Mozzi and B. E. Warren, J. Appl. Crystallogr., 1969, 2, 164–172 CrossRef CAS.
- R. L. Mozzi and B. E. Warren, J. Appl. Crystallogr., 1970, 3, 251–257 Search PubMed.
- Y. Onodera, Y. Takimoto, H. Hijiya, T. Taniguchi, S. Urata, S. Inaba, S. Fujita, I. Obayashi, Y. Hiraoka and S. Kohara, NPG Asia Mater., 2019, 11, 1–11 CrossRef.
- J. Kalahe, Y. Onodera, Y. Takimoto, H. Hijiya, M. Ono, K. Miyatani, S. Kohara, S. Urata and J. Du, J. Non-Cryst. Solids, 2022, 588, 121639 Search PubMed.
- D. L. Price, S. Susman, K. J. Volin and R. J. Dejus, Phys. B, 1989, 156–157, 189–191 Search PubMed.
- P. S. Salmon and A. Zeidler, Phys. Chem. Chem. Phys., 2013, 15, 15286–15308 RSC.
- M. C. Wilding, C. J. Benmore and J. K. R. Weber, J. Phys. Chem. B, 2010, 114, 5742–5746 Search PubMed.
- Y. Xiang, J. Du, M. M. Smedskjaer and J. C. Mauro, J. Chem. Phys., 2013, 139, 44507 CrossRef PubMed.
- D. M. Zirl and S. H. Garofalini, J. Am. Ceram. Soc., 1990, 73, 2848–2856 CrossRef CAS.
- C. L. Farrow, P. Juhas, J. W. Liu, D. Bryndin, E. S. Božin, J. Bloch, T. Proffen and S. J. L. Billinge, J. Phys.: Condens. Matter, 2007, 19, 335219 Search PubMed.
- W. H. Baur, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 1195–1215 CrossRef CAS.
- I. Levin, V. Krayzman, G. Cibin, M. G. Tucker, M. Eremenko, K. Chapman and R. L. Paul, Sci. Rep., 2017, 7, 15620 CrossRef CAS PubMed.
- M. Edén, J. Magn. Reson. Open, 2023, 16–17, 100112 CrossRef.
- G. Czjzek, J. Fink, F. Götz, H. Schmidt, J. M. D. Coey, J. P. Rebouillat and A. Liénard, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23, 2513 Search PubMed.
- S. K. Lee, J. F. Stebbins, S. Keun Lee and J. F. Stebbins, Geochim. Cosmochim. Acta, 2003, 67, 1699–1709 CrossRef CAS.
- X. Xue and J. F. Stebbins, Phys. Chem. Miner., 1993, 20, 297–307 CrossRef CAS.
- M. B. Ghasemian, A. Rawal, Z. Shahrbabaki, Q. Zhang, T. Lu, Y. Liu and D. Wang, J. Mater. Chem. A, 2020, 8, 8731–8739 RSC.
- K. E. Johnston, J. M. Griffin, R. I. Walton, D. M. Dawson, P. Lightfoot and S. E. Ashbrook, Phys. Chem. Chem. Phys., 2011, 13, 7565–7576 Search PubMed.
- A. Santoro, M. Marezio, R. S. Roth and D. Minor, J. Solid State Chem., 1980, 35, 167–175 Search PubMed.
- S. J. Skinner, I. J. E. Brooks and C. N. Munnings, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, i37–i39 Search PubMed.
- J. Darriet, A. Maazaz, J. C. Bouloux and C. Delmas, J. Inorg. Gen. Chem., 1982, 485, 115–121 CAS.
- O. B. Lapina, D. F. Khabibulin, K. V. Romanenko, Z. Gan, M. G. Zuev, V. N. Krasil’nikov C and V. E. Fedorov, Solid State Nucl. Magn. Reson., 2005, 28, 204–224 Search PubMed.
- M. C. Foster, D. J. Arbogast, P. Photinos, R. M. Nielson and S. C. Abrahams, J. Appl. Crystallogr., 1999, 32, 421–425 Search PubMed.
- J. P. Cummings and S. H. Simonsen, Am. Mineral., 1970, 55, 90–97 Search PubMed.
- Y. Yoneda, R. Aoyagi, D. Fu, Y. Yoneda, D. Fu and S. Kohara, J. Phys.: Conf. Ser., 2014, 502, 012022 CrossRef CAS.
- Y. Yoneda, K. Ohara and H. Nagata, Jpn. J. Appl. Phys., 2018, 57, 11UB07 CrossRef.
- J. F. Li, K. Wang, F. Y. Zhu, L. Q. Cheng and F. Z. Yao, J. Am. Ceram. Soc., 2013, 96, 3677–3696 CrossRef CAS.
- K.-J. Range, M. Wildenauer and A. M. Heyns, Angew. Chem., Int. Ed. Engl., 1988, 27, 969–971 CrossRef.
- J. H. Faleiro, N. O. Dantas, A. C. A. Silva, H. P. Barbosa, B. H. S. T. da Silva, K. D. O. Lima, G. de, F. Silva, R. R. Gonçalves, R. Falci, Y. Messadeq, I. D. de O. Branco, B. M. Cerrutti, H. Bradtmüller, H. Eckert and J. L. Ferrari, J. Non-Cryst. Solids, 2023, 605, 122173 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ma00082c |
| This journal is © The Royal Society of Chemistry 2025 |