
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN/O co-doped porous carbon derived from polyester waste for electrochemical production of H2O2†
Mingsheng
Luo
a,
Chupeng
Wang
a,
Shiqi
Song
a,
Maochong
Tang
a,
Xiao Xia
Wang
 *b and
Min
Wu
c
*b and
Min
Wu
c
aSchool of Materials Science and Engineering, East China University of Science and Technology, Shanghai 200237, China
bSchool of Mechanical and Power Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: wangxiaoxia@ecust.edu.cn
cThe Tibetan-Plateau Scientific Research Center of Lhasa, Lhasa 850000, China
First published on 13th March 2025
Abstract
Electrochemical synthesis of hydrogen peroxide (H2O2) has been considered as an ideal alternative to traditional anthraquinone techniques due to its convenient operation and environmental friendliness. However, the development of catalysts with high activity, selectivity, and stability is still an important challenge. In this study, waste polyethylene terephthalate (PET) was converted into a high-value carbon-based catalyst for the electrochemical production of H2O2. Nitrogen–oxygen co-doped porous carbon (NOPC) was obtained by pyrolysis of PET with addition of melamine as a nitrogen source and magnesium oxide (MgO) as a templating agent. The cross-linking reaction during the pyrolysis process tightly bound nitrogen (N) and oxygen (O) active sites via amide group formation. These groups were uniformly distributed within the carbon matrix, accompanied by numerous defects, thereby enhancing both activity and selectivity. The optimized PET-derived NOPC catalysts exhibited a H2O2 selectivity of approximately 85% with an electron transfer number close to 2.3, and demonstrated long-term stability for over 10 hours in acidic solution. The electrocatalytic H2O2 yield in a custom-built cell reached 261.23 mmol gcatalyst−1 h−1 with faradaic efficiency above 80%. This work presents an efficient synthesis method for 2-electron oxygen reduction reaction (2e− ORR) electrocatalysts and provides a sustainable strategy for recycling waste plastics into valuable carbon materials.
Introduction
Electrochemical synthesis of H2O2 by 2e− oxygen reduction reaction (ORR) is considered as an environmentally friendly way for the development of green and sustainable chemistry.1 The 4-electron ORR process generating water is the main competitive reaction in the electrocatalytic preparation of H2O2.2 Consequently, electrocatalysts with high activity and selectivity for 2e− ORR have gained increasing attention in recent years.3 Precious metal (Pt, Pd, Au) alloy catalysts such as Pt–Hg, Pd–Hg, and Ag–Hg for H2O2 production have been reported,4,5 which exhibit excellent catalytic properties due to the weakened binding energy of the intermediates (*OOH).6 Unfortunately, the high cost and scarcity of precious metals limit their wide application on a large scale.7 On the other hand, carbon-based materials are more promising electrocatalysts for 2e− ORR due to their advantages of low cost, abundant resources, good conductivity, and stability.8 To date, various carbon materials have been explored for the electrochemical production of H2O2.9,10 It has been reported that the microstructure and composition of carbon will affect the catalytic activity and selectivity.11,12 The defect structures, oxygen-containing functional groups, and heteroatomic doping could increase the catalytic activity of carbon materials.3,13,14Generally, the modification of the pore structure of carbon materials is an effective strategy for enhancing their electrochemical performance. The mesoporous structures improve mass transfer and optimize electron transfer pathways, promoting H2O2 production during the 2e− ORR.15 However, precise regulation of the pore structure remains challenging due to factors such as synthesis methods, heat treatment processes, and material properties.7,16,17 Effective pore-making strategies, including templating, activation, and modifications of synthesis conditions, have been developed to address these difficulties.16,18,19
Additionally, heterogeneous atom doping (N,20 P,21 S,22etc.) has been found to adjust the microstructure of carbon materials, thereby modifying the electronic structure of active sites and improving catalytic performance. Kinetic analysis and theoretical calculation revealed that the synergistic interaction between nitrogen and oxygen active sites facilitated the conversion of *OOH to H2O2 by reducing *OOH adsorption and retaining O–O bonds.23 Currently, most attention has been paid to carbon-based electrocatalysts for electrosynthesis of H2O2 in alkaline media. However, H2O2 is more stable under acid conditions than in bases, and is more widely used in acid media with stronger oxidation ability. Consequently, the development of carbon-based catalysts for H2O2 production under acid conditions is of great importance.
Polyethylene terephthalate (PET) is one of the most commonly used polymers for packaging and beverage containers in our daily lives. However, owing to its superior stability, PET waste resists biodegradation under natural conditions, which leads to serious environmental and health issues. Recently, the conversion of PET to high-value carbon materials for different applications in the renewable energy field has been explored. Early research on the synthesis of high surface area porous carbon nanosheets through the catalytic carbonization of PET and KOH activation showed that this carbon material served as an ideal electrode for double-layer supercapacitors,24 exhibiting excellent capacitive performance. Mohd Ubaidullah and co-workers promoted this research by converting PET to metal–organic frameworks (MOF-5), and then producing nitrogen-doped mesoporous carbon for supercapacitor applications.18 Nitrogen-doped microporous carbon produced by the controlled carbonization of PET using molten salts also demonstrated exceptional CO2 adsorption capacity.16 Hard carbon anode materials with constructed closed-pore structures derived from PET have been applied in sodium-ion batteries.25 In addition, layered porous nitrogen-doped carbon derived from PET has also been applied as an efficient electrocatalytic carrier for hydrogen evolution reaction (HER) under alkaline conditions.19 These studies demonstrate that the trash-to-treasure strategy of recycling plastic waste into energy materials might be a promising way to resolve the issue of white pollution. Although PET-derived carbon materials have been applied in many fields, few studies have been reported for the development of electrocatalysts, especially for the electrosynthesis of hydrogen peroxide. Actually, the pyrolysis of PET disrupts weak oxygen-containing bonds, leading to partial collapse of the carbon skeleton and resulting in low-porosity, low-activity carbon materials.16 Therefore, controlled carbonization of PET to obtain carbon materials with high electrocatalytic activity remains significantly challenging.
Herein, we propose a method to prepare nitrogen–oxygen co-doped porous carbon (NOPC) by carbonization of PET with melamine and MgO. It was found that melamine could react with the intermediates during the PET pyrolysis process to form a stable cross-linked structure. MgO was used as a template agent to modulate the pore structure of the obtained PET-derived carbon. N,O synergistic carbon catalysts with mesoporous structures have been successfully synthesized. The optimized catalyst demonstrated favourable 2e− ORR activity and excellent H2O2 selectivity under acidic conditions. Moreover, the catalyst maintained high selectivity for at least 10 hours in acidic electrolytes. This work provides a promising and sustainable method to convert waste PET to NOPC for the electrochemical production of hydrogen peroxide (Scheme 1).
 | ||
| Scheme 1 Schematic illustration of the conversion process of PET to NOPC for hydrogen peroxide production. | ||
Results and discussion
Material synthesis and characterization
MgO was mixed with PET in different mass ratios, and the mixture was then heated, in order to regulate the additive amount of MgO. The obtained carbon materials after the carbonization process were studied. Fig. S2 (ESI†) shows the SEM images and EDS results of the samples of OPC1-800. After acid leaching, irregular macroporous structures were clearly observed on the surface of the samples (Fig. S2a–c, ESI†). During heat treatment, PET initially fused and MgO nanoparticles dispersed within the melted PET, leading to molecular rearrangement around the MgO particles. At the same time, some MgO nanoparticles agglomerated and their particle size increased. MgO particles initially encapsulated by carbonation products were removed during HCl leaching, resulting in the formation of a macroporous structure. The EDS result of OPC1-800 exhibited a trace amount of Mg (0.84 at%) and Cl (1.08 at%), indicating that most of MgO had been removed by HCl leaching (Fig. S2d, ESI†). The XRD pattern of OPC1-800 showed a broad peak at 2θ = 23.22° owing to the characteristic diffraction peak of the graphite (002) plane, which suggested an amorphous structure in the PET-derived carbon and confirmed the effective removal of MgO again (Fig. S3, ESI†).Nitrogen adsorption–desorption curves and pore size distribution for different PET-derived carbon materials are shown in Fig. S4 (ESI†), which exhibits a typical type-IV isotherm. The low adsorption capacity at a lower relative pressure (P/P0 < 0.1) indicated less microporous structure in the obtained carbon. The significant hysteresis loops indicated that all materials had mesoporous structures (Fig. S4a, ESI†). The specific surface areas for all OPC samples were about 700 m2 g−1 as listed in Table S1 (ESI†), which demonstrated that MgO played an important role in forming the mesoporous structure. The pore size distribution curves indicated that the pore size in the carbon materials was in the range of 3–20 nm (Fig. S4b, ESI†). Especially, the pore size at about 10–20 nm was in accordance with the diameter of MgO nanoparticles. The mesopore volume increased with the addition of MgO and reached the maximum value of 1.22 cm3 g−1 when M/P was 1, and then dropped to 1.09 cm3 g−1 for OPC2-800 (Table S1, ESI†).
The linear scanning voltammogram (LSV) curves of different OPC samples are shown in Fig. S5a (ESI†). It could be observed that all the samples showed ORR catalytic activities, while only OPC2-800 reached a diffusion-limited current plateau. The H2O2 selectivity trends and the number of transferred electrons (n) derived from LSV are displayed in Fig. S5b and c (ESI†). OPC1-800 demonstrated optimal hydrogen peroxide selectivity, reaching 75% at 0 V (Fig. S5b, ESI†), with the number of transferred electrons of about 2.5 (Fig. S5c, ESI†). These results confirmed that an optimal MgO content promoted a well-developed mesoporous structure, facilitating mass transfer and enhancing selective H2O2 production.
To further understand the effect of temperature on the electrochemical properties of carbon materials, the mixture of MgO and PET with a mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was carbonized at different temperatures under a N2 atmosphere. As shown in Fig. S5(d)–(f) (ESI†), the LSV results indicated that the catalytic activity was correlated with carbonization temperature. With the increase of heat treatment temperature, the onset potential (the potential at a current density of 0.01 mA cm−2) increased from 0.1 V to 0.3 V (Fig. S5d, ESI†). Higher carbonization temperature enhanced the activity of the materials. The samples heated at 800 °C showed the best hydrogen peroxide selectivity with the electron transfer number being close to 2 (Fig. S5(e) and (f), ESI†). The present results verified that carbonization of PET with MgO in an appropriate mass ratio might be a promising strategy to prepare porous carbon materials for the electrosynthesis of H2O2. Based on the present results, the mass ratio of MgO/PET was fixed at 1 and the heat treatment temperature was 800 °C.
:1 was carbonized at different temperatures under a N2 atmosphere. As shown in Fig. S5(d)–(f) (ESI†), the LSV results indicated that the catalytic activity was correlated with carbonization temperature. With the increase of heat treatment temperature, the onset potential (the potential at a current density of 0.01 mA cm−2) increased from 0.1 V to 0.3 V (Fig. S5d, ESI†). Higher carbonization temperature enhanced the activity of the materials. The samples heated at 800 °C showed the best hydrogen peroxide selectivity with the electron transfer number being close to 2 (Fig. S5(e) and (f), ESI†). The present results verified that carbonization of PET with MgO in an appropriate mass ratio might be a promising strategy to prepare porous carbon materials for the electrosynthesis of H2O2. Based on the present results, the mass ratio of MgO/PET was fixed at 1 and the heat treatment temperature was 800 °C.
It has been reported that heteroatomic doping in carbon materials could help to improve their selectivity and activity for 2e− ORR.3,26,27 In order to further improve the electrochemical activity of PET-derived carbon materials, nitrogen was introduced using melamine as a nitrogen sources.28 At first, the cross-linking reaction between melamine and PET was studied at a lower heat temperature of 300 °C. Fig. 1(a) shows the photographs of PET, melamine, and the PET–melamine mixture before and after heat treatment. Obviously, PET transformed from dispersed particles into an aggregated solid. Since PET was a kind of thermoplastic resin, it melted when heated at 300 °C and then solidified at room temperature.29,30 However, melamine exhibited no observable changes, indicating its stability at 300 °C. Significantly, the PET–melamine mixture turned dark brown after heat treatment, suggesting that cross-linking reactions had occured.26 TGA results also confirmed the interaction of melamine and PET during the process of pre-treatment (Fig. S6, ESI†). As shown, PET exhibited rapid mass loss between 400 and 520 °C, primarily due to ester bond cleavage and chain rearrangement, while the weight loss of melamine began at about 300 °C, indicating its lower carbonization temperature than PET. The mixtures of PET and melamine started to display significant mass loss at 300 °C, similar to the case of melamine, suggesting accelerated degradation of PET (Fig. S6a, ESI†). The differential thermal analysis (DTA) also demonstrated that the mixture of PET and melamine began to degrade at a lower temperature than PET, showing two main endothermic peaks (Fig. S6b, ESI†). These results indicated that melamine facilitated the breaking of ester bonds and was beneficial for the cross-linking reactions with PET chain segments.
 | ||
| Fig. 1 (a) Photographs of PET, melamine, and the PET–melamine mixture before and after heat treatment at 300 °C, (b) FT-IR spectra of PET-300, melamine-300, PET–melamine-300. | ||
To further verify the cross-linking process between PET and melamine, Fourier-transform infrared (FT-IR) spectroscopy was used to analyse the functional groups of the products after heated at 300 °C (Fig. 1(b)).31 In the infrared spectrum of PET-300, the peak at 3435 cm−1 represented the O–H stretching vibration, and the peaks at 2968 cm−1 and 2904 cm−1 corresponded to the asymmetric and symmetric stretching vibrations of C–H in H–C–H, respectively. The peak at 1728 cm−1 indicated the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in the carboxyl group. Meanwhile, the peaks at 1573, 1508, 1469, and 1411 cm−1 showed the vibration of the aromatic ring. The peak at 1240 cm−1 revealed the (CO)–C stretching vibration, while the peak at 1095 cm−1 corresponded to the C–O vibration.
O in the carboxyl group. Meanwhile, the peaks at 1573, 1508, 1469, and 1411 cm−1 showed the vibration of the aromatic ring. The peak at 1240 cm−1 revealed the (CO)–C stretching vibration, while the peak at 1095 cm−1 corresponded to the C–O vibration.
In the spectrum of melamine-300, the peak at 3334 cm−1 reflected the asymmetric stretching vibration of N–H, while the peak at 3132 cm−1 corresponded to the symmetric stretching vibration of N–H. The peak at 1651 cm−1 represented the CN stretching vibration, the peaks at 1184 and 1024 cm−1 corresponded to the C–N stretching vibration, and the peak at 813 cm−1 belonged to the N–H out-of-plane bending vibration.16
For the spectrum of PET–melamine-300, the peak at 1674 cm−1 represented the CO bond of the amide group, the peak at 1381 cm−1 represented the C–N bond of the amide group, and the peak at 1261 cm−1 belonged to the stretching vibration of amide III.32 The formation of amide groups indicated that the carboxyl groups that existed in the degradation products of PET had reacted with the amino groups in melamine through an amide reaction.33 These results confirmed that melamine could introduce nitrogen into PET-derived carbon by cross-linking with the PET degradation products.
XRD patterns of NOPC samples after HCl/HNO3 washing revealed two broad peaks at 2θ = 23.22° and 43.8° (Fig. S3, ESI†), corresponding to the (002) and (100) diffraction planes of graphitic carbon, respectively.34 TEM analysis was conducted to investigate the micromorphology of NOPC. Fig. 2(a) and (b), and Fig. S7 (ESI†) show the obvious porous structure of PET-derived carbon. Moreover, the carbon exhibited a low degree of graphitization. High-angle annular dark field-scanning transmission microscopy (HAADF-STEM) and EDS analysis confirmed the plenty of pores and the uniform distribution of C, N, and O elements within the materials (Fig. 2(c)–(f)). N2 adsorption–desorption isotherms for NOPC samples are shown in Fig. 3(a). NOPC-0.05 and NOPC-0.1 displayed similar curve shapes to those without melamine addition (OPC), showing hysteresis loops at a relative pressure of 0.4 < P/P0 < 1.0 (Fig. 3(a)). On the other hand, with the increase of melamine, NOPC-0.3 and NOPC-0.5 exhibited a larger N2 uptake at a low relative pressure (P/P0 < 0.1) and almost no hysteresis loops, which indicated the presence of more micropores and fewer mesopores. The pore size distribution curves confirmed this change (Fig. 3b). More pores with a size of 3–5 nm formed in the samples of NOPC-0.3 and NOPC-0.5, while there were almost no pores in the range of 10–30 nm in these samples. The reason for this might be that more melamine decomposed without cross-linking with PET degradation products when a higher mass ratio of melamine was applied. Moreover, the specific surface areas of NOPC-0.3 and NOPC-0.5 in Table S1 (ESI†) were 718.8 m2 g−1 and 696.5 m2 g−1, respectively, which were much higher than those of NOPC-0.05 (409.26 m2 g−1) and NOPC-0.1 (389.77 m2 g−1), confirming more microporous structures again. The average pore size concluded in Table S1 (ESI†) indicated that in the absence of melamine, the pyrolysis of PET with MgO resulted in molecular chain breakage and rearrangement, with pore formation primarily attributed to the removal of MgO. Consequently, OPCs exhibited a relatively stable average pore size. However, with the addition of a small amount of melamine, the average pore size increased, likely due to melamine acting as a chain extender, thereby altering the cleavage and rearrangement of PET molecular chains. With the further increase of melamine content, the pore size decreased. The reason for this might be that more micropores formed, making the average pore size decrease. Additionally, the denser crosslinked network reduced the average pore size.
 | ||
| Fig. 2 (a) TEM and (b) HRTEM images of NOPC-0.1. (c) HAADF-STEM image and STEM-EDS mapping profiles for (d) C, (e) N, and (f) O of NOPC-0.1. | ||
 | ||
| Fig. 3 (a) N2 adsorption–desorption isotherms, (b) pore size distribution, (c) Raman spectra, (d) FT-IR spectra corresponding to different NOPC samples. | ||
In Raman spectroscopy (Fig. 3(c)), the peaks at 1350 cm−1 and 1576 cm−1 corresponded to the D-band and G-band, respectively.16,35 The D-band was associated with the introduction of sp3–C bonds and structural defects at edges or basal planes. The G-band was related to graphitic carbon.36 The intensity ratio of the D-band to the G-band (ID/IG) was usually used to describe the relative defect content in carbon materials.13 The ID/IG values for NOPC-0.05, NOPC-0.1, NOPC-0.3, and NOPC-0.5 were 1.41, 1.45, 1.37, and 1.2, respectively, indicating that the defect content dropped with the increase of melamine. These abundant defects were associated with sp3–C and edge sites in mesoporous carbon materials,7 which could act as active sites for the adsorption or reaction during the electrocatalytic process.
The infrared spectrum analysis (FT-IR) showed the peak at 1571 cm−1 in NOPC-0.1, which was attributed to the N–H stretching vibration of the amide group (Fig. 3(d)). The peak at 1205 cm−1 was assigned to the amide III band, while the peak at 1095 cm−1 corresponded to the C–O stretching vibration of the amide group. The peaks at 1396 cm−1 and 3426 cm−1 were associated with C–N–C symmetric stretching and N–H asymmetric stretching, respectively. These functional groups clearly demonstrated the presence of amide groups in the melamine-doped carbonized products. The appearance of amide groups further suggested that the N and O active sites were tightly bonded in the PET-derived carbon materials.37
XPS was used to characterize the surface elements and functional groups, which showed the presence of C, N, and O elements in all NOPC samples (Fig. S8, ESI†). The content of nitrogen increased with the increase of melamine addition (Table S2, ESI†), demonstrating that nitrogen originated from melamine and the effective cross-linking reaction between melamine and PET. The N 1s spectra of the NOPC samples were deconvoluted into four peaks (Fig. 4): pyridinic N (398.48 eV), pyrrolic N (400.11 eV), graphitic N (401.03 eV), and oxidized N (403.53 eV),38,39 and their relative atomic percentages are concluded in Table S3 (ESI†). NOPC-0.1 exhibited a high percent of pyrrolic N, which was reported to be responsible for the optimal selectivity for 2e− ORR.40 Generally, more nitrogen doping would contribute to the 4e− ORR process.41,42 Compared to OPC1-800, the oxygen content also increased for NOPC samples, resulting from the acid leaching with HNO3. The high-resolution C 1s spectra of the NOPC samples shown in Fig. S9 (ESI†) could be divided into four distinct peaks: graphite carbon (284.80 eV), CN (286.00 eV), CO (288.13 eV), and π → π* (290.73 eV).13,16 NOPC-0.1 was found to contain the highest amount of CO (Table S4, ESI†). It has been reported that CO bonds from carboxyl and carbonyl groups were considered as the key active sites for 2e− ORR.43 In addition, the high electronegativity of nitrogen atoms caused charge redistribution, which changed the adsorption strength of intermediates on carbon atoms and ultimately affected 2e− ORR performance.39,44,45
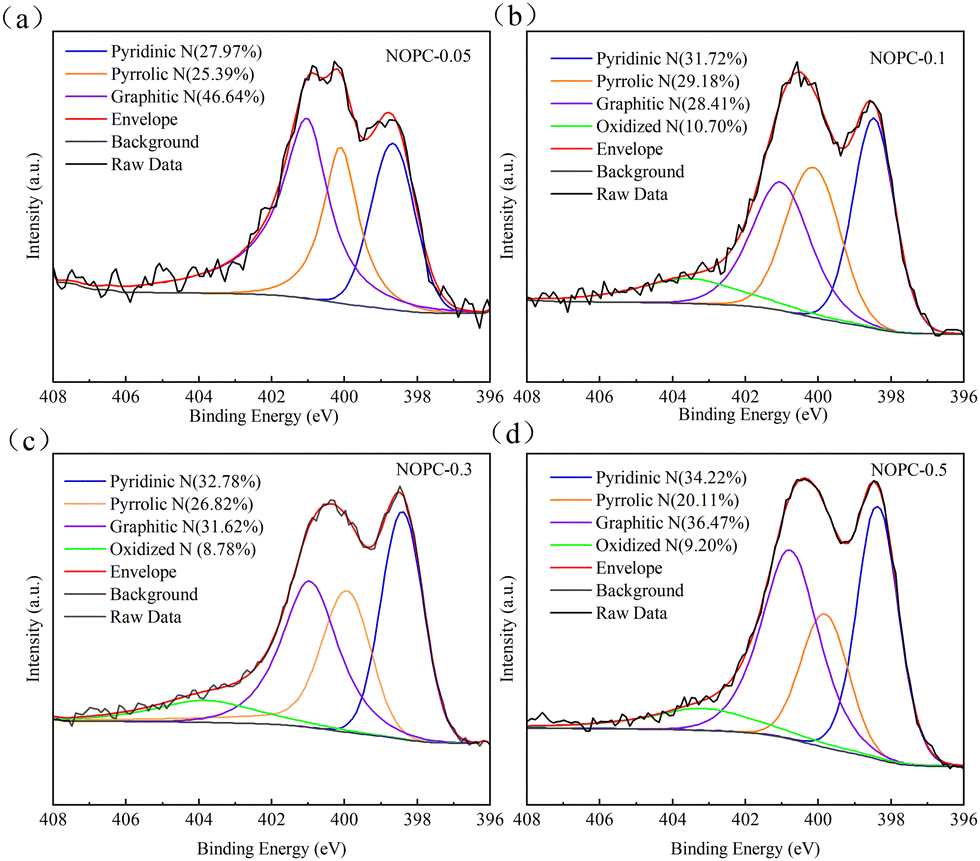 | ||
| Fig. 4 High-resolution N 1s XPS spectra of different NOPC samples: (a) NOPC-0.05, (b) NOPC-0.1, (c) NOPC-0.3, and (d) NOPC-0.5. | ||
Electrochemical characterization
To investigate the electrochemical activity of the catalysts, cyclic voltammetry (CV) curves were measured under N2 and O2 atmospheres.46 No significant peaks were observed under N2-saturated conditions for all catalysts. However, distinct reduction peaks were observed in the O2-saturated electrolyte, indicating the active oxygen reduction capabilities of all catalysts (Fig. S10, ESI†). The ORR catalytic activities of different catalysts were evaluated using linear sweep voltammetry (LSV). As displayed in Fig. 5(a), the LSV curves for all NOPC samples shifted positively to higher potential compared to that of OPC1-800, indicating that the nitrogen doping in carbon materials facilitated enhancement of the ORR catalytic activity. With the increase of melamine amount, an increased oxygen reduction current was observed and the onset potential shifted to higher potentials. The onset potential of OPC1-800 was measured to be 0.259 V, while that of NOPC-0.05, 0.1, 0.3, and 0.5 was determined to be 0.396 V, 0.401 V, 0.444 V, and 0.472 V, respectively. These results indicated that the activities of the NOPC catalysts were significantly enhanced after doping with a small amount of nitrogen.9 | ||
| Fig. 5 Electrochemical test of the catalysts in 0.5 M H2SO4. (a) RRDE polarization curves of different catalysts. (b) H2O2 selectivity and (c) electron transfer number of different catalysts. (d) Tafel slope of different catalysts. (e) Capacitive current density at 0.59 V as a function of scan rate. (f) Stability test of NOPC-0.1 at a fixed disk potential of 0.3 V (vs. RHE). | ||
The H2O2 selectivity of different samples is shown in Fig. 5(b). As shown, within the potential range of 0–0.3 V, the H2O2 selectivity of NOPC samples was higher than that of OPC1-800. This demonstrated that nitrogen incorporation resulted in an enhanced oxygen reduction activity coupled with improved H2O2 selectivity. Among the NOPC catalysts, NOPC-0.1 exhibited the highest H2O2 selectivity of 80–84.7% in the potential range of 0–0.3 V. Moreover, the NOPC-0.1 catalyst showed a lower electron transfer number of about 2.3, which was also confirmed by the results calculated from the K–L equation (Fig. 5(c) and Fig. S11(a) and (b), ESI†). Catalysts with higher melamine doping (NOPC-0.3, NOPC-0.5) exhibited lower H2O2 selectivity compared to NOPC-0.1. This suggested that moderate N doping helped to improve the H2O2 selectivity, but excessive N content facilitated the ORR by the 4e− pathway with the formation of water. As reported, excessive N doping led to the aggregation of isolated active sites, which contributed to the breaking of the O–O bond and favoured the 4e− ORR pathway. However, appropriate N doping limited the number of active sites, favouring the 2e− ORR.9
Notably, there was no significant positive relationship between H2O2 selectivity and specific surface area. For NOPC-0.1, it had a large average pore size (13.77 nm) and the lowest specific surface area (389.77 m2 g−1), but it exhibited the highest selectivity (84.77%). However, a positive correlation between defect levels and selectivity was found. NOPC-0.1 with the highest ID/IG value showed the best electrocatalytic activity for H2O2 production, since the formation of defects provided active sites for adsorption or reactions, facilitating OOH* accumulation and increasing H2O2 selectivity.36 Consequently, for PET-derived N,O-doped carbon materials, comprehensive effects of microstructure and composition on the catalytic activity and selectivity for the 2e− ORR catalysts should be considered.
The Tafel slopes of the catalysts are shown in Fig. 5(d).47 The Tafel slopes of NOPC-0.05, NOPC-0.1, NOPC-0.3, and NOPC-0.5 were 200.04 mV dec−1, 189.26 mV dec−1, 248.32 mV dec−1, and 286.13 mV dec−1, respectively. It was noteworthy that NOPC-0.1 had the lowest Tafel slope, indicating the optimum electrical conductivity and the lowest charge transfer resistance.3,13
The electrochemical double-layer capacitance (Cdl) was often used to characterize the electrochemical specific surface area.13 The Cdl values for NOPC-0.05, NOPC-0.1, NOPC-0.3, and NOPC-0.5 were measured from CV curves at different scanning rates (Fig. S12, ESI†), which were 15.04 mF cm−2, 42.17 mF cm−2, 26.44 mF cm−2, and 14.44 mF cm−2, respectively (Fig. 5(e)). Generally, catalysts exhibiting high electrochemical double-layer capacitance values possessed more active sites exposed for O2 adsorption, leading to enhancement of catalytic activity.48 Electrochemical capacitance evaluation of NOPC indicated a linear correlation between the double-layer capacitance and the ring current at 0 V (Fig. S13, ESI†). This suggested that NOPC-0.1 with a more mesoporous structure could favour more active sites exposed to the electrolyte and facilitate the 2e− oxygen reduction reaction for hydrogen peroxide generation. The stability of the catalysts under acidic conditions was evaluated by the amperometric i–t curve test.49 The H2O2 ring current of NOPC-0.1 remained stable during 10 h of operation, indicating its good electrochemical stability and promising potential for practical application (Fig. 5(f)).
H2O2 production demonstration
The optimized PET-derived carbon materials, NOPC-0.1, were evaluated for H2O2 production at different applied potentials in the bulk electrolyte. Based on the standard concentration curve of Ce4+ (Fig. S14, ESI†), the accumulated amount of H2O2 concentration normalized by catalyst loading amount versus reaction time in O2-saturated 0.5 M H2SO4 is shown in Fig. 6(a). It could be observed that the H2O2 production amount increased as the potential shifted negatively in the potential range of −0.1–0.3 V. The plots of accumulated H2O2 concentration reflected an almost linear relationship between H2O2 production amount and reaction time. At −0.1 V, the NOPC-0.1 electrocatalyst achieved a high H2O2 generation rate of 261.23 mmol gcatalyst−1 h−1, comparable with those catalysts reported recently.9 As shown in Fig. 6(b), the faradaic efficiency of NOPC-0.1 was about 86.45% under a potential of −0.1 V, higher than that under the potential of 0.1 and 0.3 V. The faradaic efficiency decreased firstly and then stabilized relatively at about 76.38% and 65.27% when the potential was 0.1 V and 0.3 V, respectively. As anticipated, NOPC-0.1 exhibited a favourable faradaic efficiency for H2O2 production. As summarized in Table S5 (ESI†), the present NOPC-0.1 exhibited comparable or even better performance than those previously reported catalysts, further confirming the excellent electrochemical properties of NOPC-0.1. The practical demonstration suggested that the PET-derived porous carbon materials might be a promising catalyst for the electrochemical synthesis of H2O2. | ||
| Fig. 6 (a) H2O2 production amount normalized by catalyst loading amount over the reaction time at different voltages, (b) the corresponding faradaic efficiencies (FE%). | ||
Conclusions
In conclusion, a N,O co-doped porous carbon material was successfully synthesized, which was achieved through the cross-linking carbonization strategy of PET with melamine and MgO. The prepared NOPC exhibited an abundant mesoporous structure with N doping. The cross-linking carbonization process tightly bound the active sites of the catalysts through the formation of a large number of amide groups consisting of nitrogen and oxygen elements. The well-developed NOPC-0.1 catalyst showed a high H2O2 selectivity of 84.77% and long-time stability of 10 hours. Moreover, a high-yield electrosynthesis of H2O2 was achieved, with a production rate of 261.23 mmol gcatalyst−1 h−1 and an acceptable faradaic efficiency of 86.45%. Additionally, this study provided a promising approach for converting low-cost waste polyester into high-value carbon-based catalyst materials, as well advanced the development of waste-to-value strategies.Author contributions
M. Luo designed experiments, synthesized materials, performed electrochemical characterization, and wrote the manuscript. C. Wang contributed to data collection, analysis, and manuscript review. S. Song organized data and reviewed the manuscript. M. Tang assisted with data organization. X. Wang managed the project, reviewed and edited the manuscript. M. Wu reviewed the manuscript.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was funded by Xizang Autonomous Region Science and Technology Plan: XZ202402ZY0014, XZ202401ZY0067.References
- C. Ni, Z. Fan, N. Deng and X. Huang, Inhibiting Joule Heating to Enhance the Hydrogen Peroxide Electrosynthesis Efficiency at Industrial Level Current Density, ACS Appl. Energy Mater., 2024, 7, 3116–3124 CrossRef CAS.
- K. S. Exner, Steering Selectivity in the Four-Electron and Two-Electron Oxygen Reduction Reactions: On the Importance of the Volcano Slope, ACS Phys. Chem. Au, 2023, 3, 190–198 CrossRef CAS PubMed.
- M. Qin, S. Fan, L. Wang, G. Gan, X. Wang, J. Cheng, Z. Hao and X. Li, Oxygen and nitrogen co-doped ordered mesoporous carbon materials enhanced the electrochemical selectivity of O2 reduction to H2O2, J. Colloid Interface Sci., 2020, 562, 540–549 CrossRef CAS PubMed.
- H. Li, P. Wen, D. S. Itanze, Z. D. Hood, S. Adhikari, C. Lu, X. Ma, C. Dun, L. Jiang, D. L. Carroll, Y. Qiu and S. M. Geyer, Scalable neutral H2O2 electrosynthesis by platinum diphosphide nanocrystals by regulating oxygen reduction reaction pathways, Nat. Commun., 2020, 11, 3928 CrossRef CAS PubMed.
- X. Huang, M. Song, J. Zhang, T. Shen, G. Luo and D. Wang, Recent Advances of Electrocatalyst and Cell Design for Hydrogen Peroxide Production, Nano-Micro Lett., 2023, 15, 86 CrossRef CAS PubMed.
- G.-L. Chai, Z. Hou, T. Ikeda and K. Terakura, Two-Electron Oxygen Reduction on Carbon Materials Catalysts: Mechanisms and Active Sites, J. Phys. Chem. C, 2017, 121, 14524–14533 CrossRef CAS.
- S. Chen, Z. Chen, S. Siahrostami, T. R. Kim, D. Nordlund, D. Sokaras, S. Nowak, J. W. F. To, D. Higgins, R. Sinclair, J. K. Nørskov, T. F. Jaramillo and Z. Bao, Defective Carbon-Based Materials for the Electrochemical Synthesis of Hydrogen Peroxide, ACS Sustainable Chem. Eng., 2017, 6, 311–317 CrossRef.
- H. He, S. Liu, Y. Liu, L. Zhou, H. Wen, R. Shen, H. Zhang, X. Guo, J. Jiang and B. Li, Review and perspectives on carbon-based electrocatalysts for the production of H2O2via two-electron oxygen reduction, Green Chem., 2023, 25, 9501–9542 RSC.
- Y. Sun, I. Sinev, W. Ju, A. Bergmann, S. Dresp, S. Kühl, C. Spöri, H. Schmies, H. Wang, D. Bernsmeier, B. Paul, R. Schmack, R. Kraehnert, B. Roldan Cuenya and P. Strasser, Efficient Electrochemical Hydrogen Peroxide Production from Molecular Oxygen on Nitrogen-Doped Mesoporous Carbon Catalysts, ACS Catal., 2018, 8, 2844–2856 CrossRef CAS.
- Y. Xia, X. Zhao, C. Xia, Z. Y. Wu, P. Zhu, J. Y. T. Kim, X. Bai, G. Gao, Y. Hu, J. Zhong, Y. Liu and H. Wang, Highly active and selective oxygen reduction to H2O2 on boron-doped carbon for high production rates, Nat. Commun., 2021, 12, 4225 CrossRef CAS PubMed.
- W. Ju, A. Bagger, G. P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Understanding activity and selectivity of metal-nitrogen-doped carbon catalysts for electrochemical reduction of CO2, Nat. Commun., 2017, 8, 944 Search PubMed.
- I. Masood ul Hasan, L. Peng, J. Mao, R. He, Y. Wang, J. Fu, N. Xu and J. Qiao, Carbon-based metal-free catalysts for electrochemical CO2 reduction: Activity, selectivity, and stability, Carbon Energy, 2020, 3, 24–49 Search PubMed.
- L. Sun, L. Sun, L. Huo and H. Zhao, Promotion of the Efficient Electrocatalytic Production of H2O2 by N,O- Co-Doped Porous Carbon, Nanomaterials, 2023, 13, 1188 CrossRef CAS PubMed.
- Z. Lu, G. Chen, S. Siahrostami, Z. Chen, K. Liu, J. Xie, L. Liao, T. Wu, D. Lin, Y. Liu, T. F. Jaramillo, J. K. Nørskov and Y. Cui, High-efficiency oxygen reduction to hydrogen peroxide catalysed by oxidized carbon materials, Nat. Catal., 2018, 1, 156–162 CAS.
- J. Park, Y. Nabae, T. Hayakawa and M.-A. Kakimoto, Highly Selective Two-Electron Oxygen Reduction Catalyzed by Mesoporous Nitrogen-Doped Carbon, ACS Catal., 2014, 4, 3749–3754 CrossRef CAS.
- C. Song, B. Zhang, L. Hao, J. Min, N. Liu, R. Niu, J. Gong and T. Tang, Converting poly(ethylene terephthalate) waste into N-doped porous carbon as CO2 adsorbent and solar steam generator, Green Energy Environ., 2022, 7, 411–422 CrossRef CAS.
- C.-Y. Wang, H.-Y. Chu and C.-C. Wang, Converting waste PET plastics to high value-added MOFs-based functional materials: A state of the art review, Coord. Chem. Rev., 2024, 518, 216106 CrossRef CAS.
- M. Ubaidullah, A. M. Al-Enizi, T. Ahamad, S. F. Shaikh, M. A. Al-Abdrabalnabi, M. S. Samdani, D. Kumar, M. A. Alam and M. Khan, Fabrication of highly porous N-doped mesoporous carbon using waste polyethylene terephthalate bottle-based MOF-5 for high performance supercapacitor, J. Energy Storage, 2021, 33, 102125 CrossRef.
- C. Juan, B. Lan, C. Zhao, H. Zhang, D. Li and F. Zhang, From waste plastics to layered porous nitrogen-doped carbon materials with excellent HER performance, Chem. Commun., 2023, 59, 6187–6190 RSC.
- H. Guo, L. Zhou, K. Huang, Y. Li, W. Hou, H. Liao, C. Lian, S. Yang, D. Wu, Z. Lei, Z. Liu and L. Wang, Nitrogen-Rich Carbon Dot-Mediated n → π* Electronic Transition in Carbon Nitride for Superior Photocatalytic Hydrogen Peroxide Production, Adv. Funct. Mater., 2024, 34, 2402650 CrossRef CAS.
- J. Cao, H. Wang, Y. Zhao, Y. Liu, Q. Wu, H. Huang, M. Shao, Y. Liu and Z. Kang, Phosphorus-doped porous carbon nitride for efficient sole production of hydrogen peroxide via photocatalytic water splitting with a two-channel pathway, J. Mater. Chem. A, 2020, 8, 3701–3707 RSC.
- V. Perazzolo, C. Durante, R. Pilot, A. Paduano, J. Zheng, G. A. Rizzi, A. Martucci, G. Granozzi and A. Gennaro, Nitrogen and sulfur doped mesoporous carbon as metal-free electrocatalysts for the in situ production of hydrogen peroxide, Carbon, 2015, 95, 949–963 CrossRef CAS.
- Y. Wu, A. Muthukrishnan, S. Nagata and Y. Nabae, Kinetic Understanding of the Reduction of Oxygen to Hydrogen Peroxide over an N-Doped Carbon Electrocatalyst, J. Phys. Chem. C, 2019, 123, 4590–4596 CrossRef CAS.
- Y. Wen, K. Kierzek, J. Min, X. Chen, J. Gong, R. Niu, X. Wen, J. Azadmanjiri, E. Mijowska and T. Tang, Porous carbon nanosheet with high surface area derived from waste poly(ethylene terephthalate) for supercapacitor applications, J. Appl. Polym. Sci., 2019, 137, 48338 Search PubMed.
- C. Qiu, A. Li, D. Qiu, Y. Wu, Z. Jiang, J. Zhang, J. Xiao, R. Yuan, Z. Jiang, X. Liu, X. Chen and H. Song, One-Step Construction of Closed Pores Enabling High Plateau Capacity Hard Carbon Anodes for Sodium-Ion Batteries: Closed-Pore Formation and Energy Storage Mechanisms, ACS Nano, 2024, 18, 11941–11954 CAS.
- E. T. Zanoni, R. Benavides, L. Da Silva, M. D. Morales-Acosta and D. Morales-Acosta, N-Doped Mesoporous Carbon from Melamine-Modified Resorcinol-Formaldehyde Polymer Resin as Catalytic Support for Electrochemical Investigation of Formic acid Oxidation Reaction, J. Inorg. Organomet. Polym. Mater., 2023, 34, 1977–1987 Search PubMed.
- E. Chen, M. Bevilacqua, C. Tavagnacco, T. Montini, C.-M. Yang and P. Fornasiero, High surface area N/O co-doped carbon materials: Selective electrocatalysts for O2 reduction to H2O2, Catal. Today, 2020, 356, 132–140 CAS.
- Y. Wang, H. Xuan, G. Lin, F. Wang, Z. Chen and X. Dong, A melamine-assisted chemical blowing synthesis of N-doped activated carbon sheets for supercapacitor application, J. Power Sources, 2016, 319, 262–270 CAS.
- M. H. Ghasemi, N. Neekzad, F. B. Ajdari, E. Kowsari and S. Ramakrishna, Mechanistic aspects of poly(ethylene terephthalate) recycling-toward enabling high quality sustainability decisions in waste management, Environ. Sci. Pollut. Res. Int., 2021, 28, 43074–43101 CAS.
- F. Cao, L. Wang, R. Zheng, L. Guo, Y. Chen and X. Qian, Research and progress of chemical depolymerization of waste PET and high-value application of its depolymerization products, RSC Adv., 2022, 12, 31564–31576 CAS.
- A. B. D. Nandiyanto, R. Ragadhita and M. Fiandini, Interpretation of Fourier Transform Infrared Spectra (FTIR): A Practical Approach in the Polymer/Plastic Thermal Decomposition, Indones. J. Sci. Technol., 2022, 8, 113–126 Search PubMed.
- Y. Ji, X. Yang, Z. Ji, L. Zhu, N. Ma, D. Chen, X. Jia, J. Tang and Y. Cao, DFT-Calculated IR Spectrum Amide I, II, and III Band Contributions of N-Methylacetamide Fine Components, ACS Omega, 2020, 5, 8572–8578 CAS.
- I. V. Komarov, A. Y. Ishchenko, A. Hovtvianitsa, V. Stepanenko, S. Kharchenko, A. D. Bond and A. J. Kirby, Fast Amide Bond Cleavage Assisted by a Secondary Amino and a Carboxyl Group-A Model for yet Unknown Peptidases?, Molecules, 2019, 24, 572 Search PubMed.
- Y. Quan, C. Hu, P. Feng, Y. Song, K. Liang, X. Jian and J. Xu, Modulation of Free Carbon Structures in Polysiloxane-Derived Ceramics for Anode Materials in Lithium-Ion Batteries, Molecules, 2024, 29, 4461 CAS.
- Z. Wu and X.-b Zhang, N,O-codoped porous carbon nanosheets for capacitors with ultra-high capacitance, Sci. China Mater., 2016, 59, 547–557 CAS.
- Y. Liu, X. Quan, X. Fan, H. Wang and S. Chen, High-yield electrosynthesis of hydrogen peroxide from oxygen reduction by hierarchically porous carbon, Angew. Chem., Int. Ed., 2015, 54, 6837–6841 CAS.
- W. Si, Q. Xie, Y. Shen and Z. Wang, Melamine–Formaldehyde Resin Derived Carbon Catalysts with Abundant Intrinsic Defects to Afford Superior Oxygen Reduction Activity, Catal. Lett., 2023, 154, 1819–1827 Search PubMed.
- S. N. Faisal, E. Haque, N. Noorbehesht, W. Zhang, A. T. Harris, T. L. Church and A. I. Minett, Pyridinic and graphitic nitrogen-rich graphene for high-performance supercapacitors and metal-free bifunctional electrocatalysts for ORR and OER, RSC Adv., 2017, 7, 17950–17958 CAS.
- L. Yang, Y.-P. Zhu, A.-J. Wang, X. Weng and J.-J. Feng, Simple pyrolysis of graphene-wrapped PtNi nanoparticles supported on hierarchically N-doped porous carbon for sensitive detection of carbendazim, Microchim. Acta, 2023, 190, 211 CAS.
- L. Li, C. Tang, Y. Zheng, B. Xia, X. Zhou, H. Xu and S. Z. Qiao, Tailoring Selectivity of Electrochemical Hydrogen Peroxide Generation by Tunable Pyrrolic-Nitrogen-Carbon, Adv. Energy Mater., 2020, 10, 2000789 CAS.
- Y. Pang, H. Xie, Y. Sun, M.-M. Titirici and G.-L. Chai, Electrochemical oxygen reduction for H2O2 production: catalysts, pH effects and mechanisms, J. Mater. Chem. A, 2020, 8, 24996–25016 CAS.
- D. Wang, S. Li, X. Zhang, B. Feng, Y. Pei, Y. Zhu, W. Xu, Z.-H. Li, M. Qiao and B. Zong, Pyrolyzed polydopamine-modified carbon black for selective and durable electrocatalytic oxygen reduction to hydrogen peroxide in acidic medium, Appl. Catal., B, 2022, 305, 121036 CrossRef CAS.
- W. Zhou, L. Xie, J. Gao, R. Nazari, H. Zhao, X. Meng, F. Sun, G. Zhao and J. Ma, Selective H2O2 electrosynthesis by O-doped and transition-metal-O-doped carbon cathodes via O2 electroreduction: A critical review, Chem. Eng. J., 2021, 410, 128368 CrossRef CAS.
- B. Li, L. Zhang, J. Zhang and Y. Su, Recent Insight in Transition Metal Anchored on Nitrogen-Doped Carbon Catalysts: Preparation and Catalysis Application, Electrochem, 2022, 3, 520–537 CrossRef CAS.
- L. M. Ombaka, P. G. Ndungu, J. Kibet and V. O. Nyamori, The effect of pyridinic- and pyrrolic-nitrogen in nitrogen-doped carbon nanotubes used as support for Pd-catalyzed nitroarene reduction: an experimental and theoretical study, J. Mater. Sci., 2017, 52, 10751–10765 CrossRef CAS.
- G. Li, L. Anderson, Y. Chen, M. Pan and P.-Y. Abel Chuang, New insights into evaluating catalyst activity and stability for oxygen evolution reactions in alkaline media, Sustainable Energy Fuels, 2018, 2, 237–251 RSC.
- Y. Wu, A. Muthukrishnan, S. Nagata and Y. Nabae, Tafel Slope Analysis from Inherent Rate Constants for Oxygen Reduction Reaction Over N-doped Carbon and Fe–N-doped Carbon Electrocatalysts, Catal. Surv. Asia, 2022, 27, 84–94 CrossRef.
- Z. Xie, W. Qu, E. A. Fisher, J. Fahlman, K. Asazawa, T. Hayashi, H. Shirataki and H. Murase, Capacitance Determination for the Evaluation of Electrochemically Active Surface Area in a Catalyst Layer of NiFe-Layered Double Hydroxides for Anion Exchange Membrane Water Electrolyser, Materials, 2024, 17, 556 CAS.
- S. S. Kocha, K. Shinozaki, J. W. Zack, D. J. Myers, N. N. Kariuki, T. Nowicki, V. Stamenkovic, Y. Kang, D. Li and D. Papageorgopoulos, Best Practices and Testing Protocols for Benchmarking ORR Activities of Fuel Cell Electrocatalysts Using Rotating Disk Electrode, Electrocatalysis, 2017, 8, 366–374 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, computational details and figures. See DOI: https://doi.org/10.1039/d5ma00111k |
| This journal is © The Royal Society of Chemistry 2025 |