
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic performance of Cu–Co/metakaolinite: role of textural and structural properties in the partial oxidation of glycerol†
Laura C.
Paredes-Quevedo
ab,
Catherine
Batiot-Dupeyrat
b and
Mauricio
Velasquez
 *a
*a
aUniversidad Nacional de Colombia-Bogotá, Facultad de Ciencias, Departamento de Química, Estado Sólido y Catálisis Ambiental (ESCA), carrera 30 No. 45-03, Bogotá, Código postal 111321, Colombia. E-mail: lmvelasquezma@unal.edu.co
bUniversité de Poitiers, Institut de Chimie des Milieux et Matériaux de Poitiers (IC2MP), ENSIP-bâtiment B1 rue Marcel Doré, TSA41105, 86073, Poitiers Cedex 9, France
First published on 22nd April 2025
Abstract
Cu–Co catalysts were synthesized using microporous and mesoporous metakaolinite and calcined at 400, 600, and 800 °C. The effect of calcination temperature on the textural and structural properties was investigated. Characterization of the solids revealed the formation of a CuCo2O4 spinel structure when the catalyst was calcined at 400 °C. Increasing the calcination temperature led to structural modifications in the metallic phase due to the interaction of Cu with Al atoms in the support, which favored the formation of another spinel phase (CuAl2O4). Additionally, thermal treatment reduced the specific surface area and pore volume, suggesting an enhanced dispersion of the metal phase. The catalysts were evaluated for their performance in glycerol oxidation, using H2O2 as the oxidizing agent in a 0.54 M aqueous glycerol solution at 80 °C under atmospheric pressure. Although all catalysts demonstrated activity, a distinct trend was observed with calcination temperature. The catalyst calcined at 800 °C exhibited the highest activity, achieving complete conversion after 4 hours of reaction and a selectivity of 92% toward dihydroxyacetone. The structural modification of the catalyst at elevated calcination temperatures also resulted in reduced leaching of Cu and Co, indicating improved stability. X-ray Photoelectron Spectroscopy (XPS) analysis revealed a surface enrichment in tetrahedral copper attributed to the formation of the spinel CuAl2O4 at high calcination temperature. The beneficial role of copper associated with cobalt was evidenced experimentally and DRIFT analysis indicates that metakaolinite plays a fundamental role in the activation of the secondary OH in glycerol.
Introduction
The circular economy emphasizes reducing waste and promoting the sustainable use of natural resources through designing, reusing, and recycling processes to create other valuable products. Transforming biomass-derived feedstocks into high-value building blocks for fine and polymer chemistry or essential commodity products has garnered significant interest. As a result, policies supporting renewable energy and biobased products have heightened the importance of biomass feedstocks in the European Union.1 Among the biomass feedstocks, glycerol, a by-product of the transesterification reaction for biodiesel production,2 has attracted growing interest from the catalytic community.The rising demand for biodiesel has generated an oversupply of glycerol, leading to economic, industrial, and environmental challenges related to its disposal. To address this, upcycling glycerol through catalytic transformations has become necessary to enhance the economic viability of biodiesel production. Oxidation in aqueous media is one of the most extensively studied pathways to produce promising compounds that serve as essential intermediates or valuable fine chemicals.
However, these chemicals are not yet widely applied at a large scale due to high production costs and time-intensive processes.3 For example, dihydroxyacetone is used as a synthon in organic chemistry and as a tanning agent in cosmetics, while glyceric acid is an intermediate in amino acid synthesis, and glyceraldehyde is used in polyester and adhesive production. These applications highlight the importance of glycerol oxidation and its potential industrial benefits.4
Recently, researchers have concentrated on glycerol oxidation via conventional catalytic processes, employing H2O2 as an oxygen source.5–8 Copper–cobalt-based catalysts have been extensively studied for this purpose due to their high activity, selectivity, and cost-effectiveness.5,6,9–14 However, the extent of leaching of Cu and Co during the reaction is rarely addressed in these studies. Despite their catalytic efficiency under oxidizing conditions, copper and cobalt exhibit limited stability in acidic media, as indicated by Pourbaix diagrams.15,16 Therefore, this limitation underscores the need to develop highly stable heterogeneous catalysts to prevent the deactivation by leaching.17
The performance of heterogeneous catalysts can be improved by enhancing the interaction between the active phase and the support material. Aluminosilicates are particularly attractive supports due to their capacity to disperse active phases, their acid–base properties, high thermal stability, and affordability.18,19 Traditional catalyst preparation methods, such as impregnation, enable the distribution of the metallic phase on the surface but can reduce surface area.20 Moreover, controlling the calcination temperature of the freshly impregnated solids is critical, as high-temperature sintering can adversely affect the distribution of the metallic phase.18
Calcination temperature also influences the structural properties of catalysts and their metal–support interactions.21 For instance, strong copper–support interactions may induce the formation of new crystalline phases, such as CuAl2O4 or aluminates.17,20,22–25 A study has shown that increasing calcination temperatures reduces copper leaching but can lower catalytic activity due to insufficient Cu in the homogeneous phase to initiate the reaction.17 Conversely, using porous supports can enhance metal dispersion, reduce sintering at elevated temperatures, and strengthen metal–support interactions, thereby improving overall catalyst performance.
Natural and modified clay-type aluminosilicates, like metakaolinite, have long been studied as supports in heterogeneous catalysis. Natural kaolinite is a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 type clay mineral;26 and its modification through acid and thermal treatment significantly enhances its surface area by increasing its micro- and mesoporosity, which improves active phase dispersion.27 In prior work, CuCo2O4 supported on dealuminized metakaolinite (calcined at 400 °C) exhibited high activity and selectivity for glycerol oxidation. However, the catalyst suffered from significant metal leaching after extended reaction times.6 Thus, strategies to enhance the stability and catalytic performance of this material remain a critical area of investigation. This study aims to evaluate the relationship between the textural and structural changes of Cu–Co particles supported on porous metakaolinite and their catalytic performance in partial glycerol oxidation. Catalysts with low metal loading (∼5 wt%) were synthesized via wet impregnation and calcined at 400, 600, and 800 °C. Their performance was assessed under atmospheric pressure, using hydrogen peroxide as the oxidizing agent.
:1 type clay mineral;26 and its modification through acid and thermal treatment significantly enhances its surface area by increasing its micro- and mesoporosity, which improves active phase dispersion.27 In prior work, CuCo2O4 supported on dealuminized metakaolinite (calcined at 400 °C) exhibited high activity and selectivity for glycerol oxidation. However, the catalyst suffered from significant metal leaching after extended reaction times.6 Thus, strategies to enhance the stability and catalytic performance of this material remain a critical area of investigation. This study aims to evaluate the relationship between the textural and structural changes of Cu–Co particles supported on porous metakaolinite and their catalytic performance in partial glycerol oxidation. Catalysts with low metal loading (∼5 wt%) were synthesized via wet impregnation and calcined at 400, 600, and 800 °C. Their performance was assessed under atmospheric pressure, using hydrogen peroxide as the oxidizing agent.
Materials and methods
Materials and chemicals
The catalytic support (DMx) was prepared following the procedure described by Torres-Luna et al.27 Natural kaolinite (Sigma-Aldrich) was ground, sieved through a 100-ASTM mesh (particle size <150 μm), and calcined at 900 °C for 2 hours to dehydroxylate the mineral. The resulting solid was treated with 3 M HCl under reflux for 2 hours to remove aluminum atoms, thereby generating micro- and mesoporosity. Afterwards, the suspension was washed, dried at 60 °C for 24 hours, calcined at 400 °C for 2 hours, and sieved through the same mesh.The catalysts were synthesized using the wet impregnation method. Aqueous solutions of Cu(NO3)2·3H2O (Alfa Aesar, 99%) and Co(NO3)2·6H2O (Sigma-Aldrich, 98%) were prepared and mixed to maintain a Cu:Co molar ratio of 1:2. These solutions were combined with a dispersion of the support in deionized water (5 g of support in 40 mL of solution) with a target loading of 5 w/w%. The mixture was stirred and heated until the solvent completely evaporated, dried at 60 °C for 24 hours, and calcined in static air at 5 °C min−1 for 2 hours. The resulting catalysts were designated as DMx (Cu–Co 5%) x, where x corresponds to the calcination temperature: 400, 600, or 800 °C.
Characterization
Elemental analysis was performed via inductively coupled plasma (ICP) spectroscopy using an Agilent 5110 ICP-OES spectrometer. Samples were dissolved in HNO3 and HCl, and the solutions were diluted to a final volume of 50 mL with ultrapure water. No additional treatment was required for liquid samples.X-ray diffraction (XRD) patterns were recorded using a Bruker AXS D8 diffractometer with Cu-Kα radiation (λ = 1.54056 Å). Measurements were taken in the 2θ range of 5° to 70°, with a step interval of 0.02° and a step time of 10 s at room temperature. Raman spectroscopy was performed at room temperature using a HORIBA Jobin Yvon LabRAM HR 800 UV confocal Raman microscope equipped with a CCD detector. A 532 nm laser diode provided the excitation source, with 0.2 mW delivered to the sample. Textural properties were analyzed through N2 physisorption using a Micromeritics ASAP 2020 instrument. Samples were degassed overnight at 350 °C before measurement. Scanning electron microscopy (SEM) images were acquired with a JSM-6700F field emission microscope operating at 3 kV, using secondary electron imaging. Transmission electron microscopy (TEM) was conducted with a JEOL 2100 instrument at 200 kV coupled with an Energy Dispersive X-ray Spectrometer (EDS). For TEM, samples were suspended in ethanol and applied to clean nickel grids before air-drying.
X-ray photoelectron spectroscopy (XPS) spectra were recorded with a Kratos Axis Ultra DLD spectrometer. Monochromatic Al-Kα radiation was used, with 160 eV for general spectra and 20 eV for high-resolution spectra. Binding energies were calibrated using the C1s signal at 284.6 eV as a reference. Spectra were analyzed with CasaXPS software, employing 30% Gaussian and 70% Lorentzian deconvolution with a Shirley baseline algorithm.
In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was used to study isopropanol and acetone adsorption and desorption as probe molecules. Measurements were performed with a Shimadzu IRTracer-100 spectrometer equipped with a Harrick reaction chamber and ZnSe windows. Approximately 60 mg of catalyst with a particle size of 45 μm was pretreated under N2 flow (30 mL min−1) at 120 °C for 1.5 hours before analysis. Adsorption was conducted by passing isopropanol or acetone vapor through the catalyst at 80 °C for 30 minutes, followed by N2 purging.
Catalytic tests
The partial oxidation of glycerol was performed under reflux at atmospheric pressure. For each reaction, 10 g of 0.54 M glycerol solution and 200 mg of catalyst were placed in a 50 mL two-neck round-bottom flask. The reaction mixture was stirred at 500 rpm and heated in an oil bath at 80 °C. Hydrogen peroxide (1.21 M solution) was continuously added at 2.5 mL h−1 using a Metrohm 765 dosimat until a glycerol:H2O2 molar ratio of 1:2 was reached. To avoid photocatalytic effects, all reactions were conducted in dark conditions. After the reaction, the mixture was filtered (Millipore 0.45 μm) before HPLC analysis. Catalyst recyclability was evaluated through filtration and reuse without additional treatment. Reaction products were identified and quantified using certified standard compounds and analyzed by HPLC equipped with UV and refractive index detectors.
Results and discussion
Characterization of solids
The SiO2/Al2O3 ratio of dealuminized metakaolinite was calculated based on the Si and Al contents obtained through ICP analysis. As shown in Table 1, the observed ratio confirms dealumination compared to the theoretical value for natural kaolinite.26 This demonstrates that the thermal and acid treatments effectively facilitated aluminum dissolution and removal by washing.27 However, the SiO2/Al2O3 ratio of 14.5 was lower than the literature-reported value of 27.3 for similar kaolinites, suggesting greater stability of the raw material and less Al removal.6,27 ICP analysis was also employed to quantify Cu and Co in the catalysts. For all solids, the total metal content (Cu + Co) and the Cu/Co molar ratio (∼0.5) aligned closely with the intended nominal values, validating the incorporation of metals onto the support.| Support | SiO2/Al2O3 | |||
|---|---|---|---|---|
| Kaolinite (theoretical) | 1.20 | |||
| DM | 14.5 | |||
| Catalyst | Cu + Co (%) | Cu + Co (%)Theoretical | Cu/Co | Cu/CoTheoretical |
| DM400 (Cu–Co 5%) 400 | 5.00 ± 0.08 | 5.01 | 0.47 | 0.50 |
| DM600 (Cu–Co 5%) 600 | 4.94 ± 0.12 | 5.01 | 0.45 | 0.50 |
| DM800 (Cu–Co 5%) 800 | 4.87 ± 0.18 | 5.02 | 0.46 | 0.50 |
The X-ray diffraction (XRD) patterns (Fig. 1(a)) of the starting kaolinite material showed the characteristic peaks of a kaolinite structure (Triclinic, space group P1, Hermann–Mauguin notation).28 Including prominent signals at 2θ = 12.5° and 25.1°, corresponding to planes (001) and (002), respectively.19,26 XRD patterns of kaolins vary depending on the origin and source of the clay, which explains the differences in crystallinity compared to previous reports in the literature.6,27 The peaks at 2θ = 9.0° and 26.7° correspond to illite and quartz, respectively, which are common clay mineral impurities in kaolin.26,27 Thermal treatment at 900 °C produced amorphous metakaolinite by dehydroxylating the structure and eliminating hydrogen bonds, as evidenced by the loss of long-range structural order in the XRD profile of DM400.27
 | ||
| Fig. 1 (a) X-ray diffraction patterns of commercial kaolinite and the modified solid by thermal-acid treatment (k = kaolinite, i = illite, q = quartz). (b) X-ray diffraction profiles for Cu–Co/dealuminized metakaolinite catalysts varying the calcination temperature. (c) amplified 35–40° 2θ ranges (Black dot: spinel structure). (d) Raman spectra (λ = 532 nm) of the catalysts, Co3O4 and CuCo2O4 spinel synthesized as a reference. Nitrogen adsorption–desorption isotherms (at 77 K) of (e) the supports and (f) the catalysts. | ||
For catalysts (Fig. 1(b)), spinel-type structures were identified. The spinel materials belong to a group of compounds with cubic symmetry (space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, Hermann–Mauguin notation) with eight A2BO4 units per cell.29,30 However, perhaps due to the high dispersion in the catalysts, the diffraction peaks are not easily discernible.31 The most intense signal in the spinel structure corresponds to the plane (311), located around 37.0° 2θ.32 Then, in Fig. 1(c), the signal at 36.9° could be associated with the formation of CuCo2O4 or aluminate spinels such as CuAl2O4 or CoAl2O4 due to the interaction with the support. Additionally, the increasing intensity of this signal with higher calcination temperatures indicated improved crystallinity and no additional oxide phases, such as CuO or CoO, were detected.
m, Hermann–Mauguin notation) with eight A2BO4 units per cell.29,30 However, perhaps due to the high dispersion in the catalysts, the diffraction peaks are not easily discernible.31 The most intense signal in the spinel structure corresponds to the plane (311), located around 37.0° 2θ.32 Then, in Fig. 1(c), the signal at 36.9° could be associated with the formation of CuCo2O4 or aluminate spinels such as CuAl2O4 or CoAl2O4 due to the interaction with the support. Additionally, the increasing intensity of this signal with higher calcination temperatures indicated improved crystallinity and no additional oxide phases, such as CuO or CoO, were detected.
Raman spectroscopy provided complementary structural insights (Fig. 1(d)). The spectra confirmed the spinel structure, showing five active Raman bands at ∼190, 480, 520, 620, and 680 cm−1 corresponding to the symmetries F2g, Eg, F2g, F2g and A1g.29,33
For the DM400 (Cu–Co 5%) 400 catalyst, calcination at 400 °C favored CuCo2O4 formation. The broadening of the A1g band suggested Cu2+ substitution at Co2+ in octahedral sites, leading to unit cell expansion.34 Therefore, this structural distortion causes a change in the local environment of the octahedral positions occupied by the Co3+ ions that interact with the Cu2+ ions.35,36 Higher calcination temperatures resulted in sharper peaks, indicating enhanced structural organization.
The absence of the characteristic peaks of CuO (Ag + 2Bg symmetries) confirms that there is not segregation of Cu.37 Likewise, the formation of CoAl2O4 is also ruled out due to the low intensity of the F2g (520 cm−1) mode in the spectra. Literature reports that this F2g signal is quite intense for this spinel since it is associated with the Co–O vibrations of the tetrahedral sites.30 Therefore, the results suggest that the solids calcined at 600 °C and 800 °C, the Cu–Co spinel structure is conserved but with a higher degree of organization.
In Fig. 1(e) and (f), textural analyses revealed that all solids exhibited microporosity and mesoporosity (IUPAC classification, type Ib and IVa isotherms).38 The textural values in Table 2 show a significant decrease in the area for the support calcined at 800 °C, probably due to the collapse of some microporous. The reduced surface area and pore volume in catalysts compared to the support suggested metal deposition within pores and on external surfaces, particularly at higher calcination temperatures.
| Solid | Surface area (m2 g−1) | Pore volumen (cm3 g−1) | ||
|---|---|---|---|---|
| Microporea (t-plot) | Mesoporeb | Microporea | Totalc | |
| a The Harkins–Jura equation was used for statistical thickness. b De Boer's method. c According to Gurvitsch's rule. | ||||
| DM400 | 418 | 5 | 0.15 | 0.20 |
| DM600 | 416 | 11 | 0.15 | 0.22 |
| DM800 | 354 | 18 | 0.13 | 0.19 |
| DM400(Cu–Co 5%) 400 | 349 | 16 | 0.12 | 0.16 |
| DM600(Cu–Co 5%) 600 | 332 | 15 | 0.12 | 0.15 |
| DM800(Cu–Co 5%) 800 | 236 | 11 | 0.08 | 0.12 |
Alcohol oxidation is a redox process in which metals (Co3+, Co2+, Cu2+, and Cu+) participate in multiple cycles of reduction and oxidation. H2O2 plays a crucial role in maintaining the structural integrity of the catalyst during the reaction.5,39 The performance of these catalysts can be directly linked to their reducibility. The normalized TPR profiles of the catalysts, shown in Fig. 2(a), reveal reduction temperatures below 400 °C. Literature indicates that the reduction events correspond to Cu2+ → Cu0 and Co3+ → Co2+ → Co0 transitions.40,41
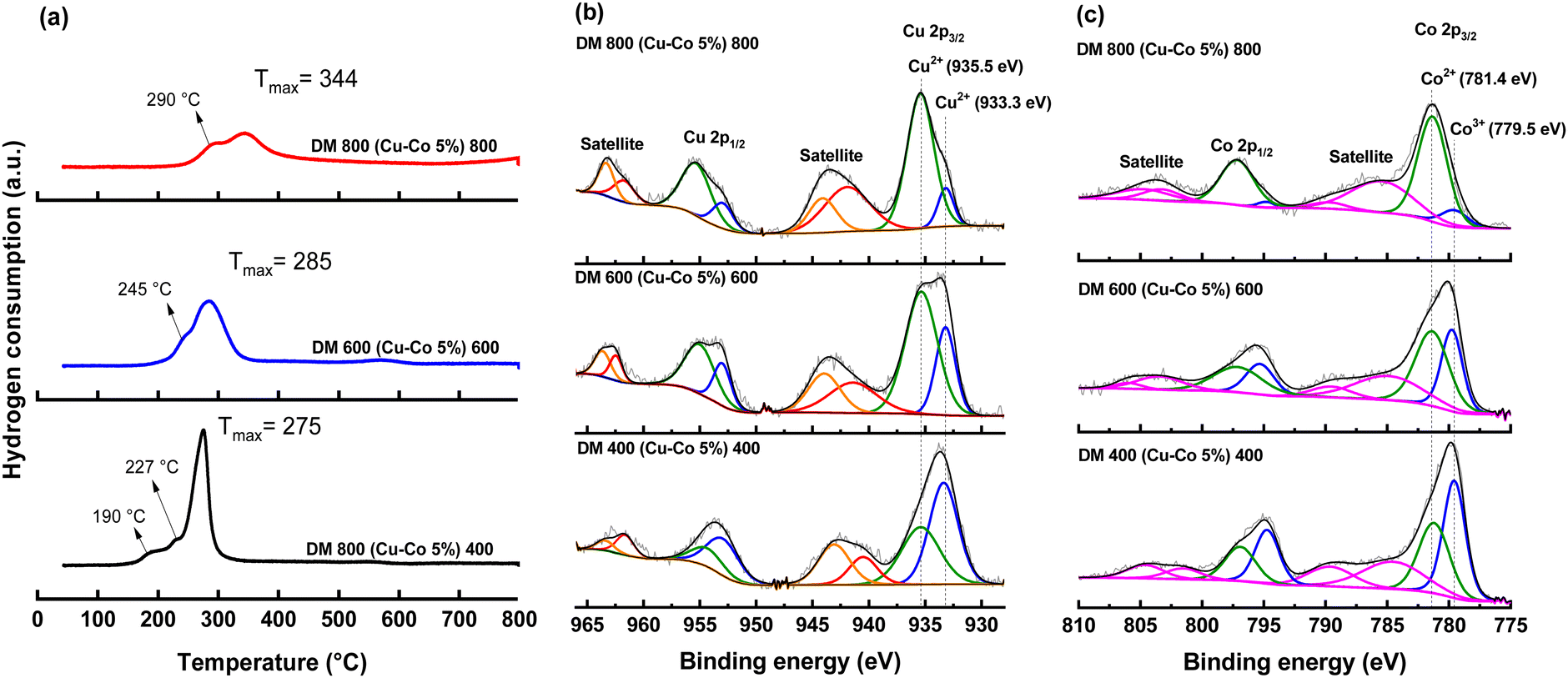 | ||
| Fig. 2 H2-TPR profiles (a) and HR-XPS spectra of (b) Cu 2p and, (c) Co 2p for the Cu–Co/dealuminized metakaolinite catalysts. | ||
For the catalyst calcined at 400 °C, two small shoulders are observed at 190 °C and 227 °C, along with a prominent peak at 275 °C. The shoulders correspond to weak interactions between the CuCo2O4 spinel and the support, while the main peak indicates stronger interactions. At higher calcination temperatures, the peaks shift to higher reduction temperatures, suggesting stronger metal–support interactions and reduced hydrogen consumption. This trend supports the hypothesis of spinel structure formation involving aluminum and copper. The possible formation of CuAl2O4 likely reduces the number of reducible sites due to strong Cu–Al interactions.
X-ray photoelectron spectroscopy (XPS) was employed to support this analysis. The Cu 2p spectrum (Fig. 2(b)) exhibits two predominant peaks, 2p3/2 and 2p1/2, with a spin–orbit interaction of 20.0 eV. These peaks, along with their satellite signals, serve as definitive indicators of the presence of Cu2+. Following the deconvolution of the 2p3/2 core level, two distinct peaks were identified, confirming the presence of Cu2+ in two different spinel structures. The literature extensively reports that calcination at higher temperatures (>700 °C) facilitates the incorporation of Cu and Al into a spinel structure, characterized by a binding energy of approximately 935 eV.42,43 Similarly, previous XPS studies of Cu have associated the signal at 933.3 eV with Cu2+ in the CuCo2O4 spinel structure.6 Consequently, the signal observed at 933.3 eV is characteristic of Cu2+ in the inverse spinel CuCo2O4, whereas the peak at 935.5 eV corresponds to Cu2+ located in CuAl2O4.
The observed increase in the peak at 935.5 eV with rising calcination temperature correlates with a higher presence of Cu2+ due to the formation of CuAl2O4. In contrast, the decrease in the peak area at 933.3 eV suggests the migration of Cu2+ from CuCo2O4 to CuAl2O4. The area ratio between copper located in these two binding energies, as presented in Table 3, indicates a compositional transformation within the spinel structure, with an increasing proportion of CuAl2O4 at elevated temperatures. A similar pattern was observed in the deconvoluted signal corresponding to the 2p1/2 peak.
| Solid | Cu2+ in 935.5 eV/Cu2+ in 933.3 eV |
|---|---|
| DM400 (Cu–Co 5%) 400 | 0.75 |
| DM600 (Cu–Co 5%) 600 | 4.87 |
| DM800 (Cu–Co 5%) 800 | 7.63 |
In the Co 2p spectrum (Fig. 2(c)), the spin–orbit coupling of 15.1 eV between the 2p3/2 and 2p1/2 core levels confirms the presence of cobalt within spinel structures. The presence of satellite peaks indicates cobalt exists in two distinct oxidation states, Co2+ and Co3+. In the deconvolution of the 2p3/2 core level, the peak at 779.5 eV corresponds to Co3+, while the peak at 781.4 eV is attributed to Co2+. The identification of cobalt in these two oxidation states further supports the formation of the CuCo2O4 inverse spinel, characterized by the partial substitution of Cu2+ ions. The reduced area of the Co3+ peak in DM800 (Cu–Co 5%) at 800 °C suggests the incorporation of Cu2+ into the support, which promotes the migration of Co2+ to the surface. This process facilitates the subsequent formation of Co3O4, as corroborated by Raman spectroscopy.
Scanning electron microscopy (SEM) analysis (Fig. 3) reveals that the support consists of aggregates with a platelet morphology. The platelets range in size from 50 nm to 200 nm, with thicknesses of 50 to 100 nm.
 | ||
| Fig. 3 SEM images (a) and (e) DM400, (b) and (f) DM400 (Cu–Co 5%) 400, (c) and (g) DM600 (Cu–Co 5%) 600 and, (d) and (h) DM800 (Cu–Co 5%) 800. | ||
For all three catalysts, the metallic phase is well-distributed, predominantly in the form of nanoparticles. Energy-dispersive X-ray (EDX) analysis of the metakaolinite confirms the heterogeneity of Si and Al in the solid.
To further investigate the potential impact of particle size on catalytic activity, transmission electron microscopy (TEM) analysis was performed (Fig. 4). The metallic particles exhibited irregular shapes, making precise size distribution histograms impractical. However, it was confirmed that particle sizes ranged between 3 nm and 35 nm on the widest side. Smaller particles likely exist within the pores, but their observation was limited by magnification constraints. A reduction in pore volume due to the formation of particles smaller than 2 nm within micropores was observed with increasing calcination temperature, as evidenced in micrographs (Fig. S13–S15, ESI†). In areas where metallic particles were not visible, EDX spectra detected Cu and Co in lower amounts relative to Si and Al content, indicating partial occupancy within the pores.
 | ||
| Fig. 4 TEM analysis (a) DM400 (Cu–Co 5%) 400. (b) DM600 (Cu–Co 5%) 600 (c) DM800 (Cu–Co 5%) 800. | ||
Catalytic tests
The results, presented in Table 4, demonstrate significant differences in glycerol conversion with varying calcination temperatures and reaction times. After 2 hours, DM400 (Cu–Co 5%) 400 achieved only 5% conversion, whereas DM600 (Cu–Co 5%) 600 and DM800 (Cu–Co 5%) 800 reached 21% and 59% respectively (entry 1–3). Complete conversion was obtained after 4 hours with DM800 (Cu–Co 5%) 800. The positive correlation between glycerol conversion and calcination temperature can be attributed to changes in the nanoparticle composition, specifically the interaction between copper and aluminum in the support and between copper and cobalt as evidenced by XPS analysis.| Entry | Solid | Time (h) | Conversion (%) | Selectivity (%) | Carbon balance (%) | ||
|---|---|---|---|---|---|---|---|
| Dihydroxyacetone | Glyceraldehyde | Glyceric acid | |||||
| 1 | DM400 (Cu–Co 5%)400 | 2 | 5 | 79 | 19 | <1 | 99 |
| 4 | 29 | 94 | 5 | 0 | 99 | ||
| 2 | DM600 (Cu–Co 5%)600 | 2 | 21 | 88 | 12 | 1 | 100 |
| 4 | 59 | 87 | 12 | 1 | 100 | ||
| 3 | DM800 (Cu–Co 5%)800 | 2 | 58 | 93 | 6 | 1 | 100 |
| 4 | 99 | 92 | 7 | 1 | 100 | ||
| 4 | DM800 (Cu 5%)800 | 4 | 22 | 91 | 4 | 4 | 100 |
| 5 | DM800 (Co 5%)800 | 4 | 10 | 84 | 16 | 0 | 100 |
| 6 | CuCo2O4 (10 mg) | 4 | 10 | 89 | 6 | 4 | 99 |
This interaction likely increased the cobalt content on the surface, altering the electronic properties of the spinel structure and enhancing glycerol oxidation. This assumption is corroborated by the low catalytic activity of the pure cobalt and copper-based catalysts DM800 (Cu5%) 800 and DM800 (Co 5%) 800 (entry 4 and 5). The bulk CuCo2O4 spinel has very low glycerol oxidation activity (entry 6) proving that the metakaolinite as catalytic support plays a significant role for glycerol oxidation.
Selectivity remained unaffected by calcination temperature, with all catalysts showing high selectivity toward dihydroxyacetone (DHA) after two hours of reaction. When the reaction is extended to 4 h, the selectivity towards DHA is not affected proving that no further transformation occurs.
It is thus possible to propose a classical reaction scheme for the transformation of glycerol in which two paths in parallel are involved, the main one (with a thick arrow) leading to dihydroxyacetone and the less favorable path (fine arrow) to glyceraldehyde (Fig. 5).
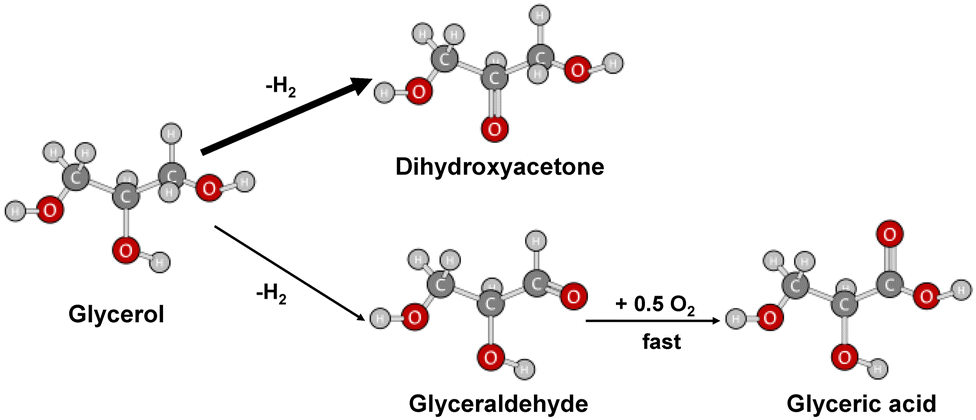 | ||
| Fig. 5 Reaction scheme of partial glycerol oxidation. | ||
When comparing the results at iso-conversion for DM600 (Cu–Co 5%) 600 at 4 hours and DM800 (Cu–Co 5%) 800 at 2 hours (59% and 58%, respectively), DM800 (Cu–Co 5%) 800 was the most active and selective catalyst. The carbon balance indicated that DHA, glyceraldehyde, and glyceric acid accounted for all glycerol transformation products.
Although glycerol oxidation has been studied since 1993,44 the explanation for selectivity toward DHA or glyceraldehyde remains debated. Studies on noble metal-based catalysts (Table S1, ESI†), such as Pt and Au, reveal differing pathways: Pt catalysts (conversion: 74–92%) favor glyceric acid (selectivity: 87%) at 50–60 °C and reaction times between 6 and 16 h,45,46 whereas Au catalysts (conversion: 58–91%) favor DHA formation (selectivity: 70–83%) at 50–60 °C with reaction times between 5–6 h, linked to oxygen vacancies.47–49 Likewise, a recent investigation reported the use of PtBi/ZrO2 catalyst and revealed that dihydroxyacetone tends to be easily desorbed, preventing C–C cleavage in other products.50 Cu- and Co-based catalysts, however, do not exhibit a clear product preference. The comparison of this work with existing studies shows that DM800 (Cu–Co 5%) 800 stands out for its remarkable catalytic activity and DHA selectivity.
Chemisorption and desorption studies
In situ chemisorption and desorption studies of isopropanol (IPA) and acetone have been performed using spectroscopic techniques to investigate glycerol oxidation on noble and non-noble metal-based catalysts.50,51 IPA, a secondary alcohol, served as a probe molecule to evaluate the adsorptive activation of glycerol through its secondary hydroxyl group to produce DHA.The difference infrared spectra of IPA chemisorption and desorption over time are shown for bulk catalyst (CuCo2O4 calcined at 400 °C), the supported catalysts, and the support, in Fig. 6. High intensities of IR bands post-desorption confirm that IPA forms strongly adsorbed species. Likewise, significant differences in IPA adsorption behavior between the bulk catalyst and the supported catalysts were observed. For the bulk catalyst, Fig. 6(a), the signals at 2975 cm−1 and 2890 cm−1 correspond to symmetric (νs(C–H)) and antisymmetric (νas(C–H)) stretching vibrations,51 while signals at 1465 cm−1 and 1378 cm−1 are attributed to symmetric (δs(CH3)) and antisymmetric (δas(CH3)) bending vibrations.50,51 Therefore, the adsorption occurs through two modes: dissociative and non-dissociative, as described for other metal oxides (Fig. 7(a)–(c)).52–54 For the support (Fig. 6(b)) and supported catalysts (Fig. 6(c) and (d)), C–H stretching and bending frequencies remained like free IPA, but hydroxyl functionalities exhibited marked differences.
 | ||
| Fig. 6 In situ DRIFT spectra of IPA adsorption followed by desorption at 0, 5, 10, 20 and 30 min (from top to bottom) on (a) CuCo2O4 (b) DM (c) DM400 (Cu–Co 5%) 400, (d) DM600 (Cu–Co 5%) 600, (e) DM800 (Cu–Co 5%) 800 and (f) comparison between DM400, DM800 (Cu–Co 5%)800 and CuCo2O4. | ||
 | ||
| Fig. 7 Schematic representation of IPA adsorption modes on the bulk catalyst (a) monodentate, (b) bidentate, (c) non-dissociative, and (d) hydrogen bonds on the support. | ||
For the support, the O–H vibration intensity of sharp hydroxyl stretching bands (3750–3580 cm−1) appears in negative absorbance.53,55 This suggests that OH of IPA interacts with Si–OH and Al–OH groups on metakaolinite.
The same band is observable for the supported Cu–Co/metakaolinite materials, while for the bulk CuCo2O4 no negative band is observed. Additionally, a notable distinction is evidenced in the absence of C–O vibrational modes (1250–1100 cm−1) for both the support and the supported catalysts (Fig. 6(f)). This observation indicates that the mechanism of adsorption of IPA diverges from that the bulk CuCo2O4, implying that the interactions involving M–OH (Fig. 7(a)–(c)) may be present, albeit with diminished intensity attributable to the pronounced dispersion of metal nanoparticles. Nonetheless, the predominant mechanism of adsorption appears to occur via hydrogen bonding on the support (Fig. 7(d)). Consequently, these variances underscore the critical function of metakaolinite in the activation of the secondary hydroxyl group within the glycerol molecule.
Acetone, formed through IPA dehydrogenation, was monitored for adsorption and desorption behavior. In Fig. 8, infrared bands at 1735 cm−1 and 1637 cm−1 were attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations, representing physically and chemically adsorbed acetone.51 Desorption studies revealed changes in the time of acetone desorption under the operation temperature (80 °C). DM400(Cu–Co5%) 400 (Fig. 8(a)) exhibited slower and uniform desorption, while DM600(Cu–Co 5%) 600 followed a similar trend with significant desorption after 10 minutes (Fig. 8(b)). It is interesting to observe that after 2 minutes, the bands have decreased significantly for DM800(Cu–Co5%) 800 showing faster acetone desorption than the other materials (Fig. 8(c)). For the bulk CuCo2O4 (Fig. 8(d)), significant desorption occurs after 6 minutes and the bulk metakaolinite support show the intensity of the bands decreasing slowly with time (Fig. 8(e)).
O stretching vibrations, representing physically and chemically adsorbed acetone.51 Desorption studies revealed changes in the time of acetone desorption under the operation temperature (80 °C). DM400(Cu–Co5%) 400 (Fig. 8(a)) exhibited slower and uniform desorption, while DM600(Cu–Co 5%) 600 followed a similar trend with significant desorption after 10 minutes (Fig. 8(b)). It is interesting to observe that after 2 minutes, the bands have decreased significantly for DM800(Cu–Co5%) 800 showing faster acetone desorption than the other materials (Fig. 8(c)). For the bulk CuCo2O4 (Fig. 8(d)), significant desorption occurs after 6 minutes and the bulk metakaolinite support show the intensity of the bands decreasing slowly with time (Fig. 8(e)).
 | ||
| Fig. 8 In situ DRIFT spectra of acetone adsorption followed by desorption at 0, 5, 10, 20 and 30 min at 80 °C (from top to bottom) on (a) DM400 (Cu–Co 5%) 400, (b)DM600 (Cu–Co 5%) 600, (c) DM800 (Cu–Co 5%) 800, (d) CuCo2O4 and (e) DM400. | ||
These observed trends align with the structural modifications associated with the calcination temperature, suggesting that the catalytic cycle of DM800 (Cu–Co 5%) at 800 °C is faster than other solids, thereby elucidating the elevated catalytic activity and selectivity exhibited by the solid subjected to calcination at 800 °C.
Mechanistic insights from in situ spectroscopy
In situ DRIFT spectroscopy of isopropanol adsorption–desorption provided insights into glycerol activation mechanisms. For all catalysts, strong isopropanol adsorption was observed, involving dissociative and non-dissociative modes.Signals associated with C–O vibrations confirmed the formation of isopropoxy species for the bulk CuCo2O4, while for supported catalyst hydroxyl vibrations indicated OH-surface interactions on the supported catalysts.
On the other hand, desorption studies revealed temperature-dependent behavior. Catalysts calcined at 800 °C showed faster desorption rates for acetone (the dehydrogenation product of IPA), explaining the high DHA selectivity in the glycerol dehydrogenation reaction. Slower desorption in DM400 (Cu–Co 5%) 400 is correlated with lower catalytic activity.
Catalyst stability
Stability is a critical parameter for aqueous-phase reactions, with metal leaching serving as an indirect measure. Table 5 shows that calcination temperature significantly influenced leaching. DM800 (Cu–Co 5%) 800 exhibited minimal Cu and Co leaching due to strong metal–support interactions, confirmed by characterization techniques.Despite low leaching levels, homogeneous catalysis contributions from dissolved Cu2+ and Co3+ ions were evaluated. DM400 (Cu–Co 5%) 400, which exhibited the highest leaching, showed the lowest glycerol conversion, indicating negligible homogeneous catalysis contributions. Recyclability tests further demonstrated the stability of DM800 (Cu–Co 5%) 800 catalyst (Fig. 9(a)). At least up to the first four reaction cycles under identical conditions (80 °C, 4 hours), only a slight decrease in glycerol conversion was observed, while selectivity remained unaffected (Fig. 9(b)). XRD profiles of fresh and post-reaction catalysts (Fig. 9(c) and (d)) highlighted structural changes. For DM400 (Cu–Co 5%) 400 the characteristic spinel peak (311) disappeared after 4 hours, confirming significant Co and Cu leaching. In contrast, catalysts calcined at higher temperatures retained their spinel structure, confirming enhanced metal–support interaction.
 | ||
| Fig. 9 Recycling stability of DM800 (Cu–Co 5%) 800. (a) Conversion, (b) selectivity, XRD profiles of fresh (c) and post reaction catalysts. (d) k: kaolinite, black dot: spinel structure. | ||
Conclusions
A set of Cu–Co catalysts with low metal loading (∼5 wt%) was successfully prepared and characterized. The results demonstrated that calcination temperature strongly influenced the structural and textural properties of the catalysts, as well as their catalytic performance in glycerol oxidation. The formation of CuCo2O4 and CuAl2O4 phases depended on the calcination temperature, with higher temperatures promoting stronger Cu–Al interactions and Co ion migration to the surface, favoring dehydrogenation reactions.All synthesized catalysts were active for the partial oxidation of glycerol, exhibiting high selectivity toward dihydroxyacetone (DHA). The best-performing catalyst, DM800 (Cu–Co 5%) 800, achieved complete glycerol conversion in 4 hours with excellent selectivity. In situ DRIFT spectroscopy revealed that glycerol activation involved strong interactions between alcohol molecules and the catalyst surface, with faster desorption of reaction products at higher calcination temperatures contributing to enhanced activity and selectivity.
Moreover, increasing the calcination temperature improved catalyst stability by reducing Cu and Co leaching. Recyclability tests confirmed that DM800 (Cu–Co 5%) 800 catalyst retained its activity and selectivity over multiple cycles. These findings highlight the potential of thermally treated Cu–Co/metakaolinite catalysts for efficient glycerol upcycling into high-value-added products.
Author contributions
L. Paredes-Quevedo: investigation, methodology, writing – original draft; C. Batiot-Dupeyrat: conceptualization, validation, supervision, resources, project administration, and writing – review & Editing and M. Velasquez: conceptualization, validation, supervision, resources, project administration, visualization, and writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the research projects HERMES Cod 51190 from Universidad Nacional de Colombia-Sede Bogotá and GLYPLAS between La Région Nouvelle-Aquitaine and the Université de Poitiers, France. This work was carried out in the IC2MP (Institut de Chimie des Milieux et Matériaux de Poitiers) and ESCA (Estado Sólido y Catálisis Ambiental) Universidad Nacional de Colombia-Bogotá.References
- T. A. Ewing, N. Nouse, M. van Lint, J. van Haveren, J. Hugenholtz and D. S. van Es, Green Chem., 2022, 24, 6373–6405 RSC.
- S. Tantisriyanurak, H. N. Duguid, L. Peattie and R. Dawson, ACS Appl. Polym. Mater., 2020, 2, 3908–3915 CrossRef CAS.
- X. Hu, J. Lu, Y. Liu, L. Chen, X. Zhang and H. Wang, Environ. Chem. Lett., 2023, 21, 2825–2861 CrossRef CAS.
- D. T. Johnson and K. A. Taconi, Environ. Prog., 2007, 26, 338–348 CrossRef CAS.
- P. N. Amaniampong, Q. T. Trinh, J. J. Varghese, R. Behling, S. Valange, S. H. Mushrif and F. Jérôme, Green Chem., 2018, 20, 2730–2741 RSC.
- L. C. Paredes-Quevedo, J. G. Carriazo and M. Velasquez, Catal. Commun., 2023, 178, 106676 CrossRef CAS.
- T. Jedsukontorn, V. Meeyoo, N. Saito and M. Hunsom, Chem. Eng. J., 2015, 281, 252–264 CrossRef CAS.
- X. Wang, C. Shang, G. Wu, X. Liu and H. Liu, Catalysts, 2016, 6, 101 CrossRef.
- X. Deng, G. Dodekatos, K. Pupovac, C. Weidenthaler, W. N. Schmidt, F. Schüth and H. Tüysüz, ChemCatChem, 2015, 7, 3832–3837 CrossRef CAS.
- X. Jin, M. Zhao, C. Zeng, W. Yan, Z. Song, P. S. Thapa, B. Subramaniam and R. V. Chaudhari, ACS Catal., 2016, 6, 4576–4583 CrossRef CAS.
- S. Schünemann, F. Schüth and H. Tüysüz, Catal. Sci. Technol., 2017, 7, 5614–5624 RSC.
- Z. Zhong, J. Yan, B. Chen, Q. Zhang, Y. Zhu, S. Xu, A. Xie and S. Luo, J. Solid State Chem., 2024, 332, 124574 CrossRef CAS.
- T. G. Vo, P. Y. Tsai and C. Y. Chiang, J. Catal., 2023, 424, 64–73 CrossRef CAS.
- Z. Xi, H. Zhou, Y. Liu and C. Xu, Electrochim. Acta, 2023, 470, 143285 CrossRef CAS.
- K. Rekab, C. Lepeytre, F. Goettmann, M. Dunand, C. Guillard and J. M. Herrmann, J. Radioanal. Nucl. Chem., 2015, 303, 131–137 CrossRef CAS.
- Y.-J. Oh, G.-S. Park and C.-H. Chung, J. Electrochem. Soc., 2006, 153, G617 CrossRef CAS.
- N. A. Sacco, M. E. Lovato, F. A. Marchesini and A. V. Devard, J. Hazard. Mater. Lett., 2022, 3, 100059 CrossRef CAS.
- G. Busca, in Advances in Catalysis, ed. F. Jentoft, Academic Press, 2014, vol. 57, pp. 319–404 Search PubMed.
- A. K. Panda, B. G. Mishra, D. K. Mishra and R. K. Singh, Colloids Surf., A, 2010, 363, 98–104 CrossRef CAS.
- C. Sánchez-Trinidad, G. del Angel, G. Torres-Torres, A. Cervantes-Uribe, A. A. S. Pavón, Z. Guerra-Que, J. C. Arévalo-Pérez and F. J. Tzompantzi-Morales, ChemistryOpen, 2019, 8, 1143–1150 CrossRef PubMed.
- B. Yang, X. Chen, L. Guo and L. Zhang, J. Mater. Chem. A, 2024, 19861–19884 RSC.
- H. Yahiro, K. Nakaya, T. Yamamoto, K. Saiki and H. Yamaura, Catal. Commun., 2006, 7, 228–231 CrossRef CAS.
- A. Devard, P. Brussino, F. A. Marchesini and M. A. Ulla, J. Environ. Chem. Eng., 2019, 7, 103201 CrossRef CAS.
- A. M. Bahmanpour, F. Héroguel, M. Klllç, C. J. Baranowski, L. Artiglia, U. Röthlisberger, J. S. Luterbacher and O. Kröcher, ACS Catal., 2019, 9, 6243–6251 CrossRef CAS.
- A. M. Bahmanpour, B. P. Le Monnier, Y. P. Du, F. Héroguel, J. S. Luterbacher and O. Kröcher, Chem. Commun., 2021, 57, 1153–1156 RSC.
- A. K. Chakraborty, Phase Transformation of Kaolinite Clay, Springer, New Delhi, Kolkata (India), 2014 Search PubMed.
- J. A. Torres-Luna and J. G. Carriazo, Solid State Sci., 2019, 88, 29–35 Search PubMed.
- E. Volkova, A. K. Narayanan Nair, J. Engelbrecht, U. Schwingenschlögl, S. Sun and G. Stenchikov, J. Phys. Chem. C, 2021, 125, 24126–24136 Search PubMed.
- W. B. White and B. A. DeAngelis, Spectrochim. Acta, Part A, 1967, 23, 985–995 CrossRef CAS.
- V. D’Ippolito, G. B. Andreozzi, D. Bersani and P. P. Lottici, J. Raman Spectrosc., 2015, 46, 1255–1264 Search PubMed.
- M. F. Luo, P. Fang, M. He and Y. L. Xie, J. Mol. Catal. A: Chem., 2005, 239, 243–248 CrossRef CAS.
- S. W. Oh, H. J. Bang, Y. C. Bae and Y. Sun, J. Powder Sources, 2007, 173, 502–509 Search PubMed.
- E. Alsharaeh, Y. Mussa, F. Ahmed, Y. Aldawsari, M. Al-Hindawi and G. K. Sing, J. Phys. C: Solid State Phys., 1988, 21, 199–201 Search PubMed.
- G. Ercolino, A. Grodzka, G. Grzybek, P. Stelmachowski, S. Specchia and A. Kotarba, Top. Catal., 2017, 60, 333–341 CrossRef CAS.
- Z. H. He, N. Li, K. Wang, W. T. Wang and Z. T. Liu, Mol. Catal., 2019, 470, 120–126 CrossRef.
- J. Su, Z. Zhang, D. Fu, D. Liu, X. C. Xu, B. Shi, X. Wang, R. Si, Z. Jiang, J. Xu and Y. F. Han, J. Catal., 2016, 336, 94–106 Search PubMed.
- A. Chauhan, R. Verma, K. M. Batoo, S. Kumari, R. Kalia, R. Kumar, M. Hadi, E. H. Raslan and A. Imran, J. Mater. Res., 2021, 36, 1496–1509 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- Z. Zhong, J. Yan, B. Chen, Q. Zhang, Y. Zhu, S. Xu, A. Xie and S. Luo, J. Solid State Chem., 2024, 332, 124574 CrossRef CAS.
- X. Zhuge, J. Zhou, Z. Chen, S. Liu and K. Du, Colloids Surf., A, 2023, 677, 132340 CrossRef CAS.
- J. Zeng, H. Xie, G. Zhang, X. Cheng, G. Zhou and Y. Jiang, Ceram. Int., 2020, 46, 21542–21550 CrossRef CAS.
- Y. Liu, S. Qing, X. Hou, F. Qin, X. Wang, Z. Gao and H. Xiang, Catal. Sci. Technol., 2017, 7, 5069–5078 RSC.
- I. S. Zhidkov, A. A. Belik, A. I. Kukharenko, S. O. Cholakh, L. S. Taran, A. Fujimori, S. V. Streltsov and E. Z. Kurmaev, JETP Lett., 2021, 114, 556–560 CrossRef CAS.
- H. Kimura and K. Tsuto, Appl. Catal., A, 1993, 96, 217–228 CrossRef CAS.
- X. Huang, Z. Long, Z. Wang, S. Li, P. Zhang and Y. Leng, Chem. Eng. J., 2023, 470, 144037 CrossRef CAS.
- H. Yan, M. Zhao, X. Feng, S. Zhao, X. Zhou, S. Li, M. Zha, F. Meng, X. Chen, Y. Liu, D. Chen, N. Yan and C. Yang, Angew. Chem., Int. Ed., 2022, 61(21), e202116059 CrossRef CAS PubMed.
- Y. Wang, W. Liu, J. Zhao, Z. Wang and N. Zhao, Appl. Catal., A, 2024, 671, 119578 CrossRef CAS.
- Y. Li, X. Wei, R. Pan, Y. Wang, J. Luo, L. Li, L. Chen and J. Shi, Energy Environ. Sci., 2024, 17, 4205–4215 RSC.
- M. Zhao, H. Yan, R. Lu, Y. Liu, X. Zhou, X. Chen, X. Feng, H. Duan and C. Yang, AIChE J., 2022, 68, 1–15 Search PubMed.
- P. Luo, J. Wang, W. Rui, R. Xu, Z. Kuai, D. Yang, X. Wan, C. Zhou, Y. Yang and Y. Dai, Chem. Eng. J., 2023, 467, 143502 CrossRef CAS.
- Z. An, H. Ma, H. Han, Z. Huang, Y. Jiang, W. Wang, Y. Zhu, H. Song, X. Shu, X. Xiang and J. He, ACS Catal., 2020, 10, 12437–12453 CrossRef CAS.
- P. F. Rossi, G. Busca, V. Lorenzelli, O. Saur and J. Lavalley, Langmuir, 1987, 3, 52–58 Search PubMed.
- A. Rodrigues, J. M. Tatibouët and E. Fourré, Plasma Chem. Plasma Process., 2016, 36, 901–915 CrossRef CAS.
- C. Barakat, P. Gravejat, O. Guaitella, F. Thevenet and A. Rousseau, Appl. Catal., B, 2014, 147, 302–313 Search PubMed.
- T. Armaroli, T. Bécue and S. Gautier, Oil Gas Sci. Technol., 2004, 59, 215–237 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ma00113g |
| This journal is © The Royal Society of Chemistry 2025 |