
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective production of aldehydes: from traditional alternatives to alcohol photo-oxidation using g-C3N4-based materials
M. A.
Quintana
a,
D.
Rodriguez-Padrón
b,
P.
Jiménez-Calvo
c,
M.
Calero
a,
R. R.
Solís
*a and
M. J.
Muñoz-Batista
 *a
*a
aDepartment of Chemical Engineering, University of Granada, 18074 Granada, Spain. E-mail: rafarsolis@ugr.es; mariomunoz@ugr.es
bDipartimento di Scienze Molecolari e Nanosistemi, Università Ca’ Foscari di Venezia, 30123 Venezia, Italy
cChemistry of Thin Film Materials, IZNF, Friedrich-Alexander-Universität Erlangen-Nürnberg, Cauerstraße 3, Erlangen, 91058, Germany
First published on 5th May 2025
Abstract
Once largely overlooked, selective photocatalysis has recently seen rapid development and now includes several new applications. One of the most sought-after applications is the photocatalytic oxidation of alcohols to their corresponding aldehydes. There is significant interest in the selective production of compounds such as benzaldehyde, cinnamaldehyde, and vanillin using this technology, among various other valuable components. This work examines common production methods and alternative synthesis routes for aldehydes from their alcohols, focusing on g-C3N4-based photocatalytic schemes. The discussion includes an analysis of g-C3N4 structures that have shown significant results in the selective oxidation of alcohols and provides a critical review of the mechanism, highlighting the importance of reporting quantum yields of the reaction. Literature data suggest that photocatalysis is a viable alternative to traditional aldehyde production methods, with maximum selectivity values towards the product of interest (>99%), but catalytic activity remains limited, with relatively low quantum yields, restricting the transition beyond the laboratory scale. However, the charge handling in g-C3N4-based advanced materials is adequate for this application, and their implementation under solar illumination conditions is promising.
 M. A. Quintana | Dr M. Alejandra Quintana earned her degree from the Universidad de Bogotá Jorge Tadeo Lozano in 2019 and completed her Master's degree (2022) and PhD (2024) at the University of Granada. She is currently a research scientist at the University of Granada. Her work focuses on the design, synthesis, and characterization of new photocatalytic materials for applications such as the organic synthesis of high-value compounds and the degradation of liquid-phase pollutants. She has also developed a strong interest in waste valorization, exploring sustainable and environmentally relevant processes. |
 D. Rodriguez-Padrón | Daily Rodríguez-Padrón is a Marie-Curie Post Doc researcher at University Ca’ Foscari Venezia, Italy. Her research has been focused on mechanochemistry and biomass valorization, with 87 articles and 2 patents published. She serves on the Early Career Advisory Board of ChemCatChem. Her achievements include the Dan David Prize 2019 from Tel Aviv University, Israel and the Green Talent Award 2020 from the German Federal Ministry of Education and Research. She was recognized as part of the Green Chemistry Emerging Investigators Series 2024 in Green Chemistry, the Sustainability Talents Special Collection 2024 in ChemSusChem, and the Catalysis Talents Class of 2023 in ChemCatChem. |
 P. Jiménez-Calvo | Pablo Jiménez-Calvo earned his PhD in Materials Chemistry and Physics from the University of Strasbourg (2019) and completed postdoctoral research at the University of Paris-Saclay (2020) and Max Planck Institute of Colloids and Interfaces (2022). He is a European Marie Skłodowska-Curie Postdoctoral Fellow at Friedrich-Alexander-Universität Erlangen-Nürnberg, Germany. His research concerns photoelectrocatalysis, electrode design, and device engineering, with emphasis on carbon nitride interfaces for electrolyzers and photoreactors aimed at Solar-to-X applications such as water splitting, CO2 reduction, and ammonia oxidation. He received the Rising Star of Materials Today Catalysis award (2023) and was a finalist for the European Young Chemist Award (2020). |
 M. Calero | M. Calero is a Full Professor of Chemical Engineering at the University of Granada, Spain and is responsible for the Research Group “Waste Recovery Technologies and Catalytic Processes (RNM-152). Its main lines of research focus on the characterization and valorization of waste from diverse sources to obtain compounds of interest. Some of the main technologies studied focus on thermochemical recycling processes, such as pyrolysis and gasification, and the development of catalysts for the different technologies. Finally, she collaborates in environmental impact studies through life cycle analysis. |
 R. R. Solís | Rafael Rodríguez Solís is a professor working in the Department of Chemical Engineering at the University of Granada (Spain). He has a BSc, MSc and PhD in Chemical Engineering from the University of Extremadura (Spain). His PhD was focused on Advanced Oxidation Technologies based on photocatalysis, ozone and peroxides for water treatment. He performed a postdoctoral stay at the University of Cincinnati (USA) and then joined the Autonomous University of Madrid. In 2021 he joined the University of Granada where he researches novel materials with photocatalytic properties, and the valorization of residues with energy and environmental applications. |
 M. J. Muñoz-Batista | Mario J. Muñoz-Batista is a Professor at the University of Granada. He obtained his PhD in Applied Chemistry from the Autonomous University of Madrid – Institute of Catalysis and Petrochemistry (CSIC). His research expertise lies in the development of innovative methods for material preparation and mathematical modelling of energy-efficient and environmentally sustainable chemical processes. His work spans a broad range of applications, from effluent treatment to novel pathways for chemical production, fuel generation, and the development of emerging energy vectors, with a strong emphasis on catalytic processes and waste valorization. He has authored more than 110 SCI-indexed articles (h-index = 37), holds two patents, and has contributed to several books and book chapters. He also serves in editorial roles for various leading journals in his field. |
1. Introduction: the importance of aromatic aldehydes
Organic synthesis is a fundamental discipline of the modern chemical industry, responsible for producing versatile feedstock molecules for numerous applications. These compounds play a critical role as intermediates in the manufacture of end-use products, contributing to technological advancement and improving the quality of life for a growing population.1–3 In modern chemistry, aldehydes have particular importance due to their high reactivity, which stems from the presence of the carbonyl functional group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). This group features sp2 hybridization with a trigonal planar geometry, enabling dipolar motion. Additionally, the polarized carbon–oxygen double bond, resulting from the significant electronegativity difference between the two atoms, makes the carbonyl carbon a strong electrophile, prone to nucleophilic addition reactions.4–6
O). This group features sp2 hybridization with a trigonal planar geometry, enabling dipolar motion. Additionally, the polarized carbon–oxygen double bond, resulting from the significant electronegativity difference between the two atoms, makes the carbonyl carbon a strong electrophile, prone to nucleophilic addition reactions.4–6
Aldehydes can be classified as aliphatic or aromatic. Aliphatic aldehydes are characterized by linear or branched carbon chains, while aromatic aldehydes contain an aromatic ring in their structure. The aromatic ring enhances intermolecular interactions due to electron delocalization and inductive effects from substituents.6–8 These properties, coupled with their synthetic diversity, make aldehydes highly valuable as raw materials for high added-value product synthesis, such as dyes, pesticides, pharmaceuticals, cleaning agents, flavorings, and fragrances.9–11
The broad range of applications of aromatic aldehydes has garnered significant attention in the global market. In 2022, the market for these compounds reached an estimated value of $6.3 billion, with projections suggesting growth to $8.5 billion by 2031, at a compound annual growth rate of 3.9%.12 The most widely used aromatic aldehydes in the industry are benzaldehyde, cinnamaldehyde, and vanillin. Benzaldehyde stands out as the simplest aromatic aldehyde, consisting of a benzene ring with a carbonyl substituent.13 It is widely used with an annual consumption of approximately 20 tons and a market price of around $260 per kilogram.14 Cinnamaldehyde, which occurs naturally as trans-cinnamaldehyde, has a chemical structure comprising a phenyl group attached to an unsaturated aldehyde.15,16 This aldehyde has gained interest in the world market, reaching values of $189 million in 2021, in addition to having a projection of $241.2 million in 2028.17 Meanwhile, vanillin is one of the most widely used flavors globally. It has a structure composed of a phenol substituted with an aldehyde and a methoxy group.18 Market forecasts project that the global vanillin market will reach $706 million by 2027, underscoring its growing relevance across multiple industries.19
Due to their recognized presence in the market, aldehydes have been considered compounds of great interest over the years, as they have applications in a wide range of fields, from food additives, plastics, cosmetics, and fragrances to pharmaceuticals. Consequently, both industry and the scientific community have focused on different ways to produce these organic compounds.
2. Obtaining aromatic aldehydes: from natural to catalytic schemes
To meet the aromatic aldehyde consumption, diverse processes have been implemented to produce them from their alcohol or acid derivatives, as summarized in Fig. 1. The most popular synthesis routes mainly include methodologies, such as direct extraction of natural products or redox processes based on biocatalytic processes, microbial synthesis, and chemical production. | ||
| Fig. 1 Schematic representation of alternatives to produce selectively high-added value aldehydes. | ||
2.1. Natural extraction
Some aromatic aldehydes of interest are found in natural extracts, for example, benzaldehyde, which is mainly present in almonds. It is also in the seeds of some fruits, such as apricots, peaches, and cherries. The hydrodistillation of peach and cherry leaves has been stated as favourable to obtaining benzaldehyde. Hence, under a temperature of 130 °C for 2 h, the aldehyde yielded 95.5% and 99.7% for peach laurel and cherry laurel, respectively. However, pilot-scale production has become a challenge, as it has been documented that 200 to 300 kg of leaves are required to obtain just 1 kg of benzaldehyde. This, in turn, makes industrial-scale production difficult.20Cinnamaldehyde is primarily extracted from the bark of the evergreen aromatic trees of the Cinnamomum genus. The typical procedure involves steam distillation, where a stream of steam acts on the crushed cinnamon to release the essential oil, obtaining a mixture of cinnamaldehyde and other compounds. The oil is recovered from the mixture by condensation and decantation, allowing the collection of an essential oil rich in cinnamaldehyde.21,22 Although this aldehyde is obtained naturally, it presents difficulties since it is time-consuming and energy-consuming, resulting in scarce yields. For instance, in a study carried out by R. Yitbarek et al., oil was obtained by employing the steam distillation method on 600 g of cinnamon leaves in a time of ∼175 min and an extraction temperature of 105 °C. Finally, the yield of cinnamon leaf essential oil was 2.9%, containing 34.6% of cinnamaldehyde. This data suggests that the cinnamaldehyde extraction process is not yet ready to be applied at an industrial level due to its economic disadvantages. Alternatively, the cinnamaldehyde obtained by chemical synthesis through the reaction between benzaldehyde and acetaldehyde presents problems of unwanted parallel reactions leading to derivatives and stereoisomers, which challenges the purification of the targeted cinnamaldehyde.23 Vanillin is obtained from the pods of Vanilla planifolia, a member of the orchid family. This process requires intensive labour because of the crops' low natural pollination rate. As a result, vanilla represents only 1–2% (w/w) of the cured pods and supplies just 1% of the global vanillin market, with selling prices reaching around $25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 per kilogram.24 Natural extraction techniques result in excessive consumption of plant material and prolonged production times, making product prices excessively high and the natural production of vanillin unaffordable.
000 per kilogram.24 Natural extraction techniques result in excessive consumption of plant material and prolonged production times, making product prices excessively high and the natural production of vanillin unaffordable.
2.2. Biocatalytic processes
Biocatalysis involves two different approaches. Firstly, the de novo synthesis approach under microbial action, which uses natural or engineered organisms to metabolize simple substrates, generates the desired final product. Secondly, the biotransformation or bioconversion of substrates, where various enzymes convert the precursor to the product through one or several steps.25 The biosynthesis of benzaldehyde occurs under the action of enzymes such as phenylalanine ammonia-lyase, which can generate cinnamic acid, subsequently transformed into benzaldehyde via β-oxidative and non-β-oxidative pathways, the latter being either dependent on coenzyme A (Co-A) or not. The CoA-independent non-β-oxidative pathway generates benzaldehyde and glyoxylic acid from cinnamic acid through the direct cleavage of its double bond by the dioxygenase enzyme. Alternatively, the attack can occur at the double bond of the branched chain of cinnamic acid, which undergoes hydration to form 3-hydroxy-3-phenylpropanoic acid as an intermediate, subsequently producing benzaldehyde and acetate via a hydratase/lyase enzyme.13The biocatalytic method for cinnamaldehyde production has not been extensively studied yet. A pathway for synthesizing cinnamaldehyde studied by R. Zhang et al. involves the generation of cinnamyl alcohol, with cinnamaldehyde as an intermediate. In this case, artificial biosynthetic pathways (in vitro assays) are used, where the substrate (L-phenylalanine) is treated with phenylalanine ammonia-lyase 2, producing cinnamic acid. Finally, carboxylic acid reductase is used to convert cinnamic acid into cinnamaldehyde.26 The biocatalytic synthesis of vanillin includes the use of a versatile enzyme called feruloyl esterase obtained from microorganisms such as Aspergillus niger, to extract an intermediate like ferulic acid from wheat bran. This reaction conducted in a packed column reactor, followed by an adsorption step on Amberlite resin, has reported 99.5% of vanillin using methanol as a reusable eluent. However, the process is highly dependent on parameters such as enzyme load, reaction time, pH, and temperature to obtain a good amount of ferulic acid, the precursor of vanillin, necessitating strict control of different parameters, making the process economically unviable.27
The biocatalytic processes belong to a developing field that displays advantages such as the reduction of chemical solvents and competitive selectivity toward the aldehyde. However, the high costs of enzymes and external cofactors, the non-reusability of enzymes, and the low stability of biocatalytic intermediates when exposed to different temperature and pH conditions make this methodology difficult to apply in the industry.28
2.3. Microbial synthesis
The microbial synthesis of benzaldehyde is carried out by various microbes such as Lactobacillus plantarum, Streptococcus thermophilus, and L. helveticus, capable of producing phenylmethanal through amino acid catabolism.29 The method used by Y. Takakura et al. for benzaldehyde production involves the use of Escherichia coli to which four different enzymes are added, such as L-amino acid deaminase, 4-hydroxymandelate synthase, (S)-mandelate dehydrogenase, and benzoylformate decarboxylase, using L-phenylalanine as the precursor source. This method presents low yields due to the accumulation of intermediates such as phenylpyruvic acid. The process has been enhanced by modifying the 4-hydroxymandelate synthase from Actinoplanes teichomycetus, obtaining a mutant that demonstrated 2.4 times higher activity than the E. coli strain.30In a study conducted by H. B. Bang et al., cinnamaldehyde was produced by designing Escherichia coli using three biosynthetic enzymes, such as phenylalanine ammonia-lyase, 4-coumarate: CoA ligase, and cinnamoyl-CoA reductase. Additionally, in this study, the metabolic pathways of this microorganism were modified to increase the intracellular accumulation of the main substrate of cinnamaldehyde, which is L-phenylalanine. The procedure was carried out in three steps involving enzymatic interference. The first step consists of the deamination of L-phenylalanine into cinnamic acid by phenylalanine ammonia-lyase, followed by the acid–thiol ligation of cinnamic acid to cinnamoyl-CoA by 4-coumarate: CoA ligase, and finally, the reduction of cinnamoyl-CoA to cinnamaldehyde by cinnamoyl-CoA reductase.21
In a study conducted by F. Luziatelli et al. to obtain vanillin, genetic engineering was used to optimize the E. coli strain and make it more stable, integrating genes from Pseudomonas that encode feruloyl-CoA synthetase and enoyl-CoA hydratase/aldolase into the chromosome. These enzymes allow, in the first step, the production of feruloyl-CoA mediated by feruloyl-CoA synthetase and, in the second step, the conversion of this compound into vanillin and acetyl-CoA by enoyl-CoA hydratase/aldolase. To optimize the procedure, a fed-batch operation mode was used in which ferulic acid was encapsulated and released in a controlled manner. Additionally, an optimal process temperature of 30 °C and a pH of 9 was established, and resting cells were used, favouring substrate entry into the cell and inhibiting endogenous enzymes responsible for vanillin reduction.29,31 Some drawbacks of the microbial synthesis of aldehydes include the bioaccumulation of intermediates and the low genetic stability of microorganisms, preventing the effective formation of the product of interest. To reduce process failures, genetic engineering is currently under research, which often requires complicated and expensive procedures. Additionally, sometimes it is difficult to maintain the genetic modifications over time, making the application of this methodology at an industrial scale challenging.
2.4. Chemical production
Most benzaldehyde production worldwide takes place through the oxidation of toluene with air, either in the vapour or liquid phase. This process, which requires high temperature and pressure conditions, employs metal-based catalysts. However, it yields low selectivity and efficiency due to the formation of multiple byproducts.32 Additionally, several studies indicate that other commonly used methods, such as the hydrolysis of benzyl chloride, tend to generate chlorine-containing waste. Cinnamaldehyde is commonly synthesized through the Perkin reaction, which requires an aldol addition using benzaldehyde and acetaldehyde under alkaline conditions at room temperature. The reaction is conducted in the presence of a polyethylene glycol-based ion exchange resin catalyst, yielding efficiencies between 75% and 85%.33,34 The predominant method for industrial vanillin synthesis, accounting for 85% of global production, is oxidative decarboxylation. The process includes two steps: (I) the reaction of guaiacol with glyoxylic acid, starting with base-promoted condensation, followed by (II) the oxidative decarboxylation of vanillylmandelic acid to vanillin, catalyzed by Cu2+ in an alkaline medium at 80–130 °C. The vanillin is then purified by vacuum distillation and recrystallization.35 Another approach is the alkaline aerobic oxidation of Kraft or sulfonated lignin, a macromolecule found in woody biomass. Several parameters have been studied in this reaction, such as temperature, oxygen pressure, and NaOH concentration.36 For example, Y. Zhu et al. reported a maximum vanillin yield of 21% under 160 °C, 1 h, 7.5 wt% NaOH, and 1 MPa O2.37 This oxidation process is followed by precipitation using an acid, which generates undesired byproducts, imposing solvent-based extraction. A study by A. Kaufmann et al. evaluated the effect of various solvents, such as esters, ethers, terpenes, and terpenoids, achieving a 95% extraction efficiency with butyl acetate at either 25 °C or 50 °C.38The aldehyde industry has leaned toward chemical production due to its economic advantages for large-scale enterprises. A striking example is natural vanillin, which sells at $25000 per kg, compared to synthetic vanillin at $15 per kg. This stark difference has limited natural vanillin extraction to less than 1% of the 16000 tons sold annually.39 While chemical methods provide significant utility for aldehyde synthesis, they face challenges such as racemic product generation, demanding purification processes, hazardous intermediates handling, and certain environmental concerns. In response, efforts have focused on developing more versatile and sustainable processes aligned with market demands and green chemistry. A promising advancement is the selective photocatalytic oxidation of alcohols, which aligns with green chemistry principles and offers an alternative to conventional approaches.40
2.5. Photocatalytic synthesis
In recent years, heterogeneous photocatalysis has garnered significant attention for addressing environmental and energy challenges. This technology offers attractive operating conditions, including the application of light to drive chemical reactions, under room or mild temperatures and pressures, and reduced use of chemical reagents.41–45Photocatalysis operates on the principle that when a semiconductor is irradiated with a photon of energy equal to or greater than the bandgap (Eg), typically below 3.0 eV,46 it generates a charge pair consisting of an excited electron (e−) and a positive hole (h+).47 These charge carriers can dissociate and migrate to the catalyst's surface, where they may drive redox reactions. However, some excitons undergo undesirable recombination, either in the bulk or on the surface, reducing efficiency.48 The process relies on semiconductor materials with distinct valence and conduction bands, where the energy gap between them determines the photocatalytic activity.49–51 Although this mechanism has been successfully evaluated for selective photocatalytic oxidation of alcohols, offering an intriguing alternative for aldehyde production, challenges persist.52 These include unfavourable semiconductor band positioning, which shortens the lifetime of e− and h+, leading to charge recombination. During recombination, excited electrons quickly return to the valence band, reducing the formation of reactive species and hindering selective reactions. Addressing this issue requires strategies such as photogenerated charge trapping or defect engineering to develop photocatalysts that mitigate recombination and possess a bandgap that effectively absorbs photons to drive desirable chemical transformations.45,53–55
To date, photocatalytic systems composed mainly of metal oxides, ferrites, metal–organic frameworks (MOFs), and sulfur compounds (CdS and MoS2), among others, have been predominantly employed. These materials have different energy bands and bandgap widths,56 which gives them interesting electronic and photocatalytic properties, see Fig. 2. A widely studied archetypical metal oxide is titanium dioxide (TiO2),57 a benchmark semiconductor due to its high thermal and chemical stability, great availability, and large-scale synthetic process.58,59
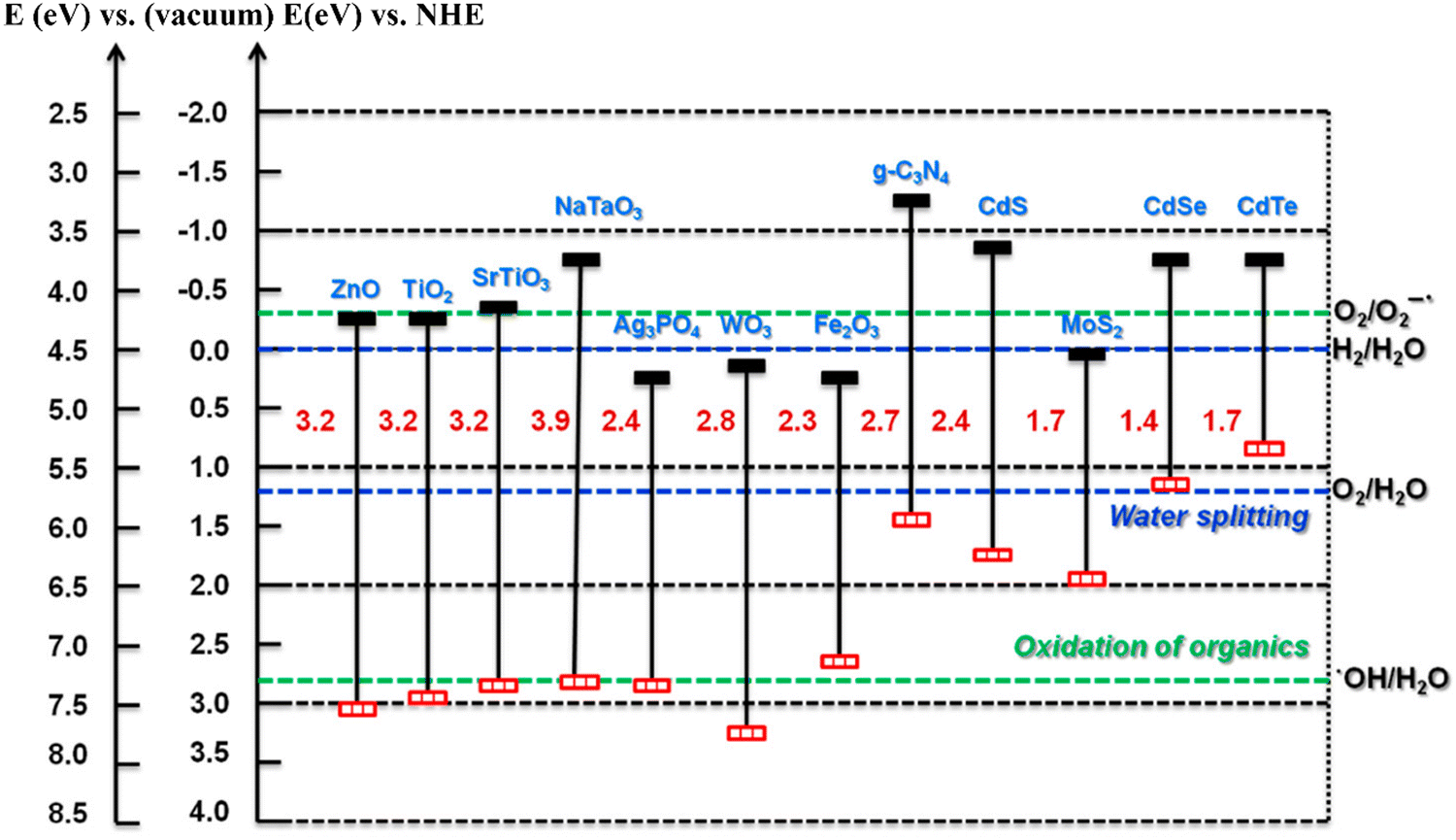 | ||
| Fig. 2 Band alignment of different semiconductors. Reproduced with permission from ref. 56. | ||
The photocatalytic activity of TiO2 has been largely explored. For example, in a study conducted by X. Lang et al., TiO2 was used with peroxo species laterally anchored and loaded with small amounts of Brønsted–Lowry acids on the surface. Upon irradiation, this system weakens the O–O bonds, and through the O2 transfer pathway, generates oxidizing species of interest, such as superoxide radicals, resulting in improved catalytic activity without affecting selectivity.57 Similarly, C. Xu et al. employed carbide/titanium dioxide nanowire (Ti3C2/TiO2) photocatalysts for the selective oxidation of aromatic alcohols, achieving benzyl alcohol conversions of 92%, attributed to the introduction of carbide, which in turn contributes to the reduction of the bandgap and improves light absorption. Additionally, charge separation is enhanced due to the Schottky barrier between Ti3C2 and TiO2 nanowires.60 C. M. Crombie et al. used bimetallic (Pd–Fe) catalysts supported on TiO2 for the synthesis of benzaldehyde, where H2O2 was generated in situ from molecular H2 and O2, obtaining alcohol oxidation rates higher than those observed in materials composed of Pd–Au or Pd.61 However, this material presents some difficulties related to the bandgap, which reaches a value of 3.2 eV,59,62 limiting the material's activity to only under ultraviolet light, resulting in low reaction efficiency and selectivity, and it does not act alone with molecular oxygen, suggesting that this type of catalyst has limited catalytic activity.63,64
Meanwhile, H. Li et al. have worked with other semiconductors such as BiOCl, which has been modified with Au and used for the oxidation of benzyl alcohol, under O2 as the electron acceptor. This material proved to be effective as it contains oxygen vacancies that allow electron capture and transfer. This, in turn, facilitates interaction with adsorbed oxygen, generating superoxide radicals (O2˙−).65 WO3 has also been widely studied. For instance, a study conducted by O. Tomita et al. includes the use of this catalyst loaded with PdO2 as a co-catalyst, reporting a selectivity of 80%, improved values compared to catalysts such as platinum supported on tungsten trioxide, and palladium and platinum supported on titanium dioxide (Pt/WO3, Pd/TiO2 and Pt/TiO2). The study reveals a particular advantage in containing molecular O2, offering the opportunity to function in aqueous environments. The use of ferrites in the oxidation of alcohols to aldehydes has been studied by X. Zhu et al., where they synthesized copper ferrite (CuFe2O4) nanoparticles stable to oxygen and easily reusable due to their magnetic properties. Ferrites were particularly attractive due to their ability to act in an aqueous medium, without requiring the addition of any base, ligand, or additive.66 A study conducted by S. Gyu Lee et al. includes the synthesis of CdS-based materials, modified with Au and S for the photocatalytic oxidation of benzyl alcohol. The catalysts were prepared through a cation exchange procedure, and a selectivity of 99% was obtained, influenced by the exchanged cationic species and their improved transfer of photogenerated electrons and holes.67
Other materials such as MOFs have been used. For example, W. Jumpathong et al. started with iron acetylacetonate to incorporate iron into the Zr node of MOF-808, performing an exchange of the –OH group in the Zr node and the acetylacetonate ligand, obtaining a metal–organic framework based on iron (Fe-MOF-808) for the oxidation of benzyl alcohol. Finally, conversions and selectivity of 99% and 96%, respectively, were obtained, which are high values compared to those of materials such as metal–organic frameworks based on iron, derived from UiO-66 (Fe-UiO-66).68,69
The use of metal semiconductors other than TiO2 presents difficulties. These materials require meticulous control during synthesis to generate vacancies in the crystal structure and obtain the necessary morphologies. Additionally, synthesis requires the use of pricey metallic reagents. Similarly, metal–organic frameworks (MOFs) face difficulties related to cycles of reuse, as they are prone to leaching of their metallic species. This hinders the development of effective photocatalytic and quantum reactions, significantly obstructing their practical applications. Therefore, there is an urgent need for a more robust alternative photocatalytic system. In this context, graphitic carbon nitride g-C3N4 emerges as a promising option since it represents a metal-free photocatalyst that can be prepared from abundant natural resources such as melamine or urea.69
3. Carbon nitride: a non-metal semiconductor
Graphitic carbon nitride (g-C3N4) is one example of the earliest known polymers, typically featuring a stoichiometric composition of 3C:4N, with sp2-bonded nitrogen forming aromatic ring structures.70,71 It was first synthesized in 1834 by Jöns Jacob Berzelius and Justus Von Liebig, who named it “melon”. The approximate elemental composition was defined by a stoichiometric ratio between carbon and nitrogen 3:4.72,73 In the 1980s, g-C3N4 gained importance after a theoretical article was published proposing the hypothesis that cubic carbon nitride could contain a solid structure, like or superior to that of diamond. Therefore, during this decade, efforts were focused on obtaining this ordered solid, but this objective was not achieved.74 Consequently, the study took another direction, focusing on the synthesis of carbon nitrides (CxNyHz).75 Subsequently, a study based on computational simulations, conducted by Liu and Cohen, demonstrated the existence of five different phases of carbon nitride, focusing on a graphitic phase, which they identified as one of the most stable allotropes under ambient conditions.76 In the mid-2000s, g-C3N4 began to be used in the field of heterogeneous catalysis, and later, Lotsch et al. experimentally discovered the exact structure of melon, describing it as melamine (tri-s-triazine) monomers with NH bridges linked with hydrogen in a zigzag geometry, naming it polymeric carbon nitride (PCN).77,78 It was not until 2009 that a polymeric photocatalyst study conducted by Wang et al. made a breakthrough in the field. Thermally polymerized g-C3N4 undergoes water-splitting half reactions under visible light in the presence of sacrificial agents. Since then, there has been an increase in the number of articles globally containing the keywords metal-free and inorganic conjugated polymers.77,79
g-C3N4 is employed in photocatalytic processes due to its remarkable thermal, optical, electrical, structural, and chemical properties. Its unique electronic structure gives it excellent thermal stability, withstanding temperatures up to 600 °C. This material possesses outstanding optoelectronic properties, related to its medium optical band gap value estimated at 2.7 eV and its valence and conduction band potentials of +1.6 eV and −1.1 eV vs. normal hydrogen electrode (NHE), respectively. These values make the material sensitive to visible light, specifically at a wavelength lower than 460 nm, allowing catalytic activity to be achieved with lower energy input.77,80
The morphology of g-C3N4 can be tuned, allowing the formation of nanosheets, nanotubes, and quantum dot nanostructures. Additionally, this material can be synthesized through simple and low-cost methods, starting from abundant carbon- and nitrogen-rich precursors. This reduces synthesis costs and, being metal-free, ensures its benign decomposition, making it environmentally friendly and biocompatible.32,81
g-C3N4 has emerged as a highly versatile material capable of addressing critical challenges in chemical transformation, the development of environmentally friendly processes, and advancing sustainability goals. Its unique properties, including a tunable electronic structure, high chemical stability, and metal-free composition, make it particularly attractive for a wide range of applications. As a photocatalytic material, g-C3N4 has gained vast attention due to its ability to harness solar energy for processes such as water splitting, CO2 reduction, N2 fixation, and pollutant degradation. In fact, the number of publications involving it has risen exponentially to over ten thousand articles in the last decade, reflecting substantial progress in understanding and optimizing its photocatalytic behaviour. This growing body of research highlights g-C3N4's potential to drive innovations in renewable energy, environmental remediation, and sustainable chemical production, making it a key material for addressing global energy and environmental challenges.
3.1. Structural properties
g-C3N4 is a polymeric material containing sp2-hybridized carbon and nitrogen atoms with covalent bonds organized in a flat pseudohexagonal network. This structure comprises trigonal planar geometry and the formation of delocalized π–π bonds, endowing it with favourable electrical conductivity and optical characteristics.82 This polymer is of great interest due to its graphite-like layered structure. Each layer is composed of building blocks, either s-triazine (C3N3) or tris-s-triazine (C6N7) units or even a mixture of them, see Fig. 3. Tri-s-triazine, also known as heptazine, is the most stable phase at room temperature. Intraplanar, these units are linked by tertiary amines, while the layers are held together by van der Waals forces, forming a three-dimensional porous structure like a honeycomb. Graphitic carbon nitride comprises interlayer distances ranging from 0.315 nm to 0.335 nm, facilitating the homogeneous insertion of various chemical compounds on its surface.83–85 Although this material possesses unique properties, it still presents some disadvantages that limit its efficiency in the photoproduction of aldehydes, such as its low surface area and high charge recombination rate. Therefore, modifications are necessary to optimize its characteristics. The strategies include various methods such as heterojunction construction, morphology control, functionalization, and defect creation. | ||
| Fig. 3 (A) Triazine and (B) tri-s-triazine (heptazine) structures of g-C3N4. Reproduced with permission from ref. 77. | ||
3.2. Modifications of graphitic carbon nitride
 | ||
| Fig. 4 Scheme of four different possibilities when coupling medium and large band gap SCs. Heterojunctions type I and II, Z-scheme dynamics, and Schottky junction. Reproduced with permission from ref. 88. | ||
This approach provides key advantages, such as enabling photosensitization by extending the light absorption of the wide-bandgap semiconductor into the visible range and enhancing charge transfer kinetics. g-C3N4 is characterized by forming heterojunctions with other semiconductors through non-covalent bonds. The development of these strategies results in improved energy bands, charge separation, and energy level modification, obtaining materials with excellent redox properties. A study conducted by Z. Tong et al. describes the formation of an isotopic tubular g-C3N4 heterojunction through a two-step synthesis that includes a hydrothermal process (molecular self-assembly) followed by calcination. The procedure begins with the hydrothermal treatment of melamine and cyanuric acid, with the set of hexagonal prismatic crystals. In the second step, it is calcined to finally form the tubular g-C3N4 heterojunction. The results show that the heterojunction design improved the transfer of photogenerated electrons and holes, resulting in better performance.90
In another study, Q. Shi et al., prepared covalent heterojunctions in the form of hierarchical microflowers of g-C3N4/TiO2(B), denoted as CNT-x (where x represents the nominal mass percentage of g-C3N4). These materials were synthesized using an in situ hydrothermal exfoliation and assembly method, as shown in Fig. 5A, and were employed for the photooxidation of benzyl alcohol under visible light. The results demonstrated improved catalytic activity, attributed to excellent charge separation, which favoured the selective formation of benzaldehyde without additional byproducts. The conversion of benzyl alcohol after 3 hours with pristine TiO2 was negligible, while g-C3N4 achieved 12%. In contrast, the CNT-x compounds showed significantly superior performance, achieving a maximum conversion of 100% in the CNT-0.14 sample.91
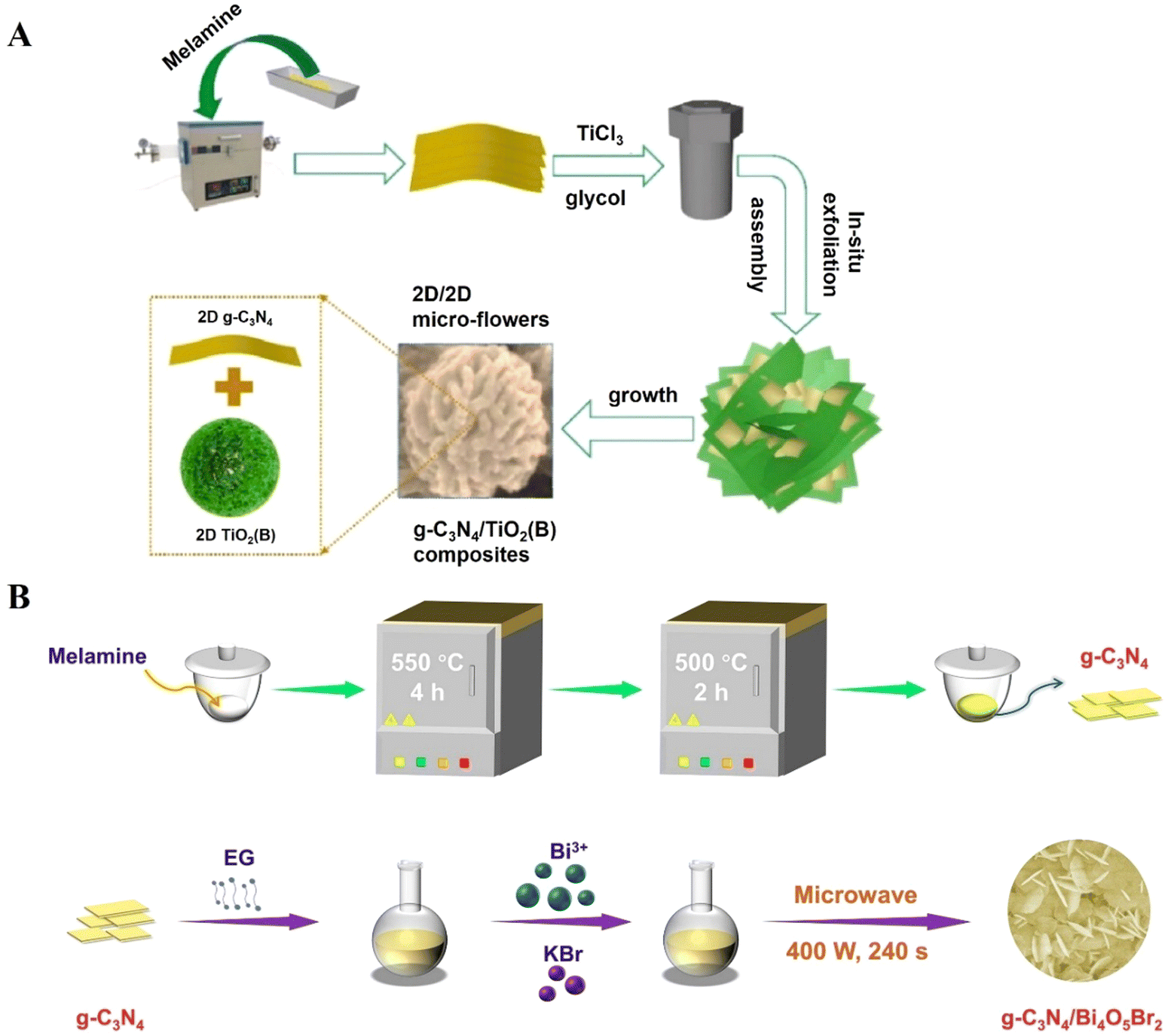 | ||
| Fig. 5 (A) Schematic synthesis procedure of the gC3N4/TiO2(B) covalent heterojunction, reproduced with permission from ref. 91, and (B) the g-C3N4/Bi4O5Br2 noncovalent heterojunction, reproduced with permission from ref. 92. | ||
The formation of heterojunctions through non-covalent interactions was studied by C. Li et al., where graphitic carbon nitride/bismuth oxybromide (g-C3N4/Bi4O5Br2) was synthesized using a microwave-assisted method (see Fig. 5B), resulting in a Z-scheme heterojunction held together by weak van der Waals interactions. This material demonstrated great potential in the selective photocatalytic conversion of benzyl alcohol, achieving reaction rates 9.2 and 5.3 times higher than those observed for pure g-C3N4 and Bi4O5Br2, respectively. Additionally, a maximum selectivity of 99% was achieved. The radical formation mechanism was investigated through electron spin resonance and radical quenching, showing that the main reactive species involved in the alcohol oxidation reaction were superoxide radicals (O2˙−) and holes (h+).92
 | ||
| Fig. 6 Overview of traditional structure-control of the g-C3N4-based system. | ||
Quantum dots are structures with sizes smaller than 10 nm. In recent years, they have been implemented due to their morphology, which allows for effective performance in various photocatalytic applications. These tiny particles can efficiently absorb light and easily generate electron–hole pairs. Additionally, their high specific surface area maximizes the availability of active sites for chemical reactions.94 In a study conducted by R. Bayan et al., oxygenated graphitic carbon nitride quantum dots were synthesized, supported on bio-based, metal-free hyperbranched polyurethane. This material was used as a bifunctional catalyst in organic transformations such as the oxidation of benzyl alcohol to benzaldehydes and the reduction of benzaldehydes to benzyl alcohol. The synthesized material demonstrated improved photocatalytic performance, achieving up to 90% productivity in both reactions under sunlight. Besides its enhanced catalytic activity, the photocatalyst proved to be attractive due to its reusability in multiple cycles, promoting sustainable and environmentally friendly processes.95
Furthermore, the formation of g-C3N4 nanotubes leads to hollow structures that offer unique advantages, such as increased light absorption and scattering capacity, as well as a lower recombination rate of photogenerated electron–hole pairs. These structures can be obtained by hard-template, soft-template, and self-template methods.96 For example, in a study carried out by Y. Ding et al., a heterojunction was synthesized between g-C3N4 nanotubes and Cs3Bi2Br9 nanoparticles (CBB-NP@-tube-CN) for the selective oxidation of benzyl alcohol. The material synthesis procedure involved hydrothermal treatment, calcination, and the incorporation of Cs3Bi2Br9 (CBB) into the g-C3N4 nanotubes, as shown in Fig. 7A. The tubular morphology, with abundant pores in the walls, was confirmed through TEM, SEM, and HAADF-STEM imaging, as illustrated in Fig. 7B–E. The optimized heterojunction, containing 15 wt% CBB, demonstrated high photo-oxidation potential, a Z-scheme charge migration pathway, and structural advantages that resulted in accelerated kinetics. This enabled a benzyl alcohol conversion rate of 19, 14, and 3 times higher than that of g-C3N4 nanotubes, CBB, and CBB with g-C3N4, respectively.97
 | ||
| Fig. 7 (A) Schematic diagram for the synthesis of the CBB-NP@P-tube-CN composite. (B) SEM, and (C) and (D) TEM and HAADF-STEM images of the tube-CN sample. (E) TEM image. Reproduced with permission from ref. 97. | ||
The formation of two-dimensional nanosheets is a strategy of particular interest as it generates materials with high specific areas, superior electronic mobility, and the creation of a greater number of active surface sites, resulting in improved photocatalytic performance.98 In a study conducted by W. Zhang et al., porous g-C3N4 nanosheets (PCNS) were synthesized by thermal polymerization followed by thermal oxidation etching. The obtained PCNS presented an average pore diameter of 24.3 nm and a thickness of approximately 5 nm. The synthesized materials showed high surface areas and excellent separation of photogenerated charge carriers. The material was evaluated in benzaldehyde production, and it was observed that PCNS accelerated the conversion of alcohol to aldehyde, obtaining an almost quantitative yield of 99% of benzaldehyde in 12 hours. PCNS was then compared with pure g-C3N4, and it was found that the latter material is usually less effective in terms of conversion, obtaining conversions close to 50% of benzyl alcohol.99
Similarly, X. Li et al. synthesized a heterostructure composed of lanthanum vanadate and g-C3N4 nanosheets (LaVO4/CN) by a two-step process, including a calcination and hydrothermal method. These heterostructures were used as bifunctional materials in the photocatalytic evolution of H2 and the production of furfural from biomass-derived furfuryl alcohol, see Fig. 8A. The latter reaction was carried out to consume the h+ present in the photocatalytic system and thus avoid the rapid recombination of the induced photocarriers, which tends to disrupt the effective conversion of solar energy to hydrogen.100 According to the obtained results, it could be observed that the LaVO4/CN heterojunction achieved a three times higher H2 evolution rate (0.287 mmol g−1 h−1) and a higher furfural production rate (0.95 mmol g−1 h−1) than that of pure graphitic carbon nitride (CN). Finally, an apparent quantum efficiency (AQE) of H2 evolution of 22.16% irradiated at 400 nm was obtained.100
 | ||
| Fig. 8 (A) Schematic illustration of fabricating the 2D/2D LaVO4/CN heterostructure for efficient photocatalytic H2-evolution coupled with high value-added furfural production. Reproduced with permission from ref. 100 and (B) photocatalytic H2 production coupled with benzyl alcohol oxidation. Reproduced with permission from ref. 102. | ||
The formation of spheres has also reported excellent performance. The structural arrangement of the spheres fully exposes the active sites on the semiconductor surface, shortens the electron transfer distance, and eventually improves light utilization efficiency, allowing incident light to be reflected and refracted multiple times within the sphere. For example, in a study conducted by G. Sun et al., a bifunctional photocatalyst composed of g-C3N4 nanospheres, on which cadmium zinc sulfide (CdZnS) quantum dots were deposited, was employed, resulting in an inorganic–polymeric S-scheme structure. This material was used for H2 evolution and furfural production. The results showed that the H2 evolution rate improved and the conversion of furfuryl alcohol increased to 84.2%. The S-scheme heterojunction was confirmed by electron paramagnetic resonance (EPR) and theoretical calculations (DFT), highlighting advantages such as charge separation and enhanced redox capacity of the carrier.101 In another study, F. Zhang et al. developed similar morphological structures, synthesizing hierarchical mesoporous spheres of g-C3N4 doped with phosphorus and sulfur. The sulfur-doped spheres offered notable advantages due to their bifunctionality, see Fig. 8B, facilitating the simultaneous production of hydrogen and benzaldehyde. This material achieved a hydrogen production rate 13.2 times higher than pure g-C3N4 and a benzaldehyde production rate of 3.87 μmol h−1. These properties significantly stand out compared to non-doped materials, making them an ideal combination for H2 generation and the chemical production of high-value-added organic compounds.102
Oxidation reactions are carried out to introduce oxygen functional groups. This procedure is performed using oxidants that react with g-C3N4. Common oxidants include NaOH, oxalic acid, CrO3, K2CrO7, H2SO4, and HNO3. This strategy was employed by G. Dong et al., where oxygen-functionalized graphitic carbon nitride was obtained using H2O2, which proved effective in anoxic photo-oxidation, increasing its mineralization and contaminant degradation constants by approximately 18 and 7 times under visible light, respectively.104
The incorporation of functional groups other than oxygen has proven to be a promising strategy to improve charge separation. The inclusion of –SO3H conducted by Quintana et al. into g-C3N4 prepared from melamine calcination with chlorosulfonic acid improved the kinetics of benzyl alcohol oxidation in aqueous solution and boosted the selectivity toward benzaldehyde. Terminal sulfonic groups were anchored to N2C positions, leading to better separation of photogenerated charges and a minor recombination rate.105
Amide functionalization is achieved by amidation reactions on the surface of graphitic carbon nitride through an amide bond (NNH–CO). A study conducted by S. Zhao et al. demonstrated the covalent bonding between graphitic carbon nitride nanosheets and carbon nanotubes through the amidation reaction. This functionalization showed efficient catalytic performance for the photocatalytic production of H2O2 in the presence of formic acid under visible light.106 The non-covalent interaction technique is an attractive approach due to its accessibility and ease of forming physical bonds and weak molecular interactions. Therefore, it is widely used to functionalize g-C3N4. A study conducted by E. Da Silva et al. reported the synthesis of porphyrin-sensitized carbon nitride through impregnation via non-covalent interactions, to be evaluated in hydrogen generation from water splitting. Different types of porphyrins with various carboxyl substituents and porphyrin positions on the periphery were used to observe the influence of these parameters on the photocatalytic H2 production performance. As a result, the best performance was obtained with the meso-tetrakis(meta-carboxyphenyl)porphyrin (mTCPP) hybrids, generating 326 and 48.4 μmol of H2 under 6 hours of UV-vis and visible light irradiation, respectively.107
In a study conducted by X. Bao et al., g-C3N4 with nitrogen vacancies and a KOH mass of y: 1, 5, 7 or 10 mg (CNx−y), was synthesized and its performance in the photocatalytic oxidation of benzyl alcohol was evaluated. Theoretical and experimental results demonstrated that the presence of nitrogen vacancies (NV) promotes the formation of electron traps, which facilitates the adsorption and activation of benzyl alcohol. Additionally, the hypothesis was confirmed that O2, essential for the formation of more selective radicals such as superoxide, are adsorbed at the NV sites. For this purpose, Pd/CNx−5 was prepared using the photoreduction method, where Pd particles were selectively deposited at the NV sites. The results showed that the Pd-modified material significantly inhibited photocatalytic performance, achieving conversions that were twice as low as the CNx−5 sample.109 Similarly, a study conducted by J. Goclon et al. investigated the influence of nitrogen vacancies on the selective oxidation of aromatic alcohols using materials with and without defects. The DFT method was used to understand the nature of the active sites, the structures of the intermediates, and the transition states. The results showed that the existence of vacancies favoured the initial interaction of alcohol and oxygen molecules with the surface through the formation of covalent bonds. Additionally, it was found that vacancy defects narrowed the band gap and thus improved the production of photogenerated charge carriers. In the case of the defective material, the surface interaction with O2 and ˙OOH occurred through covalent bonds, while in defect-free g-C3N4 these interactions were limited to van der Waals forces on the surface. A transition state barrier value of 1.65 eV was obtained for defect-free g-C3N4, approximately 1 eV higher than the sample with nitrogen vacancies, indicating that the defective catalyst facilitates the formation of benzaldehyde more efficiently.110
Clusters are the aggregation of defects, especially when the chemical agent used tends to form aggregates rather than being homogeneously distributed across the graphitic layers. In a study conducted by K. Trivedi et al., a ternary heterojunction of reduced graphene oxide functionalized with graphitic carbon nitride and deposited bismuth vanadate (B/CN@rGO) was synthesized for the selective photo-oxidation of benzyl alcohol driven by visible light and an eco-friendly oxidant like H2O2. High-resolution transmission electron microscopy (HR-TEM) revealed the formation of BiVO4 clusters, which were crucial for achieving 100% selectivity towards benzaldehyde. This catalyst demonstrated excellent thermal stability and could be reused for more than five cycles without affecting the selectivity of the product.111 A study conducted by S. Hu et al. reports on the anchorage of phosphorus through substitutions and modifications at interstitial sites in g-C3N4 using diammonium hydrogen phosphate and dicyandiamide and ionic liquid (BMIM-PF6) as precursors. The material was evaluated for the oxidation of Rhodamine B under visible light, and it was found that the material modified at the interstitial sites was more effective if compared to the substitutionally modified material.112 These types of strategies could also be suitable for developing systems aimed at the selective production of aldehydes.
The formation of interstitial sites involves the introduction of atoms into the spaces between the atoms of the original lattice, creating lattice strains and additional energy states, which result in the modification of charge movement. As schematized in Fig. 9, the two possible interstitial sites where the modification of graphitic carbon nitride based on tri-s-triazine could be made are shown. Moreover, atomic substitutions occur when an atom in the crystal lattice is replaced, driven by the difference in ion size or charge, which in turn generates deformations in the crystal lattice and modifies the charge distribution. As stated in Fig. 9, the five possible substitution sites in the structure of graphitic carbon nitride based on tri-s-triazine are shown, labelled as N1, N2, N3, C1, and C2.
 | ||
| Fig. 9 Five substitutional sites and two interstitial sites in tri-s-triazine-based g-C3N4. Reproduced with permission from ref. 113. | ||
Surface oxygenation has been shown to reduce photocatalytic activity in terms of alcohol conversion, but to increase selectivity toward aldehyde production. In this sense, Marcì et al. reported this behaviour when treating g-C3N4 with H2O2 during hydrothermal synthesis evidenced by a poor mobility of carriers.116 Boron has been largely used as a dopant agent in graphitic carbon formulas. For instance, K2B4O7·4H2O and KBH4 were used as boron precursors to modify the g-C3N4 structure by Lou et al.117 The better production of benzaldehyde reported by these authors was attributed not only to boron linked to the sp2 N positions but also to the nitrogen vacancies formed on the surface after doping. In another study with B doping published by Quintana et al., B-induced defects exchanging C and N positions of g-C3N4 depending on the nature of the B precursor.118 Thus, the elemental B had a preference to exchange positions with N atoms, whereas the use of NaBH4 prefers the insertion of B into the C positions. This difference significantly affected the photocatalytic activity during the selective oxidation of benzyl alcohol, with the latter showing the highest effectiveness. The S doping also has reported beneficial results. The modification with Na2S2O3 of g-C3N4 formulas suggests a better charge separation of the photogenerated charges after S incorporation in N2C positions, minimizing the recombination rate of photogenerated charges.119
3.2.5.1. Metal doping. Metal doping involves the inclusion of metal atoms into the lattice to serve as novel active catalytic sites, which modifies the charge distribution on the surface of g-C3N4 and creates two possible energy levels: one where donor levels are formed and another where acceptor levels are formed. These changes improve the mobility of charge carriers and reduce the band gap. This results in enhanced light absorption capacity, increased adsorption of reactants on the surface, and reduced recombination of photogenerated charges.120 Metal doping of g-C3N4 can be carried out through various processes, such as photo-deposition, hydrothermal treatments, chemical reduction approaches, and high-temperature condensation of individual molecule precursors or inorganic metal salts.121 The metals used for doping include transition metals, rare earth metals, alkali metals, and alkaline earth metals, such as iron, zinc, ruthenium, cobalt, nickel, titanium, and copper.122
Within metal doping, transition metals and rare earth metals stand out due to their strong interaction with g-C3N4. Rare earth metals are of particular interest because they possess unoccupied 4s and 5s orbitals, which can act as electron capture centres, allowing control over the formation of highly reactive radicals that negatively impact the reaction. Doping with transition and rare earth metals results in reduced band gaps and a red-shifted absorption edge.123 The use of transition metals was developed by J. Gao et al., with Fe-doped g-C3N4 nanosheets through a one-step synthesis procedure. In this process, NH4Cl was used as a dynamic gas template and FeCl3 as the iron source, as shown in Fig. 10. The incorporation of Fe3+ was achieved through the formation of bonds with N atoms, resulting in better visible light absorption and a reduced band gap. A specific surface area of 236.5 m2 g−1 was achieved, a value 2.5 times greater than that of g-C3N4 nanosheets. As a result, higher catalytic performance was achieved with the sample modified with 0.5 wt% FeCl3, capable of splitting water under visible light.124
 | ||
| Fig. 10 Synthesis process of Fe-doped g-C3N4 nanosheets. Reproduced with permission from ref. 124. | ||
However, M. Ismael performed Ni doping of g-C3N4 assisted by ultrasound in a single step, intending to improve the photocatalytic hydrogen evolution. Different doping percentages with Ni (1.5–20%) were applied, using nickel acetate as the precursor. An improvement in catalytic activity was observed for the sample modified with 2.4 wt% Ni, resulting in greater visible light absorption and enhanced charge transfer.125 A recent study by Juan Carlos Colmenares et al. explores the effect of copper incorporation into graphitic carbon nitride to elucidate the influence in the selective photocatalytic production of benzaldehyde. Loading 3 wt% of Cu significantly enhances the activity under both UV and visible light and results in nearly 100%, with a high yield-to-power ratio (0.35 mmol g−1 h−1 W−1). Ab initio simulations indicate that Cu+ species act as hole traps through the formation of mid-gap states, promoting the selective oxidative reactions.126
Another attractive option for metal doping is using alkali and alkaline earth metal ions such as Mg+, Li+, K+, and Na+, among others. This group of metals presents low toxicity and costs and they have a high capacity to donate electrons. These electrons overlap with the upper and lower layers of g-C3N4, creating electron or hole traps, resulting in a lower charge recombination rate. These modifications lead to electron loss and delocalization, directly affecting the energy band structure of the catalyst. Specifically, for Li+ and Na+, the modifications occur in the N-plane voids due to their small atomic radius and weak metallic properties.123 In a study conducted by S. Wu et al., sodium-doped g-C3N4 nanotubes (Na-CNT) were synthesized in two steps, as shown in Fig. 11. In the first step, a sodium-doped melamine cyanurate intermediate was formed through self-assembly between melamine and cyanuric acid in a NaOH solution via hydrothermal treatment. In the second step, the intermediate was subjected to microwave-assisted thermal treatment to produce Na-CNT. It was found that sodium improved the attraction force between the layers and induced various structural defects.127
 | ||
| Fig. 11 Illustration of the formation process of Na-CNTs. Reproduced with permission from ref. 127. | ||
3.2.5.2. Non-metal doping. Non-metal doping emerged as an effective alternative to overcome some limitations associated with metal doping, such as leaching, thermal instability, self-degradation, and corrosion. Non-metal elements have proven to be of great interest due to their intrinsic properties, such as high ionization energy and high electronegativity, making them highly compatible with g-C3N4. It is important to highlight the significance of using a dopant with a lower electronegativity value than the substituted element, as this effectively reduces the band gap.128,129 These properties favour the incorporation of atoms into the crystal lattice through covalent interactions, promoting the delocalization of conjugated π electrons and integrating these atoms without excessively distorting the structure. This modification leads to the formation of new energy levels and modifies properties such as conductivity and electrical mobility, which in turn improves the performance of redox reactions.129,130 Some non-metal heteroatoms studied for the modification of g-C3N4 include oxygen, sulfur, phosphorus, boron, fluorine, iodine, and bromine, along with self-doping with elements such as carbon and nitrogen. These elements are introduced from various precursors and under different types of atmospheres.120,123,131
The use of non-metallic atoms in photocatalytic applications has gained significant attention, with photocatalytic alcohol oxidation being a prominent example. A notable study by M. Bellardita et al. involved the synthesis of pure and phosphorus-doped graphitic carbon nitride (g-C3N4) through the thermal condensation of melamine, urea, or thiourea. These materials were evaluated in the selective oxidation of benzyl alcohol, 4-methoxybenzyl alcohol, and piperonyl alcohol under both ultraviolet and visible irradiation. In general, it was observed that the presence of phosphorus in the g-C3N4 structure enhanced aldehyde selectivity, while alcohol conversion decreased slightly. Additionally, the partial oxidation reaction followed the order 4-MBA > PA > BA, indicating that the presence of the methoxy substituent in the aromatic ring facilitates the partial photocatalytic oxidation of the aromatic alcohol to aldehyde.130
3.2.5.3. Multi-doped strategies of carbon nitride. Co-doping with two or more atoms has been employed as an excellent strategy to increase surface area, reduce charge recombination rates, and decrease the bandgap energy as well.132 For example, in a study by Huang et al., g-C3N4 co-doped with phosphorus and oxygen was prepared, where phosphorus atoms substituted carbon sites and oxygen atoms substituted nitrogen sites. This resulted in improved properties such as a reduced bandgap, better charge separation, and an increased specific surface area. Similarly, D. Sheng et al. synthesized a bimetallic Bi–Mo photocatalyst, denoted as Bi–Mo/CN-x, where x represents the amount of Na2MoO4 added, which was used in the selective photooxidation of benzyl alcohol under visible light in an aqueous solution, using air as the oxidizing agent. The optimal catalyst, Bi–Mo/CN-2, showed benzyl alcohol conversions of 51% and a maximum selectivity of 99.9%. During the reaction, O2 is reduced on the surface, generating superoxo (
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Bi–OO˙), peroxo (Bi–OOH), and peroxo (Mo–O2) radicals, which then selectively oxidize benzyl alcohol through the hydrogen abstraction pathway.133
Bi–OO˙), peroxo (Bi–OOH), and peroxo (Mo–O2) radicals, which then selectively oxidize benzyl alcohol through the hydrogen abstraction pathway.133
4. Activity and selectivity evaluation of the photocatalytic oxidation of alcohols
4.1. Reaction rates and selectivity to aldehyde
Table 1 compares several critical parameters to evaluate the efficiency of various photocatalysts, including conversion (XA %), selectivity (SAD %), and quantum efficiency (QE %). The type of illumination (UV vs. visible light) significantly impacts photocatalyst efficiency, with UVA generally showing high conversion and selectivity, while some photocatalysts under visible light (or solar-type schemes), like Ru/CN-x,134 achieve selectivity over 99%. This type of result is common, and in several instances, the decrease in activity under visible light conditions is accompanied by an increase in selectivity. As is well-known, the conversion parameter is influenced by factors such as the initial reactant concentration, making it difficult to properly compare catalytic results obtained under different operating conditions. Quantum efficiency (QE), defined as the number of reaction events (reaction rate) per absorbed photon (photon rate), is crucial for standardized comparison, considering both reaction and photon absorption efficiency.105,118,135–137 Besides, selectivity analysis highlights its importance in aldehyde reactions due to the 1:1 stoichiometry of alcohol to aldehyde conversion.135 In these cases, it is not necessary to include the selectivity factor when a different number of photons are required per mole of the produced/consumed component. The calculation of QE, recommended by IUPAC,138 provides a common basis for comparison, considering both the reaction rate and photon absorption rate. The Local Volumetric Rate of Photon Absorption (LVRPA) can be calculated from the Radiative Transfer Equation, considering the spatial distribution of light intensity and absorption by the photocatalyst.139–141 The development of a strict radiation model is not a simple task, and in liquid phase and suspension catalyst systems, its strict determination may require a multidimensional approach that is not always developed by researchers conducting photocatalytic studies. Comprehensive analysis based on QE data is essential for optimizing photocatalytic processes and developing more efficient photocatalysts for practical applications. The sample with the best QE is P/CN with a value of 0.206%, which represents an increment factor of approximately 6.87 compared to pure CN.142 It should also be noted that the QE values reported are relatively low, which limits the possibility of scaling up this application beyond initial research (laboratory scale). Therefore, further work is needed to achieve efficiencies that make this technology potentially scalable.
| Structure | Photocatalyst | Radiation source, initial concentration alcohol, product | XA (%) | SAD (%) | QE (%) | Ref. |
|---|---|---|---|---|---|---|
| Pristine | g-C3N4 | UVA (365 nm), 0.5 mM, benzaldehyde | 14.6 | 33.0 | 0.030 | 143 |
| Nitride carbon graphitic bulk (M/g-C3N4) | UVA (365 nm), 0.5 mM, cinnamaldehyde | 81.0 | 13.0 | n.d. | 116 | |
| Metallic doping | Fe/g-C3N4 | UVA (365 nm), 1.5 mM, benzaldehyde | 20.0 | 70.0 | n.d. | 144 |
| Cu/g-C3N4 | visible light (465 nm), 1 mM, benzaldehyde | 25 | 96 | n.d. | 126 | |
| Ru/g-C3N4-x | Visible light (λ 420nm), 95 mM, benzaldehyde |
n.d. | >99 | 0.480 | 134 | |
| NaOH/g-C3N4 | UVA (365 nm), 0.5 mM, 4-methoxybenzaldehyde | 61.0 | 62.0 | n.d. | 145 | |
| Au/M g-C3N4 (melamine precursor) | UVA (365 nm), 24.7 mM, benzaldehyde | 65.0 | 60.0 | n.d. | 146 | |
| Au/U g-C3N4 (urea precursor) | UVA (365 nm), 24.7 mM, benzaldehyde | 75.0 | 86.0 | n.d. | 146 | |
| Non-metallic doping | B/g-C3N4 | UVA (365 nm), 0.7 mM, benzaldehyde | 15.0 | 30.0 | 0.046 | 118 |
| P/g-C3N4 | UVA (365 nm), 1 mM, cinnamaldehyde | 53.0 | 49.2 | 0.206 | 147 | |
| P/g-C3N4 | Visible light (λ > 400nm), 1 mM, cinnamaldehyde |
25.5 | 16.1 | n.d. | 147 | |
| S/g-C3N4 | UVA (365 nm), 0.5 mM, benzaldehyde | 53.0 | 63.6 | 0.124 | 105 | |
| S/U g-C3N4 (urea precursor) | UVA (365 nm), 1 mM, vanillin | n.d. | 5.0 | n.d. | 119 | |
| CSA/g-C3N4; modified with camphor sulfonic acid | Visible light (λ > 400 nm), 1670 mM, benzaldehyde | 100 | 99.2 | n.d. | 148 | |
| DMF/g-C3N4; modified with N,N-dimethylformamide | UVA (365 nm), 0.5 mM, 4-methoxybenzaldehyde | 43.0 | 79.0 | n.d. | 145 | |
| M g-C3N4/TE-H2O2; hydrogen peroxide adduct | UVA (365 nm), 0.5 mM, 4-methoxy benzaldehyde | 82.0 | 92.0 | n.d. | 116 | |
| Co-doped | NaB/g-C3N4 | UVA (365 nm), 0.7 mM, benzaldehyde | 29.0 | 36.0 | 0.153 | 118 |
| NaB/g-C3N4/Ru | UVA (365 nm), 0.5 mM, benzaldehyde | 24.3 | 67.5 | 0.130 | 149 | |
| Heterojunction | CdS/g-C3N4 | visible light (λ > 420 nm), 25.5 mM, p-methoxybenzaldehyde | 55.0 | 97.0 | n.d | 150 |
| I-TiO2/g-C3N4 | Visible light (λ ≥ 400 nm), 33.3 mM, benzaldehyde | 25.0 | >99.0 | n.d. | 151 | |
| WOx/g-C3N4 | Visible light (λ 420nm), 100 mM, benzaldehyde |
32.1 | 98.1 | n.d. | 152 | |
| BiOCl/P-g-C3N4 | Visible light (λ 420nm), 50 mM, benzaldehyde |
55.2 | >99 | n.d. | 153 | |
| BiOCl/RGO/P g-C3N4 | Visible light (λ 420nm), 50 mM, benzaldehyde |
71.6 | >99 | n.d. | 153 | |
| FP/g-C3N4; modified with ZnFe2O4 and TCPP | visible light (λ > 420 nm), 40 mM, benzaldehyde | 96.0 | 99.0 | n.d. | 154 | |
| Co3O4/TS-g-C3N4; triple-shelled carbon nitride | solar light irradiation, 100 mM, benzaldehyde | 85.0 | >99 | n.d. | 155 |
4.2. Mechanistic discussion
The photocatalytic oxidation of alcohols is a complex process influenced by reactive oxygen species (ROS) and the photocatalyst's surface properties. Upon light absorption by g-C3N4, electrons are excited from the valence band to the conduction band, generating e−/h+ pairs. These pairs, along with ROS like hydroxyl radicals (HO˙), superoxide anion (O2˙−), and hydroperoxyl radical (˙OOH), drive the redox reactions necessary for selective alcohol oxidation.156–159 The oxidation of alcohols to aldehydes typically follows two main pathways. In the first, HO˙ radicals or photoinduced holes abstract hydrogen, creating radical intermediates like benzyloxy radicals (Ph-CH2O˙) or alkyl radicals, which further react with O2 or other ROS to form transient intermediates such as diols that dehydrate into aldehydes. In the second, the alcohol's hydroxyl group is deprotonated to form an alkoxide anion, which reacts with photoinduced holes to create a transient anionic radical. This radical is subsequently oxidized by photogenerated holes or O2˙−, resulting in aldehyde formation.99,133,160The adsorption of alcohol molecules on the g-C3N4 surface is crucial for the reaction pathway, with hydrogen bonding and electrostatic interactions enhancing it and increasing the local alcohol concentration at the catalyst surface. In this context, doping could improve e−/h+ pair separation by introducing localized electronic states, reducing recombination, and boosting ROS generation efficiency.
The alcohol oxidation process is often challenged by competing side reactions, such as overoxidation to carboxylic acids or complete mineralization to CO2. Achieving selective oxidation to aldehydes requires carefully optimizing reaction conditions and regulating ROS activity to minimize these undesired pathways. The use of g-C3N4 offers a promising route, leveraging superior solar light absorption for greener, more sustainable photocatalysis.161
In the context of photocatalysis, the electronic structure and interfacial charge dynamics of g-C3N4 have been the subject of extensive study. For example, Solís et al. investigated the photocatalytic oxidation of benzyl alcohol (BA) using NaBCN-1Ru, prepared by mixing g-C3N4 and NaBH4 in a 1:1 mass ratio, grinding the mixture in a mortar, and subsequently calcining it at 450 °C (heating rate: 10 °C min−1, for 1 hour). The study used chemical scavengers to pinpoint which reactive oxygen species drive the photocatalytic oxidation of benzyl alcohol. Removing dissolved O2 (by N2 bubbling) reduced the reaction rate by 44%, while p-benzoquinone (p-BQ) cut it by 81%. However, since p-BQ also reacts with hydroxyl radicals (HO˙) and is photolyzed under UVA radiation, tiron was used instead, reducing the rate by 36% and confirming that superoxide radicals (O2˙−) play an important role, not exclusive though. HO˙ had a minor impact—tert-butyl alcohol (TBA) reduced the rate by only 18% while scavenging photogenerated holes (h+) with oxalic acid lowered the rate by 64%, identifying h+ as the primary reactive species. Moreover, scavengers for O2˙− slightly increased benzaldehyde selectivity, whereas suppressing h+ decreased it, underscoring the pivotal role of h+ in selective oxidation.149
Furthermore, Solís et al. analyzed the band alignment of B-doped g-C3N4, with and without 1% of metallic Ru, to clarify ROS behaviour. XPS data revealed that the presence of Ru lowered the bandgap from 2.6 eV (NaBCN) to 2.3 eV (NaBCN-1Ru), stabilizing the valence band and improving the redox potential of photogenerated holes. This modification aligned with the scavenger study findings, supporting the role of Ru in enhancing photocatalytic performance.162 Their proposed mechanism links band structure optimization and ROS activity, highlighting Ru's role in stabilizing holes and enhancing selective benzaldehyde production.149
Moreover, the work of M. Alejandra Quintana et.al. also focuses on the photocatalytic oxidation of benzyl alcohol to benzaldehyde, this time using the metal-free boron-modified g-C3N4 counterpart and emphasizing the influence of reaction media on selectivity and activity. The study examines the trade-offs between selectivity and sustainability when using organic solvents like acetonitrile (ACN) versus water. ACN enhances selectivity by reducing the formation of non-selective HO˙ and O2˙−, which are more selective. Additionally, ACN increases the dissolved O2 compared to water, further driving the desired reaction toward benzaldehyde formation. While ACN (20%) improves selectivity, reaching 82%, it negatively impacts reaction rates due to the scavenging effect on HO˙, inhibiting further oxidation. In contrast, water yields higher reaction rates for both benzyl alcohol degradation and benzaldehyde formation, despite lower selectivity. Mechanistic insights into ROS were gained also through scavenger studies using methanol, oxalic acid, and nitrogen purging. Methanol, a scavenger of hydroxyl radicals, slightly altered the benzyl alcohol degradation profile but significantly improved benzaldehyde selectivity, indicating that HO˙ negatively impacts benzaldehyde formation. O2 purging with N2 reduced benzyl alcohol conversion by 50%, highlighting the essential role of O2˙−. Oxalic acid, used to trap photogenerated holes (h+), accelerated alcohol degradation, achieving complete conversion in four hours. Aldehyde selectivity initially increased to 63% but declined to 23% as benzyl alcohol conversion progressed, due to oxalate decomposition generating electrons that enhanced charge separation but led to overoxidation at longer reaction times. This study highlights the balance between reaction media and ROS activity to optimize selectivity and efficiency in photocatalysis.118
In another example, Shifu Chen and colleagues investigated g-C3N4/ZnIn2S4 (CN/ZIS) composite photocatalysts prepared via a two-step method (Fig. 12) in the oxidation of benzyl alcohol. The photocatalytic activity showed an “M” type trend with increasing g-C3N4 content, attributed to a p–n junction influencing photogenerated charge carrier migration under visible light. The band gaps of ZIS (2.31 eV) and g-C3N4 (2.75 eV) enabled distinct charge transfer pathways. When ZIS dominated, a p-CN/n-ZIS junction directed electrons from the CB of g-C3N4 to ZIS and holes from the VB of ZIS to g-C3N4, enhancing aromatic alcohol oxidation and O2˙− formation on ZIS. When g-C3N4 predominated, a Z-scheme mechanism emerged, where VB holes in ZIS showed strong oxidizing power, and CB electrons in g-C3N4 reduced O2 to O2˙−. Higher ZIS content improved activity via O2˙− radical production, while dominant g-C3N4 leveraged the Z-scheme for strong redox performance. This work offers insights into optimizing heterojunction photocatalysts for selective oxidation.163
 | ||
| Fig. 12 Proposed transfer mechanisms of the photogenerated charge carriers over the CN/ZIS composite sample under visible light irradiation. The main constituent of the composite photocatalyst is the ZIS sample (A) and the main constituent of the composite photocatalyst is the CN sample (B). Reproduced with permission from ref. 163. | ||
Expanding on this, Palmisano et al. investigated the photocatalytic performance of bare and phosphorus-doped g-C3N4 (P-g-C3N4) for selectively oxidizing benzyl alcohol, 4-methoxybenzyl alcohol (4-MBA), and piperonyl alcohol (PA) to their respective aldehydes. They observed that phosphorus doping improved the selectivity of the reaction towards benzaldehyde under UV light irradiation, although the conversion of benzyl alcohol decreased. Conversely, under visible light irradiation, the presence of phosphorus was detrimental to both conversion and selectivity. This reduction in photocatalytic performance under visible light was attributed to the electron-trapping behaviour of the phosphorus species within the g-C3N4 framework, which reduced the availability of electrons in the conduction band, thus impairing the formation of superoxide radical species, crucial for oxidation. The authors also noted that the modification of the photocatalyst surface area and the interaction between the material and the substrate played a significant role in these results.130
In addition to this, Afsaneh Rashidizadeh's study explored the photocatalytic oxidation of various substituted benzyl alcohols to aldehydes using iodine-doped titanium oxide (I-TiO2) and g-C3N4 nanosheets (CNNSs) as photocatalysts. The results revealed that benzyl alcohols with electron-donating groups, such as 2,4-dimethoxy, p-OH, p-OCH3, and p-CH3, showed higher conversion rates compared to those with electron-withdrawing groups like p-Cl, p-NO2, o-Cl, and p-F. The increased conversion of alcohol with electron-donating substituents was attributed to the enhanced electron density in the –CH2OH group, facilitating active hole and radical formation, which drives the oxidation reaction. Based on these findings, the authors proposed a mechanism for the photo-oxidation of benzyl alcohols over I-TiO2/(20 wt%)CNNSs, which involves several steps under visible light illumination. Initially, CNNSs are excited, generating electrons and holes. To enhance charge separation, electrons from the conduction band of CNNSs transfer to the CB of I-TiO2, while holes from the valence band of I-TiO2 migrate to the VB of CNNSs. The electrons in the CB of I-TiO2 reduce O2 to O2˙−. These radicals scavenge protons from benzyl alcohol, producing ˙OOH and an alkoxide anion as intermediates. The basic nature of CNNSs promotes the deprotonation of benzyl alcohol. The alkoxide anions are oxidized by holes from the VB of I-TiO2 to form radicals, which react with the ˙OOH radical to yield the corresponding aldehyde. Alternatively, the alkoxide anion can react with ˙OOH to form an anion radical and H2O2, with the final oxidation of the anion to benzaldehyde by holes from I-TiO2. To validate the mechanism, various scavengers were tested. Adding p-BQ significantly reduced the conversion of benzyl alcohol to 5%. Introducing potassium persulfate (K2S2O8), which traps photoexcited electrons, led to a slight increase in conversion to 7%, highlighting O2˙− importance. N2 bubbling to purge O2 lowered conversion to 11%, emphasizing the role of O2. The use of ammonium oxalate, a hole scavenger, and tert-butanol reduced conversions to 14% and 16%, respectively, indicating that HO˙ had a minor role. These findings confirm that O2˙−, e−, and holes were the primary active species in the oxidation process,151 in accordance with similar reports in the literature for g-C3N4/TiO2 composites.91
Furthering this understanding, a study by Shifu Chen et al. offered valuable insights into the photocatalytic mechanism for the selective oxidation of alcohols together with the reduction of nitrobenzene using a CdS/g-C3N4 composite photocatalyst. Under visible light, both g-C3N4 and CdS are photo-excited, generating electrons in their conduction bands and holes in their valence bands. Thanks to their well-matched band structures and close interface contact, the photoexcited electrons from the CB of g-C3N4 transfer to the CB of CdS, while holes from the VB of CdS move to the VB of g-C3N4. This efficient charge separation reduces recombination and enhances the photocatalyst's performance. In an N2-purging anaerobic atmosphere, nitrobenzene reduction proceeds without interference from oxidation reactions. The reduction mechanism follows a six-electron transfer process, converting nitrobenzene to aniline. At the same time, the photogenerated holes oxidize alcohols by abstracting hydrogen atoms, resulting in aldehyde formation. The coupling mechanism dictates that for every mol of nitrobenzene reduced, six moles of electrons and six moles of holes are required, leading to the production of three moles of aldehyde. However, experimental results show slight deviations from these theoretical values due to system lag effects. To confirm the efficient separation of photogenerated charges, photoluminescence (PL) spectra were analyzed. The PL spectra exhibited a strong emission peak around 450 nm, corresponding to the recombination of electron–hole pairs in g-C3N4. Importantly, the PL intensity of the CdS (10.0 wt%)/g-C3N4 composite was significantly lower than that of pure g-C3N4, indicating reduced recombination and enhanced charge separation in the composite photocatalyst.150
Furthermore, Benxia Li and colleagues have made significant progress in engineering interfacial active sites in metal-modified semiconductor photocatalysts to enhance their performance in photocatalytic reactions. They developed a Ru/g-C3N4−x photocatalyst, comprising Ru nanoparticles (NPs) anchored on defective g-C3N4−x nanoflakes with nitrogen vacancies (VNs), for hydrogen evolution and benzyl alcohol oxidation reactions (Fig. 13A and B). Mechanistic studies revealed that both water and benzyl alcohol were essential for the redox coupling reaction. Control experiments showed negligible hydrogen or benzaldehyde formation in pure water, while methanol enhanced hydrogen production but could not drive the full reaction. Scavenger tests identified holes and hydroxyl radicals as critical for benzyl alcohol oxidation, with electrons essential for H2 evolution. Radical-initiated pathways were confirmed using TEMPO, supported by in-situ EPR measurements detecting carbon-cantered and HO˙. In addition, density functional theory (DFT) calculations provided insights into the active sites. VN defects on g-C3N4−x enhanced adsorption and activation of water and benzyl alcohol, while interfacial Ru sites strongly interacted with reactants. The VN defects facilitated selective oxidation, and Ru NPs promoted hydrogen evolution.134
 | ||
| Fig. 13 Schematic diagrams of A: photoinduced charge transfer/separation at the Ru/g-C3N4−x interface and B: photocatalytic mechanism of H2 evolution coupled with selective benzyl-alcohol oxidation over the Ru/g-C3N4−x catalyst. Reproduced with permission from ref. 134. C: proposed mechanisms of the photocatalytic selective oxidation of BA to BAD over N-deficient g-C3N4. Reproduced with permission from ref. 109. | ||
Besides, Xiaolei Bao and colleagues have investigated the role of nitrogen vacancies in graphitic carbon nitride (g-C3N4) to enhance the photocatalytic selective oxidation of benzyl alcohol (Fig. 13C). They developed nitrogen-deficient g-C3N4 samples (CNx−5) that demonstrated superior conversion efficiency and product selectivity under visible light irradiation compared to pristine g-C3N4. Combining experimental characterization and DFT calculations, it was revealed that VNs significantly enhanced carrier separation and served as active sites for the adsorption and activation of reactants. The authors found that VNs play a pivotal role in the adsorption and activation of O2 molecules. Theoretical analyses indicated that O2 is preferentially adsorbed at VNs sites due to the similar atomic radius and geometry of nitrogen and oxygen atoms. To confirm this, they synthesized a Pd-modified nitrogen-deficient g-C3N4 photocatalyst (Pd/CNx−5) via a photoreduction method. The photogenerated electrons in CNx−5 were localized at VN sites, enabling selective deposition of Pd nanoparticles, which subsequently blocked the VNs. This led to a notable decrease in O2 adsorption and a corresponding reduction in conversion efficiency, validating the critical role of VNs in photocatalytic performance. Further analysis revealed that VNs modulate the electronic structure of CNx−5, resulting in a red-shifted absorption spectrum and a slightly reduced band gap, e.g. 2.78 eV for CNx−5vs. 2.80 eV for pristine g-C3N4. These changes enhance the oxidation potential of valence band holes, accelerating the conversion of benzyl alcohol to activated benzyl alcohol radicals. In situ FTIR spectroscopy supported these findings, showing stronger alcohol adsorption on CNx−5, with shifts in the VC–O bond indicating VN-assisted activation. To elucidate the role of VNs in charge carrier dynamics, the authors conducted photoluminescence (PL), time-resolved fluorescence (TRPL), and photoelectrochemical measurements. CNx−5 displayed weaker PL emission, shorter fluorescence lifetimes, and higher photocurrent responses compared to pristine g-C3N4, indicating suppressed electron–hole recombination and enhanced carrier separation. Electrochemical impedance spectroscopy (EIS) and Mott–Schottky analysis further confirmed the improved charge carrier density and transport efficiency in CNx−5. Bao and colleagues proposed a detailed mechanism for the selective oxidation of benzyl alcohol on CNx−5. Both benzyl alcohol and O2 molecules are adsorbed at NV sites, which enhances O2 activation and O2˙−. The strong oxidation power of valence band holes oxidizes BA to alcohol cation radicals, which react with O2˙− to form the aldehyde. Thus, VNs play dual roles by enhancing the adsorption and activation of O2 and boosting the oxidation capability of valence band holes, accelerating the reaction.109
Moreover, a novel approach was presented by Sergei G. Zlotin and colleagues, regarding the use of g-C3N4 photocatalysts in supercritical carbon dioxide (scCO2) for efficient, green oxidation of benzyl alcohol to benzaldehyde and benzoic acid derivatives (Fig. 14). Supercritical CO2 is an effective green solvent with unique properties that enhance photo-induced oxidation reactions, such as its transparency to visible and near-UV light and high miscibility with molecular oxygen, which increases oxygen concentration. Notably, scCO2 can induce in situ exfoliation of g-C3N4 catalysts at pressures above 10 MPa, eliminating the need for a separate exfoliation step in catalyst preparation. These properties make scCO2 a highly attractive medium for photochemical reactions involving g-C3N4, providing both environmental and practical benefits. In this case, the mechanism involves a hybrid photocatalytic and autocatalytic radical process driven by reactive oxygen species. The process begins with g-C3N4 photocatalytically activating molecular oxygen to form superoxide, which abstracts a proton from benzyl alcohol, creating a hydroperoxide radical and alcoholate intermediate. The alcoholate is further oxidized by g-C3N4's photoinduced holes, generating an O-centered radical that undergoes hydrogen atom transfer with the hydroperoxide radical, forming hydroperoxide and benzaldehyde. The reaction is self-accelerating, with water facilitating the formation of ROS like hydroperoxide and hydroxyl radicals, which further oxidize benzyl alcohol and transform benzaldehyde into benzoic acid. Experimental confirmation showed that adding water increased conversion rates and maintained high selectivity by generating ROS that accelerated the reaction. ROS scavenging studies provided further evidence for in situ formation of active species like hydroperoxide radicals. The combined photocatalytic and autocatalytic pathways ensure efficient conversion while preventing overoxidation. This method leverages g-C3N4 catalysis, ROS generation, and scCO2 as a sustainable medium, offering an efficient and environmentally sustainable approach to green chemical synthesis.164
 | ||
| Fig. 14 Plausible hybrid autocatalytic mechanism of benzyl alcohol photo-oxidation on g-C3N4 in scCO2 medium. Reproduced with permission from ref. 164. | ||
Equally important, the oxidation of cinnamyl alcohol (CA) has also been investigated using g-C3N4 photocatalysts. In more detail, the photocatalytic oxidation mechanism of CA, using phosphorus-doped g-C3N4, has been investigated by M. Alejandra Quintana, focusing on the role of reactive oxygen species. The presence of O2˙− in the reaction was confirmed using p-BQ and tiron, with tiron reducing the CA oxidation rate by 50%. A N2 atmosphere also slowed the oxidation rate, highlighting the critical role of superoxide radicals. The involvement of photogenerated holes was studied by adding oxalic acid, which decreased the reaction kinetics, indicating their importance. Additional tests with EDTA, a hole scavenger, further confirmed the role of photogenerated holes in CA oxidation. The mechanism was concluded to involve holes creating organic radicals, which are oxidized to cinnamaldehyde by holes or superoxide radicals. LC-ESI(+)-QTOF analysis identified cinnamaldehyde and cinnamic acid as the main products, with minimal formation of epoxide or other by-products (see Fig. 15).147
 | ||
| Fig. 15 Proposed degradation mechanism of cinnamyl alcohol over P-CN-4%. Reproduced with permission from ref. 147. | ||
Expanding on this, Hongbing Ji and colleagues recently reported the selective oxidation of cinnamaldehyde to benzaldehyde using boron-doped graphitic carbon nitride as a photocatalyst, see Fig. 16. Boron was introduced from either potassium borohydride (KBH4) or potassium tetraborate tetrahydrate (K2B4O7·4H2O), with KBH4-doped g-C3N4 (KBCN) demonstrating superior catalytic performance. The proposed mechanism for cinnamaldehyde oxidation under visible light involves the synergistic effects of boron dopants in KBCN. Upon light irradiation, photoexcited electrons are generated, which are captured by adsorbed oxygen, producing superoxide radicals. Simultaneously, holes interact with the π electrons of the carbon–carbon double bond in cinnamaldehyde, forming carbocations and carbon-centered radicals. These radicals then react with O2˙− to form peroxide intermediates, which undergo ring-opening cleavage to yield benzaldehyde. The boron dopants in KBCN also contribute to the generation of singlet oxygen (1O2), enhancing the reaction efficiency and contributing to side reactions.117
 | ||
| Fig. 16 The proposed mechanism underlying the photocatalytic action of KBCN in oxygen-guided selective oxidation of cinnamaldehyde under visible light irradiation. Reproduced with permission from ref. 117. | ||
Another interesting example was presented by Qing Yang and colleagues, introducing a Ni–Au-modified g-C3N4 photocatalyst (Ni–Au/CN) for simultaneous hydrogen (H2) production and furfural synthesis via selective oxidation of furfuryl alcohol in aqueous solution (Fig. 17). The Ni–Au/CN photocatalyst showed enhanced performance due to the synergy between plasmonic Au nanoparticles (NPs) and Ni species. Au NPs enhanced visible light absorption and promoted electron–hole separation, while Ni sites acted as electron traps and active sites for proton reduction, promoting H2 evolution. Upon light irradiation, g-C3N4 nanosheets generated electron–hole pairs, and Au NPs produced “hot charge carriers” through their localized surface plasmon resonance (LSPR) effect. The generated electrons migrated to Ni sites, where they were trapped and used in proton reduction to release H2. At the same time, photogenerated holes remained on g-C3N4 and oxidized hydroxyl ions and furfural alcohol molecules, generating ROS like HO˙ and carbon-centered radicals, further facilitating the oxidation of furfural alcohol to produce furfural. Control experiments and in situ electron spin resonance (ESR) studies confirmed the roles of HO˙ and holes in the reaction. Suppression of these species led to a significant decrease in both H2 production and furfural synthesis. Fluorescence experiments with coumarin confirmed superior HO˙ production by Ni–Au/CN compared to pure g-C3N4, further supporting the enhanced photocatalytic activity. The mechanism was further investigated using linear sweep voltammetry (LSV), which revealed that Ni–Au/CN had a higher current density and lower overpotential for proton reduction, indicating the high activity of Ni sites. The LSPR effect of Au NPs increased electron density at Ni sites, boosting H2 evolution. Additionally, the dissociation of H2O and furfural alcohol provided protons and hydroxyl ions, supporting both H2 production and selective oxidation. This dual-function system demonstrates the synergy between LSPR-enhanced electron dynamics, Ni–Au interfacial interaction, and ROS-driven oxidation, enabling efficient and sustainable H2 production and furfural synthesis.165
 | ||
| Fig. 17 Photocatalytic mechanism for the simultaneous production of H2 and furfural from a furfural-alcohol aqueous solution over the Ni–Au/CN photocatalyst. Reproduced with permission from ref. 165. | ||
5. Summary and perspectives
The extrapolation of photocatalysis to organic oxidation reactions, such as the oxidation of alcohols to aldehydes, leads to opportunities for real applications following the green chemistry principles. The process does not require consumable chemicals, only the presence of an active solid and radiation that can be harvested from the solar spectrum. The use of g-C3N4-based photocatalysts is an attractive reaction platform for the oxidation of alcohols, even in a water medium, due to an appropriate band alignment of these semiconductors, limiting the release of HO˙ from water molecule oxidation, therefore, and the overoxidation reported by other active photocatalysts. The modification of the structure to enhance the photo-generated charge separation has been demonstrated to be effective in the improvement of bare graphitic carbon formulas. Hence, the modification of textural, optical, and electronic properties results in a positive photocatalytic response. The non-metal doping, metal deposition, and heterojunction strategies are suitable strategies with demonstrated promising results. However, the development of bimetallic or trimetallic photocatalysts, and their plausible synergistic effects, have been tested little in this kind of reaction.The pivotal role played by the superoxide radical and photo-generated holes in g-C3N4-based materials has been widely investigated. However, most research is focused on chemical scavenger tests, whose interpretation is rather controversial and a challenge in most cases due to the lack of selectivity toward a specific ROS, their plausible interaction in adsorption and photolysis steps. In this sense, the in situ characterization may help to better elucidate and corroborate the explored mechanism. Paramagnetic resonance would enforce the detection of ROS. In addition, the in situ evaluation of the interaction onto the surface with techniques such as operando FTIR, Raman, and X-ray absorption spectroscopy would enforce the mechanical aspects of the interaction of the alcohols, ROS, and the surface of the photocatalyst. Furthermore, whether the changes are reversible or not, the characterization of the reaction course is of paramount relevance to comprehend the more affected properties of the reaction media or the influence on the oxidative pathway.
To date, photocatalytic production of aldehydes from the oxidation of their alcohols has been conducted at the lab-scale, focusing on the assessment of the modification of a target photocatalyst. As with other applications involving photocatalysts, the bottle-neck from which the technology suffers is regarding the design of a photoreactor with efficient photon uptake which requires the evaluation of the novel photocatalyst under well-defined conditions and calculation of photon fluence and absorption. Besides, the recovery of the solid after use requires further implementation, such as magnetization, or immobilization of the photocatalyst. The stability of the solid after multiple reuse cycles is commonly evaluated based on its photocatalytic activity, complemented by post-reaction characterization of the materials to detect potential structural changes, sintering, or lixiviation phenomena. In addition, the performance assessment under realistic operating conditions needs further development. In this sense, artificial intelligence is a powerful tool to be explored to develop a rational design of the process, for which a powerful storage database is essential.
This work provides a basic overview of the main strategies for aldehyde production, along with a detailed analysis of photocatalytic approaches for synthesizing valuable aldehydes such as vanillin, benzaldehyde, and cinnamaldehyde. The aim is to help systematize the production process based on a critical assessment of the current methodologies, highlighting both their potential and limitations. By offering a structured perspective, this study seeks to guide future developments toward more efficient, selective, and sustainable pathways for aldehyde synthesis.
Data availability
This review article does not contain new data, software or code that has been generated by the authors. All the data referenced in this paper are available in the original publications that are cited in the reference list.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from PID2022-139014OB-I00/SRA (State Research Agency)/10.13039/501100011033.501100011033. D. R.-P. received funding from the European Union′s Horizon2020 research and innovation program under the Marie Sklodowska-Curie Cofound Grant Agreement No. 945361. P.J-C. received funding from the European Union's Horizon Europe research and innovation program under the Marie Skłodowska-Curie grant agreement no. 101068996.References
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS
.
- K. C. Nicolaou, Proc. R. Soc. London, Ser. A, 2014, 470, 20130690 CAS
- K. R. Campos, P. J. Coleman, J. C. Alvarez, S. D. Dreher, R. M. Garbaccio, N. K. Terrett, R. D. Tillyer, M. D. Truppo and E. R. Parmee, Science, 2019, 363, eaat0805 CrossRef CAS
- S. Vijayraghavan and N. Saini, Chem. Res. Toxicol., 2023, 36, 983–1001 Search PubMed
- A. Meza, M. E. Campbell, A. Zmich, S. A. Thein, A. M. Grieger, M. J. McGill, P. H. Willoughby and A. R. Buller, ACS Catal., 2022, 12, 10700–10710 Search PubMed
- R. M. LoPachin and T. Gavin, Chem. Res. Toxicol., 2014, 27, 1081–1091 Search PubMed
- A.-S. Paschke, D. Selishchev, M. Lyulyukin and D. Kozlov, Mol. Catal., 2022, 524, 112263 Search PubMed
- M. A. Floss, T. Fink, F. Maurer, T. Volk, S. Kreuer and L. M. Müller-Wirtz, Molecules, 2022, 27, 5258 Search PubMed
- E. A. Verochkina, N. V. Vchislo and I. B. Rozentsveig, Molecules, 2021, 26, 4297 Search PubMed
- H. Zhao, H. P. Caldora, O. Turner, J. J. Douglas and D. Leonori, Angew. Chem., Int. Ed., 2022, 61, e202201870 CrossRef CAS
- A. Böhme, N. Ulrich and G. Schüürmann, Chem. Res. Toxicol., 2023, 36, 1055–1070 Search PubMed
- Straits Research Pvt. Ltd, Aldehyde Market Size, Share|Growth Report 2031, https://straitsresearch.com/press-release/global-aldehyde-market-share, (accessed 5 February 2025).
- X.-Q. Huang, R. Li, J. Fu and N. Dudareva, Nat. Commun., 2022, 13, 1352 Search PubMed
-
A. Paula Dionísio, G. Molina, D. Souza de Carvalho, R. dos Santos, J. L. Bicas and G. M. Pastore, Natural Food Additives, Ingredients and Flavourings, Elsevier, 2012, pp. 231–259 Search PubMed
- C. M. Cova, A. Zuliani, M. J. Muñoz-Batista and R. Luque, Green Chem., 2019, 21, 4712–4722 RSC
- M. Ramasamy, J.-H. Lee and J. Lee, Colloids Surf., B, 2017, 160, 639–648 CrossRef CAS PubMed
- 24chemicalresearch, Cinnamaldehyde Market – Global Outlook and Forecast 2022–2028, https://www.24chemicalresearch.com/reports/129834/global-cinnamaldehyde-market-2022-2028-133, (accessed 5 February 2025).
- N. J. Gallage and B. L. Møller, Mol. Plant, 2015, 8, 40–57 CrossRef CAS
- F. Liaqat, L. Xu, M. I. Khazi, S. Ali, M. U. Rahman and D. Zhu, Ind. Crops Prod., 2023, 204, 117372 CrossRef CAS
- P. Dočolomanský, V. Sitkey and I. Čičová, Eur. Pharm. J., 2022, 69, 34–41 CrossRef
- H. B. Bang, Y. H. Lee, S. C. Kim, C. K. Sung and K. J. Jeong, Microb. Cell Fact., 2016, 15, 16 CrossRef
- H. G. Lee, Y. Jo, K. Ameer and J. H. Kwon, Food Sci. Biotechnol., 2018, 27, 1607–1617 CrossRef CAS
- R. M. Yitbarek, H. Admassu, F. M. Idris and E. G. Fentie, Appl. Biol. Chem., 2023, 66, 1–13 Search PubMed
- E. Hofmann, P. Degot, D. Touraud, B. König and W. Kunz, Food Chem., 2023, 417, 135944 CrossRef CAS
- D. Ribeaucourt, B. Bissaro, F. Lambert, M. Lafond and J.-G. Berrin, Biotechnol. Adv., 2022, 56, 107787 CrossRef CAS PubMed
- R. Zhang, W.-Q. Liu, S. Ling and J. Li, J. Agric. Food Chem., 2023, 71, 8551–8557 CrossRef CAS
- N. Gopalan and K. M. Nampoothiri, Biocatal. Agric. Biotechnol., 2018, 15, 304–310 CrossRef
- N. J. Claassens, S. Burgener, B. Vögeli, T. J. Erb and A. Bar-Even, Curr. Opin. Biotechnol, 2019, 60, 221–229 CrossRef CAS
- R. Maurya, H. Patel, D. Bhatt, S. Shakhreliya, N. Gohil, G. Bhattacharjee, N. L. Lam, K. J. Alzahrani, E. Gyanchander and V. Singh, Recent Adv. Food Biotechnol., 2022, 139–159 Search PubMed
- Y. Takakura, T. Ono, K. Danjo and H. Nozaki, Biosci., Biotechnol., Biochem., 2022, 86, 1718–1725 CrossRef PubMed
- F. Luziatelli, L. Brunetti, A. G. Ficca and M. Ruzzi, Front. Bioeng. Biotechnol., 2019, 7, 482112 Search PubMed
- M. J. Lima, A. M. T. Silva, C. G. Silva and J. L. Faria, J. Catal., 2017, 353, 44–53 CrossRef CAS
- J. Son, J. H. Jang, I. H. Choi, C. G. Lim, E. J. Jeon, H. Bae Bang and K. J. Jeong, Microb. Cell Fact., 2021, 20, 145 CrossRef CAS PubMed
- M. Edwards, P. M. Rourk, P. G. Riby and A. P. Mendham, Tetrahedron, 2014, 70, 7245–7252 CrossRef CAS
- R. Ciriminna, A. Fidalgo, F. Meneguzzo, F. Parrino, L. M. Ilharco and M. Pagliaro, ChemistryOpen, 2019, 8, 660–667 CrossRef CAS
- N. Lee, Y. T. Kim and J. Lee, Polymers, 2021, 13, 364 CrossRef CAS
- Y. Zhu, Y. Liao, W. Lv, J. Liu, X. Song, L. Chen, C. Wang, B. F. Sels and L. Ma, ACS Sustainable Chem. Eng., 2020, 8, 2361–2374 CrossRef CAS
- A. Kaufmann, L. Maier and M. Kienberger, Sep. Purif. Technol., 2024, 340, 126780 CrossRef CAS
- A. M. Kunjapur, Y. Tarasova and K. L. J. Prather, J. Am. Chem. Soc., 2014, 136, 11644–11654 CrossRef CAS
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS
- A. S. Sharma, H. Kaur and D. Shah, RSC Adv., 2016, 6, 28688–28727 RSC
- J. K. Mobley and M. Crocker, RSC Adv., 2015, 5, 65780–65797 RSC
- H. G. Jin, P. C. Zhao, Y. Qian, J. D. Xiao, Z. S. Chao and H. L. Jiang, Chem. Soc. Rev., 2024, 53, 9378–9418 RSC
- U. Caudillo-Flores, M. J. Muñoz-Batista, M. Fernández-García and A. Kubacka, Catal. Rev.: Sci. Eng., 2024, 66, 531–585 CrossRef CAS
- A. Kubacka, M. Fernández-García and G. Colón, Chem. Rev., 2012, 112, 1555–1614 CrossRef CAS PubMed
- A. Balapure, J. Ray Dutta and R. Ganesan, RSC Appl. Interfaces, 2024, 1, 43–69 RSC
- K. Takanabe, ACS Catal., 2017, 7, 8006–8022 CrossRef CAS
- P. Jiménez-Calvo, V. Caps and V. Keller, Renewable Sustainable Energy Rev., 2021, 149, 111095 CrossRef
- F. Mohamadpour and A. M. Amani, RSC Adv., 2024, 14, 20609–20645 RSC
- S. N. Ahmed and W. Haider, Nanotechnology, 2018, 29, 342001 CrossRef
- S. Zhu and D. Wang, Adv. Energy Mater., 2017, 7, 1700841 CrossRef
- D. Friedmann, A. Hakki, H. Kim, W. Choi and D. Bahnemann, Green Chem., 2016, 18, 5391–5411 RSC
- S. Bai, J. Jiang, Q. Zhang and Y. Xiong, Chem. Soc. Rev., 2015, 44, 2893–2939 RSC
- D. Tang, G. Lu, Z. Shen, Y. Hu, L. Yao, B. Li, G. Zhao, B. Peng and X. Huang, J. Energy Chem., 2023, 77, 80–118 CrossRef CAS
- M. J. Muñoz-Batista, M. M. Ballari, A. Kubacka, O. M. Alfano and M. Fernández-García, Chem. Soc. Rev., 2019, 48, 637–682 RSC
- S. Kumar, S. Karthikeyan and A. F. Lee, Catalysts, 2018, 8, 74 CrossRef
- X. Lang, W. Ma, C. Chen, H. Ji and J. Zhao, Acc. Chem. Res., 2014, 47, 355–363 CrossRef CAS
- M. Ramalingam, D. Kamaraj, S. R. R. Senthil Kumar, R. Raghavi and D. Rajalinggam, Mater. Today Proc., 2023 DOI:10.1016/j.matpr.2023.07.331
- M. Ge, Z. Hu, J. Wei, Q. He and Z. He, J. Alloys Compd., 2021, 888, 161625 CrossRef CAS
- C. Xu, F. Yang, B. Deng, Y. Zhuang, D. Li, B. Liu, W. Yang and Y. Li, J. Catal., 2020, 383, 1–12 CrossRef CAS
- C. M. Crombie, R. J. Lewis, R. L. Taylor, D. J. Morgan, T. E. Davies, A. Folli, D. M. Murphy, J. K. Edwards, J. Qi, H. Jiang, C. J. Kiely, X. Liu, M. S. Skjøth-Rasmussen and G. J. Hutchings, ACS Catal., 2021, 11, 2701–2714 CrossRef CAS
- Y. Cui, Z. Ding, P. Liu, M. Antonietti, X. Fu and X. Wang, Phys. Chem. Chem. Phys., 2012, 14, 1455–1462 RSC
- S. Singh, R. Singhal, R. K. Yadav and N. K. Gupta, Diamond Relat. Mater., 2024, 149, 111609 CrossRef CAS
- M. A. Alotaibi, A. I. Alharthi, T. F. Qahtan, S. Alotibi, I. Ali and Md. A. Bakht, J. Alloys Compd., 2024, 978, 173388 CrossRef CAS
- H. Li, F. Qin, Z. Yang, X. Cui, J. Wang and L. Zhang, J. Am. Chem. Soc., 2017, 139, 3513–3521 CrossRef CAS PubMed
- X. Zhu, D. Yang, W. Wei, M. Jiang, L. Li, X. Zhu, J. You and H. Wang, RSC Adv., 2014, 4, 64930–64935 RSC
- S. G. Lee, M. J. Kang, M. Park, K. Kim, H. Lee and H. S. Kim, Appl. Catal., B, 2022, 304, 120967 CrossRef CAS
- W. Jumpathong, T. Pila, Y. Lekjing, P. Chirawatkul, B. Boekfa, S. Horike and K. Kongpatpanich, APL Mater., 2019, 7, 111109 CrossRef
- G. Kumar, G. Bhargava, S. Kumar, J. K. Rajput, B. Singh, P. Singh and R. Kumar, Inorg. Chim. Acta, 2024, 563, 121935 CrossRef CAS
- V. W. Lau and B. V. Lotsch, Adv. Energy Mater., 2022, 12, 2101078 CrossRef CAS
-
I. F. Teixeira and P. Jiménez-Calvo, Renewable Energy Production and Distribution, Elsevier, 2023, vol. 2, pp. 145–180 Search PubMed
- F. Fina, S. K. Callear, G. M. Carins and J. T. S. Irvine, Chem. Mater., 2015, 27, 2612–2618 CrossRef CAS
- P. Sun, C. He, C. Zhang, H. Xiao and J. X. Zhong, Phys. B, 2019, 562, 131–134 CrossRef CAS
- N. Lopez-Salas, J. Kossmann and M. Antonietti, Acc. Mater. Res., 2020, 1, 117–122 CrossRef CAS
- C.-Y. Wang, K. Maeda, L.-L. Chang, K.-L. Tung and C. Hu, Carbon, 2022, 188, 482–491 CrossRef CAS
- G. S. Priyanga, G. Pransu and S. Sampath, Chem. Phys. Impact, 2024, 8, 100408 CrossRef
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed
- I. J. Budiarso, V. A. Dabur, R. Rachmantyo, H. Judawisastra, C. Hu and A. Wibowo, Mater. Adv., 2024, 5, 2668–2688 RSC
- A. Torres-Pinto, C. G. Silva, J. L. Faria and A. M. T. Silva, Catal. Today, 2023, 424, 113868 CrossRef CAS
- M. Ismael, J. Alloys Compd., 2023, 931, 167469 CrossRef CAS
- E. O. Oseghe, S. O. Akpotu, E. T. Mombeshora, A. O. Oladipo, L. M. Ombaka, B. B. Maria, A. O. Idris, G. Mamba, L. Ndlwana, O. S. Ayanda, A. E. Ofomaja, V. O. Nyamori, U. Feleni, T. T. I. Nkambule, T. A. M. Msagati, B. B. Mamba and D. W. Bahnemann, J. Mol. Liq., 2021, 344, 117820 CrossRef CAS
- W. K. Darkwah and Y. Ao, Nanoscale Res. Lett., 2018, 13, 388 CrossRef PubMed
- S. A. Thomas, M. R. Pallavolu, M. E. Khan and J. Cherusseri, J. Energy Storage, 2023, 68, 107673 CrossRef
- N. Rono, J. K. Kibet, B. S. Martincigh and V. O. Nyamori, Crit. Rev. Solid State Mater. Sci., 2021, 46, 189–217 CrossRef CAS
- M. Ismael, J. Alloys Compd., 2020, 846, 156446 CrossRef CAS
- I. F. Silva, C. Pulignani, J. Odutola, A. Galushchinskiy, I. F. Teixeira, M. Isaacs, C. A. Mesa, E. Scoppola, A. These, B. Badamdorj, M. Ángel Muñoz-Márquez, I. Zizak, R. Palgrave, N. V. Tarakina, S. Gimenez, C. Brabec, J. Bachmann, E. Cortes, N. Tkachenko, O. Savateev and P. Jiménez-Calvo, J. Colloid Interface Sci., 2025, 678, 518–533 CrossRef CAS PubMed
- P. Jiménez-Calvo, V. Caps and V. Keller, Renewable Sustainable Energy Rev., 2021, 149, 111095 CrossRef
- T. Su, Q. Shao, Z. Qin, Z. Guo and Z. Wu, ACS Catal., 2018, 8, 2253–2276 CrossRef CAS
- J. Li, P. Jiménez-Calvo, E. Paineau and M. N. Ghazzal, Catalysts, 2020, 10, 89 CrossRef CAS
- Z. Tong, D. Yang, Y. Sun, Y. Nan and Z. Jiang, Small, 2016, 12, 4093–4101 CrossRef CAS PubMed
- Q. Shi, X. Zhang, X. Liu, L. Xu, B. Liu, J. Zhang, H. Xu, Z. Han and G. Li, Carbon, 2022, 196, 401–409 CrossRef CAS
- C. Li, Y. Fan, S. Gu, Y. Xiao, X. Zhao, J. Nan and X. Xiao, Appl. Surf. Sci., 2023, 636, 157809 CrossRef CAS
- J. Joy, E. George, P. Poornima Vijayan, S. Anas and S. Thomas, J. Ind. Eng. Chem., 2024, 133, 74–89 CrossRef CAS
- M. Zhang, Y. Zhang, L. Tang, G. Zeng, J. Wang, Y. Zhu, C. Feng, Y. Deng and W. He, J. Colloid Interface Sci., 2019, 539, 654–664 CrossRef CAS
- R. Bayan and N. Karak, Appl. Surf. Sci., 2020, 514, 145909 CrossRef CAS
- Z. Mo, H. Xu, Z. Chen, X. She, Y. Song, J. Wu, P. Yan, L. Xu, Y. Lei, S. Yuan and H. Li, Appl. Catal., B, 2018, 225, 154–161 CrossRef CAS
- Y. Ding, C. Wang, S. Bandaru, L. Pei, R. Zheng, Y. Hau Ng, D. Arenas Esteban, S. Bals, J. Zhong, J. Hofkens, G. Van Tendeloo, M. B. J. Roeffaers, L. H. Chen and B. L. Su, J. Colloid Interface Sci., 2024, 672, 600–609 CrossRef CAS PubMed
- X. Zhao, Q. Liu, X. Li, H. Ji and Z. Shen, Chin. Chem. Lett., 2023, 34, 108306 CrossRef CAS
- W. Zhang, J. Wang, Z. Liu, Y. Pi and R. Tan, Green Energy Environ., 2022, 7, 712–722 CrossRef CAS
- X. Li, J. Hu, T. Yang, X. Yang, J. Qu and C. M. Li, Nano Energy, 2022, 92, 106714 CrossRef CAS
- G. Sun, Z. Tai, J. Zhang, B. Cheng, H. Yu and J. Yu, Appl. Catal., B, 2024, 358, 124459 CrossRef CAS
- F. Zhang, J. Li, H. Wang, Y. Li, Y. Liu, Q. Qian, X. Jin, X. Wang, J. Zhang and G. Zhang, Appl. Catal., B, 2020, 269, 118772 CrossRef CAS
- M. Majdoub, Z. Anfar and A. Amedlous, ACS Nano, 2020, 14, 12390–12469 CrossRef CAS
- G. Dong, Z. Ai and L. Zhang, RSC Adv., 2014, 4, 5553 RSC
- M. A. Quintana, R. R. Solís, G. Blázquez, M. Calero and M. J. Muñoz-Batista, Appl. Surf. Sci., 2024, 656, 159717 CrossRef CAS
- S. Zhao, T. Guo, X. Li, T. Xu, B. Yang and X. Zhao, Appl. Catal., B, 2018, 224, 725–732 CrossRef CAS
- E. S. Da Silva, N. M. M. Moura, M. G. P. M. S. Neves, A. Coutinho, M. Prieto, C. G. Silva and J. L. Faria, Appl. Catal., B, 2018, 221, 56–69 CrossRef CAS
- Y. Li, Z. He, L. Liu, Y. Jiang, W.-J. Ong, Y. Duan, W. Ho and F. Dong, Nano Energy, 2023, 105, 108032 CrossRef CAS
- X. Bao, X. Lv, Z. Wang, M. Wang, M. Liu, D. Dai, L. Zheng, Z. Zheng, H. Cheng, P. Wang, Y. Liu, Y. Dai, Y. Fan and B. Huang, Int. J. Hydrogen Energy, 2021, 46, 37782–37791 CrossRef CAS
- J. Goclon and K. Winkler, Mol. Catal., 2020, 482, 110747 CrossRef CAS
- K. Trivedi, B. Yadav, R. Shrivastav and C. K. Modi, ChemPhotoChem, 2024, 8, e202400171 CrossRef CAS
- S. Hu, L. Ma, J. You, F. Li, Z. Fan, F. Wang, D. Liu and J. Gui, RSC Adv., 2014, 4, 21657–21663 RSC
- B. Zhu, L. Zhang, B. Cheng and J. Yu, Appl. Catal., B, 2018, 224, 983–999 CrossRef CAS
- X.-L. Song, L. Chen, L.-J. Gao, J.-T. Ren and Z.-Y. Yuan, Green Energy Environ., 2024, 9, 166–197 CrossRef CAS
- F. Yang, P. Hu, F. Yang, X.-J. Hua, B. Chen, L. Gao and K.-S. Wang, Tungsten, 2024, 6, 77–113 CrossRef
- G. Marcì, E. I. García-López, F. R. Pomilla, L. Palmisano, A. Zaffora, M. Santamaria, I. Krivtsov, M. Ilkaeva, Z. Barbieriková and V. Brezová, Catal. Today, 2019, 328, 21–28 CrossRef
- X. Luo, B. Li, T. Su, X. Xie, Z. Qin and H. Ji, ACS Catal., 2025, 15, 1097–1111 CrossRef CAS
- M. Alejandra Quintana, R. R. Solís, M. Ángeles Martín-Lara, G. Blázquez, F. Mónica Calero and M. J. Muñoz-Batista, Sep. Purif. Technol., 2022, 298, 121613 CrossRef CAS
- M. A. Quintana, J. Aguirre, M. Á. Martín-Lara, M. Calero, M. J. Muñoz-Batista and R. R. Solís, Sustainable Mater. Technol., 2024, 42, e01123 CrossRef CAS
- J. Xing, N. Wang, X. Li, J. Wang, M. Taiwaikuli, X. Huang, T. Wang, L. Zhou and H. Hao, J. Environ. Chem. Eng., 2022, 10, 108782 CrossRef CAS
- P. Jiménez-Calvo, C. Marchal, T. Cottineau, V. Caps and V. Keller, J. Mater. Chem. A, 2019, 7, 14849–14863 Search PubMed
- J. Wang and S. Wang, Coord. Chem. Rev., 2022, 453, 214338 Search PubMed
- Z. Miao, G. Wu, Q. Wang, J. Yang, Z. Wang, P. Yan, P. Sun, Y. Lei, Z. Mo and H. Xu, Mater. Rep.: Energy, 2023, 3, 100235 Search PubMed
- J. Gao, Y. Wang, S. Zhou, W. Lin and Y. Kong, ChemCatChem, 2017, 9, 1708–1715 Search PubMed
- M. Ismael, Inorg. Chem. Commun., 2023, 151, 110607 CrossRef CAS
- H. Sudrajat, S. A. Wella, J. Phanthuwongpakdee, D. Lisovytskiy, K. Sobczak and J. C. Colmenares, Nanoscale, 2024, 16, 14813–14830 RSC
- S. Wu, Y. Yu, K. Qiao, J. Meng, N. Jiang and J. Wang, J. Photochem. Photobiol., A, 2021, 406, 112999 CrossRef CAS
- S. Patnaik, D. P. Sahoo and K. Parida, Carbon, 2021, 172, 682–711 CrossRef CAS
- M. A. Khan, S. Mutahir, I. Shaheen, Y. Qunhui, M. Bououdina and M. Humayun, Coord. Chem. Rev., 2025, 522, 216227 CrossRef CAS
- M. Bellardita, E. I. García-López, G. Marcì, I. Krivtsov, J. R. García and L. Palmisano, Appl. Catal., B, 2018, 220, 222–233 CrossRef CAS
- R. Singh, M. Chauhan, P. Garg, B. Sharma, P. Attri, R. K. Sharma, D. Sharma and G. R. Chaudhary, J. Cleaner Prod., 2023, 427, 138855 CrossRef CAS
- B. L. Phoon, C. C. Ong, K. C. Lee, G. T. Pan, B. F. Leo, S. Chong and K. L. Pan, Catalysts, 2022, 12, 586 CrossRef CAS
- D. Sheng, Q. He, Z. H. Cao, L. B. Chen, S. H. Wu, Y. Liu, H. T. Ren and X. Han, Ind. Eng. Chem. Res., 2024, 63, 9038–9049 CrossRef CAS
- Q. Yang, T. Wang, Z. Zheng, B. Xing, C. Li and B. Li, Appl. Catal., B, 2022, 315, 121575 CrossRef CAS
- M. J. Muñoz-Batista, A. Kubacka, A. B. Hungría and M. Fernández-García, J. Catal., 2015, 330, 154–166 CrossRef
- P. Jimenéz-Calvo, M. J. Muñoz-Batista, M. Isaacs, V. Ramnarain, D. Ihiawakrim, X. Li, M. Ángel Muñoz-Márquez, G. Teobaldi, M. Kociak and E. Paineau, Chem. Eng. J., 2023, 459, 141514 CrossRef
- M. J. Muñoz-Batista, M. M. Ballari, A. Kubacka, O. M. Alfano and M. Fernández-García, Chem. Soc. Rev., 2019, 48, 637–682 RSC
- S. E. Braslavsky, A. M. Braun, A. E. Cassano, A. V. Emeline, M. I. Litter, L. Palmisano, V. N. Parmon and N. Serpone, Pure Appl. Chem., 2011, 83, 931–1014 CrossRef CAS
- J. Marugán, R. van Grieken, C. Pablos, M. L. Satuf, A. E. Cassano and O. M. Alfano, Chem. Eng. J., 2013, 224, 39–45 CrossRef
- J. Marugán, R. van Grieken, A. E. Cassano and O. M. Alfano, Appl. Catal., B, 2008, 85, 48–60 CrossRef
- O. Fontelles-Carceller, M. J. Muñoz-Batista, J. C. Conesa, A. Kubacka and M. Fernández-García, Mol. Catal., 2018, 446, 88–97 CrossRef CAS
- M. A. Quintana, A. Picón, M. A. Á. Martín-Lara, M. Calero, M. J. Muñoz-Batista and R. R. Solís, Appl. Catal., A, 2024, 674, 119607 CrossRef CAS
- R. R. Solís, M. A. Quintana, G. Blázquez, M. Calero and M. J. Muñoz-Batista, Catal. Today, 2023, 423, 114266 CrossRef
- K. Cerdan, W. Ouyang, J. C. Colmenares, M. J. Muñoz-Batista, R. Luque and A. M. Balu, Chem. Eng. Sci., 2019, 194, 78–84 CrossRef CAS
- T. Montalvo-Herrera, D. Sánchez-Martínez, E. I. García-López and G. Marcì, Res. Chem. Intermed., 2023, 49, 1197–1212 CrossRef CAS
- L. Hervé, S. Heyte, M. Marinova, S. Paul, R. Wojcieszak and J. Thuriot-Roukos, Solids, 2024, 5, 172–192 Search PubMed
- M. A. Quintana, A. Picón, M. Á. Martín-Lara, M. Calero, M. J. Muñoz-Batista and R. R. Solís, Appl. Catal., A, 2024, 674, 119607 Search PubMed
- S. Hosseini and N. Azizi, Photochem. Photobiol., 2024, 100, 1214–1234 CrossRef CAS PubMed
- R. R. Solís, M. A. Quintana, G. Blázquez, M. Calero and M. J. Muñoz-Batista, Catal. Today, 2023, 423, 114266 CrossRef
- X. Dai, M. Xie, S. Meng, X. Fu and S. Chen, Appl. Catal., B, 2014, 158–159, 382–390 CrossRef CAS
- B. Akhtar, H. Ghafuri and A. Rashidizadeh, Mol. Catal., 2021, 506, 111527 CrossRef CAS
- F. He, Q. Xiao, Y. Chen, H. Wang and X. Wang, Appl. Catal., B, 2024, 343, 123525 CrossRef CAS
- J. Xue, X. Li, S. Ma, P. Xu, M. Wang and Z. Ye, J. Mater. Sci., 2019, 54, 1275–1290 CrossRef CAS
- M. Heidari-Golafzani, M. Rabbani and R. Rahimi, Inorg. Chem. Commun., 2024, 168, 112967 CrossRef CAS
- M. Tayebnia, B. Akhlaghinia and M. N. Mahmoodabadi, Catal. Lett., 2024, 154, 5045–5068 CrossRef CAS
- J. Cui, X. Lu, M. Guo, M. Zhang, L. Sun, J. Xiong, R. Zhang, X. Li, Y. Qiao, D. Li, M. Guo and Z. Yu, Catal. Sci. Technol., 2023, 13, 940–957 RSC
- R. Nejat, Z. Najminejad, F. Fazlali, S. Shahraki and Z. Khazaee, Inorg. Chem. Commun., 2021, 132, 108842 CrossRef CAS
- M. R. Pradhan, D. Rath, R. Sethi, B. B. Nanda and B. Nanda, Inorg. Chem. Commun., 2021, 130, 108717 CrossRef CAS
- X. Chu, H. Liu, H. Yu, L. Bai, F. Yang, L. Zhao, Z. Zhao, Y. Jiao, W. Li, G. Zhang and L. Jing, Mater. Today Energy, 2022, 25, 100963 CrossRef CAS
- W. Wang, X. Cong, X. Zhang, W. Xu, J. Diao, Y. Bao and F. Guo, J. Environ. Chem. Eng., 2025, 13, 115552 CrossRef CAS
- D. A. Giannakoudakis, A. Qayyum, M. Barczak, R. F. Colmenares-Quintero, P. Borowski, K. Triantafyllidis and J. C. Colmenares, Appl. Catal., B, 2023, 320, 121939 Search PubMed
- M. J. Lima, P. B. Tavares, A. M. T. Silva, C. G. Silva and J. L. Faria, Catal. Today, 2017, 287, 70–77 CrossRef CAS
- X. Ye, T. Zhu, Z. Hui, X. Wang, J. Wei and S. Chen, J. Catal., 2021, 401, 149–159 CrossRef CAS
- V. G. Merkulov, O. V. Turova, E. A. Ivanova, I. V. Kuchurov, M. N. Zharkov and S. G. Zlotin, ChemPhotoChem, 2024, e202400227 Search PubMed
- Q. Yang, T. Wang, F. Han, Z. Zheng, B. Xing and B. Li, J. Alloys Compd., 2022, 897, 163177 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2025 |