DOI:
10.1039/D5MA00247H
(Paper)
Mater. Adv., 2025, Advance Article
Cinnamic acid-derived carbon dots by microwave irradiation synergise the ciprofloxacin effect against Staphylococcus aureus and promote its skin permeability†
Received
19th March 2025
, Accepted 12th May 2025
First published on 14th May 2025
Abstract
Aqueous solubility and biocompatibility have encouraged the development of carbon dots (CDs) for multiple applications. Considering the significance of natural molecules as precursors, cinnamic acid (CA) was used to develop carbon dots (CA-CDs) using an efficient microwave irradiation method which gave a higher quantum yield in comparison with other conventional methods. The positively charged CA-CDs showed a broad spectrum of action against Gram-positive (S. aureus) and Gram-negative (E. coli) bacteria. Unlike ciprofloxacin (CIP), CA-CDs showed potent inhibition of clinically resistant S. aureus (SA-DR) while reducing biofilm production, leading to synergy with CIP. CA-CDs also acted as carriers to enhance the skin-permeability of CIP through their ionic complex (CIP-CA-CDs). The complex retained the antimicrobial properties and showed the ability to gradually release CIP like a reservoir. Considering the poor permeability of CIP as a limitation, this work can encourage the translational application of CIP for topical application. This work provides a novel method of developing CDs for antimicrobial properties and can encourage their use as a complementary therapy or as carriers for further application.
1. Introduction
Nanomaterials based on carbon or carbon dots have tunable properties, which have attracted the attention of biomedical research groups for different applications capitalizing on their low toxicity, optical properties, and novel biological activities.1 Judicious selection of the method of synthesis and precursor has allowed optimum core and surface properties to meet specific biomedical requirements.2 Although carbon dots inherit the biological properties of their precursor and often potentiate activity in many instances, there are examples where new biological activities are associated with the carbon dots.3 Thus, there is continued interest in exploring novel methods and precursors to develop carbon dots with interesting biomedical applications.
Bacterial infections continue to challenge humanity with growing incidences of resistance and fewer discoveries of novel antimicrobials. In this scenario, nanomaterials widely reported with antimicrobial properties have gained significant research attention. Compared to other nanomaterials, including polymeric nanoparticles, metallic nanoparticles, and metal oxides, carbon dots have a distinct advantage in terms of low cytotoxicity.4,5 Although the specific mechanisms of many of these carbon dots are unknown, the antimicrobial action is generally attributed to the following properties.6–9 The small size of CDs promotes diffusion into bacteria, altering the membrane and its intracellular components. Furthermore, the surface charges of CDs promote electrostatic adsorption, leading to membrane disruption. Typically, bacterial membranes have a net negative charge because of peptidoglycans rich in amine moieties. Accordingly, CDs with positive charge have been prioritized for exploring antimicrobial properties.10 However, there is no definite quantitative correlation between CDs' surface charge and antimicrobial potency. Thus, there is further scope to explore novel CDs for antimicrobial properties.
Precursors rich in amine moieties have generally been used to develop CDs with a positive charge. The amine groups on the surface impart CDs a net positive zeta potential. Contrary to this, the precursor's carboxylic acidic groups (–COOH) contribute to the negative charge. However, carbonisation is known to induce the removal of oxygen-containing groups, which may reduce the negative charge. Thus, the presence of acidic groups in the precursor may not be a deterrent to developing positively charged carbon dots. trans-Cinnamic acid (CA) is an unsaturated acid with a natural origin and diverse biological activities, including effects against cancer,11 oxidative stress,12 and inflammation.13 However, its antimicrobial effects are relatively poor.14,15 Nonetheless, carbonisation of CA may minimise its negative charge leading to CDs (CA-CDs) with potential antimicrobial properties. As shown in Fig. 1, CA-CDs were synthesized from CA. The microwave irradiation method was the most effective. The carbonisation imparted a net positive zeta potential and the CA-CDs inhibited both Gram-positive and Gram-negative bacteria. Taking Staphylococcus aureus (SA) as a model microbe for topical infection, CA-CDs were screened against this and its resistant strain (SA-DR). It showed high potency against SA-DR and reduced its capacity to form biofilms. Since biofilm production is a mode of resistance of SA to ciprofloxacin (CIP), CA-CDs showed synergism with CIP against SA-DR. To extend the application of CA-CDs as nano-carriers of CIP to overcome its poor biopharmaceutical properties (BCS-IV drug), their ionic complex was prepared, which significantly enhanced the skin permeability of CIP while retaining efficacy. This work provides an opportunity to develop safe CA-CDs as novel antimicrobial agents, adjuvants and carriers for CIP, which can be extended for potential antimicrobial application.
 |
| | Fig. 1 Schematic representation of steps involved in the synthesis of CA-CDs and their purification and activity against a biofilm. The process illustrates the production pathway of cinnamic acid derived carbon dots and their application in biofilm inhibition. | |
2. Experimental
2.1. Materials
trans-Cinnamic acid (Cat. No. 29955-500G) was procured from SRL Pvt. Ltd, Mumbai, India. An EZcount™ MTT cell assay kit (Cat. No. CCK003) was procured from Himedia Laboratories Pvt. Ltd, Mumbai, India. Bacterial strains, including Staphylococcus aureus (SA) MTCC 96 and Escherichia coli (EC) MTCC 443, were procured from MTCC, India. The drug-resistant isolates SA-DR were obtained from the Department of Microbiology, All India Institute of Medical Sciences, Bhubaneswar, Odisha. The SA-DR were cultured in mannitol salt agar (Cat. No. M118-500 G). Bacterial isolates were cultured in respective culture media and maintained in a nutrient broth. For further investigations, each test bacterium was transferred to nutrient agar or a Mueller–Hinton broth separately for overnight culture. Bacterial culture media and reagents, including Nutrient Broth (NB, Cat. No. M002-500 G), Mueller–Hinton Broth (MHB, Cat. No. M391-500 G), soybean casein digest medium (pH at 25 °C: 7.3 ± 0.2), Luria–Bertani broth (LB broth, Cat. No. M124-500 G), crystal violet (Cat. No. TC510-25 G) and the antibiotic ciprofloxacin (Cat. No. CMS 1891), were procured from Himedia Laboratories Pvt. Ltd, Mumbai, India. Glutaraldehyde solution (Cat. No. 111-30-8) and sulphuric acid (Cat. No. DE3D730780-500 mL) were procured from Sigma (India). Glacial acetic acid (Cat. No. 027017) was procured from SRL Pvt. Ltd, Mumbai, India. All the chemicals used were of analytical grade.
2.2. Synthesis of CA-CDs
Microwave-assisted synthesis of CA-CDs was achieved using a microwave synthesizer (Monowave 400, Anton Paar). Briefly, 15 mg of cinnamic acid was transferred into a G 30 vial and DMSO (200 μL) was added. The reaction mixture was kept inside the microwave synthesizer at different temperatures (270–290 °C) and times (3–10 min) at 800 rpm with a cooling temperature of 55 °C. For the hydrothermal method,16 2 g of cinnamic acid was mixed with 2 mL of DMSO and de-ionized (DI) water (20 mL) in a 50 mL Teflon-lined stainless-steel vessel and kept in a hot air oven (180–260 °C for 5–8 h). For the muffle furnace method, 2 g of cinnamic acid was transferred to a china dish and heated at different temperatures (280–360 °C) and times (0.5–2 h).17 A domestic microwave oven was also used to synthesize 2 g of cinnamic acid in a 50 mL beaker (200–700 W for 10–45 min).18
Following synthesis, 20 mL of DI water was added to the reaction vial, and it was sonicated for 5–10 min to promote extraction of CA-CDs from the reaction vial. Furthermore, it was centrifuged for 20 min (1118 G-force). The centrifugation was repeated thrice while the residue was removed during each cycle. Subsequently, the supernatants were filtered through a nylon syringe filter with a pore size of 0.22 microns. For further purification, it was subjected to dialysis for 24 h. The smaller nanosized CA-CDs passing through this membrane were collected by volume reduction and stored at 4 °C until further use.19
2.3. Quantum yield measurement
The relative quantum yield of the CA-CDs was calculated following the established method20 with 375 nm as the excitation wavelength and 457 nm as the emission wavelength.21 Quinine sulfate (quantum yield 54%) in 0.1 M H2SO4 solution was used as the reference standard for the calculation of the relative quantum yield using the following equation:22| |
 | (1) |
where QCD represents the quantum yield of CD, QR is the quantum yield of a reference material (R) with a known quantum yield in a specific solvent, ICD is the integrated fluorescence intensity of the carbon dots (area under its emission spectrum), IR is the integrated fluorescence intensity of the reference material (area under its emission spectrum), AR is the absorbance of the reference material at the excitation wavelength, ACD is the absorbance of the CD at the excitation wavelength, ηCD2 is the refractive index of the solvent used for CDs and ηR2 is the refractive index of the solvent used for the reference material (R).22
2.4. Characterization of carbon dots
The CA-CDs were kept under a UV lamp (364 nm) to have a preliminary assessment of their fluorescence. Furthermore, a fluorescence spectrometer (Shimadzu RF-600, Kyoto, Japan) was employed to assess the fluorescence of the CA-CDs. The sizes of the CA-CDs were determined using a Nano-ZS Zetasizer (Malvern, UK). FTIR (FT-IR-4600, JASCO) spectra were recorded to assess the functional groups associated with the CA-CDs. The CA-CDs with optimum fluorescence yield and size were further tested for purity by HPLC (Prominence, Shimadzu, Japan) and analysed by transmission electron microscopy (TEM) to confirm the size and morphology (JEOL, JEM-2100PLUS, ELECTRON MICROSCOPE). The crystalline nature was assessed by X-ray diffraction (XRD) (Bruker, Germany. D8 ADVANCE), and the zeta potential was confirmed with the Nano-ZS Zetasizer (Malvern, UK).
2.5. In vitro cytotoxicity study
MTT assay was performed to determine the cytotoxicity of CA-CDs using EZcount™ MTT cell assay in Vero cells according to the manufacturer's protocol.23 About 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells were seeded in 96-well plates (Corning) 24 h before the experiment. Once the cells reached 70% confluency, they were exposed to varying concentrations of the drugs, with water and DMSO serving as a reagent control. Then, 10 μL of MTT reagent (5 mg mL−1) was added to the cells 24 h post-drug treatment and incubated for 1 h at 37 °C in a CO2 incubator. The medium was then discarded, and 100 μL of solubilization buffer was added into each well, followed by a 15 min incubation at 37 °C to dissolve the formazan crystals. Finally, the absorbance was measured at 570 nm using a multimode plate reader, and the % of metabolically active cells was compared to the control cells to determine cytotoxicity.
000 cells were seeded in 96-well plates (Corning) 24 h before the experiment. Once the cells reached 70% confluency, they were exposed to varying concentrations of the drugs, with water and DMSO serving as a reagent control. Then, 10 μL of MTT reagent (5 mg mL−1) was added to the cells 24 h post-drug treatment and incubated for 1 h at 37 °C in a CO2 incubator. The medium was then discarded, and 100 μL of solubilization buffer was added into each well, followed by a 15 min incubation at 37 °C to dissolve the formazan crystals. Finally, the absorbance was measured at 570 nm using a multimode plate reader, and the % of metabolically active cells was compared to the control cells to determine cytotoxicity.
2.6. Assessment of CA-CDs for antibacterial activity
The antimicrobial activities of CA-CDs, CIP, and CA were evaluated against the reference standard strain of SA (MTCC 96), EC (MTCC 443), and SA-DR. The strains were cultured and maintained in MHB. The stock bacterial suspension was diluted to 0.5 McFarland, equal to 1 × 108 colony-forming units (CFU per mL). It was diluted to a final concentration of 1 × 105 CFU per mL for antibacterial screening.
The minimum inhibitory concentration (MIC) was estimated using the microdilution method as per established protocol.24,25 Briefly, in a 96-well plate, 50 μL of MHB was placed in each well. The CA-CDs (10 μg mL−1), CA (2000 μg mL−1), and CIP (10 μg mL−1) were serially diluted and added to the wells in triplicate. Then, 100 μL of bacterial suspension (1 × 105 CFU per mL−1) was added to each well and incubated at 37 °C for 18 h. Following incubation, 10 μL of alamarBlue™ dye was added to each well and incubated at 37 °C for 1 h. The MIC was determined as the minimum concentration that shows a blue colour.
2.7. Antibiofilm effect of CA-CDs against SA-DR
The biofilm inhibition of SA-DR was conducted as per the established protocol.26 Briefly, an inoculum of SA-DR was inoculated in 5 mL of nutrient broth and incubated at 37 °C overnight. After incubation, the culture was measured at 600 nm to evaluate the antibiofilm activity. Then, 100 μL of culture containing 1 × 105 cells per well in LB + 2% glucose was added to the experimental wells. Then different concentrations of CA-CDs (10 μg mL−1 to 0.02 μg mL−1) were serially diluted in the experimental 96 wells. The experimental plate was incubated for 48 h at 37 °C, and the sub-population biomass was estimated by using the staining method. After incubation, the fluid contents were decanted and washed with PBS (pH 7). 0.1% (w/v) crystal violet solution was added for 30 min of incubation. The stained solution was discarded and washed with PBS solution. Before quantitative analysis, wells were air dried, 30% glacial acetic acid was added, and absorbance was recorded at 570 nm using a microplate spectrophotometer (BioTek Epoch 2). The identical approach was repeated three times and recorded in an Excel file for subsequent analysis interpretation.
A scanning electron microscope (SEM, JEOI JSM-7610F) was used to examine morphological changes on the cell surface; bacterial inoculum was used as a control for SEM analysis. Sterile LB + 2% glucose medium was added into a 12-well plate containing sterile coverslips and a bacterial inoculum comprising 1 × 105 cells per well, treated with sub-MBIC doses of CA-CDs for 24 h. The biofilm-covered coverslips were retrieved and rinsed with distilled water and 1× PBS. The samples were then fixed with 2.5% glutaraldehyde solution for 20 min at 4 °C. The coverslips were washed with 1× PBS, dehydrated with varying ethanol concentrations (30%, 50%, 70%, 90%, and 100%), and air-dried at room temperature. The prepared samples were processed for SEM imaging analysis.26
2.8. Checkerboard assay
To evaluate the antibacterial activity of different combinations, an established checkerboard dilution method based on the MIC was followed.24 Briefly, combinations of CA-CDs and CIP (based on their individual MIC) were used against SA (MTCC 96) and SA-DR. Following serial dilution, the test compounds were added to the wells for antibacterial effect screening as described earlier. The MIC of the combinations was noted. The fractional inhibitory concentration index (FICI) analysis was performed to determine the type of interaction: synergistic (≤0.5), additive (>0.5–≤1), no difference (>1–≤4), or antagonism (>4)27 using the following equation:| |
 | (2) |
A1 is the concentration of CIP in the combination showing MIC, A2 is the MIC of CIP alone, B1 is the concentration of CA-CDs in the combination showing MIC and B2 is the MIC of CA-CDs alone.
2.9. Conjugation of CIP onto CA-CDs
CIP was reversibly conjugated to the CA-CDs through ionic interaction by capitalizing on the pKa of CIP (6.09), pH (8) of the reaction medium, and cationic nature of the CA-CDs. Briefly, 1 mg of CIP was transferred to 10 mL of water, and the pH was adjusted to 8 with NaOH (0.5% w/v). 1 mg of CA-CDs was added gradually while stirring the mixture. The formation of the ionic complex was monitored using TLC at different time points (0, 15, and 30 min) using a mobile phase comprising chloroform:methanol:ammonia (40:60:1) with the retardation factor (Rf) of CA-CDs and CIP being 0 and 0.6 respectively. The lack of spots for CIP or CA-CDs in TLC was considered a completion of the complexation process (Fig. S1, ESI†). This was purified using a dialysis membrane (12000–14000 MW cut-off).28 Furthermore, the ionic complex (CIP-CA-CDs) was characterized by UV-vis spectroscopy, FTIR, spectrofluorimetry, TEM and zeta potential measurements as discussed in Section 2.4. The nuclear magnetic resonance (NMR) spectroscopy study was conducted (Bruker ASCEND 600) to further analyse the structural characteristics of the CIP-CA-CDs.
2.10. In vitro drug release study
An in vitro drug release study was conducted per established protocol,28 using a 10 mL aliquot of the CIP-CA-CD sample, placed in a dialysis bag with a molecular weight cut-off of 12000–14000 MW. This bag was suspended in 52 mL of PBS at pH 7.4, maintained at 37 °C with stirring at 250 rpm. At regular intervals, 3 mL of the release medium was withdrawn and replaced with an equal fresh solution. The amount of CIP released was measured using the validated UV-visible spectrophotometric method at 274 nm to assess the release profile under physiological conditions. The DDSolver software was used to determine the best-fit release models for drug release profiles. The best model was identified by evaluating the residual sum of squares (RSS), Akaike information criterion (AIC), and coefficient of determination (R2).29
2.11. Ex vivo permeability studies
Ex vivo skin permeability studies of CIP and CIP-CA-CDs were carried out by following reported methods using a Franz diffusion cell mounted with ear pinna skin from a goat.30 Briefly, 3 mL of the CIP-CA-CD solution (1 mg mL−1) was kept in the donor chamber while maintaining the receiver chamber with PBS7.4 (52 mL capacity) and constant temperature (37 ± 1 °C). Stirring at 250 rpm was continued to ensure the homogeneity of the receiver chamber. The study used a CIP solution (1 mg mL−1) in distilled water as a control. At predetermined time intervals (0.25, 0.75, 1.25, 2.25, 3.75, 7.75, 16.25, 18.75, 21.25, 24 and 26 h), 3 mL of the sample was withdrawn, and an equal volume of fresh PBS (37 °C) was replaced to maintain sink conditions. The drug concentration was estimated at 274 nm using the verified UV method. The percentage of CIP permeated was estimated considering the release versus time curves, which were plotted and compared with the CIP solution. The drug permeability profiles were analysed using the DDSolver software.31 The steady-state flux (J) across goat ear pinna skin and the permeation coefficient (P) were calculated using eqn (3) and (4), respectively, and were then compared with the control solution.29| |
 | (3) |
Q represents the cumulative drug amount crossing the goat ear pinna skin, t denotes the exposure time and A signifies the available surface area for diffusion.where C0 is the initial drug concentration in the donor compartment.
2.12. Antibacterial activity of CIP-CA-CDs
The MIC was estimated using the microdilution method per the established protocol.24,25 Briefly, in a 96-well plate, 50 μL of MHB was placed in each well. The CIP-CA-CDs (5 μg mL−1) were serially diluted and added to the wells in triplicate. Then, 100 μL of bacterial suspension (1 × 105 CFU per mL) was added to each well and incubated at 37 °C for 18 h. Following incubation, 10 μL of alamarBlue™ dye was added to each well and incubated at 37 °C for 1 h. The MIC was determined as the minimum concentration that shows a blue colour. Furthermore, the time-dependent bacterial growth curve of SA and SA-DR was studied per established protocol26,32 at various time intervals (0, 3, 6, 9, 12, 15, 18, and 21 h) using MIC, ½ MIC and ¼th MIC.
2.13. Statistical analysis
All experiments were performed in triplicate. The results were expressed as mean ± standard deviation. The data were analysed and/or graphically represented using Microsoft Excel, Origin, and ImageJ software.33,34
3. Results and discussion
3.1. Synthesis of carbon dots by different methods and characterization
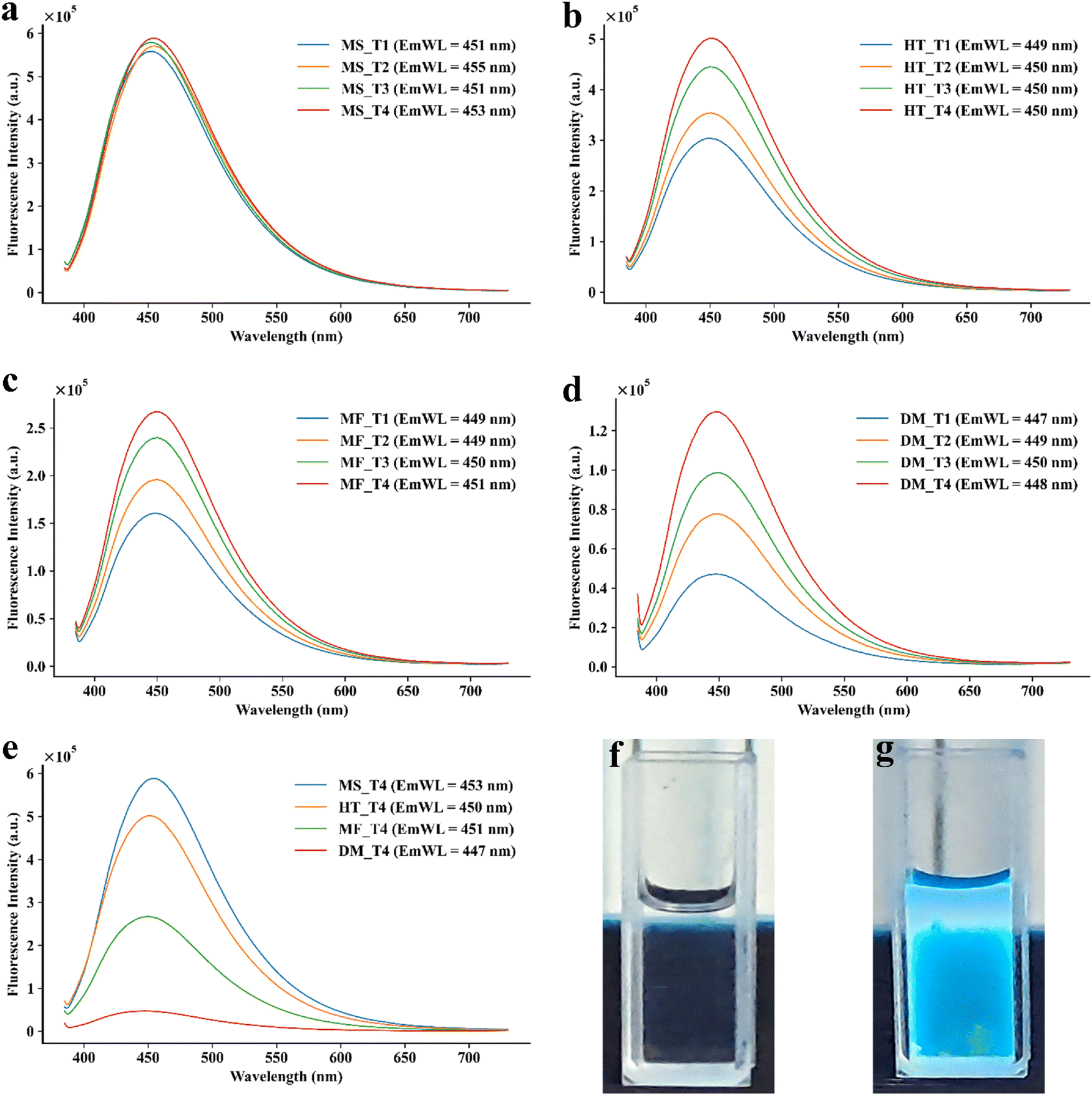
Multiple methods have been reported for the synthesis of CDs.1 Accordingly, different methods were investigated for the synthesis of CA-CDs. Conventionally, the synthesis efficiency of CDs is determined by the quantum yield of fluorescence (Fig. 2) and the size of the CDs. Accordingly, the quantum yield was monitored for CA-CDs prepared by microwave irradiation, the hydrothermal method, or the muffle furnace method (Table 1). Since fluorescence yield has been inversely correlated to the size of the CDs,35 the size of CDs prepared by each method was analysed using Zetasizer (Fig. 3). CA-CDs prepared with a domestic microwave oven exhibited inferior quantum yield and higher size (Table 1). Dielectric heating is the key to the effectiveness of the microwave irradiation36 and domestic microwaves have been used to develop carbon dots.35 However, lower irradiation power and non-uniformity in heating are the limiting factors for their efficiency.36 This can partly explain the poor yield of CA-CDs prepared by this method.
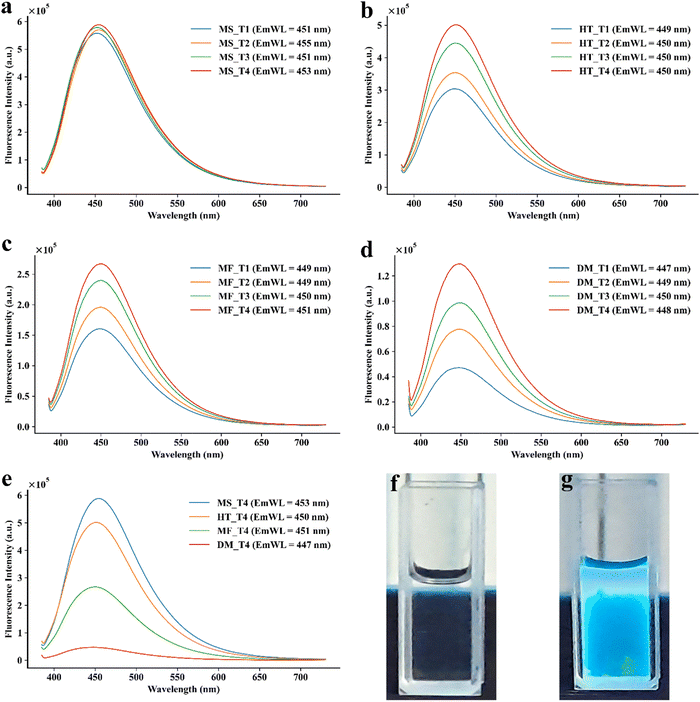 |
| | Fig. 2 Illustration of fluorescence intensity of CA-CDs synthesized with different trials using (a) microwave irradiation, (b) hydrothermal, (c) muffle furnace, and (d) microwave oven methods. (e) Comparison of higher fluorescence intensity obtained from different methods of CA-CD synthesis. Photographs of visual observation of fluorescence of (f) CA and (g) CA-CDs under UV light (long wavelength – 364 nm). | |
Table 1 Comparative study of CA-CD synthesis using different methods
| Methods |
Quantity |
Temperature |
Time |
Quantum yield (%) |
| Microwave synthesizer |
15 mg |
270 °C |
3 min |
50.39 |
| 290 °C |
5 min |
51.84 |
| 290 °C |
8 min |
52.28 |
| 290 °C |
10 min |
53.85 |
| Hydrothermal synthesis |
2 g |
180 °C |
8 h |
27.27 |
| 200 °C |
7 h |
31.92 |
| 240 °C |
6 h |
40.13 |
| 260 °C |
5 h |
45.24 |
| Muffle furnace method |
2 g |
280 °C |
30 min |
14.49 |
| 300 °C |
1 h |
17.67 |
| 330 °C |
1.5 h |
21.63 |
| 360 °C |
2 h |
24.09 |
| Domestic microwave oven synthesis |
2 g |
200 W |
10 min |
6.29 |
| 360 W |
15 min |
7.02 |
| 700 W |
30 min |
8.92 |
| 700 W |
45 min |
11.67 |
Muffle furnaces and hydrothermal techniques are popularly used for the preparation of CDs. Under optimum conditions, the highest yield was 24.09% and 45.24% for CA-CDs prepared with a muffle furnace and hydrothermal technique, respectively. However, the CDs were not uniform in size. Compared to these, the highest quantum yield for fluorescence was observed for CA-CDs prepared by microwave irradiation in the microwave synthesizer. Consequently, these CA-CDs showed a narrow size distribution with an average size of about 3 ± 0.3 nm (Fig. 3(a)). This agrees with the phenomenon of “quantum confinement”, which happens because of restricted electron movement in very low-size carbon dots, leading to higher energy emission and better fluorescence.37 This suggests that microwave irradiation or dielectric heating is preferable for preparing CA-CDs. This is supported by the fact that CA is a polarised molecule able to couple with microwave energy.38 High-quality CA-CDs were prepared with microwave irradiation for 10 min (290 °C and 800 rpm). Under conditions of 270 °C and 3 min exposure, the quantum yield was about 50%. The yield increased gradually with an increase in temperature and exposure. However, the enhancement in the quantum yield was not very significant. Considering the instrumental limitation (30 bar and 300 °C), CA-CDs obtained with 53.85% quantum yield were close enough to the reference standard (QS). Hence, these CA-CDs were taken forward for further investigation.
 |
| | Fig. 3 Boxplot of DLS (zeta sizer) analysis showing the particle size distribution. Size distribution of CA-CDs synthesized using (a) microwave irradiation (at 290 °C for 10 min), (b) a hydrothermal method (at 160 °C for 5 h), (c) a muffle furnace method (at 330 °C for 1.5 h), and (d) a domestic microwave oven method (at 700 W for 45 min). | |
3.2. Characterization of CA-CDs
TLC of the purified CA-CDs showed no spots corresponding to the precursor (CA), indicating carbonisation (Fig. 4(a)). Unlike CA, its CDs showed a single fluorescent spot at a longer wavelength (364 nm), which is in agreement with its fluorescent properties (Fig. 4(b)). Its relatively higher polarity than CA was indicated by poor migration and high retention on the silica of the TLC plate (Fig. 4(b)). This also suggested its purity, which was further evident from the 3D image of its HPLC chromatogram (Fig. S2, ESI†) supported by its UV-spectrum (Fig. 4(c)).
 |
| | Fig. 4 Characterization of CA and CA-CDs. Photographs of TLC of CA and CA-CDs at (a) a short wavelength (254 nm) and (b) a long wavelength (364 nm), (c) UV/vis absorption spectra of CA (276 nm) and CA-CDs (208 nm), (d) comparative FTIR spectra of CA and CA-CDs, (e) EDX spectra for elemental analysis of CA-CDs and (f) comparative zeta potentials of CA and CA-CDs. | |
The FT-IR spectra of CA showed a peak at 3065.3 cm−1 for O–H stretching supported by C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1670.05 cm−1 and C–O stretching at 976.769 cm−1. Furthermore, peaks at 3021.91 cm−1 for aromatic C–H stretching and at 1625.7 cm−1 for CC stretching were observed. The O–H peak of CA was shifted to 3058.55 cm−1 in its CDs. The CO peak shifted to 1675.84 cm−1, whereas the C–O peak shifted to 1029.8 cm−1. Overall, the intensities of the peaks were reduced. This change in peaks suggests the change in the structural organization of the CDs (Fig. 4(d)). The elemental analysis also supported carbonisation. While CA has 72.97% of carbon and 21.62% of oxygen, its CDs showed 89.36% of carbon and 12.76% of oxygen (Fig. 4(e)). The enhanced carbon proportion confirms the carbonisation, while the lower oxygen proportion supports the loss of functional groups in support of the carbonisation. The reduction in functional groups bearing oxygen can influence the zeta potential of the molecule because of the reduced proportion of electronegative elements. Accordingly, compared to cinnamic acid (−6.96 ± 0.5 mV) (Fig. 4(f)), its CA-CDs showed a relatively positive zeta potential (3.17 ± 0.8 mV) (Fig. 4(f)). Further support of carbonisation was evident from the XRD profile of the CA-CDs (Fig. 5(a)). It revealed intense peaks at 2θ = 22.9° and 25.46° with a lattice spacing of 0.38 and 0.35 nm respectively. This is close to the (002) plane of graphitic structures.39–41 Although these peaks indicate the crystallinity of the material, the XRD pattern displaying multiple small diffraction peaks indicates a disordered carbon arrangement.42 Accordingly, the degree of crystallinity was found to be about 69%. Carbonisation is expected to disrupt the highly ordered arrangement of the crystalline molecule, which can lead to some degree of amorphization.43 Since this can promote solubility, CA-CDs were found to be more polar than CA. However, a high degree of amorphous nature or a very low degree of crystallinity of solid molecules can have mechanical issues during further processing or application. Hence, CA-CDs with 69% crystallinity can be considered suitable for further application.44 The crystalline morphology was also supported by an HR-TEM image (Fig. 5(b)) that showed uniform dispersion without apparent aggregation. The HR-TEM image of a single particle with a lattice spacing of 0.3 nm (Fig. 5(c)) closely matched with those obtained from the XRD data (Fig. 5(a)) and supported the crystalline nature. Furthermore, the selected area electron diffraction (SAED) pattern in Fig. 5(d) showed diffuse rings, suggesting the polycrystalline nature of the CA-CDs. The particles were spherical, with an average diameter of approximately 1.52 nm (Fig. 5(e)). The nano-size, positive charge, and apparent crystalline nature of CA-CDs can be capitalized for further application. The MTT assay in Vero cells showed 107%, 104%, and 77.8% cell viability at 100 μg mL−1, 500 μg mL−1, and 1000 μg mL−1 concentrations, respectively (Fig. 5(f)). Thus, CA-CDs are relatively noncytotoxic, which supports their potential for biological application.
O stretching at 1670.05 cm−1 and C–O stretching at 976.769 cm−1. Furthermore, peaks at 3021.91 cm−1 for aromatic C–H stretching and at 1625.7 cm−1 for CC stretching were observed. The O–H peak of CA was shifted to 3058.55 cm−1 in its CDs. The CO peak shifted to 1675.84 cm−1, whereas the C–O peak shifted to 1029.8 cm−1. Overall, the intensities of the peaks were reduced. This change in peaks suggests the change in the structural organization of the CDs (Fig. 4(d)). The elemental analysis also supported carbonisation. While CA has 72.97% of carbon and 21.62% of oxygen, its CDs showed 89.36% of carbon and 12.76% of oxygen (Fig. 4(e)). The enhanced carbon proportion confirms the carbonisation, while the lower oxygen proportion supports the loss of functional groups in support of the carbonisation. The reduction in functional groups bearing oxygen can influence the zeta potential of the molecule because of the reduced proportion of electronegative elements. Accordingly, compared to cinnamic acid (−6.96 ± 0.5 mV) (Fig. 4(f)), its CA-CDs showed a relatively positive zeta potential (3.17 ± 0.8 mV) (Fig. 4(f)). Further support of carbonisation was evident from the XRD profile of the CA-CDs (Fig. 5(a)). It revealed intense peaks at 2θ = 22.9° and 25.46° with a lattice spacing of 0.38 and 0.35 nm respectively. This is close to the (002) plane of graphitic structures.39–41 Although these peaks indicate the crystallinity of the material, the XRD pattern displaying multiple small diffraction peaks indicates a disordered carbon arrangement.42 Accordingly, the degree of crystallinity was found to be about 69%. Carbonisation is expected to disrupt the highly ordered arrangement of the crystalline molecule, which can lead to some degree of amorphization.43 Since this can promote solubility, CA-CDs were found to be more polar than CA. However, a high degree of amorphous nature or a very low degree of crystallinity of solid molecules can have mechanical issues during further processing or application. Hence, CA-CDs with 69% crystallinity can be considered suitable for further application.44 The crystalline morphology was also supported by an HR-TEM image (Fig. 5(b)) that showed uniform dispersion without apparent aggregation. The HR-TEM image of a single particle with a lattice spacing of 0.3 nm (Fig. 5(c)) closely matched with those obtained from the XRD data (Fig. 5(a)) and supported the crystalline nature. Furthermore, the selected area electron diffraction (SAED) pattern in Fig. 5(d) showed diffuse rings, suggesting the polycrystalline nature of the CA-CDs. The particles were spherical, with an average diameter of approximately 1.52 nm (Fig. 5(e)). The nano-size, positive charge, and apparent crystalline nature of CA-CDs can be capitalized for further application. The MTT assay in Vero cells showed 107%, 104%, and 77.8% cell viability at 100 μg mL−1, 500 μg mL−1, and 1000 μg mL−1 concentrations, respectively (Fig. 5(f)). Thus, CA-CDs are relatively noncytotoxic, which supports their potential for biological application.
 |
| | Fig. 5 Structural and biological characterization of CA-CDs. (a) XRD pattern showing crystalline nature, (b) HRTEM image revealing morphology, (c) lattice fringe measurements indicating the crystalline structure, and (d) electron diffraction pattern. (e) The size distribution histogram was derived from the TEM analysis and (f) cell viability. Values represented as mean ± S.D. (n = 3). | |
3.3. Determination of the minimum inhibitory concentration of CA-CDs
Positively charged CDs have been reported to promote antimicrobial properties, possibly through interaction with bacterial membranes rich in negatively charged peptidoglycans.10 So, CA-CDs were investigated against SA and EC strains. This revealed MICs of 700 μg mL−1 and 500 μg mL−1 against SA and EC, respectively, for CA. Compared to this, MICs of 0.07 μg mL−1 and 0.625 μg mL−1 were observed against SA and EC, respectively, for CA-CDs. As expected, the positive control (CIP) showed low MICs against SA (0.07 μg mL−1) and EC (0.039 μg mL−1) because of its well-known broad antimicrobial action. In agreement with the poor antimicrobial properties of CA against both SA45 and EC,15 it was found to have high MICs (Table 2), similar to its reported values against these microbes. While the antimicrobial potency of CA-CDs was similar to CIP against SA, it was relatively less against EC. Nevertheless, the low MIC against these Gram-positive and negative strains suggests the potential for broad antimicrobial properties of CA-CDs.
Table 2 Anti-microbial activities (MIC in μg mL−1) of test compounds
| Compounds |
Staphylococcus aureus |
E. coli |
Staphylococcus aureus DR |
| Cinnamic acid |
700 |
500 |
1500 |
| CA-CDs |
0.07 |
0.625 |
1.25 |
| Ciprofloxacin |
0.07 |
0.039 |
3.5 |
To assess the effect against drug resistant strains, the SA-DR which is less susceptible to CIP was used as a model strain. Because of the resistant nature of SA-DR, the CIP control showed a relatively high MIC (3.5 μg mL−1), which is very close to its resistance profile (4 μg mL−1).46 Interestingly, the CA-CDs were more potent than CIP against SA-DR with an MIC of 1.25 μg mL−1 (Table 2).
3.4. Biofilm inhibition by CA-CDs
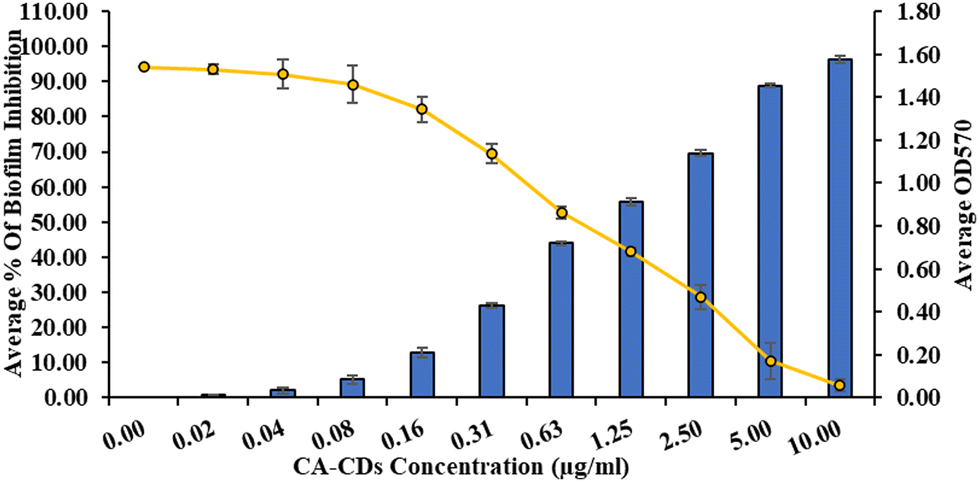
The high MIC (3.5 μg mL−1) of CIP against SA-DR indicates that a relatively high CIP concentration is required to overcome the poor susceptibility.46 Biofilm production by SA is a primary mechanism for developing resistance to CIP.47 Considering the higher potency of CA-CDs against SA-DR, we hypothesized that CA-CDs may show antibiofilm activity and investigated the same. It showed that CA-CDs significantly inhibited the total biofilm formation of SA-DR (Fig. 6). The inhibition was dose-dependent, and at the MIC of CA-CDs, there was >50% inhibition in biofilm production. The inhibition was >90% beyond the 5 μg mL−1 concentration. This was also evident from the SEM analysis (Fig. 7), which revealed a remarkable reduction in thickness and aggregation of biofilm in comparison with the bacterial inoculums.
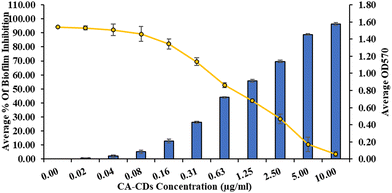 |
| | Fig. 6 Biofilm inhibition of CA-CDs against S. aureus DR. values represented as mean ± S.D. (n = 3). | |
 |
| | Fig. 7 SEM images of (a) S. aureus DR bacterial inoculum served as the control and (b) CA-CD biofilm inhibition of S. aureus DR. | |
3.5. Checkerboard assay of CA-CDs
Since CA-CDs were found to inhibit the biofilm production of SA remarkably, they are likely to complement CIP and enhance susceptibility. A checkerboard assay was carried out to test this. Three combinations showed complementary benefits. While the additive effect was evident in the combination of CIP (1.75 μg mL−1) and CA-CDs (0.156 μg mL−1), synergy was displayed by the other two combinations. Out of these, the combination of CIP (0.437 μg mL−1) and CA-CDs (0.312 μg mL−1) showed the lowest FIC (Fig. 8). Thus, following combination, there were more than 8-fold and 4-fold enhancements in the potency of CIP and CA-CDs, respectively, against SA-DR. Since SA is the predominating pathogen of skin infection,48 this combination can be most beneficial for the management of topical infections involving SA-DR.
 |
| | Fig. 8 Checkerboard assay of CA-CDs in combination with CIP on S. aureus DR to study the synergism between CIP and CA-CDs. | |
3.6. Conjugation of CIP to CA-CD
Poor permeability is a deterrent for skin application49 of CIP. There has been growing interest in using CDs as carriers for drug delivery.50 Thus, it was worthwhile to investigate CA-CDs as carriers for CIP. Although multiple combinations of CIP and CA-CDs showed synergism, approximately 1:1 w/w ratio of CIP (0.437 μg mL−1) and CA-CDs (0.312 μg mL−1) was relatively more synergistic against SA-DR. Hence, it was thought worthwhile to develop a reversible complex of similar composition. CIP is a BCS-class IV drug with poor solubility and permeability. The high solubility of CA-CDs may aid in higher solubilization of CIP through this complex. Furthermore, the nano-size of CA-CDs may promote its permeability. With these objectives, CIP was reversibly conjugated to the CA-CDs. The medium was maintained at pH 8 to ensure that the CIP anionic form interacted with the cationic CA-CDs. The endpoint of complexation was noted through TLC (Fig. 9(a) and (b)). Because of higher aqueous solubility, the CA-CDs did not migrate (Rf, 0) in the plate, whereas poor aqueous solubility ensured a high Rf (0.6) for CIP. The lack of spots corresponding to CIP and CA-CDs and the appearance of new spots with Rf 0.07 suggested complexation. This also shows that all of the CIP is consumed and there are no unbound residues of CIP. Hence, CIP is efficiently loaded onto the CA-CDs. The formation of the ionic complex was also supported by zeta potential measurements. Unlike the relatively high negative potential of CIP (−7.70 ± 1 mV) (Fig. 9(g)), the complex showed low potential (−0.30 ± 0.1 mV) (Fig. 9(g)). This can be attributed to the interaction between anionic CIP and cationic CA-CDs. FTIR data also supported the interaction (Fig. 10(a)). The characteristic peaks for O–H and CO stretching of CIP were shifted to lower wavelengths. Furthermore, there was peak broadening in the hydrogen index region of the complex, unlike that of CIP, which supports the complexation. This was also supported by 1H NMR data (Fig. S3, ESI†). Two doublets (7.34, 7.35 ppm and 7.51, 7.52 ppm) for aromatic C–H and multiplets (3.45–4.01 ppm) for aliphatic C–H corresponding to the tricyclic ring and the piperazine ring indicated characteristic structural features of CIP and matched with that of the reported NMR of CIP.51 Additionally, a reduction in fluorescence intensity following capping also suggested the formation of the complex (Fig. 9(c)).37,52 Although CIP has some fluorescence properties53 and CA-CDs are fluorescent (Fig. 9(d)), their complex seems to have undergone some fluorescence (Fig. 9(e)) quenching following complexation.52
 |
| | Fig. 9 TLC of CA-CDs, CIP and CIP-CA-CDs at (a) a short wavelength (254 nm) and (b) a long wavelength (364 nm). (c) Comparative fluorescence intensity before and after conjugation of CA-CDs with CIP (CIP-CA-CDs). Photographs of (d) CA-CDs and (e) CIP-CA-CDs under UV light (long wavelength – 365 nm). Zeta analysis for (f) size (CIP-CA-CDs) and (g) potential of CIP and CIP-CA-CDs. | |
 |
| | Fig. 10 Comparative FTIR spectra of (a) CA-CDs, CIP-CA-CDs and CIP. (b) TEM image of CIP-CA-CDs. (c) The size distribution histogram was derived from the TEM images and (d) cell viability of CIP-CA-CDs. | |
A narrow size distribution was indicated by Zetasizer analysis, suggesting the mono-dispersibility of this complex (Fig. 9(f)). This was also evident from the TEM data that showed an average size of 4.52 nm for the complex (Fig. 10(b)). Nanoparticles with sizes between 4 and 20 nm have been shown to permeate both intact and damaged skin.54 Hence, despite a moderate increase in size, the complex is small enough to achieve higher permeability. The mono-dispersibility and ultra-size can favour higher permeation with uniform distribution and reduced self-diffusion barriers.55 Furthermore, at concentrations of 10 μg mL−1, 50 μg mL−1, and 100 μg mL−1, CIP-CA-CDs showed 99%, 96%, and 85% cell viability, respectively (Fig. 10(d)). Thus, like CA-CDs, the CIP-CA-CDs are relatively noncytotoxic.
3.7. In vitro drug release study
The reversibility of the ionic complex between CIP and CA-CDs is critical to ensure its application as a carrier system. Thus, an in vitro drug release study was conducted per the established protocol.28 Considering its potential for skin application, the release study was carried out while maintaining the pH of the medium at 7.4. The amount of CIP released was measured using a validated UV-visible spectrophotometric method at 274 nm. At equivalent concentrations, CIP showed (Fig. S4(a), ESI†) characteristic UV spectrum.56 While CIP-CA-CDs showed relatively lower absorbance (Fig. S4(b), ESI†) with a different spectrum and higher (306 nm) λmax, CA-CDs’ absorbance at these wavelengths was insignificant. This indicated the suitability of estimating CIP from this matrix at 274 nm. The spectra of the drug release study showed characteristic peaks of CIP (Fig. S4(c), ESI†), indicating the diffusibility of CIP across the dialysis bag. The lack of peaks for CIP-CA-CDs in these further supported the suitability of the method. Accordingly, the validated method (Fig. S4(d), ESI†) was used and concentrations above the limit of quantitation (LOQ) were considered for the release study. Till 2 h, the release of CIP was insignificant. However, beyond this and until 12 h, a steady release of CIP was observed from the complex with a cumulative release of more than 60%. The release order is better suited to zero orders (Table S2, ESI†). Furthermore, >90% release was observed by 26 h (Fig. 11(a)). This shows that CIP is released slowly but steadily over a long period, which may be a desirable feature for topical application of this complex as it can potentially act as a reservoir for sustained efficacy.
 |
| | Fig. 11 (a) In vitro cumulative release profile of the CIP-CA-CD solution. Values represented as mean ± S.D. (n = 3). (b) The Franz-diffusion apparatus was used to perform an ex vivo permeation profile of CIP solution (3 mg mL−1 of CIP) in PBS (pH 7.4) at 37 °C. Values represented as mean ± S.D. (n = 3) and (c) ex vivo permeation profile of the CIP-CA-CD solution (3 mg mL−1 of CIP-CA-CDs) in PBS (pH 7.4) at 37 °C. Values represented as mean ± S.D. (n = 3). | |
3.8. Ex vivo permeability studies
To assess the utility of this complex as a permeation enhancer, skin permeability studies were performed on the ear pinna of the goat skin to determine the ex vivo permeation of CIP from the CIP-CA-CD solution. The control (only CIP solution) showed a cumulative permeation of 1.783% (Fig. 11(b)) with an average permeability coefficient of 0.00049 cm h−1. A similar low permeability coefficient for CIP has also been reported using Caco-2 cells,57 which is in agreement with its properties as a BCS-IV drug with poor permeability. Compared to this, the cumulative permeability of CIP from CIP-CA-CDs was 31.15% (Fig. 11(c)). Thus, a permeability coefficient of 0.0172 cm h−1 for CIP-CA-CDs suggests a 34.9-fold increase in the permeability. Hence, the CA-CDs seem to act as carriers for promoting the permeability of CIP.
3.9. Effect of CIP-CA-CDs on SA
The release studies revealed insignificant CIP release in the first few hours. Thus, unless the CIP-CA-CD complex retains the antimicrobial activity, this may be ineffective during the initial application stages. Therefore, the antimicrobial effects of CIP-CA-CD were investigated against both susceptible and resistant strains of SA. Interestingly, the CIP-CA-CD showed higher potency as indicated by lower MICs against susceptible (0.0087 μg mL−1) as well as resistant SA (0.035 μg mL−1). This shows that complexation has not adversely affected the antimicrobial properties. Thus, despite minimal CIP release during initial hours, the CIP-CA-CDs can manage infection.
The bacterial growth kinetic study provides additional support to the antibacterial studies. The bacterial growth curve (Fig. S5, ESI†) and Fig. 12) based on optical density (OD) showed characteristic phases of bacterial growth kinetics.58 At MIC levels, CA-CDs and CIP inhibited bacterial growth (Fig. S5, ESI†) both in SA and SA-DR. Similarly, the CIP-CA-CDs showed inhibition of growth of SA and SA-DR (Fig. 12). In agreement with the MIC data, reduction in the concentration of CIP-CA-CDs to half or one-fourth of MIC reduced these effects. This suggests a concentration-dependent effect against both SA and SA-DR. Moreover, it also indicates that the complex CIP-CA-CDs themselves are capable of significantly inhibiting the bacterial growth.
 |
| | Fig. 12 Growth kinetics analysis showing antimicrobial efficacy at MIC, ½ and ¼th of the MIC of (a) CIP-CA-CDs against S. aureus and (b) CIP-CA-CDs against S. aureus DR (values represented as mean ± S.D. (n = 3)). | |
4. Conclusion
In this work, cinnamic acid, a natural molecule with diverse biological activities but poor antimicrobial properties, was selected as a precursor. Contrary to the common practice of using precursors with amine functional groups for developing positively charged CDs for antimicrobial properties, this work relied on the process of carbonisation to minimise negative charge. This also demonstrated the advantage of microwave irradiation for synthesising CDs with a high quantum yield using polar precursor. The positively charged CDs showed a potent effect against both Gram-positive (SA) and Gram-negative (EC) bacteria. Moreover, it showed a low MIC against clinically resistant SA, unlike CIP. Biofilm is well-known to reduce the susceptibility of CIP to SA. CA-CDs showed the ability to inhibit biofilms induced by resistant SA. Interestingly, multiple combinations of CA-CDs showed synergy against resistant strains of CA. Other than enhancing the susceptibility of SA to CIP, these CDs also promoted the skin-permeability through the formation of their ionic complex (CIP-CA-CDs) while retaining the antimicrobial properties. Moreover, the complex showed the ability to gradually release CIP like a reservoir. Considering, the poor permeability of CIP as a limitation, this work can encourage the translational application of CIP for topical application. However, further validation in the infection model in vivo is necessary to validate the findings. Nonetheless, this work provides a novel method of developing CDs for antimicrobial application as well as using them as complementary therapy or carriers for further application.
Author contributions
Conceptualization: TKV and BBS; data curation: TKV and AR; formal analysis: TKV, VRR, and AR; investigation: TKV; methodology: TKV, VRR, BSD, AS, AKP, and BBS; resources: BBS and SC; project administration: BBS; resources: BBS; software: VRR; supervision: AKP, SC, and BBS; validation: TKV and BBS; visualization: TKV, VRR, and MG; writing – original draft: TKV and BBS; and writing – review & editing: TKV, VRR, BSD, AS, MG, AR, SC, AKP, and BBS.
Data availability
The data supporting this article have been included as part of the ESI.† Additional data required to understand and verify the research are available from the authors upon reasonable request.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work is partly funded by the Department of Biotechnology (DBT), Ministry of Science and Technology, New Delhi, India (Grant Id: BT/PR42322/TRM/120/525/2021). MG is supported by a fellowship funded by the Department of Biotechnology (DBT), Ministry of Science and Technology, New Delhi, India (Grant Id: BT/INF/22/SP45078/2022). VRR was supported by a fellowship funded by Siksha ‘O’ Anusandhan (Deemed to be) University. The authors would like to acknowledge Prof. (Dr) Ashoka Mahapatra, Department of Microbiology, All India Institute of Medical Sciences, Bhubaneswar, India for providing SA-DR strain for the antibacterial study.
References
- D. Ozyurt, M. Al Kobaisi, R. K. Hocking and B. Fox, Carbon Trends, 2023, 12, 100276 CrossRef CAS.
- A. A. Madhavan, R. G. Moulick and J. Bhattacharya, Carbon Quantum Dots for Sustainable Energy and Optoelectronics, Elsevier, 2023, pp. 225–237 Search PubMed.
- A. Salvi, S. Kharbanda, P. Thakur, M. Shandilya and A. Thakur, Carbon Trends, 2024, 17, 100407 CrossRef CAS.
- W. Su, H. Wu, H. Xu, Y. Zhang, Y. Li, X. Li and L. Fan, Mater. Chem. Front., 2020, 4, 821–836 RSC.
- X. Du, M. Zhang, Y. Ma, Y. Zhang, W. Li, T. Hu, Y. Liu, H. Huang and Z. Kang, J. Mater. Chem. B, 2024, 12, 2346–2353 RSC.
- S. Huang, Y. Song, J. Zhang, X. Chen and J. Zhu, Small, 2023, 19(31), e2207385 CrossRef PubMed.
- J. Du, N. Xu, J. Fan, W. Sun and X. Peng, Small, 2019, 15(32), e1805087 CrossRef PubMed.
- X. Chu, M. Wang, S. Shi, B. Sun, Q. Song, W. Xu, J. Shen and N. Zhou, J. Mater. Sci., 2022, 57, 12752–12781 CrossRef CAS.
- C. Li, G. Chen, Y. Zhang, F. Wu and Q. Wang, J. Am. Chem. Soc., 2020, 142, 14789–14804 CrossRef CAS PubMed.
- W. Zhao, K. Liu, Y. Wang, F. Li, R. Guo, S. Song and C. Shan, Adv. Healthcare Mater., 2023, 12(23), e2300324 CrossRef PubMed.
- E. L. Niero and G. M. Machado-Santelli, J. Exp. Clin. Cancer Res., 2013, 32(1), 31 CrossRef PubMed.
- Y.-C. Hseu, M. Korivi, F.-Y. Lin, M.-L. Li, R.-W. Lin, J.-J. Wu and H.-L. Yang, J. Dermatol. Sci., 2018, 90, 123–134 CrossRef CAS PubMed.
- K. Jia, P. Shi, L. Zhang, X. Yan, J. Xu and K. Liao, J. Nutr. Biochem., 2025, 135, 109769 CrossRef CAS PubMed.
- S. Yilmaz, M. Sova and S. Ergün, J. Appl. Microbiol., 2018, 125(6), 1714–1727 CrossRef CAS PubMed.
- J. D. Guzman, Molecules, 2014, 19, 19292 CrossRef PubMed.
- J. Wang, Y. H. Ng, Y.-F. Lim and G. W. Ho, RSC Adv., 2014, 4, 44117–44123 RSC.
- C. Wang, Y. Chen, T. Hu, Y. Chang, G. Ran, M. Wang and Q. Song, Nanoscale, 2019, 11, 11967–11974 RSC.
- Y. Hu and Z. Gao, J. Hazard. Mater., 2020, 382, 121048 CrossRef CAS PubMed.
- J. D. Robertson, L. Rizzello, M. Avila-Olias, J. Gaitzsch, C. Contini, M. S. Magoń, S. A. Renshaw and G. Battaglia, Sci. Rep., 2016, 6, 27494 CrossRef CAS PubMed.
- R. E. de Araujo and C. T. Dominguez, Methods Mol. Biol., 2020, 2135, 37–51 CrossRef CAS PubMed.
- A. Tiwari, S. Walia, S. Sharma, S. Chauhan, M. Kumar, T. Gadly and J. K. Randhawa, J. Mater. Chem. B, 2023, 11, 1029–1043 RSC.
- V. N. Mehta, S. Jha and S. K. Kailasa, Mater. Sci. Eng. C, 2014, 38, 20–27 CrossRef CAS PubMed.
- R. N. Dash, A. Ray, P. Mamidi, S. De, T. K. Mohapatra, A. K. Moharana, T. Mukherjee, S. Ghosh, S. Chattopadhyay, B. B. Subudhi and S. Chattopadhyay, ACS Omega, 2024, 9, 146–156 CrossRef CAS PubMed.
- A. D. R. Nurcahyanti, J. Lady and M. Wink, Curr. Microbiol., 2023, 80, 260 CrossRef CAS PubMed.
- I. Wiegand, K. Hilpert and R. E. W. Hancock, Nat. Protoc., 2008, 3, 163–175 CrossRef CAS PubMed.
- A. Sarangi, B. S. Das, A. Sahoo, B. Jena, G. Patnaik, S. Giri, D. Chattopadhyay and D. Bhattacharya, Curr. Microbiol., 2024, 81, 245 CrossRef CAS PubMed.
- R. Sari, L. Pratiwi and P. Apridamayanti, J. Pharmacopunct., 2022, 25, 101–105 CrossRef PubMed.
- A. S. Krishna, C. Radhakumary, S. S. Priya, R. M. Ramesan and K. Sreenivasan, RSC Adv., 2016, 6, 56313–56318 RSC.
- V. P. R. Regu, D. Behera, S. P. Sunkara, V. Gohel, S. Tripathy, R. P. Swain and B. B. Subudhi, J. Pharm. Sci., 2023, 112, 2494–2505 CrossRef CAS PubMed.
- N. Ahmad, R. Ahmad, T. Mohammed Buheazaha, H. Salman AlHomoud, H. A. Al-Nasif and M. Sarafroz, Saudi J. Biol. Sci., 2020, 27, 1024–1040 CrossRef CAS PubMed.
- Y. Zhang, M. Huo, J. Zhou, A. Zou, W. Li, C. Yao and S. Xie, AAPS J., 2010, 12, 263–271 CrossRef CAS PubMed.
- H. Wang, F. Lu, C. Ma, Y. Ma, M. Zhang, B. Wang, Y. Zhang, Y. Liu, H. Huang and Z. Kang, J. Mater. Chem. B, 2021, 9, 125–130 RSC.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- M. D. Abràmoff, P. J. Magalhães and S. J. Ram, Biophotonics Int., 2004, 11, 36–41 Search PubMed.
- W. Guo, S. Ma, H. Wang, L. Qiao, L. Chen, C. Hong, B. Liu, X. Zheng and H. Peng, Aggregate, 2024, 5(1), e415 CrossRef CAS.
- T. V. de Medeiros, J. Manioudakis, F. Noun, J.-R. Macairan, F. Victoria and R. Naccache, J. Mater. Chem. C, 2019, 7, 7175–7195 RSC.
- H.-L. Yang, L.-F. Bai, Z.-R. Geng, H. Chen, L.-T. Xu, Y.-C. Xie, D.-J. Wang, H.-W. Gu and X.-M. Wang, Mater. Today Adv., 2023, 18, 100376 CrossRef CAS.
- T. Mohorič and U. Bren, J. Mol. Liq., 2018, 266, 218–228 CrossRef.
- S. K. Tammina, D. Yang, S. Koppala, C. Cheng and Y. Yang, J. Photochem. Photobiol., B, 2019, 194, 61–70 CrossRef CAS PubMed.
- K. Damarla, S. Mehra, T. S. Kang, S. Yadav, A. Mishra and A. Kumar, Mater. Adv., 2020, 1, 3476–3482 RSC.
- P. Kadyan, M. Kumar, A. Tufail, A. Ragusa, S. K. Kataria and A. Dubey, Mater. Adv., 2025, 6, 805–826 RSC.
- K. Sekioka, N. Mosleh, D. Boice, R. Hailstone and X. Sun, Mater. Adv., 2025, 6, 2875–2884 RSC.
- A. Pal, M. P. Sk and A. Chattopadhyay, Mater. Adv., 2020, 1, 525–553 RSC.
- M. Rams-Baron, R. Jachowicz, E. Boldyreva, D. Zhou, W. Jamroz and M. Paluch, Amorphous Drugs, Springer International Publishing, Cham, 2018, pp. 107–157 Search PubMed.
- S. O. G. Almawla, L. Dali, Y. M. Ragheb, A. I. Edan, M. M. Ahmed, A. H. Abdulkareem and A. I. Waheeb, J. Appl. Nat. Sci., 2024, 16, 356–363 CAS.
- M. E. Jones, M. R. Visser, M. Klootwijk, P. Heisig, J. Verhoef and F. J. Schmitz, Antimicrob. Agents Chemother., 1999, 43, 421–423 CrossRef CAS PubMed.
- Z.-X. Luo, Y. Li, M.-F. Liu and R. Zhao, Front. Microbiol., 2023, 14, 1328947 CrossRef PubMed.
- M. S. Linz, A. Mattappallil, D. Finkel and D. Parker, Antibiotics, 2023, 12, 557 CrossRef CAS PubMed.
- D. A. Volpe, AAPS PharmSci, 2004, 6, e13 CrossRef PubMed.
- S. K. Kailasa, J. R. Bhamore, J. R. Koduru and T. J. Park, Biomedical Applications of Nanoparticles, Elsevier, 2019, 295–317 Search PubMed.
- S. Gaia, S. Carenzi, A. L. Barilli, E. Bugianesi, A. Smedile, F. Brunello, A. Marzano and M. Rizzetto, J. Hepatol., 2011, 54, 64–71 CrossRef PubMed.
- R. F. Chen, Anal. Lett., 1986, 19, 963–977 CrossRef CAS.
- P. Rana, D. Dabur and H.-F. Wu, Mater. Today Chem., 2024, 38, 102074 CrossRef CAS.
- F. Larese Filon, M. Mauro, G. Adami, M. Bovenzi and M. Crosera, Regul. Toxicol. Pharmacol., 2015, 72, 310–322 CrossRef CAS PubMed.
- J. Xu, M. Song, Z. Fang, L. Zheng, X. Huang and K. Liu, J. Controlled Release, 2023, 353, 699–712 CrossRef CAS PubMed.
- M. Thakur, S. Pandey, A. Mewada, V. Patil, M. Khade, E. Goshi and M. Sharon, J. Drug Delivery, 2014, 2014, 282193 Search PubMed.
- D. El-Sabawi, R. Abu-Dahab, A. G. Al Bakri and I. I. Hamdan, Trop. J. Pharm. Res., 2019, 18, 377 CrossRef CAS.
- K. Theophel, V. J. Schacht, M. Schluter, S. Schnell, C.-S. Stingu, R. Schaumann and M. Bunge, Front. Microbiol., 2014, 10(5), 544 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Varaprasad Rao Regu
a,
Varaprasad Rao Regu