
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A multilayered supramolecular chelate of diclofenac-appended AgI-hydrazide as an efficient anti-inflammatory, anticancer, and nanometal dispersing material, in comparison to analogous ZnII-hydrazide†
Qurrat-ul-Ain *a,
Sammer Yousufb,
Summaiyah Bibia,
Irum Hamida,
Shazia Shaha and
Sumaira Khurshidc
*a,
Sammer Yousufb,
Summaiyah Bibia,
Irum Hamida,
Shazia Shaha and
Sumaira Khurshidc
aDepartment of Chemistry, University of Karachi, Karachi 75270, Pakistan. E-mail: qurrat_chem@uok.edu.pk
bH.E.J. Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi, Karachi 75270, Pakistan
cDepartment of Chemistry, Federal Urdu University of Arts, Science and Technology, Gulshan-e-Iqbal Campus, Karachi-75300, Pakistan
First published on 20th May 2025
Abstract
We herein present new AgI and ZnII chelating materials based on diclofenac (dicf)-appended hydrazide (dh), along with their comparative structural, crystal, solution, anti-inflammatory, and cytotoxic properties. The spectral (FT-IR, NMR, FAB-MS, and UV-vis), physicochemical, SEM, and single-crystal X-ray studies ensure the AgI complex as [Ag(dh)NO3] (1), where neutral dh is a bidentate N,O donor, while nitrate is a monodentate O donor. Chelate 1 bears a distorted trigonal planar geometry and a triclinic (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) crystal. It assembles into a unique supramolecular multilayered (sheet) framework stabilized by N–H⋯O, C–H⋯O, X–H⋯π, Ag⋯π/O/N, and Cl⋯O/H contacts, evidenced by Hirshfeld surface, QTAIM and NCI analyses. ZnII forms a microcrystalline stable tetrahedral complex [Zn(dh)SO4] (2) with two chelate rings from bidentately attached dh and sulfate. Intracellular oxidative burst (ROS) suppression-based anti-inflammatory studies manifest chelate 1 as the strongest ROS inhibitor (IC50 = 2.6 μg mL−1), with the cumulative effect of precursors that makes chelate 1 3-fold superior to reference drugs, dicf and ibuprofen (ibup). Molecular docking validates empirical results and cyclooxygenase-2 (COX-2) inhibition by these compounds as a dominant anti-inflammatory mechanism. The stabilization based on the number of H-bonding/hydrophobic/ionic interactions within the COX-2 binding pocket exhibits the following order: chelate 1 > dh > chelate 2 > dicf > ibup. Chelate 1 shows a high selectivity towards COX-2 comparable to celecoxib, while chelate 2 shows non-selective non-competitive COX-2 inhibition. Replacing nitrate in chelate 1 with bio-anions (Cl−, HCO3−, H2PO4−, and HSO4−) does not significantly alter its COX-2 inhibitory mechanism. Chelate 1 exhibits strong cytotoxicity on cancer cells (PC3 and H460) compared to that on normal BJ fibroblasts, being most selective against PC3 (IC50 = 3.5 μg mL−1, twelve-fold better than cisplatin), while chelate 2 shows low cytotoxicity overall. Chelate 1 also acts as a precursor for the controlled production of stable silver colloids (AgNPs) and uniform thin reflective films (TRFs) in methanol through concentration-dependent selective intramolecular chemical reduction and deposition, not shown by chelate 2 for zinc. This study indicates chelate 1 as a promising future anti-inflammatory/anticancer drug candidate and a plausible single-molecule source for AgNPs and TRFs.
) crystal. It assembles into a unique supramolecular multilayered (sheet) framework stabilized by N–H⋯O, C–H⋯O, X–H⋯π, Ag⋯π/O/N, and Cl⋯O/H contacts, evidenced by Hirshfeld surface, QTAIM and NCI analyses. ZnII forms a microcrystalline stable tetrahedral complex [Zn(dh)SO4] (2) with two chelate rings from bidentately attached dh and sulfate. Intracellular oxidative burst (ROS) suppression-based anti-inflammatory studies manifest chelate 1 as the strongest ROS inhibitor (IC50 = 2.6 μg mL−1), with the cumulative effect of precursors that makes chelate 1 3-fold superior to reference drugs, dicf and ibuprofen (ibup). Molecular docking validates empirical results and cyclooxygenase-2 (COX-2) inhibition by these compounds as a dominant anti-inflammatory mechanism. The stabilization based on the number of H-bonding/hydrophobic/ionic interactions within the COX-2 binding pocket exhibits the following order: chelate 1 > dh > chelate 2 > dicf > ibup. Chelate 1 shows a high selectivity towards COX-2 comparable to celecoxib, while chelate 2 shows non-selective non-competitive COX-2 inhibition. Replacing nitrate in chelate 1 with bio-anions (Cl−, HCO3−, H2PO4−, and HSO4−) does not significantly alter its COX-2 inhibitory mechanism. Chelate 1 exhibits strong cytotoxicity on cancer cells (PC3 and H460) compared to that on normal BJ fibroblasts, being most selective against PC3 (IC50 = 3.5 μg mL−1, twelve-fold better than cisplatin), while chelate 2 shows low cytotoxicity overall. Chelate 1 also acts as a precursor for the controlled production of stable silver colloids (AgNPs) and uniform thin reflective films (TRFs) in methanol through concentration-dependent selective intramolecular chemical reduction and deposition, not shown by chelate 2 for zinc. This study indicates chelate 1 as a promising future anti-inflammatory/anticancer drug candidate and a plausible single-molecule source for AgNPs and TRFs.
1 Introduction
Inflammation, pain, and fever are interconnected defensive alarms that occur as part of the body's natural immune reaction to injury or harmful stimuli.1 This involves the discharge of various inflammatory biomarkers and mediators, including cytokines, prostaglandins (PGs), leukotrienes, and reactive oxygen/nitrogen species (ROS/RONS), resulting in symptoms such as pain, warmness, erythema, edema, asthma, and allergic reactions.1,2 Although inflammation is a defense mechanism, chronic or excess inflammation can cause several illnesses such as cancer, arthritis, cardiovascular diseases (CVDs), sepsis, and Alzheimer's dementia.1 These diseases are recognized as a significant burden worldwide, severely impacting the quality of life.3The inflammatory disorders are widely treated with nonsteroidal anti-inflammatory drugs (NSAIDs); however, long-term therapy can lead to adverse gastrointestinal (GI), cardiac, renal, hematologic, and hepatic side effects.4 Diclofenac (dicf) is one of the top-ranked most effective medications belonging to the phenylalkanoic acid family of NSAIDs.5 It is widely prescribed for postoperative pain, rheumatoid arthritis, osteoarthritis, and dysmenorrhea.6,7 Antibacterial and antitumor effects of dicf are also documented. Despite its effectiveness, dicf also has drawbacks (like all NSAIDs) such as GI ulceration or bleeding effects and increased heart stroke risk.6 Developing powerful and safer anti-inflammatory agents is thus a great challenge.8
The dicf and other NSAIDs primarily function by blocking the PG biosynthesis through inhibition of cyclooxygenases (COXs).4 The GI side effects of classical NSAIDs are mainly attributed to their non-selective COX inhibition or COX-1 selectivity. This is because the widely expressed COX-1 regulates gastric mucosal integrity and hence GI health with other physiological processes, while COX-2 is the main isoform responsible for the formation of PGs, induced as a mediated inflammatory response.6 Thus, COX-2 selective inhibitors such as rofecoxib, celecoxib, and etoricoxib, making a subclass of NSAIDs with the capability to selectively suppress COX-2 while rescuing COX-1, are recognized better in reducing inflammation with reduced GI adverse effects.4
Another key factor causing GI damage related to carboxylic acid family NSAIDs is the topical effect with local irritation due to the interaction between the acidic COOH moiety and GI mucosa.8 To mitigate these effects, various synthetic approaches involving chemical modification of the carboxyl group of drugs with less acidic organic scaffolds (hydrazones, amides, azoles, etc.) have been pursued to form novel NSAID derivatives (NSAIDDs) with reduced ulcerogenicity, enhanced anti-inflammatory activity, and often additional potency against cancer.5,9–13
Modifying NSAIDs with metals to develop new complexes has also been extensively researched to improve or broaden therapeutic potency and reduce drug toxicity.14–16 The M–NSAID complexes can provide enhanced drug stability, solubility, and pharmacokinetics through synergy, unique molecular architectures, charge neutralization, or ligand exchange/redox reactivity.17–19 The carboxylate NSAIDs, typically behaving as mono- or bi-dentate (chelating or bridged) O-donor ligands, coordinate a number of metals (transition, rare earth, alkali, alkaline earth, or post-transition) to form M–NSAID complexes with fascinating structures and diverse nuclearities, which have attracted much attention from inorganic chemists.14–16,20–24 The M–NSAID complexes of dicf and other carboxylate NSAIDs (such as aspirin, ibuprofen, diflunisal, indomethacin, niflumic acid, naproxen, and piroxicam) with CuII, CoII, NiII, MnII, CdII, MgII, CaII, BaII, SrII, FeIII, VOII, EuIII, GdIII, TbIII, InIII, RuII, PtII, SnIV, ZnII, and Ag1 ions have been recently reviewed, indicating their bioefficacies against inflammation, pain, fever, bacteria, proliferation, cancer, gastric ulcer, lipoxygenase (LOX), and ROS.14–16,25 ZnII–NSAIDs can also act as metallovesicles for multi-drug delivery.26
Another important class of modified NSAIDs are the metal complexes of NSAIDDs (M–NSAIDDs), which are relatively new and less frequently documented, mostly containing the aroyl hydrazone-Schiff base (HSB) moiety in the derivatized drug.19,27,28 Examples include a trinuclear CuII complex of naproxen salicyl-HSB with DNA binding ability,29 antibacterial tetracoordinate metal complexes of naproxen 5-bromo-salicyl-HSB,30 anticancer organoselenides of different NSAIDs,31 and anti-inflammatory metallogelators based on 3-pyridyl amide derivatives of ibuprofen, sulindac, and flurbiprofen.32
The adjustment of an attached ligand, hence, can readily fine tune properties such as architecture, kinetics, thermodynamics, toxicology, and the use of a metal complex.18,19 Designing new metal complexes with NSAIDD ligands bearing biologically active moieties such as the hydrazide group could make them more potent and safer. Hydrazides are a versatile class of N,O containing ligands (R-CO-NH-NH2), with enormous scope in coordination chemistry, catalysis, and therapeutics.33–39 We previously reported square-planar PdII-(R-benzohydrazide) complexes in the mono- or bidentate mode with antimicrobial, anti-ROS, antiglycation, and enzyme (phosphodiesterase, urease, α-glucosidase, carbonic anhydrase, LOX, and butyrylcholinesterase) inhibiting effects.40–43 ZnII-hydrazide complexes have shown various activities, including antimicrobial,44 anticancer,45 antioxidant,38 and hydrogel formation.34 SilverI complexes of hydrazides46 and the effect of metal bonding to NSAID hydrazides on their biological47 or other properties have received limited research interest. Concomitantly, the anti-inflammatory role of complexes of zinc27,28,32,48–50 and silver,32,51–54 made from drugs or other classes of ligands, is well established. These considerations have prompted us to synthesize, characterize, and analyze the anti-inflammatory and cytotoxic potential of unexplored AgI and ZnII complexes of dicf-derived hydrazide (dh). The fate of the AgI–dh complex in methanol towards the formation of Ag nanoparticles and thin films through selective intra-molecular redox activity has also been exploited as an additional new application of silverI-hydrazide materials.
2 Experimental
2.1 Materials and physicochemical measurements
The syntheses and analyses were executed utilizing reagents of analytical grade, mostly procured from Merck, Sigma Aldrich, Alfa Aesar, or BDH, without further purification. Different solvents such as ethanol and methanol were distilled by applying standard procedures before use. Elemental (C,H,N,S) contents were analyzed through a 2400 (II) analyzer, PerkinElmer. FT-IR spectra (400–4000 cm−1) of solid samples (prepared as KBr disks) were recorded on a 560 IR spectrophotometer (Shimadzu). The 1H NMR and 13C{1H} NMR spectra of fresh DMSO-d6 solutions (room temperature) were obtained through an AVANCE NEO 500-MHz spectrometer, utilizing the internal standard tetramethylsilane. FAB/EI mass data of compounds were collected from JEOL 600H-2/1 apparatus. The molar conductance of compounds in 1 mM DMSO solutions was estimated using a calibrated Hanna conductivity meter at room temperature. The metal content in complexes was determined by complexometric (zinc) or argentometric (silver) procedures from the literature.55 The atomic absorption data for Zn in the respective complex were also obtained using a Thermo Scientific atomic absorption spectrophotometer Model-AA301. Electronic spectra were recorded using a BK-UV1800PC spectrophotometer. The surface morphology and dimensions of synthesized metal compounds were determined using SEM (model JSM-6380A, JEOL, Japan); solid samples were loaded onto carbon-coated copper grids, and images were taken at appropriate magnification applying an accelerating voltage of 20 kV.2.2 Synthesis of ligand and metal chelates
The diclofenac-derived hydrazide, 2-[2-(2,6-dichlorophenylamino)phenyl]acetohydrazide (dh), was prepared as reported previously.13 The silver1 and ZnII complexes (chelates) of dh were synthesized according to Scheme 1. | ||
| Scheme 1 Synthetic pathway for metal complexes of dh: 1 (Ag1) and 2 (ZnII). | ||
Experimental data: yield: 86%. Anal. Calcd for C14H13N4O4Cl2Ag (MW, 480.0 g mol−1): C, 35.0; H, 2.7; N, 11.7; Ag, 22.5. Found: C, 35.1; H, 2.7; N, 11.7; Ag, 22.4%. UV-vis, λmax (DMSO)/nm 280 (ε/dm3 mol−1 cm−1 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600); λmax (MeCN)/nm 196 and 275 (ε/dm3 mol−1 cm−1 59600 and 15300). FTIR (KBr cm−1): 3269, 3208 (νNH/NH2), 3044 (νCHar), 2926 (νasCH2), 2862 (νsCH2), 1663 (νC
600); λmax (MeCN)/nm 196 and 275 (ε/dm3 mol−1 cm−1 59600 and 15300). FTIR (KBr cm−1): 3269, 3208 (νNH/NH2), 3044 (νCHar), 2926 (νasCH2), 2862 (νsCH2), 1663 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1583 (δNH2), 1506 (CC), 1450 (δCH2), 1400 (νNO3), 1302 (νC–N), 1034 (νN–N), 831 (δNO3), 532 (νAg–N). 1H NMR (500 MHz, DMSO-d6, Me4Si) δ (ppm) = 3.53 (s, 2H, CH2), 4.81 (s, 2H, NH2), 6.29 (d, 1H, H-7, J = 8.0 Hz), 6.84 (t, 1H, H-9, J = 7.5 Hz), 7.03 (t, 1H, H-8, J = 7.8 Hz), 7.16 (m, 2H, H-4/H-10), 7.50 (d, 2H, H-3/H-5, J = 8.0 Hz), 8.37 (s, 1H, CONH), 9.74 (s, 1H, Ar–NH–Ar). 13C{1H} NMR (125 MHz, DMSO-d6, Me4Si) δ (ppm) = 37.62 (CH2), 116.00 (C-7), 120.69 (C-9), 125.02 (C-8), 125.06 (C-4), 127.31 (C-10), 129.17 (C-3/C-5), 129.37 (C-11), 130.34 (C-2/C-6), 137.12 (C-1), 142.91 (C-12), 170.93 (CO). FAB(+)-MS m/z: 418 [M + 2 − NO3]+. FAB(−)-MS m/z: 479 [M + 2 − H]−. Molar conductivity (DMSO): 85.5 Ω−1 cm2 mol−1.
O), 1583 (δNH2), 1506 (CC), 1450 (δCH2), 1400 (νNO3), 1302 (νC–N), 1034 (νN–N), 831 (δNO3), 532 (νAg–N). 1H NMR (500 MHz, DMSO-d6, Me4Si) δ (ppm) = 3.53 (s, 2H, CH2), 4.81 (s, 2H, NH2), 6.29 (d, 1H, H-7, J = 8.0 Hz), 6.84 (t, 1H, H-9, J = 7.5 Hz), 7.03 (t, 1H, H-8, J = 7.8 Hz), 7.16 (m, 2H, H-4/H-10), 7.50 (d, 2H, H-3/H-5, J = 8.0 Hz), 8.37 (s, 1H, CONH), 9.74 (s, 1H, Ar–NH–Ar). 13C{1H} NMR (125 MHz, DMSO-d6, Me4Si) δ (ppm) = 37.62 (CH2), 116.00 (C-7), 120.69 (C-9), 125.02 (C-8), 125.06 (C-4), 127.31 (C-10), 129.17 (C-3/C-5), 129.37 (C-11), 130.34 (C-2/C-6), 137.12 (C-1), 142.91 (C-12), 170.93 (CO). FAB(+)-MS m/z: 418 [M + 2 − NO3]+. FAB(−)-MS m/z: 479 [M + 2 − H]−. Molar conductivity (DMSO): 85.5 Ω−1 cm2 mol−1.
Experimental data: yield: 84%. Anal. Calcd for C14H13N3O5SCl2Zn (MW, 471.6 g mol−1): C, 35.7; H, 2.8; N, 8.9; S, 6.8; Zn, 13.9%. Found: C, 35.6; H, 2.75; N, 9.0; S, 6.8; Zn, 13.8%. UV-vis, λmax (DMSO)/nm 280 (ε/dm3 mol−1 cm−1 14500). FTIR (KBr cm−1): 3335, 3269, 3192 (νNH/NH2), 3057 (νCHar), 2922 (νasCH2), 2861 (νsCH2), 1653 (νCO), 1597 (δNH2), 1508 (νCC), 1450 (δCH2), 1294 (νC–N), 1109 (νN–N, νSO4), 617 (δasSO4), 528 (νZn–N). 1H NMR (500 MHz, DMSO-d6, Me4Si) δ (ppm) = 3.54 (s, 2H, CH2), 4.66 (s, 2H, NH2), 6.28 (d, 1H, H-7, J = 8.0 Hz), 6.83 (t, 1H, H-9, J = 7.0 Hz), 7.03 (t, 1H, H-8, J = 7.8 Hz), 7.16 (m, 2H, H-4/H-10), 7.50 (d, 2H, H-3/H-5, J = 8.0 Hz), 8.31 (s, 1H, CONH), 9.77 (s, 1H, Ar–NH–Ar). 13C{1H} NMR (125 MHz, DMSO-d6, Me4Si) δ (ppm) = 37.29 (CH2), 115.97 (C-7), 120.68 (C-9), 124.95 (C-8), 125.10 (C-4), 127.29 (C-10), 129.15 (C-3/C-5), 129.51 (C-11), 130.33 (C-2/C-6), 137.17 (C-1), 142.93 (C-12), 171.28 (CO). FAB(+)-MS m/z: 474 [M + 2 + 3H]+. FAB(−)-MS m/z: 470 [M + 2 − H]−. Molar conductivity (DMSO): 20.7 Ω−1 cm2 mol−1.
2.3 X-ray crystallographic studies
The single crystal of complex 1 of appropriate size (0.12 × 0.14 × 0.24 mm3) was subjected to X-ray diffraction for structural elucidation employing a Bruker diffractometer (D8 Venture) coupled with a graphite-monochromated microfocus radiation source (Cu-Kα, 1.54178 Å) and a Photon-100 CMOS detector at 273(2) K. To integrate and reduce data, the Bruker SAINT program was used. Absorption correction of data was accomplished using SADABS (multi-scan method). The Bruker SHELXTL software package was applied to solve and refine the structure under direct and full-matrix least-squares on F2 procedures using SHELXT-2014/556 and SHELXL-2018/357 programs, respectively. The refinement of nonhydrogen atoms was made anisotropically, while H-atoms were positioned at ideal geometry with refinement using the riding model. Three unsubstituted carbon atoms of the dichlorobenzene ring of one of the two molecules of complex 1 were disordered, which were refined using the free variable value and EADP constraint on a displacement parameter. Molecular graphics related to structure, interactions, and packing in the crystal lattice were produced using Mercury 2023.3.0.58 Crystal data for complex 1 have been deposited at the Cambridge Crystallographic Data Center (CCDC number 2260170).To characterize, illustrate and quantify non-covalent intermolecular interactions, Hirshfeld surface analysis (HSA) of complex 1 crystal structure was conducted using Crystal Explorer 21.559 and the CIF file, and the respective 2D fingerprint plots were created between di and de, the distance from the surface to the nearest interior and exterior atoms, respectively. The contact enrichment ratio (EXY) for an element pair (X⋯Y) was computed from HSA information by comparing the proportion of actual contacts (CXY) with the theoretical proportion of equivalently distributed random contacts (RXY) in the crystal (EXY = CXY/RXY).60 To comprehend the topology of intra- and inter-molecular interactions in complex 1 crystals, the Bader's quantum theory of atoms-in-molecules (QTAIM) and non-covalent interaction (NCI) analyses were executed on parts of self-assembled supramolecular structure using Multiwfn 3.8 software.61 The Multiwfn output files were applied to render the reduced density gradient (RDG) isosurfaces using the VMD 1.9.4a53 program.62 Molecular electrostatic potential (MEP) analysis was carried out using the GaussView 6/Gaussian 16 W program after geometry optimization using the DFT-B3LYP approach with GenECP mixed basis sets [LanL2DZ for metals and 6-311+G(2d,p) for other atoms].63
2.4 Anti-inflammatory assay
A luminol-enhanced chemiluminescence method involving oxidative burst as used by Mesaik et al. (2012) was performed with slight alterations to determine the anti-inflammatory activity of synthesized compounds.64 This method utilized 2 mg mL−1 serum opsonized zymosan (SOZ) [Fluka, Switzerland] as an activator and 7 × 10−5 M luminol (Research Organics, Cleveland, USA) as an intracellular ROS (reactive oxygen species) detecting probe. Initially, 25 μL of sample solution of varying concentrations (1–100 μg mL−1), in triplicates, was mixed with the same volume of human whole blood diluted to 1 × 106 cells mL−1 in Hank's Balanced Salt Solution (HBSS++, having chloride salts of CaII and MgII) [Sigma, St. Louis, USA], taken in 96-well white 1/2-area microplates (Costar, NY, USA). The resulting suspension was incubated in the luminometer thermostat (Labsystems, Helsinki, Finland) at 37 °C for 15 minutes. Then, all the wells (except wells for blank, containing only HBSS++) were treated with 25 μL of each of SOZ and luminol. After 50 minutes, the RLU (relative light units) were recorded using a luminometer as a measure of the ROS level. Control wells contained no sample compounds but only cells and HBSS++. The ROS (oxidative burst) inhibition percentage was estimated using the given formula:64,65
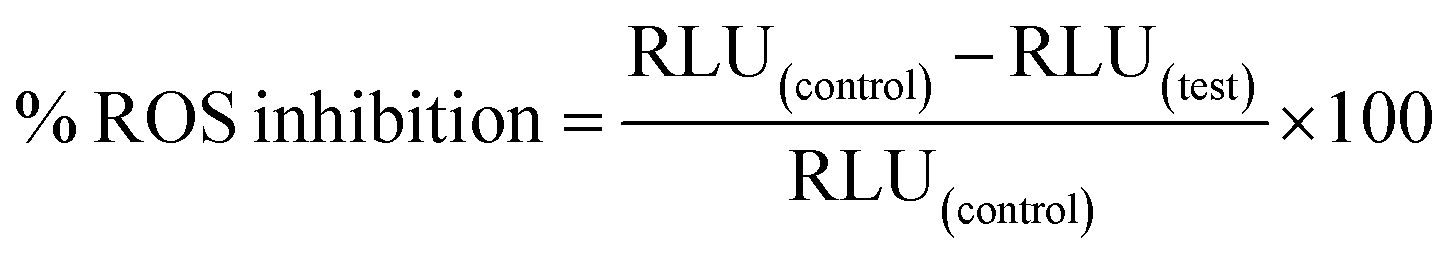 | (1) |
The ibuprofen and diclofenac were used as standard or reference drugs for the anti-inflammatory assay. The results of the anti-inflammatory assay were presented as mean ± SD for triplicates.
2.5 Cytotoxicity assay
The cytotoxic potential of complexes (1 and 2), their precursors (free metals and the dh ligand) and the standard (cisplatin) was evaluated using spectrophotometric MTT [3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyl-tetrazolium bromide] assay67 on human normal and cancerous cell lines from ATCC: BJ (normal foreskin fibroblast, CRL-2522), H460 (lung carcinoma, HTB-177), and PC3 (prostate adenocarcinoma, CRL-1435). Dulbecco's Modified Eagle Medium (for BJ and H460) and Gibco RPMI-1640 medium (for PC3), supplemented with penicillin (100 IU mL−1), streptomycin (100 μg mL−1), L-glutamine (2 mM), and fetal bovine serum (5%), were used for culturing of cells at 37 °C incubation temperature under 5% CO2. The harvested cells at 5 × 104 cells per mL, counted using a haemocytometer, were seeded in 96-well plates (100 μL per well) and incubated overnight to adhere. The medium was then removed, and the cells were treated with 200 μL of test compounds at 0.1–25 μg mL−1 concentration for 48 h. Afterwards, 200 μL of MTT solution (0.5 mg mL−1) was added to each well, and the cells were incubated for another 3 h, followed by addition of 100 μL of DMSO to dissolve the reduced MTT product (blue formazan). The mixture was gently mixed for 15 min, and absorbance was measured at 550 nm using a microplate reader (Spectra Max plus, Molecular Devices, CA, USA). The cytotoxicity (cell growth inhibition) percent was computed using the mean optical density of triplicate test samples compared to positive and negative control groups. Data processing for IC50 values, the concentration causing 50% cell growth inhibition, was conducted on SoftMax Pro software (Molecular Device, USA).3 Results and discussion
3.1 Materials design
The chelates of silverI (1) and zincII (2) were prepared in good yields by reacting metal salts with diclofenac hydrazide (dh) in equimolar amounts at RT (Scheme 1). Gray single crystals of complex 1 were grown for 2–3 days in a static dark environment, while complex 2 instantly appeared as a white microcrystalline solid. Methanol produced smaller crystals of complex 1 compared to ethanol. Prolonged stirring, heat, or higher L/M mole ratios complicated complex 1 crystallization with a high yield of silver side products. Thus, the M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :L molar ratio of 1:1, ethanol, and static normal conditions were optimum for high-quality crystal growth of complex 1. The complexes were characterized using spectroscopic, physicochemical, and microscopic methods, with analytical data detailed in Section 2.2. Complex 1 was also confirmed by X-ray crystallography, aligning with other analytical results.
:L molar ratio of 1:1, ethanol, and static normal conditions were optimum for high-quality crystal growth of complex 1. The complexes were characterized using spectroscopic, physicochemical, and microscopic methods, with analytical data detailed in Section 2.2. Complex 1 was also confirmed by X-ray crystallography, aligning with other analytical results.
3.2 Spectral studies
O bond of dh was shifted by 25 cm−1 for complex 1 and 15 cm−1 for complex 2, suggesting additional coordination with carbonyl oxygen.37 A new medium-intensity M–N stretching band (at 528–532 cm−1)38 and a significant spectral deviation in N–N stretching (a red shift of 54 cm−1 for 1 and a blue shift of 21 cm−1 for 2) relative to free dh also verified the coordination of NH2 nitrogen to the metal ions in the complexes. The IR absorption signals for the carbonyl and amine groups of complexes also experienced a significant broadness compared to free dh, indicating the participation of NH2 hydrogen and CO oxygen in intra- or inter-molecular hydrogen bonding in the solid state, likely due to the attachment of electropositive metal ions to these groups.
 | ||
| Fig. 1 Comparative vibrational spectra of complex 1 (red), complex 2 (green), and parent ligand dh (blue). | ||
Two distinct bands at 1109 and 617 cm−1 (for sulfate stretching and asymmetric bending)68,69 indicated the presence of sulfate in complex 2. The existence of the nitrate ion in complex 1 was evidenced by characteristic bands at 1400 and 831 cm−1 due to the asymmetric stretching of the N–O bond and out-of-plane deformation of nitrate, respectively.70,71
C–NH proton envisaged that the dh ligand remains protonated (neutral) in polar solvents, suggesting that the metals do not bond directly with CONH nitrogen.35,41 Other proton signals of dh, ascribed to CH2 (3.51 ppm), Ar–NH–Ar (9.48 ppm), and aromatic protons (6.28–7.52 ppm), showed nearly similar chemical shifts after metal complexation. The peaks of carbon atoms in the 13C{1H} NMR spectrum of dh and its complexes matched predicted values. The aromatic carbon signals for free dh (16.01–142.93 ppm) showed minimal shifts (Δ = 0–0.04 ppm) after complexing with AgI and ZnII due to changes in conjugation. The carbonyl carbon signal for free dh at 170.76 ppm was shifted downfield to 170.93 ppm for complex 1 (Δ = 0.17 ppm) and 171.28 ppm for complex 2 (Δ = 0.52 ppm). These shifts indicate metal ion binding to the CO oxygen in both complexes.44The dh ligand (calcd mass = 309) in complexes was verified by the [dh − H]− peak at m/z 308 (negative ion mode) or the [dh + H]+ peak at m/z 310 (positive ion mode). Positive ion FAB for complex 1 produced fragment peaks at m/z 278, 214, 180, and 167, corresponding to the sequential release of –NH–NH2, [CO + Cl], second Cl, and CH2 groups from dh. Complex 2 displayed dh fragments at m/z 214 and 180, with an additional signal at m/z 295 attributed to [dh − N]+. For complex 1, free [Ag]+ was detected at m/z 107 and 109 in the positive ion mode,75 with prominent negative ion fragments at m/z 416, 402, 323, 206, and 179, conforming to [Ag(dh) + 2 − 2H]−, [Ag(dh) + 2 − NH2]−, [Ag2(Cl)3 + 4]−, [Ag(Cl)(NO3) + 2]−, and [Ag(Cl)2 + 2]−. Complex 2 exhibited fewer fragment ions, including [M − Cl2 − NH3]+ (m/z = 382) in the positive ion mode, while [M + 2 − N2H4 − 3H]− (m/z = 436), [M − Cl2 − H2O − H]− (m/z = 380), and [Zn(SO4)(Cl)]− (m/z = 195) in the negative ion mode, showing better stability than complex 1. The sulfate in complex 2 was strongly bound to ZnII, as the free metal ion was not detected.
The silver ions freed from complex 1 formed new complex ions with the matrix at metal/glyc molar ratios of 1:1, 1:2, and 1:3, such as [Ag(glyc)]+ (m/z 199), [Ag(glyc)2]+ (m/z 291), and [Ag(glyc)3 − H]+ (m/z 382) in the positive ion mode, and [Ag(glyc)(O2) + 2H]− (m/z 233) and [Ag(glyc)2 + 2H]− (m/z 293) in the negative ion mode.76–78 This reflects the high ligand-exchange reactivity of complex 1, with glyc coordinating Ag1 more effectively than DMSO. The glyc also appeared to trap the anions from ionizable metal complexes, indicated by signals at m/z 127, 154, 219, and 246, corresponding to [glyc + Cl]−, [glyc + NO3]−, [2glyc + Cl]−, and [2glyc + NO3]− ions.
 | ||
| Fig. 2 UV-vis spectral characterization of complexes 1 and 2. (a) Electronic spectra of complex 1 and precursors (the dh ligand and the metal salt AgNO3) in ACN solution (25 μM), (b) electronic spectra of complex 1, complex 2, and the respective ligand (dh) in DMSO solution (25 μM). | ||
In DMSO, band I of complex 1 and dh could not be accessed due to solvent spectral interference, while band II (λmax = 280 nm) exhibited a 5 nm bathochromic shift (Fig. 2b) compared to ACN due to negative solvatochromism. A hyperchromic shift in band II of complex 1 with a 1.6-fold increase in its ε compared to free dh in DMSO further supported its formation. The solubility issues of complex 2 limited its investigation to DMSO, where its band II λmax (280 nm) matched that of complex 1 and dh (Fig. 2b). Band II of complex 2 had a 1.4-fold higher ε (hyperchromic shift) compared to free dh, thus confirming its formation. The absence of d–d transitions in the visible region was expected for complexes due to d10 metal ions. The complexes and ligand remained stable in DMSO and ACN, with no changes in their electronic spectra from 4 minutes to 24 hours.
3.3 X-ray crystal structure of complex 1
A large crystal of complex 1, suitable for X-ray diffraction, was slowly grown from an ethanolic (equimolar) mixture of AgNO3 and dh at RT under placid dark conditions. ESI,† Table S1 outlines the crystal data collection and refinement criteria. The molecular structure is shown in Fig. 3a, while Table S2 (ESI†) details the important bond angles and lengths. Complex 1 crystallized in a triclinic P system, comprising two independent molecules (A and A′) in the asymmetric unit, with the formula [Ag(dh)NO3] and a distorted trigonal planar coordination geometry (Fig. 3b). Each AgI atom was bound to one dh ligand through a NO chelate system and one oxygen atom from the nitrate counter anion, forming an ONO coordination plane. A long Ag⋯Ag separation of 5.16 Å excluded any direct intermetallic interaction.
 | ||
| Fig. 3 (a) Crystal structure (asymmetric unit) of complex 1 at 30% ellipsoid probability, showing intramolecular N–H⋯O interactions (turquoise dashed lines) by two symmetry-independent molecules, A (left) and A′ (right), excluding passive hydrogen atoms for clarity, (b) perspective view of a distorted trigonal planar (extended Y) coordination environment around silver centers Ag1 and Ag1′ in complex 1. | ||
Molecules A and A′, excluding their middle benzene rings (cg1/cg1′), were almost planar. The cg1/cg1′ rings formed a second plane that intersected the main molecular plane at CH2 carbon and Ar–NH nitrogen, with dihedral angles of 65.5° for A and 70.9° for A′ (Fig. S10, ESI†). Chlorine atoms and nitrato oxygen (O4) in A were slightly displaced, while Ag1′ and O4′ in A′ deviated more from their molecular planes. The best-fit main molecular planes of A and A′ intersected at 56.7°, while the planes of cg1 and cg1′ crossed at 58.4° (Fig. S10, ESI†). In the unit cell of complex 1, the main molecular planes of A and A′ were parallel and separated by 13.1 Å and 6.9 Å, respectively, forming a parallelogram centered in the unit cell (Fig. S11, ESI†). The cg1 and cg1′ rings hung from opposite sides of this parallelogram and pointed inward with alternate ring arrangements along the sides. This setup enabled clustering of hydrophobic moieties surrounded by polar layers, resulting in a unique layered structure for complex 1, discussed in detail later.
The structural geometry of metal complexes is crucial in coordination chemistry as it governs their properties. Tri-coordinate geometry can be characterized by angles α, β, and γ around the metal center, with the sum of angles (∑∡'s) ≥ 354° (ideally 360°) for planarity.80 The ∑∡'s for Ag1 and Ag1′ was 355 and 347°, respectively, based on their α/β/γ values: 143/142/70° (Ag1) and 146/130/71° (Ag1′). Ag1 met the criteria for being classified as a relatively symmetric monoclinic planar ‘extended Y’ shaped center. Specifically, it matched a ‘5/12 constricted γ-dominant Y’ configuration, with a γ constriction level of 41.7%. In contrast, the Ag1′ center was less symmetric, with α ≠ β ≠ γ and its ∑∡'s deviating by 7° from minimum planarity, orienting 0.42 Å away from the basal ONO plane. Ag1′ geometry can be assigned as an ‘acute triclinic pyramid,’ derived from a folding distortion (13° or 18% α fold) of the ‘5/12 constricted γ-dominant Y’ projection. The acute pyramid type is confirmed if the sum of any two apex angles exceeds 180°.80 The bond lengths of Ag1 and Ag1′ were similar, with dative dh bonds (Ag–O/Ag–N) slightly weaker than Ag–O(nitrato) bonds,81 likely due to chelation. The bond lengths were consistent with previously reported Ag–O25,82,83 and Ag–N70,80,84,85 bonds for tri-coordinate AgI complexes.
The small deviation from planarity at the Ag1′ center of A′ may be linked to positional disorder in the unsubstituted carbon atoms of the cg2′ ring, a weak Ag1′⋯O4′(nitrato) secondary interaction suggested by 2.96 Å separation, resonance equivalence of nitrato oxygen atoms, and O4′ tilt towards Ag1′ eased by O4′⋯H4–C4(cg1) hydrogen bonding, not found analogously in A (Fig. S10 and S11, ESI†). A similar out-of-plane distortion of AgI caused by non-coordinated carboxyl oxygen has been previously reported in mixed tri-coordinate carboxylate-phosphine complexes.25 The ‘extended Y’ coordination geometry in complex 1 (Fig. 3b) compared to the ideal ‘trigonal planar’ geometry resulted from the chelate effect of bidentate dh, stabilizing molecules through a five-membered ring (Fig. 3a). This chelation constrains the γ angle around the AgI center to 70–71°, allowing the α and β angles to exceed 120°, consistent with the observed structure.
The AgI–hydrazide–nitrate complexes are scarcely documented in the literature. A recent work conducted by Selvam et al. (2022) on a hydrazide-hydrazone ligand (hh) is taken for comparison.46 Unlike our complex 1, the AgI-hh complex crystallized in the P21/c system as a 1D polymer with the formula [Ag(hh)NO3]n and distorted tetrahedral (seesaw) geometry, where the hh bonding mode was quite similar to dh but nitrate acted as a bridging ligand to link two Ag atoms. The dative bond lengths and γ angle of two complexes were consistent. However, the hh was almost planar, and its AgI complex formed fewer hydrogen bonds than complex 1. The coordination geometry and bond parameters of complex 1 were in good agreement with [Ag(pc)NO3] (pc = pyridine-2-carboxaldoxime) reported by Abu-Youssef et al. (2010) except that the pc ligand formed an NN chelate.81
| D—H⋯A | D—H (Å) | H⋯A (Å) | D⋯A (Å) | D—H⋯A (°) |
|---|---|---|---|---|
| Symmetry codes: #1: x − 1, y, z; #2: −x, −y, −z; #3: −x + 1, −y, −z; #4: −x + 1, −y + 1, −z; #5: x + 1, y, z. | ||||
| N1–H1⋯O3#1 | 0.86 | 2.48 | 2.973(5) | 118 |
| N1–H1⋯O4#1 | 0.86 | 2.37 | 3.207(6) | 165 |
| N2–H2A⋯O3#1 | 0.89 | 2.42 | 2.884(5) | 113 |
| N2–H2A⋯O2′#2 | 0.89 | 2.40 | 2.938(5) | 120 |
| N2–H2B⋯O3#3 | 0.89 | 2.15 | 3.037(5) | 171 |
| N3–H3⋯O1 | 0.86 | 2.43 | 2.927(4) | 117 |
| N3–H3⋯O1#4 | 0.86 | 2.60 | 3.412(4) | 158 |
| C2–H2D⋯O4#4 | 0.97 | 2.65 | 3.309(5) | 125 |
| C4–H4⋯O4′ | 0.93 | 2.59 | 3.271(6) | 130 |
| N1′–H1′⋯O4′#5 | 0.86 | 2.18 | 3.006(5) | 161 |
| N1′–H1′⋯O3′#5 | 0.86 | 2.51 | 2.988(5) | 116 |
| N2′–H2A′⋯O2#3 | 0.89 | 2.13 | 3.002(5) | 166 |
| N2′–H2B′⋯O1 | 0.89 | 2.33 | 3.099(5) | 144 |
| N3′–H3′⋯O1′ | 0.86 | 2.33 | 2.929(5) | 127 |
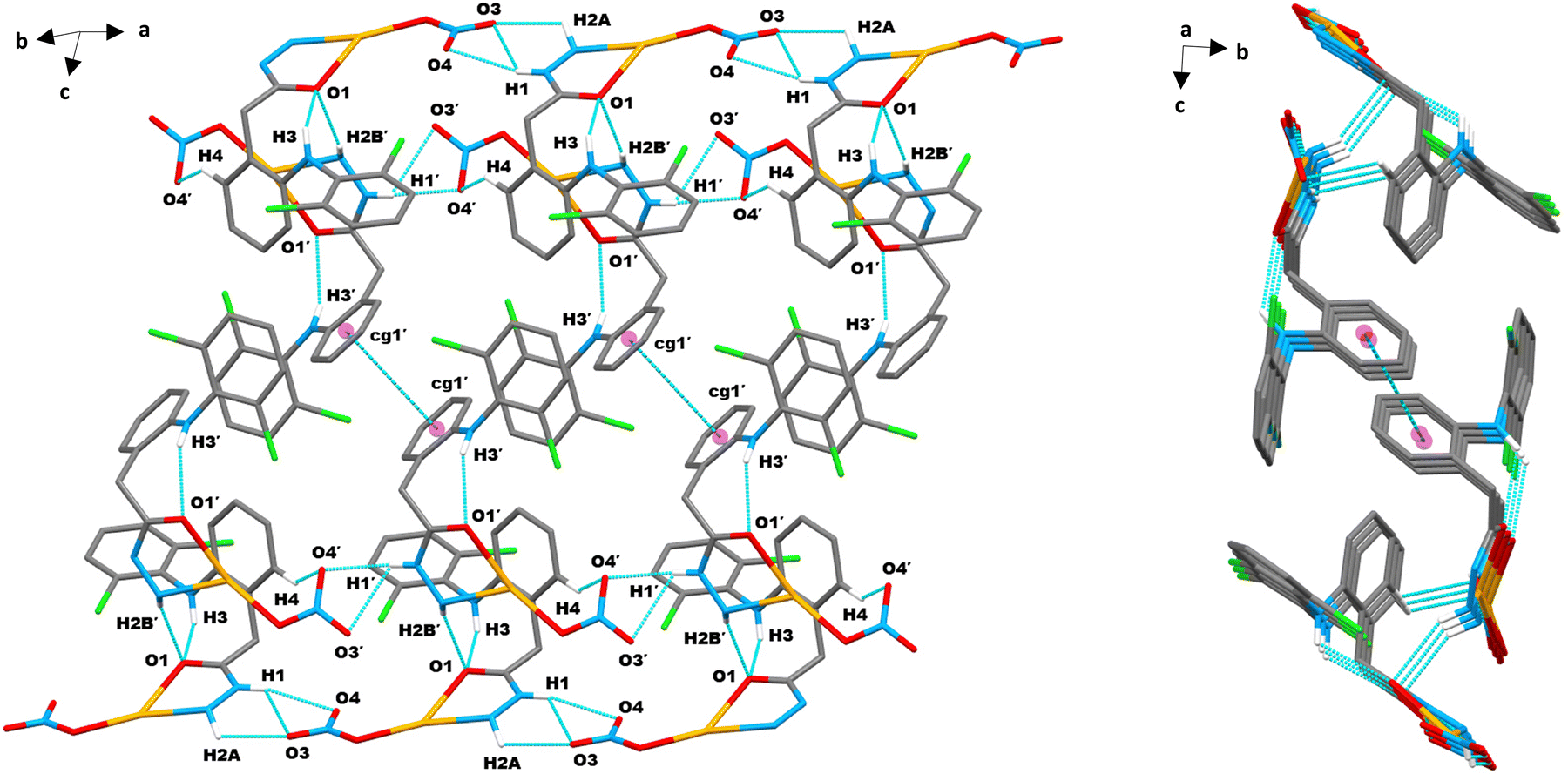 | ||
| Fig. 4 Molecular packing of complex 1 in the crystallographic a direction showing intra- and inter-molecular hydrogen bonds and π⋯π (cg1′–cg1′) contacts (turquoise dashed lines): a closed (expanded) view showing an infinite conveyer chain-like 1D layered structure (left) and the respective view along the bc-plane showing parallelogram shaped stacked layers (right). Only a few important hydrogen atoms are shown for clarity. | ||
 | ||
| Fig. 5 2D supramolecular layered assembly of complex 1 generated through intermolecular X–H⋯π interactions (C–H⋯π and N–H⋯π, magenta dashed lines) and hydrogen bonds (C–H⋯O and N–H⋯O, turquoise dashed lines). | ||
 | ||
| Fig. 6 (a) Supramolecular multilayer infinite 3D framework mediated by multiple intermolecular N–H⋯O/C–H⋯O hydrogen bond linkages (turquoise dashed lines) in complex 1, (b) twisting candy wrap-shaped 3D perspective layered structure of complex 1, layers added towards the b-axis and interconnected through intermolecular hydrogen bonds from a/c packing, and (c) birds flying-shaped 3D perspective layered structure of complex 1. | ||
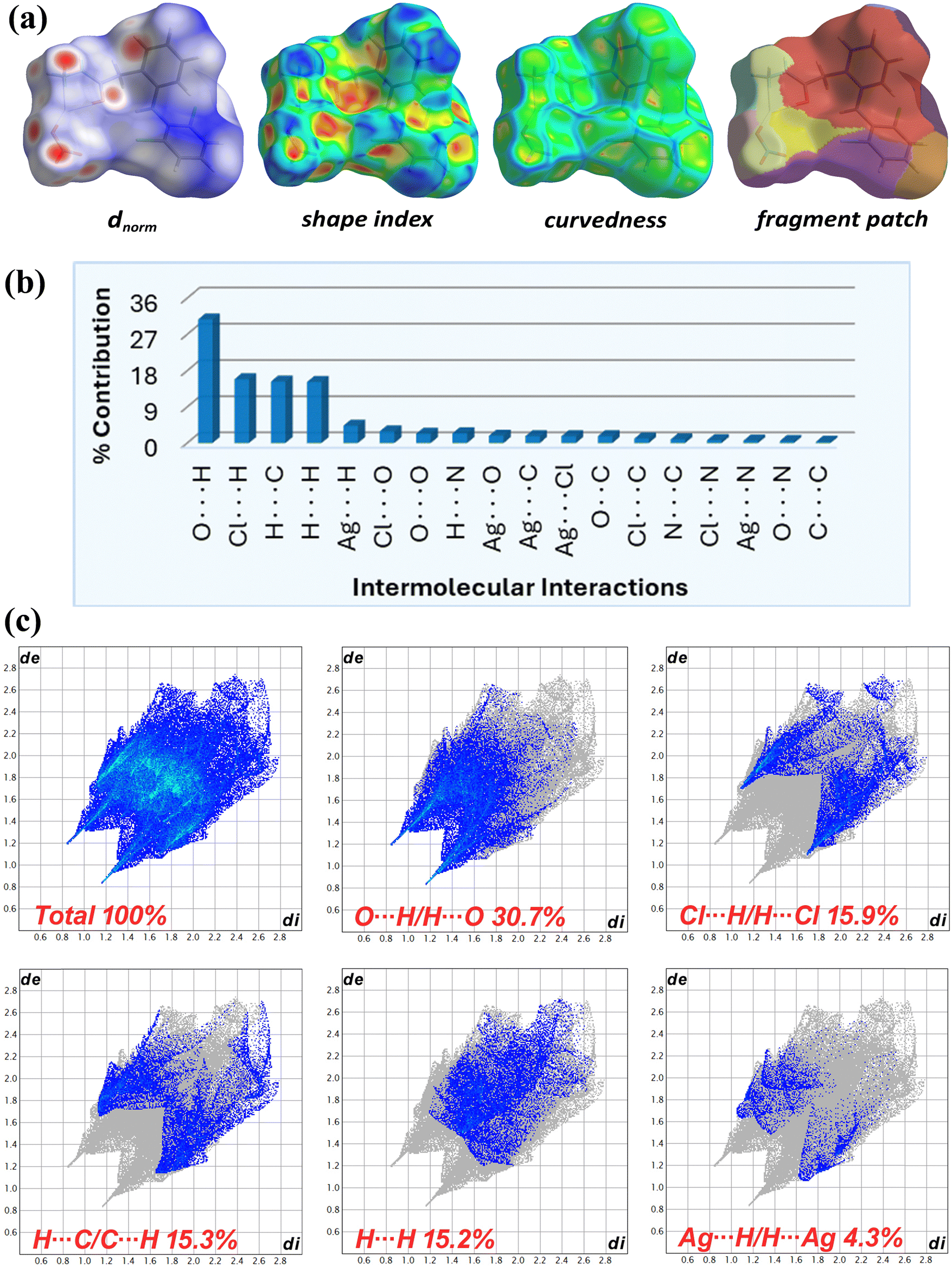 | ||
| Fig. 7 (a) Hirshfeld surfaces of complex 1 mapped over dnorm, shape index, curvedness, and fragment patch, (b) graphical overview of relative contribution of various intermolecular contacts of complex 1, and (c) 2D fingerprint plots of complex 1 displaying percent contribution of full and decomposed major intermolecular contacts. | ||
Fig. 7b displays the relative contact contribution from the decomposed 2D fingerprint plots (Fig. 7c and Fig. S15, ESI†) of complex 1 HS. In the complementary acceptor (di > de) and donor (de > di) fingerprint regions, the dominant spikes at di + de ∼ 2.0 Å (less than the O/H rvdW sum of 2.7 Å) denoted the most prominent intermolecular O⋯H/H⋯O close interactions (30.7%), followed by sharp spikes of intermolecular Cl⋯H/H⋯Cl close contacts (15.9%) among Cl1(cg2) and C4–H4(cg1) atoms at di + de ∼ 2.7 Å (less than the Cl/H rvdW sum of 2.9 Å). The intense complementary wings corresponded to C⋯H/H⋯C interactions (15.3%), validating the supplementary role of C–H⋯π and N–H⋯π contacts in the self-assembled supramolecular crystal framework. Hydrophobic H⋯H contacts from aromatic hydrogens, seen in the central plot region at di + de ∼ 2.8 Å, contributed 15.8% to the entire HS area. The contribution of intermolecular Ag⋯H/H⋯Ag, Cl⋯O/O⋯Cl, O⋯O, N⋯H/H⋯N, Ag⋯O/O⋯Ag, Ag⋯Cl/Cl⋯Ag, Ag⋯C/C⋯Ag, and O⋯C/C⋯O contacts to HS was low, in falling order from 4.3% to 1.7%. The contribution of Cl⋯C/C⋯Cl (1.1%), N⋯C/C⋯N (0.9%), Cl⋯N/N⋯Cl (0.7%), Ag⋯N/N⋯Ag (0.6%), and O⋯N/N⋯O (0.5%) contacts to the overall HS area was minor. The π⋯π stacking interaction delivered the least (0.3%) to the HS area.
The enrichment ratios (EXY) for element pairs (X⋯Y) were also calculated (Table S3, ESI†) using HSA information to identify privileged (EXY > 1) and disfavoured (EXY < 1) contacts in crystal packing.60 The EOH value (1.47) for most HS imparting O⋯H contacts indicated their high prevalence due to high abundance of hydrogen (49.5%) and oxygen (21.1%) on the molecular surface. The minor Ag⋯N and C⋯N linkages, corresponding to Ag1/N3(Ar) and C9(cg2)/N2(Hz) atoms, were most over-represented (EAgN = 2.33 and ECN = 1.66). The Ag⋯π/Cl (EAgC = 1.58, EAgCl = 1.51), π⋯H (ECH = 1.45), Cl⋯H (EClH = 1.44), and Cl⋯N (EClN = 1.23) contacts were also enriched, while H⋯H bonds were less prevalent (EHH = 0.62). Other minor interactions were mostly under-represented (EXY < 1), with π–π contacts being least favored (ECC = 0.26). The self-contacts of N, Cl and Ag were fully avoided (EXY = 0). Overall, the hydrogen bonds were found to be vital for the crystal stability of complex 1.
 | ||
| Fig. 8 AIM topological images illustrating non-covalent intra- and inter-molecular interaction paths (magenta), bond critical points (black) and ring critical points (orange) for complex 1. The modeled structures are self-assembled dimers of two types: AA′ (a and c) and A2 (b). | ||
 | ||
| Fig. 9 (a–c) NCI isosurface density plots for complex 1. The intermolecular interactions are indicated by black arrows. | ||
 | ||
| Fig. 10 (a–c) 2D scatter [RDG vs. sign(λ2)ρ] plots for complex 1. | ||
3.4 Surface morphology
The fine crystals of complex 1 and the powder of complex 2 were examined using scanning electron microscopy (SEM) to analyze their morphology. Fig. 11a reveals well-developed flat crystals of complex 1 as microsheets with a thickness of less than 50 μm. This is in line with the layered molecular structure of complex 1 evident from the X-ray crystallographic study. The other dimensions of the crystals of complex 1 varied widely, ranging from micro- to macro-sizes. Among the different crystals of complex 1, an irregular outline/boundary pattern was observed, and adjacent crystal facets exhibited diverse intersecting angles. This irregular outline arrangement may be associated with the triclinic crystal system of complex 1. Unlike complex 1, complex 2 showed polyhedral microcrystals of varying shapes and sizes, similar to river rocks (Fig. 11b). A few of these microcrystals seemed to have a truncated or distorted tetrahedral shape. | ||
| Fig. 11 SEM imaging of crystals of (a) complex 1 (Ag1) and (b) complex 2 (ZnII). | ||
3.5 Physicochemical measurements
The synthesized metal complexes were soluble in coordinating solvents like DMF and DMSO. Complex 2 was insoluble in other solvents, while complex 1 could dissolve slowly in methanol/ethanol and instantly in acetonitrile. The molar conductivity in fresh 1 mM DMSO solution was 85.5 Ω−1 cm2 mol−1 for complex 1 and 20.7 Ω−1 cm2 mol−1 for complex 2. The low conductance and poor solubility in polar media for complex 2 supported its non-electrolytic nature. Complex 1 with high conductivity, however, couldn't be ionic as the other analytical data conformed to a neutral nature. The conductivity of complex 1 may arise from replacing weakly coordinated nitrate with DMSO, forming ionic species [Ag(dh)(dmso)]NO3,90 consistent with ligand exchange activity evident from FAB-MS studies. Moreover, complex 1 solubility in polar organic solvents can also be allied to their ability to exchange coordinated nitrate, indicated by its high conductivities (35–165 Ω−1 cm2 mol−1) in ethanol, methanol, and acetonitrile.The CHN/S, metal content, and conductivity analysis results were consistent with the spectral studies, predicting the formula of complex 1 as [Ag(dh)NO3] and 2 as [Zn(dh)SO4]. Both complexes thus exhibited a 1:1 metal-to-ligand molar ratio, with neutral dh acting as a bidentate N,O donor ligand that forms a 5-membered chelate ring using amino nitrogen and carbonyl oxygen from the hydrazide moiety,34 irrespective of the nature of the metal. The nitrate was bound to Ag1 in a monodentate fashion as an O donor, while the sulfate was expected to bind strongly to ZnII bidentately, resulting in an additional 4-membered chelate ring, as previously reported.91,92 Complex 1 had trigonal planar geometry consistent with X-ray crystal studies, while complex 2 was suggested to be tetrahedral, a common stable geometry for ZnII complexes due to the full valence d orbital.26,93
3.6 Electrostatic potential analysis
The color-coded molecular electrostatic potential (MEP) surfaces, based on electronic charge distribution, are important for predicting and visualizing electron donor and acceptor regions, relevant to hydrogen bonding sites crucial for chemical and biological reactivity.87 The MEP maps for complex 1 and complex 2 (Fig. 12), calculated at the B3LYP/GenECP level, revealed the N-hydrazine (–NH–NH2) moiety of the dh ligand as the most electron-deficient site (blue color), with acidic hydrogen atoms linked to nucleophilic reactions. In contrast, the carbonyl oxygen and hydrazinic hydrogen atoms of free ligand were electron-rich, favoring electrophilic attacks (Fig. S16, ESI†). This indicates the sharing of the negative charge of the hydrazide moiety by silver and zinc metal centers through complexation. The coordinated O-nitrate in complex 1 and O-sulfate in complex 2 showed the highest nucleophilic character (red color), with their oxygen atoms acting as potential sites for electrophilic attacks. The remaining molecular part comprising the dh aromatic system exhibited minimal charge distribution. | ||
| Fig. 12 Molecular electrostatic potential (MEP) surface maps of (a) complex 1 and (b) complex 2. | ||
3.7 Anti-inflammatory activity
Reactive oxygen species (ROS) generated in phagocytic defense actions are some of the key inflammation mediators and signaling molecules.1,2 Thus, ROS suppression is a propitious approach in identifying and controlling inflammation-related chronic syndromes.94 Currently available non-steroidal anti-inflammatory drugs (NSAIDs) suffer from adverse gastrointestinal or cardiovascular effects, demanding the exploration of new anti-inflammatory agents with minimal or no adverse effects.4,12 The chemical modulation of the –COOH functionality of NSAIDs with groups that are less acidic5,9–11,13 or complexing these drugs with metal ions14–16 can enhance their anti-inflammatory potentiality with lowered ulcerogenicity and gastrointestinal (GI) toxicities. Considering all these aspects, the NSAID diclofenac (dicf) was modified into a nitrogen–oxygen containing hydrazide (dh) and complexed with AgI (1) and ZnII (2) to screen for anti-inflammatory potential and ascertain how structural alteration, particularly coordination, affects inflammation.The anti-inflammatory potential of compounds was assessed using an oxidative-burst luminol-enhanced chemiluminescence test.64 The oxidative burst mechanism primarily entailed the activation of human whole blood phagocytes, which produced intracellular ROS (H2O2, O2˙−, etc.). Zymosan was utilized as an activator of phagocytosis, while luminol (3-aminophthalhydrazide) served as a probe for intracellular ROS detection. The degree of ROS was measured in terms of RLU (relative light units). The compound's anti-inflammatory activity was demonstrated by the decrease in chemiluminescence, based on its inhibition of intracellular ROS.65 Table 2 compares the % ROS inhibition at a concentration of 25 μg mL−1 synthesized complexes and the corresponding free ligand and metal ions. For cases where the percentage of ROS inhibition (at 25 μg mL−1) was higher than 50%, the IC50 values (μg mL−1, equal to 50% ROS suppression by the compounds) were also computed and provided in Table 2. Two underivatized anti-inflammatory drugs, dicf and ibuprofen (ibup), were applied as the reference (standards) in this study.
| Compounds | Anti-inflammatory activity | Cytotoxic activity (MTT assay) | ||||||
|---|---|---|---|---|---|---|---|---|
| ROS | BJ (normal skin fibroblasts) | H460 (lung cancer cells) | PC3 (prostate cancer cells) | |||||
| % inhibitiona | IC50b (μg mL−1) | % inhibitionc | IC50b (μg mL−1) | % inhibitionc | IC50b (μg mL−1) | % inhibitionc | IC50b (μg mL−1) | |
| NA = not applicable.a % inhibition of reactive oxygen species (ROS) at 25 μg mL−1.b Results in mean ± SD (n = 3).c % inhibition of cells at 30 μg mL−1. | ||||||||
| dh | 70.9 | 5.5 ± 1.2 | 10.7 | NA | 24.9 | NA | 23.2 | NA |
| Complex 1 | 90.8 | 2.6 ± 0.8 | 45.9 | NA | 88.6 | 13.1 ± 0.3 | 95.6 | 3.5 ± 0.0 |
| Complex 2 | 37.6 | NA | 24.5 | NA | 20.0 | NA | 3.4 | NA |
| AgI salt | 39.7 | NA | 83.4 | 1.0 ± 0.8 | 89.7 | 2.4 ± 0.1 | 95.9 | 0.5 ± 0.0 |
| ZnII salt | −15.2 | NA | 7.9 | NA | 13.0 | NA | 0.0 | NA |
| Standard drugs | ||||||||
| Diclofenac | 73.0 | 8.3 ± 0.5 | — | — | — | — | — | — |
| Ibuprofen | 73.2 | 8.0 ± 1.9 | — | — | — | — | — | — |
| Cisplatin | — | — | 58.1 | 6.0 ± 0.0 | 96.4 | 1.1 ± 0.0 | 39.9 | 42.1 ± 1.2 |
The chemical modification of the parent drug dicf (73.0% inhibition, IC50 = 8.3 μg mL−1) with the hydrazide moiety forming dh (70.9% inhibition, IC50 = 5.5 μg mL−1) did not significantly affect anti-inflammatory activity in terms of suppression of oxidative burst from the human whole blood cells. However, this derivatization of dicf –COOH hydrogen could be beneficial for reducing gastrointestinal complications associated with NSAID therapy because of the less acidic –NH–NH2 group that may also contribute to other anti-inflammatory pathways, such as inhibitory interactions at the active sites of cyclooxygenases (COXs) through nitrogen.
Among the tested compounds, the AgI–dh complex (1) with the nitrate co-ligand was discovered as the most powerful anti-inflammatory agent. Complex 1 showed a much higher ROS inhibition (90.8%, IC50 = 2.6 μg mL−1) than its parent free components, dh (70.9%) and AgI nitrate (39.7%). This demonstrates an excellent cumulative effect of AgI and dh in improving parent drug activity against inflammation, validating the crucial role of complexation in biology. The ROS inhibitory response (IC50) of complex 1 was about 3-fold superior to reference drugs, dicf and ibup. Owing to reduced solubility, increased lipophilicity, and stabilization into a 3D architecture, complex 1 could have reduced cell toxicity and improved biocompatibility compared to that exhibited by the parent drug or AgNO3 salt.15,17,18,51 Consistent with our results, previously reported AgI complexes of ligands including 3-py-ibuprofen,32 N-heterocyclic carbene,53 chloro-thioamide-phosphine,54 ibuprofen-caffeine,95 and naproxen96 showed synergistically enhanced activity against inflammation. Data on the IC50 or percent anti-inflammation for earlier AgI complexes under similar assay conditions are lacking in the literature. However, Table S5 (ESI†) compares various silver compounds analyzed through different anti-inflammatory assays, such as RBC membrane stabilization, protein anti-denaturation, intracellular IL-I/TNF-α/NO/PGE2 suppression, and COX-1/COX-2 inhibition. Table S5 (ESI†) shows that complex 1 exhibits a lower IC50 or higher percent anti-inflammatory activity at lower concentrations compared to previously reported silver compounds (IC50 ≥ 26.5 μg mL−1, activity ≤82% at ≥100 μg mL−1), indicating complex 1's superior effectiveness against inflammation.32,53,54,95–97
The ZnII–dh complex (2) also demonstrated anti-inflammation potential with a ROS inhibition of 37.6%, which was lower than free dh but better than the precursor salt ZnII sulfate (−15.2%). The negative percent inhibition can be expected from two aspects: (i) if new ROS, e.g., intracellularly generated persulfate SO4˙−,98 superoxide, or peroxide radicals probably from sulfate ions, are more dominantly formed than suppression of intracellular ROS in the applied assay or (ii) if net chemiluminescence of the adopted assay system is enhanced compared to control because of the interference arising from the chemiluminescent properties of any sample. Free ZnII shows proven bioluminescence upon interaction with certain proteins or enzymes through facile coordination.99 Such bioluminescence response may also arise by the interaction of free ZnII with the whole blood proteins used in the assay. Thus, the relatively lower apparent ROS inhibition activity of complex 2 compared to the ligand may be associated with a negative response of the zinc or sulfate moiety under the applied assay conditions. Accordingly, ZnII and its complex (2) must also be evaluated through any other (non-chemiluminescence) assay to verify their anti-inflammatory ability. The potential of complex 2 against inflammation by other mechanisms, such as coordination by ZnII to inflammation-associated biochemicals or inhibition of related enzymes through the stabilized metal–ligand 3D scaffold cannot be overlooked,19,30 as ZnII is an essential nutrient with catalytic (co-factor), structural and immune-modulatory functions, and its depletion may contribute to inflammatory disorders.49,50 Previously, Kasare et al. (2019) observed no anti-inflammatory activity by a ZnII complex (from zincII acetate) compared to its parent ligand azo-azomethine in the in vitro protein denaturation assay,100 while in a similar assay, Kumar et al. (2023) found the enhanced anti-inflammatory potential of the ZnII-hydrazone complex (from the same salt) compared to the respective free ligand.48 Many earlier studies also reported equivalent or enhanced anti-inflammatory activity of ZnII complexes compared to parent ligands in a variety of anti-inflammatory assays.19,27,28,32
Hence, the observed ROS inhibitory activity of complexes is the cumulative effect of the individual activity of precursors. The metal, ligand, and co-ligand in a complex all can affect the anti-inflammatory activity that must be considered in structural optimization for developing related metallodrugs. Complex 1 is highlighted as the most promising metal-based anti-inflammatory drug candidate for further studies.
 | ||
| Fig. 13 Comparative anti-inflammatory effect of the uncoordinated dh ligand and its metal complexes in terms of their % ROS inhibition (in vitro) and COX-2 (5kir) binding energy (in silico). | ||
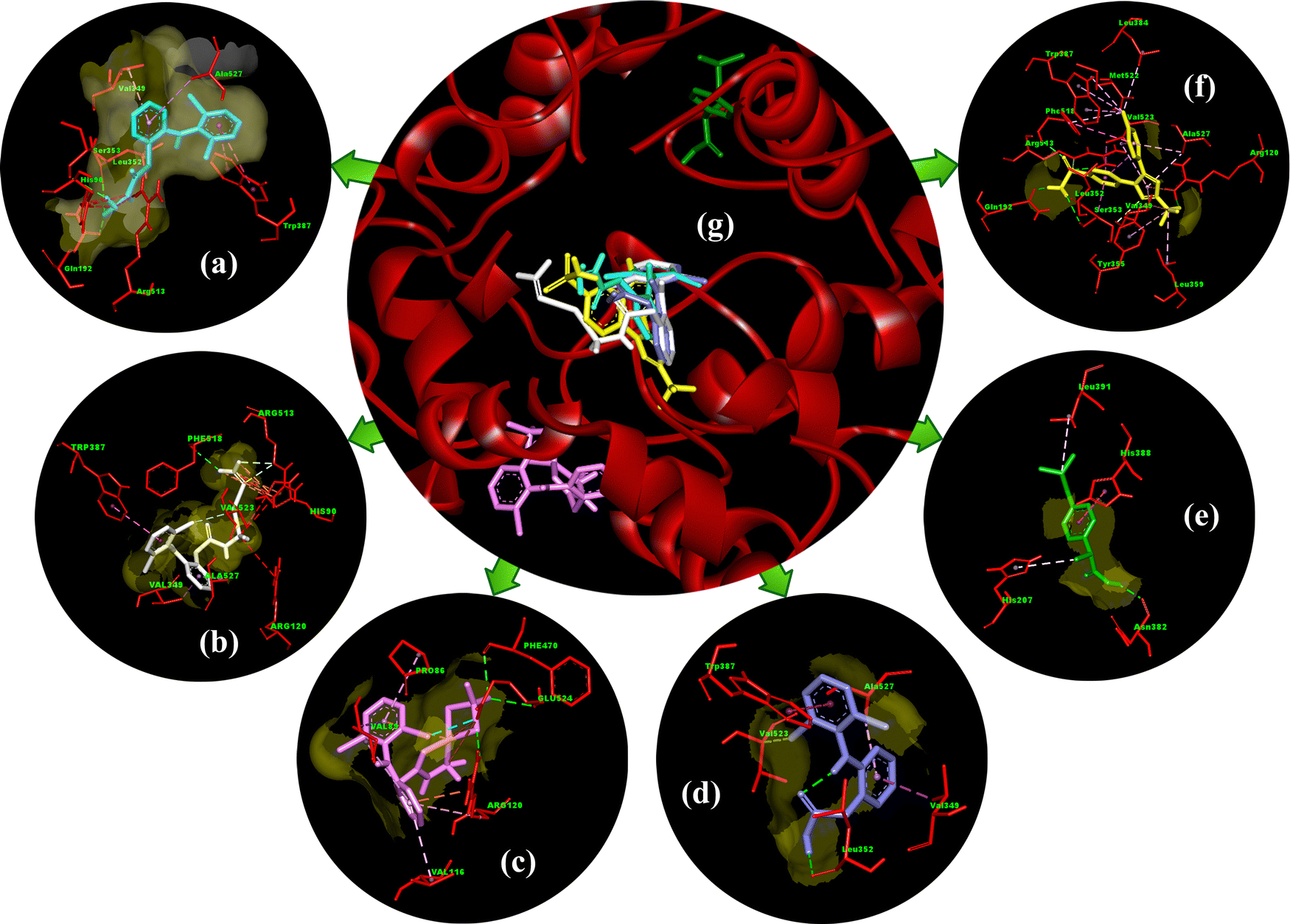 | ||
| Fig. 14 Molecular docking 3D interactions of best-docked poses of (a) the free dh ligand, (b) complex 1, (c) complex 2, (d) diclofenac, (e) ibuprofen, and (f) celecoxib, with (g) superimposed relative docked view of compounds (a)–(f) in the binding pocket of human COX-2 protein (PDB ID: 5kir). | ||
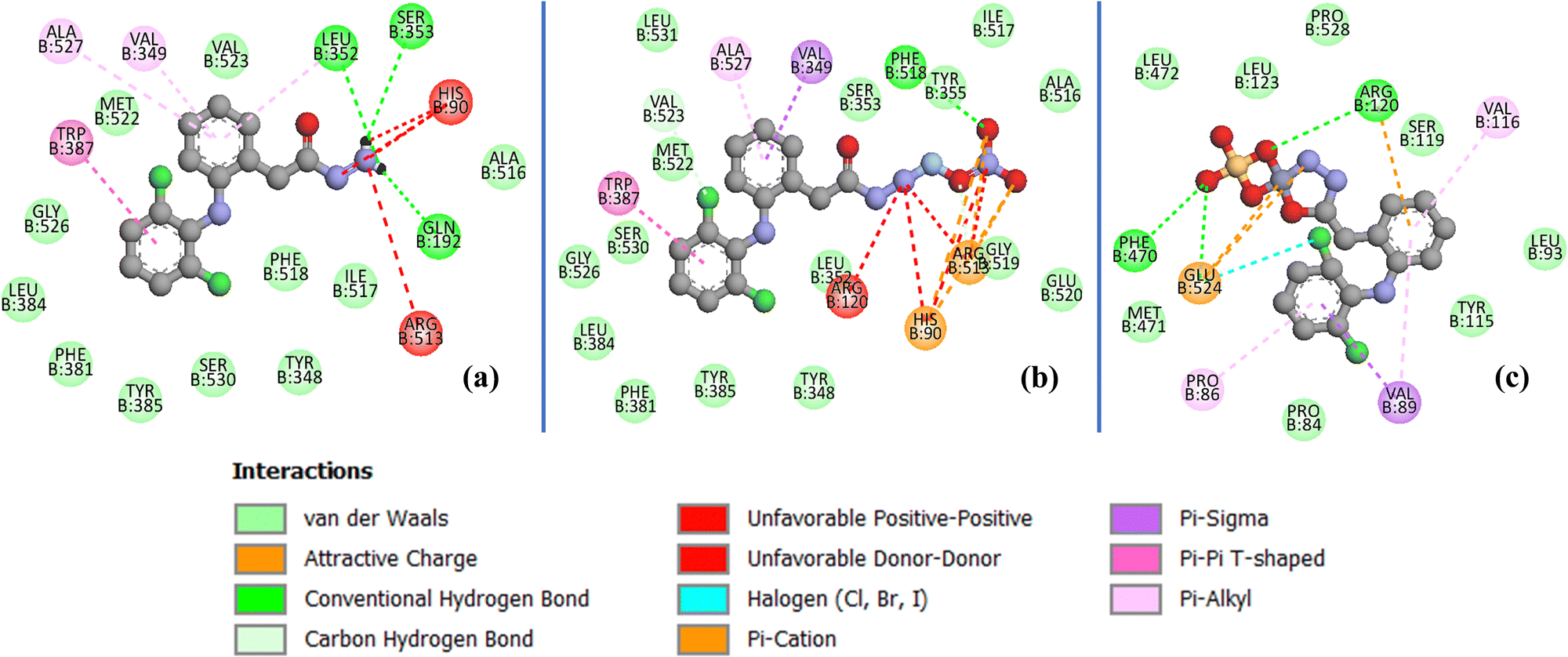 | ||
| Fig. 15 2D docking interactions of (a) the free dh ligand, (b) complex 1, and (c) complex 2 in the binding pocket of human COX-2 protein (PDB ID: 5kir). | ||
The stabilization of compounds within the COX-2 binding pocket, judged by low free binding energies (high affinities), followed the order: celecoxib > complex 1 > dh > complex 2 > dicf > ibup. This is in good agreement with experimental ROS inhibition results except for complex 2 (Fig. 13). The sufficiently low binding energy of celecoxib (−14.36 kcal mol−1) compared to dicf and ibup (−9.78 to −9.20 kcal mol−1) is in full agreement with previously known relative COX-2 selectivity profile of standards. It validates the docking analysis and serves as a benchmark to predict the selectivity of our compounds. The active site selectivity pocket of COX-2 contains Val523, Arg513, Ser353, His90, Gln192, Ile517, Phe518, and Leu352 as key catalytic residues; the initial two amino acids are absent or replaced with Ile523 (bulkier) and His513 while others are common in COX-1.101,102 Finding interactions and stabilization within the COX-2 selectivity pocket by compounds can further confirm their enzyme selectivity. Consistent with the ROS inhibition findings, complex 1 emerged as the strongest COX-2 inhibitor (Fig. 13), whose binding energy (−12.36 kcal mol−1) was comparable to celecoxib and lower than all others. Like celecoxib (Fig. S17c and Table S7, ESI†), complex 1 generated hydrogen bonding with Phe518 (through a noncoordinate nitrate oxygen) and Arg 513 (through noncoordinate and AgI bonded nitrate oxygen atoms) in the selectivity pocket (Table S6 and Fig. 15b, ESI†). Three residues Trp387, Val349, and Ala527 from the COX-2 hydrophobic binding pocket, producing π–π (T-shaped), π–σ, and π-alkyl contacts, respectively, with phenyl rings of complex 1, were also common to celecoxib. One of the Cl atoms of the 2,6-dichlorobenzene moiety of complex 1 created halogen and carbon–hydrogen bonds in the secondary side pocket margined by Val523, unlike celecoxib, which develops hydrophobic linkages with Val523. Several electrostatic interactions (not shown by celecoxib) were also revealed by complex 1 with basic residues, namely the π–cation linkage of nitrate nitrogen with the His90 imidazole ring and four attractive charge interlinkages (two from each noncoordinate nitrate oxygen) with Arg513 guanidino nitrogen (cationic), in addition to some positive charge repulsions caused by cationic Arg513, Arg120 and His90 nitrogen with amino or nitrate nitrogen of complex 1. The orientation of the best-docked conformer of complex 1 was similar to celecoxib, with excellent overlap of –CO–N2H4–Ag–NO3 (1) and –C6H4–SO2NH2 (celecoxib) scaffolds in the selectivity pocket (Fig. 16a). The binding affinity, stabilization by key residue contacts, and orientation relative to celecoxib strongly advocate a very high selectivity of complex 1 for COX-2 inhibition. Consonantly, an AgI complex of N-heterocyclic carbene has been previously found more selective towards COX-2 than COX-1 in vitro.53 The second most potent COX-2 inhibitor relative to standards was the dh ligand, which is also positioned similarly to celecoxib in the enzyme binding pocket (Fig. 16b); however, dh acquired a slightly lower stabilization than complex 1, indicated by higher binding energy and a smaller number of interactions for COX-2 (Table S6 and Fig. 15a, ESI†). The dh ligand established three hydrogen bonding interactions (through both of its NH2 hydrogen atoms) with Gln192, Leu352, and Ser353 inside the selectivity pocket, and five hydrophobic linkages involving two π–π (Trp387) and three π–alkyl (Val349, Leu352, Ala527) contacts, where all residues were common to celecoxib. Thus, dh also demonstrated good selectivity towards COX-2 inhibition. Surprisingly, complex 2 (the weakest ROS inhibitor) revealed COX-2 inhibition better than ibup and dicf (Fig. 13), based on binding energy and the number of favorable interactions: hydrogen bonds, electrostatic attractive charge/π–cation, halogen, and hydrophobic π–sigma/π–alkyl (Table S6 and Fig. 15c, ESI†). Noticeably, Glu524 formed four different linkages, including a unique attractive charge interaction with ZnII of complex 2. However, the relative orientation of docked complex 2 within COX-2 was quite different and far from the selectivity pocket than other compounds and standards (Fig. 14g). This shows non-selective and non-competitive type inhibition of COX-2 by complex 2. Unlike celecoxib or our compounds, dicf and ibup induced only fewer hydrogen bonds (1–2) and hydrophobic (three) connections with COX-2 (Table S6, ESI†). The dicf docked much closer to celecoxib in the selectivity pocket, but the docked ibup was located far away from the selectivity pocket (Fig. 14g). This indicates poor selectivity of dicf for COX-2 than celecoxib, complex 1, and dh, all showing the competitive-type inhibitory mechanism. In contrast, ibup is expected to show non-selective and non-competitive inhibition for COX-2, somewhat similar but punier to complex 2. Thus, the degree of binding affinity and selectivity of compounds towards COX-2 showed a good correlation to their hydrogen bonding ability.
 | ||
| Fig. 16 COX-2 (5kir) receptor surface close view of the orientation of most selective inhibitors relative to celecoxib (yellow): (a) complex 1 (white) and (b) dh ligand (cyan). | ||
Since the weakly coordinated nitrate in complex 1 may undergo ligand exchange in biological systems, we also examined in silico effects of replacing nitrate with four common small bio-monoanions (Cl−, HCO3−, H2PO4−, and HSO4−) on COX-2, while retaining the tricoordinate geometry and electroneutrality of the complex. As shown in Table S8 (ESI†), the docking scores for the three oxygen-containing anions (OAs) derived from carbonate, phosphate and sulfate (−12.73 to −11.94 kcal mol−1) were comparable to nitrate/complex 1 (−12.57 kcal mol−1), while the chloride-exchanged complex had a higher binding energy (−10.07 kcal mol−1) than OAs-complexes and dh yet better than dicf and ibup. Despite fewer interactions, the chloride-exchanged complex retained all interactions common to celecoxib and complex 1 (parent or OAs-exchanged) in the COX-2 selectivity pocket (Arg513/Val523) and hydrophobic binding pocket (Trp387/Ala527) (Table S8 and Fig. S18, ESI†). The carbon–hydrogen or halogen bonds formed between the chloride-exchanged complex and carbon atoms of COX-2 specific key catalytic residues Arg513 and Val523 suggested a strong COX-2 selectivity of this complex compared to complex 2, dh, dicf and ibup. Furthermore, the relative orientations of all forms of anion-exchanged complex 1 were nearly similar to that of the parent complex (Fig. 17). This implies that replacing nitrate in complex 1 with OAs or other bio-monoanions would not significantly alter its COX-2 selectivity or inhibition efficiency against inflammation.
 | ||
| Fig. 17 3D (a) ribbon view and (b) receptor surface view of COX-2 (5kir) with superimposed orientations of docked complex 1 (white) and its four anion-exchanged forms, where its nitrate is replaced with bio-monoanions: Cl− (pink), HCO3−(green), H2PO4− (blue), and HSO4− (orange). | ||
3.8 Cytotoxicity
To evaluate the safety and impact of our metal complexes on human cells, MTT cytotoxicity assay67 was conducted on healthy skin fibroblasts (BJ) and two cancer cell lines: H460 (lung cancer) and PC3 (prostate cancer). The cell growth inhibition percent (at 25 μg mL−1) and the corresponding IC50 values (for cases where cell growth inhibition ≥50% at 25 μg mL−1) are provided in Table 2 for complexes, precursors and the standard anticancer drug cisplatin. Both complexes showed low cytotoxic effects on BJ fibroblasts (24–46% inhibition), compared to cisplatin (58.1% BJ growth inhibition). The role of complexation was particularly notable in reducing the cytotoxicity of free silver (AgI), which inhibited normal fibroblast growth by 83.4%. Conversely, complex 2 was more toxic to normal fibroblasts than its free metal (ZnII), which showed 7.9% BJ growth inhibition. Complex 1 displayed significantly higher inhibition of cancer cell lines (88–96%) compared to normal BJ fibroblasts (45.9%), while complex 2 was less effective against cancer cells (3–20% inhibition) than normal fibroblasts (24.5% BJ inhibition). The free silverI was equipotent against cancer cell lines as its complex. In contrast, the dh ligand alone showed a low cytotoxic effect on normal fibroblasts (10.7% inhibition) and cancer cells (23–25% inhibition). These results suggested the high selectivity of complex 1 for cancer cells over healthy cells. Notably, complex 1 was found to be strongly selective against the prostate cancer PC3 cell line (95.6% inhibition, IC50 = 3.5 μg mL−1), with IC50 twelve-fold better than cisplatin (39.9% inhibition, IC50 = 42.1 μg mL−1). A comparison with literature IC50 values (μg mL−1) revealed that complex 1 is superior or comparable to many previously reported potent anticancer AgI complexes, for example, [Ag3(μ-nap)3(3-pic)2] (IC50 = 14.1 on MDA-MB-453, breast cancer),103 [(Metronidazole)2AgNO3] (IC50 = 3.9 on HepG2, liver cancer),104 Na[(SO3-NHC)AgCl] (IC50 = 3.6 on HeLa, cervical cancer),105 and [(4-OHMePy)2Ag]NO3 (IC50 = 3.4 on 1.2B4, pancreatic cancer).106 This study highlights complex 1 as a safe metallodrug with significant anticancer potential.3.9 Silver NPs and thin reflective films in methanol (solution behavior)
While studying the solution stability of complexes in different solvents, pure crystalline complex 1 surprisingly showed the spontaneous formation of silver nanoparticles (AgNPs) and thin reflective films (TRFs) in methanol under normal conditions, depending upon the incubation time and initial concentration of precursor complex, as verified by related electronic spectra (Fig. 18 and 19) and photographic images (Fig. 20). The silver0 formation from complex 1 is expected through an intramolecular redox (IMR) reaction of Ag1 with the coordinated ligand dh acting as a reducing agent in methanol, a nucleophilic solvent with a high content of naturally dissolved oxygen. Kim et al. (2023) recently reviewed the related role of metal complexes as precursors for the controlled formation of metal nanoclusters but involving some external incitement.107 The dh in complex 1 can be oxidized to the respective methyl ester or carboxylic acid during the IMR mechanism in methanol.108 This IMR process was initiated by the immediate entry of a methanol molecule into the coordination sphere in place of nitrate as indicated by high molar conductivities of complex 1 in fresh methanolic solution. Complex 1 in non-alcoholic solvents while complex 2 in all solvents remained stable, with no such IMR activity. | ||
| Fig. 18 Concentration-controlled formation of silver NPs from intramolecular redox reaction of precursor complex 1 studied at 3–3950 μM concentration in methanol. Electronic spectra are shown for equilibrium (3 day old) solutions. | ||
 | ||
| Fig. 19 Electronic spectral changes as a function of time during the formation of silver NPs by the IMR reaction of the AgI complex (1) in methanolic solution (417 μM). | ||
 | ||
| Fig. 20 Some real photographs showing the formation of silver NPs and thin reflective films: (a) from pure complex 1 crystals at controlled varying concentrations in methanol, showing no Ag NP formation at 116 μM or below (stable colorless solution of complex 1), stable yellow solution of Ag NPs (concentration >116 μM and ≤417 μM), or uniform Ag thin films (concentration >1350 μM) after an equilibrium incubation period of 1–3 days and (b) and (c) uncontrolled co-formation of reflective thin films of silver on glass (b) or plastic (c) vessels during complex 1 crystallization in methanol at an L/M molar ratio of >1. | ||
As shown in Fig. 18 and 20a, Complex 1 remained stable at a critical concentration of 116 μM or below in methanol, with no IMR activity and a colorless solution, showing only IL n–σ* and π–π*/n–π* transition bands around 203 nm and 275 nm, respectively. Concentrations of complex 1 above the critical point up to 417 μM represented a range for AgNP formation with high colloidal stability and no agglomeration, characterized by an additional surface plasmon resonance (SPR) band of silver around 417 nm and a transparent yellow solution even after several days. For a certain concentration of complex 1 within the colloidal stability range, the intensity of the color and respective SPR band enhanced with time reaching a maximum value and attaining an equilibrium within 1–2 days (Fig. 19). The broadness in the SPR band from the vertex towards a longer wavelength with time and increasing concentration suggests uneven distribution and increased size of AgNPs in the colloidal solution.109 The residual dh and/or its possible oxidized products at certain levels in solution were likely responsible for the high protection and stabilization of AgNPs within the colloidal stability range. Higher concentrations of complex 1 (beyond 417 μM) in methanol initially demonstrated colloidal solutions of AgNPs for a few hours, which lost their stability with time due to agglomeration and started depositing from saturated colloidal solutions on glass or plastic container walls/substrates as intriguing uniform and well adherent TRFs in 1–2 days under static normal conditions (Fig. 20a). The TRFs covered the full area of the vessel that was in direct contact with the liquid. After 2 days, the residual solutions from complex 1 (>417 μM) showed no SPR bands, indicating no remnant silver colloids due to the deposition of almost whole silver0 as TRFs (Fig. 18).
The redox (reducing) ability of dh for Ag1 was also apparent as an uncontrolled co-reaction occurring to varying extent during complex 1 synthesis in methanol, particularly dominant when the ligand was taken in excess with an initial L/M molar ratio greater than 1. For example, constant stirring of a methanolic mixed solution of Ag1 nitrate and dh in a 1:2 molar ratio at 45 °C revealed a characteristic pale-yellow color of the solution after 1–2 h, which got darker with time and gradually resulted in deposition of blackish-grey precipitates of silver (1a). The supernatant after separating 1a produced low-quality (somewhat porous) TRFs of silver on plastic or glass containers upon storage for a few days (Fig. 20b and c), with inadequate co-crystallization of complex 1. Product 1a was later characterized as well-dispersed spherical silver nanoclusters of size below 100 nm (by SEM, Fig. 21), capped by diclofenac acid, the oxidized carboxylic acid end-product of dh (verified by FT-IR, 1H NMR, 13C{1H} NMR, and EI-MS spectra, ESI,† Fig. S19–S22).
 | ||
| Fig. 21 SEM microphotograph of diclofenac-capped silver NPs (1a) obtained by direct reaction of silverI nitrate and dh (molar ratio 1:2) in methanol upon stirring at 45 °C. | ||
Thus, silver NPs and TRFs can also be obtained through the direct reaction of Ag1 nitrate and dh at higher initial L/M ratios, but in an uncontrolled manner in terms of stability and quality due to the co-formation of the Ag1 complex (1). However, pure complex 1, acting as an intermediate, was a better source for the controlled formation of stable colloidal silver NPs and high-quality uniform TRFs applying an appropriate initial concentration of a pure single-molecule precursor. The given strategy for the controlled formation of silver NPs and TRFs provides additional benefits of easy to apply and low-cost methods without the need for usual high pressure, temperature, energy, toxic passivator reagents (to prevent aggregation), and sophisticated expensive synthetic approaches (electrochemical, photoinduced, ultrasonic, or heat evaporation, etc.).109,110 The uniform TRFs of silver thus generated would be explored further for future applications as a protective coat, mirror, supercapacitor, solar cell, optical sensor, gas sensor, etc.
4 Conclusions
This study explores new chelates of AgI (1) and ZnII (2) with a hydrazide ligand (dh) derived from an NSAID diclofenac (dicf), offering interesting material, coordination, anti-inflammatory, anticancer, and solution properties. Spectroscopy, CHN/S and metal analyses, conductivity, SEM, and X-ray crystallography successfully identify the compounds. Complexes exhibit the general formula [M(dh)(A)] (1, M = AgI, A = NO3−; 2, M = ZnII, A = SO42−). Irrespective of the metal type, the neutral dh acts as a strong bidentate N,O donor and forms a 5-membered chelate ring using the NH2 nitrogen and CO oxygen of the hydrazide moiety. In complex 1, the nitrate counterion (monodentate O donor) is stable in the solid state but is immediately replaced by solvent molecules such as DMSO, methanol, ethanol, acetonitrile, and glycerol in solution, as verified by conductivity or FAB-MS spectra. In nucleophilic solvents with enough dissolved oxygen like methanol, complex 1 endures an intramolecular redox (IMR) reaction, forming Ag0 colloids and thin reflective films (TRFs), with stability and quality controlled by the initial concentration. The sulfate anion in the bidentate (O,O) mode forms an additional 4-member chelate ring, providing high stability to tetrahedral complex 2, with no obvious ligand exchange or IMR reactivity in solutions. The single crystal X-ray study reveals a distorted trigonal planar ‘extended Y’ geometry around two non-interacted AgI centers in the asymmetric unit. An intriguing repeated orientation of parallel and intersecting molecular planes from complex 1 crystal packing results in elaborate multilayered supramolecular 3D frameworks, facilitated by inclusive non-covalent intermolecular contacts. The HSA, QTAIM, and RDG approaches characterize these intermolecular contacts in the following order of strength: Ag⋯π > N–H⋯O > C–H⋯O > Ag⋯O/N ≈ N⋯O > C–H⋯π ≈ Cl⋯H > N⋯π. Complex 1 offers remarkable anti-ROS activity superior to standards (dicf and ibup), with strong interaction (stabilization) within the COX-2 selectivity pocket, comparable to celecoxib, highlighting the importance of complexing AgI with an N,O containing NSAID derivative to improve anti-inflammatory activity. Swapping of weakly bonded nitrate in complex 1 with common bio-monoanions, such as Cl−, HCO3−, H2PO4−, and HSO4−, does not impact its COX-2 selectivity or anti-inflammatory efficacy. Complex 1 is less toxic to normal BJ fibroblasts than cisplatin and is twenty-fold more effective against the prostate cancer cell line (PC3), recommending further research into its anticancer mechanism of action. Material 1 has been identified as a single-molecule reserve for AgNPs/TRFs and a future NSAID contender with an allied anti-ROS/anti-COX-2/anticancer profile.
Author contributions
Q.-A.: writing – discussion – reviewing and editing, result interpretation, software, in silico modeling and graphics, data curation and formal analysis (electronic and SEM), partial material preparation (crystallization), conceptualization, overall supervision, funding acquisition, resources; SY: writing, data curation and formal analysis (X-ray crystallography); SB: partial material preparation and characterization; IH: data collection (spectral and biological); SS: conceptualization, organic ligand chemistry and provision; and SK: writing – reviewing.Data availability
The data supporting the manuscript (NMR/mass/FT-IR spectra, crystal unit cell/fitted planes/hydrogen bonds/HSA-2D fingerprints, ligand MEP map, interactions in docked reference drugs/anion-exchanged complex, and tables of crystallographic structure refinement, bond parameters, HSA-enrichment ratios, QTAIM parameters, and anti-inflammatory literature comparison) are available as part of the ESI.†Conflicts of interest
The authors declare no competing interests.Acknowledgements
Partial research support from the Pakistan Council of Scientific & Industrial Research (PCSIR), Pakistan under access to the Scientific Instrumentation Program is gratefully acknowledged. Q-A (PI) thanks Higher Education Commission (HEC), Pakistan for software support under the Institutional Strengthening Program (Grant No. 38-2/ISULL/ACAD/HEC/18/614). Any possible facilitation by the Department of Chemistry and Dean Faculty of Science, University of Karachi is also appreciated.References
- W. L. Stone, H. Basit and B. Burns, Pathology, Inflammation, StatPearls Publishing, Treasure Island (FL), 2024 Search PubMed.
- L. Kiss, Pathol. Oncol. Res., 2021, 27, 1610136 CrossRef PubMed.
- GBD 2019 IMID Collaborators, EClinicalMedicine, 2023, 64, 102193 CrossRef PubMed.
- I. Ghlichloo and V. Gerriets, Nonsteroidal Anti-Inflammatory Drugs (NSAIDs), StatPearls Publishing, Treasure Island (FL), 2024 Search PubMed.
- F. Hafeez, A. F. Zahoor, S. Ahmad, M. Ahmad and S. Faiz, Synth. Commun., 2019, 49(3), 325–350 CrossRef CAS.
- R. A. Alfaro and D. D. Davis, Diclofenac, StatPearls Publishing, Treasure Island (FL), 2024 Search PubMed.
- R. A. Moore and S. Derry, Prog. Pain Res. Manage., 2018, 2018, 9493413 Search PubMed.
- A. Amanullah, A. Upadhyay, R. Dhiman, S. Singh, A. Kumar, D. K. Ahirwar, R. K. Gutti and A. Mishra, Cancers, 2022, 14(18), 4385 CrossRef CAS PubMed.
- A. Alshargabi, J. Drug Delivery Sci. Technol., 2024, 95, 105544 CrossRef CAS.
- N. Nedeljkovic, M. Nikolic, P. Canovic, M. Zaric, R. Z. Zaric, J. Boskovic, M. Vesovic, J. Bradic, M. Andic, A. Kocovic, M. Nikolic, V. Jakovljevic, Z. Vujic and V. Dobricic, Pharmaceutics, 2024, 16, 1 CrossRef CAS PubMed.
- S. R. Desai, V. G. Desai and R. R. Pissurlenkar, Bioorg. Chem., 2022, 120, 105595 CrossRef CAS PubMed.
- A. Sava, F. Buron, S. Routier, A. Panainte, N. Bibire and L. Profire, Biomed. Pharmacother., 2021, 139, 111678 CrossRef CAS PubMed.
- M. M. Ibrahim, T. Elsaman and M. Y. Al-Nour, Int. J. Med. Chem., 2018, 139786 Search PubMed.
- A. C. F. Santos, L. P. G. Monteiro, A. C. C. Gomes, F. Martel, T. M. Santos and B. J. M. L. Ferreira, Int. J. Mol. Sci., 2022, 23(5), 2855 CrossRef CAS PubMed.
- H. Y. Khan, S. Parveen, I. Yousuf, S. Tabassum and F. Arjmand, Coord. Chem. Rev., 2022, 453, 214316 CrossRef.
- C. N. Banti and S. K. Hadjikakou, Eur. J. Inorg. Chem., 2016,(19), 3048–3071 CrossRef CAS.
- S. R. Munnangi, A. A. A. Youssef, N. Narala, P. Lakkala, S. Narala, S. K. Vemula and M. Repka, Pharm. Res., 2023, 40, 1519–1540 CrossRef CAS PubMed.
- I. Yousuf, M. Bashir, F. Arjmand and S. Tabassum, Coord. Chem. Rev., 2021, 445, 214104 CrossRef CAS.
- C.-H. Leung, S. Lin, H.-J. Zhong and D.-L. Ma, Chem. Sci., 2015, 6, 871–884 RSC.
- G. Psomas, Coord. Chem. Rev., 2020, 412, 213259 CrossRef CAS.
- M. S. Refat, G. G. Mohamed, M. Y. Ibrahim, H. M. Killa and H. Fetooh, Russ. J. Gen. Chem., 2013, 83, 2479–2487 CrossRef CAS.
- W. A. Zordok, S. A. Sadeek and W. H. El-Shwiniy, J. Coord. Chem., 2012, 65(2), 353–369 CrossRef CAS.
- A. T. M. Fiori, W. R. Lustri, A. Magalhaes and P. P. Corbi, Inorg. Chem. Commun., 2011, 14(5), 738–740 CrossRef CAS.
- P. Christofis, M. Katsarou, A. Papakyriakou, Y. Sanakis, N. Katsaros and G. Psomas, J. Inorg. Biochem., 2005, 99(11), 2197–2210 CrossRef CAS PubMed.
- C. N. Banti, A. D. Giannoulis, N. Kourkoumelis, A. M. Owczarzak, M. Kubicki and S. K. Hadjikakou, J. Inorg. Biochem., 2015, 142, 132–144 CrossRef CAS PubMed.
- S. Bera, A. Chowdhury, K. Sarkar and P. Dastidar, Chem. – Asian J., 2020, 15, 503 CrossRef CAS PubMed.
- A. M. Abbas, H. H. Nasrallah, A. Aboelmagd, S. M. Kishk, W. C. Boyd, H. Kalil and A. S. Orabi, Int. J. Mol. Sci., 2024, 25, 3558 CrossRef CAS PubMed.
- S. K. Gopinath, M. Pari, A. M. Rudrannagari, I. B. Kattebasaveshwara and S. K. Halappa, Biointerface Res. Appl. Chem., 2021, 11(4), 11390–11403 CAS.
- P. Bhattacherjee, M. Roy, A. Naskar, H.-C. Tsai, A. Ghosh, N. Patra and R. P. John, Appl. Organomet. Chem., 2022, 36(1), e6459 CrossRef CAS.
- M. A. Shaheen, S. Feng, M. Anthony, M. N. Tahir, M. Hassan, S.-Y. Seo, S. Ahmad, M. Iqbal, M. Saleem and C. Lu, Molecules, 2019, 24, 1237 CrossRef PubMed.
- S. Ramos-Inza, E. Almagro, M. F. I. Encio, D. Plano, C. Sanmartin, R. Sirera and E. Lizarraga, J. Therm. Anal. Calorim., 2024, 149, 1015–1028 CrossRef CAS.
- K. Sarkar, S. Khasimbi, S. Mandal and P. Dastidar, ACS Appl. Mater. Interfaces, 2018, 10(36), 30649–30661 CrossRef CAS PubMed.
- M. Bashiri, M. Hosseini-Sarvari and S. Fakhraee, Mol. Syst. Des. Eng., 2024, 9, 112–139 RSC.
- H. Ju, Q. L. Zhu, M. Zuo, S. Liang, M. Du, Q. Zheng and Z. L. Wu, Chem. – Eur. J., 2023, 29(38), e202300969 CrossRef CAS PubMed.
- K. Karami, N. Jamshidian, M. Zakariazadeh, A. A. Momtazi-Borojeni, E. Abdollahi, Z. Amirghofran, A. Shahpiri and A. K. Nasab, Comput. Biol. Chem., 2021, 91, 107435 CrossRef CAS PubMed.
- L. M. Sousa, D. M. S. Araujo, K. M. Oliveira, L. P. De Oliveira, P. I. S. Maia, V. M. Deflon, A. A. Batista, A. E. H. Machado, W. Guerra and G. V. Poelhsitz, J. Coord. Chem., 2020, 73(10), 1605–1618 CrossRef CAS.
- V. S. Sergienko, T. V. Koksharova, M. D. Surazhskaya, T. V. Mandzii and A. V. Churakov, Russ. J. Inorg. Chem., 2019, 64, 1396–1404 CrossRef CAS.
- M. Jabeen, S. Ahmad, K. Shahid, A. Sadiq and U. Rashid, Front. Chem., 2018, 6, 55 CrossRef PubMed.
- M. A. Rodrigues, I. M. Marzano, G. H. Ribeiro, L. Colina-Vegas, M. Pivatto, A. P. S. Fontes, C. M. Ribeiro, F. R. Pavan, K. J. de Almeida, A. A. Batista, E. C. Pereira-Maia and W. Guerra, Polyhedron, 2015, 98, 146–153 CrossRef CAS.
- Qurrat-ul-Ain, A. Abid, M. Lateef, N. Rafiq, S. Eijaz and S. Tauseef, Biomed. Pharmacother., 2022, 146, 112561 CrossRef CAS PubMed.
- Qurrat-ul-Ain, U. Ashiq, R. A. Jamal, M. Saleem and M. Mahroof-Tahir, Arabian J. Chem., 2017, 10(4), 488–499 CrossRef CAS.
- Qurrat-ul-Ain, S. Rasheed, M. Mahroof-Tahir, U. Ashiq, R. A. Jamal, S. Khurshid and S. Mustafa, J. Chem. Soc. Pak., 2016, 38, 864 CAS.
- Qurrat-ul-Ain, U. Ashiq, R. A. Jamal and M. Mahroof-Tahir, Spectrochim. Acta, Part A, 2013, 115, 683–689 CrossRef PubMed.
- S. E.-S. Saeed, B. A. Alomari, A. N. Al-Hakimi, M. M. A. El-Hady, J. S. Alnawmasi, H. H. Elganzory and W. A. El-Sayed, Egypt. J. Chem., 2023, 66(5), 315–329 Search PubMed.
- J.-Q. Qu, L. Qu, Q.-H. Yang and L.-F. Wang, Chem. Pap., 2009, 63, 426–431 CAS.
- P. Selvam, S. Antharjanam, K. Srinivasan and T. Premkumar, J. Phys. Chem. Solids, 2022, 160, 110368 CrossRef CAS.
- C. M. Manzano, F. R. G. Bergamini, A. L. B. Formiga, P. P. Corbi, A. L. T. G. Ruiz, E. C. S. de Oliveira, W. R. Lustri and M. A. Ribeiro, J. Mol. Struct., 2020, 1208, 127912 CrossRef CAS.
- B. Kumar, J. Devi, A. Dubey, A. Tufail and B. Taxak, Sci. Rep., 2023, 13, 15906 CrossRef CAS PubMed.
- M. Jarosz, M. Olbert, G. Wyszogrodzka, K. Mlyniec and T. Librowski, Inflammopharmacology, 2017, 25(1), 11–24 CrossRef CAS PubMed.
- H. Summersgill, H. England, G. Lopez-Castejon, C. B. Lawrence, N. M. Luheshi, J. Pahle, P. Mendes and D. Brough, Cell Death Dis., 2014, 5, e1040 CrossRef CAS PubMed.
- S. Medici, M. Peana, V. M. Nurchi and M. A. Zoroddu, J. Med. Chem., 2019, 62(13), 5923–5943 CrossRef CAS PubMed.
- S. Medici, M. Peana, G. Crisponi, V. M. Nurchi, J. I. Lachowicz, M. Remelli and M. A. Zoroddu, Coord. Chem. Rev., 2016, 327–328, 349–359 CrossRef CAS.
- M. A. Iqbal, M. I. Umar, R. A. Haque, M. B. K. Ahamed, M. Z. B. Asmawi and A. M. S. A. Majid, J. Inorg. Biochem., 2015, 146, 1–13 CrossRef CAS PubMed.
- L. Kyros, N. Kourkoumelis, M. Kubicki, L. Male, M. B. Hursthouse, I. I. Verginadis, E. Gouma, S. Karkabounas, K. Charalabopoulos and S. K. Hadjikakou, Bioinorg. Chem. Appl., 2010, 2010, 386860 CrossRef PubMed.
- G. H. Jaffery, J. Bassett, J. Mendham and R. C. Denney, Vogel's Textbook of Quantitative Chemical Analysis, Thames Polytechnic, London, 5th edn, 1989 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, SHELXL-2018 Program for Crystal Structure Refinement, University of Gottingen, Gottingen, 2018 Search PubMed.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- C. Jelsch, K. Ejsmont and L. Huder, IUCrJ, 2014, 1, 119–128 CrossRef CAS PubMed.
- T. Lu, J. Chem. Phys., 2024, 161, 082503 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- L. H. Abdel-Rahman, M. T. Basha, B. S. Al-Farhan, M. R. Shehata and E. M. Abdalla, Appl. Organomet. Chem., 2022, 36(1), e6484 CrossRef CAS.
- M. A. Mesaik, A. Jabeen, S. A. Halim, A. Begum, A. S. Khalid, M. Asif, B. Fatima, Z. Ul-Haq and M. I. Choudhary, Chem. Biol. Drug Des., 2012, 79(3), 290–299 CrossRef CAS PubMed.
- V. M. Thadhani, M. A. Mesaik, M. Asif, V. Karunaratne and I. M. Choudhary, Int. J. Pharmcol. Pharm. Sci., 2015, 7(11), 144–147 CAS.
- B. J. Orlando and M. G. Malkowski, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2016, 72, 772–776 CrossRef CAS PubMed.
- P. Price and T. J. McMillan, Cancer Res., 1990, 50, 1392–1396 CAS.
- A. Periasamy, S. Muruganand and M. Palaniswamy, Rasayan J. Chem., 2009, 2(4), 981–989 CAS.
- P. H. de O. Santiago, E. de A. Duarte, E. C. M. Nascimento, J. B. L. Martins, M. S. Castro and C. C. Gatto, Appl. Organomet. Chem., 2022, 36, e6461 CrossRef.
- A. Castineiras, N. Fernandez-Hermida, I. Garcia-Santos, J. L. Perez-Lustres and I. Rodriguez-Gonzqlez, Dalton Trans., 2012, 41, 3787–3796 RSC.
- S. Kintzel, K. Eckhardt, J. Getzschmann, V. Bon, J. Grothe and S. Kaskel, Eur. J. Inorg. Chem., 2020, 3167–3173 CrossRef CAS.
- M. I. Bruce and M. J. Liddell, Appl. Orqanomet. Chem., 1987, 1, 191–226 CrossRef CAS.
- R. Jirasko and M. Holcapek, Mass Spectrom. Rev., 2011, 30, 1013–1036 CrossRef CAS PubMed.
- V. M. Filip, D. P. Stevan, D. B. Silvana, V. D. Borislava, M. Z. Branko, S. M. Milovan, K. J. Zeljko and V. R. Suzana, Arabian J. Chem., 2023, 16, 104461 CrossRef CAS.
- J. M. Miller and K. Balasanmugam, Can. J. Chem., 1989, 67, 1496–1500 CrossRef CAS.
- R. L. Cerny, B. P. Sullivan, M. M. Bursey and T. J. Meyer, Anal. Chem., 1983, 55(12), 1954–1958 CrossRef CAS.
- R. Isobe, T. Komori, F. Abe and T. Yamauchi, Biomed. Environ. Mass Spectrom., 1986, 13(11), 585–594 Search PubMed.
- G. Szekely and J. Allison, J. Am. Soc. Mass Spectrom., 1997, 8(4), 337–351 CrossRef CAS.
- K. Angela, A. V. Lucica, C. Nicoleta, R. Ileana and S. Nicolae, J. Serb. Chem. Soc., 2010, 75(2), 229–242 CrossRef.
- T. L. Davis, J. L. Watts, K. J. Brown, J. S. Hewage, A. R. Treleven, S. Lindeman and J. R. Gardinier, Dalton Trans., 2015, 44, 15408–15412 RSC.
- M. A. Abu-Youssef, S. M. Soliman, V. Langer, Y. M. Gohar, A. A. Hasanen, M. A. Makhyoun, A. H. Zaky and L. R. Ohrstrom, Inorg. Chem., 2010, 49(21), 9788–9799 CrossRef CAS PubMed.
- S. Soliman and A. El-Faham, Crystals, 2017, 7(6), 160 CrossRef.
- P. Ristic, T. R. Todorovic, V. Blagojevic, O. R. Klisuric, I. Marjanovic, B. B. Hollo, P. Vulic, M. Gulea, M. Donnard, M. Monge, M. Rodriguez-Castillo, J. M. Lopez-de-Luzuriaga and N. R. Filipovic, Cryst. Growth Des., 2020, 20(7), 4461–4478 CrossRef CAS.
- F. Hung-Low and K. K. Klausmeyer, Inorg. Chim. Acta, 2008, 361(5), 1298–1310 CrossRef CAS.
- S. A. Cotton, Silver and gold, Chemistry of Precious Metals, Springer, Dordrecht, 1st edn, 2012, p. 285 Search PubMed.
- D. Tsering, P. Dey, K. K. Kapoor and S. K. Seth, ACS Omega, 2024, 9, 36242–36258 CAS.
- S. Celik, S. Yurdakul and B. Erdem, J. Mol. Struct., 2022, 1261, 132902 CrossRef CAS.
- P. Dey, S. Islam, P. Das and S. K. Seth, J. Mol. Struct., 2024, 1296, 136820 CrossRef CAS.
- A. P. Prakasham, S. K. Patil, C. Nettem, S. Dey, G. Rajaraman and P. Ghosh, ACS Omega, 2023, 8, 6439–6454 CrossRef CAS PubMed.
- I. Ali, W. A. Wani and K. Saleem, Synth. React. Inorg. Met.-Org. Chem., 2013, 43(9), 1162–1170 CrossRef CAS.
- V. P. Singh, S. Singh and A. Katiyar, J. Enzyme Inhib. Med. Chem., 2009, 24(2), 577–588 CrossRef CAS PubMed.
- K. L. Zhong, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66(2), m131 CrossRef CAS PubMed.
- T. A. Saiyed, J. O. Adeyemi and D. C. Onwudiwe, Open Chem., 2021, 19, 974–986 CrossRef CAS.
- S. Yaqoob, N. Nasim, R. Khanam, Y. Wang, A. Jabeen, U. Qureshi, Z. Ul-Haq, H. R. El-Seedi, Z.-H. Jiang and F.-A. Khan, Molecules, 2021, 26(5), 1272 CrossRef CAS PubMed.
- A. Borowka, A. Sieroslawska, A. Baier, A. Rymuszka and E. Olszewska, Molecules, 2024, 29, 506 CrossRef CAS PubMed.
- M. S. Hasan and N. Das, Alexandria J. Med., 2017, 53(2), 157–165 CrossRef.
- B. V. Kumari, R. Mani, B. R. Asokan, K. Balakrishnan, A. Ramasamy, R. Parthasarathi, C. Kandasamy, R. Govindaraj, N. Vijayakumar and S. Vijayakumar, J. Compos. Sci., 2023, 7, 453 CrossRef.
- Q. Lin and Y. Deng, Environ. Sci. Technol., 2021, 55(22), 15010–15012 CrossRef CAS PubMed.
- E. G. Matveeva, A. K. Stoddard, H.-H. Zeng, G. Franke, L. Bourne, C. A. Fierke and R. B. Thompson, Int. J. Mol. Sci., 2022, 23(23), 14936 CrossRef CAS PubMed.
- M. S. Kasare, P. P. Dhavan, B. L. Jadhav and S. D. Pawar, Synth. Commun., 2019, 49(23), 3311–3323 CrossRef CAS.
- A. Munir, A. Khushal, K. Saeed, A. Sadiq, R. Ullah, G. Ali, Z. Ashraf, E. U. Mughal, M. S. Jan, U. Rashid, I. Hussain and A. Mumtaza, Bioorg. Chem., 2020, 104, 104168 CrossRef CAS PubMed.
- M. J. Alam, O. Alam, S. A. Khan, M. J. Naim, M. Islamuddin and G. S. Deora, Drug Des., Dev. Ther., 2016, 10, 3529–3543 Search PubMed.
- S. Caglar, A. Altay, B. Harurluoglu and B. Caglar, Maced. J. Chem. Chem. Eng., 2021, 40(2), 171–180 CrossRef.
- L. Radko, S. Stypula-Trebas, A. Posyniak, D. Zyro and J. Ochocki, Molecules, 2019, 24(10), 1949 CrossRef CAS PubMed.
- S. Yasar, T. K. Koprulu, S. Tekin and S. Yasar, Appl. Organomet. Chem., 2018, 32(2), e4016 CrossRef.
- D. Zyro, A. Sliwinska, I. Szymczak-Pajor, M. Strek and J. Ochocki, Cancers, 2020, 12(12), 3848 CrossRef CAS PubMed.
- J. S. Kim, H. Chang, S. Kang, S. Cha, H. Cho, S. J. Kwak, N. Park, Y. Kim, D. Kang, C. K. Song, J. Kwag, J.-S. Hahn, W. B. Lee, T. Hyeon and J. Park, Nat. Commun., 2023, 14, 3201 CrossRef CAS PubMed.
- R. I. J. Amos, B. S. Gourlay, C. H. Schiesser, J. A. Smith and B. F. Yates, Chem. Commun., 2008, 1695–1697 RSC.
- N. M. Shinde, A. C. Lokhande and C. D. Lokhande, J. Photochem. Photobiol., B, 2014, 136, 19–25 CrossRef CAS PubMed.
- E. Acosta, Thin films/properties and applications, Thin Films, IntechOpen, London, 2021 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2260170. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ma00265f |
| This journal is © The Royal Society of Chemistry 2025 |