
Synthesis and anti-tumor activity of new benzofuran-based chalcone derivatives as potent VEGFR-2 inhibitors†
Chunfei
Zhang‡
a,
Yixin
Liu‡
a,
Xiao
Zhang
a,
Chunping
Wan
*ab and
Zewei
Mao
 *a
*a
aSchool of Chinese Materia Medica, School of Clinical Medicine, Yunnan University of Chinese Medicine, Kunming 650500, PR China. E-mail: wanchunping1012@163.com; maozw@ynucm.edu.cn
bChuxiong Autonomous Prefecture Hospital of Traditional Chinese Medicine, Chuxiong 675000, PR China
First published on 16th October 2024
Abstract
Cancer is one of the most significant public health problems worldwide, and the discovery and development of efficient VEGFR-2 inhibitors has been a research hotspot in cancer treatment. In the present work, a series of novel benzofuran-based chalcone derivatives have been prepared, and in vitro anti-tumor activities of them have been evaluated. The results indicated that the compounds displayed potent anticancer activity against HCC1806, HeLa and A549 cell lines. The preliminary mechanism study showed that 4g could effectively induce the apoptosis of HCC1806 cells, and showed inhibitory effect on VEFGR-2. The molecular docking study indicated that 4g had an obvious binding site with the target VEGFR-2 (PDB ID: 4BSK). Therefore, the benzofuran-based chalcone derivatives could be considered as potent VEGFR-2 inhibitors.
Introduction
Cancer, a genetic disease caused by genetic defects or mutations that cause uncontrolled cell growth, is one of the most significant public health problems worldwide, which has become a serious threat to the health of residents, economic and social development.1 The International Agency for Research on Cancer (IARC) has reported the latest global cancer statistics for 2022, with nearly 20 million new cancer cases and 9.7 million deaths from cancer.2,3 Over the past decades, various new technologies and drugs for the treatment of cancers have been developed with the concerted efforts of the scientific community. However, traditional methods such as surgery, chemotherapy and radiotherapy can no longer meet the requirements of individualized and precise treatment of cancers in clinical practice. As the incidence of cancer continues to rise and the problem of drug resistance becomes more prominent, drug treatment of cancer still faces great challenges. Therefore, there is an urgent need for exploring highly effective anti-tumor drugs.Recently, small molecule targeted anti-tumor drugs have been widely studied by researchers and gradually applied in clinics. The latest studies have shown that overexpression of tyrosine kinases is closely related to cancer proliferation and/or metastasis.4,5 Among them, the vascular endothelial growth factor (VEGF) is a functional glycoprotein with high biological activity, and is the most important angiogenesis stimulator.6,7 VEGF inhibitors can inhibit cancer angiogenesis so as to achieve the effect of treating cancers. There are more than 15 anti-tumor drugs targeting VEGF or its receptor in the world.8 VEGFR-2, a member of the protein tyrosine kinase receptor superfamily, could inhibit the formation of new blood vessels for cancer treatment by blocking the VEGF/VEGFR-2 signaling pathway. Therefore, the discovery and development of efficient VEGFR-2 inhibitors has become a hotspot.
Natural active ingredients come from a wide range of sources, and they are a shortcut to drug exploration in modifying the structure and eventually developing them into drugs with better efficacy and pharmacokinetic properties.9 Benzofurans are one of the typical oxygen-containing heterocyclic compounds, which are widely present in plants and animals, such as Dorsmerunin A–C and Moracin A–Z (Scheme 1).10 A large number of studies have indicated that benzofurans showed good pharmacological effects, including anti-tumor, anti-HIV, antibacterial, anti-inflammatory, antioxidant and anti-cardiovascular aging properties.11–13 To date, more than 30 benzofuran drugs have been used in clinical applications. For example, fruquintinib, amiodarone and benzbromarone are used for colorectal cancer, cardiac arrhythmias and hyperuricemia, respectively (Scheme 1). In former works, we have carried out a series of studies on the structure–activity relationship of benzofuran derivatives, and found that benzofuran compounds bearing heterocyclic fragments showing the characteristics of high anticancer activity and low toxicity.14–16
 | ||
| Scheme 1 Natural benzofuran products and drugs. | ||
In recent years, there have been a lot of studies on benzofurans worldwide, including natural products and derivatives. Among them, benzofuran-chalcone hybrids are a class of compounds with a special structure, containing both benzofuran and chalcone structural units, which have been proved to show pharmacological activities, such as anti-tumor, antibacterial, anti-inflammatory and anti-tubulin activities.17–19 In the present work, we have designed new benzofuran-chalcone hybrids based on fragment-based drug design (FBDD) by recombination of active benzofuran and chalcone fragments (Scheme 2), and the active unit of the anticancer drugs fruquintinib or gefitinib was introduced into the molecule. Moreover, the in vitro anti-tumor activity, mechanism and docking study of hybrids have been carried out. The results indicated that the benzofuran-based chalcone derivatives could be considered as potent VEGFR inhibitors.
 | ||
| Scheme 2 Design of new benzofuran-based chalcone derivatives. | ||
Results and discussion
Chemistry
The new benzofuran-based chalcone derivatives (4a–4r) have been prepared by a general procedure as shown in Scheme 3. Firstly, benzofuran 1 was formed by substitution and condensation of 2-hydroxy-5-nitrobenzaldehyde with chloroacetone in CH3OH. Then, reduction of the nitro group with Fe to an amino group gave compound 2 in reflux saturated ammonium chloride solution in high yield. Subsequently, compound 2 was substituted with 4-chloro-6,7-dimethoxy-quinazoline to obtain the key intermediate 3 in reflux isopropanol. Finally, a series of benzofuran-chalcone derivatives were synthesized by the aldol reaction with aldehydes in 22–95% yields (Table 1). However, during the preparation of the title compounds, we found that the bases (KOH, piperidine and t-BuOK) affected the aldol reaction greatly. The effect of piperidine and t-BuOK catalyzed reaction was poor; even if the reaction time was prolonged or the reaction temperature was increased, the yield of the product did not enhance obviously. When piperidine is used as the catalyst, the yield of the reaction is the best compared to other bases. In addition, in order to investigate the effects of different chalcone fragments on the anti-tumor activity of the title hybrids, we introduced different types of aldehydes (Ar–CHO or Het–CHO) into the molecules. Under the optimized reaction conditions, 18 new benzofuran-chalcone hybrids (4a–4r) have been obtained (Table 1). All prepared new benzofuran-based chalcone derivatives were characterized by 1H NMR and 13C NMR, and some compounds were detected by HRMS analysis (ESI†). | ||
| Scheme 3 The synthetic route of benzofuran-chalcone derivatives. | ||














Biological assay
| Compound | IC50/(μmol L−1) | ||
|---|---|---|---|
| HCC1806 | HeLa | A549 | |
| 4a | >25 | >25 | >25 |
| 4b | >25 | >25 | >25 |
| 4c | >25 | >25 | >25 |
| 4d | 17.13 | >25 | >25 |
| 4e | >25 | >25 | >25 |
| 4f | >25 | >25 | >25 |
| 4g | 5.93 | 5.61 | >25 |
| 4h | >25 | >25 | >25 |
| 4i | >25 | >25 | >25 |
| 4j | 18.45 | 24.79 | >25 |
| 4k | >25 | >25 | >25 |
| 4l | 6.60 | 6.19 | 6.27 |
| 4m | >25 | >25 | >25 |
| 4n | 7.03 | 3.18 | 21.63 |
| 4o | 6.40 | 6.36 | >25 |
| 4p | 16.58 | >25 | >25 |
| 4q | 9.52 | 4.95 | >25 |
| 4r | >25 | >25 | >25 |
| DDP | 2.65 | 7.10 | 2.59 |
In addition, compound 4l exhibited the highest cytotoxicity against HeLa (IC50 = 6.19 μmol L−1), A549 (IC50 = 6.27 μmol L−1) and HCC1806 (IC50 = 6.60 μmol L−1). Compound 4n also showed good cytotoxic activity against three tumor cell lines, and its inhibitory activity was HeLa (IC50 = 3.18 μmol L−1), HCC1806 (IC50 = 7.03 μmol L−1), and A549 (IC50 = 21.63 μmol L−1), respectively. Among all the derivatives, 5 compounds showed better cytotoxic activity against HeLa, and the inhibitory activity was better than that of DDP (IC50 = 7.10 μmol L−1). The compounds with their IC50 values were 4g (IC50 = 5.61 μmol L−1), 4l (IC50 = 6.19 μmol L−1), 4n (IC50 = 3.18 μmol L−1), 4o (IC50 = 6.36 μmol L−1), and 4q (IC50 = 4.95 μmol L−1), respectively.
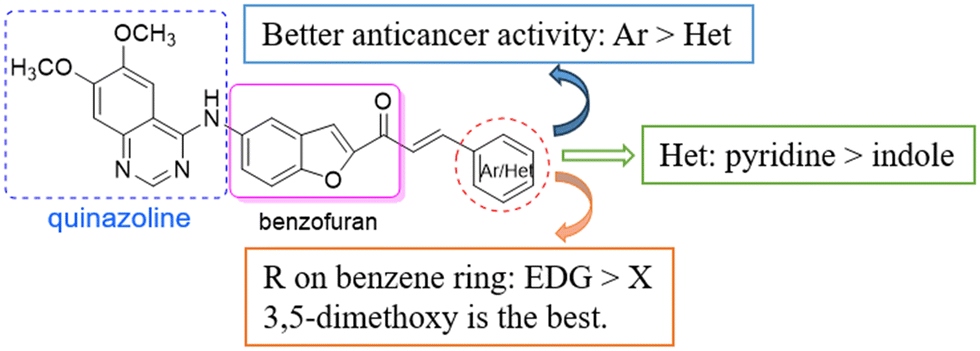
The preliminary structure–activity relationship is shown in Scheme 4. The structures of the chalcones have an obvious effect on the anti-tumor activity of the derivatives. In general, aryl chalcones displayed better cytotoxic activity than heterocyclic chalcones. In addition, when there were EDGs on the benzene ring, the compounds showed good anticancer activity against cancer cell lines, such as CH3O and OH. Among heterocyclic derivatives, pyridine was more conducive to the activity than indole. The SAR results provided a basis for further structural optimization and mechanism research of benzofuran-chalcone compounds.
 | ||
| Scheme 4 Preliminary SAR analysis of derivatives. | ||
 | ||
| Fig. 1 Compound 4g induced apoptosis of HCC1806. | ||
 | ||
| Fig. 2 Inhibitory rate of 4g on VEGFR-2 kinase. | ||
 | ||
| Fig. 3 Molecular docking of compound 4g with the target protein 4BSK. | ||
Conclusions
In the present work, a series of novel benzofuran-based chalcone derivatives have been prepared, which were screened for their in vitro anti-tumor activities. The results indicated that most compounds showed good cytotoxic activity against HCC1806, HeLa and A549 cell lines. In particular, compound 4g showed the best anti-tumor activity against HCC1806 and HeLa with IC50 of 5.93 μmol L−1 and 5.61 μmol L−1, respectively. The preliminary mechanism assessment showed that compound 4g could effectively trigger the apoptosis of HCC1806 cells in a dose dependent manner. Moreover, the molecular docking study showed that compound 4g had an obvious binding site with the target VEGFR-2 (PDB ID: 4BSK). Therefore, the benzofuran-chalcone derivatives may be considered as new potent VEGFR-2 inhibitors.Experimental
General information
All chemical reagents and solvents were commercially available. NMR data was recorded on a Bruker AV 400 spectrometer (Bruker), with TMS as the standard. HRMS data was obtained on a Triple TOF 5600+ spectrometer (AB Sciex). Human cervical cancer cells (HeLa), human lung cancer cells (A549), and human mammary squamous cancer cells (HCC1806) were purchased from Guangzhou Cellcook Biotech Co., Ltd. Biological data was obtained using a BSM-120.4 electronic balance, carbon dioxide incubator and flow cytometer. Thin-layer chromatography (TLC) analysis was performed on silica gel plates GF254.General preparation of compound (1)
To a stirred solution of 5-nitrosalicylaldehyde (1.67 g, 10 mmol) in 50 mL methanol, potassium hydroxide (0.56 g, 10 mmol) was added and left to react at room temperature for half an hour, and the reaction system was stirred in an ice water bath for 10 min. Then chloracetone (12 mmol) was slowly added to the reaction mixture and heated to 75 °C in an oil bath for 5–6 h. When the reaction was completed (detected by TLC), the mixture was cooled, and poured into a separating funnel. Then the mixture was extracted with methylene chloride (100 mL) and washed with water for three times, and the organic phase was dried by anhydrous sodium sulfate and concentrated by vacuum. The residue was subjected to silica gel column chromatography to give a yellow solid (1.30 g, 63% yield).Synthesis of compound (2)
To a stirred solution of iron powder (1.41 g, 25 mmol) and 30 mL of saturated ammonium chloride aqueous, the solution of compound 1 (1.30 g, 6.3 mmol) in 8 mL of anhydrous ethanol was slowly added and left to react at 100 °C for 5 h. After the reaction was completed by TLC, the reaction mixture was cooled and then extracted with ethyl acetate (50 mL) for three times. The organic phase was combined and dried with Na2SO4, concentrated in vacuo and purified by column chromatography to afford an orange solid (0.91 g, 82% yield).Synthesis of key intermediate (3)
To a stirred solution of compound 2 (0.91 g. 5.19 mmol) and 4-chloro-6,7-dimethoxyquinazoline (1.17 g, 5.19 mmol) in isopropyl alcohol (25 mL), the mixture was heated in reflux overnight. After the reaction was completed, the mixture was filtered and washed with isopropyl alcohol (15 mL) for 3–4 times to afford a pale yellow solid after drying.General synthetic procedure for title derivatives (4a–4r)
To a stirred solution of compound 3 (145.3 mg, 0.4 mmol) and various aldehydes (0.48 mmol) in ethanol (5 mL), piperidine (2 d) was added and the mixture was refluxed for 5 h. After completion of the reaction as indicated by TLC, the mixture was concentrated in vacuo, and the residue was purified by column chromatography to give the corresponding products.Compound 4b. 1H NMR(400 Hz, DMSO-d6) δ: 9.76(s, 1H), 8.49(s, 1H), 8.27(d, J = 2.0 Hz, 1H), 8.22(s, 1H), 7.85–7.91(m, 1H), 7.51–7.79(m, 4H), 7.68(d, J = 15.6 Hz, 1H), 7.21(s, 1H), 7.03(d, J = 8.9 Hz, 2H), 3.98(s, 3H), 3.95(s, 3H), 3.76(t, J = 4.6 Hz, 4H), 3.27–3.30(m, 4H); 13C NMR(100 MHz, DMSO-d6) δ: 178.72, 157.17, 154.84, 154.78, 153.29, 153.12, 153.53, 149.45, 144.49, 135.95, 131.16, 127.63, 125.21, 124.81, 118.00, 117.09, 114.56, 112.41, 109.17, 107.22, 102.54, 66.36, 56.74, 56.32, 47.54; HRMS-ESI calcd for C31H29N4O5 (M + H)+ 537.2133, found 537.2129.
Compound 4c. 1H NMR(400 Hz, DMSO-d6) δ: 9.80(s, 1H), 8.64(d, J = 2.5 Hz, 1H), 8.50(s, 1H), 8.35(d, J = 0.5 Hz, 1H), 8.30(d, J = 2.0 Hz, 1H), 8.07(d, J = 8.8 Hz, 2H), 7.99(d, J = 8.8 Hz, 2H), 7.90–7.94(m, 3H), 7.78–7.89(m, 2H), 7.20(s, 1H), 6.60–6.61(m, 1H), 3.98(s, 3H), 3.94(s, 3H); 13C NMR(100 MHz, DMSO-d6) δ: 178.76, 157.18, 154.86, 154.39, 153.04, 152.73, 149.47, 142.99, 142.11, 141.59, 136.04, 132.58, 130.90, 128.53, 127.53, 125.65, 122.07, 118.87, 117.24, 115.79, 112.51, 109.15, 108.90, 107.09, 102.54, 56.75, 56.33.
Compound 4d. 1H NMR(400 Hz, DMSO-d6) δ: 9.66(s, 1H), 8.48(s, 1H), 8.29(d, J = 1.7 Hz, 2H), 7.54–8.25(m, 7H), 7.49(t, J = 8.1 Hz, 1H), 7.34(t, J = 7.6 Hz, 2H), 7.20(s, 1H), 3.98(s, 3H), 3.94(s, 3H); 13C NMR(100 MHz, DMSO-d6) δ: 183.09, 161.77, 160.33, 159.48, 158.85, 158.12, 158.01, 157.81, 157.39, 154.14, 152.05, 141.02, 135.39, 133.45, 132.45, 132.35, 130.36, 128.93, 127.57, 126.68, 121.73, 120.38, 119.06, 117.24, 116.64, 114.01, 112.35, 107.15, 61.45, 61.03, 60.12.
Compound 4e. 1H NMR(400 Hz, DMSO-d6) δ: 9.64(s, 1H), 8.47(s, 1H), 8.29(d, J = 2.0 Hz, 1H), 8.23(d, J = 0.6 Hz, 1H), 7.93–8.03(m, 1H), 7.83–7.89(m, 3H), 7.49(d, J = 9.0 Hz, 1H), 7.73(d, J = 3.3 Hz, 1H), 7.53(d, J = 15.4 Hz, 1H), 7.20–7.23(m, 2H), 3.98(s, 3H), 3.94(s, 3H); 13C NMR(100 MHz, DMSO-d6) δ: 183.09, 161.77, 160.33, 159.48, 158.85, 158.12, 158.01, 157.81, 157.39, 154.14, 152.05, 141.02, 135.39, 133.45, 132.45, 132.35, 130.36, 128.93, 127.57, 126.68, 121.73, 120.38, 119.06, 117.24, 116.64, 114.01, 112.35, 107.15, 61.45, 61.03, 60.12.
Compound 4f. 1H NMR(400 Hz, DMSO-d6) δ: 9.63(s, 1H), 8.46(s, 1H), 8.25(d, J = 2.0 Hz, 1H), 8.10(d, J = 0.7 Hz, 1H), 7.87–7.90(m, 2H), 7.78(d, J = 9.0 Hz, 1H), 7.61(d, J = 15.4 Hz, 1H), 7.39(d, J = 15.4 Hz, 1H), 7.19(s, 1H), 7.04(d, J = 3.3 Hz, 1H), 6.36–6.37(m, 1H), 3.97(s, 3H), 3.93(s, 3H), 2.41(s, 3H); 13C NMR(100 MHz, DMSO-d6) δ: 183.26, 161.76, 159.45, 159.13, 157.19, 154.93, 154.11, 152.06, 140.91, 135.13, 132.38, 129.98, 125.15, 121.93, 121.62, 119.29, 117.14, 115.32, 114.01, 112.35, 107.14, 61.43, 61.00.
Compound 4g. 1H NMR(400 Hz, DMSO-d6) δ: 9.88(s, 1H), 8.50(s, 1H), 8.38(d, J = 0.6 Hz, 1H), 8.33(d, J = 2.0 Hz, 1H), 7.95(s, 1H), 7.85–7.91(m, 2H), 7.73–7.79(m, 2H), 7.21(s, 1H), 7.10(d, J = 2.2 Hz, 2H), 6.62(t, J = 4.6 Hz, 1H), 3.99(s, 3H), 3.94(s, 3H), 3.83(s, 6H); 13C NMR(100 MHz, DMSO-d6) δ: 183.56, 166.00, 161.97, 159.67, 159.03, 157.64, 157.52, 154.25, 148.78, 141.51, 140.77, 132.23, 130.44, 127.59, 122.00, 120.84, 117.26, 113.89, 112.07, 111.67, 108.30, 107.41, 61.56, 61.09, 60.70, 57.25.
Compound 4h. 1H NMR(400 Hz, DMSO-d6) δ: 9.69(s, 1H), 8.47(s, 1H), 8.30–8.32(m, 2H), 7.85–7.89(m, 2H), 7.77–7.79(m, 3H), 7.55(d, J = 2.0 Hz, 1H), 7.41–7.44(m, 1H), 7.20(s, 1H), 7.07(d, J = 8.4 Hz, 1H), 3.98(s, 3H), 3.94(s, 3H), 3.89(s, 3H), 3.84(s, 3H); 13C NMR(100 MHz, DMSO-d6) δ: 183.54, 161.84, 159.51, 159.29, 157.98, 157.33, 156.79, 154.29, 154.15, 151.90, 149.17, 140.88, 132.42, 132.29, 132.14, 130.10, 129.42, 124.67, 121.72, 120.01, 117.21, 116.87, 116.00, 114.00, 112.26, 107.18, 61.47, 61.05, 60.98, 60.87; HRMS-ESI calcd for C29H26N3O6 (M + H)+ 512.1816, found 512.1816.
Compound 4i. 1H NMR(400 Hz, DMSO-d6) δ: 9.97(s, 1H), 8.52(s, 1H), 8.34(d, J = 0.7 Hz, 1H), 8.29(d, J = 2.0 Hz, 1H), 7.91–7.97(m, 3H), 7.83–7.90(m, 1H), 7.78(d, J = 9.3 Hz, 2H), 7.56(d, J = 8.5 Hz, 2H), 7.20(s, 1H), 3.99(s, 3H), 3.94(s, 3H); 13C NMR(100 MHz, DMSO-d6) δ: 183.45, 162.03, 159.75, 159.00, 157.57, 154.30, 147.20, 140.65, 140.59, 138.58, 135.86, 134.26, 132.22, 130.55, 127.82, 122.18, 120.77, 117.24, 113.79, 111.28, 107.53, 61.59, 61.12.
Compound 4j. 1H NMR(400 Hz, DMSO-d6) δ: 10.06(s, 1H), 8.54(s, 1H), 8.35(d, J = 0.6 Hz, 1H), 8.28(d, J = 2.0 Hz, 1H), 7.99(s, 1H), 7.86–7.94(m, 4H), 7.78–7.82(m, 2H), 7.71(d, J = 8.5 Hz, 2H), 7.22(s, 1H), 3.99(s, 3H), 3.95(s, 3H); 13C NMR(100 MHz, DMSO-d6) δ: 183.48, 162.15, 159.87, 159.03, 157.66, 154.38, 147.34, 140.51, 138.91, 137.20, 136.07, 132.24, 130.68, 129.52, 127.87, 122.37, 120.79, 117.30, 113.71, 110.93, 107.61, 61.62, 61.16.
Compound 4k. 1H NMR(400 Hz, DMSO-d6) δ: 9.69(s, 1H), 8.73(d, J = 4.6 Hz, 1H), 8.48(s, 1H), 8.27–8.29(m, 2H), 8.15(d, J = 15.4 Hz, 1H), 7.80–7.94(m, 6H), 7.46–7.49(m, 1H), 7.20(s, 1H), 3.98(s, 3H), 3.94(s, 3H); 13C NMR(100 MHz, DMSO-d6) δ: 183.80, 161.81, 159.52, 158.83, 157.65, 157.45, 154.16, 147.79, 142.54, 140.96, 132.31, 130.70, 130.48, 130.31, 130.00, 121.85, 117.29, 113.98, 112.26, 107.14, 61.45, 61.05.
Compound 4l. 1H NMR(400 Hz, DMSO-d6) δ: 10.21(s, 1H), 9.70(s, 1H), 8.47(s, 1H), 8.29(d, J = 2.0 Hz, 2H), 8.24(s, 2H), 7.87–7.90(m, 4H), 7.65–7.79(m, 1H), 7.20(s, 1H), 6.89(d, J = 8.6 Hz, 2H), 3.98(s, 3H), 3.94(s, 3H); 13C NMR(100 MHz, DMSO-d6) δ: 178.83, 160.92, 157.06, 154.70, 154.59, 153.32, 152.49, 149.35, 147.24, 144.43, 136.12, 131.64, 127.57, 125.96, 125.27, 124.00, 122.50, 118.77, 116.97, 116.40, 114.91, 112.40, 109.25, 107.55, 102.43, 56.71, 56.27; HRMS-ESI calcd for C27H21N3O5 (M + H)+ 468.1554, found 468.1545.
Compound 4n. 1H NMR(400 Hz, DMSO-d6) δ: 9.72(s, 1H), 8.48(s, 1H), 8.30–8.33(m, 2H), 7.85–7.92(m, 6H), 7.79(d, J = 9.0 Hz, 1H), 7.48–7.49(m, 3H), 7.19(s, 1H), 3.98(s, 3H), 3.93(s, 3H); 13C NMR(100 MHz, DMSO-d6) δ: 178.83, 157.06, 154.74, 154.27, 153.21, 152.64, 149.38, 147.00, 143.93, 136.16, 134.84, 131.36, 129.46, 129.43, 127.49, 125.56, 122.32, 117.06, 115.86, 112.46, 109.21, 107.39, 102.43, 56.71, 56.27.
Compound 4o. 1H NMR(400 Hz, DMSO-d6) δ: 10.73(s, 1H), 8.72(d, J = 6.0 Hz, 2H), 8.66(s, 1H), 8.44(d, J = 0.5 Hz, 1H), 8.26(d, J = 2.0 Hz, 1H), 8.16(d, J = 12.8 Hz, 1H), 7.89–8.09(m, 1H), 7.87–7.89(m, 3H), 7.77–7.84(m, 2H), 7.28(s, 1H), 4.01(s, 3H), 3.97(s, 3H); 13C NMR(100 MHz, DMSO-d6) δ: 183.32, 162.76, 160.56, 158.87, 158.22, 155.63, 154.82, 146.82, 145.83, 139.70, 132.20, 131.36, 127.81, 117.52, 113.18, 108.46, 108.30, 61.86, 61.38.
Compound 4p. 1H NMR(400 Hz, DMSO-d6) δ: 9.75(s, 1H), 8.49(s, 1H), 8.37(d, J = 24.8 Hz, 1H), 8.16(s, 1H), 8.14(t, J = 10.0 Hz, 1H), 7.90(s, 3H), 7.78–7.82(m, 3H), 7.61(q, J = 8.9 Hz, 1H), 7.21(s, 1H), 3.99(s, 3H), 3.95(s, 3H); 13C NMR(100 MHz, DMSO-d6) δ: 178.79, 154.81, 154.19, 153.15, 152.76, 149.43, 141.75, 136.32, 127.44, 117.20, 116.28, 112.53, 109.17, 102.43, 56.72, 56.32;
Compound 4q. 1H NMR(400 Hz, DMSO-d6) δ: 12.06(s, 1H), 10.15(s, 1H), 8.56(s, 1H), 8.30(d, J = 1.9 Hz, 1H), 8.23(s, 1H), 8.13–8.17(m, 3H), 8.04(s, 1H), 7.80–7.87(m, 2H), 7.65(d, J = 15.6 Hz, 1H), 7.52–7.54(m, 1H), 7.27–7.29(m, 2H), 7.24(s, 1H), 4.00(s, 3H), 3.96(s, 3H); 13C NMR(100 MHz, DMSO-d6) δ: 178.87, 157.47, 155.15, 155.07, 152.63, 152.41, 149.65, 139.21, 138.11, 135.44, 134.65, 127.81, 125.50, 125.13, 123.36, 121.80, 121.05, 117.50, 115.46, 113.72, 113.21, 113.05, 112.46, 108.93, 102.97, 56.90, 56.42; HRMS-ESI calcd for C29H23N4O4 (M + H)+ 491.1714, found 491.1710.
Compound 4r. 1H NMR(400 Hz, DMSO-d6) δ: 12.17(s, 1H), 9.66(s, 1H), 8.48(s, 1H), 8.23–8.36(m, 4H), 8.13(d, J = 15.6 Hz, 1H), 7.79–7.90(m, 3H), 7.62(d, J = 15.6 Hz, 1H), 7.50(d, J = 8.6 Hz, 1H), 7.41(d, J = 8.4 Hz, 1H), 7.21(s, 1H), 3.99(s, 3H), 3.95(s, 3H); 13C NMR(100 MHz, DMSO-d6) δ: 178.95, 157.06, 154.84, 154.69, 153.40, 152.37, 149.35, 147.40, 138.28, 136.73, 136.03, 135.08, 127.74, 127.32, 125.96, 124.85, 122.97, 116.90, 116.32, 114.94, 114.54, 114.21, 112.77, 112.39, 109.28, 107.68, 102.39, 56.70, 56.28; HRMS-ESI calcd for C29H22BrN4O4 (M + H)+ 569.0819, found 569.0815.
In vitro anti-tumor activity
The anticancer activity of the synthetic derivatives (4a–4r) was tested by the MTT method, with cisplatin as the positive control drug, and HeLa (human cervical cancer cell), A549 (human lung cancer cell) and HCC1806 (human mammary squamous cancer cell) were selected as the tumor strains to be detected. At first, a constant temperature incubator (37 °C, 5% CO2) was used to provide a culture environment. When cells were found at the bottom of the petri dish, passage culture was started for later experiments. On a 96-well plate, tumor cells with a logarithmic growth stage (5 × 104 cells per mL) were inoculated, cultured overnight, and then compounds were added to them, and five concentration gradients of 100 μmol L−1, 50 μmol L−1, 25 μmol L−1, 12.5 μmol L−1 and 6.25 μmol L−1 were set. Each concentration has 3 compound pores. After 48 hours of reaction, 20 μL and 5 mg ml−1 of MTT were added to each well and cultured continuously for 4 h. Finally, it was centrifuged at 3000 rpm for 10 min, the culture solution was absorbed and discharged, 150 μL DMSO was added to finish the reaction, and the reaction was shaken in the shaker for 10 min. The absorption was measured at 570 nm wavelength by an enzymograph device, the absorbance was determined, and the IC50 value was calculated.Apoptosis test
The logarithmic growth phase HCC1806 (human breast squamous carcinoma cell) cells were digested for cell suspension and the cell density was diluted to 4 × 105 cells per mL. 1 mL of cell suspension was added to each six-well plate, and then the six-well plate was cultured at 37 °C in an incubator with 5% CO2 for 24 h. One day later (24 h), 1 mL of the sample solution with a certain concentration gradient (3 μmol L−1, 6 μmol L−1) was added to the six-well plate, and the culture was continued for 24 h at 37 °C. A day later (24 h), the collected cells were immersed and washed twice with cold PBS, and finally centrifuged at 2000 rpm for 5 min, then the supernatant was absorbed and discarded, and precipitated in an ice bath at 0 °C. 500 μL Annexin binding buffer was gently suspended into each well. Then the 5 μL red fluorescent dye (PI) and 5 μL green fluorescent dye (Annexin V) were added into the mixture, mixed slowly and evenly, and placed in an ice water bath for 10 minutes away from light. The results were detected by flow cytometry.VEGFR-2 test
The inhibitory effect of compound 4g on VEGFR-2 kinase was determined using a VEGFR-2 (KDR) kinase assay kit. The compound was prepared at 100-fold the highest desired concentration in 100% DMSO and then diluted 10-fold in 1× kinase buffer 1 to prepare the highest concentration of the 10-fold intermediate dilutions. For positive and negative controls, 10% DMSO was prepared in 1× kinase buffer 1 (v/v) so that all wells contained the same amount of DMSO (diluent solution). Then 25 μL of the master mix and 5 μL of the compound solution were added to each well. 20 μL of 1× kinase buffer 1 and 5 μL of diluent solution were added to the “blank” and “positive control” wells as well. Subsequently, the reaction was initiated by adding 20 μL of diluted VEGFR-2 kinase to the wells designated “positive control” and “test inhibitor.” Then the mixture was incubated at 30 °C for 45 min and reacted for 45 min, and 50 μL of Kinase-Glo™ MAX reagent was added. The plate was covered with aluminum foil and incubated at room temperature for 15 min, and were immediately read using a luminometer or microplate reader capable of reading luminescence.Molecular docking experiment
The 2D planar structure of the desired compound was drawn in ChemDraw 2D and saved as a mol format file. The three-dimensional crystal structure of VEGFR-1 in PDB format was obtained from the PDB database (https://www.rcsb.org/), and the 4BSK fragment was selected as the intended target protein of the synthesized drug. The obtained protein was pre-treated with water removal and hydrogen replenishment in Discovery Studio and saved as a PBD format file. Docking between small molecules and target proteins was carried out in Discovery Studio, the scoring results were obtained, the docking results were visualized, and interaction diagrams of the docking sites were obtained.Data availability
Data for this article, including experimental details and spectra data, are available as part of the ESI.†Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
This work was financially supported by the Key Project of Applied Basic Research of Yunnan Province (202401AS070007), and the Yunnan Provincial Science and Technology Department-Applied Basic Research Joint Special Funds of Yunnan University of Chinese Medicine (202001AZ070001-007).Notes and references
- B. Han, R. Zheng, H. Zeng, S. Wang, K. Sun, R. Chen, L. Li, W. Wei and J. He, J. Natl. Cancer Cent., 2024, 4, 47–53 Search PubMed.
- F. Bray, M. Laversanne, H. Sung, J. Ferlay, R. L. Siegel, I. Soerjomataram and A. Jemal, Ca-Cancer J. Clin., 2024, 74, 229–263 CrossRef.
- N. Jokhadze, A. Das and D. S. Dizon, Ca-Cancer J. Clin., 2024, 74, 224–226 CrossRef.
- S. Hussain, M. Mursal, G. Verma, S. M. Hasan and M. F. Khan, Eur. J. Pharmacol., 2024, 970, 176484 CAS.
- D. Özmen, D. D. Alpaydın, M. A. Saldoğan and A. E. Eşkazan, Expert Opin. Drug Saf., 2024, 23, 411–423 CrossRef PubMed.
- D. R. Aboshouk, M. A. Youssef, M. S. Bekheit, A. R. Hamed and A. S. Girgis, RSC Adv., 2024, 14, 5690–5728 RSC.
- M. Malekan, N. K. Haass, G. R. Rokni, N. Gholizadeh, M. A. Ebrahimzadeh and A. Kazeminejad, Life Sci., 2024, 345, 122563 CrossRef CAS PubMed.
- J. Zeng, Q. Deng, Z. Chen, S. Yan, Q. Dong, Y. Zhang, Y. Cui, L. Li, Y. He and J. Shi, Bioorg. Chem., 2024, 146, 107278 CrossRef CAS.
- R. J. Nevagi, S. N. Dighe and S. N. Dighe, Eur. J. Med. Chem., 2015, 97, 561–581 CrossRef CAS PubMed.
- R. Naik, D. S. Harmalkar, X. Xu, K. Jang and K. Lee, Eur. J. Med. Chem., 2015, 90, 379–393 CrossRef CAS.
- H. Khanam and Shamsuzzaman, Eur. J. Med. Chem., 2015, 97, 483–504 CrossRef CAS PubMed.
- Z. Xu, S. Zhao, Z. Lv, L. Feng, Y. Wang, F. Zhang, L. Bai and J. Deng, Eur. J. Med. Chem., 2019, 162, 266–276 CrossRef CAS PubMed.
- J. Farhat, L. Alzyoud, M. Alwahsh and B. Al-Omari, Cancers, 2022, 14, 2196 CrossRef CAS.
- H. Gao, X. Zhang, X. J. Pu, X. Zheng, B. Liu, G. X. Rao, C. P. Wan and Z. W. Mao, Bioorg. Med. Chem. Lett., 2019, 29, 806–810 CrossRef CAS.
- Y. Liu, N. Zou, M. Li, C. Wan and Z. Mao, Curr. Org. Synth., 2024, 21, 928–940 CrossRef CAS.
- Y. Liu, Z. Wu, M. Li, H. Gao, C. Wan and Z. Mao, Bioorg. Med. Chem. Lett., 2023, 91, 129378 CrossRef CAS.
- M. Mostofi, G. Z. Mohammadi, M. Mahdavi, A. Moradi, H. Nadri, S. Emami, H. Alinezhad, A. Foroumadi and A. Shafiee, Eur. J. Med. Chem., 2015, 103, 361–369 CrossRef CAS PubMed.
- T. Constantinescu and C. N. Lungu, Int. J. Mol. Sci., 2021, 22, 11306 CrossRef CAS PubMed.
- D. Coskun, M. Erkisa, E. Ulukaya, M. F. Coskun and F. Ari, Eur. J. Med. Chem., 2017, 136, 212–222 CAS.
- C. Cooksey, Biotech. Histochem., 2021, 96, 401–407 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR and HRMS spectra. See DOI: https://doi.org/10.1039/d4md00621f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |