
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploiting the DCAF16–SPIN4 interaction to identify DCAF16 ligands for PROTAC development†
Isabella A.
Riha
 a,
Miguel A.
Campos
ab,
Xiaokang
Jin
a,
Fiona Y.
Wang
a,
Chenlu
Zhang
a,
Sara F.
Dunne
c,
Benjamin F.
Cravatt
d and
Xiaoyu
Zhang
*abefg
a,
Miguel A.
Campos
ab,
Xiaokang
Jin
a,
Fiona Y.
Wang
a,
Chenlu
Zhang
a,
Sara F.
Dunne
c,
Benjamin F.
Cravatt
d and
Xiaoyu
Zhang
*abefg
aDepartment of Chemistry, Northwestern University, Evanston, IL 60208, USA. E-mail: zhang@northwestern.edu
bChemistry of Life Processes Institute, Northwestern University, Evanston, IL 60208, USA
cHigh Throughput Analysis Laboratory, Northwestern University, Evanston, IL 60208, USA
dDepartment of Chemistry, The Scripps Research Institute, La Jolla, CA 92037, USA
eInternational Institute for Nanotechnology, Northwestern University, Evanston, IL 60208, USA
fRobert H. Lurie Comprehensive Cancer Center, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA
gCenter for Human Immunobiology, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA
First published on 6th December 2024
Abstract
Traditional small molecule drugs often target protein activity directly, but challenges arise when proteins lack suitable functional sites. An alternative approach is targeted protein degradation (TPD), which directs proteins to cellular machinery for proteolytic degradation. Recent studies have identified additional E3 ligases suitable for TPD, expanding the potential of this approach. Among these, DCAF16 has shown promise in facilitating protein degradation through both PROTAC and molecular glue mechanisms. In this study, we developed a homogeneous time resolved fluorescence (HTRF) assay to discover new DCAF16 binders. Using an in-house electrophile library, we identified two diastereomeric compounds, with one engaging DCAF16 at cysteines C177–179 and another reducing its expression. We demonstrated that the compound covalently engaging DCAF16 can be transformed into a PROTAC capable of degrading FKBP12.
Introduction
Traditional small molecule drugs typically work by directly interfering with the activity of proteins. However, designing effective drugs can be challenging when proteins lack suitable functional sites.1,2 An emerging alternative strategy is the use of small molecules to direct proteins to cellular machinery for proteolytic degradation, which leads to the complete removal of the target proteins.3 This targeted protein degradation (TPD) strategy employs two main types of small molecules: 1) heterobifunctional compounds known as PROTACs (proteolysis-targeting chimeras), which connect E3 ligase ligands to substrate ligands through a variable linker;4 and 2) monofunctional compounds named molecular glues, which form complexes involving specific E3 ligases and neo-substrate proteins.5 The TPD approach offers several advantages, including the ability to convert inactive protein-binding small molecules into active protein degraders, thereby expanding the range of druggable proteins. Moreover, this approach can operate catalytically,6 potentially reducing the required drug concentrations for therapeutic efficacy.An increasing number of proteins have been shown to be susceptible to ligand-induced degradation.3 Most of these degradations are mediated by two E3 ligases, cereblon (CRBN) and von Hippel–Lindau (VHL), which have demonstrated notable capabilities in the TPD field.7,8 Nevertheless, CRBN and VHL exhibit limitations, such as a restricted substrate scope.9–11 Recent studies have identified additional E3 ligases with diverse properties that support TPD, expanding its potential applications in both biological research and therapeutic settings.12 Among these, DCAF16, a DDB1 and CUL4-associated factor (DCAF), was identified through a screening of electrophilic PROTACs designed to recruit E3 ligases.13 Subsequent studies have demonstrated that DCAF16 supports the degradation of various proteins through both PROTAC14,15 and molecular glue16–18 mechanisms. Notably, DCAF16 contains multiple ligandable cysteines, including C58,17 C119,19 and a cluster of cysteines (C177–179),13 which contribute to protein degradation when engaged by electrophilic small molecule degraders.
In the context of PROTACs, the DCAF16 recruiter KB02 is a fragment electrophile that interacts with a wide range of ligandable cysteines across the proteome at concentrations required for inducing protein degradation.13 This broad engagement restricts the applicability of this first-generation recruiter in biological studies and therapeutic applications. Here, we present the development of a homogeneous time resolved fluorescence (HTRF) assay with potential for identifying additional DCAF16 recruiters. Using this assay, we screened an in-house electrophilic library and identified several hits, including two diastereomeric compounds. Validation studies indicated that one diastereomer potentially engages DCAF16 at cysteines C177–179, while the other reduces DCAF16 expression. Finally, we demonstrated that the compound directly engaging DCAF16 can be converted into a PROTAC capable of degrading the protein FKBP12.
Results
Development of an HTRF assay for measuring the DCAF16–SPIN4 interaction
The HTRF assay is a quantitative and high-throughput screening (HTS)-compatible technology used to measure analytes in a homogeneous format.20 In the HTRF assay, a signal is generated through fluorescence resonance energy transfer (FRET) between a donor and an acceptor molecule when they are in close proximity. Consequently, HTRF is frequently employed to measure protein–protein interactions.21 We previously identified SPIN4, a histone trimethylation binder protein, as a native substrate and binder of DCAF16.22 We hypothesize that the interaction between DCAF16 and SPIN4 could be utilized to discover electrophilic ligands that bind to DCAF16 by the disruption of this interaction.We first generated HEK293T cells stably expressing FLAG-tagged SPIN4 and HA-tagged DCAF16. These epitope tags can be recognized by high-affinity immunoglobulin G conjugated with terbium (Tb) cryptate as the donor fluorophore and d2 as the acceptor. When DCAF16 and SPIN4 interact, the proximity of the epitope tags and their binding antibodies generates an HTRF signal that can be detected by a plate reader (Fig. 1a). We tested two pairs of antibodies – HA-d2 and FLAG-Tb, and HA-Tb and FLAG-d2 – and found that the combination of HA-d2 (recognizing DCAF16) and FLAG-Tb (recognizing SPIN4) produced a higher signal compared to the other pair (Fig. 1b). This HTRF signal was not observed when SPIN1 or SPIN2A, two other SPIN family proteins, were co-expressed with DCAF16 (Fig. 1c and d). Moreover, as we previously demonstrated that SPIN4 exhibits a significantly increased interaction with DCAF16 in the presence of the proteasome inhibitor MG132,22 this increased interaction was also observed in the HTRF assay (Fig. 1c). These results collectively suggest the effectiveness of the HTRF assay in measuring the interaction between DCAF16 and SPIN4.
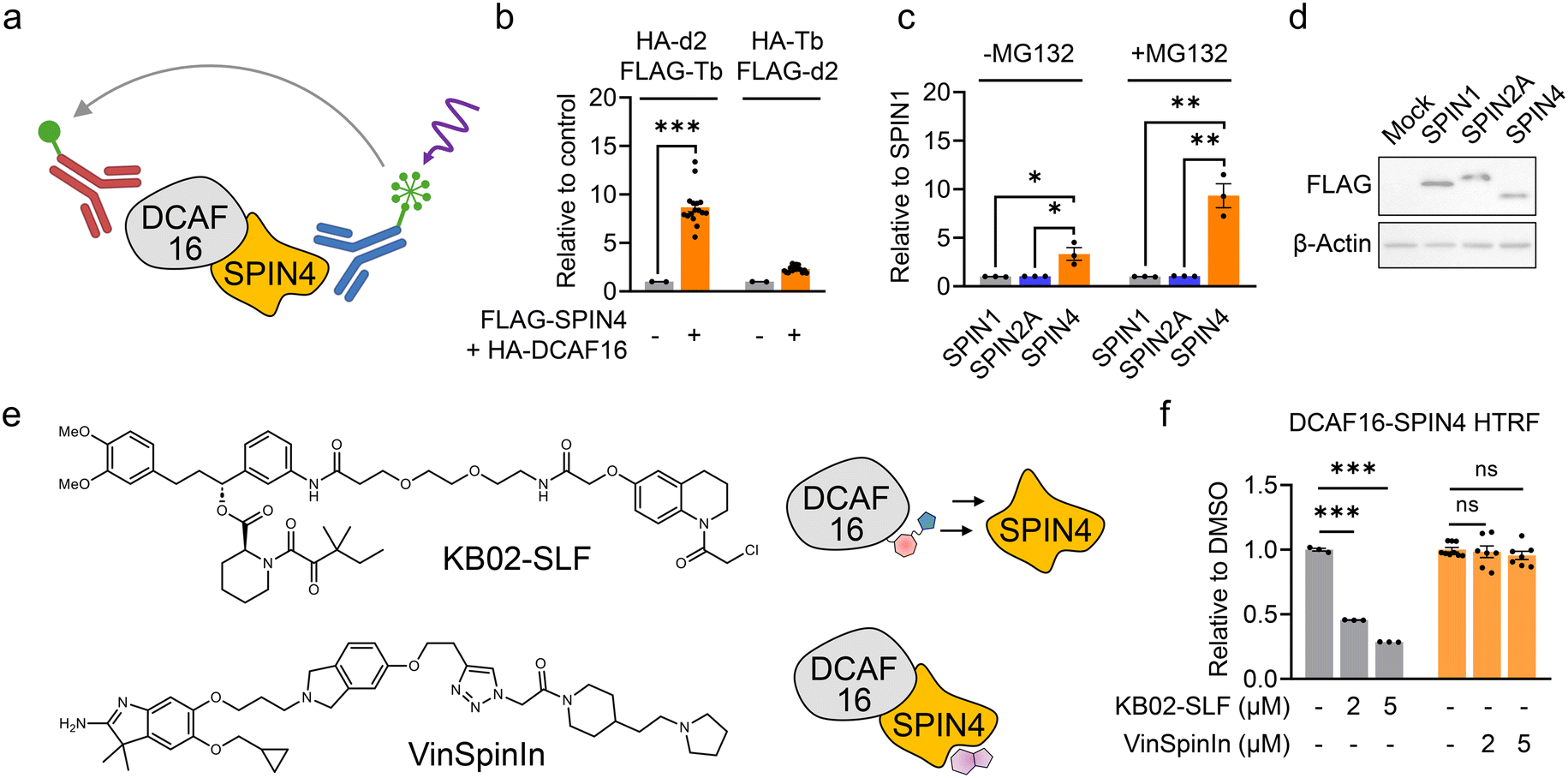 | ||
| Fig. 1 Development of an HTRF assay for measuring the DCAF16–SPIN4 interaction. a. Schematic representation of an HTRF assay for measuring the interaction between DCAF16 and SPIN4. b. HTRF assay measuring the interaction between HA-DCAF16 and FLAG-SPIN4 using anti-HA-d2 and anti-FLAG-Tb, or anti-HA-Tb and anti-FLAG-d2. Data are presented as the mean values ± S.E.M. (n = 16 for FLAG-SPIN4 and HA-DCAF16-expressing HEK293T cells; n = 2 for HEK293T parental cells). Data are normalized to the HTRF signal from HEK293T parental cells to obtain ‘relative to control’ values. Statistical significance was evaluated through unpaired two-tailed Student's t-tests, comparing FLAG-SPIN4 and HA-DCAF16-expressing cells to parental cells. ***P < 0.001. c. HTRF assay measuring the interaction between HA-DCAF16 and FLAG-SPIN1, FLAG-SPIN2A, or FLAG-SPIN4, with or without MG132 treatment (1 μM, 2 hours). Data are presented as the mean values ± S.E.M. (n = 3). Data are normalized to the HTRF signal from SPIN1-expressing cells to obtain ‘relative to SPIN1’ values. Statistical significance was evaluated through unpaired two-tailed Student's t-tests, comparing FLAG-SPIN4-expressing cells to FLAG-SPIN1 or FLAG-SPIN2A-expressing cells. *P < 0.05; **P < 0.01. d. Western blot analysis of FLAG-SPIN1, FLAG-SPIN2A, and FLAG-SPIN4 expression levels in cells used in the HTRF assay. The result is representative of two independent experiments with similar results. e. Structures of KB02-SLF and VinSpinIn. The schematic on the right illustrates the proposed model, showing that DCAF16 ligands disrupt the DCAF16–SPIN4 interaction, while SPIN4 ligands interacting with the histone trimethylation binding pocket do not affect this interaction. f, HTRF assay measuring the interaction between HA-DCAF16 and FLAG-SPIN4 in the presence of KB02-SLF or VinSpinIn. Data are presented as the mean values ± S.E.M. (n = 3 for KB02-SLF-treated cells; n = 7 for VinSpinIn-treated cells). Data are normalized to the HTRF signal from DMSO-treated cells to obtain ‘relative to DMSO’ values. Statistical significance was evaluated through unpaired two-tailed Student's t-tests, comparing compound-treated to DMSO-treated cells. ***P < 0.001; ns, not significant. | ||
Next, we aimed to determine whether the previously reported covalent DCAF16 engager, KB02-SLF13 (Fig. 1e), could disrupt the DCAF16–SPIN4 interaction. KB02-SLF is known to modify one of the three clustered cysteines on DCAF16 (C177–179), thereby inducing a ternary complex with FKBP12 and promoting its proteasomal degradation.13 We observed a concentration-dependent decrease in the HTRF signal following a 2 hour treatment with KB02-SLF (Fig. 1f). This result suggests that the region of DCAF16 modified by KB02-SLF may be involved in the interaction with SPIN4. Conversely, we tested VinSpinIn (Fig. 1e), a reported inhibitor with nanomolar binding potency to SPIN4,23 and observed that it did not inhibit the HTRF signal at concentrations of 2 and 5 μM (Fig. 1f). We surmise that the binding pocket for this inhibitor, which is also involved in histone trimethylation binding,23 is distant from the interaction interface between DCAF16 and SPIN4 (Fig. 1e). This suggests that the HTRF assay measuring the DCAF16–SPIN4 interaction may be used to identify DCAF16 ligands that decrease the HTRF signal, rather than SPIN4 ligands that bind the histone trimethylation binding pocket.
High-throughput screening to identify electrophilic small molecules that disrupt the DCAF16–SPIN4 interaction
Using the HTRF assay, we aimed to identify electrophilic small molecules that disrupt the DCAF16–SPIN4 interaction, thereby providing candidate compounds for further validation as DCAF16 binders. We screened a focused in-house library of 640 structurally elaborated electrophiles. For the screening, HEK293T cells expressing FLAG-SPIN4 and HA-DCAF16 were seeded in a 384-well plate and co-treated with MG132 and each candidate compound for 1 hour. Following this, the cells were lysed and analyzed using the HTRF assay (Fig. 2a). Each compound was tested in duplicate at a concentration of 5 μM. The initial screen identified 45 compounds that exhibited ≥30% inhibition of the HTRF signal (Fig. 2b and ESI† Table S1). We then conducted a second round of dose-dependent assays with these 45 compounds at concentrations of 0.625, 1.25, 2.5, 5, and 10 μM, in triplicate. Compounds showing less than 30% inhibition at 10 μM were excluded. This filtering process resulted in 23 hit compounds (Fig. 2c, S1, and Table S1†).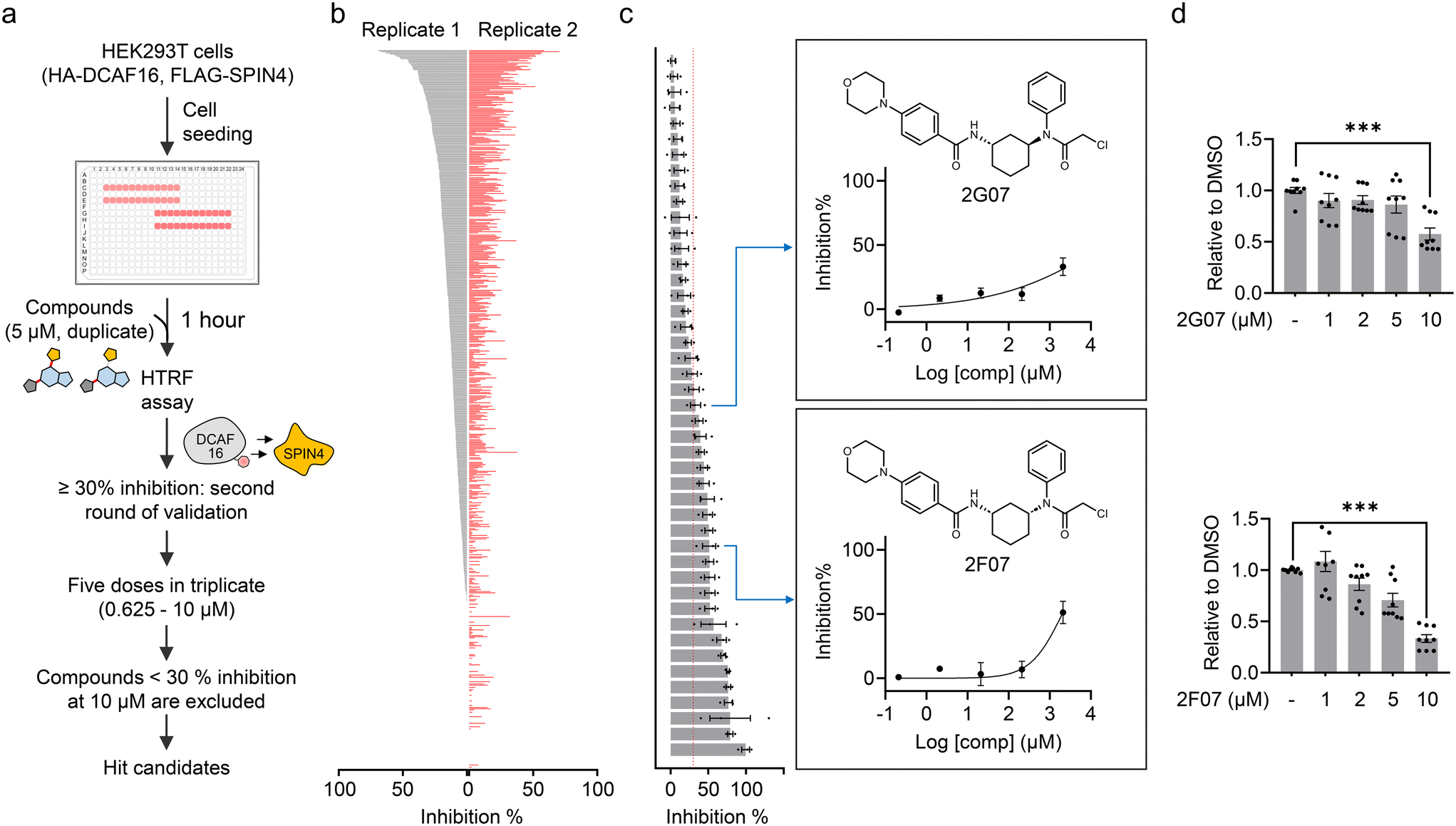 | ||
| Fig. 2 High-throughput screening to identify electrophilic small molecules that disrupt the DCAF16–SPIN4 interaction. a. Schematic representation of the workflow for using the HTRF assay to identify electrophilic small molecules that disrupt the DCAF16–SPIN4 interaction. b. Initial compound screening at a single concentration (5 μM) in duplicate. The bar graph represents the inhibition of each compound on the HTRF signal for the DCAF16–SPIN4 interaction. c. The second round of validation using 45 compounds that showed ≥30% inhibition in the initial screen. The bar graph represents inhibition by these compounds at 10 μM. Data are presented as mean values ± S.E.M. (n = 3). The right panel shows the structures of 2G07 and 2F07, along with the inhibition curves for 2G07 and 2F07 at five concentrations (0.625, 1.25, 2.5, 5, and 10 μM). d. HTRF assay measuring the interaction between HA-DCAF16 and FLAG-SPIN4 in the presence of re-synthesized 2G07 or 2F07. Data are presented as the mean values ± S.E.M. (n = 9 for both compounds). Data are normalized to the HTRF signal from DMSO-treated cells to obtain ‘relative to DMSO’ values. Statistical significance was evaluated through unpaired two-tailed Student's t-tests, comparing 10 μM compound-treated to DMSO-treated cells. ***P < 0.001. | ||
While we are actively investigating all 23 hit compounds to evaluate their potential as DCAF16 recruiters for TPD applications, in this study, we were particularly intrigued by the identification of two diastereomers, 2G07 and 2F07 (Fig. 2c). Recent studies have demonstrated the utility of stereochemistry in chemical probe development, enabling the identification of stereochemically defined probes with specific target engagement and phenotypic effects.24–27 Consequently, we decided to further investigate these two diastereomers to determine if they both function as covalent DCAF16 ligands, and whether the stereochemistry is not essential for this functionality. We re-synthesized these compounds and confirmed their ability to disrupt the DCAF16–SPIN4 interaction using the HTRF assay (Fig. 2d).
2G07 and 2F07 inhibit the HTRF signal for the DCAF16–SPIN4 interaction through distinct mechanisms
Using 2G07 and 2F07, we sought to determine whether they inhibit the HTRF signal from the DCAF16–SPIN4 interaction by directly engaging DCAF16. We employed cysteine-directed activity-based protein profiling (ABPP), a chemical proteomics technique for examining electrophilic small molecule-cysteine interactions across the proteome.28 We incubated 2G07, 2F07, and a DMSO control with HEK293T cell lysates. Target engagement of these compounds was assessed using a broad-spectrum iodoacetamide probe, which reacts with thousands of cysteines29,30 (Fig. 3a). We identified two cysteine-containing peptides on DCAF16: one with C119 and the other with a cysteine cluster spanning C177–179 (Fig. 3b and ESI† Table S2). Previous studies indicated that α-chloroacetamide-containing PROTACs interact with the C177–179 cluster in DCAF16, leading to targeted protein degradation.13 Our results revealed that 2G07 exhibited approximately 60% engagement with DCAF16_C177–179 at 10 μM, but no engagement with DCAF16_C119, which is distant from the C177–179 cluster (Fig. 3b–d). In contrast, 2F07 did not show engagement with either C119 or C177–179 (Fig. 3b and c). This lack of engagement is unlikely due to decomposition of 2F07, as both 2F07 and 2G07 engage several shared reactive cysteines from additional proteins, with 2F07 engaging even more cysteines than 2G07 (Fig. S2 and Table S2†). In addition, we accessed off-target proteins bound by 2G07 through the ABPP experiment. Five proteins demonstrated stronger engagement with 2G07 compared to DCAF16 (Fig. S2 and Table S2†). Among these, two proteins were engaged at their catalytic cysteines (CASP8_C360 and THIOM_C90), while the remaining three were engaged at previously reported hyperreactive cysteines.29,30 These cysteines exhibit increased nucleophilicity towards electrophiles and are frequently identified as off-target sites.31 To evaluate the impact of 2G07 and 2F07 on cell viability and confirm that the concentrations used for HTRF and ABPP experiments do not induce significant toxicity, we conducted cell viability assays. The results demonstrated IC50 values of 23.2 μM for 2G07 and 26.3 μM for 2F07 (Fig. S3†), confirming that the concentrations employed for HTRF (1–10 μM) and ABPP (10 μM) did not lead to notable cytotoxic effects.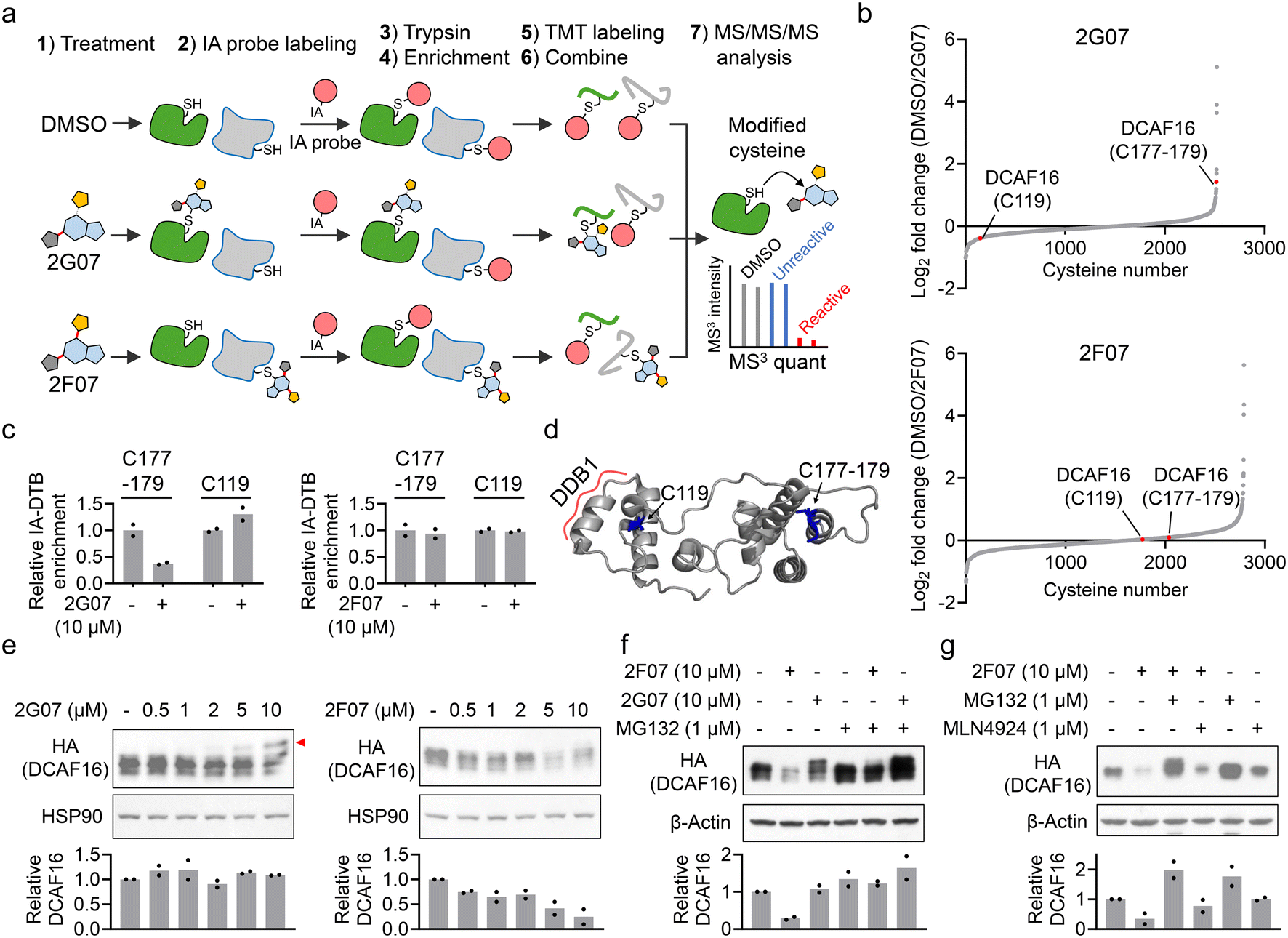 | ||
| Fig. 3 2G07 and 2F07 inhibit the HTRF signal for the DCAF16–SPIN4 interaction through distinct mechanisms. a. Schematic representation of cysteine-directed ABPP for identifying targets of 2G07 and 2F07. b. Waterfall plot showing the ratio values of IA probe-enriched peptides in DMSO-treated versus compound-treated samples. A higher ratio indicates potential modification of cysteine-containing peptides by the compound. Ratio values are shown as mean values (n = 2). c. Bar graph showing the quantification of two IA-DTB-modified peptides in DCAF16 treated with 2G07 or 2F07. Data are presented as the mean values (n = 2). d. DCAF16 component from a cryo-EM structure (PDB: 8G46), with cysteines C119 and C177–179 highlighted in blue. e. Western blot analysis of HA-DCAF16 in cells treated with 0.5–10 μM of 2G07 or 2F07 for 2 hours. The red arrow indicates a higher molecular weight form of DCAF16, potentially modified by the compound. The bar graph represents quantification of the HA-DCAF16/HSP90 protein content. Data are presented as the mean values (n = 2). f. Western blot analysis of HA-DCAF16 in cells treated with 2G07 or 2F07, with or without co-treatment with MG132, for 2 hours. The bar graph represents quantification of the HA-DCAF16/β-actin protein content. Data are presented as the mean values (n = 2). g. Western blot analysis of HA-DCAF16 in cells treated with 2F07, with or without co-treatment with MG132 or MLN4924, for 2 hours. The bar graph represents quantification of the HA-DCAF16/β-actin protein content. Data are presented as the mean values (n = 2). | ||
Since our proteomics analysis did not identify all the cysteines on DCAF16, it remains unclear whether 2F07 directly engages DCAF16 through other cysteines. To assess the binding of electrophilic small molecules to DCAF16, an alternative approach is to use Western blot analysis to identify a high-molecular weight form of DCAF16, which would appear as a band-shift.13 We treated HEK293T cells expressing HA-DCAF16 with both compounds, lysed the cells, and then analyzed HA-DCAF16 using Western blot. Consistent with the ABPP results, 2G07 induced a dose-dependent band-shift in DCAF16 (Fig. 3e, left panel). In contrast, 2F07 did not produce a band-shift in DCAF16 but instead caused a dose-dependent reduction in DCAF16 expression (Fig. 3e, right panel). The reduction in DCAF16 expression induced by 2F07 was blocked by co-treatment with the proteasome inhibitor MG132 (Fig. 3f and g), but not by the NEDDylation inhibitor MLN4924 (Fig. 3g). Recent studies suggest that MG132's ability to block small molecule-induced protein reduction may indicate either proteasomal degradation13,22 or a post-transcriptional mechanism such as suppression of splicing.32 Thus, further mechanistic studies are needed to elucidate how 2F07 reduces DCAF16 expression. Nevertheless, these findings suggest that 2G07 likely directly interacts with DCAF16 at the interface between DCAF16 and SPIN4, disrupting their protein–protein interaction. Conversely, 2F07 appears to decrease DCAF16 expression, which leads to the depletion of one component in the HTRF assay, thereby resulting in a loss-of-signal outcome.
Development of a 2G07-based PROTAC for targeted protein degradation
Our findings suggest that 2G07 could be a DCAF16 ligand for developing heterobifunctional PROTACs. To explore this, we synthesized a PROTAC, 2G07-SLF, by conjugating 2G07 to a widely used FKBP12 ligand33via a PEG linker (Fig. 4a). We tested 2G07-SLF in HEK293T cells overexpressing HA-DCAF16 and nucleus-localized FKBP12 (FKBP12_NLS) and observed a dose-dependent reduction in FKBP12_NLS (Fig. 4b). This effect was not observed in HEK293T cells lacking overexpressed HA-DCAF16, indicating that the reduction is dependent on DCAF16. Additionally, we observed a band-shift pattern in DCAF16 in cells treated with 2G07-SLF (Fig. 4b), further supporting direct engagement of DCAF16 by 2G07-SLF. The 2G07-SLF-induced reduction in DCAF16 was blocked by MG132, MLN4924, and SLF (Fig. 4c), suggesting a degradation mechanism for FKBP12 that involves both the proteasome and cullin–RING ligase pathways. We further compared 2G07-SLF with the first-generation DCAF16-based FKBP12 degrader, KB02-SLF,13 and observed similar potency in inducing FKBP12 degradation under the conditions tested (Fig. S4†).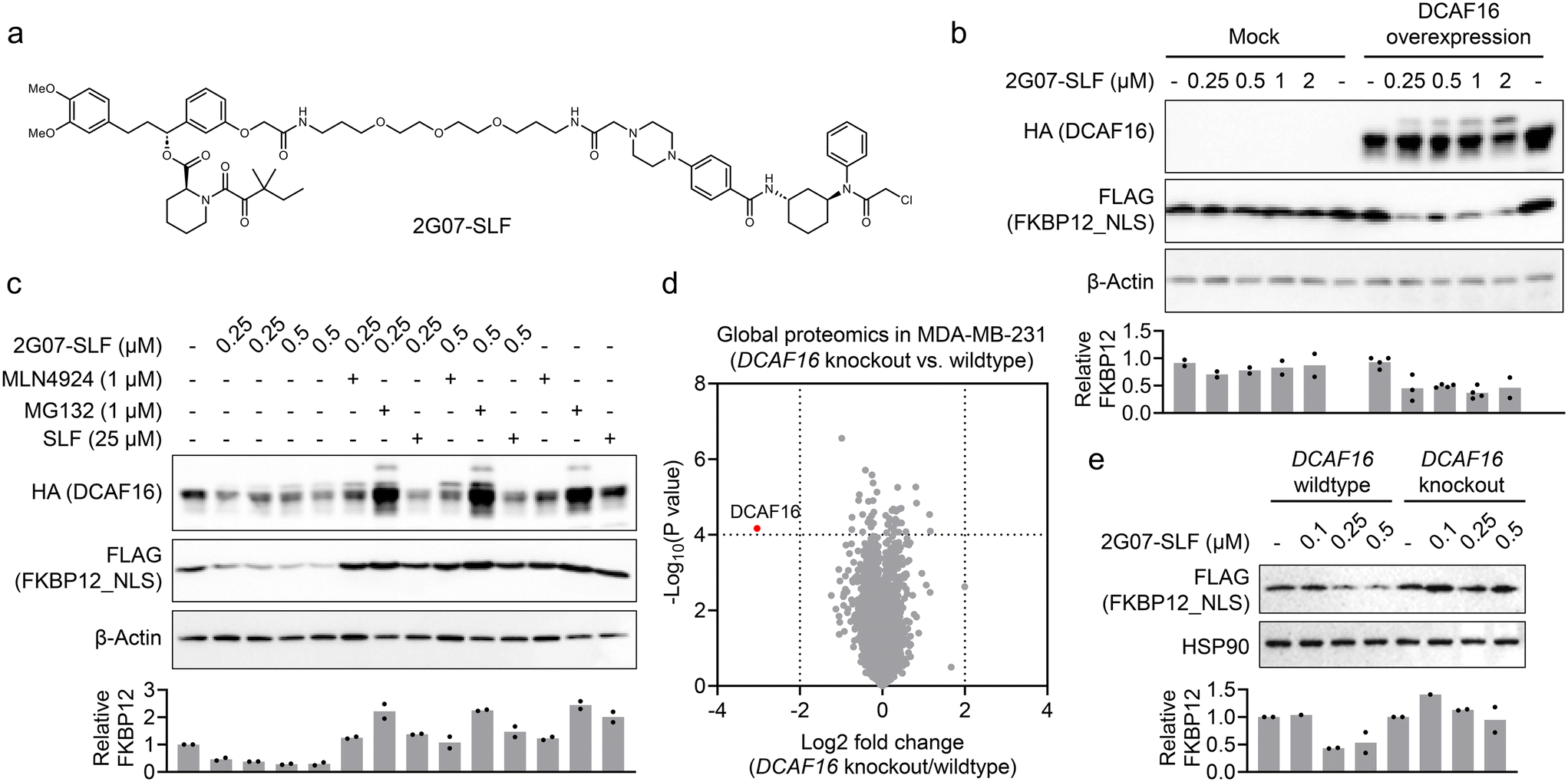 | ||
| Fig. 4 Development of a 2G07-based PROTAC for targeted protein degradation. a. Structure of 2G07-SLF. b. 2G07-SLF-induced FKBP12_NLS degradation is dependent on overexpressed DCAF16. HEK293T cells stably expressing FLAG-FKBP12_NLS and HA-DCAF16, or an empty vector, were treated with 0.25–2 μM 2G07-SLF for 8 hours. The bar graph represents quantification of the FLAG-FKBP12_NLS/β-actin protein content. Data are presented as the mean values (n = 2 for treatment in HEK293T cells without DCAF16 overexpression and HEK293T expressing HA-DCAF16 treated with 2 μM of 2G07-SLF; n = 3 for HEK293T expressing HA-DCAF16 treated with 0.25 μM of 2G07-SLF; n = 4 for HEK293T expressing HA-DCAF16 treated with DMSO, 0.5 or 1 μM of 2G07-SLF). c. 2G07-SLF-induced FKBP12_NLS degradation is blocked by MG132 (1 μM), MLN4924 (1 μM), or SLF (25 μM). HEK293T cells stably expressing FLAG-FKBP12_NLS and HA-DCAF16 were treated with 2G07-SLF, with or without co-treatment with MG132, MLN4924, or SLF, for 24 hours. The bar graph represents quantification of the FLAG-FKBP12_NLS/β-actin protein content. Data are presented as the mean values (n = 2). d. Volcano plot showing the global proteomic analysis of MDA-MB-231 parental versus DCAF16 knockout cells (n = 3). P values were calculated by two-sided t-test and adjusted using Benjamini–Hochberg correction for multiple comparisons. e. 2G07-SLF induced FLAG-FKBP12_NLS degradation in MDA-MB-231 parental but not DCAF16 knockout cells. MDA-MB-231 parental and DCAF16 knockout cells were treated with 0.1–0.5 μM 2G07-SLF for 8 hours. The bar graph represents quantification of the FLAG-FKBP12_NLS/HSP90 protein content. Data are presented as the mean values (n = 2). | ||
Next, we investigated whether 2G07-SLF can engage endogenous DCAF16 to promote the degradation of FKBP12_NLS. We chose MDA-MB-231 cells, which express functional endogenous DCAF16 for targeted protein degradation,13 and created DCAF16 knockout cells. The knockout of DCAF16 was confirmed by global proteomics (Fig. 4d and Table S3†). We then stably expressed FLAG-FKBP12_NLS in both MDA-MB-231 parental and DCAF16 knockout cells and tested 2G07-SLF for its ability to degrade FKBP12_NLS. The results showed that 2G07-SLF effectively degraded FKBP12_NLS in DCAF16 wildtype cells, but not in DCAF16 knockout cells (Fig. 4e). These findings collectively suggest that 2G07 can be utilized as a DCAF16 recruiter in the development of PROTACs for targeted protein degradation.
2G07-SLF induces a ternary complex involving DCAF16 and FKBP12 by engaging DCAF16_C177–179
We then assessed whether 2G07-SLF can induce the formation of a ternary complex involving DCAF16, FKBP12, and the compound. HEK293T cells expressing HA-DCAF16 and FLAG-FKBP12_NLS were treated with 2G07-SLF and MG132. The results revealed that HA-DCAF16 co-immunoprecipitated with FLAG-FKBP12_NLS in the presence of 2G07-SLF and MG132 (Fig. 5a), supporting the formation of a ternary complex involving 2G07-SLF, DCAF16, and FKBP12_NLS. To comprehensively evaluate the impact of 2G07-SLF on the FKBP12_NLS interactome, we employed an enrichment proteomic approach to identify proteins co-immunoprecipitating with FLAG-FKBP12_NLS from HEK293T cells treated with 2G07-SLF. This experiment identified DCAF16 and DDB1, a component of the DCAF16-associated cullin–RING complex, as proteins recruited by 2G07-SLF (Fig. 5b and Table S4†). Four additional proteins, CKB, RCC2, TXNRD1, and GPX4, were enriched as FKBP12 interacting proteins upon treatment with 2G07-SLF. Among these, GPX4 and TXNRD1 contain selenocysteine in their active sites, which exhibit enhanced nucleophilicity towards electrophiles.34 Additionally, all four proteins harbor previously identified hyperreactive cysteines that are prone to reacting with electrophiles.29,30 Therefore, we surmise that the α-chloroacetamide moiety of 2G07-SLF interacts with these reactive cysteines and/or selenocysteine, leading to their enrichment in the FKBP12 interactome study. In addition, we performed a parallel enrichment proteomics experiment using KB02-SLF at the same concentration (5 μM), which also recruited DCAF16 and DDB1 to form a complex with FKBP12_NLS (ESI† Table S4). Comparative analysis revealed that 2G07-SLF enriched greater amounts of DCAF16 and DDB1 compared to KB02-SLF (Fig. 5c), demonstrating the enhanced potency of 2G07-SLF in binding to DCAF16 compared to the first-generation compound KB02-SLF.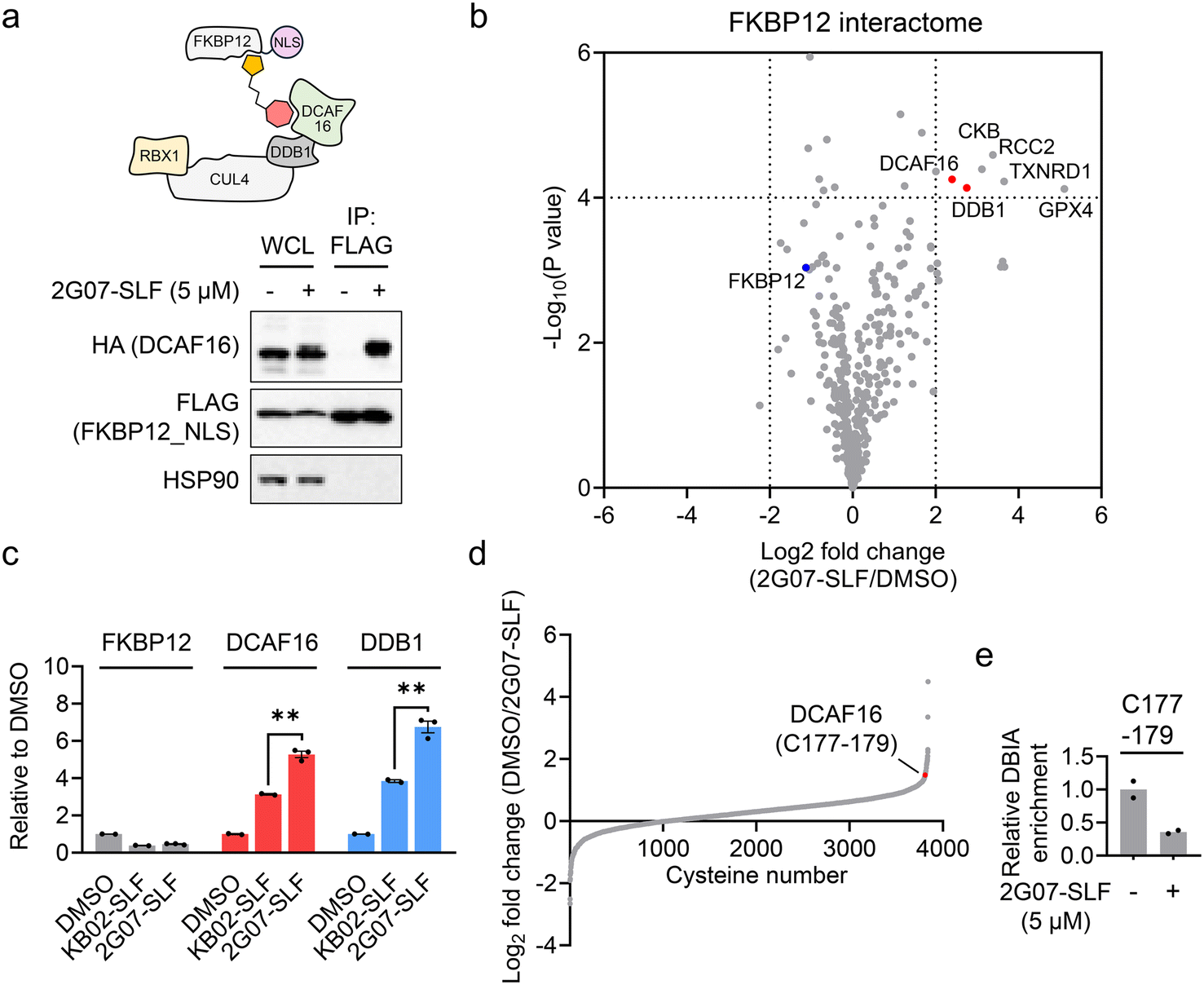 | ||
| Fig. 5 2G07-SLF induces a ternary complex involving DCAF16 and FKBP12 by engaging DCAF16_C177–179. a. Co-immunoprecipitation assay showing that HA-DCAF16 co-immunoprecipitated with FLAG-FKBP12_NLS in the presence of 2G07-SLF (5 μM) and MG132 (5 μM). The top cartoon panel illustrates a model where 2G07-SLF induces a complex involving FKBP12_NLS and the CUL4–DDB1–DCAF16 complex. WCL, whole cell lysates; IP, immunoprecipitation. b. Volcano plot showing FKBP12 interactome analysis comparing 2G07-SLF-treated versus DMSO-treated cells, both in the presence of 5 μM of MG132 (n = 3 for 2G07-SLF-treated samples; n = 2 for DMSO-treated samples). P values were calculated by two-sided t-test and adjusted using Benjamini–Hochberg correction for multiple comparisons. c. Bar graph quantification of FKBP12, DCAF16 and DDB1 protein content in FKBP12_NLS interactome samples treated with DMSO, KB02-SLF (5 μM), or 2G07-SLF (5 μM), all in the presence of 5 μM of MG132. Data are presented as the mean values ± S.E.M. (n = 3 for 2G07-SLF-treated samples; n = 2 for DMSO-treated and KB02-SLF-treated samples). Data are normalized to the signal from DMSO-treated cells to obtain ‘relative to DMSO’ values. Statistical significance was evaluated through unpaired two-tailed Student's t-tests, comparing 2G07-SLF-treated to KB02-SLF-treated cells. **P < 0.01. d. Waterfall plot showing the ratio values of IA probe-enriched peptides in DMSO-treated versus 2G07-SLF-treated samples. A higher ratio indicates potential modification of cysteine-containing peptides by 2G07-SLF. Ratio values are shown as mean values (n = 2). e. Bar graph showing the quantification of DBIA-modified C177–179-containing peptide in DCAF16 treated with 2G07-SLF. Data are presented as the mean values (n = 2). | ||
Finally, we employed cysteine-directed ABPP to assess the engagement of 2G07-SLF in DCAF16. The results revealed that 2G07-SLF exhibited approximately 65% engagement on the cysteine cluster (C177–179) of DCAF16 at 5 μM (Fig. 5d, e and ESI† Table S5). This data suggests that converting the initial DCAF16 ligand 2G07 into the heterobifunctional PROTAC 2G07-SLF retains its interaction with the DCAF16 cysteine cluster. Notably, 30 proteins exhibit stronger engagement with 2G07-SLF at 5 μM compared to DCAF16 (ESI† Table S5). These interactions involve catalytic cysteines (e.g., THIO_C32 and TXD17_C43) and previously reported hyperreactive cysteines (e.g., HMOX2_C265, HMOX2_C282, and CKAP4_C100).29,30 Compared to KB02-SLF, a first-generation DCAF16-based FKBP12 degrader that engages 39 cysteines at 2 μM with the same engagement threshold (>65%),13 2G07-SLF demonstrates enhanced selectivity. Nonetheless, further optimization of 2G07's proteome-wide selectivity as a DCAF16 recruiter remains necessary, presenting a valuable avenue for future investigation.
Discussion
In this study, we developed an HTRF assay to measure the interaction between DCAF16 and SPIN4. We observed that this interaction can be disrupted by electrophilic compounds that target a cysteine cluster C177–179 within DCAF16. Based on this finding, we conducted a compound screening to identify electrophilic small molecules that reduce the HTRF signal of the DCAF16–SPIN4 interaction. These small molecules have potential as DCAF16 ligands for applications such as PROTAC development. Our screen revealed two diastereomers, 2G07 and 2F07, both of which reduced the HTRF signal. Subsequent biochemical validation showed that 2G07 covalently binds to DCAF16, while 2F07 appears to down-regulate DCAF16 expression. Notably, 2G07 can be converted into a PROTAC that targets FKBP12 for degradation, indicating its potential for developing additional PROTACs for targeted protein degradation. When employing this method to screen for additional electrophilic protein–protein interaction inhibitors for TPD applications, an important consideration is whether the candidate E3 ligases contain ligandable cysteines in positions that are suitable for effective substrate degradation. To this end, the enriched chemical proteomics datasets would provide valuable information for assessing the ligandability of E3 ligases.12 Published structures and/or AlphaFold models of E3 ligases can also provide insight into this information. Nevertheless, one caveat is the possibility that the hit compounds may bind to the substrates rather than the E3 ligases. Therefore, thorough biochemical validation, such as a cysteine-directed ABPP approach, is essential to confirm the interaction with E3 ligases, enabling further investigation of these ligands in TPD applications.One of the questions from this study is the mechanism by which 2F07 reduces DCAF16 expression. Our results suggest that the reduction of DCAF16 by 2F07 is not blocked by the NEDDylation inhibitor MLN4924, which may rule out cullin–RING ligase-mediated degradation. Although the proteasome inhibitor MG132 can block the reduction of DCAF16 induced by 2F07, it remains unclear whether this effect involves proteasomal degradation or a post-transcriptional mechanism, such as inhibition of splicing, as suggested by a recent study.32 Investigating the molecular mechanism by which 2F07 reduces DCAF16 expression would be an interesting avenue for further research, and the ABPP results with 2F07 and 2G07 in this study could provide insights into the potential targets involved in this effect (Fig. 3a, b and S2†).
Previous studies have shown that low fractional engagement (10–40%) of E3 ligases by electrophilic PROTACs is sufficient to support targeted protein degradation.13,35,36 The results of this study are consistent with these findings. Our ABPP data indicate approximately 65% engagement of DCAF16 by 2G07-SLF at 5 μM. As this compound degrades FKBP12_NLS at lower concentrations (0.25 μM), it suggests that even lower fractional engagement of DCAF16 by 2G07-SLF may be sufficient to support target degradation. Low stoichiometric engagement with the E3 ligase for target degradation can be advantageous for electrophilic PROTACs, as it may minimize perturbation of the native function of the E3 ligase by only partially engaging it.
It is important to note that 2G07 contains an α-chloroacetamide group, which may exhibit high reactivity and lead to off-target effects in the proteome, as identified in our ABPP and interactome studies (Fig. 5b and d). Therefore, optimizing 2G07 to enhance its potency and selectivity remains a crucial objective for future work. Additionally, assessing the versatility of the DCAF16 recruiter 2G07 for degrading additional proteins will be an important future avenue to gain a comprehensive understanding of the compatibility of various proteins as substrates for DCAF16-based PROTACs. Moreover, we are interested in investigating other hit compounds identified in this screen for their potential as DCAF16 recruiters in PROTAC development. From a molecular glue perspective, another important direction for investigation is whether these DCAF16 ligands can alter the E3 surface and promote the degradation of neo-substrates. Finally, using the HTRF assay of the DCAF16–SPIN4 interaction to screen additional compound libraries could uncover additional DCAF16 ligands, therefore expanding the toolbox in the TPD field.
Methods
Reagents
The anti-HA (clone C29F4, cat. 3724, dilution 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5000) and horse radish peroxidase (HRP)-link anti-heat-shock protein 90 (HSP90; clone C45G5, cat. 79641, dilution 1:5000) antibodies were purchased from Cell Signaling Technologies. KB02-2-SLF (cat. 914738), anti-FLAG affinity gel (clone M2, cat. A2220), and anti-FLAG HRP antibody (clone M2, cat. A8592, dilution 1:5000) were purchased from Sigma-Aldrich. The anti-β-actin (clone C4, cat. Sc-47778, dilution 1:5000) antibody was purchased from Santa Cruz Biotechnology. HTRF MAb anti-HA-d2 (cat. 610HADAA), HTRF MAb anti-HA-Tb cryptate (cat. 610HATAF), HTRF MAb anti-FLAG-d2 (cat. 61FGBDLF), and HTRF MAb anti-FLAG-Tb (cat. 61FGBTLF) were purchased from Revvity. The antibiotics blasticidin (cat. Ant-bl-05) and puromycin (cat. Ant-pr-1) were purchased from InvivoGen. MG132 (cat. S2619) was purchased from Selleck Chemicals. MLN4924 (cat. 15217) and SLF (cat. 10007974) were purchased from Cayman Chemical. Polyethylenimine (molecular weight 40000, cat. 24765-1) was purchased from Polysciences. Enzyme-linked chemiluminescence (ECL) (cat. 32106), ECL plus (cat. 32132), tandem mass tag (TMT) isobaric label reagent (cat. 90110), and streptavidin agarose (cat. 20349) were purchased from Thermo Fisher Scientific. FuGene6 (cat. E2692) transfection reagent and sequencing-grade modified trypsin (cat. V5111) were purchased from Promega.
:5000) and horse radish peroxidase (HRP)-link anti-heat-shock protein 90 (HSP90; clone C45G5, cat. 79641, dilution 1:5000) antibodies were purchased from Cell Signaling Technologies. KB02-2-SLF (cat. 914738), anti-FLAG affinity gel (clone M2, cat. A2220), and anti-FLAG HRP antibody (clone M2, cat. A8592, dilution 1:5000) were purchased from Sigma-Aldrich. The anti-β-actin (clone C4, cat. Sc-47778, dilution 1:5000) antibody was purchased from Santa Cruz Biotechnology. HTRF MAb anti-HA-d2 (cat. 610HADAA), HTRF MAb anti-HA-Tb cryptate (cat. 610HATAF), HTRF MAb anti-FLAG-d2 (cat. 61FGBDLF), and HTRF MAb anti-FLAG-Tb (cat. 61FGBTLF) were purchased from Revvity. The antibiotics blasticidin (cat. Ant-bl-05) and puromycin (cat. Ant-pr-1) were purchased from InvivoGen. MG132 (cat. S2619) was purchased from Selleck Chemicals. MLN4924 (cat. 15217) and SLF (cat. 10007974) were purchased from Cayman Chemical. Polyethylenimine (molecular weight 40000, cat. 24765-1) was purchased from Polysciences. Enzyme-linked chemiluminescence (ECL) (cat. 32106), ECL plus (cat. 32132), tandem mass tag (TMT) isobaric label reagent (cat. 90110), and streptavidin agarose (cat. 20349) were purchased from Thermo Fisher Scientific. FuGene6 (cat. E2692) transfection reagent and sequencing-grade modified trypsin (cat. V5111) were purchased from Promega.
Cell lines
Human embryonic kidney (HEK) 293 T cells and MDA-MB-231 cells were purchased from American Type Culture Collection. Both cells were cultured in DMEM (Corning) supplemented with 10% (v/v) fetal bovine serum (Omega Scientific), 2 mM (v/v) L-glutamine (Gibco), and 100 units per mL of 1:1 penicillin/streptomycin (Gibco). All cell lines tested negative for mycoplasma contamination.
Cloning and mutagenesis
Human FKBP12 with an N-terminal FLAG-tag and C-terminal nuclear localization sequence (NLS, PKKKRKV) was cloned into pCDH-CMV-MCS-EF1-Puro vector using EcoRI and BamHI sites. Human DCAF16 with an N-terminal HA-tag was cloned into pCDH-CMV-MCS-EF1-Blast vector using EcoRI and NotI sites.Generation of FKBP12 and DCAF16 stably expressed cells by lentivirus transduction
FLAG-FKBP12 and HA-DCAF16 containing lentivirus were generated by co-transfection of FLAG-FKBP12/HA-DCAF16, psPAX2, and pMD2.G into HEK293T cells using FuGene 6 transfection reagent. The lentivirus was filtered using a 0.45 μM Millex-HV sterile syringe filter unit (MilliporeSigma, cat. SLHV013SL) and used to transduce HEK293T cells in the presence of 10 μg mL−1 polybrene. Stably expressed cells were selected 48 hours after transduction by the addition of 2 μg mL−1 puromycin or 10 μg mL−1 blasticidin for 7 days.Generation of DCAF16 knockout in MDA-MB-231 cells
To generate MDA-MB-231 cells with DCAF16 knockout, a 4D-Nucleofector (Lonza Bioscience) was used for the electroporation of Cas9–sgRNA ribonucleoprotein (RNP) complex. The DCAF16 targeting sgRNA (TCTGACAAGTGGTCAGGAGA) was used for forming the RNP complex. To confirm the knockout of DCAF16, genomic DNA was extracted using a PureLink genomic DNA mini kit (Thermo Fisher Scientific). The PCR amplified DCAF16 gene was confirmed by DNA Sanger sequencing. The sequencing primers for DCAF16 were AATTGCAGTGCTCCATTTAGAGTG (forward) and GCCACAAGCATACTTTATGTGTGT (reverse).Homogenous time resolved fluorescence high throughput assay
In a Corning white 384 well plate, 2 × 104 cells were plated in 40 μL of DMEM media. The cells were incubated at 37 °C for 24 hours. The compounds were dispensed at 5 μM with DMSO added to the wells and co-treated with 5 μM MG132. The compounds were dispensed by Echo Liquid Handler and incubated for 1 hour at 37 °C. The media was removed and 10 μL of 1% Triton NP-40 prepared in HTRF buffer was added to all wells and incubated at room temperature for 30 minutes. 10 μL of diluted anti-FLAG-M2-Tb (100×) and anti-HA-d2 (62.5×) was added to each well and incubated at room temperature for 1 hour. The fluorescence signal was measured on a Perkin Elmer M1000 Infinite Plate reader with 620 nm (emission bandwidth 10 nm; excitation 340, bandwidth 20 nm; manual gain 220; Z-position manual 25000 μm) and 665 nm (emission bandwidth 10 nm; excitation 340, bandwidth 20 nm; manual gain 245; Z-position manual 25000 μm) with a lag time of 60 μs. The averaged fluorescence across the control values per wavelength were subtracted from each sample fluorescence. The resulting sample signal at 665 nm was then divided by signal at 620 nm.
Compound validation using homogenous time resolved fluorescence assay
In a clear Corning 12-well plate, 1 × 105 cells were plated in 1 mL of DMEM media and incubated at 37 °C for 24 hours. The cells were co-treated with 1–10 μM of compound and 5 μM MG132 at 37 °C for 2 hours. Cell pellets were collected and lysed in 60 μL of PBS with 0.1% BSA (Sigma Aldrich, cat. 03117057001) and sonicated at 40% intensity for three rounds of ten pulses. 10 μL of the lysates were added to white Cisbio HTRF 96-well low volume plates (cat. 66PL96025). 10 μL of diluted anti-FLAG-M2-Tb (100×) and anti-HA-d2 (62.5×) was added to each well and incubated at room temperature for 1 hour. The fluorescence signal was measured on a CLARIOstar Plus microplate reader (BMG LABTECH) at 620 nm and 665 nm (excitation Ex TR; gain 2400) with a lag time of 60 μs. The averaged fluorescence across the control values per wavelength were subtracted from each sample fluorescence. The resulting sample signal at 665 nm was then divided by signal at 620 nm.Cell lysis and western blotting
The cells were lysed in radioimmunoprecipitation lysis buffer (RIPA, Thermo Fisher Scientific, cat. 89900) containing 25 mM Tris–HCl pH 7.6, 150 mM NaCl, 1% sodium deoxycholate, 0.1% SDS and supplemented with cOmplete protease inhibitor cocktail (MilliporeSigma, cat. 11873580001). The suspension mixture was sonicated for five cycles with four pulses per cycle at 40% power. The cell lysis was then centrifuged at 16000g at 4 °C for 10 minutes. The protein concentration was determined by the BCA assay (Thermo Fisher Scientific, cat. 23225). The normalized protein lysate was then combined with Laemmli Sample Buffer (Bio-Rad, cat. 1610737EDU) and heated at 95 °C for 5 minutes. The proteins were separated using 4–20% Novex Tris-Glycine mini gels (Thermo Fisher Scientific, cat. XP04205BOX). The proteins were transferred onto a 0.2 μM polyvinylidene fluoride (PVDF) membrane (Bio-Rad, cat. 1620177) and incubated with a solution of 5% nonfat milk in TBST buffer (0.1% Tween 20, 20 mM Tris–HCl pH 7.6, and 150 mM NaCl) at room temperature for 1 hour. Antibodies were diluted in 5% nonfat milk in TBST buffer and applied to the membrane at a dilution of 1:5000. After incubation, the membrane was washed three times with TBST buffer. The chemiluminescence signal on the membrane was developed using ECL western blotting detection reagent. The signal was captured using ChemiDoc MP (Bio-Rad). Band intensities were quantified using ImageJ (version 1.54j).
Immunoprecipitation of FLAG-FKBP12_NLS
A solution of NP-40 lysis buffer (25 mM Tris–HCl pH 7.4, 150 mM NaCl, 10% glycerol, and 1% Nonidet P-40) was supplemented with cOmplete protease inhibitor cocktail for the suspension and lysis of cells. The suspension was incubated on ice for 10 minutes before centrifugation at 16000g at 4 °C for 10 minutes. The resulting supernatant was collected for immunoprecipitation. For immunoprecipitation, 25 μL of FLAG affinity gel slurry per sample was added to the collected protein lysates and rotated at 4 °C for 2 hours. The affinity gel was than washed 4 times with immunoprecipitation washing buffer (0.2% NP-40, 25 mM Tris–HCl pH 7.4, and 150 mM NaCl). The resulting solution was then mixed with Laemmli sample buffer and heated at 95 °C for 10 minutes. The resulting supernatant was collected and used for western blot analysis.
Mass spectrometry-based global proteomic analysis
Cell pellets were lysed in 100 μL of PBS through sonication (three rounds, eight pulses at 40% intensity). Protein concentration was determined by the DC assay (Bio-Rad, cat. 5000112) and adjusted to 1 mg mL−1. 100 μL of lysates containing 100 mg proteasome was denatured in 8 M urea. The proteins were then reduced with 5 μL of 200 mM DTT (in water) at 65 °C for 15 minutes. Alkylation was performed by the addition of 5 μL of 400 mM iodoacetamide (in water) in the dark at 37 °C for 30 minutes. 300 μL of PBS was then added to the alkylated sample. To achieve digestion, 2 μg of trypsin was added, and the solution was incubated for 12 hours at 37 °C. For TMT labeling, approximately 8.5 μg of each peptide solution in 35 μL was incubated with 9 μL of CH3CN and 5 μL of TMT tags. TMT labeling was conducted at room temperature for 1 hour. The TMT labeling was quenched by the addition of 6 μL of 5% hydroxylamine and incubated at room temperature for 15 min. 2.5 μL of formic acid was added to each sample before pooling. The desalted peptides were separated and collected in 12 distinct fractions using the Thermo Vanquish UHPLC fractionator. The peptide fractions were then analyzed by liquid chromatography–mass spectrometry (LC–MS) using an Orbitrap Eclipse Tribrid MS coupled with Vanquish Neo UHPLC System. The peptides were introduced to the EASY-Spray HPLC column (C18, 2 μm particle size, 75 μm inner diameter and 250 mm length) and eluted at a 0.25 μL min−1 flow rate with the gradient: 5% buffer B (80% acetonitrile with 0.1% formic acid) in buffer A (water with 0.1% formic acid) from 0 to 15 min, 5% to 45% buffer B from 15–155 min and 45% to 100% buffer B from 155–180 min. The voltage of the nano-LC electrospray ionization source was set to 1.5 kV. The analysis started with a MS1 master scan (Orbitrap analysis; resolution, 120000; m/z range 375–1600; RF lens 30%; standard automatic again control (AGC) target; auto maximum injection time). In the MS2 analysis, initial precursor ions were isolated by the quadrupole with an isolation window of 0.7 and then subjected to higher-energy collisional dissociation (HCD) in the ion trap (stand AGC; collision energy 30%; maximum injection time, 35 ms). After each MS2 spectrum, synchronous precursor selection (SPS) chose up to ten MS2 fragment ions for MS3 analysis. These precursors were fragmented by HCD and analyzed by the Orbitrap (collision energy, 55%; AGC, 250%; maximum injection time, 200 ms; resolution, 50000). The raw data was collected using Xcalibur (version 4.5.445.18) and analyzed in Proteome Discoverer 2.5.
Activity-based protein profiling of cysteine reactivity
Cell pellets were lysed in 500 μL of PBS through sonication (three rounds, eight pulses at 40% intensity). Protein concentration was determined by the DC assay and adjusted to 1 mg mL−1. 500 μL of the normalized lysate was labeled with 100 μM of iodoacetamide alkyne (IA-alkyne) or desthiobiotin iodoacetamide (DBIA) at room temperature for 1 hour. For IA-alkyne labeling, a copper(I)-catalyzed alkyne–azide cycloaddition was performed following a previously reported method.13 Protein clean-up was performed by adding 100 μL of 1:1 hydrophobic and hydrophilic Sera-Mag SpeedBeads per sample. The cell lysates were incubated with the beads at room temperature for 5 minutes, with rotation at 1000 rpm in a thermomixer. The lysate-bead mixture was then incubated with 1 mL of ethanol at room temperature for 5 minutes, with rotation at 1000 rpm in a thermomixer. The supernatant was removed after beads had settled using the DynaMag-2 magnet. Resuspend the beads in 1 mL 80% ethanol and wash for a total of three times. Resuspend the beads in 500 μL of 2 M urea. Add 25 μL of 200 mM DTT and incubate at 65 °C for 15 minutes. Subsequently, 25 μL of 400 mM iodoacetamide was added and incubated with the lysate-bead mixture at 37 °C for 30 minutes. The beads were then washed in 1 mL of 80% ethanol three times, resuspended in 200 μL of PBS and digested with 2 μg of trypsin at 37 °C for 12 hours. After digestion, the supernatant was collected and incubated with 300 μL of wash buffer (50 mM TEAB, 150 mM NaCl, 0.2% NP-40) containing 50 μL of streptavidin agarose. The streptavidin–peptide mixture was rotated at room temperature for 2 hours. The beads were washed three times with 1 mL wash buffer, three times with 1 mL PBS, and three times with 1 mL HPLC water in a pre-washed filter spin column. Peptides were eluted from the beads by adding 300 μL of 50% acetonitrile with 0.1% formic acid. The eluted peptides were dried with a SpeedVac vacuum concentrator (Thermo Fisher Scientific). The subsequent steps for TMT labeling and LC–MS analysis were carried out as described above. The raw data was collected using Xcalibur (version 4.5.445.18) and analyzed in Proteome Discoverer 2.5.
FLAG-FKBP12_NLS interactome
A solution of NP-40 lysis buffer (25 mM Tris–HCl pH 7.4, 150 mM NaCl, 10% glycerol, and 1% Nonidet P-40) was supplemented with cOmplete protease inhibitor cocktail for the suspension and lysis of cells. The suspension was incubated on ice for 10 minutes, then centrifuged at 16000g at 4 °C for 10 minutes. The resulting supernatant was collected for immunoprecipitation. 25 μL of FLAG affinity gel slurry per sample were mixed with the protein lysates and rotated at 4 °C for 2 hours. The affinity gel was washed 4 times with immunoprecipitation washing buffer, followed by two washes with PBS. FLAG-FKBP12_NLS and associated interacting proteins were eluted by adding 8 M urea dissolved in PBS and heating at 65 °C for 10 minutes. The eluted proteins were reduced with 12.5 mM DTT at 65 °C for 15 minutes. The proteins were alkylated with 25 mM iodoacetamide at 37 °C for 12 hours. The protein solution was diluted with PBS to a final concentration of 2 M urea and then digested with 2 μg of trypsin at 37 °C for 12 hours. Subsequently, 6 μL of TMT tags were added, and the reaction was allowed to proceed at room temperature for 1 hour. The reaction was quenched by adding 6 μL of 5% hydroxylamine solution and 2.5 μL of formic acid. The samples were pooled and subjected to desalting using Sep-Pak C18 cartridge (Waters, cat. WAT054955). The eluted solution was dried with a SpeedVac vacuum concentrator. Peptides were analyzed by LC–MS using the described above. The raw data was collected using Xcalibur (version 4.5.445.18) and analyzed in Proteome Discoverer 2.5.
Cell viability assay
Cells were seeded in a 96-well clear-bottom white plate (Corning) at a density of 10000 cells per well in 100 μL of DMEM complete medium and incubated for 24 hours. After incubation, the cells were treated with various concentrations of 2G07 or 2F07 in 100 μL of DMEM complete medium for an additional 72 hours. Following treatment, 50 μL of CellTiter-Glo reagent (Promega) was added to each well and incubated for 10 minutes at room temperature. Luminescence was then measured using a CLARIOstar Plus microplate reader (BMG Labtech).
Statistical analysis
Quantitative data were depicted using scatter plots, displaying the mean accompanied by the standard error of the mean (SEM) represented as error bars. Differences between two groups were assessed using an unpaired two-tailed Student's t-test. Significance levels were denoted as follows: *P < 0.05, **P < 0.01 and ***P < 0.001. Statistical significance was defined for P values < 0.05.Synthetic procedures
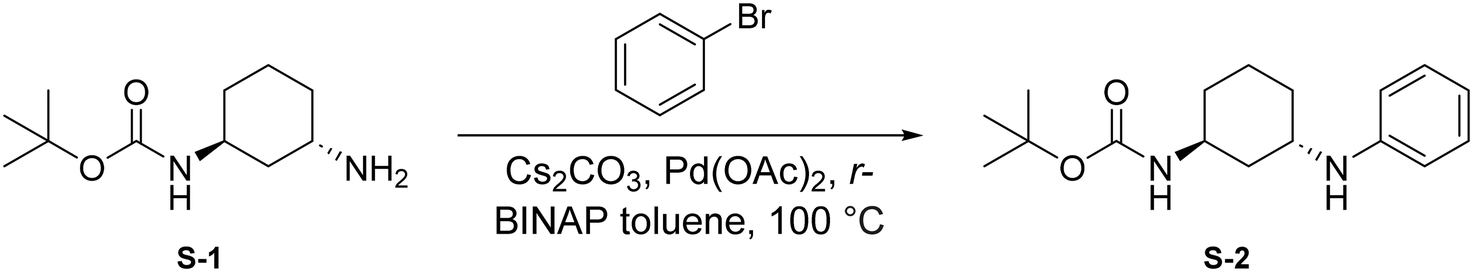
Synthesis of 2G07
To a round bottom flask was dissolved S-1 (250 mg, 1.17 mmol, 1.0 eq.), bromobenzene (184 μL, 1.75 mmol, 1.5 eq.), Cs2CO3 (760 mg, 2.33 mmol, 2 eq.), Pd(OAc)2 (52.4 mg, 0.233 mmol, 0.2 eq.), and r-BINAP (145 mg, 0.233 mmol, 0.2 eq.) in dry toluene (4 mL). The combined reaction mixture was purged three times with N2 and was allowed to stir at 100 °C for 18 hours under N2 atmosphere. Upon completion, the reaction mixture was filtered through Celite using EtOAc (20 mL) and was concentrated under reduced pressure. The crude mixture was then purified by flash chromatography (EtOAc in hexanes; 20%) to afford S-2 as a light yellow solid (287.8 mg, 0.991 mmol, 85%).
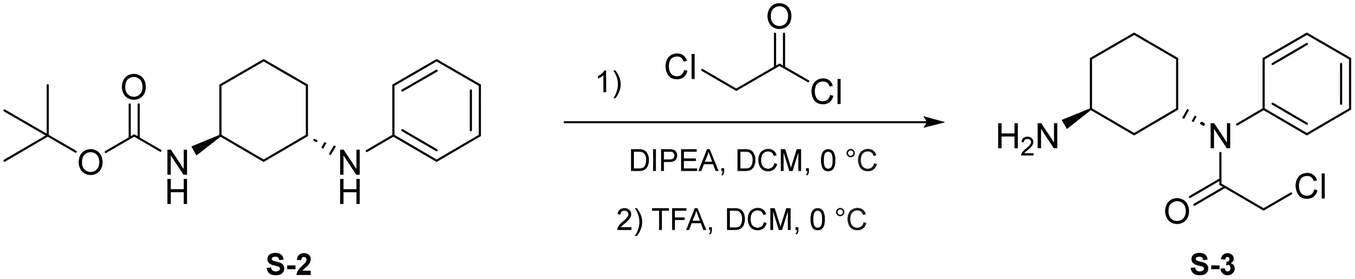
Step 1: to a round bottom flask was dissolved S-2 (250 mg, 0.861 mmol, 1.0 eq.) and DIPEA (225 μL, 1.29 mmol, 1.5 eq.) in anhydrous DCM (3 mL). After allowing to stir for 15 minutes at 0 °C, chloroacetyl chloride was added (137 μL, 1.72 mmol, 2 eq.) and the resulting mixture was allowed to stir at 0 °C for 1 hour under N2 atmosphere. Upon completion, the reaction mixture was quenched with saturated NH4Cl and washed with brine (10 mL). The crude product was extracted with DCM (3 × 5 mL). The combined organic layers were dried with Na2SO4, concentrated under reduced pressure, and the resulting light brown solid was used without further purification.
Step 2: to a round bottom flask was dissolved the above light brown solid and TFA (1.5 mL) in DCM (1.5 mL). The resulting mixture was then allowed to stir at 0 °C for 2 hours. Upon completion, the mixture was quenched with MeOH (5 × 10 mL) and concentrated to yield a light yellow solid (S-3) that was used without further purification.
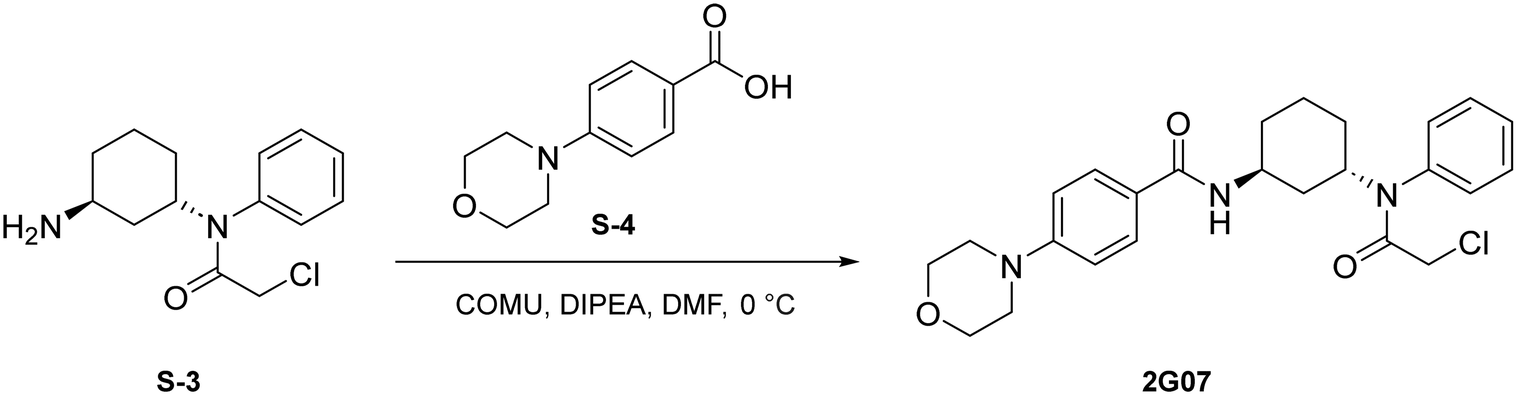
To a round bottom flask was dissolved S-4 (214 mg, 1.03 mmol, 1.2 eq.), COMU (443 mg, 1.03 mmol, 1.2 eq.), and DIPEA (451 μL, 2.59 mmol, 3 eq.) in DMF (2.5 mL). After allowing to stir at 0 °C for 30 minutes, S-3 (230 mg, 0.862 mmol, 1.0 eq.) was added. The resulting mixture was allowed to stir at 0 °C for 1 hour. Upon completion, the reaction was quenched with saturated NH4Cl and washed with brine (1 × 10 mL). The crude product was extracted with DCM (3 × 5 mL). The combined organic layers were washed with water (3 × 10 mL), dried with Na2SO4, and concentrated under reduced pressure. A portion of resulting red-orange residue (∼100 mg) was purified via preparatory TLC to yield 2G07 as a light yellow solid (29.9 mg, 0.0657 mmol, 62%).
1H NMR (500 MHz, CDCl3) δ 7.73 (d, J = 8.5 Hz, 2H), 7.49–7.42 (m, 3H), 7.18–7.12 (m, 2H), 6.89 (d, J = 8.5 Hz, 2H), 6.37 (d, J = 7.4 Hz, 1H), 4.89–4.81 (m, 1H), 4.47–4.36 (m, 1H), 3.87 (t, J = 4.4 Hz, 4H), 3.70 (s, 2H), 3.25 (t, J = 4.4 Hz, 4H), 2.09–2.02 (m, 1H), 2.00–1.92 (m, 1H), 1.90–1.82 (m, 1H), 1.76–1.68 (m, 1H), 1.68–1.56 (m, 1H), 1.39–1.29 (m, 2H), 1.19–1.09 (m, 1H).
13C-NMR (126 MHz, CDCl3) δ 166.87, 166.05, 153.55, 137.17, 130.38, 130.15, 129.94, 129.82, 129.47, 128.56, 125.12, 114.30, 66.80, 50.71, 48.29, 45.86, 42.67, 35.20, 30.84, 28.98, 20.74.
HRMS (ESI+) m/z calcd for C25H31ClN3O3+ [M + H]+: 456.2048, found 456.2082.
Synthesis of 2F07
To a round bottom flask was dissolved S-5 (250 mg, 1.17 mmol, 1.0 eq.), bromobenzene (184 μL, 1.75 mmol, 1.5 eq.), Cs2CO3 (760 mg, 2.33 mmol, 2 eq.), Pd(OAc)2 (52.4 mg, 0.233 mmol, 0.2 eq.), and r-BINAP (145 mg, 0.233 mmol, 0.2 eq.) in dry toluene (4 mL). The combined reaction mixture was purged three times with N2 and was allowed to stir at 100 °C for 18 hours under N2 atmosphere. Upon completion, the reaction mixture was filtered through celite using EtOAc (20 mL) and was concentrated under reduced pressure. The crude mixture was then purified by flash chromatography (EtOAc in Hexanes; 20%) to afford S-6 as a light yellow solid (165.5 mg, 0.570 mmol, 49%).
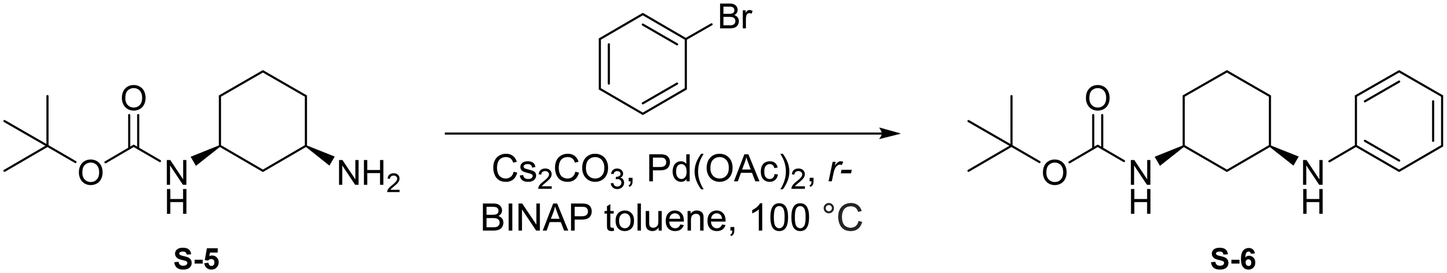
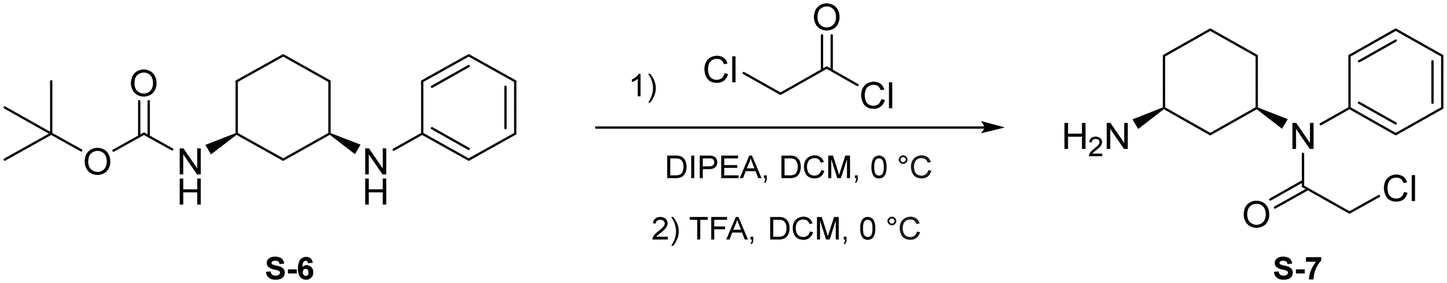
Step 1: to a round bottom flask was dissolved S-6 (144 mg, 0.496 mmol, 1.0 eq.) and DIPEA (129 μL, 0.743 mmol, 1.5 eq.) in anhydrous DCM (2 mL). After allowing to stir for 15 minutes at 0 °C, chloroacetyl chloride was added (78.8 μL, 0.991 mmol, 2 eq.) and the resulting mixture was allowed to stir at 0 °C for 1 hour under N2 atmosphere. Upon completion, the reaction mixture was quenched with saturated NH4Cl and washed with brine (10 mL). The crude product was extracted with DCM (3 × 5 mL). The combined organic layers were dried with Na2SO4, concentrated under reduced pressure, and the resulting light brown solid was used without further purification.
Step 2: to a round bottom flask was dissolved the above light brown solid and TFA (1 mL) in DCM (1 mL). The resulting mixture was then allowed to stir at 0 °C for 2 hours. Upon completion, the mixture was quenched with MeOH (5 × 10 mL) and concentrated to yield a light yellow solid (S-7) that was used without further purification.
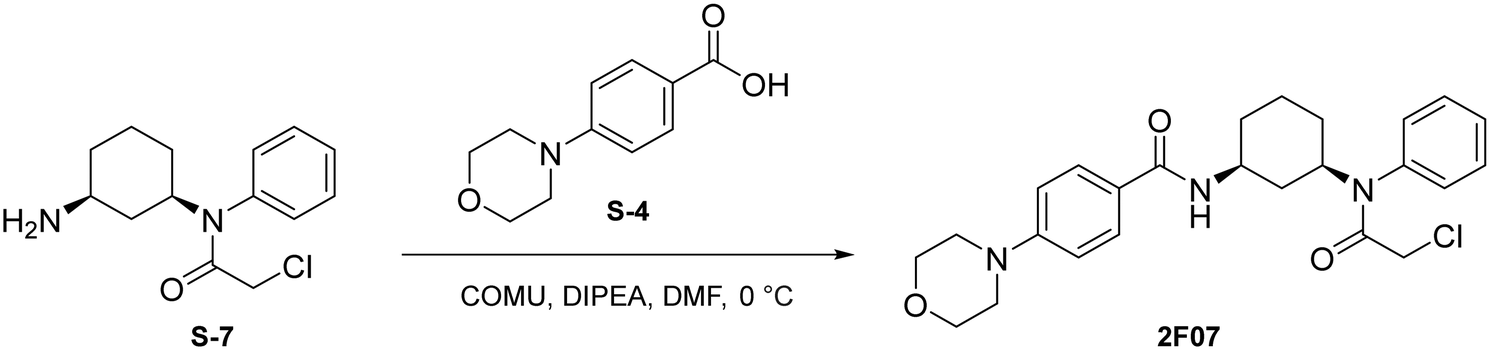
To a round bottom flask was dissolved S-4 (123 mg, 0.594 mmol, 1.2 eq.), COMU (255 mg, 0.595 mmol, 1.2 eq.), and DIPEA (259 μL, 1.487 mmol, 3 eq.) in DMF (2.5 mL). After allowing to stir at 0 °C for 30 minutes, S-7 (132 mg, 0.495 mmol, 1.0 eq.) was added. The resulting mixture was allowed to stir at 0 °C for 1 hour. Upon completion, the reaction was quenched with saturated NH4Cl and washed with brine (1 × 10 mL). The crude product was extracted with DCM (3 × 5 mL). The combined organic layers were washed with water (3 × 10 mL), dried with Na2SO4, and concentrated under reduced pressure. A portion of resulting red-orange residue (∼70 mg) was purified via preparatory TLC to yield 2F07 as a light yellow solid (14.9 mg, 0.0327 mmol, 37%).
1H NMR (500 MHz, CDCl3) δ 7.73 (d, J = 9.0 Hz, 2H), 7.48–7.41 (m, 3H), 7.19–7.14 (m, 2H), 6.85 (d, J = 8.3 Hz, 2H), 5.75 (d, J = 7.9 Hz, 1H), 4.67–4.58 (m, 1H), 4.13–4.03 (m, 1H), 3.84 (t, J = 4.9 Hz, 4H), 3.67 (s, 2H), 3.22 (t, J = 4.9 Hz, 4H), 2.27–2.21 (m, 1H), 2.03–1.90 (m, 2H), 1.85–1.78 (m, 1H), 1.58–1.47 (m, 1H), 1.11–0.85 (m, 3H).
13C-NMR (126 MHz, CDCl3) δ 166.33, 165.72, 153.46, 138.08, 130.21, 130.07, 129.83, 129.25, 128.39, 125.00, 114.17, 66.77, 54.28, 48.24, 48.18, 42.59, 37.75, 32.32, 30.10, 23.18.
HRMS (ESI+) m/z calcd for C25H31ClN3O3+ [M + H]+: 456.2048, found 456.2080.
Synthesis of 2G07-SLF
Step 1: HATU (161 mg, 0.42 mmol, 1.2 eq.) was added to a stirred solution of S-8 (200 mg, 0.35 mmol, 1.0 eq.) and S-9 (159 mg, 0.70 mmol, 2.0 eq.) in DCM (5 mL) at room temperature. DIPEA (174 μL, 1.05 mmol, 3.0 eq.) was added to the reaction and stirred for 4 h. Upon completion, the reaction was diluted with EtOAc (30 mL) and washed with saturated NaHCO3 (1 × 30 mL), and brine (1 × 30 mL). The organic layer was dried by Na2SO4, concentrated, and the resulting residue was used without further purification.
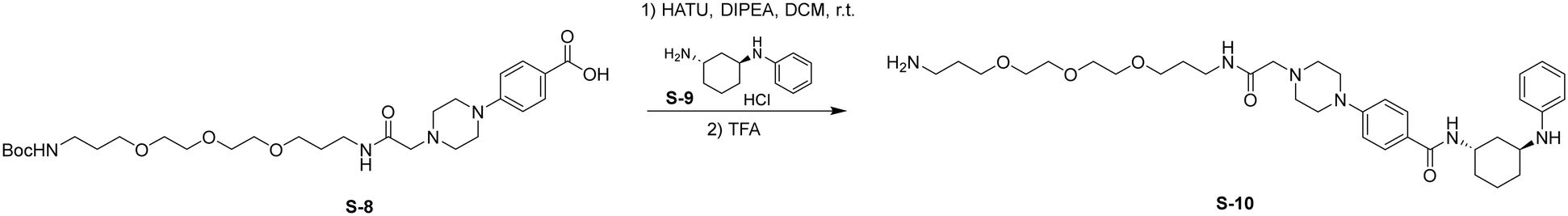
Step 2: TFA (1 mL) was added to a stirred solution of the above residue in DCM (5 mL) and the reaction was stirred for 2 h. The mixture was then concentrated, and the residue was purified by flash chromatography to provide S-10 as a colorless oil (145 mg, 65% over 2 steps).
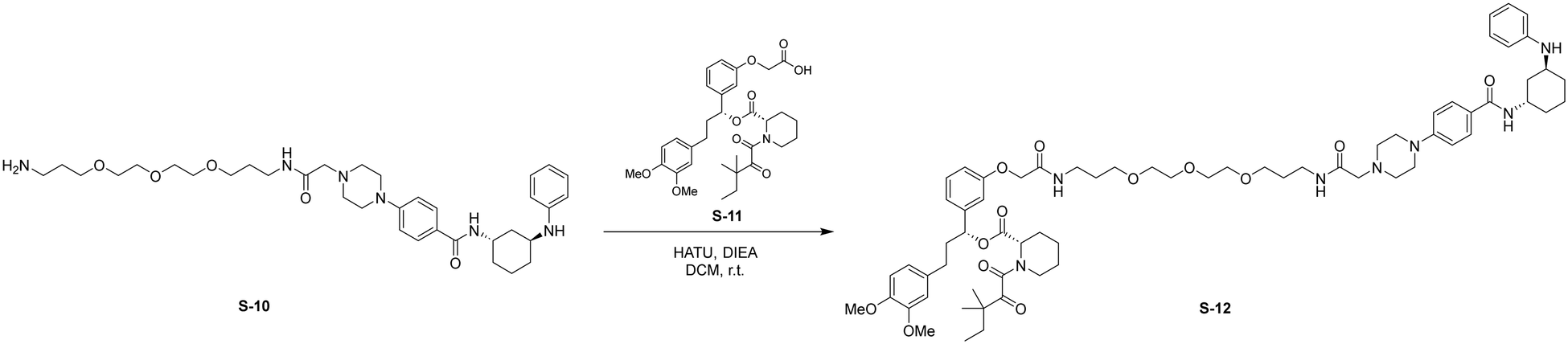
HATU (24 mg, 0.062 mmol, 1.2 eq.) was added to a stirred solution of S-10 (50 mg, 0.078 mmol, 1.5 eq.) and S-11 (30 mg, 0.052 mmol, 1.0 eq.) in DCM (5 mL) at room temperature. DIPEA (26 μL, 0.16 mmol, 3.0 eq.) was added to the reaction and stirred for 2 h. Upon completion, the reaction was diluted with EtOAc (30 mL) and washed with brine (1 × 30 mL). The organic layer was dried by Na2SO4, concentrated, and the resulting residue was purified by flash chromatography to provide S-12 as a white solid (10 mg, 0.0083 mmol, 16%).
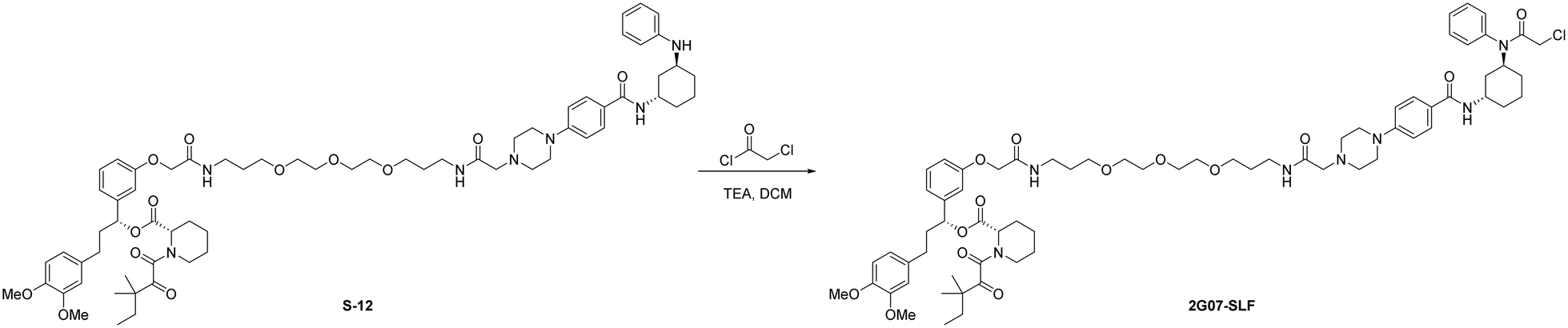
Add chloroacetyl chloride (2.0 μL, 0.025 mmol, 3.0 eq.) to a solution of S-12 (10 mg, 0.0083 mmol, 1.0 eq.) and triethylamine (3.5 μL, 0.025 mmol, 3.0 eq.) in anhydrous DCM (1.0 mL) at 0 °C under N2 atmosphere. Stir the mixture at room temperature for 5 hours. Upon completion, the reaction was diluted with DCM (10 mL) and washed with brine (1 × 10 mL). The organic layer was dried by Na2SO4, concentrated, and the resulting residue was purified by flash chromatography to provide 2G07-SLF as a white solid (9.1 mg, 0.0071 mmol, 86%).
1H NMR (400 MHz, CDCl3) δ 7.74 (d, J = 8.8 Hz, 2H), 7.48–7.46 (m, 3H), 7.33–7.31 (m, 1H), 7.16–7.13 (m, 2H), 7.00–6.99 (m, 1H), 6.94 (s, 1H), 6.90 (d, J = 8.8 Hz, 2H), 6.88–6.86 (m, 1H), 6.80–6.78 (m, 1H), 6.69–6.68 (m, 2H), 6.43 (d, J = 6.8 Hz, 1H), 5.79–5.76 (m, 1H), 5.31 (d, J = 4.8 Hz, 1H), 4.87 (s, 1H), 4.49 (s, 2H), 4.44 (s, 1H), 3.87–3.86 (m, 6H), 3.71 (s, 2H), 3.61–3.52 (m, 12H), 3.46–3.40 (m, 8H), 3.21–3.17 (m, 4H), 2.87 (br s, 2H), 2.60–2.55 (m, 2H), 2.38 (d, J = 12.4 Hz, 1H), 2.25–2.23 (m, 1H), 2.09–2.05 (m, 2H), 1.98–1.95 (m, 2H), 1.84–1.76 (m, 12H), 1.48–1.33 (m, 4H), 1.22 (d, J = 7.2 Hz, 6H), 1.17–1.12 (m, 2H), 0.89 (t, J = 7.2 Hz, 3H).
13C NMR (126 MHz, CDCl3) δ 208.04, 169.81, 168.18, 167.44, 166.66, 166.08, 165.76, 157.65, 149.05, 147.54, 142.00, 141.87, 138.10, 133.49, 130.17, 129.85, 129.27, 128.51, 120.29, 120.17, 114.97, 114.34, 113.52, 111.88, 111.48, 76.64, 70.62, 70.36, 70.34, 69.56, 67.60, 56.82, 56.07, 56.00, 54.31, 51.41, 48.24, 46.84, 44.30, 42.64, 38.98, 38.35, 38.14, 37.73, 37.26, 32.70, 32.61, 32.32, 31.40, 30.14, 27.85, 26.57, 25.08, 24.60, 23.70, 23.60, 23.32, 23.21, 21.34, 21.16, 8.91.
HRMS (ESI+) m/z calcd for C69H95ClN7O14+ [M + H]+: 1280.6620, found 1280.6634.
Data availability
The data supporting this article have been included in the ESI.† The mass spectrometry proteomics data were deposited to the ProteomeXchange Consortium through the PRIDE37 partner repository with the dataset identifier PXD055063.Author contributions
I. A. R. designed and conducted biochemical and cellular experiments to demonstrate target engagement and degradation; M. A. C. synthesized and characterized the compounds; C. Z. and I. A. R. conducted proteomics studies; X. J., F. W., and I. A. R. developed the HTRF assay; S. F. D. conducted the high-throughput screening and performed data analysis; B. F. C. provided the compound library and advised on hit selection; X. Z. supervised the project and, with contributions from all authors, wrote the manuscript.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
We gratefully acknowledge the support of the NIH R00 CA248715 (X. Z.), NSF GRFP (I. A. R.), and NIH T32 GM149439 (M. A. C.).References
- X. Huang and V. M. Dixit, Drugging the undruggables: exploring the ubiquitin system for drug development, Cell Res., 2016, 26(4), 484–498 CrossRef CAS.
- D. De Nardo, K. R. Balka, Y. Cardona Gloria, V. R. Rao, E. Latz and S. L. Masters, Interleukin-1 receptor-associated kinase 4 (IRAK4) plays a dual role in myddosome formation and Toll-like receptor signaling, J. Biol. Chem., 2018, 293(39), 15195–15207 CrossRef CAS.
- A. C. Lai and C. M. Crews, Induced protein degradation: an emerging drug discovery paradigm, Nat. Rev. Drug Discovery, 2017, 16(2), 101–114 CrossRef CAS.
- M. Békés, D. R. Langley and C. M. Crews, PROTAC targeted protein degraders: the past is prologue, Nat. Rev. Drug Discovery, 2022, 21(3), 181–200 CrossRef.
- S. L. Schreiber, Molecular glues and bifunctional compounds: Therapeutic modalities based on induced proximity, Cell Chem. Biol., 2024, 31(6), 1050–1063 CrossRef CAS PubMed.
- D. P. Bondeson, A. Mares, I. E. Smith, E. Ko, S. Campos and A. H. Miah, et al. Catalytic in vivo protein knockdown by small-molecule PROTACs, Nat. Chem. Biol., 2015, 11(8), 611–617 CrossRef CAS.
- A. Hanzl and G. E. Winter, Targeted protein degradation: current and future challenges, Curr. Opin. Chem. Biol., 2020, 56, 35–41 CrossRef CAS PubMed.
- M. Schapira, M. F. Calabrese, A. N. Bullock and C. M. Crews, Targeted protein degradation: expanding the toolbox, Nat. Rev. Drug Discovery, 2019, 18(12), 949–963 CrossRef CAS.
- H. T. Huang, D. Dobrovolsky, J. Paulk, G. Yang, E. L. Weisberg and Z. M. Doctor, et al. A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-kinase Degrader, Cell Chem. Biol., 2018, 25(1), 88–99 e6 CrossRef CAS PubMed.
- D. P. Bondeson, B. E. Smith, G. M. Burslem, A. D. Buhimschi, J. Hines and S. Jaime-Figueroa, et al. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead, Cell Chem. Biol., 2018, 25(1), 78–87 e5 CrossRef CAS.
- K. A. Donovan, F. M. Ferguson, J. W. Bushman, N. A. Eleuteri, D. Bhunia and S. Ryu, et al. Mapping the Degradable Kinome Provides a Resource for Expedited Degrader Development, Cell, 2020, 183(6), 1714–1731 e10 CrossRef CAS.
- B. P. Belcher, C. C. Ward and D. K. Nomura, Ligandability of E3 Ligases for Targeted Protein Degradation Applications, Biochemistry, 2023, 62(3), 588–600 CrossRef CAS.
- X. Zhang, V. M. Crowley, T. G. Wucherpfennig, M. M. Dix and B. F. Cravatt, Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16, Nat. Chem. Biol., 2019, 15(7), 737–746 CrossRef CAS PubMed.
- C. Pu, Y. Tong, Y. Liu, S. Lan, S. Wang and G. Yan, et al. Selective degradation of PARP2 by PROTACs via recruiting DCAF16 for triple-negative breast cancer, Eur. J. Med. Chem., 2022, 236, 114321 CrossRef CAS PubMed.
- C. Pu, Y. Liu, R. Deng, Q. Xu, S. Wang and H. Zhang, et al. Development of PROTAC degrader probe of CDK4/6 based on DCAF16, Bioorg. Chem., 2023, 138, 106637 CrossRef CAS PubMed.
- O. Hsia, M. Hinterndorfer, A. D. Cowan, K. Iso, T. Ishida and R. Sundaramoorthy, et al. Targeted protein degradation via intramolecular bivalent glues, Nature, 2024, 627(8002), 204–211 CrossRef CAS PubMed.
- Y. D. Li, M. W. Ma, M. M. Hassan, M. Hunkeler, M. Teng and K. Puvar, et al. Template-assisted covalent modification underlies activity of covalent molecular glues, Nat. Chem. Biol., 2024, 20, 1640–1649 CrossRef CAS PubMed.
- A. G. Shergalis, V. L. Marin, D. Y. Rhee, S. Senaweera, R. L. McCloud and J. A. Ronau, et al. CRISPR Screen Reveals BRD2/4 Molecular Glue-like Degrader via Recruitment of DCAF16, ACS Chem. Biol., 2023, 18(2), 331–339 CrossRef CAS.
- M. Lim, T. D. Cong, L. M. Orr, E. S. Toriki, A. C. Kile and J. W. Papatzimas, et al. DCAF16-Based Covalent Handle for the Rational Design of Monovalent Degraders, ACS Cent. Sci., 2024, 10(7), 1318–1331 CrossRef CAS PubMed.
- F. Degorce, A. Card, S. Soh, E. Trinquet, G. P. Knapik and B. Xie, HTRF: A technology tailored for drug discovery – a review of theoretical aspects and recent applications, Curr. Chem. Genomics, 2009, 3, 22–32 CrossRef CAS PubMed.
- S. Gul and K. Hadian, Protein-protein interaction modulator drug discovery: past efforts and future opportunities using a rich source of low- and high-throughput screening assays, Expert Opin. Drug Discovery, 2014, 9(12), 1393–13404 CrossRef CAS PubMed.
- X. Zhang, M. Thielert, H. Li and B. F. Cravatt, SPIN4 Is a Principal Endogenous Substrate of the E3 Ubiquitin Ligase DCAF16, Biochemistry, 2021, 60(9), 637–642 CrossRef CAS.
- V. Fagan, C. Johansson, C. Gileadi, O. Monteiro, J. E. Dunford and R. Nibhani, et al. A Chemical Probe for Tudor Domain Protein Spindlin1 to Investigate Chromatin Function, J. Med. Chem., 2019, 62(20), 9008–9025 CrossRef CAS.
- M. R. Lazear, J. R. Remsberg, M. G. Jaeger, K. Rothamel, H. L. Her and K. E. DeMeester, et al. Proteomic discovery of chemical probes that perturb protein complexes in human cells, Mol. Cell, 2023, 83(10), 1725–1742 e12 CrossRef CAS.
- Y. Wang, M. M. Dix, G. Bianco, J. R. Remsberg, H. Y. Lee and M. Kalocsay, et al. Expedited mapping of the ligandable proteome using fully functionalized enantiomeric probe pairs, Nat. Chem., 2019, 11(12), 1113–1123 CrossRef CAS.
- E. V. Vinogradova, X. Zhang, D. Remillard, D. C. Lazar, R. M. Suciu and Y. Wang, et al. An Activity-Guided Map of Electrophile-Cysteine Interactions in Primary Human T Cells, Cell, 2020, 182(4), 1009–1026 e29 CrossRef CAS PubMed.
- E. Njomen, R. E. Hayward, K. E. DeMeester, D. Ogasawara, M. M. Dix and T. Nguyen, et al. Multi-tiered chemical proteomic maps of tryptoline acrylamide-protein interactions in cancer cells, Nat. Chem., 2024, 16, 1592–1604 CrossRef CAS.
- X. Zhang and B. F. Cravatt, Chemical Proteomics-Guided Discovery of Covalent Ligands for Cancer Proteins, Annu. Rev. Cancer Biol., 2024, 8, 155–175 CrossRef.
- K. M. Backus, B. E. Correia, K. M. Lum, S. Forli, B. D. Horning and G. E. Gonzalez-Paez, et al. Proteome-wide covalent ligand discovery in native biological systems, Nature, 2016, 534(7608), 570–574 CrossRef CAS PubMed.
- E. Weerapana, C. Wang, G. M. Simon, F. Richter, S. Khare and M. B. Dillon, et al. Quantitative reactivity profiling predicts functional cysteines in proteomes, Nature, 2010, 468(7325), 790–795 CrossRef CAS PubMed.
- A. M. Roberts, C. C. Ward and D. K. Nomura, Activity-based protein profiling for mapping and pharmacologically interrogating proteome-wide ligandable hotspots, Curr. Opin. Biotechnol., 2017, 43, 25–33 CrossRef CAS.
- K. A. Scott, H. Kojima, N. Ropek, C. D. Warren, T. L. Zhang and S. J. Hogg, et al. Covalent Targeting of Splicing in T Cells, Cell Chem. Biol., 2025, 32, 1–18 Search PubMed.
- J. F. Amara, T. Clackson, V. M. Rivera, T. Guo, T. Keenan and S. Natesan, et al. A versatile synthetic dimerizer for the regulation of protein-protein interactions, Proc. Natl. Acad. Sci. U. S. A., 1997, 94(20), 10618–10623 CrossRef CAS.
- J. C. Peeler and E. Weerapana, Chemical Biology Approaches to Interrogate the Selenoproteome, Acc. Chem. Res., 2019, 52(10), 2832–2840 CrossRef CAS PubMed.
- A. A. Basu, C. Zhang, I. A. Riha, A. Magassa, M. A. Campos and A. G. Caldwell, et al. A CRISPR activation screen identifies FBXO22 supporting targeted protein degradation, Nat. Chem. Biol., 2024, 20, 1608–1616 CrossRef CAS.
- X. Zhang, L. M. Luukkonen, C. L. Eissler, V. M. Crowley, Y. Yamashita and M. A. Schafroth, et al. DCAF11 Supports Targeted Protein Degradation by Electrophilic Proteolysis-Targeting Chimeras, J. Am. Chem. Soc., 2021, 143(13), 5141–5149 CrossRef CAS PubMed.
- Y. Perez-Riverol, J. Bai, C. Bandla, D. Garcia-Seisdedos, S. Hewapathirana and S. Kamatchinathan, et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences, Nucleic Acids Res., 2022, 50(D1), D543–D552 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00681j |
| This journal is © The Royal Society of Chemistry 2025 |