
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProdrugs and their activation mechanisms for brain drug delivery
Ida Aaberg
Lillethorup
,
Andreas Victor
Hemmingsen
and
Katrine
Qvortrup
 *
*
Department of Chemistry, Technical University of Denmark, 2800, Kongens Lyngby, Denmark. E-mail: kaqvo@kemi.dtu.dk
First published on 17th January 2025
Abstract
Prodrugs are masked drugs that first become pharmacologically active after undergoing a structural change in vivo. They are designed to improve physicochemical/biopharmaceutical drug properties and increase site specificity. The prodrug approach is important when developing brain-targeting drugs due to the presence of the brain barriers that seriously limit the brain entry of highly polar, multifunctional drug entities. While several excellent reviews summarize the structural modifications facilitating transport across the brain barriers, a summary of mechanisms used for the activation of the prodrug in the brain is missing. Given the high need for innovative discoveries in brain drug development, we here review the most important tools being developed since 2000 for CNS prodrug activation.
 Ida Aaberg Lillethorup | Ida Aaberg Lillethorup received her cand. scient. pharm. degree in 2021 having completed 5 years of study in pharmaceutical sciences focusing within medicinal chemistry at the University of Copenhagen. In 2022, she started her PhD studies at the Technical University of Denmark under the supervision of Assoc. Prof. Katrine Qvortrup, where her research focuses on developing linker strategies for brain-specific drug release and activation. |
 Andreas Victor Hemmingsen | Andreas Victor Hemmingsen earned his Master of Science in Engineering from the Technical University of Denmark in 2022, specializing in medicinal chemistry and organic synthesis. Later that year, he began his Ph.D. under the supervision of Associate Professor Katrine Qvortrup, focusing on the development of novel peptide–drug conjugates designed for targeted delivery to the central nervous system. |
 Katrine Qvortrup | Katrine Qvortrup completed her PhD at the University of Copenhagen (Denmark) in 2012. After a postdoctoral stay at Princeton University (USA) and Cambridge University (UK), she started her independent career in 2018 at the Technical University of Denmark. In the Qvortrup lab, they combine tools from chemistry and biology for the study and manipulation of biological systems. The aims are to develop novel (bio)chemical methods and to provide molecular-level insight into biochemical pathways. She is interested in understanding health and disease mechanisms and to utilize the knowledge in the development of new and improved treatment strategies. |
Introduction
Classical prodrugs are compounds modified with a so-called promoiety, where the compounds first become pharmacologically active when the promoiety is removed in vivo by either enzymatic or chemical reactions or a combination of the two.1 However, to date, other design approaches have been used, making it relevant to make a broader definition of prodrugs to be pharmacologically inactive chemical compounds that first become pharmacologically active drugs after undergoing a structural change in vivo.2Prodrugs are used to overcome constraints with respect to drug absorption, distribution, metabolism, and excretion (ADME), hereby improving biopharmaceutical or pharmacokinetic performance. Thus, prodrugs are “masked” compounds, by which unfavourable physicochemical and/or pharmacokinetic properties of drugs can be temporarily altered.3
While the prodrug concept was first formulated in 1958,4 the first prodrugs can be dated back to 1899. Early prodrugs include the antibacterial prodrug methenamine (or hexamine) and the anti-inflammatory prodrug aspirin (acetylsalicylic acid), launched by Schering and Bauer, respectively. Since then, the prodrug concept has been well utilized, accounting for approximately 12% of new small molecular entities approved by the U.S. Food and Drug Administration (FDA).1
Nevertheless, the design and development of prodrugs is not simple. An efficient prodrug must be stable in circulation to avoid off-target toxicity but show an optimal conversion rate to liberate the active parent drug in an efficient and/or controlled manner when reaching the desirable site(s) of conversion. Several factors must be fulfilled, including adequate aqueous solubility of both the prodrug and drug. In addition, the prodrug and the promoiety must be safe and show no undesirable pharmacological effects and rapid excretion from the body.
Prodrugs targeting the brain
Brain diseases are poorly treated diseases. In addition to the complexity of brain diseases, the brain barriers are great hurdles. The limited permeability across the barriers seriously limits the number of drugs that can reach the brain. For a molecule to cross the blood–brain barrier (BBB) via lipid-mediated free diffusion, it must have a molecular weight of <400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Da and forms <8 hydrogen bonds, which are chemical properties lacking in most small molecule drugs. In fact, it is estimated that more than 98% of small-molecular weight drugs developed for CNS diseases do not readily cross the BBB.5,6
Da and forms <8 hydrogen bonds, which are chemical properties lacking in most small molecule drugs. In fact, it is estimated that more than 98% of small-molecular weight drugs developed for CNS diseases do not readily cross the BBB.5,6
The low brain barrier penetration and insufficient drug delivery into the brain lead to low drug efficacy, necessitating higher doses. Unfortunately, this may increase the risk of side effects, due to drug accumulation in other organs and tissues.7 The prodrug approach by transient chemical modification of the drug has been successfully employed to increase the brain delivery of poorly penetrating therapeutic agents. This includes masking the polar and ionized functionalities by lipophilic promoieties, hereby increasing hydrophobicity to allow free diffusion across the BBB. Alternatively, prodrugs that take advantage of carrier-mediated transport mechanisms offer intriguing targets in CNS drug design. Prodrugs modified with a promoiety that structurally resembles endogenous substrates can be carried across cell membranes by transporters responsible for intake of crucial polar endogenous nutrients.8,9
Common to the above prodrug structures is the need for rapid bioconversion back to the parent drug in the brain, by exploiting specific chemical conditions and/or enzymes. For a prodrug to be successful for the treatment of brain diseases, it is important that the active drug is only formed in the brain and not in peripheral circulation (Fig. 1).
 | ||
| Fig. 1 Graphical illustration of the prodrug design for brain disorders. Created in https://BioRender.com. | ||
While several excellent reviews summarize the structural modifications facilitating transport across the BBB, a summary of mechanisms used for the activation of the prodrug in the brain is missing. Given the high need for innovative discoveries in brain drug development, we here review the most important tools being developed since 2000 for CNS prodrug activation. We hope that this will stimulate scientists to develop novel and improved prodrug structures for better and selective treatment of CNS diseases.
Ester prodrugs
Many bioactive compounds contain polar alcohols, phenols or carboxylates required for high-affinity target engagement.10–12 These polar functionalities are challenging for passive diffusion across the BBB due to charge repulsion at the barrier surface and/or the unfavourable electrostatic energetics of the cellular membrane potential.13 (Bio)isosteric replacement of the anionic carboxylates can be investigated to improve BBB penetration, but this may also affect the intrinsic potency and selectivity, leading to lower efficacy and/or undesired and adverse drug effects.14–16Temporary masking of the polar carboxylate or hydroxy moiety by an ester prodrug strategy has been heavily used in brain drug delivery to introduce lipophilicity and improve passive crossing of the BBB.17–19 However, the use of the ester prodrug design in CNS drug delivery is compromised by the presence of plasma esterases and/or susceptible to chemical hydrolysis in the gastric environment, which may lower brain selectivity (Fig. 2).20
 | ||
| Fig. 2 The ester prodrug design is compromised by esterase in the periphery and/or unspecific hydrolysis. Created in https://BioRender.com. | ||
Enzymatic and chemical stability can be modulated by introducing a larger and/or branched alkyl ester,10,21–26 which at the same time can modulate the hydrophobicity of the prodrug and thereby provide a tool to increase their ability to passively cross the BBB (Fig. 3). For example, esters of the lipophilic tricyclodecane cage-shaped compound adamantane were found to substantially improve the BBB permeability of poorly absorbed drugs.24 The adamantane-esters undergo rapid enzymatic hydrolysis in the brain, leading to attainable brain concentrations.27
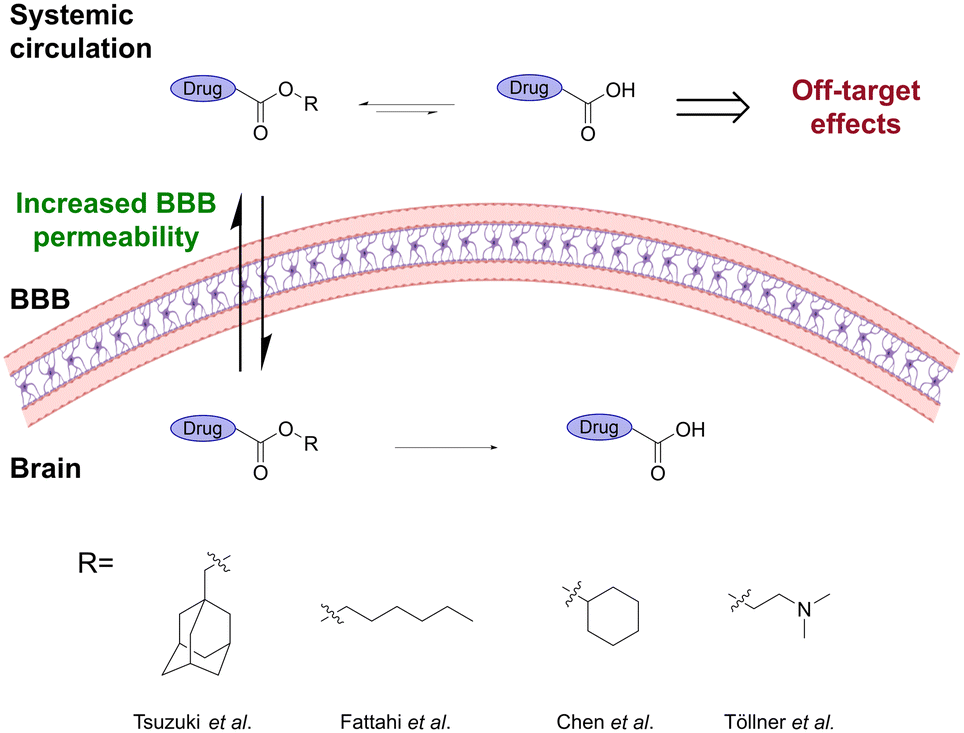 | ||
| Fig. 3 Ester prodrugs containing a larger and/or branched alkyl ester show better BBB penetration and lower unspecific hydrolysis. Inspired from ref 10 and 24–26. Created partly in https://BioRender.com. | ||
Methotrexate (MTX) is a widely used anticancer drug but has poor brain barrier penetration, limiting its application in the treatment of CNS cancers. Fattahi et al.25 synthesized various diester prodrugs of MTX, a hydrophilic anticancer drug, widely utilized in the treatment of autoimmune diseases and brain cancer. Their results showed that the larger dihexyl MTX ester decreased the unspecific hydrolysis of the prodrug, leading to a significantly higher brain:plasma ratio. This resulted in a 6-fold decrease in the IC50 value and a reduction in off-target effects. A related study showed that the highly polar ZL006, decorated with both phenolic hydroxyls, a secondary amine, and a carboxyl, has significantly higher permeability across the BBB and a longer duration time when the carboxyl group was esterified with cyclohexanol.26 Similarly, Töllner et al.10 showed that the pivaloyloxymethyl and N,N-dimethylaminoethyl ester prodrugs of bumetanide resulted in significantly higher brain levels of bumetanide than when administrating the parent drug.
Dual ester prodrugs have also been developed.28–30 Weitman et al.29 developed a dual prodrug, which, after hydrolysis in the brain, released benactyzine and GABA that acted synergistically to protect the CNS from organophosphate poisoning. Thatcher and coworkers30 explored the mutual prodrug design by studying a construct incorporating GABA-mimetic and NO-mimetic mechanisms connected through a carbamate. LC-MS/MS measurements were used to confirm BBB penetration, and its relevant hydrolytic metabolites were also observed in the brain.
Ester prodrugs have also been used to introduce a targeting moiety that recognizes a BBB-expressed carrier. Transporters that facilitate the delivery of nutrients to CNS have been of high interest, with the glucose transporter type 1 (GLUT1),31–40 the sodium-dependent vitamin C transporter 2 (SVCT2),34,35 organic cation/carnitine transporter type 2 (OCNT2),41 and L-type amino acid transporter 1 (LAT1)42–46 being extensively implicated in carrier-mediated brain-targeting prodrug design.
A LAT1 targeting perforin inhibitor prodrug was developed to improve the drug transport across the BBB and also into the brain parenchymal cells, including neurons, astrocytes, and microglia.44 Although the brain uptake was increased, the brain specificity was still limited due to the prodrug being bioconverted to the parent drug by both acetylcholinesterase (AChE) and carboxylesterases (CES), CES1b and CES2 (Fig. 4).47
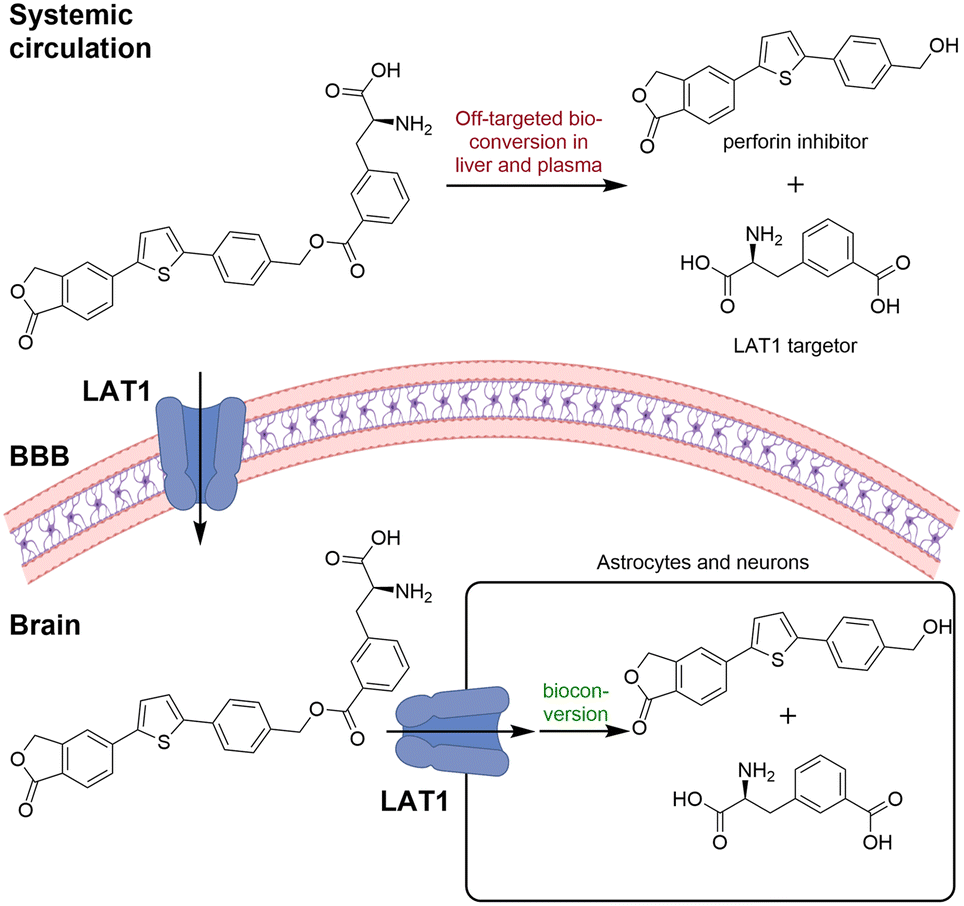 | ||
| Fig. 4 Although the LAT1 targeting perforin inhibitor prodrug showed improved brain uptake of the prodrug, the unspecific bioconversion reduces the brain specificity. Inspired from ref. 47. Created partly in https://BioRender.com. | ||
It is unfortunately often the case that the mechanism of drug release from ester prodrugs includes a combination of both chemical and enzymatic hydrolysis.48 This will unavoidably limit the selectivity of drug release. Further restricting selectivity, the enzymes responsible for prodrug hydrolysis are often not specific and may involve several esterases, including CES, AChE, butyrylcholinesterase (BuChE), paraoxonase, and arylesterase.49,50 Furthermore, oral administration of ester prodrugs may suffer from preliminary hydrolysis due to metabolism while traversing the gastrointestinal tract or in plasma, prior to BBB penetration.51 If drug release happens before the prodrug reaches its target destination, undesired or adverse drug effects may occur.
Unfortunately, esterification to increase hydrophobicity of a polar drug not only increases BBB permeability but generally increases barrier crossing, which is accompanied by an increase in uptake by peripheral tissues and cells. Due to the unspecific cleavage mechanism of ester prodrugs, this may potentially cause undesired side effects. This inspired the investigation of amide prodrugs, as more stable hydrolysable prodrugs, hereby increasing the stability to prevent premature activation and drug release.
Amide prodrugs
To mask the highly polar carboxylic acid-52 or amine-53 functionalized drugs, amide prodrugs have been intensively studied due to their high plasma stability, allowing them to reach the CNS.54Similar to ester prodrugs, amide prodrugs have been used to introduce a targeting moiety that recognizes a BBB-expressed carrier, including GLUT1,31,55,56 glutathione (GSH)57–59 and LAT1 transporters.43,53,54,60,61
Selective regeneration of the active drugs in the brain can however be difficult with amide prodrugs. It is challenging to identify amidases that are specifically expressed in the brain, in addition to being difficult to achieve selective cleavage by one specific amidase. Therefore, many amide prodrugs are also cleaved by several different and unspecific amidases, which will unavoidably lead to a distribution of the active drug between the brain and the periphery.62–64
Higher prodrug activation is however often seen in the brain compared to the periphery, due to the higher percentage and/or other distribution of microsomal amidases, glutamine transaminases, and ω-amidases in the brain.64 Structural design of drugs/prodrugs can also affect the brain:plasma ratio.
To enhance the brain barrier penetration of MTX, Singh et al.65 conjugated MTX to two lysine residues through the carboxylic acid functionalities to enhance CNS delivery via the LAT1 transporter. The prodrug displayed high plasma stability (half-life ≈ 3.2 h) and a sustained release in brain homogenate (half-life ≈ 2.0 h). They suggested that the prodrug hydrolysis in the brain was caused by brain-specific forms of cytochrome P enzymes and amidases.63,65
Hugele et al.66 investigated the enzymes responsible for the bioconversion of LAT1-targeting amide prodrugs. They evaluated a handful of different LAT1 prodrugs containing both aliphatic and aromatic LAT1 substrates (Fig. 5). The aromatic amino acid prodrugs released their parent drug both in vitro and in vivo. In the majority of cases, the brain-selective aminopeptidase B was found to be responsible for the bioconversion. The salicylic acid prodrug was, however, not bioconverted by aminopeptidase B but exhibited selective drug release in the brain with 80% bioconversion via an unknown release mechanism.66 Further investigation of the mechanism responsible for this bioconversion is highly relevant in order to develop brain-targeting drug delivery systems.
 | ||
| Fig. 5 Aminopeptidase B is responsible for the bioconversion of the majority of the aromatic amino acid prodrugs targeting LAT1. Blue represents the drug while green/orange/red represent the LAT-1 recognition parts. Inspired from ref. 66. | ||
Differences in plasma and brain amidase levels are also seen from research on dual amide-prodrugs, where different cleavage profiles are observed in plasma and the brain.67 Dalpiaz et al.68 proposed a dual amide-prodrug consisting of dopamine and an A2A antagonist connected via a succinic spacer. In human blood, the amide bond to the A2A antagonist was the main cleavage site (half-life ≈ 2.7 h) whereas in rat brain homogenates, the prodrug was cleaved at the amide bond to dopamine (half-life over 8 h).
While important steps have been taken, the potential of utilizing the differences in brain and peripheral amidase levels to develop brain-specific prodrugs has still to be further investigated before a brain-specific amidase-cleavable prodrug can be developed. To achieve higher brain selectivity and limit off-target side effects, increasing focus has been given to target-specific prodrug activation. For amide prodrugs, brain selectivity was partly achieved by targeting the fatty acid amide hydrolase (FAAH).
Fatty acid amide hydrolases
FAAH is a serine hydrolase that catalyses the hydrolysis of a diverse array of fatty acid amide signalling molecules with higher expression in the brain, liver and testis compared to other organs.69–72 Structure–activity relationship (SAR) studies have shown that FAAH is also capable of hydrolysing a diverse set of synthetic arachidonoyl amides, including substituted derivatives of ethylamine and aniline as well as synthetic luciferin derivatives.73,74Scanlan and coworkers75,76 developed a prodrug of the carboxylate-containing sobetirome. The prodrugs were synthesized as ethanolamino ester prodrugs that readily underwent O,N-acyl migration to form the thermodynamically favored amides. The N-isopropyl amide provided the greatest brain:serum ratio, but it showed very low brain levels of the free drug, suggesting that the FAAH-mediated hydrolysis was too slow. A subsequent study revealed the N-methyl amide of sobetirome to be the optimal amide for delivering sobetirome to the CNS while minimizing the peripheral conversion (Fig. 6).77 SAR studies have shown a clear preference for a “less-is-more” principle with small, nonpolar amide modifications giving a higher brain:serum ratio in vivo.78
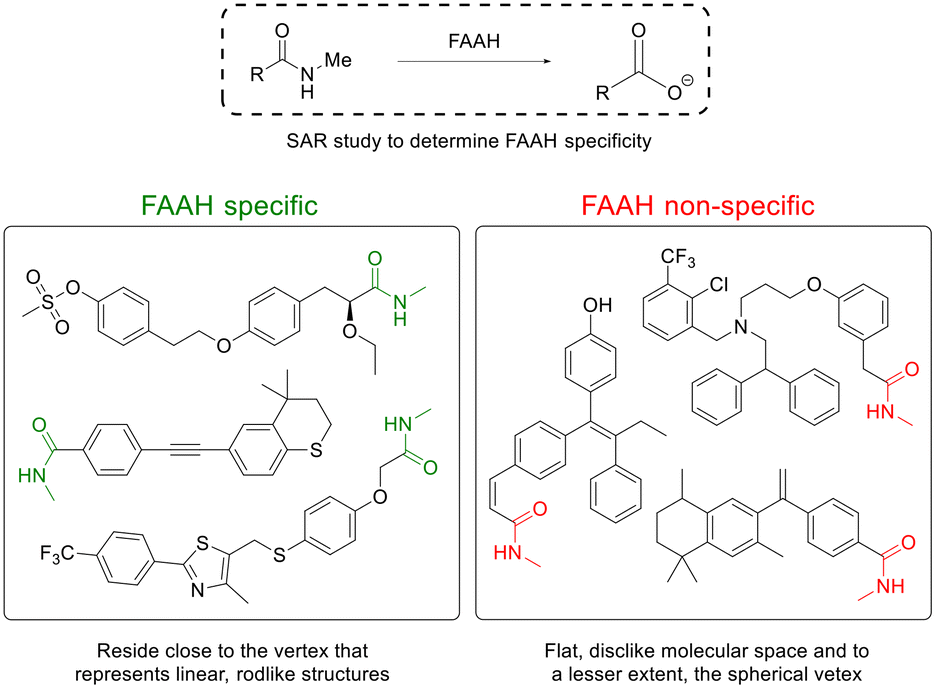 | ||
| Fig. 6 Linear, rod-like N-methylamides are good substrates for the brain-selective enzyme FAAH. Inspired from ref. 79. | ||
Ferrara and Scanlan79 showed the delivery of a variety of carboxylic acid-containing drug structures, by converting the drug's carboxylic acid functionality into N-methyl amides, imparting beneficial BBB penetration via passive diffusion. The prodrugs were substrates for the FAAH that cleaved the prodrug into the carboxylate-containing parent drug. Co-administration of a FAAH inhibitor significantly reduced the prodrug-to-drug conversion, confirming that FAAH was the critical hydrolase responsible for prodrug cleavage. Interestingly, the prodrugs with the highest brain:serum ratio had among the slowest FAAH hydrolysis rates, which suggests that prodrugs that survive hydrolysis by peripheral-expressed FAAHs during circulation allow for a greater fraction of prodrugs to enter the CNS for cleavage by CNS-expressed FAAH.79 A SAR study revealed that prodrugs displaying CNS selectivity all represent linear, rod-like structures, while prodrugs not cleaved appreciably by FAAH deviated from linearity and spread closer toward the flat, disc-like molecular space and, to a lesser extent, the spherical vertex (Fig. 6).79
To increase brain selectivity, other cleavage mechanisms have also been exploited.
Redox homeostasis of the brain
Redox homeostasis is recognized to be involved in all aspects of CNS development, function, aging, and disease. A diverse nature of redox reactions and homeostasis contribute to brain physiology, and when dysregulated, they cause pathological consequences. CNS redox processes involve both the nonspecific nature of oxidative damage and the very specific oxidation/reduction reactions that are involved in redox signaling and the regulation of a myriad of neurological processes such as neurotransmission, homeostasis, and degeneration.80Oxidoreductases
Oxidoreductases are a group of enzymes that play a crucial role in the brain by facilitating the transfer of electrons between molecules. These enzymes are essential for various metabolic processes and help maintain cellular homeostasis.80The oxidoreductase short-chain dehydrogenase/reductase (SDR) plays a significant role in the estrogen antioxidative cycle where the phenolic A-ring of estrogens can be regenerated from their corresponding quinols in the brain tissue.81 Prokai et al.82 developed a 17β-estradiol (E2) prodrug for brain-selective hormone treatment. The prodrug, 10β,17β-dihydroxyestra-1,4-dien-3-one (DHED), does not contain a classical “promoiety” but is transformed into an active drug via transient chemical alteration within the structure. Upon entering the brain, DHED is reduced by a NADPH-dependent SDR enzyme to E2 through a hydride transfer from the coenzyme to the C1 position of the C1–C2 double bond of DHED's A-ring that is conjugated to the 3-carbonyl group (Fig. 7). This is followed by spontaneous water elimination involving the 10(β)-OH. LC-MS/MS-based bioassay in vivo showed no bioconversion in the periphery while a fast bioconversion to E2 (≈ 20 min) after entering the brain indicated the high brain specificity of the prodrug.82 Tschiffely et al.83 also reported positive neurobiochemical effects of the prodrug in an Alzheimer's disease (AD) mouse model. The prodrug upon release of E2 in the brain showed a decrease in APP/Aβ levels and cognitive benefits without uterotrophic side effects. A similar bioactivation mechanism was utilized to activate the bioprecursor prodrug of α-E2, α-DHED (Fig. 7).84
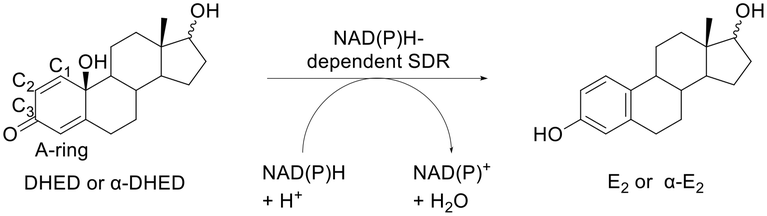 | ||
| Fig. 7 NAD(P)H-dependent short-chain dehydrogenase/reductase (SDR) bioconversion of the prodrugs DHED/α-DHED to the active drugs E2/α-E2. Inspired from ref. 82 and 84. | ||
NAD(P)H/quinone oxidoreductase (NQO1) is an antioxidant enzyme that plays an important role in controlling the cellular redox state. The expression of NQO1 is increased in the brain tissues of patients suffering from neurodegenerative diseases and has therefore been used to design a prodrug for treatment of the neurodegenerative disease AD.85 Schäfer et al.86 developed NQO1-targeting prodrugs of bexarotene to avoid unwanted peripheral side effects. The conjugates consisted of an indolequinone moiety with the drug attached to C-3. Upon entering the brain, the quinone was reduced resulting in the release of the drug (Fig. 8). HPLC studies showed that only 13% unspecific cleavage was observed over 24 h using porcine liver esterase while 50% of bexarotene was cleaved within 2 h by NQO1. Introducing a methyl group at R1 led to a complete loss of recognition by the enzyme.
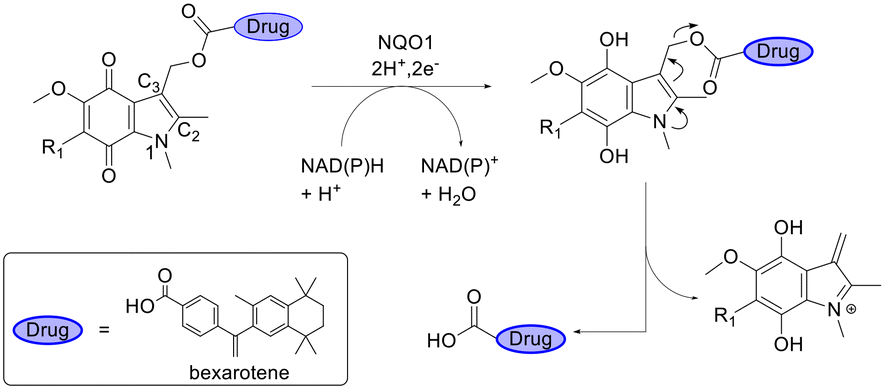 | ||
| Fig. 8 NAD(P)H-dependent NAD(P)H/quinone oxidoreductase (NQO1) bioconversion of a bexarotene prodrug. Inspired from ref. 86. | ||
Brain-specific activation has also been demonstrated for disulfide-based prodrugs. This strategy utilizes that apolar disulfides can enter the brain and once in the brain, they are readily reduced by brain reductases to release two thiol-functionalized drug molecules.87 This was utilized to develop BBB-permeable prodrugs of a murine brain aminopeptidase (APA) inhibitor EC33 for the treatment of hypertension.88–91 The prodrugs consisted of two EC33 molecules connected via disulfide bridges that upon brain entry were reduced to release two active thiol-functionalized EC33 molecules.88 Sterically hindered disulfide bridges were shown to give higher plasma stability (less than 10% ester hydrolysis after 100 h) while still allowing reduction to release the drugs in the brain (half-life = 8.8 h), hereby increasing brain specificity.92,93
AChE inhibitors (AChEIs) have been highly investigated for treatment of AD,94 with all marketed AChEIs containing a tertiary or primary amine that are mostly protonated at physiological pH. The active protonated form is in equilibrium with the brain-permeable free base form.95
Levacher and coworkers95–97 synthesized a library of prodrug analogues based on the AChEI rivastigmine structure. The aromatic ring of rivastigmine was expanded to a 1,4-dihydroquinoline structure that possessed a non-protonated enamine nitrogen at physiological pH. The 1,4-dihydroquinoline prodrugs crossed the BBB by passive mechanisms while quickly being oxidized in the brain to the corresponding BBB-impermeable quaternary quinolium salt acting as an AChEI (Fig. 9).
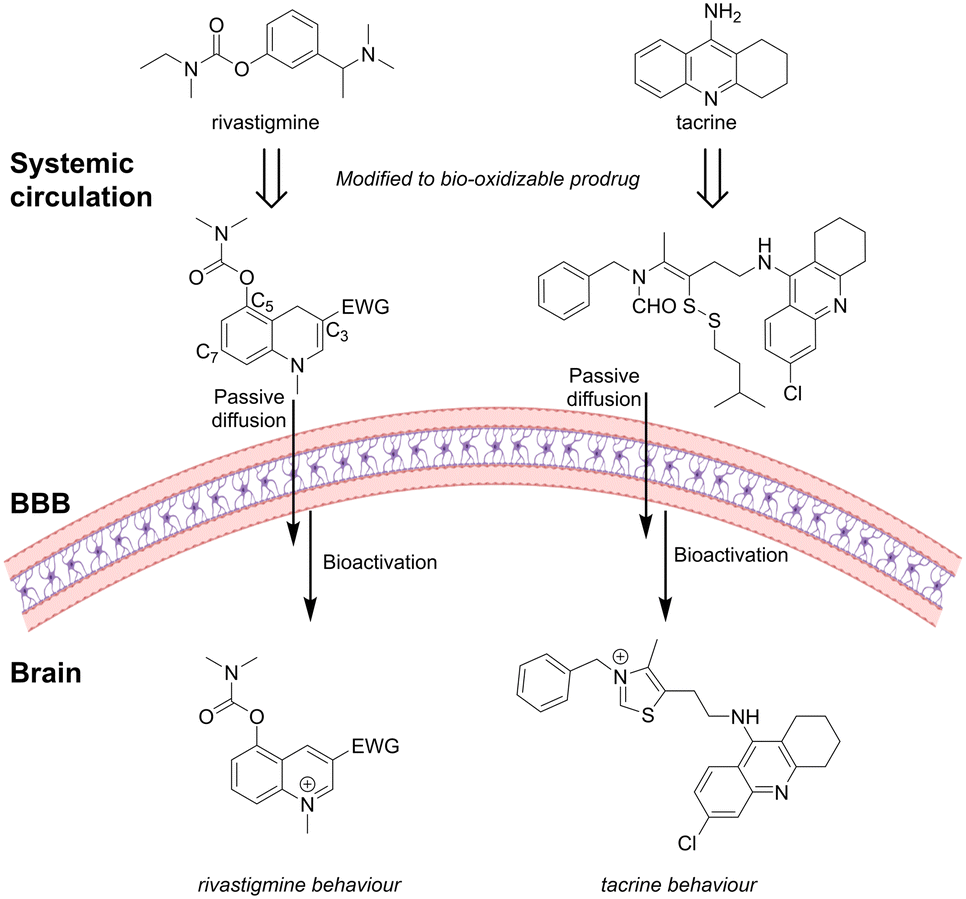 | ||
| Fig. 9 The AChEI rivastigmine and tacrine inspired prodrugs: in vivo redox activation initiates a structural rearrangement to form active AChEIs. Inspired from ref. 95 and 98. Created partly in https://BioRender.com. | ||
They found that it was favorable to install the enzyme recognition site, a carbamate, at the C-5 position compared to the C-7 position, with a N,N-dimethylcarbamate giving higher potency than an N-ethylcarbamate.95 Introduction of an electron-withdrawing group (EWG) at the C-3 position ensured a good balance between stability of the enamine moiety in the prodrug and AChE inhibition of the parent drug. An EWG amide group gave higher stability than the corresponding ester analogues.95,99In vivo, the brain and peripheral bioactivation were compared. Brain activation indicated by max hypothermia was observed after 1 h. Peripheral activation, evidenced by salivation, occurred only within the first 15 min, in contrast to the sustained effects seen with tacrine. This suggests rapid elimination of the prodrug from peripheral circulation, thereby minimizing off-target effects.96
A similar concept was used to develop prodrugs of the AChEI donepezil. The piperidine ring of donepezil was modified to a non-protonated 1,4-dihydropyridine ring, which in the brain, was converted to the active pyridinium analogue. In vitro, the prodrug showed good stability in the periphery (3% unspecific oxidation in plasma after 3 h) while 35% was oxidized in the mice brain homogenate after 3 h.100 Modification of the 1,4-dihydropyridine C-3 position improved the stability of the prodrug. Furthermore, installation of an aromatic moiety via a polyethylene glycol (PEG)-spacer in the C-3 position introduced a secondary interaction via the peripheral anionic site of AChE.101 Similar to the studies with the rivastigmine structure, the activity was significantly decreased when larger N-substituents were installed at the AChE recognition carbamate.
Based on the structure of donepezil and tacrine, Liu et al.98 designed substituted formamide disulfide prodrugs. In the brain, the disulfide bond was reduced by the abundant GSH to release the free thiol, which rapidly cyclized with the formamide, followed by elimination of water to form the N-substitute thiazole salt as a good AChEI (Fig. 9). They investigated different disulfides and the rate of GSH-mediated drug formation. The benzyl disulfide substituted prodrug formed the active AChEI too rapidly, with the risk of causing drug release in the periphery before entering CNS. Prodrugs containing larger sterically hindered isobutyl or isoamyl disulfides were able to gradually generate the thiazolate AChEI faster than the ethyl disulfide-functionalized prodrug.
Reactive oxygen species
As the most metabolically active organ in the body, the brain consumes around 20% of the total basal oxygen.102 This is accompanied by a high production of reactive oxygen species (ROS).103,104 The high levels of ROS in the brain compared to other tissues have been exploited for brain-selective drug activation.Liu et al. designed a self-immolative ROS-responsive prodrug consisting of two drugs, a non-steroidal anti-inflammatory drug (NSAID), ibuprofen, and an AChE inhibitor, tacrine, for treatment of AD. The prodrug released the two drugs at the pathologically high oxidative environment of AD upon stimulation by ROS followed by a 1,6-elimination reaction (Fig. 10).105
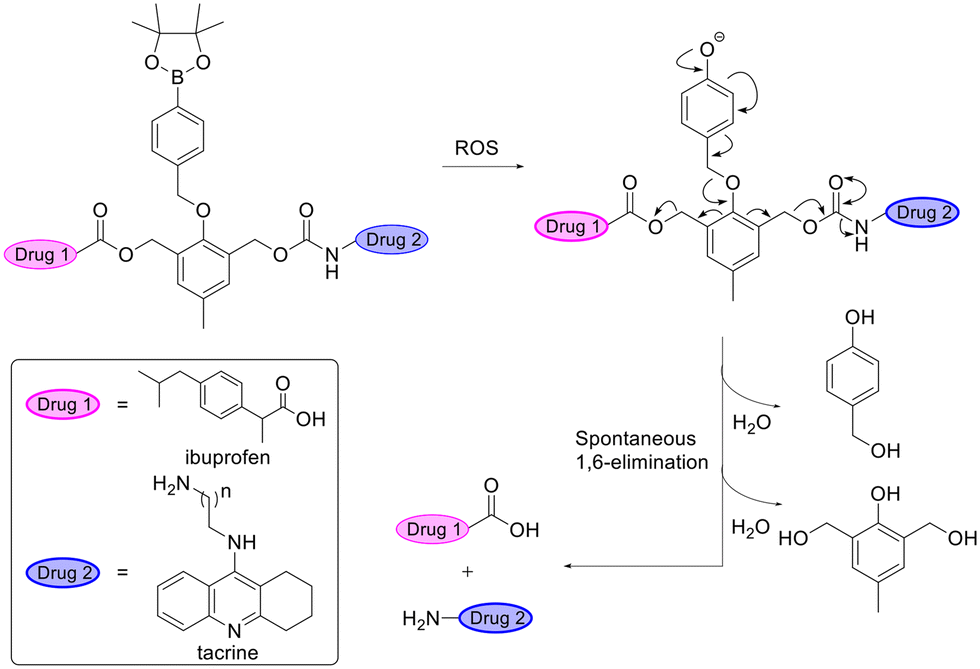 | ||
| Fig. 10 Dual reactive oxygen species (ROS)-responsive prodrug leading to the release of two drugs after 1,6-elimination. Inspired from ref. 105. | ||
‘Lock’ the prodrug in the brain
A subgroup of prodrugs is designed to contain promoieties that ‘lock’ the prodrug in the brain before the release of the active drug, hereby increasing the brain selectivity (Fig. 11). In pioneering studies, the ‘lock-in-the-brain’ system was based on the 1,4-dihydrotrigonelline/trigonelline system.106–111 The hydrophobic 1,4-dihydropyridine form can pass the brain barrier via passive mechanisms, but once inside the brain, it is readily converted by the NAD(P)H/NA(P)+ system into its hydrophilic quaternary form, which is impermeable to the barrier. | ||
| Fig. 11 Graphical illustration of the “lock-in the brain” prodrug design. Created in https://BioRender.com. | ||
The 1,4-dihydropyridine system
Sharpe et al.112 developed a ‘lock-in-the-brain’ prodrug for targeting glioma cells. The prodrug MP-MUS was based on 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) that was selectively activated by the brain selective monoamine oxidase B (MAO-B). The prodrug was developed based on the knowledge that a non-toxic compound, MPTP, is oxidized into the neurotoxin MPP+ by MAO-B through an MDP+ intermediate. A covalent conjugation to a cytotoxic DNA-alkylating moiety, such as nitrogen mustard (MUS), made the MTP substrates act as anti-glioma prodrugs. Firstly, the MP-MUS was oxidized to MD+-MUS by MAO-B, followed by further oxidation into the mature drug, P+-MUS.112Learning from the 1,4-dihydropyridine system, ‘lock-in-the-brain’ peptide prodrugs were developed. Replacing the central basic His of thyrotropin-releasing hormone (TRH) tripeptide with pyridinium derivatives introduced a permanent positive charge to the peptides in the brain (Fig. 12). The pyridinium moiety further improved the lipophilicity to increase BBB penetration. In vitro, the conversion to the pyridinium derivative was faster in the brain (half-life = 6 min) compared to plasma (half-life = 20 min).113,114
 | ||
| Fig. 12 Replacing the His or PGlu in the tripeptide TRH with substituted pyridinium leads to increased uptake in the brain due to the ‘lock’ in effect. Inspired from ref. 114 and 115. Created partly in https://BioRender.com. | ||
Also, replacing the Glu of the TRH peptide with a pyridinium moiety led to analogues with an improved brain selectivity and good anti-depressive properties as well as reduced analeptic side effects (Fig. 12), while the attachment of the pyridinium unit to the peptide via a spacer resulted in a fast oxidation in brain homogenates.115
The high susceptibility to oxidation and/or hydration of the enamine 5,6-double bond of the dihydropyridine ring116–118 inspired the development of the more stable 1,4-dihydroquinolines as ‘lock-in-the-brain’ promoieties, where annelation of the 1,4-dihydropyrdine ring protects the enamine 5,6-double bond from hydration. A study by Bodor et al.119 found that the 1,2-dihydroisoquinoline moiety was sufficiently stable in both aqueous media and air. The 1,2-dihydroisoquinoline moiety showed reasonable oxidation rates in biological fluids with the possibility of further tuning redox properties by functionalization of the phenyl ring.120,121
Gourand et al.122 utilized the ‘lock-in-the-brain’ system to deliver radiolabeled m-iodobenzylguanidine (MIBG) for imaging of catecholamine-secreting tumors. Installation of a self-immolative spacer between the ‘lock-in-the-brain’ moiety and the cargo improved the brain uptake by increasing hydrophobicity. The system underwent fast oxidation upon brain entry followed by hydrolysis of the resulting quinolinium salt to release the spacer–MIBG intermediate that could undergo cyclization to release the MIBG along with a γ-lactam by-product (Fig. 13).
 | ||
| Fig. 13 “Lock-in the brain” design based on the 1,4-dihydroquinoline system combined with a cyclization spacer. Inspired from ref. 122. Created partly in https://BioRender.com. | ||
The ‘lock-in-the-brain’ concept was also utilized for a dual prodrug consisting of a dihydroquinoline AChEI structure123 with a dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) inhibitor, INDY-3 anchored through a carbonate group at the C-3 position without loss of activity. The prodrug was able to cross the brain barrier and, in the brain, underwent bio-oxidation to the corresponding quinolinium species. The DYRK1A inhibitor was released due to hydrolysis of the carbonate linker. The quinolinium salt exhibited good AChE and BuChE inhibition, while no inhibitory effect was observed for the prodrug. Also, no inhibition of DYRK1A was observed before the release of INDY.124
Thiamine system
Another prodrug design, taking advantage of the ‘lock-in-the-brain’ concept, is the thiamine disulfide system (TDS).33,35,125 Upon entering the brain, these prodrugs are reduced by disulfide reductases leading to the formation of the free sulfide which can then undergo a ring-closure reaction forming thiazolium (Fig. 14). The charged thiazolium is locked inside the brain and the given drug can then be released by ester hydrolysis.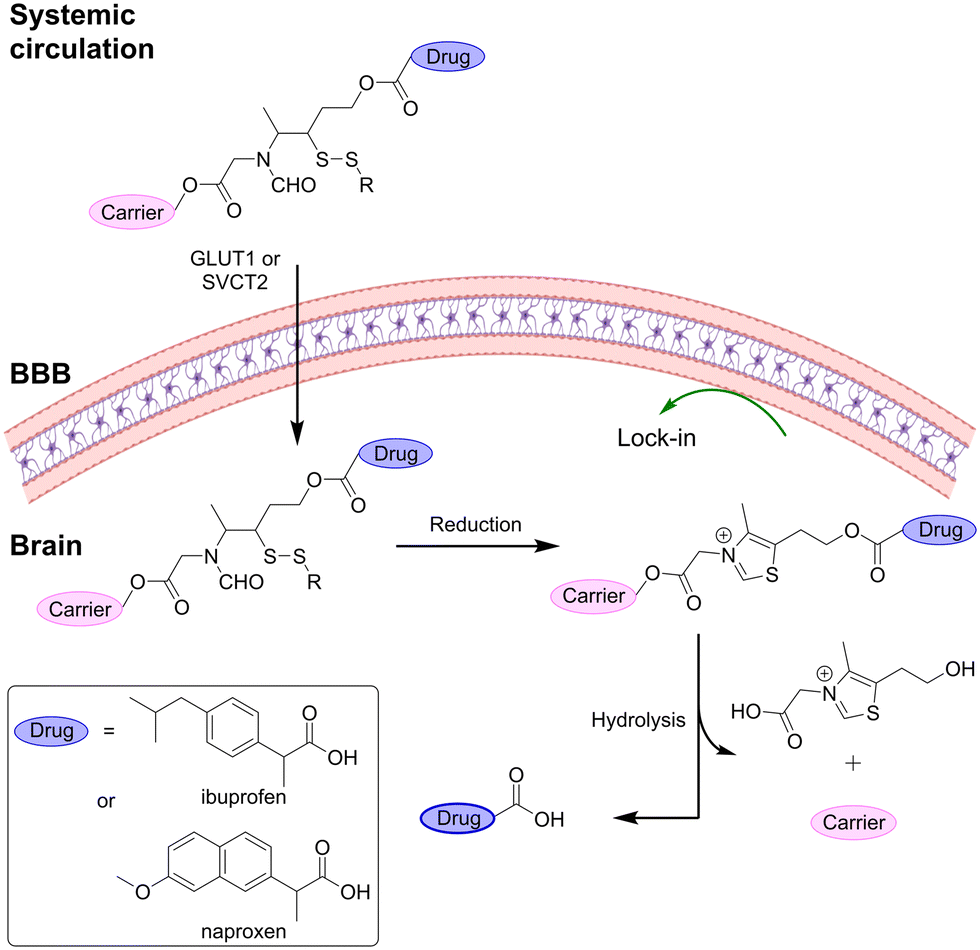 | ||
| Fig. 14 “Lock-in the brain” design based on the disulfide to thiamine conversion. Inspired from ref. 33 and 35. Created partly in https://BioRender.com. | ||
Fan et al.33 applied this concept to transport naproxen into the brain. The prodrug consisted of the TDS attached to a spacer through an ester bond and a glucose moiety at the C-6 position. The glucose moiety took advantage of GLUT1 to transport the system into the brain where it was readily reduced to lock it in the brain. Introduction of steric hindrance by decorating the TDS prodrugs with alkyl substituents around the disulfide bond of the prodrug was found to improve brain delivery since it was poorly reduced by reductases in plasma (half-life ≈ 15 min vs. 2 min), hereby increasing the circulation time.33,126
Similarly, Zhao et al.35 developed a TDS prodrug of ibuprofen using L-ascorbic acid as the carrier. The prodrug showed good stability in plasma, allowing it to reach the brain, where it was rapidly reduced. In the brain, the prodrug was slowly hydrolyzed to release ibuprofen.
Conclusions
The present review provides a systematic up-to-date summary of progress in the development of activation mechanisms for prodrugs targeting the brain, by putting specific emphasis on discussing strategies to improve brain selectivity.We revealed that despite many attempts to develop brain-targeting (pro)drugs, there are several factors limiting the rational development of truly brain-specific prodrugs. While many studies focus on solving the challenge of crossing the brain barriers, a larger effort should also be given to identify brain-selective bioactivation mechanisms of prodrugs. A prodrug can be widely distributed throughout the body but if it is only or predominantly activated at the desired site, site-selectivity will be increased. Therefore, there is a need for knowledge about tissue differences in bio-converting enzymes, their localizations, expression levels, and activities or the investigation of tissue-specific conditions.
We hope that the current review can stimulate work in the field and suggest a combination of a transporter-mediated uptake, a ‘lock-in-brain’ strategy, and utilization of a brain-selective prodrug-activation mechanism, to develop ‘triple-targeted’ approaches.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Ida Aaberg Lillethorup: conceptualization, investigation, validation, and writing – original draft. Andreas Victor Hemmingsen: conceptualization, investigation, and writing – review & editing. Katrine Qvortrup: conceptualization, investigation, supervision, funding acquisition, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the independent Research Fund Denmark (1032-00302B) and the Novo Nordisk Foundation (NNF21OC0071381) for financial support of the work.References
- J. Rautio, N. A. Meanwell, L. Di and M. J. Hageman, Nat. Rev. Drug Discovery, 2018, 17, 559–587 CrossRef CAS PubMed.
- P. Ettmayer, G. L. Amidon, B. Clement and B. Testa, J. Med. Chem., 2004, 47, 2393–2404 CrossRef CAS PubMed.
- S. Yanni and D. R. Thakker, in Prodrugs: Challenges and Rewards Part 1, ed. V. J. Stella, R. T. Borchardt, M. J. Hageman, R. Oliyai, H. Maag and J. W. Tilley, Springer New York, 2007, pp. 1043–1081 Search PubMed.
- A. Albert, Nature, 1958, 182, 421–423 CrossRef CAS PubMed.
- B. Pavan, A. Dalpiaz, N. Ciliberti, C. Biondi, S. Manfredini and S. Vertuani, Molecules, 2008, 13, 1035–1065 CrossRef CAS PubMed.
- W. M. Pardridge, J. Cereb. Blood Flow Metab., 2012, 32, 1959–1972 CrossRef CAS PubMed.
- M. N. Pangalos, L. E. Schechter and O. Hurko, Nat. Rev. Drug Discovery, 2007, 6, 521–532 CrossRef CAS PubMed.
- E. Puris, G. Fricker and M. Gynther, Pharm. Res., 2022, 39, 1415–1455 CrossRef CAS PubMed.
- Q. R. Smith, Int. Congr. Ser., 2005, 1277, 63–74 CrossRef CAS.
- K. Töllner, C. Brandt, M. Töpfer, G. Brunhofer, T. Erker, M. Gabriel, P. W. Feit, J. Lindfors, K. Kaila and W. Löscher, Ann. Neurol., 2014, 75, 550–562 CrossRef PubMed.
- A. Trotier-Faurion, S. Dézard, F. Taran, V. Valayannopoulos, P. De Lonlay and A. Mabondzo, J. Med. Chem., 2013, 56, 5173–5181 CrossRef CAS PubMed.
- R. Pignatello, V. Pantò, S. Salmaso, S. Bersani, V. Pistarà, V. Kepe, J. R. Barrio and G. Puglisi, Bioconjugate Chem., 2008, 19, 349–357 CrossRef CAS PubMed.
- A. Seelig, J. Mol. Neurosci., 2007, 33, 32–41 CrossRef CAS PubMed.
- N. A. Meanwell, Top. Med. Chem., 2014, 9, 283–382 Search PubMed.
- C. Ballatore, D. M. Huryn and A. B. Smith, ChemMedChem, 2013, 8, 385–395 CrossRef CAS PubMed.
- N. A. Meanwell, in Topics in Med Chem 9 Tactics in Contemporary Drug Design, ed. P. R. Beinstein, A. Buschauer, G. I. Georg, J. A. Lowe, U. Stilz, C. T. Supuran and A. K. S. Saxena, Springer New York, 2015, pp. 1–391 Search PubMed.
- M. Nedelcovych, R. P. Dash, L. Tenora, S. C. Zimmermann, A. J. Gadiano, C. Garrett, J. Alt, K. R. Hollinger, E. Pommier, A. Jančařík, C. Rojas, A. G. Thomas, Y. Wu, K. Wozniak, P. Majer, B. S. Slusher and R. Rais, Mol. Pharmaceutics, 2017, 14, 3248–3257 CrossRef CAS PubMed.
- H. Danny Kao, A. Traboulsi, S. Itoh, L. Dittert and A. Hussain, Pharm. Res., 2000, 17, 978–984 CrossRef PubMed.
- A. G. Cairns, A. Vazquez-Romero, M. M. Moein, J. Ådén, C. S. Elmore, A. Takano, R. Arakawa, A. Varrone, F. Almqvist and M. Schou, ACS Chem. Neurosci., 2018, 9, 2542–2547 CrossRef CAS PubMed.
- B. M. Liederer and R. T. Borchardt, J. Pharm. Sci., 2006, 95, 1177–1195 CrossRef CAS PubMed.
- K. Prokai-Tatrai, V. Nguyen, A. D. Zharikova, A. C. Braddy, S. M. Stevens and L. Prokai, Bioorg. Med. Chem. Lett., 2003, 13, 1011–1014 CrossRef CAS PubMed.
- H. Wang, A. A. Hussain and P. J. Wedlund, Pharm. Res., 2005, 22, 556–562 CrossRef CAS PubMed.
- R. Rais, A. Jančařík, L. Tenora, M. Nedelcovych, J. Alt, J. Englert, C. Rojas, A. Le, A. Elgogary, J. Tan, L. Monincová, K. Pate, R. Adams, D. Ferraris, J. Powell, P. Majer and B. S. Slusher, J. Med. Chem., 2016, 59, 8621–8633 CrossRef CAS PubMed.
- N. Tsuzuki, T. Hama, M. Kawada, A. Hasui, R. Konishi, Y. Ochis, S. Futakis and K. Kitagawas, J. Pharm. Sci., 1994, 481–484 CrossRef CAS PubMed.
- N. Fattahi, A. Ramazani, M. Hamidi, M. Parsa, K. Rostamizadeh and H. Rashidzadeh, Int. J. Pharm., 2021, 600, 120479 CrossRef CAS PubMed.
- D. Chen, T. Zhao, K. Ni, P. Dai, L. Yang, Y. Xu and F. Li, Bioorg. Med. Chem. Lett., 2016, 26, 2152–2155 CrossRef CAS PubMed.
- L. H. A. Prins, J. L. du Preez, S. van Dyk and S. F. Malan, Eur. J. Med. Chem., 2009, 44, 2577–2582 CrossRef CAS PubMed.
- A. Dalpiaz, G. Paganetto, B. Pavan, M. Fogagnolo, A. Medici, S. Beggiato and D. Perrone, Mol. Pharmaceutics, 2012, 9, 957–968 CrossRef CAS PubMed.
- M. Weitman, A. Eisenkraft, Z. TaShma, I. Makarovsky, D. Last, D. Daniels, D. Guez, R. Shneor, Y. Mardor, A. Nudelman and A. Krivoy, Sci. Rep., 2022, 12, 18078 CrossRef CAS PubMed.
- Z. Qin, J. Luo, L. Vandevrede, E. Tavassoli, M. Fa, A. F. Teich, O. Arancio and G. R. J. Thatcher, J. Med. Chem., 2012, 55, 6784–6801 CrossRef CAS PubMed.
- C. Fernández, O. Nieto, E. Rivas, G. Montenegro, J. A. Fontenla and A. Fernández-Mayoralas, Carbohydr. Res., 2000, 327, 353–365 CrossRef PubMed.
- A. Angusti, E. Durini, S. Vertuani, A. Dalpiaz, A. Ruffo, R. Di Fabio, D. Donati, G. Pentassuglia, G. Vitulli, R. J. Barnaby and S. Manfredini, Farmaco, 2005, 60, 393–397 CrossRef CAS PubMed.
- W. Fan, Y. Wu, X. K. Li, N. Yao, X. Li, Y. G. Yu and L. Hai, Eur. J. Med. Chem., 2011, 46, 3651–3661 CrossRef CAS PubMed.
- Q. Yue, Y. Peng, Y. Zhao, R. Lu, Q. Fu, Y. Chen, Y. Yang, L. Hai, L. Guo and Y. Wu, Drug Delivery, 2018, 25, 426–434 CrossRef CAS PubMed.
- Y. Zhao, B. Qu, X. Wu, X. Li, Q. Liu, X. Jin, L. Guo, L. Hai and Y. Wu, Eur. J. Med. Chem., 2014, 82, 314–323 CrossRef CAS PubMed.
- L. Wang, L. Zhang, Y. Zhao, Q. Fu, W. Xiao, R. Lu, L. Hai, L. Guo and Y. Wu, Arch. Pharm., 2018, 351, 1700382 CrossRef PubMed.
- A. Dalpiaz, R. Filosa, P. de Caprariis, G. Conte, F. Bortolotti, C. Biondi, A. Scatturin, P. D. Prasad and B. Pavan, Int. J. Pharm., 2007, 336, 133–139 CrossRef CAS PubMed.
- F. Bonina, C. Puglia, M. G. Rimoli, D. Melisi, G. Boatto, M. Nieddu, A. Calignano, G. La Rana and P. de Caprariis, J. Drug Targeting, 2003, 11, 25–36 CAS.
- Y. Zhao, L. Zhang, Y. Peng, Q. Yue, L. Hai, L. Guo, Q. Wang and Y. Wu, Chem. Biol. Drug Des., 2018, 91, 707–716 CrossRef CAS PubMed.
- M. Gynther, J. Ropponen, K. Laine, J. Leppänen, P. Haapakoski, L. Peura, T. Järvinen and J. Rautio, J. Med. Chem., 2009, 52, 3348–3353 CrossRef CAS PubMed.
- C. Napolitano, M. Scaglianti, E. Scalambra, S. Manfredini, L. Ferraro, S. Beggiato and S. Vertuani, Molecules, 2009, 14, 3268–3274 CrossRef CAS PubMed.
- M. Gynther, K. Laine, J. Ropponen, J. Leppänen, A. Mannila, T. Nevalainen, J. Savolainen, T. Järvinen and J. Rautio, J. Med. Chem., 2008, 51, 932–936 CrossRef CAS PubMed.
- A. Montaser, M. Lehtonen, M. Gynther and K. M. Huttunen, Pharmaceutics, 2020, 12, 344 CrossRef CAS PubMed.
- M. Gynther, D. S. Pickering, J. A. Spicer, W. A. Denny and K. M. Huttunen, Mol. Pharmaceutics, 2016, 13, 2484–2491 CrossRef CAS PubMed.
- E. Puris, M. Gynther, J. Huttunen, S. Auriola and K. M. Huttunen, Eur. J. Pharm. Sci., 2019, 129, 99–109 CrossRef CAS PubMed.
- J. Tampio, S. Löffler, M. Guillon, A. Hugele, J. Huttunen and K. M. Huttunen, Int. J. Pharm., 2021, 601, 120565 CrossRef CAS PubMed.
- J. Tampio, J. Huttunen, A. Montaser and K. M. Huttunen, Mol. Neurobiol., 2020, 57, 4563–4577 CrossRef CAS PubMed.
- S. Hasabelnaby, A. Goudah, H. K. Agarwal, M. S. M. Abd Alla and W. Tjarks, Eur. J. Med. Chem., 2012, 55, 325–334 CrossRef CAS PubMed.
- M. Prabha, V. Ravi and N. Ramachandra Swamy, Indian J. Clin. Biochem., 2013, 28, 283–291 CrossRef CAS PubMed.
- T. Fukami and T. Yokoi, Drug Metab. Pharmacokinet., 2012, 27, 466–477 CrossRef CAS PubMed.
- Z. Xuan, L. Xing, G. Tao, S. Xun and Z. R. Zhang, Acta Pharmacol. Sin., 2012, 33, 279–288 CrossRef PubMed.
- A. Sinha, J. C. Chang, P. Xu, K. Gindinova, Y. Cho, W. Sun, X. Wu, Y. M. Li, P. Greengard, J. W. Kelly and S. C. Sinha, ACS Med. Chem. Lett., 2020, 11, 1973–1979 CrossRef CAS PubMed.
- L. Peura, K. Malmioja, K. Huttunen, J. Leppänen, M. Hämäläinen, M. M. Forsberg, J. Rautio and K. Laine, Pharm. Res., 2013, 30, 2523–2537 CrossRef CAS PubMed.
- M. Gynther, L. Peura, M. Vernerová, J. Leppänen, J. Kärkkäinen, M. Lehtonen, J. Rautio and K. M. Huttunen, Neurochem. Res., 2016, 41, 2797–2809 CrossRef CAS PubMed.
- C. Fernandez, O. Nieto, J. A. Fontenla, E. Rivas, M. L. de Ceballos and A. Fernandez-Mayoralas, Org. Biomol. Chem., 2003, 5, 767–771 RSC.
- C. Jiang, X. Wan, J. Jankovic, S. T. Christian, Z. B. Pristupa, H. B. Niznik, J. S. Sundsmo and W. Le, Clin. Neuropharmacol., 2004, 27, 63–73 CrossRef CAS PubMed.
- S. S. More and R. Vince, J. Med. Chem., 2008, 51, 4581–4588 CrossRef CAS PubMed.
- F. Pinnen, I. Cacciatore, C. Cornacchia, P. Sozio, A. Iannitelli, M. Costa, L. Pecci, C. Nasuti, F. Cantalamessa and A. Di Stefano, J. Med. Chem., 2007, 50, 2506–2515 CrossRef CAS PubMed.
- F. Pinnen, I. Cacciatore, C. Cornacchia, A. Mollica, P. Sozio, L. S. Cerasa, A. Iannitelli, A. Fontana, C. Nasuti and A. Di Stefano, Amino Acids, 2012, 42, 261–269 CrossRef CAS PubMed.
- M. Gynther, A. Jalkanen, M. Lehtonen, M. Forsberg, K. Laine, J. Ropponen, J. Leppänen, J. Knuuti and J. Rautio, Int. J. Pharm., 2010, 399, 121–128 CrossRef CAS PubMed.
- L. Peura, K. Malmioja, K. Laine, J. Leppänen, M. Gynther, A. Isotalo and J. Rautio, Mol. Pharmaceutics, 2011, 8, 1857–1866 CrossRef CAS PubMed.
- L. I. Giannola, V. De Caro, G. Giandalia, M. G. Siragusa and L. Lamartina, Pharmazie, 2008, 63, 704–710 CAS.
- W. M. Pardridge, Physiol. Rev., 1983, 63, 1481–1535 CrossRef CAS PubMed.
- F. Dholkawala, C. Voshavar and A. K. Dutta, Eur. J. Pharm. Biopharm., 2016, 103, 62–70 CrossRef CAS PubMed.
- V. K. Singh and B. B. Subudhi, Drug Delivery, 2016, 23, 2327–2337 CrossRef CAS PubMed.
- A. Hugele, S. Löffler, B. H. Molina, M. Guillon, A. B. Montaser, S. Auriola and K. M. Huttunen, Front. Pharmacol., 2022, 13, 1034964 CrossRef CAS PubMed.
- P. Sozio, E. D'Aurizio, A. Iannitelli, A. Cataldi, S. Zara, F. Cantalamessa, C. Nasuti and A. Di Stefano, Arch. Pharm., 2010, 343, 133–142 CrossRef CAS PubMed.
- A. Dalpiaz, B. Cacciari, C. B. Vicentini, F. Bortolotti, G. Spalluto, S. Federico, B. Pavan, F. Vincenzi, P. A. Borea and K. Varani, Mol. Pharmaceutics, 2012, 9, 591–604 CrossRef CAS PubMed.
- T. J. Phillips, J. R. K. Mootz and C. Reed, Int. Rev. Neurobiol., 2016, 126, 39–85 CrossRef CAS PubMed.
- B. F. Cravatt, D. K. Giang, S. P. Mayfield, D. L. Boger, R. A. Lerner and N. B. Gilula, Nature, 1996, 384, 83–87 CrossRef CAS PubMed.
- N. Ueda, R. A. Puffenbarger, S. Yamamoto and D. G. Deutsch, Chem. Phys. Lipids, 2000, 108, 107–121 CrossRef CAS PubMed.
- B. Q. Wei, T. S. Mikkelsen, M. K. McKinney, E. S. Lander and B. F. Cravatt, J. Biol. Chem., 2006, 281, 36569–36578 CrossRef CAS PubMed.
- W. Lang, C. Qin, S. Lin, A. D. Khanolkar, A. Goutopoulos, P. Fan, K. Abouzid, Z. Meng, D. Biegel and A. Makriyannis, J. Med. Chem., 1999, 42, 896–902 CrossRef CAS PubMed.
- D. M. Mofford, S. T. Adams, G. S. K. K. Reddy, G. R. Reddy and S. C. Miller, J. Am. Chem. Soc., 2015, 137, 8684–8687 CrossRef CAS PubMed.
- A. T. Placzek, S. J. Ferrara, M. D. Hartley, H. S. Sanford-Crane, J. M. Meinig and T. S. Scanlan, Bioorg. Med. Chem., 2016, 24, 5842–5854 CrossRef CAS PubMed.
- S. J. Ferrara, J. M. Meinig, A. T. Placzek, T. Banerji, P. McTigue, M. D. Hartley, H. S. Sanford-Crane, T. Banerji, D. Bourdette and T. S. Scanlan, Bioorg. Med. Chem., 2017, 25, 2743–2753 CrossRef CAS PubMed.
- J. M. Meinig, S. J. Ferrara, T. Banerji, T. Banerji, H. S. Sanford-Crane, D. Bourdette and T. S. Scanlan, ACS Chem. Neurosci., 2017, 8, 2468–2476 CrossRef CAS PubMed.
- J. M. Meinig, S. J. Ferrara, T. Banerji, T. Banerji, H. S. Sanford-Crane, D. Bourdette and T. S. Scanlan, ACS Med. Chem. Lett., 2019, 10, 111–116 CrossRef CAS PubMed.
- S. J. Ferrara and T. S. Scanlan, J. Med. Chem., 2020, 63, 9742–9751 CrossRef CAS PubMed.
- R. Franco and M. R. Vargas, Antioxid. Redox Signaling, 2018, 28, 1583–1586 CrossRef CAS PubMed.
- L. Prokai, K. Prokai-Tatrai, P. Perjesi, A. D. Zharikova, E. J. Perez, R. Liu and J. W. Simpkins, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11741–11746 CrossRef CAS PubMed.
- L. Prokai, V. Nguyen, S. Szarka, P. Garg, G. Sabnis, H. A. Bimonte-Nelson, K. J. Mclaughlin, J. S. Talboom, C. D. Conrad, P. J. Shughrue, D. G. Todd, A. Brodie, I. Merchenthaler, P. Koulen and K. Prokai-Tatrai, Sci. Transl. Med., 2015, 7, 297ra113 Search PubMed.
- A. E. Tschiffely, R. A. Schuh, K. Prokai-Tatrai, L. Prokai and M. A. Ottinger, Horm. Behav., 2016, 83, 39–44 CrossRef CAS PubMed.
- K. Prokai-Tatrai, V. Nguyen and L. Prokai, ACS Chem. Neurosci., 2018, 9, 2528–2533 CrossRef CAS PubMed.
- L. Yuhan, M. Khaleghi Ghadiri and A. Gorji, J. Transl. Med., 2024, 22, 4 CrossRef CAS PubMed.
- A. Schäfer, E. S. Burstein and R. Olsson, Bioorg. Med. Chem. Lett., 2014, 24, 1944–1947 CrossRef PubMed.
- M. C. Fournié-Zaluski, P. Coric, S. Turcaud, E. Lucas, F. Noble, R. Maldonado and B. P. Roques, J. Med. Chem., 1992, 35, 2473–2481 CrossRef PubMed.
- M. C. Fournie-Zaluski, C. Fassot, B. Valentin, D. Djordjijevic, A. Reaux-Le Goazigo, P. Corvol, B. P. Roques and C. Llorens-Cortes, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7775–7780 CrossRef CAS PubMed.
- L. Bodineau, A. Frugière, Y. Marc, N. Inguimbert, C. Fassot, F. Balavoine, B. Roques and C. Llorens-Cortes, Hypertension, 2008, 51, 1318–1325 CrossRef CAS PubMed.
- M. Keck, R. Hmazzou and C. Llorens-Cortes, Curr. Hypertens. Rep., 2019, 21, 50 CrossRef PubMed.
- N. Inguimbert, P. Coric, H. Dhotel, C. Llorens-Cortes, M. C. Fournié-Zaluski and B. P. Roques, J. Labelled Compd. Radiopharm., 2004, 47, 997–1005 CrossRef CAS.
- H. A. Namanja, D. Emmert, D. A. Davis, C. Campos, D. S. Miller, C. A. Hrycyna and J. Chmielewski, J. Am. Chem. Soc., 2012, 134, 2976–2980 CrossRef CAS PubMed.
- W. Lv, G. Zhang, C. Barinka, J. H. Eubanks and A. P. Kozikowski, ACS Med. Chem. Lett., 2017, 8, 510–515 CrossRef CAS PubMed.
- M. B. Colovi, D. Z. Krstic, T. D. Lazarevi-Pati, A. M. Bondi and V. M. Vasi, Curr. Neuropharmacol., 2013, 11, 315–335 CrossRef PubMed.
- P. Bohn, N. Le Fur, G. Hagues, J. Costentin, N. Torquet, C. Papamicaël, F. Marsais and V. Levacher, Org. Biomol. Chem., 2009, 7, 2612–2618 RSC.
- P. Bohn, F. Gourand, C. Papamicaël, M. Ibazizène, M. Dhilly, V. Gembus, F. Alix, M. L. Ţînţaş, F. Marsais, L. Barré and V. Levacher, ACS Chem. Neurosci., 2015, 6, 737–744 CrossRef CAS PubMed.
- F. Alix, V. Gembus, L. Coquet, M. Hubert-Roux, P. Chan, L. Truong, M. Sebban, G. Coadou, H. Oulyadi, C. Papamicaël and V. Levacher, ACS Omega, 2018, 3, 18387–18397 CrossRef CAS.
- C. Liu, M. Zou, J. Zuo, H. Xie, W. Lyu, J. Xu, F. Feng, H. Sun, W. Liu and X. Jiang, Bioorg. Chem., 2023, 139, 106702 CrossRef CAS PubMed.
- P. Bohn, F. Gourand, C. Papamicaël, M. Ibazizène, M. Dhilly, V. Gembus, F. Alix, M. L. Ţînţaş, F. Marsais, L. Barré and V. Levacher, ACS Chem. Neurosci., 2015, 6, 737–744 CrossRef CAS PubMed.
- L. Peauger, R. Azzouz, V. Gembus, M. L. Ţînţaş, J. Sopková-De Oliveira Santos, P. Bohn, C. Papamicaël and V. Levacher, J. Med. Chem., 2017, 60, 5909–5926 CrossRef CAS PubMed.
- M. L. Ţînţaş, R. Azzouz, L. Peauger, V. Gembus, E. Petit, L. Bailly, C. Papamicaël and V. Levacher, J. Org. Chem., 2018, 83, 10231–10240 CrossRef PubMed.
- K. Fonseca-Azevedo and S. Herculano-Houzel, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18571–18576 CrossRef CAS PubMed.
- S. Mitra, N. Kaushik, I. S. Moon, E. H. Choi and N. K. Kaushik, Biomedicines, 2020, 8, 348 CrossRef CAS PubMed.
- J. Peiró Cadahiá, V. Previtali, N. S. Troelsen and M. H. Clausen, MedChemComm, 2019, 10, 1531–1549 RSC.
- Z. Liu, B. Zhang, S. Xia, L. Fang and S. Gou, Eur. J. Med. Chem., 2021, 212, 112997 CrossRef CAS PubMed.
- N. Bodor, H. H. Farag and M. E. Brewster, Science, 1981, 214, 1370–1372 CrossRef CAS PubMed.
- K. W. Morin, L. I. Wiebe and E. E. Knaus, Carbohydr. Res., 1993, 249, 109–116 CrossRef CAS PubMed.
- S. K. Aggarwal, S. R. Gogu, S. R. S. Rangan and K. C. Agrawal, J. Med. Chem., 1990, 33, 1505–1510 CrossRef CAS PubMed.
- M. E. Brewster, K. Raghavan, E. Pop and N. Bodor, Antimicrob. Agents Chemother., 1994, 38, 817–823 CrossRef CAS PubMed.
- C. K. Chu, V. S. Bhadti, K. J. Doshi, J. T. Etse, J. M. Gallo, F. D. Boudinot and R. F. Schinazi, J. Med. Chem., 1990, 33, 2188–2192 CrossRef CAS PubMed.
- V. Carelli, F. Leberatore, L. Scipione, M. Impicciatore, E. Barocelli, M. Cardellini and G. Giorgioni, J. Controlled Release, 1996, 42, 209–216 CrossRef CAS.
- M. A. Sharpe, J. Han, A. M. Baskin and D. S. Baskin, ChemMedChem, 2015, 10, 621–628 CrossRef CAS PubMed.
- K. Prokai-Tatrai, P. P. Perjési, A. D. Zharikova, X. Li and L. Prokai, Bioorg. Med. Chem. Lett., 2002, 12, 2171–2174 CrossRef CAS PubMed.
- L. Prokai, K. Prokai-Tatrai, A. D. Zharikova, V. Nguyen, P. Perjesi and S. M. Stevens, J. Med. Chem., 2004, 47, 6025–6033 CrossRef CAS PubMed.
- K. Prokai-Tatrai, V. Nguyen, S. Szarka, K. Konya and L. Prokai, Pharmaceutics, 2013, 5, 318–328 CrossRef CAS PubMed.
- E. Pop, T. Loftsson and N. Bodor, Pharm. Res., 1991, 8, 1044–1049 CrossRef CAS PubMed.
- N. Bodor and J. J. Kaminski, J. Mol. Struct., 1988, 163, 315–330 CrossRef.
- E. Pop, M. E. Brewster, M.-J. Huang and N. Bodor, J. Mol. Struct., 1995, 337, 49–55 CrossRef CAS.
- N. Bodor, H. H. Farag, M. D. C. Barros, W. M. Wu and P. Buchwald, J. Drug Targeting, 2002, 10, 63–71 CrossRef CAS PubMed.
- L. Foucout, F. Gourand, M. Dhilly, P. Bohn, G. Dupas, J. Costentin, A. Abbas, F. Marsais, L. Barré and V. Levacher, Org. Biomol. Chem., 2009, 7, 3666–3673 RSC.
- F. Gourand, M. L. Ţînţaş, A. Henry, M. Ibazizène, M. Dhilly, F. Fillesoye, C. Papamicaël, V. Levacher and L. Barré, ACS Chem. Neurosci., 2017, 8, 2457–2467 CrossRef CAS PubMed.
- F. Gourand, D. Patin, A. Henry, M. Ibazizène, M. Dhilly, F. Fillesoye, O. Tirel, M. L. Tintas, C. Papamicaël, V. Levacher and L. Barré, ACS Med. Chem. Lett., 2019, 10, 352–357 CrossRef CAS PubMed.
- M. L. Ţînţaş, V. Gembus, F. Alix, A. Barré, G. Coadou, L. Truong, M. Sebban, C. Papamicaël, H. Oulyadi and V. Levacher, Eur. J. Med. Chem., 2018, 155, 171–182 CrossRef PubMed.
- A. Barré, R. Azzouz, V. Gembus, C. Papamicaël and V. Levacher, Molecules, 2019, 24, 1–14 CrossRef PubMed.
- Y. Zhao, L. Zhang, Y. Peng, Q. Yue, L. Hai, L. Guo, Q. Wang and Y. Wu, Chem. Biol. Drug Des., 2018, 91, 707–716 CrossRef CAS PubMed.
- D. Xiao, F. H. Meng, W. Dai, Z. Yong, J. Q. Liu, X. B. Zhou and S. Li, Molecules, 2016, 21, 488 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |