DOI:
10.1039/D4MD00929K
(Research Article)
RSC Med. Chem., 2025, Advance Article
Synthesis and evaluation of 6-arylaminobenzamides as positron emission tomography imaging ligands for the sphingosine-1-phosphate-5 receptor†
Received
27th November 2024
, Accepted 24th December 2024
First published on 3rd January 2025
Abstract
The sphingosine-1-phosphate-5 (S1P5) receptor is one of the five membrane G protein-coupled receptors that are activated by the lysophospholipid, sphingosine-1-phosphate, resulting in regulation of many cellular processes. S1P5 receptors are located on oligodendrocytes and are proposed to influence oligodendrocyte physiology. Understanding S1P5 modulation during processes such as remyelination could have potential applications for demyelinating CNS disorders such as multiple sclerosis (MS). Herein, we report the synthesis and preliminary evaluation of a series of fluorinated 6-arylaminobenzamides as positron emission tomography (PET) ligands of S1P5. Pharmacokinetic screening and binding evaluation using a [35S]GTPγS assay led to the discovery of TEFM78, a selective and high affinity agonist of S1P5. Radiosynthesis of [18F]TEFM78 allowed pilot PET imaging studies in an animal model, which showed that [18F]TEFM78 can cross the blood brain barrier with good uptake in rat brain and spinal cord.
Introduction
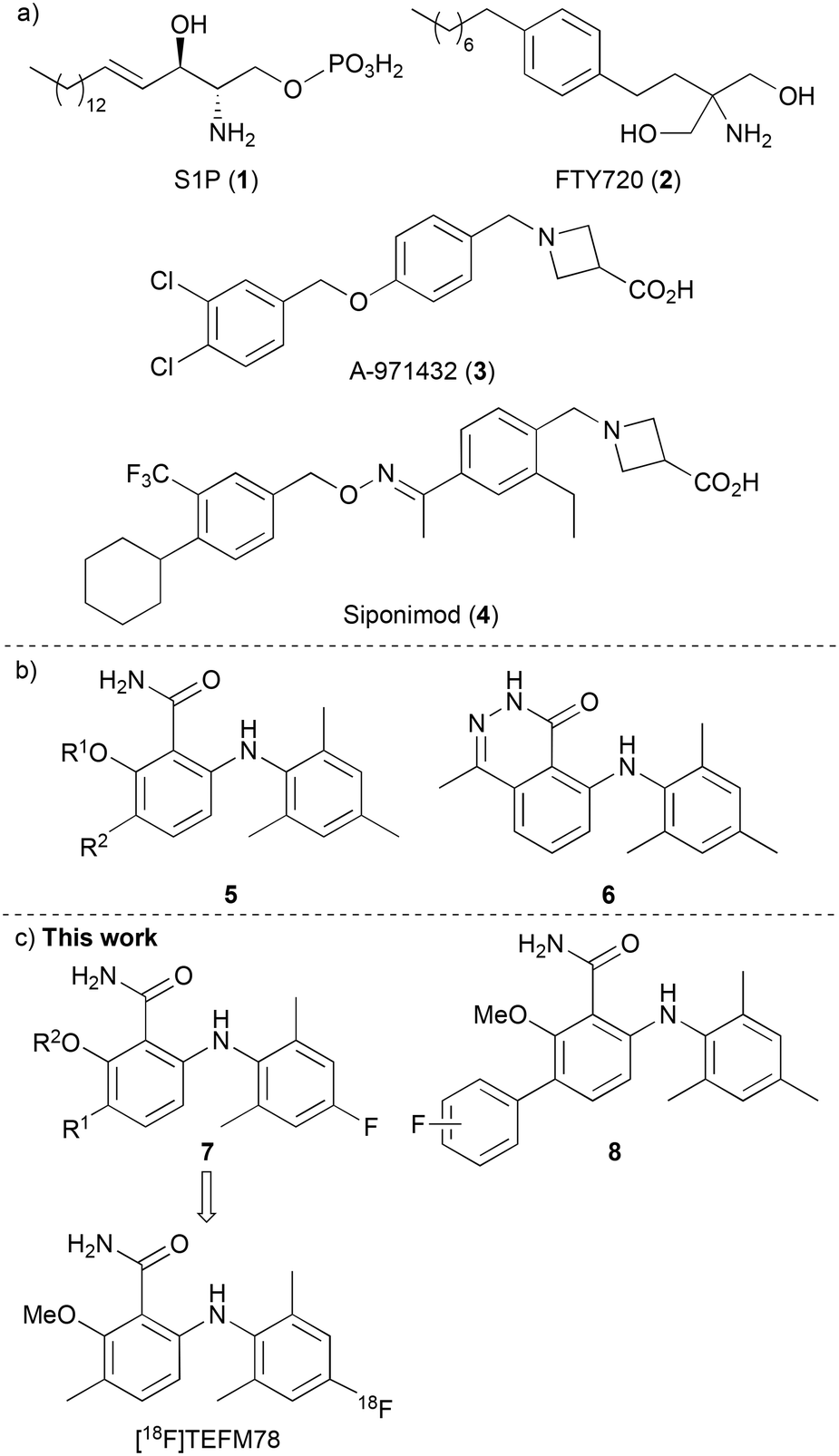
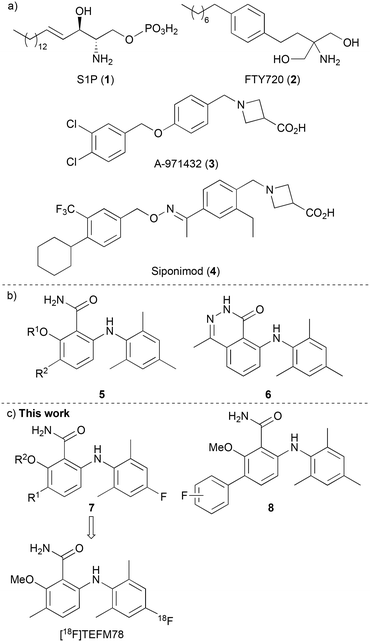
The lysophospholipid sphingosine-1-phosphate (S1P) (1) is an important signaling molecule, which plays a key role as a cellular mediator (Fig. 1a).1,2 This activity occurs through interaction with five highly specific, membrane protein-coupled receptors, S1P1–5, which are involved in cell growth and apoptosis in cells of the immune, cardiovascular and central nervous systems.3,4 Interest in the S1P1–5 receptors increased following the U.S. Food and Drug Administration approval of the S1P analogue FTY720 (Fingolimod, 2), developed for the treatment of patients with relapsing-remitting multiple sclerosis (MS).5,6 On phosphorylation, FTY720 engages with four of the S1P receptors (all except S1P2), and crucially, acts as a functional antagonist to S1P1. This results in receptor internalisation and degradation, leading to lymphocyte sequestration in lymph nodes, preventing entry into the CNS and a relapse of MS.
 |
| | Fig. 1 a) Agonists of S1P receptors. b) Selective agonists of S1P5. c) This work: assessment of fluorinated 6-arylaminobenzamides. | |
The S1P5 receptor is mainly expressed in white matter tracts of the CNS, on oligodendrocytes, brain cells that are involved in the regulation of the myelination process.7,8 Studies have shown that S1P activation of S1P5 receptors promotes the survival of myelin-forming mature oligodendrocytes,9 and this has led to the proposal that selective agonists of this receptor could also have potential applications in various demyelinating CNS disorders such as MS. In fact, selective agonists (e.g. 3, Fig. 1a)10 and antagonists11 of S1P5 have been reported. This includes Siponimod (4), a selective S1P1 and S1P5 modulator that has been recently approved for the treatment of MS, especially for the active secondary progressive form of the disease.12 However, the exact mechanism of action of compounds such as siponimod is still unclear and thus, tool compounds are required to fully understand the effects of S1P5 modulation and how this relates to neurodegenerative disease.
The radionuclide imaging technique of positron emission tomography (PET) is widely used in the biological and medical sciences for the study of biochemical processes in the living body.13 The use of fluorine-18 labelled compounds is of particular interest due to the relatively long half-life (t1/2 = 109.7 min) and low emission energy of this radioisotope, which results in high resolution images.14 In recent years, various PET radioligands have been reported as imaging tools for the S1P receptors, with particular focus on the development of carbon-11 and fluorine-18 labelled compounds for the study of S1P1.15 As far as we are aware, no fluorine-18 radioligands with S1P5 specificity have yet been reported. In 2010, Mattes and co-workers reported a series of selective and potent S1P5 agonists that included 6-arylaminobenzamides 5 and 6-arylaminophthalazinone 6 (Fig. 1b).16 Based on the high receptor selectivity and bioavailability properties of the 6-arylaminobenzamides, we proposed that these could serve as suitable scaffolds for a structure activity relationship study for the discovery of a S1P5 PET radioligand. It was believed that fluorine atom incorporation at one of the existing aromatic rings (e.g. 7) or the addition of a fluoro-(hetero)aryl group (8) would generate potential PET imaging agents for S1P5 (Fig. 1c). Here, we report the design, synthesis and pharmacokinetic properties of a series of fluorinated 6-arylaminobenzamides as selective agonists of the S1P5 receptor. We also describe the preliminary evaluation of lead compound [18F]TEFM78, which showed good brain and spinal cord uptake in rodents.
Results and discussion
Design and chemistry
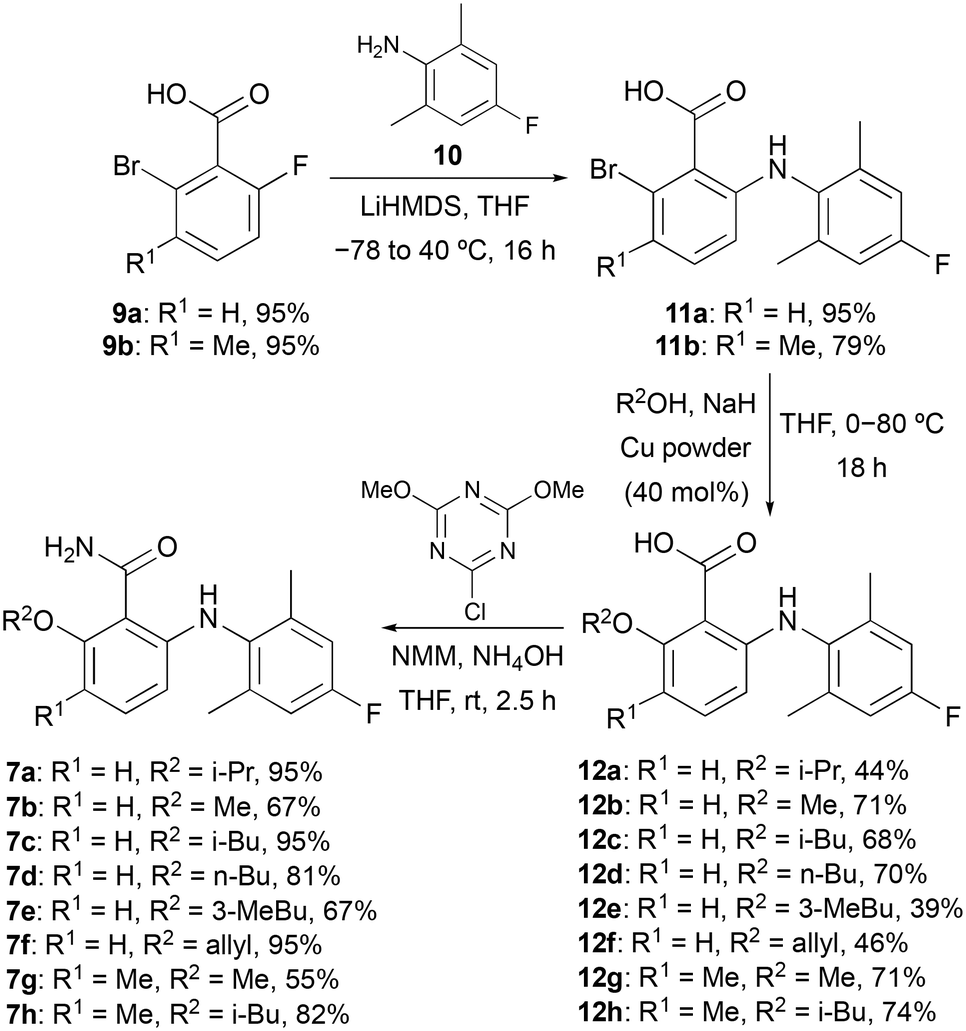
Docking studies performed by Mattes and co-workers using homology models of bovine rhodopsin and a 6-arylaminobenzamide suggested these compounds adopt a twisted conformation with the benzamide ring residing in a large hydrophobic pocket.16 In addition, smaller cavities near the ortho- and meta-positions of the benzamide ring indicated positions to develop receptor selectivity. Using this insight, our first series of fluorinated 6-arylaminobenzamides explored various alkoxy groups at the ortho-position of the benzamide, while incorporating the key fluorine atom within the aniline ring (Scheme 1). These compounds were prepared in three steps. Nucleophilic aromatic substitution of 2-bromo-6-fluorobenzoic acid (9a) with 2,6-dimethyl-4-fluoroaniline (10) in the presence of LiHMDS,17 gave 2-arylaminobenzamide 11a in 95% yield. Various alkoxy groups were then introduced at the ortho-position using a copper-catalysed Ullmann coupling of bromide 11a with a range of alcohols.18 The synthesis of the first series of targets 7a–7f was then completed by amidation using 2-chloro-4,6-dimethoxy-1,3,5-triazine, N-methylmorpholine (NMM) and ammonia.19 Further SAR studies by the Novartis group demonstrated that high S1P5 selectivity could be achieved by the incorporation of substituents at the meta-position of the benzamide ring.16 Based on this, two further compounds (7g and 7h) containing a 3-methyl group were prepared using this route from 2-bromo-3-methyl-6-fluorobenzoic acid (9b).
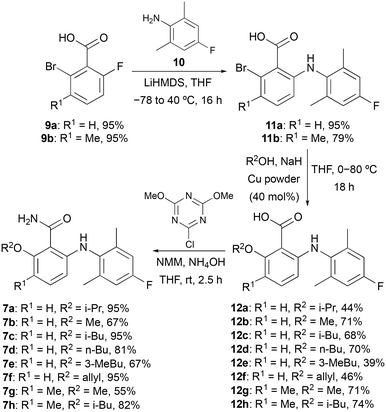 |
| | Scheme 1 Three-step synthesis of fluorinated 6-arylaminobenzamides 7a–7h. | |
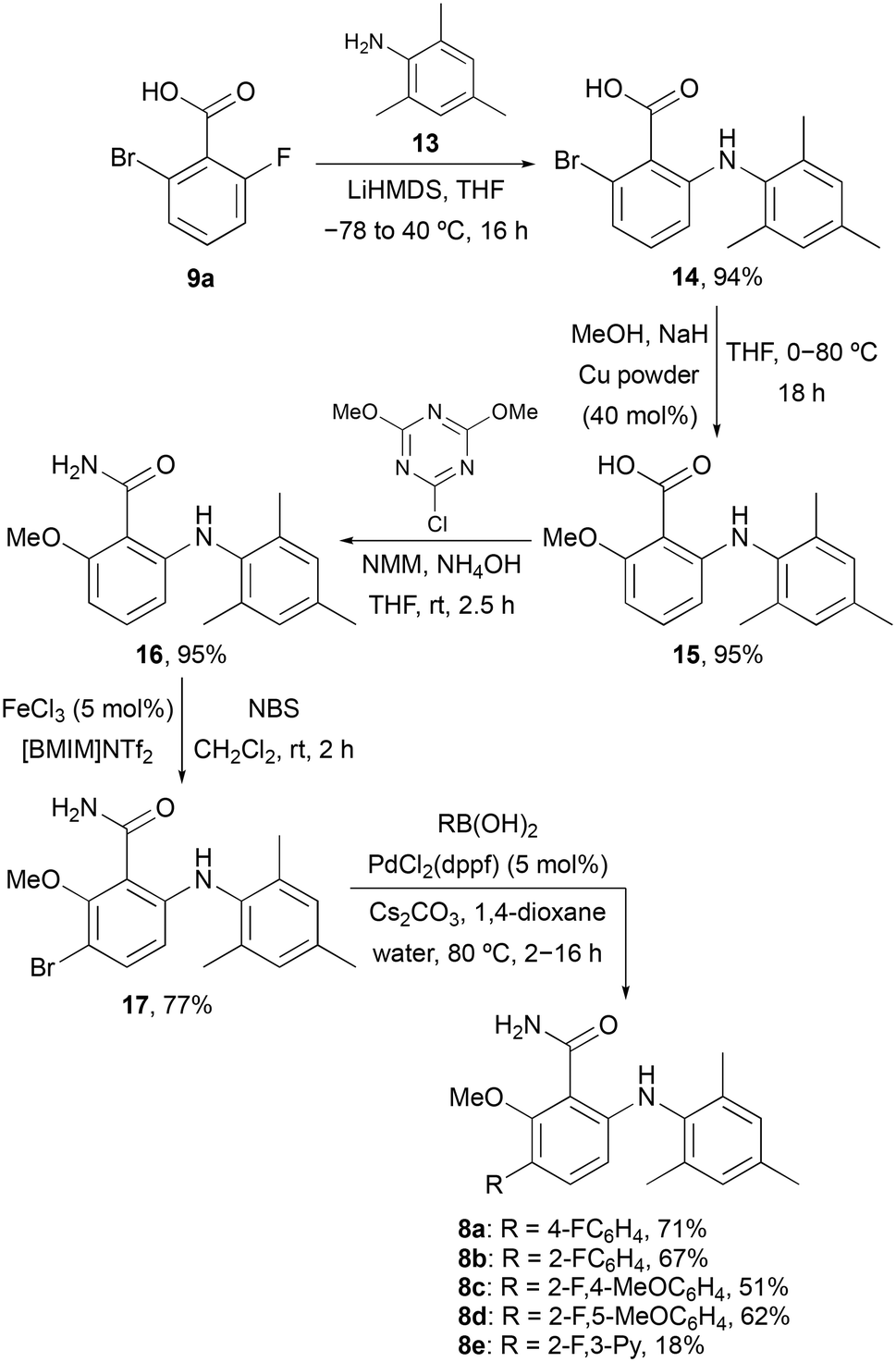
To further probe the meta-position of the benzamide ring, a second series of agonists of the S1P5 receptor was prepared by incorporation of a fluorine-containing (hetero)aryl substituent at this position (Scheme 2). The same series of reactions involving nucleophilic aromatic substitution, copper-mediated methanolysis and amidation gave 6-arylaminobenzamide 16 in 85% overall yield. Substitution of the benzamide ring was then performed using a two-step strategy. The ring was initially brominated using N-bromosuccinimide (NBS) and the super Lewis acid catalyst iron(III) triflimide (generated from FeCl3 and the ionic liquid [BMIM]NTf2),20 which allowed the regioselective synthesis of 17 in 77% yield. Introduction of the fluorinated (hetero)aryl groups was then achieved using the Suzuki–Miyaura cross-coupling reaction with various boronic acids.21 This gave compounds 8a–8e in 18–71% yields.
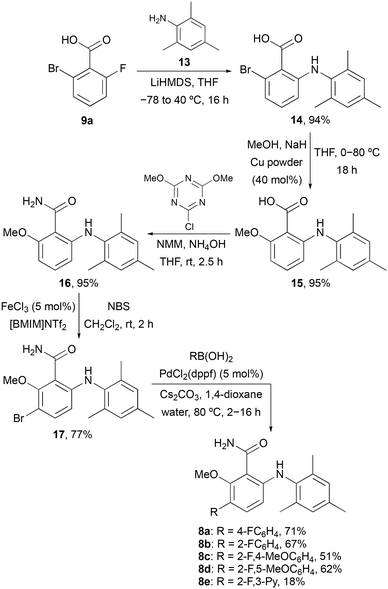 |
| | Scheme 2 Synthesis of 6-arylaminobenzamides 8a–8e. | |
Physicochemical and binding studies
The physicochemical properties of both sets of compounds was initially performed to assess their potential as brain imaging agents. The calculated partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P), the permeability (Pm), the membrane partition coefficient (Km) and the percentage of plasma protein binding (%PPB) were determined for each compound using established methods (see ESI,† Table S1).22,23 As expected, the biaryl amines showed high plasma protein binding, with calculated logP values from 3.41–5.57. The permeability and membrane partition coefficient for the majority of compounds were well within the ideal limits for these parameters (Pm < 0.5; Km < 250), which are based on known CNS imaging agents.23
P), the permeability (Pm), the membrane partition coefficient (Km) and the percentage of plasma protein binding (%PPB) were determined for each compound using established methods (see ESI,† Table S1).22,23 As expected, the biaryl amines showed high plasma protein binding, with calculated logP values from 3.41–5.57. The permeability and membrane partition coefficient for the majority of compounds were well within the ideal limits for these parameters (Pm < 0.5; Km < 250), which are based on known CNS imaging agents.23
A GTPγS assay was then conducted to evaluate receptor agonist activity of each compound at nine concentrations from 100 fM to 10 μM (n = 3).24,25 Initially, all compounds were tested for agonist activity with S1P5 and relative efficacy versus S1P (Table 1).26 Compounds which showed an EC50 value below 40 nM for S1P5, were then assessed against S1P1, S1P2 and S1P3. Previous work by the Novartis group described the importance of hydrogen bonding between the carboxamide and 2-substituent, to maintain an in-plane conformation of the amide that was critical for agonist activity against S1P receptors.16 Similar results were observed in this study. The use of relatively bulky alkoxy groups (e.g. 7a, 7c, 7d and 7h) that likely disrupt hydrogen bonding to the amide showed loss of agonist activity with S1P5. This was more pronounced for compounds with both a bulky alkoxy group and a 3-substituent, with 6-arylaminobenzamide 7h found to have the weakest activity. From this library, five compounds were found to have strong S1P5 activity and were screened against the other receptors. Compound 7f with a 2-allyloxy group was found to have the highest S1P5 agonist activity, while 8e with a 2-fluoropyridin-3-yl motif at the 2-position was also found to have good activity. However, both compounds showed high activity for the other receptors, particularly, 8e with a EC50 value of 0.336 nM against S1P1. Of the other compounds, 7b and 7g were found to possess high activity for S1P5, while showing good selectivity against the other receptors. Based on the binding studies and the physicochemical results, 6-arylaminobenzamide 7g, named as TEFM78 was chosen as the lead compound. As well as high activity and good selectivity for S1P5, TEFM78 possessed ideal Pm (0.233) and Km (70.5) values and one of the lowest logP values of 3.90.
Table 1 Binding studies of 7a–7h and 8a–8e with S1P5 and other S1P receptors (n = 3, errors in parentheses are derived from 95% confidence intervals)
| |
Efficacy (%) |
EC50 (nM) |
| S1P5 |
S1P1 |
S1P2 |
S1P3 |
| S1P (1) |
100 |
10.7 (3.56) |
— |
— |
42.6 (21.8) |
| 7a |
105 |
419 (159) |
— |
— |
— |
| 7b |
108 |
39.8 (9.92) |
>10000 |
>10000 |
>10000 |
| 7c |
137 |
180 (87.7) |
— |
— |
— |
| 7d |
140 |
234 (169) |
— |
— |
— |
| 7e |
102 |
49.0 (97.4) |
— |
— |
— |
| 7f |
164 |
6.66 (3.09) |
25.58 (1549) |
>10000 |
27.64 (106.2) |
| 7g |
183 |
22.3 (5.20) |
104 (11428) |
>10000 |
2030 (5239) |
| 7h |
154 |
2238 (3589) |
— |
— |
— |
| 8a |
129 |
72.3 (46.3) |
— |
— |
— |
| 8b |
136 |
954 (514) |
— |
— |
— |
| 8c |
120 |
29.8 (24.0) |
76.0 (691) |
>10000 |
>10000 |
| 8d |
142 |
612 (1262) |
— |
— |
— |
| 8e |
175 |
23.4 (21.3) |
0.336 (6.458) |
>10000 |
172 (102) |
Docking studies
To determine how TEFM78 (7g) binds to S1P5, docking studies were performed. Previously, Mattes and co-workers used homology models based on bovine rhodopsin for docking studies.16 However, in the last few years, structures of S1P5 with bound agonists have been reported and these have been used to understand Gi protein coupling, the mechanism of activation and drug recognition.27–29 For this study, we chose the cryo-electron microscopy (EM) structure of the S1P5-Gi heterotrimer-scFV16 complex with bound agonist, Siponimod (4) (PDB code: 7EW1).29,30 Docking studies with this complex showed that TEFM78 binds in the same orthosteric site as S1P and other agonists (Fig. 2a).27 This is composed of an upper hydrophilic pocket that binds the phosphonate group of S1P and a lower hydrophobic region, which accommodates the hydrocarbon chain. In their docking and SAR studies, Mattes and co-workers proposed that a twisted 6-arylaminobenzamide structure with hydrogen bonding between the aniline and amide was critical for affinity with S1P5. Our docking studies were found to corroborate this view. TEFM78 adopts a twisted conformation with a 72° angle between the two aryl rings and intramolecular hydrogen bonding involving the amide carbonyl and aniline NH moiety (1.95 Å), as well as the 2-methoxy substituent and amide NH2 group (1.82 Å) (Fig. 2b). Both aryl rings occupy hydrophobic pockets with multiple Van der Waals contacts. For example, the fluorophenyl moiety has close contacts with Leu186, Tyr193 and Phe116, while the benzamide moiety residues in a pocket composed of various amino acid residues such as leucine, valine and tryptophan. In comparison to other agonists, the benzamide moiety of TEFM78 occupies a similar region as the lipophilic end of Siponimod (4) (Fig. 2c). More specifically, the 2-methoxy and 3-methyl substituents of TEFM78 reside in the same lipophilic sub-pocket as the trifluoromethyl group of Siponimod. The alkoxy groups of other agonists such as Cenerimod and Ozanimod also bind in this sub-pocket.29 There is also overlap between the TEFM78 amide and the Siponimod oxime. The main difference between the two structures is caused by the twisting of the 6-arylaminobenzamide, which positions the fluorophenyl moiety in a more preferred lipophilic cavity. These docking studies give an insight into the binding of TEFM78 with S1P5 and provide a model for the design of future analogues.
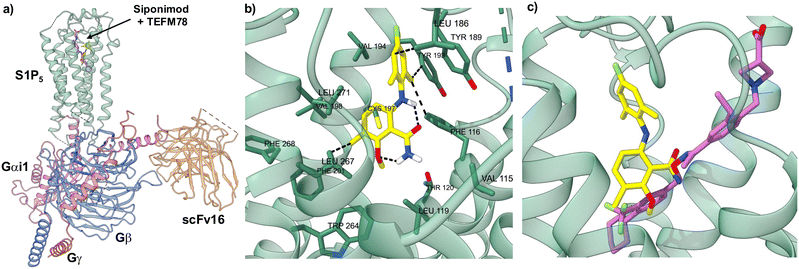 |
| | Fig. 2 Docking studies of TEFM78 (7g) with the cryo-EM structure of S1P5. a) The cryo-EM structure of S1P5-Gi heterotrimer-scFv16 complex with Siponimod (purple) and docked TEFM78 (yellow). b) Close up view of docked TEFM78 showing key interactions. c) Close up view of the Siponimod (purple structure) binding site with docked TEFM78 (yellow structure). | |
Radiochemistry
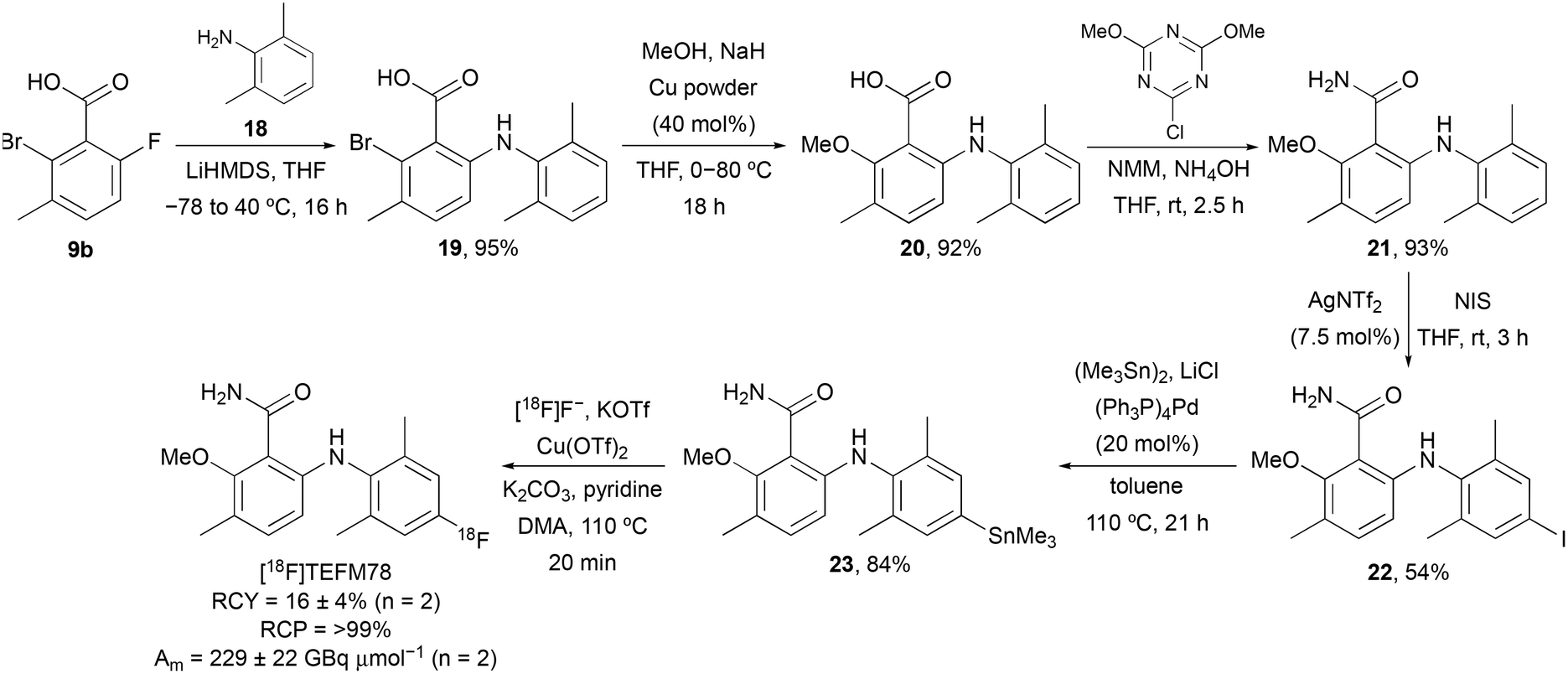
On identification of TEFM78 as the lead compound, a synthetic route to access a suitable precursor for late stage radiofluorination was developed (Scheme 3). Our approach focused on the synthesis of a 6-arylaminobenzamide that could undergo late-stage functionalisation at the 4′-position for subsequent radiofluorination. Nucleophilic aromatic substitution of 9b with 2,6-dimethylaniline (18), followed by Ullmann coupling and amidation gave benzamide 21 in 81% yield over the three steps. Activation of the electron-rich aniline ring was achieved using silver(I) triflimide-catalysed iodination.17 Following optimisation (solvent and reaction time), this gave 22 in 54% yield. Subsequent Pd(0)-catalysed stannylation gave trimethyltin precursor 23 in 84% yield. The radiosynthesis of [18F]TEFM78 was then performed using a TRACERlab FXFN automated synthesiser. No-carrier-added [18F]fluoride from the cyclotron was trapped on a carbonate-pre-conditioned quaternary methyl ammonium (QMA) cartridge, eluted into the reaction vessel and then azeotropically dried. Radiofluorination studies with 23 and using Cu(OTf)2 identified a temperature of 110 °C and a 20 minute reaction time as optimal. Following semi-preparative HPLC purification, [18F]TEFM78 was isolated in 16 ± 4% radiochemical yield (RCY), >99% radiochemical purity (RCP) and with a molar activity (Am) of 229 ± 22 GBq μmol−1, starting from 75.3 ± 2.2 GBq of [18F]fluoride. The total synthesis time of [18F]TEFM78 from delivery of [18F]fluoride to extraction of product was 62 minutes. [18F]TEFM78 was found to be highly stable by both radio-HPLC and radio-TLC with >99% RCP ten hours after production.
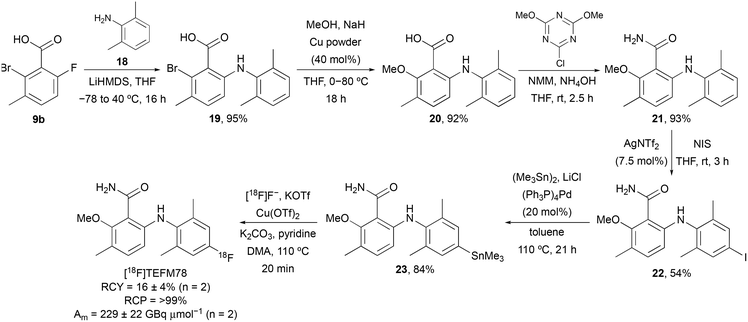 |
| | Scheme 3 Synthesis of [18F]TEFM78. | |
Pilot PET in vivo imaging studies with [18F]TEFM78
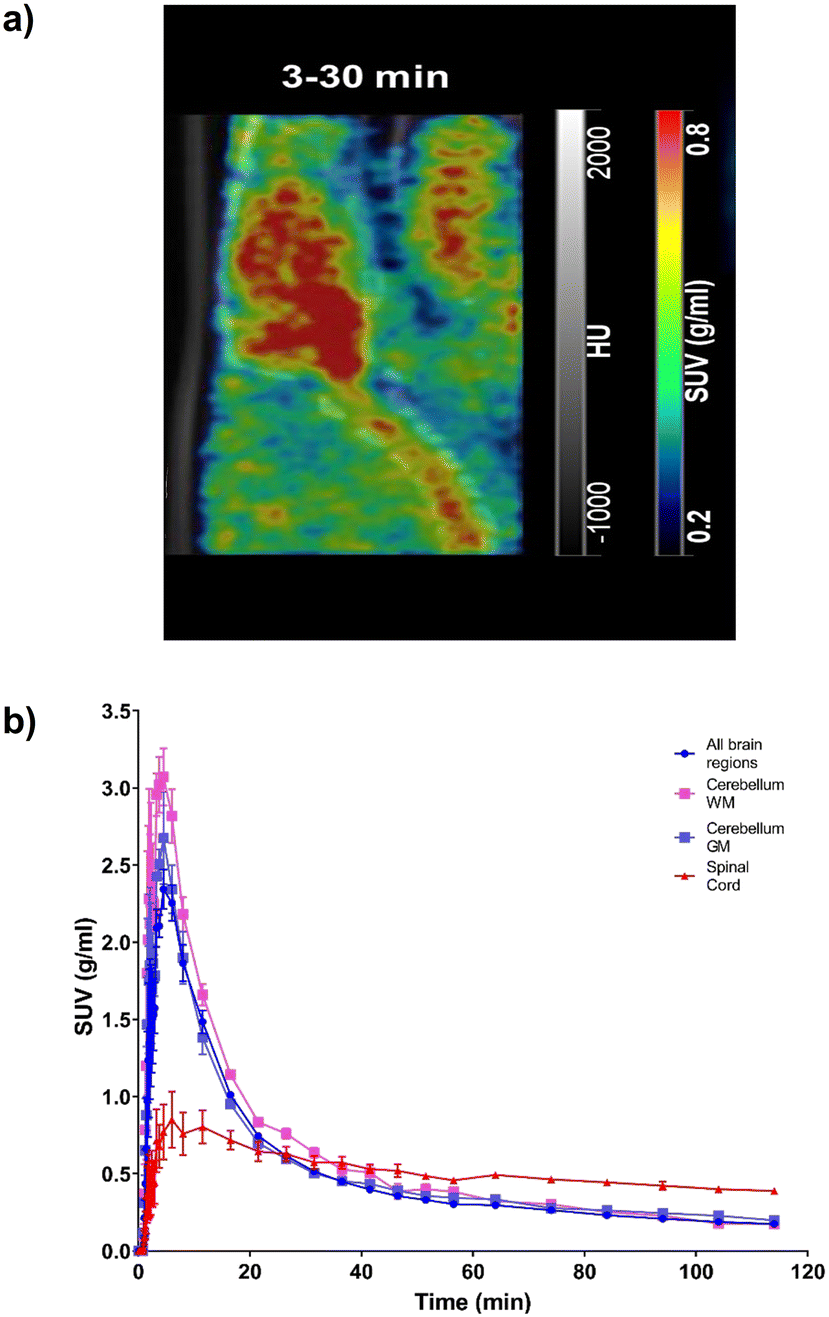
Following successful preparation of [18F]TEFM78, PET imaging with rodents was next conducted. On intravenous bolus administration of [18F]TEFM78, there was rapid uptake of the radiotracer into the rat brain (Fig. 3a), followed by clearance within 20 minutes of the scan starting (Fig. 3b). A peak standard uptake value (SUV) of 3 g mL−1 was measured in the brain, demonstrating good brain penetration (typical recommended peak SUV for successful brain PET ligands should be >1 g mL−1).31 The biodistribution and kinetics of the spinal cord were different from the brain with a lower peak radiotracer uptake but slower clearance (Fig. 3b). This initial data showed that [18F]TEFM78 can penetrate the mammalian brain and display an in vivo kinetics profile compatible with the half-lives of PET imaging radioisotopes. Typically, low free fraction of compounds can limit brain penetration. However, as shown in the PET images presented in Fig. 3, despite low free fraction, brain penetration of [18F]TEFM78 is successfully achieved. This demonstrates that, albeit low, the measured radiotracer free fraction does not preclude blood brain barrier penetration and successful brain PET imaging in rats. Notwithstanding, further preclinical studies are needed to investigate radiotracer brain kinetics in more detail and it is possible that higher free fractions would be required for successful human brain imaging.
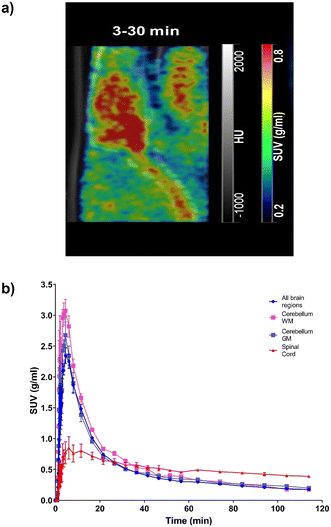 |
| | Fig. 3 Brain and spinal cord kinetics of [18F]TEFM78 in naïve rats. The molar activity of [18F]TEFM78 used in this study was 50.2 ± 14 GBq mmol−1. a) Representative SUV PET image average between 3 and 30 minutes post-intravenous bolus injection showing good brain penetration and spinal cord uptake. The PET image was overlaid onto the CT scan. b) Average [18F]TEFM78 time activity curves for whole brain, whole grey matter, whole white matter and spinal cord in rats showing faster clearance from the brain compared with the spinal cord. Data expressed as SUV and presented as mean ± SEM, n = 3. | |
Conclusions
In summary, various fluorinated 6-arylaminobenzamides have been prepared and evaluated as potential PET imaging agents of the S1P5 receptor. One series with a 4′-fluorophenylamino group examined the SAR of the 3-alkoxy substituent of the benzamide ring, while the second series investigated the incorporation of the fluorine atom through an additional 4-aryl benzamide substituent. Binding evaluation using a [35S]GTPγS assay against the main S1P receptors identified TEFM78 (7g) as a selective and high affinity agonist of S1P5. Physicochemical analysis of TEFM78 revealed that this compound possessed ideal Pm and Km values and a good logP value of 3.90. Based on these results, a synthetic route for the preparation of [18F]TEFM78 was developed, with the resulting imaging agent used in pilot PET imaging studies with rodents. These studies showed fast uptake in rat brain with clearance within 20 minutes. Based on these encouraging results, future work will focus on further in vivo assessment of [18F]TEFM78, including biodistribution studies with disease models.
Experimental
General experimental
All reagents and starting materials were obtained from commercial sources and used as received. All reactions were performed under an atmosphere of air unless otherwise stated. All dry solvents were purified using a PureSolv 500 MD solvent purification system. Brine is defined as a saturated solution of aqueous sodium chloride. Flash column chromatography was carried out using Merck Geduran Si 60 (40–63 μm). Merck aluminium-backed plates pre-coated with silica gel 60 (UV254) were used for thin layer chromatography and were visualised under ultraviolet light and by staining with KMnO4, ninhydrin or vanillin. 1H NMR and 13C NMR spectra were recorded on a Bruker DPX 400 spectrometer or Bruker 500 spectrometer with chemical shift values in ppm relative to tetramethylsilane (δH 0.00 and δC 0.0) or residual chloroform (δH 7.26 and δC 77.2) as the standard. Assignment of 1H and 13C NMR signals are based on 2-dimensional COSY, HSQC and DEPT experiments. Infrared spectra were recorded using a Shimadzu FTIR-84005 spectrometer and mass spectra were obtained using a JEOL JMS-700 spectrometer or a Bruker MicroTOFq high resolution mass spectrometer. Melting points were determined on a Gallenkamp melting point apparatus. Purity of all final compounds is >95%, as determined by HPLC.
General procedure A: nucleophilic aromatic substitution of 2-fluorobenzoic acids. The benzoic acid (1.0 equiv.) was dissolved in tetrahydrofuran (4.5 mL mmol−1) under an atmosphere of argon. The aniline (2.0 equiv.) was added and the mixture cooled to −78 °C. Lithium bis(trimethylsilyl)amide (3.0 equiv.; 1.0 M in tetrahydrofuran) was then slowly added over 0.1 h. The reaction mixture was then allowed to reach room temperature before heating to 40 °C and stirring for 16 h. The reaction was quenched with water (2.5 mL mmol−1) and acidified to pH 2 with 10% aqueous hydrochloric acid. The mixture was then extracted with ethyl acetate (3 × 10 mL), dried (MgSO4), filtered and concentrated in vacuo. The resulting residue was then purified by flash column chromatography, eluting with 20–40% ethyl acetate in petroleum ether (40–60) to afford the benzoic acid derivative.
General procedure B: Ullmann condensation of 2-bromobenzoic acids. Sodium hydride (3.0 equiv.; 60% in mineral oil) was added to a dry flask under argon. The sodium hydride was then washed with hexane (2 × 5 mL mmol−1) to remove the oil and the flask cooled to 0 °C. The required dry alcohol (3.3 mL mmol−1) was added to the reaction vessel carefully over 0.1 h and then stirred for 0.5 h. The benzoic acid derivative (1.0 equiv.) was added followed by the addition of copper powder (0.4 equiv.) and the reaction mixture heated to 80 °C for 18 h. The reaction mixture was cooled to room temperature and filtered through a pad of Celite®. The filtrate was concentrated in vacuo and dissolved in water (3.3 mL mmol−1). The mixture was acidified to pH 2 using 10% aqueous hydrochloric acid and extracted with dichloromethane (3 × 5.0 mL mmol−1). The organic layer was dried (MgSO4), filtered and concentrated in vacuo. The resulting residue was then purified by flash column chromatography, eluting with 20% ethyl acetate in petroleum ether (40–60) to afford the ether product.
General procedure C: amidation of benzoic acid derivatives. The benzoic acid derivative (1.0 equiv.) was dissolved in tetrahydrofuran (3.0 mL mmol−1) and 2-chloro-4,6-dimethoxy-1,3,5-triazine (1.2 equiv.) and N-methylmorpholine (3.0 equiv.) were added. The mixture was then stirred for 2 h at room temperature. The precipitate was filtered and ammonium hydroxide (9.0 mL mmol−1) was added to the filtrate. The reaction mixture was stirred for 0.5 h at room temperature and then filtered. 2 M sodium hydroxide (9.0 mL mmol−1) was added to the filtrate and the crude product was extracted with ethyl acetate (3 × 9.0 mL mmol−1). The organic layer was dried (MgSO4), filtered and concentrated in vacuo. The resulting residue was then purified by flash column chromatography, eluting with 10% ethyl acetate in petroleum ether (40–60) to afford the benzamide product.
General procedure D: Suzuki–Miyaura reaction of 3-bromobenzamides. To a reaction vessel containing the aryl bromide (1.0 equiv.) in 1,4-dioxane (15 mL mmol−1) and water (1.1 mL mmol−1) was added [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II) (1:1) (5 mol%), the boronic acid (1.6 equiv.) and caesium carbonate (3.0 equiv.). The reaction mixture was then sealed and degassed under argon for 0.2 h, before heating to 80 °C for 2–16 h. The reaction mixture was cooled, filtered through Celite® and washed with ethyl acetate (50 mL mmol−1). The filtrate was washed with water (3 × 50 mL mmol−1) and brine (50 mL mmol−1) and the organic layer dried (MgSO4), filtered and concentrated in vacuo. The resulting residue was then purified by flash column chromatography to afford the coupled product.
2-Bromo-6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]benzoic acid (11a). The reaction was carried out according to general procedure A and gave 2-bromo-6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]benzoic acid (11a) as a brown solid (1.45 g, 95%). Mp 148–150 °C; IR (neat) 2920, 1672, 1595, 1560, 1481, 1439, 1242, 858, 767 cm−1; 1H NMR (500 MHz, CDCl3): δ 11.19 (br s, 1H, OH), 7.78 (br s, 1H, NH), 7.00 (dd, 1H, J = 7.8, 1.4 Hz, 3-H), 6.97 (t, 1H, J = 7.8 Hz, 4-H), 6.85 (d, 2H, J = 9.0 Hz, 3′-H and 5′-H), 6.15 (dd, 1H, J = 7.8, 1.4 Hz, 5-H), 2.18 (s, 6H, 2′-CH3 and 6′-CH3); 13C{1H} NMR (126 MHz, CDCl3): δ 172.1 (C), 161.0 (C, d, 1JCF = 245.3 Hz), 150.1 (C), 138.9 (2× C, d, 3JCF = 8.9 Hz), 133.7 (CH), 132.5 (C, d, 4JCF = 2.8 Hz), 123.6 (CH), 123.2 (C), 115.1 (2× CH, d, 2JCF = 21.9 Hz), 113.2 (C), 111.9 (CH), 18.39 (CH3), 18.38 (CH3); MS (EI) m/z 337 (M+, 84%), 319 (72), 293 (58), 212 (100), 198 (37); HRMS (EI) m/z: [M]+ calcd for C15H1379BrFNO2 337.0114; found 337.0122.
2-Bromo-6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]-3-methylbenzoic acid (11b). The reaction was carried out according to general procedure A and gave 2-bromo-6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]benzoic acid (11b) as a brown solid (0.120 g, 79%). Mp 130–132 °C; IR (neat) 2924, 1696, 1606, 1491, 1215, 1129, 1019, 862, 757 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.02 (d, 1H, J = 8.5 Hz, 4-H), 6.83 (d, 2H, J = 9.0 Hz, 3′-H and 5′-H), 6.09 (d, 1H, J = 8.5 Hz, 5-H), 2.33 (s, 3H, 3-CH3), 2.17 (s, 6H, 2′-CH3 and 6′-CH3); 13C{1H} NMR (101 MHz, CDCl3): δ 173.2 (C), 160.7 (C, d, 1JCF = 245.0 Hz), 146.2 (C), 138.6 (2× C, d, 3JCF = 8.6 Hz), 134.1 (CH), 133.0 (C, d, 4JCF = 2.8 Hz), 128.1 (C), 124.2 (C), 116.8 (C), 115.0 (2× CH, d, 2JCF = 21.8), 112.0 (CH), 23.0 (CH3), 18.37 (CH3), 18.36 (CH3); MS (ESI) m/z 350 ([M–H]−, 100%); HRMS (ESI) m/z: [M–H]− calcd for C16H1479BrFNO2 350.0197; found 350.0197.
6-[(4′-Fluoro-2′,6′-dimethylphenyl)amino]-2-isopropoxybenzoic acid (12a). The reaction was carried out according to general procedure B and gave 6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]-2-isopropoxybenzoic acid (12a) as an orange solid (0.062 g, 44%). Mp 99–101 °C; IR (neat) 3250, 2982, 1697, 1578, 1458, 1252 cm−1; 1H NMR (400 MHz, CDCl3): δ 12.04 (br s, 1H, OH), 9.89 (br s, 1H, NH), 7.10 (t, 1H, J = 8.4 Hz, 4-H), 6.83 (d, J = 9.0 Hz, 3′-H and 5′-H), 6.24 (br d, 1H, J = 8.4 Hz, 3-H), 5.87 (dd, J = 8.4, 0.8 Hz, 5-H), 4.83 (sept, 1H, J = 6.1 Hz, OCH(CH3)2), 2.16 (s, 6H, 2′-CH3 and 6′-CH3), 1.51 (d, 6H, J = 6.1 Hz, OCH(CH3)2); 13C{1H} NMR (101 MHz, CDCl3): δ 169.3 (C), 160.9 (C, d, 1JCF = 244.9 Hz), 158.3 (C), 152.7 (C), 139.2 (2× C, d, 3JCF = 8.7 Hz), 134.2 (CH), 133.0 (C, d, 4JCF = 2.8 Hz), 114.8 (2× CH, d, 2JCF = 21.7 Hz), 106.8 (CH), 100.5 (CH), 100.0 (C), 74.3 (CH), 22.1 (2× CH3), 18.39 (CH3), 18.37 (CH3); MS (ESI) m/z 340 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C18H20FNNaO3 340.1319; found 340.1306.
6-[(4′-Fluoro-2′,6′-dimethylphenyl)amino]-2-methoxybenzoic acid (12b). The reaction was carried out according to general procedure B and gave 6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]-2-methoxybenzoic acid (12b) as a brown solid (0.031 g, 71%). Mp 100–101 °C; IR (neat) 3246, 2953, 1695, 1577, 1464, 1443, 1255, 1084, 806 cm−1; 1H NMR (500 MHz, CDCl3): δ 11.50 (br s, 1H, OH), 9.90 (br s, 1H, NH), 7.13 (t, 1H, J = 8.4 Hz, 4-H), 6.83 (d, 2H, J = 9.0 Hz, 3′-H and 5′-H), 6.25 (dd, 1H, J = 8.4, 0.9 Hz, 3-H), 5.91 (dd, 1H, J = 8.4, 0.9 Hz, 5-H), 4.05 (s, 3H, OCH3), 2.15 (s, 6H, 2′-CH3 and 6′-CH3); 13C{1H} NMR (126 MHz, CDCl3): δ 169.0 (C), 160.9 (C, d, 1JCF = 245.0 Hz), 159.9 (C), 152.7 (C), 139.2 (2× C, d, 3JCF = 8.9 Hz), 134.4 (CH), 132.9 (C, d, 4JCF = 2.7 Hz), 114.8 (2× CH, d, 2JCF = 21.9 Hz), 107.2 (CH), 98.8 (C), 98.0 (CH), 56.9 (CH3), 18.36 (CH3), 18.35 (CH3); MS (ESI) m/z 312 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C16H16FNNaO3 312.1006; found 312.0994.
6-[(4′-Fluoro-2′,6′-dimethylphenyl)amino]-2-isobutoxybenzoic acid (12c). The reaction was carried out according to general procedure B and gave 6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]-2-isobutoxybenzoic acid (12c) as an orange solid (0.033 g, 68%). Mp 74–76 °C; IR (neat) 3255, 2966, 1695, 1577, 1456, 1407, 1252, 1057, 756 cm−1; 1H NMR (500 MHz, CDCl3): δ 11.77 (br s, 1H, OH), 9.90 (br s, 1H, NH), 7.10 (t, 1H, J = 8.3 Hz, 4-H), 6.83 (d, 2H, J = 9.1 Hz, 3′-H and 5′-H), 6.22 (dd, 1H, J = 8.3, 0.9 Hz, 3-H), 5.89 (dd, 1H, J = 8.3, 0.9 Hz, 5-H), 4.00 (d, 2H, J = 6.4 Hz, 1′′-H2), 2.29–2.19 (m, 1H, 2′′-H), 2.16 (s, 6H, 2′-CH3 and 6′-CH3), 1.11 (d, 6H, J = 6.4 Hz, 2′′-CH3 and 3′′-H3); 13C{1H} NMR (126 MHz, CDCl3): δ 169.1 (C), 160.9 (C, d, 1JCF = 244.6 Hz), 159.5 (C), 152.6 (C), 139.2 (2× C, d, 3JCF = 8.7 Hz), 134.4 (CH), 132.9 (C, d, 4JCF = 2.8 Hz), 114.8 (2× CH, d, 2JCF = 21.6 Hz), 106.9 (CH), 98.91 (CH), 98.87 (C), 76.7 (CH2), 28.1 (CH), 19.2 (2× CH3), 18.37 (CH3), 18.36 (CH3); MS (ESI) m/z 354 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C19H22FNNaO3 354.1476; found 354.1465.
2-n-Butoxy-6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]benzoic acid (12d). The reaction was carried out according to general procedure B and gave 2-n-butoxy-6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]benzoic acid (12d) as a brown solid (0.033 g, 70%). Mp 77–79 °C; IR (neat) 3246, 2963, 1694, 1578, 1456, 1408, 1252, 1219, 754 cm−1; 1H NMR (500 MHz, CDCl3): δ 11.78 (br s, 1H, OH), 9.90 (br, 1H, NH), 7.10 (t, 1H, J = 8.3 Hz, 4-H), 6.83 (d, 2H, J = 9.2 Hz, 3′-H and 5′-H), 6.23 (dd, 1H, J = 8.3, 0.8 Hz, 3-H), 5.89 (dd, 1H, J = 8.3, 0.8 Hz, 5-H), 4.22 (t, 2H, J = 6.5 Hz, 1′′-H2), 2.15 (s, 6H, 2′-CH3 and 6′-CH3), 1.95–1.87 (m, 2H, 2′′-H2), 1.60–1.49 (m, 2H, 3′′-H2), 1.02 (t, 3H, J = 7.4 Hz, 4′′-H3); 13C{1H} NMR (126 MHz, CDCl3): δ 169.1 (C), 160.9 (C, d, 1JCF = 244.6 Hz), 159.4 (C), 152.6 (C), 139.2 (2× C, d, 3JCF = 8.9 Hz), 134.4 (CH), 132.9 (C, d, 4JCF = 2.7 Hz), 114.8 (2× CH, d, 2JCF = 21.9 Hz), 106.9 (CH), 99.0 (CH and C), 70.2 (CH2), 30.9 (CH2), 19.2 (CH2), 18.37 (CH3), 18.36 (CH3), 13.7 (CH3); MS (ESI) m/z 354 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C19H22FNNaO3 354.1476; found 354.1466.
6-[(4′-Fluoro-2′,6′-dimethylphenyl)amino]-2-(3′′-methyl-n-butoxy)benzoic acid (12e). The reaction was carried out according to general procedure B and gave 6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]-2-(3′′-methyl-n-butoxy)benzoic acid (12e) as a brown viscous oil (0.020 g, 39%). IR (neat) 3244, 2959, 1694, 1578, 1458, 1408, 1252, 1217, 1076, 754 cm−1; 1H NMR (400 MHz, CDCl3): δ 11.69 (br s, 1H, OH), 9.90 (br s, 1H, NH), 7.11 (t, 1H, J = 8.3 Hz, 4-H), 6.83 (d, 2H, J = 9.0 Hz, 3′-H and 5′-H), 6.24 (br d, 1H, J = 8.3 Hz, 3-H), 5.89 (dd, 1H, J = 8.3, 0.8 Hz, 5-H), 4.24 (t, 2H, J = 6.5 Hz, 1′′-H2), 2.16 (s, 6H, 2′-CH3 and 6′-CH3), 1.91–1.77 (m, 3H, 2′′-H2 and 3′′-H), 1.01 (d, 6H, J = 6.4 Hz, 3′′-CH3 and 4′′-H3); 13C{1H} NMR (126 MHz, CDCl3): δ 169.1 (C), 160.9 (C, d, 1JCF = 244.8 Hz), 159.5 (C), 152.7 (C), 139.2 (2× C, d, 3JCF = 8.4 Hz), 134.3 (CH), 133.0 (C, d, 4JCF = 2.7 Hz), 114.8 (2× CH, d, 2JCF = 21.9 Hz), 107.0 (CH), 99.03 (C), 98.98 (CH), 69.0 (CH2), 37.7 (CH2), 25.1 (CH), 22.5 (2× CH3), 18.37 (CH3), 18.36 (CH3); MS (ESI) m/z 368 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C20H24FNNaO3 368.1632; found 368.1620.
2-(Allyloxy)-6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]benzoic acid (12f). The reaction was carried out according to general procedure B and gave 2-(allyloxy)-6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]benzoic acid (12f) as a brown viscous oil (0.0643 g, 46%). IR (neat) 3258, 2920, 1694, 1576, 1456, 1252, 1051, 806 cm−1; 1H NMR (500 MHz, CDCl3): δ 11.60 (br s, 1H, OH), 9.89 (br s, 1H, NH), 7.10 (t, 1H, J = 8.2 Hz, 4-H), 6.83 (d, 2H, J = 9.1 Hz, 3′-H and 5′-H), 6.23 (d, 1H, J = 8.2 Hz, 3-H), 6.11 (ddt, 1H, J = 17.0, 10.5, 5.8 Hz, 2′′-H), 5.91 (d, 1H, J = 8.2 Hz, 5-H), 5.51 (dd, 1H, J = 17.0, 0.8 Hz, 3′′-HH), 5.45 (dd, 1H, J = 10.5, 0.8 Hz, 3′′-HH), 4.76 (br d, 2H, J = 5.8 Hz, 1′′-H2), 2.15 (s, 6H, 2′-CH3 and 6′-CH3); 13C{1H} NMR (126 MHz, CDCl3): δ 169.0 (C), 160.9 (C, d, 1JCF = 245.0 Hz), 159.1 (C), 152.7 (C), 139.2 (2× C, d, 3JCF = 8.3 Hz), 134.3 (CH), 132.9 (C, d, 4JCF = 2.7 Hz), 131.0 (CH), 120.7 (CH2), 114.8 (2× CH, 2JCF = 21.8 Hz), 107.3 (CH), 99.4 (CH), 99.2 (C), 71.2 (CH2), 18.4 (2× CH3); MS (ESI) m/z 338 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C18H18FNNaO3 338.1163; found 338.1151.
6-[(4′-Fluoro-2′,6′-dimethylphenyl)amino]-2-methoxy-3-methylbenzoic acid (12g). The reaction was carried out according to general procedure B and gave 6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]-2-methoxy-3-methylbenzoic acid (12g) as an orange viscous oil (0.0307 g, 71%). IR (neat) 3308, 2926, 1700, 1574, 1504, 1216, 757 cm−1; 1H NMR (500 MHz, CDCl3): δ 11.93 (br s, 1H, OH), 9.45 (br s, 1H, NH), 7.02 (d, 1H, J = 8.7 Hz, 4-H), 6.83 (d, 2H, J = 9.1 Hz, 3′-H and 5′-H), 5.98 (d, 1H, J = 8.7 Hz, 5-H), 3.93 (s, 3H, OCH3), 2.20 (s, 3H, 3-CH3), 2.15 (s, 6H, 2′-CH3 and 6′-CH3); 13C{1H} NMR (126 MHz, CDCl3): δ 168.7 (C), 160.9 (C, d, 1JCF = 244.6 Hz), 158.3 (C), 150.3 (C), 139.2 (2× C, d, 3JCF = 8.9 Hz), 137.2 (CH), 133.0 (C, d, 4JCF = 2.7 Hz), 116.5 (C), 114.8 (2× CH, d, 2JCF = 21.7 Hz), 109.8 (CH), 102.3 (C), 62.3 (CH3), 18.38 (CH3), 18.37 (CH3), 15.1 (CH3); MS (ESI) m/z 326 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C17H18FNNaO3 326.1163; found 326.1148.
6-[(4′-Fluoro-2′,6′-dimethylphenyl)amino]-2-isobutoxy-3-methylbenzoic acid (12h). The reaction was carried out according to general procedure B and gave 6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]-2-isobutoxy-3-methylbenzoic acid (12h) as an orange solid (0.073 g, 74%). Mp 130–132 °C; IR (neat) 3294, 2963, 1705, 1573, 1504, 1404, 1227, 1034, 818 cm−1; 1H NMR (400 MHz, CDCl3): δ 12.24 (br s, 1H, OH), 9.44 (br s, 1H, NH), 7.02 (d, 1H, J = 8.7 Hz, 4-H), 6.83 (d, 2H, J = 9.1 Hz, 3′-H and 5′-H), 5.97 (d, 1H, J = 8.7 Hz, 5-H), 3.76 (d, 2H, J = 6.7 Hz, 1′′-H2), 2.28–2.19 (m, 1H, 2′′-H), 2.18 (s, 3H, 3-CH3), 2.15 (s, 6H, 2′-CH3 and 6′-CH3), 1.12 (d, 6H, J = 6.7 Hz, 2′′-CH3 and 3′′-H3); 13C{1H} NMR (101 MHz, CDCl3): δ 168.7 (C), 160.9 (C, d, 1JCF = 244.8 Hz), 157.2 (C), 150.3 (C), 139.2 (2× C, d, 3JCF = 8.5 Hz), 137.1 (CH), 133.0 (C, d, 4JCF = 2.7 Hz), 116.8 (C), 114.9 (2× CH, d, 2JCF = 21.9 Hz), 109.6 (CH), 102.7 (C), 82.1 (CH2), 29.1 (CH), 19.0 (2× CH3), 18.36 (CH3), 18.35 (CH3), 15.2 (CH3); MS (ESI) m/z 368 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C20H24FNNaO3 368.1632; found 368.1619.
6-[(4′-Fluoro-2′,6′-dimethylphenyl)amino]-2-isopropoxybenzamide (7a). The reaction was carried out according to general procedure C and gave 6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]-2-isopropoxybenzamide (7a) as a pale yellow solid (0.0380 g, 95%). Mp 204–206 °C; IR (neat) 3444, 3188, 2982, 2361, 1638, 1584, 1456, 1250, 1111, 1044 cm−1; 1H NMR (400 MHz, CDCl3): δ 10.13 (br s, 1H, NH), 8.16 (br s, 1H, NH), 7.01 (t, 1H, J = 8.3 Hz, 4-H), 6.82 (d, 2H, J = 9.2 Hz, 3′-H and 5′-H), 6.20 (br d, 1H, J = 8.3 Hz, 3-H), 5.80 (dd, 1H, J = 8.3, 0.9 Hz, 5-H), 5.65 (br s, 1H, NH), 4.70 (sept, 1H, J = 6.1 Hz, OCH(CH3)2), 2.18 (s, 6H, 2′-CH3 and 6′-CH3), 1.44 (d, 6H, J = 6.1 Hz, OCH(CH3)2); 13C{1H} NMR (126 MHz, CDCl3): δ 171.5 (C), 160.6 (C, d, 1JCF = 244.0 Hz), 158.3 (C), 152.1 (C), 139.1 (2× C, d, 3JCF = 8.2 Hz), 134.0 (C, d, 4JCF = 2.7 Hz), 132.6 (CH), 114.6 (2× CH, d, 2JCF = 21.8 Hz), 105.9 (CH), 103.2 (C), 100.9 (CH), 72.0 (CH), 22.3 (2× CH3), 18.49 (CH3), 18.48 (CH3); MS (ESI) m/z 339 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C18H21FN2NaO2 339.1479; found 339.1465.
6-[(4′-Fluoro-2′,6′-dimethylphenyl)amino]-2-methoxybenzamide (7b). The reaction was carried out according to general procedure C and gave 6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]-2-methoxybenzamide (7b) as a pale yellow solid (0.0201 g, 67%). Mp 171–173 °C; IR (neat) 3462, 3186, 2924, 2359, 1638, 1580, 1458, 1250, 1128, 752 cm−1; 1H NMR (400 MHz, CDCl3): δ 10.17 (br s, 1H, NH), 7.94 (br s, 1H, NH), 7.05 (t, 1H, J = 8.4 Hz, 4-H), 6.82 (d, 2H, J = 9.0 Hz, 3′-H and 5′-H), 6.22 (dd, 1H, J = 8.4, 0.9 Hz, 3-H), 5.84 (dd, 1H, J = 8.4, 0.9 Hz, 5-H), 5.77 (br s, 1H, NH), 3.92 (s, 3H, OCH3), 2.17 (s, 6H, 2′-CH3 and 6′-CH3); 13C{1H} NMR (101 MHz, CDCl3): δ 171.3 (C), 160.6 (C, d, 1JCF = 245.0 Hz), 160.0 (C), 152.0 (C), 139.1 (2× C, d, 3JCF = 8.7 Hz), 133.9 (C, d, 4JCF = 2.6 Hz), 132.8 (CH), 114.6 (2× CH, d, 2JCF = 21.8 Hz), 106.4 (CH), 102.1 (C), 98.4 (CH), 56.0 (CH3), 18.5 (CH3), 18.4 (CH3); MS (ESI) m/z 311 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C16H17FN2NaO2 311.1166; found 311.1158.
6-[(4′-Fluoro-2′,6′-dimethylphenyl)amino]-2-isobutoxybenzamide (7c). The reaction was carried out according to general procedure C and gave 6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]-2-isobutoxybenzamide (7c) as an orange solid (0.0285 g, 95%). Mp 152–154 °C; IR (neat) 3455, 3181, 2922, 2363, 1636, 1582, 1493, 1449, 1246, 1061, 864, 754 cm−1; 1H NMR (500 MHz, CDCl3): δ 10.14 (br s, 1H, NH), 8.06 (br s, 1H, NH), 7.02 (t, 1H, J = 8.5 Hz, 4-H), 6.82 (d, 2H, J = 9.1 Hz, 3′-H and 5′-H), 6.19 (br d, 1H, J = 8.5 Hz, 3-H), 5.82 (dd, 1H, J = 8.5, 0.8 Hz, 5-H), 5.73 (br s, 1H, NH), 3.86 (d, 2H, J = 6.4 Hz, 1′′-H2), 2.21–2.13 (m, 7H, 2′-CH3, 6′-CH3 and 2′′-H), 1.08 (d, 6H, J = 6.7 Hz, 2′′-CH3 and 3′′-H3); 13C{1H} NMR (126 MHz, CDCl3): δ 171.4 (C), 160.6 (C, d, 1JCF = 243.6 Hz), 159.5 (C), 152.0 (C), 139.1 (2× C, d, 3JCF = 8.4 Hz), 133.9 (C, d, 4JCF = 2.7 Hz), 132.8 (CH), 114.6 (2× CH, d, 2JCF = 21.4 Hz), 106.1 (CH), 102.1 (C), 99.3 (CH), 75.7 (CH2), 28.3 (CH), 19.5 (2× CH3), 18.47 (CH3), 18.46 (CH3); MS (ESI) m/z 353 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C19H23FN2NaO2 353.1636; found 353.1623.
2-n-Butoxy-6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]benzamide (7d). The reaction was carried out according to general procedure C and gave 2-n-butoxy-6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]benzamide (7d) as a pale orange solid (0.0244 g, 81%). Mp 156–158 °C; IR (neat) 3460, 3181, 2932, 1645, 1570, 1491, 1445, 1248, 752 cm−1; 1H NMR (500 MHz, CDCl3): δ 10.16 (br s, 1H, NH), 8.06 (br s, 1H, NH), 7.02 (t, 1H, J = 8.5 Hz, 4-H), 6.81 (d, 2H, J = 9.1 Hz, 3′-H and 5′-H), 6.20 (dd, 1H, J = 8.5, 0.8 Hz, 3-H), 5.82 (dd, 1H, J = 8.5, 0.8 Hz, 5-H), 5.70 (br s, 1H, NH), 4.09 (t, 2H, J = 6.5 Hz, 1′′-H2), 2.17 (s, 6H, 2′-CH3 and 6′-CH3), 1.90–1.81 (m, 2H, 2′′-H2), 1.57–1.47 (m, 2H, 3′′-H2), 1.00 (t, 3H, J = 7.5 Hz, 4′′-H3); 13C{1H} NMR (126 MHz, CDCl3): δ 171.5 (C), 160.6 (C, d, 1JCF = 243.9 Hz), 159.5 (C), 152.0 (C), 139.1 (2× C, d, 3JCF = 8.4 Hz), 134.0 (C, d, 4JCF = 2.8 Hz), 132.8 (CH), 114.6 (2× CH, d, 2JCF = 21.7 Hz), 106.1 (CH), 102.2 (C), 99.3 (CH), 69.0 (CH2), 31.3 (CH2), 19.5 (CH2), 18.45 (CH3), 18.44 (CH3), 13.8 (CH3); MS (ESI) m/z 353 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C19H23FN2NaO2 353.1636; found 353.1623.
6-[(4′-Fluoro-2′,6′-dimethylphenyl)amino]-2-(3′′-methyl-n-butoxy)benzamide (7e). The reaction was carried out according to general procedure C and gave 6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]-2-(3′′-methyl-n-butoxy)benzamide (7e) as a pale yellow solid (0.014 g, 67%). Mp 143–145 °C; IR (neat) 3467, 3201, 2960, 1641, 1580, 1451, 1245, 1216, 754 cm−1; 1H NMR (500 MHz, CDCl3): δ 10.16 (br s, 1H, NH), 8.05 (br s, 1H, NH), 7.02 (t, 1H, J = 8.5 Hz, 4-H), 6.81 (d, 2H, J = 9.1 Hz, 3′-H and 5′-H), 6.21 (br d, 1H, J = 8.5 Hz, 3-H), 5.82 (dd, 1H, J = 8.5, 0.8 Hz, 5-H), 5.65 (br s, 1H, NH), 4.11 (t, 2H, J = 6.6 Hz, 1′′-H2), 2.17 (s, 6H, 2′-CH3 and 6′-CH3), 1.87–1.73 (m, 3H, 2′′-H2 and 3′′-H), 0.99 (d, 6H, J = 6.4 Hz, 3′′-CH3 and 4′′-H3); 13C{1H} NMR (126 MHz, CDCl3): δ 171.4 (C), 160.6 (C, d, 1JCF = 243.7 Hz), 159.5 (C), 152.1 (C), 139.1 (2× C, d, 3JCF = 8.8 Hz), 133.9 (C, d, 4JCF = 2.7 Hz), 132.8 (CH), 114.6 (2× CH, d, 2JCF = 21.4 Hz), 106.2 (CH), 102.2 (C), 99.3 (CH), 67.8 (CH2), 38.1 (CH2), 25.3 (CH), 22.6 (2× CH3), 18.46 (CH3), 18.45 (CH3); MS (ESI) m/z 367 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C20H25FN2NaO2 367.1792; found 367.1774.
2-(Allyloxy)-6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]benzamide (7f). The reaction was carried out according to general procedure C and gave 2-(allyloxy)-6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]benzamide (7f) as a pale yellow solid (0.0445 g, 95%). Mp 176–178 °C; IR (neat) 3443, 3181, 2920, 1647, 1605, 1582, 1451, 1238, 1053, 810 cm−1; 1H NMR (500 MHz, CDCl3): δ 10.12 (br s, 1H, NH), 7.99 (br s, 1H, NH), 7.02 (t, 1H, J = 8.2 Hz, 4-H), 6.82 (d, 2H, J = 9.1 Hz, 3′-H and 5′-H), 6.20 (br d, 1H, J = 8.2 Hz, 3-H), 6.11 (ddt, 1H, J = 17.2, 10.5, 5.6 Hz, 2′′-H), 5.84 (dd, 1H, J = 8.2, 0.8 Hz, 5-H), 5.75 (br s, 1H, NH), 5.46 (dd, 1H, J = 17.2, 1.2 Hz, 3′′-HH), 5.36 (dd, 1H, J = 10.5, 1.2 Hz, 3′′-HH), 4.63 (br d, 2H, J = 5.6 Hz, 1′′-H2), 2.17 (s, 6H, 2′-CH3 and 6′-CH3); 13C{1H} NMR (126 MHz, CDCl3): δ 171.3 (C), 160.6 (C, d, 1JCF = 244.1 Hz), 159.0 (C), 152.0 (C), 139.1 (2× C, d, 3JCF = 8.4 Hz), 133.9 (C, d, 4JCF = 2.7 Hz), 132.7 (CH), 132.4 (CH), 119.2 (CH2), 114.6 (2× CH, 2JCF = 21.4 Hz), 106.5 (CH), 102.5 (C), 99.7 (CH), 70.1 (CH2), 18.46 (CH3), 18.44 (CH3); MS (ESI) m/z 337 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C18H19FN2NaO2 337.1323; found 337.1313.
6-[(4′-Fluoro-2′,6′-dimethylphenyl)amino]-2-methoxy-3-methylbenzamide (7g). The reaction was carried out according to general procedure C and gave 6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]-2-methoxy-3-methylbenzamide (7g) as a pale yellow solid (0.016 g, 55%). Mp 136–138 °C; IR (neat) 3464, 3226, 2923, 1623, 1569, 1496, 1404, 1257, 1130, 1046, 865 cm−1; 1H NMR (500 MHz, CDCl3): δ 9.36 (br s, 1H, NH), 7.95 (br s, 1H, NH), 6.94 (d, 1H, J = 8.6 Hz, 4-H), 6.81 (d, 2H, J = 9.1 Hz, 3′-H and 5′-H), 5.91 (d, 1H, J = 8.6 Hz, 5-H), 5.74 (br s, 1H, NH), 3.78 (s, 3H, OCH3), 2.17 (s, 6H, 2′-CH3 and 6′-CH3), 2.17 (s, 3H, 3-CH3); 13C{1H} NMR (126 MHz, CDCl3): δ 170.8 (C), 160.5 (C, d, 1JCF = 243.7 Hz), 158.2 (C), 149.2 (C), 139.0 (2× C, d, 3JCF = 8.5 Hz), 135.1 (CH), 134.0 (C, d, 4JCF = 2.7 Hz), 117.7 (C), 114.6 (2× CH, d, 2JCF = 21.8 Hz), 109.0 (CH), 106.9 (C), 61.2 (CH3), 18.48 (CH3), 18.47 (CH3), 15.2 (CH3); MS (ESI) m/z 325 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C17H19FN2NaO2 325.1323; found 325.1315.
6-[(4′-Fluoro-2′,6′-dimethylphenyl)amino]-2-isobutoxy-3-methylbenzamide (7h). The reaction was carried out according to general procedure C and gave 6-[(4′-fluoro-2′,6′-dimethylphenyl)amino]-2-isobutoxy-3-methylbenzamide (7h) as a white solid (0.059 g, 82%). Mp 125–127 °C; IR (neat) 3449, 3233, 2963, 1651, 1574, 1497, 1258, 1042 cm−1; 1H NMR (400 MHz, CDCl3): δ 9.23 (br s, 1H, NH), 7.93 (br s, 1H, NH), 6.93 (d, 1H, J = 8.6 Hz, 4-H), 6.81 (d, 2H, J = 9.1 Hz, 3′-H and 5′-H), 5.92 (br s, 1H, NH), 5.90 (d, 1H, J = 8.6 Hz, 5-H), 3.62 (d, 2H, J = 6.7 Hz, 1′′-H2), 2.22–2.06 (m, 10H, 3-CH3, 2′-CH3, 6′-CH3 and 2′′-H), 1.06 (d, 6H, J = 6.7 Hz, 2′′-CH3 and 3′′-H3); 13C{1H} NMR (101 MHz, CDCl3): δ 171.0 (C), 160.5 (C, d, 1JCF = 243.7 Hz), 157.1 (C), 148.9 (C), 139.0 (2× C, d, 3JCF = 8.6 Hz), 135.0 (CH), 134.0 (C, d, 4JCF = 2.8 Hz), 118.0 (C), 114.6 (2× CH, d, 2JCF = 21.7 Hz), 108.8 (CH), 107.7 (C), 80.7 (CH2), 29.3 (CH), 19.3 (2× CH3), 18.48 (CH3), 18.47 (CH3), 15.4 (CH3); MS (ESI) m/z 367 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C20H25FN2NaO2 367.1792; found 367.1781.
2-Bromo-6-(mesitylamino)benzoic acid (14). 16The reaction was carried out according to general procedure A and gave 2-bromo-6-(mesitylamino)benzoic acid (14) as a brown solid (1.44 g, 94%). The spectroscopic data were consistent with the literature.16 Mp 169–171 °C; 1H NMR (400 MHz, CDCl3): δ 7.00–6.90 (m, 4H, 3-H, 4-H, 3′-H and 5′-H), 6.18 (dd, 1H, J = 7.8, 1.8 Hz, 5-H), 2.31 (s, 3H, 4′-CH3), 2.15 (s, 6H, 2′-CH3 and 6′-CH3); 13C{1H} NMR (101 MHz, CDCl3): δ 172.5 (C), 150.3 (C), 136.5 (C), 136.2 (2× C), 134.0 (C), 133.5 (CH), 129.3 (2× CH), 123.4 (C), 122.8 (CH), 113.1 (C), 112.2 (CH), 21.0 (CH3), 18.1 (2× CH3); MS (EI) m/z 333 (M+, 100%), 315 (55), 300 (64), 208 (25), 135 (21).
6-(Mesitylamino)-2-methoxybenzoic acid (15). The reaction was carried out according to general procedure B and gave 6-(mesitylamino)-2-methoxybenzoic acid (15) as an orange solid (1.62 g, 95%). Mp 83–85 °C; IR (neat) 3277, 3176, 2918, 1676, 1573, 1435, 1397, 1356, 1254, 1190, 1061, 771 cm−1; 1H NMR (500 MHz, CDCl3): δ 11.50 (br s, 1H, OH), 9.93 (s, 1H, NH), 7.10 (t, 1H, J = 8.3 Hz, 4-H), 6.95 (s, 2H, 3′-H and 5′-H), 6.22 (br d, 1H, J = 8.3 Hz, 3-H), 5.96 (dd, 1H, J = 8.3, 0.9 Hz, 5-H), 4.04 (s, 3H, OCH3), 2.31 (s, 3H, 4′-CH3), 2.14 (s, 6H, 2′-CH3 and 6′-CH3); 13C{1H} NMR (126 MHz, CDCl3): δ 169.1 (C), 159.9 (C), 152.9 (C), 136.5 (2× C), 136.4 (C), 134.4 (C), 134.3 (CH), 129.2 (2× CH), 107.5 (CH), 98.6 (C), 97.7 (CH), 56.8 (CH3), 21.0 (CH3), 18.1 (2× CH3); MS (ESI) m/z 284 ([M–H]−, 100%); HRMS (ESI) m/z: [M–H]− calcd for C17H18NO3 284.1292; found 284.1292.
6-(Mesitylamino)-2-methoxybenzamide (16). The reaction was carried out according to general procedure C and gave 6-(mesitylamino)-2-methoxybenzamide (16) as a white solid (1.53 g, 95%). Mp 180–182 °C; IR (neat) 3439, 3171, 2917, 1646, 1590, 1458, 1406, 1365, 1254, 1084, 806 cm−1; 1H NMR (500 MHz, CDCl3): δ 10.18 (br s, 1H, NH), 7.90 (br s, 1H, NH), 7.01 (t, 1H, J = 8.3 Hz, 4-H), 6.92 (s, 2H, 3′-H and 5′-H), 6.17 (dd, 1H, J = 8.3, 0.9 Hz, 3-H), 6.09 (br s, 1H, NH), 5.88 (dd, 1H, J = 8.3, 0.9 Hz, 5-H), 3.89 (s, 3H, OCH3), 2.29 (s, 3H, 4′-CH3), 2.15 (s, 6H, 2′-CH3 and 6′-CH3); 13C{1H} NMR (126 MHz, CDCl3): δ 171.5 (C), 159.9 (C), 152.1 (C), 136.5 (2× C), 135.6 (C), 135.4 (C), 132.6 (CH), 129.0 (2× CH), 106.6 (CH), 102.0 (C), 98.0 (CH), 55.9 (CH3), 20.9 (CH3), 18.2 (2× CH3); MS (ESI) m/z 285 (M + H+, 100%); HRMS (ESI) m/z: [M + H]+ calcd for C17H21N2O2 285.1598; found 285.1600.
3-Bromo-6-(mesitylamino)-2-methoxybenzamide (17). To a dry flask under argon was added iron(III) chloride (0.0450 g, 0.275 mmol, 5 mol%) and 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([BMIM]NTf2) (2.30 mL, 11.0 mmol) and the mixture stirred for 0.5 h. 6-(Mesitylamino)-2-methoxybenzamide (16) (1.56 g, 5.49 mmol) in dichloromethane (14 mL) was added along with N-bromosuccinimide (0.784 g, 4.72 mmol, 0.8 equiv.). After 1 h of stirring at room temperature, N-bromosuccinimide (0.196 g, 1.10 mmol, 0.2 equiv.) was added and the reaction stirred for a further 1 h. The mixture was diluted with ethyl acetate (20 mL) and filtered through a short pad of silica. The organic layer was then washed with 1 M sodium thiosulfate (2 × 50 mL), brine (50 mL) and then dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography, eluting with 20% ethyl acetate in petroleum ether (40–60) gave 3-bromo-6-(mesitylamino)-2-methoxybenzamide (17) as a white solid (1.54 g, 77%). Mp 184–186 °C; IR (neat) 3456, 3256, 2924, 1651, 1566, 1489, 1404, 1258, 1042, 818 cm−1; 1H NMR (500 MHz, CDCl3): δ 9.56 (br s, 1H, NH), 7.83 (br s, 1H, NH), 7.21 (d, 1H, J = 9.1 Hz, 4-H), 6.94 (s, 2H, 3′-H and 5′-H), 6.08 (br s, 1H, NH), 5.95 (d, 1H, J = 9.1, Hz, 5-H), 3.89 (s, 3H, OCH3), 2.30 (s, 3H, 4′-CH3), 2.14 (s, 6H, 2′-CH3 and 6′-CH3); 13C{1H} NMR (126 MHz, CDCl3): δ 170.0 (C), 156.7 (C), 150.7 (C), 136.42 (CH), 136.39 (2× C), 136.2 (C), 134.7 (C), 129.3 (2× CH), 111.0 (CH), 108.2 (C), 102.0 (C), 61.9 (CH3), 21.1 (CH3), 18.3 (2× CH3); MS (ESI) m/z 385 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C17H1979BrN2NaO2 385.0522; found 385.0512.
3-(4′′-Fluorophenyl)-6-(mesitylamino)-2-methoxybenzamide (8a). The reaction was carried out according to general procedure D and gave 3-(4′′-fluorophenyl)-6-(mesitylamino)-2-methoxybenzamide (8a) as a white solid (0.0738 g, 71%). Mp 209–211 °C; IR (neat) 3449, 3225, 2924, 1651, 1489, 1250, 1227, 1042, 818, 756 cm−1; 1H NMR (400 MHz, CDCl3): δ 9.73 (br s, 1H, NH), 8.07 (br s, 1H, NH), 7.46 (dd, 2H, J = 8.7, 5.5 Hz, 2′′-H and 6′′-H), 7.08 (t, 2H, J = 8.7 Hz, 3′′-H and 5′′-H), 7.06 (d, 1H, J = 8.8 Hz, 4-H), 6.95 (s, 2H, 3′-H and 5′-H), 6.08 (d, 1H, J = 8.8 Hz, 5-H), 5.83 (br s, 1H, NH), 3.45 (s, 3H, OCH3), 2.32 (s, 3H, 4′-CH3), 2.19 (s, 6H, 2′-CH3 and 6′-CH3); 13C{1H} NMR (101 MHz, CDCl3): δ 171.0 (C), 161.8 (C, d, 1JCF = 245.6 Hz), 157.8 (C), 150.9 (C), 136.5 (2× C), 136.0 (C), 135.1 (C), 134.7 (CH), 134.5 (C, d, 4JCF = 3.4 Hz), 130.8 (2× CH, d, 3JCF = 7.8 Hz), 129.2 (2× CH), 121.4 (C), 115.2 (2× CH, d, 2JCF = 21.2 Hz), 109.7 (CH), 106.7 (C), 61.4 (CH3), 21.1 (CH3), 18.4 (2× CH3); MS (ESI) m/z 401 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C23H23FN2NaO2 401.1636; found 401.1623.
3-(2′′-Fluorophenyl)-6-(mesitylamino)-2-methoxybenzamide (8b). The reaction was carried out according to general procedure D and gave 3-(2′′-fluorophenyl)-6-(mesitylamino)-2-methoxybenzamide (8b) as a white solid (0.0698 g, 67%). Mp 219–221 °C; IR (neat) 3464, 3264, 2916, 1620, 1481, 1396, 1242, 1034, 748 cm−1; 1H NMR (400 MHz, CDCl3): δ 9.77 (br s, 1H, NH), 8.02 (br s, 1H, NH), 7.37 (t, 1H, J = 7.5 Hz, ArH), 7.34–7.24 (m, 1H, ArH), 7.20–7.07 (m, 2H, ArH), 7.04 (d, 1H, J = 8.7 Hz, 4-H), 6.94 (s, 2H, 3′-H and 5′-H), 6.08 (d, 1H, J = 8.7 Hz, 5-H), 5.88 (br s, 1H, NH), 3.47 (s, 3H, OCH3), 2.30 (s, 3H, 4′-CH3), 2.20 (s, 6H, 2′-CH3 and 6′-CH3); 13C{1H} NMR (101 MHz, CDCl3): δ 170.9 (C), 160.0 (C, d, 1JCF = 246.4 Hz), 158.3 (C), 151.2 (C), 136.4 (2× C), 135.9 (C), 135.1 (CH, d, 4JCF = 1.8 Hz), 135.0 (C), 132.1 (CH, d, 4JCF = 3.1 Hz), 129.1 (2× CH), 128.7 (CH, d, 3JCF = 8.1 Hz), 126.0 (C, d, 2JCF = 15.7 Hz), 123.9 (CH, d, 3JCF = 3.6 Hz), 116.2 (C), 115.7 (CH, d, 2JCF = 22.8 Hz), 109.2 (CH), 106.4 (C), 61.6 (CH3), 20.9 (CH3), 18.3 (2× CH3); MS (ESI) m/z 401 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C23H23FN2NaO2 401.1636; found 401.1621.
3-(2′′-Fluoro-4′′-methoxyphenyl)-6-(mesitylamino)-2-methoxybenzamide (8c). The reaction was carried out according to general procedure D and gave 3-(2′′-fluoro-4′′-methoxyphenyl)-6-(mesitylamino)-2-methoxybenzamide (8c) as a white solid (0.0573 g, 51%). Mp 212–214 °C; IR (neat) 3466, 2913, 1624, 1489, 1256, 1040, 951, 826 cm−1; 1H NMR (400 MHz, CDCl3): δ 9.75 (br s, 1H, NH), 8.04 (br s, 1H, NH), 7.27 (t, 1H, J = 8.0 Hz, 6′′-H), 7.02 (d, 1H, J = 8.6 Hz, 4-H), 6.94 (s, 2H, 3′-H and 5′-H), 6.73 (dd, 1H, J = 8.0, 4.0 Hz, 5′′-H), 6.69 (dd, 1H, J = 11.8, 4.0 Hz, 3′′-H), 6.07 (d, 1H, J = 8.6 Hz, 5-H), 5.79 (br s, 1H, NH), 3.83 (s, 3H, 4′′-OCH3), 3.47 (s, 3H, 2-OCH3), 2.30 (s, 3H, 4′-CH3), 2.19 (s, 6H, 2′-CH3 and 6′-CH3); 13C{1H} NMR (101 MHz, CDCl3): δ 170.9 (C), 160.5 (C, d, 1JCF = 247.5 Hz), 160.0 (C, d, 3JCF = 10.8 Hz), 158.3 (C), 151.0 (C), 136.4 (2× C), 135.8 (C), 135.3 (CH, d, 4JCF = 1.6 Hz), 135.1 (C), 132.3 (CH, d, 3JCF = 5.2 Hz), 129.1 (2× CH), 118.1 (C, d, 2JCF = 16.2 Hz), 116.1 (C), 109.8 (CH, d, 4JCF = 3.0 Hz), 109.2 (CH), 106.4 (C), 101.7 (CH, d, 2JCF = 26.6 Hz), 61.5 (CH3), 55.6 (CH3), 20.9 (CH3), 18.3 (2× CH3); MS (ESI) m/z 431 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C24H25FN2NaO3 431.1741; found 431.1721.
3-(2′′-Fluoro-5′′-methoxyphenyl)-6-(mesitylamino)-2-methoxybenzamide (8d). The reaction was carried out according to general procedure D and gave 3-(2′′-fluoro-5′′-methoxyphenyl)-6-(mesitylamino)-2-methoxybenzamide (8d) as a white solid (0.0694 g, 62%). Mp 196–198 °C; IR (neat) 3456, 2934, 1647, 1564, 1489, 1404, 1248, 1206, 1038, 752 cm−1; 1H NMR (400 MHz, CDCl3): δ 9.78 (br s, 1H, NH), 8.03 (br s, 1H, NH), 7.07–7.00 (m, 2H, 4-H and 3′′-H), 6.94 (s, 2H, 3′-H and 5′-H), 6.90 (dd, 1H, J = 6.0, 3.5 Hz, 6′′-H), 6.81 (dt, 1H, J = 8.8, 3.5 Hz, 4′′-H), 6.08 (d, 1H, J = 8.8 Hz, 5-H), 5.67 (br s, 1H, NH), 3.80 (s, 3H, 5′′-OCH3), 3.51 (s, 3H, 2-OCH3), 2.31 (s, 3H, 4′-CH3), 2.19 (s, 6H, 2′-CH3 and 6′-CH3); 13C{1H} NMR (101 MHz, CDCl3): δ 170.8 (C), 158.3 (C), 155.4 (C, d, 4JCF = 2.0 Hz), 154.5 (C, d, 1JCF = 238.9 Hz), 151.3 (C), 136.4 (2× C), 135.9 (C), 135.1 (CH, d, 4JCF = 1.8 Hz), 135.0 (C), 129.1 (2× CH), 126.6 (C, d, 2JCF = 17.5 Hz), 116.7 (CH, d, 3JCF = 3.1 Hz), 116.2 (C), 116.1 (CH, d, 2JCF = 28.7 Hz), 113.7 (CH, d, 3JCF = 8.0 Hz), 109.2 (CH), 106.4 (C), 61.7 (CH3), 55.8 (CH3), 21.0 (CH3), 18.3 (2× CH3); MS (ESI) m/z 431 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C24H25FN2NaO3 431.1741; found 431.1723.
3-(2′′-Fluoropyridin-3′′-yl)-6-(mesitylamino)-2-methoxybenzamide (8e). The reaction was carried out according to general procedure D and gave 3-(2′′-fluoropyridin-3′′-yl)-6-(mesitylamino)-2-methoxybenzamide (8e) as a white solid (0.0190 g, 18%). Mp 214–216 °C; IR (neat) 3441, 3163, 1667, 1566, 1427, 1258, 1034, 825, 756 cm−1; 1H NMR (400 MHz, CDCl3): δ 9.77 (br s, 1H, NH), 8.18 (ddd, 1H, J = 4.8, 1.9, 1.1 Hz, 4′′-H), 7.93 (br s, 1H, NH), 7.85 (ddd, 1H, J = 9.5, 7.3, 1.9 Hz, 6′′-H), 7.23 (ddd, 1H, J = 7.3, 4.8, 1.7 Hz, 5′′-H), 7.06 (dd, 1H, J = 8.8, 1.3 Hz, 4-H), 6.95 (s, 2H, 3′-H and 5′-H), 6.10 (d, 1H, J = 8.8 Hz, 5-H), 5.76 (br s, 1H, NH), 3.49 (s, 3H, OCH3), 2.31 (s, 3H, 4′-CH3), 2.19 (s, 6H, 2′-CH3 and 6′-CH3); 13C{1H} NMR (101 MHz, CDCl3): δ 170.4 (C), 160.9 (C, d, 1JCF = 239.5 Hz), 158.3 (C), 151.6 (C), 146.1 (CH, d, 3JCF = 14.5 Hz), 142.3 (CH, d, 3JCF = 4.4 Hz), 136.4 (2× C), 136.1 (C), 134.7 (CH, 4JCF = 2.1 Hz), 134.7 (C), 129.2 (2× CH), 121.3 (CH, d, 4JCF = 4.3 Hz), 120.8 (C, d, 2JCF = 30.4 Hz), 114.3 (C, d, 3JCF = 4.5 Hz), 109.5 (CH), 106.5 (C), 61.9 (CH3), 21.0 (CH3), 18.3 (2× CH3); MS (ESI) m/z 402 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C22H22FN3NaO2 402.1588; found 402.1578.
2-Bromo-6-[(2′,6′-dimethylphenyl)amino]-3-methylbenzoic acid (19). The reaction was carried out according to general procedure A and gave 2-bromo-6-[(2′,6′-dimethylphenyl)amino]-3-methylbenzoic acid (19) as a brown solid (2.74 g, 95%). Mp 126–128 °C; IR (neat) 3389, 2918, 1690, 1661, 1489, 1234, 773 cm−1; 1H NMR (500 MHz, CDCl3): δ 10.29 (br s, 1H, OH), 7.17–7.07 (m, 3H, 3′-H, 4′-H and 5′-H), 7.02 (d, 1H, J = 8.5 Hz, 4-H), 6.14 (d, 1H, J = 8.5 Hz, 5-H), 2.34 (s, 3H, 3-CH3), 2.20 (s, 6H, 2′-CH3 and 6′-CH3); 13C{1H} NMR (126 MHz, CDCl3): δ 173.4 (C), 146.0 (C), 137.1 (C), 136.0 (2× C), 134.0 (CH), 128.6 (2× CH), 128.0 (C), 126.4 (CH), 124.0 (C), 116.9 (C), 112.5 (CH), 23.0 (CH3), 18.2 (2× CH3); MS (ESI) m/z 356 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C16H1679BrNNaO2 356.0257; found 356.0250.
6-[(2′,6′-Dimethylphenyl)amino]-2-methoxy-3-methylbenzoic acid (20). The reaction was carried out according to general procedure B and gave 6-[(2′,6′-dimethylphenyl)amino]-2-methoxy-3-methylbenzoic acid (20) as a brown solid (1.96 g, 92%). Mp 92–94 °C; IR (neat) 3289, 2945, 1699, 1570, 1501, 1385, 1223, 1040, 818, 770 cm−1; 1H NMR (500 MHz, CDCl3): δ 12.15 (br s, 1H, OH), 9.59 (br s, 1H, NH), 7.18–7.06 (m, 3H, 3′-H, 4′-H and 5′-H), 7.01 (d, 1H, J = 8.7 Hz, 4-H), 6.03 (d, 1H, J = 8.7 Hz, 5-H), 3.93 (s, 3H, OCH3), 2.20 (s, 3H, 3-CH3), 2.18 (s, 6H, 2′-CH3 and 6′-CH3); 13C{1H} NMR (126 MHz, CDCl3): δ 168.7 (C), 158.2 (C), 150.2 (C), 137.11 (CH), 137.09 (C), 136.7 (2× C), 128.5 (2× CH), 126.7 (CH), 116.3 (C), 110.0 (CH), 102.1 (C), 62.2 (CH3), 18.2 (2× CH3), 15.1 (CH3); MS (ESI) m/z 308 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C17H19NNaO3 308.1257; found 308.1248.
6-[(2′,6′-Dimethylphenyl)amino]-2-methoxy-3-methylbenzamide (21). The reaction was carried out according to general procedure C and gave 6-[(2′,6′-dimethylphenyl)amino]-2-methoxy-3-methylbenzamide (21) as a white solid (1.30 g, 93%). Mp 116–118 °C; IR (neat) 3443, 3184, 2936, 1645, 1570, 1497, 1366, 1261, 1047, 816, 731 cm−1; 1H NMR (400 MHz, CDCl3): δ 9.47 (br s, 1H, NH), 7.93 (br s, 1H, NH), 7.19–7.03 (m, 3H, 3′-H, 4′-H and 5′-H), 6.94 (d, 1H, J = 8.6 Hz, 4-H), 6.04 (br s, 1H, NH), 5.96 (d, 1H, J = 8.6 Hz, 5-H), 3.79 (s, 3H, OCH3), 2.21 (s, 6H, 2′-CH3 and 6′-CH3), 2.18 (s, 3H, 3-CH3); 13C{1H} NMR (101 MHz, CDCl3): δ 170.9 (C), 158.1 (C), 148.9 (C), 138.1 (C), 136.6 (2× C), 134.9 (CH), 128.3 (2× CH), 126.0 (CH), 117.5 (C), 109.3 (CH), 107.0 (C), 61.1 (CH3), 18.3 (2× CH3), 15.2 (CH3); MS (ESI) m/z 307 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C17H20N2NaO2 307.1417; found 307.1409.
6-[(4′-Iodo-2′,6′-dimethylphenyl)amino]-2-methoxy-3-methylbenzamide (22). To a solution of 6-[(2′,6′-dimethylphenyl)amino]-2-methoxy-3-methylbenzamide (21) (0.500 g, 1.76 mmol) and silver bis(trifluoromethanesulfonyl)imide (0.0512 g, 0.132 mmol, 7.5 mol%) in tetrahydrofuran (7 mL) under argon was slowly added N-iodosuccinimide (0.554 g, 2.46 mmol) in tetrahydrofuran (2.0 mL). The reaction mixture was stirred at room temperature for 3 h in the dark. The reaction mixture was filtered through a short pad of Celite®, washed with ethyl acetate and concentrated in vacuo. Purification by flash column chromatography (20–25% diethyl ether in hexane) gave 6-[(4′-iodo-2′,6′-dimethylphenyl)amino]-2-methoxy-3-methylbenzamide (22) as a white solid (0.389 g, 54%). Mp 141–143 °C; IR (neat) 3447, 3202, 2934, 1645, 1572, 1497, 1468, 1398, 1366, 1265, 1215, 1047, 851, 733 cm−1; 1H NMR (500 MHz, CDCl3): δ 9.41 (br s, 1H, NH), 7.93 (br s, 1H, NH), 7.45 (s, 2H, 3′-H and 5′-H), 6.95 (d, 1H, J = 8.6 Hz, 4-H), 5.93 (d, 1H, J = 8.6 Hz, 5-H), 5.69 (br s, 1H, NH), 3.78 (s, 3H, OCH3), 2.17 (s, 3H, 3-CH3), 2.13 (s, 6H, 2′-CH3 and 6′-CH3); 13C{1H} NMR (126 MHz, CDCl3): δ 170.8 (C), 158.3 (C), 148.5 (C), 139.2 (2× C), 138.3 (C), 137.3 (2× CH), 135.1 (CH), 118.3 (C), 109.4 (CH), 107.4 (C), 90.9 (C), 61.3 (CH3), 18.1 (2× CH3), 15.4 (CH3); MS (ESI) m/z 433 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C17H19IN2NaO2 433.0383; found 433.0385.
6-[(4′-(Trimethylstannyl)-2′-6′-dimethylphenyl)amino]-2-methoxy-3-methylbenzamide (23). An oven-dried microwave vial was flushed with argon and charged with 6-[(4′-iodo-2′,6′-dimethylphenyl)amino]-2-methoxy-3-methylbenzamide (22) (0.0800 g, 0.195 mmol) in toluene (3.0 mL). Lithium chloride (0.0413 g, 0.975 mmol) was added and the mixture was degassed under argon, while stirring for 0.2 h. Tetrakis(triphenylphosphine)palladium(0) (0.0451 g, 0.0390 mmol, 20 mol%) and hexamethylditin (0.0806 mL, 0.390 mmol) were added under argon and the reaction mixture was stirred under reflux for 21 h. After cooling to room temperature, the reaction was quenched by the addition of 30% aqueous solution of potassium fluoride (2.0 mL) and stirred at room temperature for 0.5 h. The mixture was filtered through a short pad of Celite®, washed with ethyl acetate and concentrated in vacuo. Purification by neutral alumina (Brockmann grade V) flash column chromatography, eluting with 20% diethyl ether in hexane gave 6-[(4′-(trimethylstannyl)-2′-6′-dimethylphenyl)amino]-2-methoxy-3-methylbenzamide (23) as a colourless oil (0.0734 g, 84%). IR (neat) 3445, 3188, 2918, 1651, 1576, 1497, 1267, 1258, 1049, 765 cm−1; 1H NMR (500 MHz, CDCl3): δ 9.46 (br s, 1H, NH), 7.92 (br s, 1H, NH), 7.22 (s, 2H, 3′-H and 5′-H), 6.93 (d, 1H, J = 8.6 Hz, 4-H), 5.98 (d, 1H, J = 8.6 Hz, 5-H), 5.69 (br s, 1H, NH), 3.78 (s, 3H, OCH3), 2.19 (s, 6H, 2′-CH3 and 6′-CH3), 2.17 (s, 3H, 3-CH3), 0.29 (s, 9H, Sn(CH3)3); 13C{1H} NMR (126 MHz, CDCl3): δ 170.9 (C), 158.3 (C), 149.1 (C), 139.5 (C), 138.5 (C), 136.1 (2× C), 136.0 (2× CH), 135.1 (CH), 117.7 (C), 109.6 (CH), 107.1 (C), 61.3 (CH3), 18.4 (2× CH3), 15.3 (CH3), −9.4 (3× CH3); MS (ESI) m/z 471 (M + Na+, 100%); HRMS (ESI) m/z: [M + Na]+ calcd for C20H28N2NaO2Sn 471.1065; found 471.1056.
Procedure for the [35S]GTPγS binding assay. 25The assay buffer was prepared from HEPES (20 mM), NaCl (100 mM), MgCl2 (10 mM) and fatty acid-free BSA (0.1%) at pH 7.4. ChemiScreen™ S1P1–3,5 lysophospholipid receptor membrane preparations were obtained from Eurofins. The membrane preparation was diluted to 0.4 g mL−1 with assay buffer and mixed 1:1 (v/v) with a 0.4 mg mL−1 solution of saponin in assay buffer. Nine concentrations of the compounds to be tested were prepared (10 μM, 1 μM, 100 nM, 10 nM, 1 nM, 100 pM, 10 pM, 1 pM, 100 fM). Merck™ Multiscreen® HTS 96 well filter plates were pre-wet with 70% aqueous EtOH (50 μL per well) for 30 seconds and then washed twice with assay buffer (200 μL per well). To each well was added membrane preparation (25 μL), GDP in assay buffer (25 μL, 2 μM), [35S]GTPγS in assay buffer (25 μL, 1.2 nM) and test compound (25 μL). For total binding, S1P solution in assay buffer (25 μL, 10 μM) was added and for basal, assay buffer (25 μL) was added. The assay plate was incubated at 30 °C for 0.5 h before the addition of cold assay buffer (100 μL per well) to stop the reaction. The plate was filtered using a Millipore vacuum manifold, with a pressure between 600–800 mbar and then dried at 50 °C for 1–2 h. Betaplate Scint scintillation cocktail (30 μL per well) was added and the disintegrations per minute were determined using a Wallac 1450 Microbeta Trilux liquid scintillation and luminescence counter. EC50 values were derived from non-linear regression analysis using GraphPad Prism Version 6 (GraphPad Software Inc).
Docking studies. All computational experiments were conducted on a Dell Latitude 5420 11th Gen Intel(R) Core(TM) i7-1185G7. The cryo-EM structure of siponimod-bound S1P5 in complex with Gi protein was obtained from the Protein Database (PDB code: 7EW1, http://www.rcsb.org).29 Siponimod (4) and TEFM78 (7g) were drawn using MarvinSketch 24.3.0. The three-dimensional, energetically minimised conformer of each compound was then obtained using Mercury software from the Cambridge Crystallographic Data Centre (CCDC). Using Hermes GOLD software from the CCDC, a docking was configured with the siponimod-S1P5 complex (PDB code: 7EW1) in which the bound siponimod ligand was extracted and hydrogen atoms were added to the protein. No water molecules were present within the cryo-EM structure of the protein and no further changes were made to the protein. The binding site was defined using the previously extracted siponimod ligand with a radius of 10.0 Å. The energetically minimised conformer of Siponimod (4) and TEFM78 (7g) were specified as the ligands for docking and the reference ligand was specified as the previously extracted siponimod ligand. The fitness function used to score the docking solutions was specified as ASP. The ASP scoring function was selected following a screen of all scoring functions available within the GOLD software in which the energetically minimised conformer of Siponimod (4) was redocked into S1P5. The ASP scoring function generated the highest percentage of solutions with a root-mean-square deviation (RMSD) of less than 2.0 Å than any other scoring function screened. Additional amendments to default settings were made including the number of genetic algorithm runs was set to 20 for each ligand, early termination was not allowed and search efficient was specified as >200%. The docking solutions were then analysed within Hermes and UCSF ChimeraX 1.8 software.
Radiochemistry: general experimental. No-carrier-added aqueous [18F]fluoride was produced via the 18O(p,n)18F nuclear reaction by irradiation of 18O-enriched water by a GE PETtrace 8 cyclotron. All radiofluorination reactions were carried out on a GE TRACERlab FXFN automated synthesiser. Sep-Pak QMA Carbonate Plus Light cartridges (Waters) were preconditioned with potassium trifluoromethanesulfonate (90 mg mL−1; 10 mL) and water (10 mL) prior to use. Oasis HLB Plus Light (Waters) cartridges were preconditioned with ethanol (5 mL) and then with water (10 mL) prior to use. The starting activity for calculating the radiochemical yield was determined from the GM reading taken immediately following delivery of [18F]fluoride to the synthesiser from the cyclotron. The final activity readings were recorded using a Capintec CRC-25 PET dose calibrator.
Analytical HPLC method. Analytical HPLC was carried out on a Thermo Dionex Ulimate system 3000 equipped with a Berthold FlowStar LB 513 radio flow detector and a DAD-3000 UV detector. An isocratic mobile phase of 70% acetonitrile in water was used with a Phenomenex Gemini 5 μm C18 110 Å 250 × 4.6 mm column at a rate of 1 mL min−1. The nonradioactive standards were detected using a UV wavelength of 254 nm.
Preparation of [18F]TEFM78. Immediately prior to delivering [18F]fluoride, 6-[(4′-(trimethylstannyl)-2′-6′-dimethylphenyl)amino]-2-methoxy-3-methylbenzamide (23) (5.0 mg), copper(II) trifluoromethanesulfonate (8.0 mg) and pyridine (20 μL) were dissolved in N,N-dimethylacetamide (0.70 mL) and the solution added to a vial on the synthesiser. Cyclotron target water containing [18F]fluoride was transferred to and trapped on a Sep-Pak QMA Carbonate Plus Light 46 mg cartridge. The activity was eluted into a reaction vessel using a solution of potassium trifluoromethanesulfonate (4.5 mg) and potassium carbonate (50 μg) in water (0.30 mL) and acetonitrile (0.30 mL). This solution was dried by being stirred at 100 °C under vacuum and a stream of helium gas for 2 min. This process was repeated twice using acetonitrile (2 × 1 mL). The [18F]fluoride was then completely dried by applying full vacuum for 1 min. The solution of 6-[(4′-(trimethylstannyl)-2′-6′-dimethylphenyl)amino]-2-methoxy-3-methylbenzamide (23) was added to the reaction vessel, which was sealed, and the mixture heated to 110 °C for 20 min while being stirred. The reaction mixture was cooled to 30 °C and diluted with a 50% aqueous solution of acetonitrile (2.0 mL). The reaction mixture was then transferred into the HPLC injector loop for purification. Purification was performed by semipreparative HPLC with a SYKMN S1122 solvent delivery system using a Phenomenex Luna 5 μm PFP(2) 100 Å 250 × 10 mm column and eluted using a 60% solution of acetonitrile in water at a flow rate of 4.0 mL min−1. The product fraction was identified using a gamma detector at a retention time of approximately 13 min and collected into a flask containing water (20 mL). The diluted fraction was then passed onto an Oasis HLB Plus Light cartridge, washed with water (5.0 mL), and eluted from the cartridge with ethanol (0.50 mL) and saline (4.5 mL). [18F]TEFM78 was isolated in 16 ± 4% radiochemical yield with a radiochemical purity of >99% and a molar activity of 229 ± 22 GBq μmol−1, starting from 75.3 ± 2.2 GBq of [18F]fluoride (n = 2). The total synthesis time from delivery of [18F]fluoride to extraction of the product was 62 min.
[18F]TEFM78 in vivo studies in rats. All animal procedures were performed in accordance with the guidelines for care and use of laboratory animals of Edinburgh University and experiments were approved by the Home Office under the Animals (Scientific Procedures) Act 1986, UK. Naïve adult male Sprague–Dawley rats (age: 12.92 ± 0.34 weeks, weight: 402.67 ± 19.48 g, mean ± SD, n = 3) were used for in vivo scanning. Anesthesia was induced with the use of an induction chamber with 3% isoflurane (Isoflo, APIECE) delivered in a mixture of 0.5 L min−1 of oxygen and 0.5 L min−1 nitrous oxide. Once anesthesia was induced, the rat was transferred to the bed of the PET scanner (NanoPET/CT, Mediso Medical Imaging Systems, Budapest, Hungary) ensuring the nose was in the nose cone and anesthesia was maintained with 2–2.5% isoflurane (50/50 oxygen/nitrous oxide, 1 L min−1). A rectal thermometer was inserted and a respiration monitor was placed on the chest of the rat. The tail vein was then cannulated with a 26G cannula to allow for the bolus injection of the radiotracer. PET scans were conducted on a Mediso nanoPET/CT (Mediso Medical Imaging Systems, Hungary) using a coincidence mode of 1:5. The acquisition time was set to 120 minutes emission scan. Once the PET scan was complete, a CT scan was acquired using the following parameters: a semi-circular trajectory with maximum field of view and 360 projections; a tube voltage of 50 kVp and exposure time of 300 ms were used, with a binning setting of 1:4. PET data was reconstructed using Mediso's iterative Teratomo 3D reconstruction algorithm with 4 iterations and 6 subsets, full detector model, spike filter on, normal regularization, a 400 to 600 keV energy window and a 0.4 mm voxel size. All PET data was corrected for randoms, scatter and attenuation. Emission data was re-binned as follows 18 × 10 s, 2 × 30 s, 1 × 60 s, 2 × 2 min, 10 × 5 min and 6 × 10 min. Once the PET scans were reconstructed, data was analysed using PMOD image analysis software (PMOD Technologies LLC, Zürich, Switzerland). Volumes of interest (VOIs) were created by using all regions in the rat Schiffer and SIGMA brain atlas (whole brain), whole grey matter and whole white matter regions or manually drawn in the case of the spinal cord region. Time activity curves (TACs) were created for each region and PET data expressed as standard uptake value (SUV), calculated as radioactive concentration in the VOI divided by the injected dose and animal's weight.
Data availability
The data supporting this article have been included as part of the ESI.† This includes the physicochemical properties and binding curves of 7a–7h and 8a–8e, selected analytical HPLC traces for target compounds, HPLC traces from the radiosynthesis of [18F]TEFM78, 1H and 13C NMR spectra of all compounds.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from EPSRC (studentships to T. E. F. M., EP/M506539/1, M. C. H., EP/M508056/1 and L. J. N. W., EP/T517896/1), SPRINT MS/NMD (studentship to R. C. S.), the Neurosciences Foundation and the University of Glasgow is gratefully acknowledged. H. M. acknowledges funding from MRC via the Total Body PET Infrastructure Project (MR/Y008944/1). The Carnegie Trust (Research Incentive Grant, RIG0074760), the Moray Endowment Fund Award and the Edinburgh Medical School Deanery of Clinical Sciences Funding Challenge are greatly acknowledged for providing financial support towards purchase of essential research consumables for this work. The British Heart Foundation is greatly acknowledged for providing funding towards establishment of the preclinical PET laboratory at the University of Edinburgh (RE/13/3/30183). A. A. S. T. and T. E. F. M. were funded by the British Heart Foundation (RG/16/10/32375, FS/19/34/34354). A. A. S. T. is a recipient of a Wellcome Trust Technology Development Award (221295/Z/20/Z). This publication has been made possible in part by a CZI grant (DAF2021-225273) and grant DOI https://doi.org/10.37921/690910twdfoo from the Chan Zukerberg Initiative DAF, an advised fund of Silicon Valley Community Foundation. C. J. A.-C. is supported by Edinburgh Preclinical Imaging. The authors are grateful to the Little France BVS staff for invaluable support to the project.
Notes and references
- D. Marsolais and H. Rosen, Nat. Rev. Drug Discovery, 2009, 8, 297–307 CrossRef PubMed.
- A. Cartier and T. Hla, Science, 2019, 366, eaar5551 CrossRef PubMed.
-
(a) V. A. Blaho and T. Hla, Chem. Rev., 2011, 111, 6299–6320 CrossRef PubMed;
(b) V. A. Blaho and T. Hla, J. Lipid Res., 2014, 55, 1596–1608 CrossRef PubMed.
- S. Grassi, L. Mauri, S. Prioni, L. Cabitta, S. Sonnino, A. Prinetti and P. Giussani, Front. Pharmacol., 2019, 10, 807 CrossRef PubMed.
- O. J. David, J. M. Kovarik and R. L. Schmouder, Clin. Pharmacokinet., 2012, 51, 15–28 CrossRef PubMed.
- V. Brinkmann, A. Billich, T. Baumruker, P. Heining, R. Schmouder, G. Francis, S. Aradhye and P. Burtin, Nat. Rev. Drug Discovery, 2010, 9, 883–897 CrossRef PubMed.
- D.-S. Im, J. Clemens, T. L. MacDonald and K. R. Lynch, Biochemistry, 2001, 40, 14053–14060 CrossRef PubMed.
- R. Van Doorn, M. A. Lopes Pinheiro, G. Kooij, K. Lakeman, B. van het Hof, S. M. A. van der Pol, D. Geerts, J. van Horssen, P. van der Valk, E. van der Kam, E. Ronken, A. Reijerkerk and H. E. de Vries, J. Neuroinflammation, 2012, 9, 133 CrossRef PubMed.
- C. Jaillard, S. Harrison, B. Stankoff, M. S. Aigrot, A. R. Calver, G. Duddy, F. S. Walsh, M. N. Pangalos, N. Arimura, K. Kaibuchi, B. Zalc and C. Lubetzki, J. Neurosci., 2005, 25, 1459–1469 CrossRef.
- For example, see:
(a) E. H. Demont, S. Arpino, R. A. Bit, C. A. Campbell, N. Deeks, S. Desai, S. J. Dowell, P. Gaskin, J. R. J. Gray, L. A. Harrison, A. Haynes, T. D. Heightman, D. S. Holmes, P. G. Humphreys, U. Kumar, M. A. Morse, G. J. Osborne, T. Panchal, K. L. Philpott, S. Taylor, R. Watson, R. Willis and J. Witherington, J. Med. Chem., 2011, 54, 6724–6733 CrossRef PubMed;
(b) A. D. Hobson, C. Harris, E. L. van der Kam, S. C. Turner, A. Abibi, A. L. Aguirre, P. Bousquet, T. Kebede, D. B. Konopacki, G. Gintant, Y. Kim, K. Larson, J. W. Maull, N. S. Moore, D. Shi, A. Shrestha, X. Tang, P. Zhang and K. K. Sarris, J. Med. Chem., 2015, 58, 9154–9170 CrossRef PubMed.
- B. Ma, K. M. Guckian, X.-g. Liu, C. Yang, B. Li, R. Scannevin, M. Mingueneau, A. Drouillard and T. Walzer, ACS Med. Chem. Lett., 2021, 12, 351–355 CrossRef PubMed.
- Z. T. Al-Salama, Drugs, 2019, 79, 1009–1015 CrossRef PubMed.
- S. L. Pimlott and A. Sutherland, Chem. Soc. Rev., 2011, 40, 149–162 RSC.
-
(a) D. van der Born, A. Pees, A. J. Poot, R. V. A. Orru, A. D. Windhorst and D. J. Vugts, Chem. Soc. Rev., 2017, 46, 4709–4773 RSC;
(b) R. Halder and T. Ritter, J. Org. Chem., 2021, 86, 13873–13884 CrossRef PubMed.
-
(a) E. Briard, D. Orain, C. Beerli, A. Billich, M. Streiff, M. Bigaud and Y. P. Auberson, ChemMedChem, 2011, 6, 667–677 CrossRef PubMed;
(b) J. Rokka, C. Federico, J. Jurttila, A. Snellman, M. Haaparanta, J. O. Rinne and O. Solin, J. Labelled Compd. Radiopharm., 2013, 56, 385–391 CrossRef;
(c) V. P. Prasad, S. Wagner, P. Keul, S. Hermann, B. Levkau, M. Schäfers and G. Haufe, Bioorg. Med. Chem., 2014, 22, 5168–5181 CrossRef;
(d) R. S. Shaikh, S. S. Schilson, S. Wagner, S. Hermann, P. Keul, B. Levkau, M. Schäfers and G. Haufe, J. Med. Chem., 2015, 58, 3471–3484 CrossRef PubMed;
(e) Z. Luo, J. Han, H. Liu, A. J. Rosenberg, D. L. Chen, R. J. Gropler, J. S. Perlmutter and Z. Tu, Org. Biomol. Chem., 2018, 16, 9171–9184 RSC;
(f) H. Jiang, S. Joshi, H. Liu, S. Mansor, L. Qiu, H. Zhao, T. Whitehead, R. J. Gropler, G. F. Wu, A. H. Cross, T. L. S. Benzinger, K. I. Shoghi, J. S. Permutter and Z. Tu, ACS Chem. Neurosci., 2021, 12, 3733–3744 CrossRef;
(g) L. Qiu, H. Jiang, C. Zhou, J. Wang, Y. Yu, H. Zhao, T. Huang, R. Gropler, J. S. Perlmutter, T. L. S. Benzinger and Z. Tu, J. Med. Chem., 2023, 66, 4671–4688 CrossRef PubMed;
(h) V. P. Prasad, S. Wagner, P. Keul, S. Hermann, B. Levkau, M. Schäfers and G. Haufe, Bioorg. Med. Chem., 2024, 104, 117697 CrossRef;
(i) L. Qiu, H. Jiang, C. Zhou, V. K. R. Tangadanchu, J. Wang, T. Huang, R. J. Gropler, J. S. Perlmutter, T. L. S. Benzinger and Z. Tu, Org. Biomol. Chem., 2024, 22, 5428–5453 RSC.
- H. Mattes, K. K. Dev, R. Bouhelal, C. Barske, F. Gasparini, D. Guerini, A. K. Mir, D. Orain, M. Osinde, A. Picard, C. Dubois, E. Tasdelen and S. Haessig, ChemMedChem, 2010, 5, 1693–1696 CrossRef.
- D. T. Racys, S. A. I. Sharif, S. L. Pimlott and A. Sutherland, J. Org. Chem., 2016, 81, 772–780 CrossRef.
- C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC.
- Z. J. Kamiñski, Pept. Sci., 2000, 55, 140–164 CrossRef.
-
(a) D. T. Racys, C. E. Warrilow, S. L. Pimlott and A. Sutherland, Org. Lett., 2015, 17, 4782–4785 CrossRef PubMed;
(b) M. A. B. Mostafa, E. D. D. Calder, D. T. Racys and A. Sutherland, Chem. – Eur. J., 2017, 23, 1044–1047 CrossRef PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef.
- The membrane partition coefficient (KM) is defined as the equilibrium constant for compound partitioning into fluid membranes. Permeability (PM) is directly proportional to KM but also accounts for molecular size and membrane thickness.
- A. A. S. Tavares, J. Lewsey, D. Dewar and S. L. Pimlott, Nucl. Med. Biol., 2012, 39, 127–135 CrossRef PubMed.
- C. Harrison and J. R. Traynor, Life Sci., 2003, 74, 489–508 CrossRef.
- N. W. DeLapp, W. H. Gough, S. D. Kahl, A. C. Porter and T. R. Wiernicki, in Assay Guidance Manual, ed. G. S. Sittampalam, A. Grossman and K. Brimacombe, Eli Lilly & Company and The National Center for Advancing Translational Sciences, Bethesda, 2012, pp. 217–228 Search PubMed.
- The % efficacy is the relative efficacy for each compound versus S1P. This was performed in a single 96-well plate, with the counts per minute measured for each compound against S1P5 at a single high concentration.
- F. Wunsch, T. N. Nguyen, G. Wolber and M. Bermudez, Arch. Pharm., 2023, 356, e2300387 CrossRef.
- E. Lyapina, E. Marin, A. Gusach, P. Orekhov, A. Gerasimov, A. Luginina, D. Vakhrameev, M. Ergasheva, M. Kovaleva, G. Khusainov, P. Khorn, M. Shevtsov, K. Kovalev, S. Bukhdruker, I. Okhrimenko, P. Popov, H. Hu, U. Weierstall, W. Liu, Y. Cho, I. Gushchin, A. Rogachev, G. Bourenkov, S. Park, G. Park, H. J. Hyun, J. Park, V. Gordeliy, V. Borshchevskiy, A. Mishin and V. Cherezov, Nat. Commun., 2022, 13, 4736 CrossRef PubMed.
- Y. Yuan, G. Jia, C. Wu, W. Wang, L. Cheng, Q. Li, Z. Li, K. Luo, S. Yang, W. Yan, Z. Su and Z. Shao, Cell Res., 2021, 31, 1263–1274 CrossRef PubMed.
- Various other docked poses of TEFM78 generated by GOLD are presented in the ESI† document.
- R. C. Shaw, G. D. Tamagnan and A. A. S. Tavares, Front. Neurosci., 2020, 14, 871 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Physicochemical properties and binding curves of 7a–7h and 8a–8e, selected analytical HPLC traces for target compounds, HPLC traces from the radiosynthesis of [18F]TEFM78, other docked poses of TEFM78, 1H and 13C NMR spectra of all compounds. See DOI: https://doi.org/10.1039/d4md00929k |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab,
Emma K. Granta,
Robert C. Shawbc,
Lachlan J. N. Waddella,
Martyn C. Henrya,
Holly McErlaina,
Carlos J. Alcaide-Corralb,
Sally L. Pimlottd,
Adriana A. S. Tavaresbc and
Andrew Sutherland
ab,
Emma K. Granta,
Robert C. Shawbc,
Lachlan J. N. Waddella,
Martyn C. Henrya,
Holly McErlaina,
Carlos J. Alcaide-Corralb,
Sally L. Pimlottd,
Adriana A. S. Tavaresbc and
Andrew Sutherland