
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and biological evaluation of naphthalene-1,4-dione analogues as anticancer agents†
Yao Chenga,
Tsz Tin Yu a,
Ellen M. Olzomerb,
Martina Berettab,
Alice Katena,
Jacky Sua,
John Patrick Jonesa,
David StC Blacka,
Kyle L. Hoehnb,
Frances L. Byrne*b and
Naresh Kumar*a
a,
Ellen M. Olzomerb,
Martina Berettab,
Alice Katena,
Jacky Sua,
John Patrick Jonesa,
David StC Blacka,
Kyle L. Hoehnb,
Frances L. Byrne*b and
Naresh Kumar*a
aSchool of Chemistry, University of New South Wales, Sydney, NSW 2052, Australia. E-mail: n.kumar@unsw.edu.au
bSchool of Biotechnology and Biomolecular Sciences, University of New South Wales, Sydney, NSW 2052, Australia. E-mail: frances.byrne@unsw.edu.au
First published on 19th March 2025
Abstract
The increased metabolism of glucose via aerobic glycolysis, known as the Warburg effect, is a hallmark of most cancers. Identifying molecules that disrupt the Warburg effect may allow for selective cytotoxicity towards cancer cells and reduce side effects compared to current chemotherapy agents. Our initial hit compound, BH10, which potentially targets Kelch-like ECH-associated protein 1 (Keap1), increased oxygen consumption rate and displayed increased cytotoxicity towards cancer cells over normal cells in vitro. In this project, a library of analogues based on the BH10 scaffold was prepared with the aim of improving potency and cancer-cell specificity. Among these analogues, several compounds showed notable potency, with activity (IC50) observed around 1 μM. However, when considering selectivity, the imidazole derivative, compound 44, exhibited the most optimal balance, achieving an IC50 of 6.4 μM and selectivity ratio of 3.6 which indicates greater toxicity to cancer cells vs. normal cells.
1. Introduction
A major drawback of many cancer therapies is their non-selective toxicity to healthy cells and tissues.1,2 The development of new drugs with better cancer-specific cytotoxicity would provide better treatment options and quality of life to cancer patients. A potential way to improve the selectivity of therapeutics is to target a pathway that is altered in cancer cells. One such potential target is glucose metabolism, which Hanahan and Weinberg described as a new ‘hallmark of cancer’.3 In healthy cells, glucose metabolism begins with glucose transporters (GLUTs) moving glucose from outside the cell and into the cytoplasm. Once glucose molecules are inside the cell, each glucose molecule is metabolised into two pyruvate molecules via glycolysis. Pyruvate can then be metabolised by one of two pathways depending on oxygen availability. If oxygen is present, pyruvate enters the mitochondria where it is metabolised through the tricarboxylic acid (TCA) cycle to produce reducing equivalents that feed electrons into the electron transport chain. This is required for the process of oxidative phosphorylation (aerobic respiration), which generates approximately 36 ATP molecules per glucose molecule.4 In contrast, when oxygen is absent, pyruvate remains in the cytoplasm where it is converted to lactate via anaerobic respiration, yielding only 2 ATP molecules per glucose molecule. Hence, aerobic respiration is far more efficient for cellular energy production.4 In cancer cells, however, glucose metabolism is altered significantly.3 Cancer cells primarily metabolise glucose via glycolysis and convert pyruvate to lactate even in the presence of oxygen, and this metabolic preference is known as the “Warburg effect”. Although it appears to be an inefficient means of producing cellular energy (only 4 ATP molecules produced per molecule of glucose), it can provide a selective advantage for cancer cells by multiple mechanisms (Fig. 1a). For example, the replenishment of NAD when pyruvate is converted to lactate is essential for rapidly dividing cells.4–6 | ||
| Fig. 1 (a) Glucose metabolism in differentiated tissues: glucose is metabolized through oxidative phosphorylation or anaerobic glycolysis. In cancerous tissues, glucose is predominantly metabolized through aerobic glycolysis (Warburg effect), resulting primarily in lactate fermentation.4 (b) The chemical structure of BH10. | ||
We recently developed a screening platform to identify drug-like compounds that were more toxic to cancer cells than normal cells. From this screen, we identified a quinone molecule, 2-chloro-3-[(2-morpholin-4-ylethyl)amino]naphthalene-1,4-dione (referred to as BH10 (Fig. 1b)) that showed greater cytotoxicity towards cancer cells.7 Furthermore, BH10 alters cellular glucose metabolism and increases cellular oxygen consumption rate (OCR) by targeting mitochondrial redox defence in cancer cells. This alteration increases glucose oxidation and inhibits glycolysis, which in turns induces cancer cells necrosis.7 We also identified Kelch-like ECH-associated protein 1 (Keap1) as a potential protein target of BH10 via avidin pull-down assays with biotinylated-BH10.8 Keap1 is a critical regulator of cellular defence against oxidative stress.9
BH10 is a 1,4-naphthoquinone, and this structure is important in medicinal chemistry. Furthermore, the quinone scaffold is present in many frontline chemotherapy drugs.10,11 One of the best-known examples of quinones that has been used to treat cancer is doxorubicin.11 Importantly, our study demonstrated that BH10 had better cancer cell-specific cytotoxicity than 7 other well-known anticancer quinones including doxorubicin, CDC25 phosphatase inhibitor II, vitamin K3, mitomycin C, β-lapachone, tanespimycin (17-AAG) and streptonigrin, even though these compounds show higher potency (Fig. 2).7 Thus, BH10 represents a lead molecule for future drug development.
 | ||
| Fig. 2 Chemical structure and bioactivity of BH10 and seven other well-known anticancer compounds. IC50 values (μM) indicate the cytotoxicity towards HEC1A endometrial cancer cells, while selectivity is calculated as the IC50 value for non-cancerous MAD11 cells divided by the IC50 value for HEC1A cells.7 | ||
In this study, various functional groups were introduced to the 1,4-naphthoquinone scaffold to replace the morpholine chain and aimed to increase the scaffold's rigidity by modifying the 1,4-naphthoquinone into a naphthoimidazole. Therefore, the objectives of this study were to design, synthesize and screen BH10-like molecules to identify those that are more potent than BH10 and have improved selectivity compared to BH10. Specifically, the comprehensive structure–activity relationship, and the potential anti-cancer mechanism as well as the pharmacokinetic profile of the best drug candidates were explored in this study.
2. Result and discussion
2.1. Rational design and chemical synthesis
Firstly, 3-unsubstituted naphthoquinones 5–7 were synthesized to investigate whether the chlorine atom is necessary for its bioactivity. To achieve this, naphthalene-1,4-dione 1 was reacted with different amines with the presence of Et3N or K2CO3 as the base to accelerate the reaction (Scheme 1). This provided naphthoquinones 5–7 in 25–80% yield. Moreover, the effect of replacing the 3-chloro group with a bromo or methyl group on the bioactivity of the analogues was investigated. The corresponding analogues 8–11 were synthesized via the forementioned reaction condition using 2-bromonaphthalene-1,4-dione 2 or 2-methylnaphthalene-1,4-dione 3 as the starting material. Subsequently, the effect of varying the steric nature and the electronic properties of the pendent amine group on biological activity was explored. As the morpholine ring in BH10 contains a tertiary amine and an ether functional group, altering the oxygen atom in the ring could change the hydrophilic characteristic of the ring, which might provide an insight on the importance of a hydrogen acceptor of hydrogen bonding at that position. Furthermore, modifications to the ring size or replacing the entire ring with other groups could provide information about the steric requirements of the biological target. Hence, derivatives 12–25 were synthesized using the same reaction conditions mentioned above. Finally, to explore the effect of the introduction of an amino acid ester instead of the amine side chain of the hit compound on activity and specificity, tryptophan, and phenylalanine were used in the substitution reaction to obtain analogues 26–27. | ||
| Scheme 1 Synthesis of nucleophilic substitution products. Reagents and conditions: (a) corresponding amine, Et3N or K2CO3, EtOH or Et2O, r.t., 18–72 h. | ||
There have been reports in the literature that salt compounds such as ammonium salts possess good anticancer potency, therefore salt products 29 and 30 were synthesized.12–14 Using similar conditions in Scheme 1, dichloronaphthoquinone 4 was reacted with N-Boc-L-lysine methyl ester hydrochloride to obtain 28 (Scheme 2a). Deprotection of analogues 17 and 28 was achieved using TFA in DCM at room temperature for 3 h to afford final compounds 29 and 30 in 50–60% yields (Scheme 2b).
 | ||
Scheme 2 Synthesis of salt products. Reagents and conditions: (a) corresponding amine, Et3N, EtOH, r.t., 8 h; (b) 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 TFA/DCM, r.t., 3 h. :3 TFA/DCM, r.t., 3 h. | ||
Since the triazole ring is a common heterocycle found in drugs, it is of great interest to explore the effect of introducing the triazole ring into the analogues. Through click chemistry, the alkyne group on 24 was cyclised with appropriate phenyl azides in the presence of CuSO4·5H2O and sodium ascorbate as catalysts in THF/H2O (5:1) at 90 °C for 10 h to give triazole analogues 31–33 in 16–89% yield (Scheme 3).
 | ||
| Scheme 3 Synthesis of triazole products: (a) corresponding azido compounds, CuSO4·5H2O, sodium ascorbate, THF/H2O, 90 °C, 10 h. | ||
BH10 was previously found to be unstable in the biological body and can be easily metabolised.7,8 It was postulated that this was due to the presence of a good Michael addition acceptor in the scaffold of BH10 as well as the chlorine atom at the 3-position of the molecule acting as a good leaving group. A possible solution to mitigate this limitation could be the protection of the Michael addition acceptor and the conversion of the labile chlorine into a stable amide group. To achieve this, the amide intermediates were first synthesized, following a literature method described by Ho et al., where the chloramine starting material 34 was reacted with excess alkyl anhydride in the presence of concentrated H2SO4 at room temperature for 10 h (Scheme 4).15 The desired amide compounds 35–38 were isolated in moderate to high (50–90%) yields. To expand the diversity of the analogue library, a benzyl-substituted amide was synthesized by reacting the chloramine compound 34 with phenylacetyl chloride in the presence of the Lewis acid boron trifluoride diethyl etherate in toluene at 90 °C for 1–2 h to afford 49 in moderate yield of 65% (Scheme 4).16 The synthesized chloramide intermediates 35–38 and 49 were then reacted with aminopropylpiperidine following a modified literature method by Brun et al., giving the desired products 39–43 and 50 in 30–80% yield.17
 | ||
| Scheme 4 Synthesis of amide compounds and naphthoquinoneimidazole compounds. Reagents and conditions: (a) corresponding anhydrides, concentrated H2SO4, 50 °C, 1–2 h; (b) corresponding amines, Et3N, THF, r.t., 18 h; (c) 2 M NaOH, EtOH, 50 °C, 1–2 h; (d) phenylacetyl chloride, BF3·Et2O, toluene, 90 °C, 7 h; (e) Boc2O, pyridine, THF, r.t., 24 h; (f) HCl, Et2O, r.t., 18 h. | ||
Increasing the rigidity of a molecule could increase both its selectivity and potency, as the compound might bind to fewer targets and the entropic energy cost in binding could be reduced.18 Therefore, an extra fused ring was introduced into the scaffold to investigate the effect of rigidity on anticancer activity of the analogues. The synthesis of these analogues was achieved by dehydration of amide compounds 39–43 following a literature method described by Ho et al.15 Compounds 39–43 were heated under basic conditions in EtOH for 1–2 h to produce the ring-closed imidazole compounds 44–48 in moderate to high yields of 47–100% (Scheme 4). The effect of replacing the chlorine atom in the 1,4-naphthoquinone with an amino group on the anticancer activity of the compound was also explored and the ammonium salt 53 was synthesized. The primary amino group in compound 34 was first protected using di-tert-butyl dicarbonate (Boc2O) to give compound 51, which was then reacted with aminopropylpiperidine to afford compound 52. The deprotection of the Boc-protecting group in compound 52 gave final compound 53 as a primary ammonium chloride salt.
3. In vitro cell cytotoxicity and structure–activity relationship study
To evaluate the anticancer potency and selectivity of the synthesized compounds, they were screened for cytotoxicity against human endometrial cancer (HEC1A) cells and noncancerous human endometrial stromal (MAD11) cells using the 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay (Table 1). The hit compound BH10 was used as a control molecule.P value
| Compound | HEC1A IC50 (μM) | MAD11 IC50 (μM) | Selectivity ratio | clogP |
|---|---|---|---|---|
| a IC50 values were obtained from MTT cell viability assays (n = 3 biological repeats in HEC1A and MAD11 cell lines). | ||||
| BH10 | 10.22 | 25.61 | 2.51 | 2.96 |
| 5 | >40 | >40 | — | 6.85 |
| 6 | >40 | >40 | — | 2.47 |
| 7 | 17.21 | >40 | >2.32 | 3.68 |
| 8 | 9.55 | 20.51 | 2.15 | 6.70 |
| 9 | 4.16 | 12.06 | 2.90 | 3.33 |
| 10 | 1.24 | 3.27 | 2.64 | 4.54 |
| 11 | >40 | >40 | — | 6.86 |
| 12 | 1.57 | 2.87 | 1.83 | 2.94 |
| 13 | 1.29 | 2.38 | 1.84 | 3.61 |
| 14 | 1.07 | 2.33 | 2.18 | 4.17 |
| 15 | 1.29 | 2.38 | 1.84 | 4.73 |
| 16 | 5.65 | 12.09 | 2.14 | 3.38 |
| 17 | 15.83 | 39.56 | 2.50 | 4.85 |
| 18 | 2.91 | 3.40 | 1.17 | 5.78 |
| 19 | 18.60 | 40.34 | 2.24 | 4.60 |
| 20 | 6.41 | 21.92 | 3.42 | 3.24 |
| 21 | 1.83 | 4.79 | 2.62 | 4.45 |
| 22 | 2.36 | 5.27 | 2.23 | 3.28 |
| 23 | 32.0 | >40 | >1.25 | 4.37 |
| 24 | 21.92 | 22.39 | 1.02 | 3.11 |
| 25 | 20.0 | 20.1 | 1.00 | 2.73 |
| 26 | >40 | >40 | — | 4.20 |
| 27 | >40 | >40 | — | 4.20 |
| 29 | 4.68 | 11.47 | 2.45 | 3.26 |
| 30 | 38.01 | >40 | — | 2.50 |
| 31 | 19.51 | 34.02 | 1.74 | 3.85 |
| 32 | >40 | >40 | — | 3.59 |
| 33 | 11.13 | 21.32 | 1.92 | 4.35 |
| 39 | 26.04 | >40 | — | 2.51 |
| 40 | 14.11 | 38.20 | 2.70 | 3.04 |
| 41 | 15 | >40 | — | 3.52 |
| 42 | 15 | >40 | — | 4.03 |
| 43 | >40 | >40 | — | 1.02 |
| 44 | 6.40 | 23.00 | 3.59 | 3.54 |
| 45 | 12.0 | 35.0 | 2.92 | 4.01 |
| 46 | 19 | >40 | — | 4.52 |
| 47 | 14 | 24 | 1.80 | 5.13 |
| 48 | >40 | >40 | — | 1.91 |
| 50 | 8.8 | 39.0 | 4.43 | 4.20 |
| 53 | 27 | 24 | 0.90 | 2.34 |
Upon testing of compounds 5–11, it was found that biological activity was abolished once the halogen was removed from the C3-position of the naphthoquinone ring. The unsubstituted compounds 5–6 and the 2-methyl substituted compound 11 were unable to reduce the viability of either cell line by at least 50% at the maximum tested dose of 40 μM while the IC50 of 7 also increased to 17.21 μM. In contrast, 2-bromosubstituted compounds 8–10 have better cytotoxicity profiles, with IC50 of 9.55, 4.16 and 1.24 μM, respectively, against HEC1A compared to that of BH10 which possessed IC50 of 10.22 μM against HEC1A. Compounds 8–10 also have similar selectivity ratio of 2.15, 2.90 and 2.64, respectively, compared to that of 2.51 possessed by BH10 (Table 1). These results suggest that an electron-withdrawing group such as a halogen at the C3 position of the naphthoquinone ring promotes cytotoxicity.
Interestingly, modifying the pendent amino group also had great influence on cytotoxicity (Table 1). Removal of the morpholine ring was found to have varied effects on cytotoxicity. Compound 23 with a terminal butyl chain, compound 24 with a terminal alkyne group and compound 25 with a terminal ester saw a drastic decrease in cytotoxicity, with IC50 of 32.00, 21.92 and 20.00 μM, respectively, compared to BH10 with IC50 of 10.22 μM. Moreover, the addition of an amino acid moiety in compounds 26 and 27 also resulted in a loss of potency (IC50 > 40 μM) against tumour cells. In contrast, compound 16 with a terminal 1-chloroethyl group possessed IC50 of 5.65 μM and was more potent than BH10 although the selectivity ratio of 2.14 possessed by compound 16 is slightly lower than that of 2.51 possessed by BH10. Furthermore, compounds 12 and 22 bearing a dimethylamino group showed IC50 of 1.57 and 2.36 μM, respectively, and were at least four times more potent than that of BH10. However, their selectivity of 1.83 and 2.23, respectively, is again lower than that of BH10. This suggested that a terminal amino group provides the analogues superior cytotoxicity potency while the selectivity of the analogues is reduced as a trade-off.
Similarly, when the terminal morpholine ring system of BH10 was replaced by other cyclic amine ring systems as in compounds 13–15, the potency was significantly increased while selectivity was decreased with respect to BH10 (Table 1). Amongst compounds 13–15 with different terminal cyclic amine ring size, compound 14 with a terminal six-membered piperidine ring was the most potent compound in this study, showing an IC50 of 1.07 μM and being almost 10-times more potent than BH10. On the other hand, the pyrrolidinyl derivative 13 and azapenyl derivative 15 share the same IC50 of 1.29 μM and were almost 8-times more potent than that of BH10 although they are slightly less potent than compound 14. However, all three compounds possessed lower selectivity than BH10. Given that compound 14 exhibited the highest potency amongst compounds 13–15 with different ring sizes, an additional CH2 was introduced into the terminal alkyl chain (compound 21) to assess its impact. The results showed that the IC50 of compound 21 slightly decreased to 1.83 μM compared to 1.07 μM for compound 14, suggesting that the extra CH2 in the alkyl chain is not beneficial to potency. These results highlight that the removal of the ether functionality from the morpholine ring is beneficial for potency, with the six-membered piperidine ring and ethylene linker length to be the optimum ring size and linker length, respectively, for anticancer activity. This suggested that hydrophobic terminal groups might elicit a better effect in this position of the molecule.
The incorporation of an additional nitrogen in the pendent amine ring yielded varied effects in potency. The Boc-protected intermediate 17 and indole compound 19 with a terminal indole ring showed IC50 of 15.83 and 18.60 μM, respectively, and were less potent than BH10. In contrast, the piperazinyl salt 29 with an IC50 of 4.68 μM and selectivity ratio of 2.45 was 2.2 times more potent than BH10 and have similar selectivity as BH10. However, another salt analogue 30 showed lower potency with IC50 of 38.01 μM respectively. On the other hand, the bulky analogue 18 possessed IC50 of 2.91 μM and was 3.5-times more potent than BH10, however, it was less selective with selectivity ratio of only 1.17. Therefore, large terminal groups attached to the pendent amine ring are not suitable for structural modifications aimed at simultaneously enhancing potency and selectivity.
Naphthoquinone compounds 31–33 with a 1,2,3-triazole linker and a terminal phenyl ring at the N1 position were also synthesized and tested as the triazole moiety is commonly found in bioactive compounds. However, compounds 31–33 showed moderate to low anticancer activity, with none of their potency and selectivity being better than BH10. This demonstrated that the introduction of a triazole ring into naphthoquinone is not a good structural modification strategy.
Analogues with a nitrogen-containing group substituted at the C3 position were synthesized to examine their impact on potency and specificity. This series of compounds demonstrated that replacing the halogen with an amide functionality improves specificity while retaining potency. The propionamide analogue 40, butyramide analogue 41, pentanamide analogue 42 and phenylacetamide analogue 50 showed IC50 of 14, 15, 15 and 8.8 μM, respectively, with similar potency as BH10, but the newly synthesized compounds possess higher selectivity of 2.7, 3.7, 3.9 and 4.4, respectively.
When the amide-substituted compounds were cyclised into an imidazole functionality, potency generally improved for some compounds while the specificity was retained from the amide-substituted compounds. The most potent imidazole analogue was the 2-methyl substituted analogue 44 with an IC50 of 6.4 μM, followed by the 2-ethyl substituted analogue 45 with an IC50 of 12 μM, 2-butyl substituted analogue 47 with an IC50 of 14 μM and the 2-propyl substituted analogue 46 with an IC50 of 19 μM. Upon comparing the uncyclised amide compounds 39–42 with their corresponding cyclised imidazole counterparts 44–47, the cyclised imidazole compound 44 showed a sharp increase in potency with IC50 of 6.4 μM compared to its corresponding uncyclised compound 39, which possesses IC50 of 26 μM. Cyclised imidazole compounds 45 and 47 also showed slight increases of potency with IC50 decreased to 12 and 14 μM, respectively, compared to their corresponding uncyclised amide compounds 40 and 42, which possess IC50 of 14 and 15 μM, respectively. However, when the uncyclised amide compound 41 was cyclised into the imidazole compound 46, the potency dropped slightly, with the IC50 increased from 15 μM to 19 μM. On the other hand, the good selectivity observed for the imidazole compounds could possibly be attributed to the fact that the imidazole ring is relatively stable and rigid. The above results show that the cyclisation of amide analogues into imidazole compounds generally improves the potency of compounds, while retaining specificity.
Based on the biotesting results and analysis, several structure–activity relationships (SAR) were identified (Fig. 3). Firstly, incorporating an imidazole ring into the naphthoquinones enhances both potency and selectivity. Secondly, terminal cyclic amine groups generally exhibit higher activity. Additionally, introducing an amide group at the C3 position can improve selectivity while maintaining potency. Lastly, the presence of halogen atoms is crucial for maintaining potency.
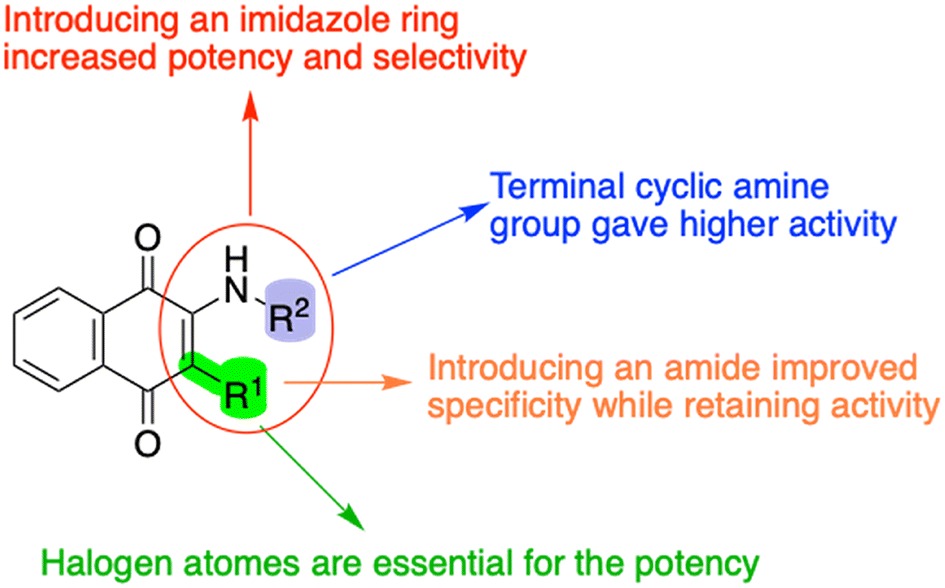 | ||
| Fig. 3 SAR conclusions of the synthesized BH10 derivatives. The incorporation of an imidazole ring enhances both potency and selectivity. Terminal cyclic amine groups generally exhibit higher activity. Introducing an amide group at the C3 position enhances selectivity while preserving potency. The presence of halogen atoms is critical for maintaining potency. | ||
To further assess the therapeutic potential and antiproliferative activity of selected potent analogues 21 and 44, additional in vitro cytotoxicity assays were performed against two human cancer cell lines (CALU-1 (lung squamous cell carcinoma) and Mia-Pa-Ca-2 (pancreatic cancer)). Among the tested compounds, compounds 21 and 44 exhibited better potency compared to BH10, especially compound 21 demonstrating significantly higher efficacy against the selected cancer cell lines (Table 2).
| Compound | Mean IC50 (μM) | |
|---|---|---|
| CALU-1 | Mia-Pa-Ca-2 | |
| a Values were obtained from MTT cell viability assays (n = 3 biological repeats in all cell lines). | ||
| BH10 | 16.67 | 19.68 |
| 21 | 4.70 | 4.75 |
| 44 | 15.38 | 14.72 |
4. Investigating the potential mechanism of action (MOA) of compounds 44 and 21
We wanted to find compounds that were more potent and had greater selectivity (cytotoxicity towards cancer cells than non-cancerous cells) than hit compound BH10. The imidazole 44 (IC50 = 6.40 μM; selectivity ratio = 3.6) and 21 (IC50 = 1.83 μM; selectivity ratio = 2.62) were selected for investigating whether their mechanism of action was similar to BH10 (Fig. 4a). | ||
| Fig. 4 (a) Chemical structures of the hit compound BH10 and its most promising analogues, 21 and 44. (b) Chemical structures of thiol-containing compounds, N-acetyl cysteine (NAC, left) and glutathione (GSH, right), with thiol functional groups highlighted in red. The thiol rescue experiment was conducted following the standard MTT assay protocol with an additional step, where HEC1A cancer cells were seeded as per the standard protocol on day 1. Cells were pre-treated with NAC or GSH (2 mM) for 40 minutes, followed by the addition of test compounds on day 2. The cells were then incubated for 48 hours according to the standard MTT assay protocol. | ||
4.1. Thiol rescue experiment
A thiol rescue experiment was performed to investigate the effect of thiol-containing compounds (antioxidants) on the cytotoxicity of BH10 analogues against cancer cells in a cellular environment. This allowed the exploration of the mechanism of action or selectivity of the newly synthesized analogues. The thiol rescue experiment followed the same protocol as the MTT assay with an additional step. HEC1A cancer cells were seeded on day 1 as normal. On day 2, the cells were pre-treated with thiol-containing compounds N-acetyl cysteine (NAC) or glutathione (GSH) (at 2 mM) for 40 min (Fig. 4b). After that, the test compounds were added to the plate and cells were incubated for 48 h, as per previous MTT protocol.8 An increase in IC50 for cells treated with thiol-containing compounds compared to the control group indicates rescue from cell death by the thiols (Table 3).In general, HEC1A cancer cells treated with compounds containing the C3 halogen (BH10, 21) were rescued by the thiol-containing compounds NAC and GSH. BH10 lost its cytotoxicity as demonstrated by the increased IC50 value of 58.00 μM and greater than 80.00 μM with NAC and GSH pre-treatment, respectively. The bioactivity also decreased 4.6 times with NAC pre-treatment. Similarly, the cytotoxicity of 21 was also partially rescued by both NAC and GSH because the IC50 values increased to 13.00 and 16.00 μM, and potency decreased by 7.1 and 8.7 times, respectively. These results suggested that compound 21 likely shares the same mechanism as BH10. In contrast, pretreating cancer cells with either NAC or GSH did not rescue the cytotoxicity of compound 44, with no significant change in IC50 values from the control group observed. An example of cellular viability curves for BH10 and 44 alone or in the presence of NAC or GSH is shown in ESI† S1.
It was hypothesized that the thiol compounds may act as scavenger molecules, reacting with the halogen-containing compounds, leading to their deactivation by preventing binding to other cellular targets. Therefore, the presence of the halogen at C3 appeared to be essential for thiol-mediated cell-rescue. Halogens are very good leaving groups, making Michael addition-type nucleophilic attack on the naphthoquinone scaffold possible. When the halogen is replaced with much poorer leaving groups, such as amides and amines, Michael addition becomes less likely. In the imidazole analogues, nucleophilic attack is very unlikely as the enone functionality is in between a fused ring system, making it no longer available to react as any reactions that occur at that position would disrupt the aromaticity of the system and hence such reactions are energetically unfavourable.
4.2. Measurements of oxygen consumption
Our initial study identified BH10 as a ‘hit’ compound based on a phenotypic screen that investigated compounds that increased cellular oxygen consumption rate (OCR).7 Therefore, oxygen consumption measurements were performed with the newly synthesized analogues using a Seahorse Extracellular Flux Analyser, which measures the OCR of cultured cells over time. High OCR can indicate a high rate of nutrient oxidation in the mitochondria caused by oxidative phosphorylation, or it can also be influenced by external mechanisms such as redox cycling.19 Although measurements of OCR do not pinpoint the location of oxygen consumption, it is a good initial indicator to compare the mechanism of action of new analogues with that of BH10. It was hypothesized that compounds without the C3 halogen and were unaffected by thiols in the cell rescue experiment might act via a different mechanism to BH10, and hence give different results in OCR.In this assay, HEC1A cells were treated with compounds BH10, 21 and 44 at various concentrations, and OCR was measured over 120 minutes. The results demonstrated that BH10 increased OCR in cells by up to 200% (Table 4 and ESI† S2), with the increase correlating with drug concentration, though 10 μM appeared to approach the maximum effect. Compound 21, another C3 halogen-containing compound, showed a similar trend to BH10, with a maximum OCR increase of approximately 280% at 20 μM. In contrast, compound 44, which lacks the C3 halogen group, induced a maximum OCR increase of 130% at doses equivalent to that of BH10 and 21, compared to the vehicle (DMSO) control. These OCR results suggest that the new analogue 44, lacking the C3 halogen, likely acts via a different mechanism compared to BH10.
| Compound | OCR (%) | |||
|---|---|---|---|---|
| DMSO | 5 μM | 10 μM | 20 μM | |
| BH10 | 100 | 125 | 200 | 200 |
| 21 | 100 | 175 | 260 | 280 |
| 44 | 100 | 100 | 130 | 130 |
5. Pharmacokinetic profiling of compound 44 in vivo
One of the major limitations of BH10 as an anticancer compound is its short half-life in vivo, with a half-life of 21.6 min when administered at 10 mg kg−1 via oral gavage in C57BL/6 mice.8 This means that the drug is rapidly metabolised, so larger doses would be required for in vivo efficacy, which in turn increases the risk of toxicity. By replacing the halogen at C3 in the hit compound BH10 to more metabolically stable functional groups such as amides and imidazole rings, it was hypothesized that the half-life would increase. To confirm this, pharmacokinetic profiling of the most potent analogue 44 was performed in mice. In such experiments, mice were administered the compound via oral gavage, and blood was drawn from their tail at regular intervals. Blood was then centrifuged, and the plasma collected was mixed with 10% MeOH in acetonitrile to precipitate protein. Mass spectrometry analyses of each sample and drug standards (drugs spiked into blood) enabled quantitation of each drug in the plasma over time to generate a pharmacokinetic profile. From this, maximum plasma concentration (Cmax) and half-life (t1/2) were determined (Table 5). The novel analogue 44, lacking the halogen at the C3 position, demonstrated a half-life of 26.8 min via oral administration (see ESI† S3), which is 5.2 min longer than BH10 (t1/2 = 21.6 min).8 Although this represents a slight improvement, further optimization is necessary to enhance the compound's half-life.| Compound | Treatment type | Dose (mg kg−1) | Mouse strain | Cmax (μM) | t1/2 (min) |
|---|---|---|---|---|---|
| a Values are the average of three runs. Cmax, maximum concentration; t1/2, half-life. | |||||
| 44 | Oral | 20 | C57BL/6 | 0.70 | 26.8 |
6. Molecular docking study
A molecular docking simulation was conducted to examine the interaction between BH10/44 and the Keap1 protein (PDB: 4XMB), aiming to elucidate their potential binding modes. The results indicated that both compounds occupy the same hydrophobic pocket within Keap1 but engage in distinct interactions. For BH10, the carbonyl group forms hydrogen bonds with key residues GLY-367, VAL-465, and VAL-606 (Fig. 5a). Additionally, the amine hydrogen forms hydrogen bonds with the carbonyl groups of GLY-367 and VAL-606, and the nitrogen in the morpholine ring forms another hydrogen bond with THR-560. In contrast, compound 44 exhibited a simpler interaction profile, with only the carbonyl group forming hydrogen bonds with residues VAL-465 and VAL-606 (Fig. 5b). Notably, compound 44 had a docking score of −7.36, closely aligned with the fitness score of −8.73 for BH10, indicating a comparable binding affinity of compound 44 for the target protein. | ||
| Fig. 5 Predicted binding model of BH10 (a) and 44 (b) with Keap1 (PDB code: 4XMB). Hydrogen bonding interactions are indicated by black dashed lines. (a) The carbonyl group of BH10 forms hydrogen bonds with residues GLY-367, VAL-465, and VAL-606 while the amine hydrogen forms hydrogen bonds with the carbonyl groups of GLY-367 and VAL-606. (b) The carbonyl group of compound 44 forms hydrogen bonds with residues VAL-465 and VAL-606. | ||
7. Conclusion
In conclusion, a series of 40 analogues based on the BH10 scaffold were successfully synthesized, accompanied by a detailed structure–activity relationship study. Several compounds exhibited promising anticancer potency with IC50 of 1–2 μM but with decreased selectivity. Among the synthesized analogues, the imidazole compound 44 demonstrated the most favourable balance, with an IC50 of 6.4 μM and a selectivity factor of 3.6. In vitro investigations into the mechanism revealed compound 44 was unaffected by thiol compounds in the cytotoxicity assay, and did not increase OCR significantly which indicates that 44 likely acts via a different mechanism to the hit compound BH10. This could indicate differences in the metabolic profiles of 44 and BH10. Pharmacokinetic profiling in vivo showed only a slight improvement in the half-life of compound 44 compared to BH10, indicating the need for further optimization to improve its pharmacokinetic properties.8. Experimental
8.1. Chemical synthesis
General synthetic method a for 5–27, 39–43 and 50. This method was based on a modification of the procedure reported by Brun et al.17 The appropriate naphthoquinone (1 equivalent) was dissolved in EtOH or Et2O (20 mL) and to the resulting mixture was added 1.0–1.2 equivalent of the appropriate amine and 1.0–1.2 equivalent of Et3N or K2CO3. The resulting mixture was stirred at r.t. for 18–72 h. The reaction mixture was then concentrated in vacuo, diluted in EtOAc and washed with water, and the organic layer was dried over anhydrous Na2SO4. The solvent was evaporated, and the resulting residue was purified via flash column chromatography (when required) in the appropriate solvent system to afford the desired product.
General synthetic method B for 29 and 30. TFA (1 mL) was added to a solution of corresponding Boc-compound (0.30 g, 0.67 mmol) in DCM (10 mL) under nitrogen on ice. The reaction mixture was stirred at r.t. for 5 h. The solvent removed in vacuo yielding corresponding products.
General synthetic method C for 31–33. Under an argon atmosphere, alkyne 24 and the corresponding azido moieties were reacted in THF: H2O (5
:1), sodium ascorbate 20 mol%, CuSO4·5H2O 10 mol% at 50 °C for 20 h. The reaction was then quenched in H2O (20 mL), and the organic layer extracted by EtOAc (2 × 30 mL) with a single brine wash (1 × 10 mL). The combined extracts were dried over Na2SO4, and the volatiles evaporated under reduced pressure. The crude product was purified using column chromatography on silica eluting with a Hexane: EtOAc solvent system to afford the target products.
General synthetic method D for amide intermediates 35–38. The appropriate excessive anhydride was added to compound 34. Concentrated sulfuric acid (2 drops) was added. The reaction mixture was stirred at r.t. for 2 h. Excess anhydride was removed by an ether wash. Column purification was performed when required.
General synthetic method E for imidazole analogues 44–48. Aqueous NaOH (2 M) was added to a solution of appropriate amide analogue 39–43 in EtOH (2–10 mL). The reaction mixture was heated at 50 °C for 1–2 h. The solvent was removed in vacuo and the residue was purified via flash column chromatography (when required) in the appropriate solvent system to afford the product.
Methyl N6-(tert-butoxycarbonyl)-N2-(3-chloro-1,4-dioxo-1,4-dihydronaphthalen-2-yl)-L-lysinate (28). Following general synthetic procedure A, N-Boc-L-lysine methyl ester hydrochloride (0.66 g, 2.2 mmol) and K2CO3 (0.10 g, 0.73 mmol) were added to a solution of 2,3-dichloro-[1,4]-naphthoquinone (0.51 g, 2.20 mmol) in 10% THF in EtOH (10 mL). The product eluted from hexane: EtOAc (5
:1) as an orange solid (0.34 g, 34%). 1H NMR (400 MHz, CDCl3) δ 8.17 (dd, J = 7.6, 1.3 Hz, 1H), 8.08 (dd, J = 7.7, 1.3 Hz, 1H), 7.76 (td, J = 7.6, 1.4 Hz, 1H), 7.67 (td, J = 7.6, 1.4 Hz, 1H), 6.34 (s, 1H), 5.25 (m, 1H), 4.55 (s, 1H), 3.82 (s, 3H), 3.13 (t, J = 6.6 Hz, 2H), 2.05–1.84 (m, 2H), 1.60 (m, 2H), 1.58–1.52 (m, 2H), 1.45 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 180.10, 176.84, 172.37, 155.95, 143.47, 134.90, 132.73, 132.31, 129.88, 126.96, 126.84, 79.25, 56.22, 52.78, 33.71, 29.79, 28.40, 22.38; IR (cm−1) 3371, 3311, 2965, 2846, 1735, 1671, 1605; HRMS (+ESI): found m/z 473.1446; [M + Na]+; C22H27ClN2O6Na required 473.1450.
N-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-yl)acetamide (35). 15Following general synthetic method D, acetic anhydride (5 mL) was added to 34 (0.21 g, 1.0 mol). Concentrated sulfuric acid (2 drops) was added. The reaction mixture was stirred at r.t. for 2 h. The resulting yellow precipitate was washed with ether and collected (1.30 g, 52%).15 1H NMR (400 MHz, DMSO-d6) δ 7.98 (d, J = 7.7 Hz, 1H), 7.84–7.66 (m, 2H), 7.60 (t, J = 7.6 Hz, 1H), 2.60 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 179.47, 172.64, 167.78, 163.39, 158.06, 135.47, 131.16, 130.33, 130.03, 125.98, 122.87, 14.17; MS (+ESI): found m/z 250.05; [M + H]+; C12H9ClNO3 required 250.03.
N-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-yl)propionamide (36). 20Following general synthetic method D, propionic anhydride (5 mL) was added to 34 (0.21 g, 1.0 mmol). Concentrated sulphuric acid (1 drop) was added. The reaction mixture was stirred well at r.t. for 2 h. The resulting yellow precipitate was washed with ether and collected (0.24 g, 86%).15,20 1H NMR (400 MHz, CDCl3) δ 8.25–8.06 (m, 2H), 7.86–7.73 (m, 2H), 7.73–7.63 (m, 1H), 2.57 (q, J = 7.5 Hz, 2H), 1.30 (t, J = 7.5 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 179.98, 177.68, 170.38, 139.13, 134.81, 134.14, 131.58, 131.10, 130.27, 127.54, 127.07, 30.52, 9.32; MS (+ESI): Found m/z 264.05; [M + H]+; C13H11ClNO3 required 264.04.
N-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-yl)butyramide (37). 20Following general synthetic method D, butyric anhydride (5 mL) was added to 34 (0.21 g, 1.0 mmol). Concentrated sulphuric acid (3 drops) was added. The reaction mixture was stirred at r.t. for 2 h. The resulting yellow precipitate was washed with ether and collected (0.18 g). The product eluted from hexane: EtOAc (5
:1) column as a yellow solid (0.15 g, 53%).20 1H NMR (400 MHz, CDCl3) δ 8.25–8.15 (m, 1H), 8.16–8.08 (m, 1H), 7.79 (dd, J = 6.7, 5.8 Hz, 2H), 7.73–7.66 (m, 1H), 2.52 (t, J = 7.4 Hz, 2H), 1.82 (q, J = 7.4 Hz, 2H), 1.07 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 179.99, 177.68, 169.64, 139.11, 134.81, 134.14, 133.19, 131.59, 130.27, 127.54, 127.06, 39.19, 18.85, 13.62; MS (+ESI): found m/z 278.05; [M + H]+; C14H13ClNO3 required 278.06.
N-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-yl)pentanamide (38). 20Following general synthetic method D, valeric anhydride (5 mL) was added to 34 (0.20 g, 0.98 mmol). Concentrated sulphuric acid (2 drops) was added. The reaction mixture was stirred well at r.t. for 2 h. The resulting yellow precipitate was washed with ether and collected (0.25 g). Product eluted from hexane: EtOAc column (5
:1) as a yellow solid (0.19 g, 65%).20 1H NMR (400 MHz, CDCl3) δ 8.26–8.05 (m, 1H), 7.87–7.65 (m, 2H), 2.53 (t, J = 7.5 Hz, 1H), 1.88–1.70 (m, 1H), 1.55–1.37 (m, 1H), 0.99 (t, J = 7.3 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 179.98, 177.67, 169.81, 139.13, 134.80, 134.13, 133.18, 131.58, 130.27, 127.53, 127.05, 37.09, 27.39, 22.25, 13.79; MS (+ESI): found m/z 292.05; [M + H]+; C15H15ClNO3 required 292.07.
N-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-yl)-2-phenylacetamide (49). 16A solution of aminochloro compound 34 (0.90 g, 4.3 mmol) in toluene (9 mL) was stirred at 90 °C. A solution of borontrifluoride diethyl etherate (600 μL, 3.2 mmol) and phenylacetyl chloride (3.0 mL, 16 mmol) in toluene (4 mL) was added dropwise over 30 min. The reaction mixture was then left stirring at 90 °C for a further 7 h. The reaction mixture was cooled to r.t. and the solvent evaporated. The resulting pale-yellow green precipitate was washed with ether and collected (0.78 g, 55%).16 1H NMR (400 MHz, DMSO-d6) δ 10.38 (s, 1H), 8.12–7.98 (m, 2H), 7.95–7.86 (m, 2H), 7.39–7.30 (m, 4H), 7.30–7.22 (m, 1H), 3.81 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 179.23, 178.06, 169.26, 141.61, 135.75, 135.31, 135.04, 135.03, 131.44, 131.30, 129.69, 128.78, 127.17, 127.12, 42.51. MS (+ESI): found m/z 326.06; [M + H]+; C18H13ClNO3 required 326.05.
2-((2-Morpholinoethyl)amino)naphthalene-1,4-dione (5). 21Following general synthetic procedure A, 1,4-naphthoquinone (0.35 g, 2.20 mmol), EtOH (20 mL), 4-(2-aminoethyl)morpholine (0.30 mL, 2.20 mmol) and Et3N (0.31 mL, 2.20 mmol) were reacted. Reaction stirred for 18 h. Product eluted with EtOAc/hexane in flash column chromatography to afford a dark orange powder (0.49 g, 79%): mp: 147–148 °C; 1H NMR (300 MHz, CDCl3) δ 8.21–8.11 (m, 1H), 8.11–8.01 (m, 1H), 7.59–7.79 (m, 2H), 7.00 (s, 1H), 4.06–3.91 (m, 2H), 3.85–3.71 (m, 4H), 2.69 (t, J = 6.0 Hz, 2H), 2.64–2.46 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 180.33, 147.00, 134.71, 132.42, 132.32, 130.05, 126.96, 126.75, 67.05, 56.65, 52.94, 41.17. IR (cm−1) 3342, 2969, 2832, 1671, 1595, 1567, 1517, 1113, 865, 724; MS (+ESI): found m/z 287.25, [M + H]+; C16H19N2O3 required 287.13.
2-((3-Morpholinopropyl)amino)-[1,4]-naphthoquinone (6). Following general synthetic procedure A, 1,4-naphthoquinone (0.35 g, 2.20 mmol), EtOH (20 mL), 3-morpholinopropylamine (0.32 mL, 2.20 mmol) and Et3N (0.31 mL, 2.20 mmol) were reacted. Reaction stirred for 18 h. Product eluted with EtOAc/hexane in flash column chromatography to afford a bright red crystals (0.42 g, 64%), mp: 129–131 °C; 1H NMR (300 MHz, CDCl3) δ 8.09 (m, 2H), 7.79–7.57 (m, 3H), 5.72 (s, 1H), 3.91 (t, J = 4.7 Hz, 4H), 3.32 (q, J = 5.8 Hz, 2H), 2.58 (s, 6H), 1.92 (s, 2H); 13C NMR (75 MHz, CDCl3) δ 182.93, 181.94, 148.64, 134.59, 133.80, 131.84, 130.69, 126.22, 126.12, 100.36, 66.64, 57.65, 53.74, 42.81, 23.37; IR (cm−1) 3258, 2963, 2852, 1677, 1592, 1562, 1519, 1111, 726; HRMS (+ESI): found m/z 301.1547, [M + H]+; C17H21N2O3 required 301.1545.
2-((3-(Piperidin-1-yl)propyl)amino)-[1,4]-naphthoquinone (7). Following general synthetic procedure A, 1,4-naphthoquinone (0.35 g, 2.20 mmol), EtOH (20 mL), 3-aminopropylpiperidine (0.35 mL, 2.20 mmol) and Et3N (0.31 mL, 2.20 mmol) were reacted. Product eluted at EtOAc/hexane = 1
:1 in flash column chromatography to afford a dark red solid (0.06 g, 10%): mp: 108–110 °C; 1H NMR (400 MHz, CDCl3): δ 8.11 (m, 2H), 7.92 (br s, 1H), 7.73 (t, J = 7.5 Hz, 1H), 7.61 (t, J = 7.6 Hz, 1H), 5.70 (s, 1H), 3.29 (q, J = 17.2 Hz, 2H), 2.53–2.45 (m, 6H), 1.90–1.70 (m, 6H), 1.53 (t, J = 4.1 Hz, 2H); 13C NMR (CDCl3): δ 182.90, 181.94, 148.88, 134.46, 133.88, 131.69, 130.79, 126.19, 126.06, 100.16, 58.10, 43.08, 30.93, 25.68, 24.47, 23.75; IR (cm−1) 3259, 3076, 2918, 2784, 1679, 1605, 1518, 725; HRMS (+ESI): found m/z 2.991754, [M + H]+; C18H23N2O2 required 299.1756.
2-Bromo-3-((2-(4-morpholinyl)ethyl)amino)-[1,4]-naphthoquinone (8). Following general synthetic procedure A, 2,3-dibromo-[1,4]-naphthoquinone (0.70 g, 2.20 mmol), Et2O (20 mL), 4-(2-aminoethyl)morpholine (0.30 mL, 2.20 mmol), Et3N (0.30 mL, 2.20 mmol). Reaction stirred for 72 h. Product eluted at EtOAc/hexane = 1
:4 in flash column chromatography to afford a red powder (0.58 g, 72%): mp: 120–122 °C; 1H NMR (300 MHz, CDCl3): δ 8.14 (d, J = 7.6 Hz, 1H), 8.04 (d, J = 8.7 Hz, 1H), 7.74 (t, J = 8.9 Hz, 1H), 7.69 (t, J = 7.9 Hz, 1H), 6.98 (br s, 1H), 4.00 (q, J = 16.8 Hz, 2H), 3.77 (t, J = 9.2 Hz, 4H), 2.69 (t, J = 11.9 Hz, 2H), 2.54 (t, J = 8.9 Hz, 4H); 13C NMR (CDCl3): δ 180.33, 134.70, 132.31, 130.52, 126.96, 126.74, 67.05, 56.65, 52.94, 41.18; IR (cm−1) 3210, 2948, 2831, 1676, 1563, 1450, 1285, 718; HRMS (+ESI): found m/z 365.0495, [M + H]+; C16H17BrN2O3 required 365.0490.
2-Bromo-3-((3-morpholinopropyl)amino)-[1,4]-naphthoquinone (9). Following general synthetic procedure A, 2,3-dibromo-[1,4]-naphthoquinone (0.70 g, 2.20 mmol), Et2O (20 mL), 3-morpholinopropylamine (0.32 mL, 2.20 mmol), Et3N (0.31 mL, 2.20 mmol). Reaction stirred for 19 h. Product eluted at EtOAc/hexane = 1
:1 in flash column chromatography to afford an orange powder (0.18 g, 22%); mp: 144–146 °C; 1H NMR (400 MHz, CDCl3): δ 8.16 (d, J = 8.6 Hz, 1H), 8.04 (d, J = 8.6 Hz, 1H), 7.73 (t, J = 16.5 Hz, 1H), 7.64 (t, J = 8.9 Hz, 1H), 4.02–4.11 (m, 2H), 3.80–3.92 (m, 4H), 2.40–2.70 (m, 6H), 1.89 (s, 2H); 13C NMR (CDCl3): δ 180.34, 147.06, 134.73, 132.51, 132.25, 129.96, 127.03, 126.81, 66.57, 57.54, 53.92, 45.50, 25.63; IR (cm−1) 3082, 2948, 2782, 1678, 1561, 1291, 1110, 721; HRMS (+ESI): found m/z 379.0652, [M + H]+; C17H19BrN2O3 required 379.0649.
2-Bromo-3-((3-(piperidin-1-yl)propyl)amino)-[1,4]-naphthoquinone (10). Following general synthetic procedure A, 2,3-dibromo-[1,4]-naphthoquinone (0.70 g, 2.20 mmol), Et2O (20 mL), N-(3-aminopropyl)piperidine (0.35 mL, 2.20 mmol), Et3N (0.31 mL, 2.20 mmol). Reaction stirred for 17 h. Product eluted at EtOAc/hexane = 1
:1 in flash column chromatography to afford a dark red solid (0.21 g, 25%): mp: 102–104 °C; 1H NMR (300 MHz, CDCl3) δ 8.19–8.16 (m, 1H), 8.07–8.03 (m, 1H), 7.72 (td, J = 7.5, 1.4 Hz, 1H), 7.63 (td, J = 7.5, 1.4 Hz, 1H), 7.56 (s, 1H), 4.04 (q, J = 6.0 Hz, 2H), 2.53 (d, J = 8.1 Hz, 6H), 1.98–1.83 (m, 2H), 1.73 (q, J = 5.6 Hz, 4H), 1.52 (d, J = 7.1 Hz, 2H); 13C NMR (76 MHz, CDCl3) δ 180.38, 161.96, 134.63, 132.54, 132.16, 131.22, 126.96, 126.79, 57.65, 54.90, 45.63, 26.02, 25.38, 24.31; IR (cm−1) 3065, 2935, 2765, 1679, 1562, 1502; HRMS (+ESI): found m/z 377.0859, [M + H]+; C18H22BrN2O2 required 377.0858.
2-Methyl-3-((2-(4-morpholinyl)ethyl)amino)-[1,4]-naphthoquinone (11). Following general synthetic procedure A, Menadione (0.38 g, 2.20 mmol), EtOH (20 mL), 4-(2-aminoethyl)morpholine (0.31 mL, 2.20 mmol). Reaction stirred for 21 h. Product eluted at EtOAc/hexane = 1
:4 in flash column chromatography to afford an orange oil that was recrystallized from 70% EtOH to afford bright red crystals. The crystals were filtered and the filtrate was further evaporated in vacuo to afford a red powder (66 mg, 9%): mp: 70–72 °C; 1H NMR (300 MHz, CDCl3) δ 7.92–7.88 (m, 1H), 7.83–7.78 (m, 1H), 7.53 (td, J = 7.5, 1.4 Hz, 1H), 7.43 (td, J = 7.5, 1.4 Hz, 1H), 6.25 (s, 1H), 3.68–3.59 (m, 4H), 3.54 (q, J = 5.6 Hz, 2H), 2.57–2.47 (m, 2H), 2.47–2.37 (m, 4H), 2.07 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 183.30, 182.39, 146.52, 134.12, 133.37, 131.74, 130.41, 126.02, 125.81, 112.46, 66.95, 57.33, 53.06, 41.35, 10.96. IR (cm−1) 2800, 1671, 1609, 1560, 1500, 1451, 1337, 1294, 1105, 715; HRMS (+ESI): found m/z 301.1547, [M + H]+; C17H21N2O3 required 301.1546.
2-Chloro-3-((2-(dimethylamino)ethyl)amino)-[1,4]-naphthoquinone (12). Following general synthetic procedure A, 2,3-dicholoro-[1,4]-naphthoquinone (0.51 g, 2.20 mmol), EtOH (20 mL), 3-(dimethylamine)-1-ethylamine (0.24 mL, 2.20 mmol) and Et3N (0.31 mL, 2.20 mmol) were reacted. Reaction stirred for 18 h. Product eluted at EtOAc/hexane = 1
:1 in flash column chromatography to afford a dark orange oil. The oil was recrystallized from 70% EtOH to afford a red powder 16 (0.41 g, 72%): mp: 80–82 °C; 1H NMR (300 MHz, CDCl3): δ 8.16 (d, J = 8.6 Hz, 1H), 8.00 (d, J = 9.0 Hz, 1H), 7.71 (t, J = 16.5 Hz, 1H), 7.61 (t, J = 16.4 Hz, 1H), 6.80 (br s, 1H), 3.97 (q, J = 17.2 Hz, 2H), 2.63 (t, J = 11.9 Hz, 2H), 2.32 (s, 6H); 13C NMR (CDCl3): δ 180.67, 176.71, 134.90, 132.89, 132.45, 130.12, 126.84, 77.57, 76.73, 58.17, 45.06, 41.98; IR (cm−1) 3222, 2940, 2821, 2773, 1677, 1607, 1573, 1488, 1281, 718, 677; HRMS (+ESI): found m/z 301.0714, [M + Na]+; C14H15C11N2O2Na required 301.0716.
2-Chloro-3-((2-(1-pyrrolidinyl)ethyl)amino)-[1,4]-naphthoquinone (13). Following general synthetic procedure A, 2,3-dichloro-[1,4]-naphthoquinone (0.51 g, 2.20 mmol), EtOH (20 mL), 1-(2-aminoethyl)pyrrolidine (0.28 mL, 2.20 mmol) and Et3N (0.31 mL, 2.20 mmol) were reacted. Reaction stirred for 18 h. Product eluted with EtOAc/hexane in flash column chromatography to afford an orange oil (0.44 g, 63%); mp: 63–64 °C; 1H NMR (300 MHz, CDCl3): δ 8.15 (d, J = 9.0 Hz, 1H), 8.04 (d, J = 9.0 Hz, 1H), 7.64 (t, J = 16.5 Hz, 1H), 7.53 (t, J = 16.4 Hz, 1H), 6.76 (br s, 1H), 3.89 (q, J = 17.5 Hz, 2H), 2.73 (t, J = 12.2 Hz, 2H), 2.52–2.59 (m, 4H), 1.62–1.75 (m, 4H); 13C NMR (75 MHz, CDCl3) δ 180.14, 176.30, 144.60, 134.63, 132.47, 132.20, 129.74, 126.52, 126.42, 54.83, 53.63, 42.92, 23.55. IR (cm−1) 3246, 2959, 2822, 1600, 1567, 1288, 1267, 717; MS (+ESI): found m/z 305.20, [M + H]+; C16H18ClN2O2 required 305.11.
2-Chloro-3-((2-(1-piperidnyl)ethyl)amino)-[1,4]-naphthoquinone (14). Following general synthetic procedure A, 2,3-dichloro-[1,4]-naphthoquinone (0.25 g, 1.10 mmol), EtOH (10 mL), 1-(2-aminoethyl)piperidine (0.16 mL, 1.20 mmol) and Et3N (0.31 mL, 2.20 mmol) were reacted. Reaction stirred for 18 h. Product eluted at EtOAc/hexane = 1
:4 in flash column chromatography to afford a dark red powder (0.20 g, 57%): mp: 108–110 °C; 1H NMR (300 MHz, CDCl3): δ 8.15 (d, J = 9.0 Hz, 1H), 8.03 (d, J = 9.5 Hz, 1H), 7.73 (t, J = 9.1 Hz, 1H), 7.63 (t, J = 9.0 Hz, 1H), 7.06 (br s, 1H), 3.97 (q, J = 17.3 Hz, 2H), 2.65 (t, J = 12.2 Hz, 2H), 2.40 (br 4H), 1.62–1.67 (m, 4H), 1.47–1.51 (m, 2H); 13C NMR (CDCl3): δ 180.67, 176.0, 144.4, 134.7, 132.3, 131.8, 130.0, 126.7, 56.6, 54.0, 41.1, 26.0, 24.3. IR (cm−1) 3208, 2923, 2847, 1674, 1599, 1568, 1267, 717; MS (+ESI): Found m/z 319.25, [M + H]+; C17H20ClN2O2 required 319.12.
2-((2-(Azepan-1-yl)ethyl)amino)-3-chloro-[1,4]-naphthoquinone (15). Following general synthetic procedure A, 2,3-dichloro-[1,4]-naphthoquinone (0.51 g, 2.20 mmol), hexamethyleneimine (0.50 mL, 4.40 mmol) and Et3N (0.31 mL, 2.20 mmol) were reacted. Reaction refluxed for 18 h. Product eluted at EtOAc/hexane = 1
:4 in flash column chromatography to afford a dark orange oil. The oil was recrystallised from 70% EtOH to afford an orange powder 24 (0.17 g, 24%): mp: 87–89 °C; 1H NMR (300 MHz, CDCl3): δ 8.05 (d, J = 7.7 Hz, 1H), 7.97 (d, J = 8.1 Hz, 1H), 7.69 (t, J = 8.9 Hz, 1H), 7.58 (t, J = 7.5 Hz, 1H), 7.14 (br s, 1H), 3.87 (q, J = 16.8 Hz, 2H), 2.76 (t, J = 11.9 Hz, 2H), 2.70–2.63 (m, 4H), 1.72–1.58 (m, 8H); 13C NMR (CDCl3): δ 180.48, 176.41, 144.86, 134.62, 132.77, 130.00, 126.58, 56.30, 54.77, 42.00, 31.56, 28.69, 26.93, 22.63; IR (cm−1) 3207, 2920, 2846, 1674, 1562, 1262, 715; HRMS (+ESI): found m/z 333.1364, [M + H]+; C18H22ClN2O2+ required 333.1367.
2-Chloro-3-((2-chloroethyl)amino)-[1,4]-naphthoquinone (16). To a suspension of 2,3-dichloro-[1,4]-naphthoquinone (1.00 g, 4.40 mmol) in MeCN (40 mL) were added 2-chloroethylamine hydrochloride (0.52 g, 4.40 mmol) and diisopropylethylamine (1.53 mL, 8.80 mmol) and the resulting light orange mixture was stirred at r.t. for 72 h. The resulting mixture was concentrated in vacuo, diluted in EtOAc and washed with water, and the organic layer was dried over anhydrous Na2SO4. The solvent was evaporated, and the resulting residue was purified via flash column chromatography (DCM/hexane = 1
:4) to afford a red powder (0.73 g, 61%): mp: 166–168 °C; 1H NMR (300 MHz, CDCl3): δ 8.14 (d, J = 7.1 Hz, 1H), 8.05 (d, J = 7.7 Hz, 1H), 7.77 (t, J = 8.9 Hz, 1H), 7.67 (t, J = 8.1 Hz, 1H), 6.33 (br s, 1H), 4.23 (q, J = 18.2 Hz, 2H), 3.79 (t, J = 11.2 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 180.20, 176.89, 143.81, 135.01, 132.74, 132.42, 129.81, 126.96, 126.94, 45.93, 44.16; IR (cm−1) 3284, 1679, 1564, 1511, 721, 678; HRMS (+ESI): found m/z 291.9903, [M + Na]+; C12H9Cl2NO2Na+ required 219.9902.
2-Chloro-3-((2-(1-bocpiperazinyl)ethyl)amino)-[1,4]-naphthoquinone (17). 2-chloro-3-((2-chloroethyl)amino)-[1,4]-naphthoquinone (0.59 g, 2.20 mmol), Boc-piperazine (1.32 g, 6.60 mmol), K2CO3 (0.33 g, 2.42 mmol), NaI (0.03 g, 0.22 mmol). Reaction heated at reflux for 22 h. The resulting mixture was concentrated in vacuo, diluted in EtOAc and washed with water, and the organic layer was dried over anhydrous Na2SO4. The solvent was evaporated, and the resulting residue was purified via flash column chromatography (EtOAc/hexane = 1
:4) to afford an orange solid (0.13 g, 14%): mp: 73–75 °C; 1H NMR (300 MHz, CDCl3) δ 8.15 (dd, J = 7.7, 1.4 Hz, 1H), 8.02 (dd, J = 7.6, 1.4 Hz, 1H), 7.73 (td, J = 7.6, 1.4 Hz, 1H), 7.63 (td, J = 7.5, 1.4 Hz, 1H), 6.89 (s, 1H), 4.03–3.91 (m, 2H), 3.53–3.42 (m, 4H), 2.69 (t, J = 6.0 Hz, 2H), 2.48 (t, J = 5.0 Hz, 4H), 1.48 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 180.58, 176.64, 171.12, 154.71, 144.63, 134.81, 132.72, 132.36, 129.93, 126.73, 126.68, 79.76, 56.41, 52.36, 40.93, 28.41, 22.64. IR (cm−1) 3286, 2970, 2810, 1679, 1609, 1420, 1236, 1129, 1005, 720, 678; HRMS (+ESI): found m/z 442.1504, [M + Na]+; C21H25ClN3NaO4+ required 442.1502.
2-Chloro-3-(4-(2-((3-chloro-1,4-dioxo-1,4-dihydronaphthalen-2-yl)amino)ethyl)piperazin-1-yl)-[1,4]-naphthoquinone (18). Following general synthetic procedure A, 2,3-dichloro-[1,4]-naphthoquinone (1.14 g, 5.00 mmol), Et2O (40 mL), 1-(2-aminoethyl)piperazine (0.66 mL, 5.00 mmol), Et3N (0.70 mL, 5.00 mmol). Reaction stirred for 63 h. Product eluted at EtOAc/hexane = 1
:9 in flash column chromatography to afford a dark brown solid (0.88 g, 29%): mp: 166–168 °C; 1H NMR (300 MHz, CDCl3): δ 8.17–8.17 (m, 2H), 8.15–8.11 (m, 2H), 7.76–7.61 (m, 4H), 7.26 (s, 1H), 4.04 (q, J = 8.0 Hz, 2H), 3.70 (t, J = 9.5 Hz, 4H), 2.77 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 180.33, 174.06, 147.03, 134.71, 132.43, 132.32, 130.06, 126.97, 126.75, 67.06, 56.65, 52.94, 41.18; IR (cm−1) 3326, 2890, 1733, 1667, 1557, 1246, 993, 718; HRMS (+ESI): found m/z 510.0982, [M + H]+; C26H22O4N3Cl2 required 510.0979.
2-((2-(1H-Indol-3-yl)ethyl)amino)-3-chloronaphthalene-1,4-dione (19). 22Following general synthetic procedure A, 2,3-dichloro-[1,4]-naphthoquinone (1.14 g, 5.00 mmol), Et2O (40 mL), tryptamine (0.9 g, 5.00 mmol), Et3N (0.70 mL, 5.00 mmol). Reaction stirred for 12 h. Product eluted at EtOAc/hexane = 1
:9 in flash column chromatography to afford a dark brown solid (1.19 g, 68%); mp: 189–190 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.86 (s, 1H), 7.97 (d, J = 7.6 Hz, 1H), 7.91 (d, J = 7.6 Hz, 1H), 7.82 (td, J = 7.5, 1.4 Hz, 1H), 7.72 (td, J = 7.5, 1.3 Hz, 1H), 7.61 (d, J = 7.7 Hz, 1H), 7.50 (s, 1H), 7.31 (d, J = 7.8 Hz, 1H), 7.19 (d, J = 2.3 Hz, 1H), 7.08–7.03 (m, 1H), 7.01–6.96 (m, 1H), 4.03 (dt, J = 8.2, 6.7 Hz, 2H), 3.08–2.97 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 180.66, 136.74, 135.30, 133.04, 127.56, 126.92, 126.20, 123.62, 121.48, 118.82, 111.86, 111.27, 46.11, 45.15. IR (cm−1) 3281, 1679, 1596, 1565, 1516, 1338, 749; MS (+ESI): found m/z 351.10, [M + H]+; C20H16ClN2O2 required 351.09.
2-Chloro-3-((3-morpholinopropyl)amino)naphthalene-1,4-dione (20). 23Follow general synthetic procedure A, 2,3-dichloro-[1,4]-naphthoquinone (0.50 g, 2.20 mmol), Et2O (20 mL), 3-morpholinopropylamine (0.30 mL, 2.20 mmol), Et3N (0.31 mL, 2.20 mmol). Reaction stirred for 17 h. Upon evaporating solvent following work-up a red solid was afforded (0.34 g, 47%): mp: 148–150 °C; 1H NMR (300 MHz, CDCl3) δ 8.19–8.14 (m, 1H), 8.14–8.01 (m, 1H), 7.74 (td, J = 7.6, 1.4 Hz, 1H), 7.68–7.61 (m, 1H), 7.55 (s, 1H), 4.05 (q, J = 6.0 Hz, 2H), 3.87 (s, 4H), 2.55 (s, 6H), 1.89 (s, 2H). 13C NMR (75 MHz, DMSO-d6) δ 180.70, 135.37, 133.07, 132.55, 126.97, 126.26, 66.46, 56.80, 53.86, 44.14, 26.81. IR (cm−1) 2952, 2785, 1680, 1601, 1565, 1512, 1456, 1331, 1297, 1264, 1111, 722; MS (+ESI): found m/z 335.20, [M + H]+; C17H20ClN2O3 required 335.11.
2-Chloro-3-((3-(piperidin-1-yl)propyl)amino)-[1,4]-naphthoquinone (21). Follow general synthetic procedure A, 2,3-dichloro-[1,4]-naphthoquinone (0.50 g, 2.20 mmol), Et2O (20 mL), N-(3-aminopropyl)piperidine (0.35 mL, 2.20 mmol), Et3N (0.31 mL, 2.20 mmol). Reaction stirred for 17 h. Product eluted at EtOAc/hexane = 1
:1 in flash column chromatography to afford a dark orange solid 27 (0.55 g, 75%): mp: 105–107 °C; 1H NMR (300 MHz, CDCl3) δ 8.18–8.13 (m, 1H), 8.05–8.01 (m, 1H), 7.72 (td, J = 7.5, 1.4 Hz, 2H), 7.61 (td, J = 7.6, 1.4 Hz, 1H), 4.10–3.92 (m, 2H), 2.52–2.40 (m, 6H), 1.90–1.76 (m, 2H), 1.69 (q, J = 5.6 Hz, 4H), 1.59–1.43 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 180.74, 134.64, 132.86, 132.11, 126.66, 126.63, 58.11, 55.02, 45.81, 25.98, 25.63, 24.51; IR (cm−1) 3065, 2921, 2817, 2765, 1679; HRMS (+ESI): found m/z 333.1364, [M + H]+; C18H22ClN2O2 required 333.1365.
2-Chloro-3-((3-(dimethylamino)propyl)amino)naphthalene-1,4-dione (22). Follow general synthetic procedure A, 2,3-dicholoro-1,4-naphthoquinome (0.51 g, 2.20 mmol), Et2O (20 mL), 3-(dimethylamine)-1-propylamine (0.28 mL, 2.20 mmol), Et3N (0.31 mL, 2.20 mmol). Reaction stirred for 19 h. Upon evaporating the solvent following the workup, the resulting orange oil was recrystallized from 70% EtOH to afford dark red crystals (0.18 g, 29%): mp: 88–92 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.83–7.99 (m, 3H), 7.78 (td, J = 7.5, 1.4 Hz, 1H), 7.69 (td, J = 7.5, 1.4 Hz, 1H), 3.78 (q, J = 6.5 Hz, 2H), 2.38 (t, J = 6.5 Hz, 2H), 2.17 (s, 6H), 1.74 (p, J = 6.6 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 180.53, 135.20, 132.94, 132.43, 126.82, 126.13, 57.23, 45.15, 44.02, 27.53; IR (cm−1) 3076, 2928, 2816, 2763, 1675, 1556, 1264; HRMS (+ESI): found m/z 315.0871, [M + Na]+; C15H17O2N2ClNa+ required 315.0872.
2-(Butylamino)-3-chloro-[1,4]-naphthoquinone (23). Following general synthetic procedure A, 2,3-dichloro-[1,4]-naphthoquinone (0.51 g, 2.20 mmol), EtOH (20 mL), butylamine (0.26 mL, 2.60 mmol), Et3N (0.31 mL, 2.20 mmol). Reaction stirred for 18 h. Product eluted at DCM/hexane = 1
:1 in flash column chromatography to afford red crystals (0.41 g, 71%): mp: 112–114 °C; 1H NMR (300 MHz, CDCl3): δ 8.12 (d, J = 1.3 Hz, 1H), 8.02 (d, J = 8.5 Hz, 1H), 7.70 (t, J = 8.9 Hz, 1H), 7.62 (t, J = 8.9 Hz, 1H), 6.04 (br s, 1H), 3.83 (t, J = 8.0 Hz, 2H), 1.75–1.66 (m, 2H), 1.50–1.42 (m, 2H), 0.99 (t, J = 12.0 Hz, 3H); 13C NMR (CDCl3): δ 180.70, 176.80, 134.94, 132.84, 132.36, 129.75, 126.85, 126.80, 44.71, 33.04, 19.83, 13.74. IR (cm−1) 3308, 2934, 2870, 1672, 1598, 1565, 1512, 1441, 1330, 1293, 719; MS (+ESI): found m/z 264.20, [M + H]+; C14H15ClNO2 required 264.08.
2-Chloro-3-(prop-2-yn-1-ylamino)naphthalene-1,4-dione (24). 24Following general synthetic procedure A, 2,3-dichloro-[1,4]-naphthoquinone (0.51 g, 2.20 mmol), EtOH (20 mL), propagylamine (0.17 mL, 2.60 mmol), Et3N (0.31 mL, 2.20 mmol). Reaction stirred for 18 h. Product eluted at EtOAc/hexane = 1
:1 in flash column chromatography to afford red solid (0.35 g, 65%): mp: 171–172 °C; 1H NMR (400 MHz, CDCl3) δ 8.17 (dd, J = 7.6, 1.3 Hz, 1H), 8.08 (dd, J = 7.7, 1.3 Hz, 1H), 7.76 (td, J = 7.6, 1.4 Hz, 1H), 7.67 (td, J = 7.6, 1.3 Hz, 1H), 6.05 (s, 1H), 4.66 (dd, J = 6.0, 2.5 Hz, 2H), 2.42 (t, J = 2.5 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 180.12, 177.00, 143.57, 134.96, 134.69, 132.79, 132.34, 129.82, 127.86, 126.93, 79.24, 73.43, 35.02. IR (cm−1) 3319, 3246, 1676, 1596, 1568, 1509, 1297, 1139, 1070, 713; MS (+ESI): found m/z 246.10, [M + H]+; C13H9ClNO2 required 246.03.
Ethyl (3-chloro-1,4-dioxo-1,4-dihydronaphthalen-2-yl)glycinate (25). 25Following general synthetic procedure A, L-glycine ethyl ester HCl (0.161 g, 1.1 mmol) and K2CO3 (35 mg, 0.26 mmol) were added to a stirred solution of 2,3-dichloro-[1,4]-naphthoquinone (0.249 g, 1.1 mmol) in 10% THF in EtOH (10 mL). Product eluted with DCM: hexane (1
:1) in flash column chromatography as an orange solid (43 mg, 13%); mp: 128–129 °C; 1H NMR (400 MHz, CDCl3) δ 8.21–8.15 (m, 1H), 8.11–8.05 (m, 1H), 7.76 (m, 1H), 7.68 (m, 1H), 6.52 (s, 1H), 4.62 (d, J = 5.5 Hz, 2H), 4.32 (q, J = 7.1 Hz, 2H), 1.35 (t, J = 7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 180.10, 176.88, 169.53, 143.89, 134.89, 132.71, 132.34, 129.89, 126.89, 126.83, 62.07, 46.28, 14.18. IR (cm−1) 3306, 2925, 2853, 1734, 1603, 1572, 1502, 1287, 1211, 725; MS (+ESI): found m/z 294.20, [M + H]+; C14H13ClNO4 required 294.05.
Methyl (3-chloro-1,4-dioxo-1,4-dihydronaphthalen-2-yl)-L-tryptophanate (26). Following general synthetic procedure A, L-tryptophan methyl ester hydrochloride (0.28 g, 1.1 mmol) and K2CO3 (37 mg, 0.27 mmol) were added to a solution of 2,3-dichloro-[1,4]-naphthoquinone (0.25 g, 1.1 mmol) in 10% THF in EtOH (5 mL). Product eluted from hexane: EtOAc (3
:2) in flash column chromatography as a red solid (0.29 g, 65%): mp: 81.8–82.0 °C; 1H NMR (300 MHz, CDCl3) δ 8.19–8.13 (m, 1H), 8.14–8.09 (m, 1H), 7.94 (d, J = 7.7 Hz, 1H), 7.72 (td, J = 7.5, 1.4 Hz, 1H), 7.65–7.55 (m, 2H), 7.39–7.29 (m, 1H), 7.19 (m, 1H), 7.16–7.07 (m, 2H), 6.36 (s, 1H), 5.56 (dt, J = 8.2, 5.9 Hz, 1H), 3.76 (s, 3H), 3.52–3.38 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 172.41, 171.42, 136.51, 134.94, 132.82, 127.35, 127.04, 126.91, 123.52, 122.82, 120.18, 118.78, 111.58, 109.35, 60.66, 56.81, 53.01, 30.03, 21.32, 14.46; IR (cm−1) 3302, 2919, 2850, 1734, 1672, 1592, 1563; HRMS (+ESI): found m/z 409.0951, [M + H]+; C22H18ClN2O4 required 409.0950.
Methyl (3-chloro-1,4-dioxo-1,4-dihydronaphthalen-2-yl)-L-phenylalaninate (27). Following general synthetic procedure A, L-phenylalanine methyl ester hydrochloride (0.24 g, 1.1 mmol) and K2CO3 (26 mg, 0.19 mmol) were added to a solution of 2,3-dichloro-[1,4]-naphthoquinone (0.25 g, 1.1 mmol) in 10% THF in EtOH (5 mL). Product eluted with hexane: EtOAc (5
:1) in flash column chromatography as an orange solid (20 mg, 5%); mp: 158.5–158.7 °C; 1H NMR (300 MHz, CDCl3) δ 8.17–8.11 (m, 1H), 8.06–7.98 (m, 1H), 7.74 (td, J = 7.5, 1.5 Hz, 1H), 7.68–7.61 (m, 1H), 7.34–7.26 (m, 3H), 7.21–7.15 (m, 2H), 6.27 (s, 1H), 5.58–5.49 (m, 1H), 3.79 (s, 3H), 3.35–3.17 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 180.07, 176.74, 171.71, 134.93, 134.83, 132.71, 129.30, 128.89, 127.58, 126.91, 126.79, 57.29, 52.69, 40.02; IR (cm−1) 3312, 2950, 1738, 1593, 1567; HRMS (+ESI): found m/z 370.0842, [M + H]+; C20H17ClNO4 required 370.0841.
2-Chloro-3-((2-(1-piperazinyl)ethyl)amino)-[1,4]-naphthoquinone salt (29). 2-Chloro-3-((2-(1-bocpiperazinyl)ethyl)amino)-[1,4]-naphthoquinone 17 (0.50 g, 1.20 mmol) was dissolved in DCM (20 mL) and to this suspension was added TFA (3 mL). The resulting orange mixture was stirred at r.t. for 3 h. The mixture was evaporated in vacuo to afford an oil which was then recrystallized from Et2O to give an orange salt (234.0 mg, 60%); mp: 198–200 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.87 (s, 2H), 7.96–8.00 (m, 2H), 7.85 (td, J = 7.5, 1.5 Hz, 1H), 7.77 (td, J = 7.5, 1.4 Hz, 1H), 7.37 (s, 1H), 3.94 (d, J = 6.3 Hz, 2H), 3.21 (s, 4H), 2.97 (s, 6H). 13C NMR (75 MHz, DMSO-d6) δ 180.57, 175.91, 158.56, 135.42, 133.31, 132.31, 126.99, 126.31, 57.02, 49.31, 42.44; IR (cm−1) 3323, 3015, 2479, 1675, 1130, 721; HRMS (+ESI): found m/z 320.1160, [M + H]+; C16H19ClN3O2 required 320.1164.
Methyl (3-chloro-1,4-dioxo-1,4-dihydronaphthalen-2-yl)-L-lysinate (30). TFA (10 mL) was added to a solution of Boc-compound 16e (0.30 g, 0.67 mmol) in DCM (10 mL) under nitrogen on ice. The reaction mixture was stirred at r.t. for 5 h. The solvent removed in vacuo yielding analogue 30 as a sticky solid (0.16 g, 50%). 1H NMR (400 MHz, DMSO-d6) δ 8.04–7.96 (m, 3H), 7.86 (td, J = 7.5, 1.4 Hz, 1H), 7.79 (td, J = 7.5, 1.3 Hz, 1H), 6.93 (d, J = 8.3 Hz, 1H), 5.08–5.02 (m, 1H), 3.71 (s, 3H), 3.57 (s, 1H), 2.78 (m, 2H), 2.06–1.86 (m, 2H), 1.56 (m, 2H), 1.43 (q, J = 7.7 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 180.43, 172.61, 158.61, 135.51, 133.58, 131.95, 130.44, 127.13, 126.37, 66.81, 56.94, 52.88, 39.02, 32.18, 27.06, 22.63; IR (cm−1) 2946, 1672, 1580, 1568; HRMS (+ESI): found m/z 351.1105, [M]+; C17H20ClN2O4 required 351.1106.
2-Chloro-3-(((1-phenyl-1H-1,2,3-triazol-4-yl)methyl)amino)naphthalene-1,4-dione (31). 24 (0.2 g, 0.8 mmol) was dissolved in 10 mL of THF
:H2O (5:1) under a nitrogen atmosphere, and azido benzene (0.19 g, 1.2 mmol), sodium ascorbate 20 mol%, and CuSO4·5H2O, 10 mol%, were added. The reaction mixture was stirred at 50 °C for 12 h and then quenched by addition of 15 mL ice water and extracted with ethyl acetate (15 mL × 3). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, and evaporated under reduced pressure. Purification was completed with flash column chromatography on silica using a DCM: MeOH solvent system to yield a red crystal (0.047 g, 16%); mp: 204.2 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.62 (s, 1H), 8.01 (dt, J = 7.6, 1.5 Hz, 2H), 7.88–7.85 (m, 2H), 7.83 (td, J = 7.5, 1.4 Hz, 1H), 7.75 (td, J = 7.6, 1.3 Hz, 1H), 7.60–7.54 (m, 3H), 7.49–7.45 (m, 1H), 5.16 (d, J = 6.6 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 180.61, 176.14, 146.77, 145.79, 137.27, 135.22, 133.19, 132.50, 130.66, 130.22, 129.00, 126.94, 126.30, 121.44, 120.62, 110.97, 40.45. HRMS (+ESI): found m/z 365.08011, [M + H]+, C19H14ClN4O2 requires 365.08053.
4-(4-(((3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)benzoic acid (32). 24 (0.2 g, 0.8 mmol) was dissolved in 10 mL of THF
:H2O (5:1) under a nitrogen atmosphere, and 4-azido benzoic acid (0.3 g, 1.8 mmol), sodium ascorbate 20 mol%, and CuSO4·5H2O, 10 mol%, were added. The reaction mixture was stirred at 50 °C for 12 h and then quenched by addition of 15 mL ice water and extracted with ethyl acetate (15 mL × 3). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, and evaporated under reduced pressure. Purification was completed with flash column chromatography on silica using a DCM: MeOH solvent system to yield as an orange powder (0.2 g, 89%); mp: 187.2 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.73 (s, 1H), 8.14–8.10 (m, 2H), 8.03–7.99 (m, 4H), 7.83 (td, J = 7.5, 1.3 Hz, 1H), 7.76 (td, J = 7.5, 1.2 Hz, 1H), 7.60 (t, J = 6.7 Hz, 1H), 5.17 (d, J = 6.6 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 180.61, 176.15, 166.72, 147.24, 145.77, 140.09, 135.23, 133.21, 132.49, 131.42, 131.30, 130.67, 126.96, 126.31, 121.57, 120.30, 111.01, 40.33. HRMS (+ESI): found m/z 409.06981, [M + H]+, C20H14ClN4O4 requires 409.07036.
Ethyl 4-(4-(((3-chloro-1,4-dioxo-1,4-dihydronaphthalen-2-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)benzoate (33). 24 (0.2 g, 0.8 mmol) was dissolved in 10 mL of THF
:H2O (5:1) under a nitrogen atmosphere, and ethyl-4-azidobenzoate (0.33 g, 1.8 mmol), sodium ascorbate 20 mol%, and CuSO4·5H2O, 10 mol%, were added. The reaction mixture was stirred at 50 °C for 12 h and then quenched by addition of 15 mL ice water and extracted with ethyl acetate (15 mL × 3). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, and evaporated under reduced pressure. Purification was completed with flash column chromatography on silica using a DCM: MeOH solvent system to yield as a dark-orange crystals (0.03 g, 8%); mp: 194.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.84 (s, 1H), 8.15–8.10 (m, 2H), 8.09–8.04 (m, 2H), 8.03–7.94 (m, 3H), 7.84 (td, J = 7.5, 1.4 Hz, 1H), 7.77 (td, J = 7.5, 1.4 Hz, 1H), 5.12 (d, J = 6.8 Hz, 2H), 4.34 (q, J = 7.1 Hz, 2H), 1.34 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 180.52, 176.14, 165.30, 147.93, 147.01, 140.21, 135.32, 133.29, 132.34, 131.38, 130.61, 128.80, 129.97, 127.02, 126.29, 121.49, 120.16, 61.56, 49.07 (MeOH), 14.60. HRMS (+ESI): found m/z 459.0830, [M + Na]+, C22HClN4O4Na requires 459.0836.
N-(1,4-Dioxo-3-((3-(piperidin-1-yl)propyl)amino)-1,4-dihydronaphthalen-2-yl)acetamide (39). Following general synthetic method A, aminopropylpiperidine (0.35 mL, 2.2 mmol) and Et3N (0.31 mL, 2.2 mmol) were added to a solution of amide 35 (0.55 g, 2.2 mmol) in diethyl ether (20 mL). The reaction mixture was refluxed for 18 h. The product purified by flash column chromatography using gradient elution (0–15% MeOH in DCM) to give a red solid (0.16 g, 21%); mp: 285–296 °C; 1H NMR (400 MHz, CDCl3) δ 8.09–7.99 (m, 2H), 7.67 (t, J = 7.6 Hz, 1H), 7.57 (t, J = 7.6 Hz, 1H), 7.33 (s, 1H), 3.58 (s, 2H), 2.43 (s, 6H), 2.22 (s, 3H), 1.78 (s, 2H), 1.70 (s, 4H), 1.50 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 182.12, 179.35, 170.83, 143.37, 134.49, 132.57, 132.08, 130.51, 126.40, 126.10, 111.49, 57.44, 54.75, 50.65, 43.22, 25.80, 25.35, 24.30, 23.41; IR (cm−1) 3337, 2928, 1662, 1605, 1600; HRMS (+ESI): found m/z 378.1784, [M + Na]+; C20H25N3O3Na required 378.1788.
N-(1,4-Dioxo-3-((3-(piperidin-1-yl)propyl)amino)-1,4-dihydronaphthalen-2-yl)propionamide (40). Following general synthetic method A, aminopropylpiperidine (0.14 mL, 0.9 mmol) and Et3N (0.12 mL, 0.9 mmol) were added to amide 36 (0.24 g, 0.9 mmol) in THF (8 mL) and ether (3 mL). The reaction mixture was refluxed for 18 h. The organic layer was collected, dried with sodium sulfate and the solvent evaporated yielding product as a red solid (0.37 g, 100%); mp: 101.1–101.8 °C; 1H NMR (400 MHz, CDCl3) δ 8.06–7.98 (m, 2H), 7.67 (t, J = 7.5 Hz, 1H), 7.60–7.54 (m, 1H), 7.49 (s, 1H), 7.35–7.30 (m, 1H), 3.58–3.51 (m, 2H), 2.48 (t, J = 7.6 Hz, 2H), 2.43–2.34 (m, 6H), 2.16 (s, 3H), 1.75 (q, J = 6.3 Hz, 2H), 1.67 (t, J = 5.7 Hz, 4H), 1.48 (q, J = 5.8 Hz, 2H), 1.25 (td, J = 7.3, 4.3 Hz, 4H); 13C NMR (101 MHz, CDCl3) δ 206.99, 182.18, 179.39, 174.40, 143.05, 134.48, 132.56, 132.09, 130.56, 126.42, 126.08, 111.54, 60.40, 57.68, 54.87, 43.37, 30.94, 30.32, 29.93, 26.02, 25.61, 24.48, 21.06, 14.20, 9.92; IR (cm−1) 3339, 2928, 1669, 1605, 1572; HRMS (+ESI): found m/z 370.2127, [M + H]+; C21H28N3O3 required 370.2125.
N-(1,4-Dioxo-3-((3-(piperidin-1-yl)propyl)amino)-1,4-dihydronaphthalen-2-yl)butyramide (41). Following general synthetic method A, aminopropylpiperidine (0.07 mL, 0.36 mmol) and Et3N (0.06 mL, 0.36 mmol) were added to a solution of amide 37 (0.10 g, 0.36 mmol) in THF (10 mL) and Et2O (10 mL). The reaction mixture was refluxed for 24 h. The product eluted from gradient column (0–20% MeOH in DCM) as a dark red solid (61 mg, 44%). 1H NMR (400 MHz, CDCl3) δ 8.11–7.99 (m, 2H), 7.69 (td, J = 7.6, 1.3 Hz, 1H), 7.60 (td, J = 7.6, 1.4 Hz, 1H), 7.43 (s, 1H), 7.36 (s, 1H), 3.60 (q, J = 5.3 Hz, 2H), 2.59–2.48 (m, 4H), 2.47 (d, J = 7.4 Hz, 3H), 1.82 (dt, J = 6.5, 2.5 Hz, 3H), 1.73 (q, J = 5.6 Hz, 4H), 1.52 (s, 2H), 1.05 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 182.26, 179.29, 173.73, 142.87, 134.48, 132.45, 132.12, 130.59, 126.98, 126.40, 126.08, 111.98, 57.13, 54.63, 54.47, 42.95, 38.79, 28.70, 25.82, 25.11, 24.12, 21.24, 19.20, 13.93; IR (cm−1) 2929, 1662, 1604, 1590; HRMS (+ESI): found m/z 384.2279, [M + H]+; C22H30N3O3 required 384.2282.
N-(1,4-Dioxo-3-((3-(piperidin-1-yl)propyl)amino)-1,4-dihydronaphthalen-2-yl)pentanamide (42). Following general synthetic method A, aminopropylpiperidine (57 μL, 0.37 mmol) and Et3N (53 μL, 0.37 mmol) were added to a solution of amide 38 (0.15 g, 0.37 mmol) in THF (6 mL) in Et2O (6 mL). The reaction mixture was refluxed for 18 h. The product eluted from gradient column (0–15% MeOH in DCM) as a dark red solid (0.10 g, 51%). 1H NMR (400 MHz, CDCl3) δ 8.08–7.97 (m, 2H), 7.70–7.63 (m, 1H), 7.57 (td, J = 7.6, 1.4 Hz, 1H), 7.31 (s, 1H), 3.58 (d, J = 5.8 Hz, 2H), 2.45 (t, J = 7.7 Hz, 4H), 1.80 (s, 2H), 1.72 (m, 6H), 1.47 (s, 2H), 1.41 (p, J = 7.4 Hz, 2H), 0.94 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 182.22, 173.74, 134.41, 132.38, 132.05, 130.56, 126.33, 126.01, 54.63, 42.95, 36.60, 27.74, 25.81, 24.13, 22.45, 13.79; IR (cm−1) 3233, 2929, 2855, 1671, 1605, 1565; HRMS (+ESI): found m/z 398.2433, [M + H]+; C23H32N3O3 required 398.2438.
N-(3-((2-Morpholinoethyl)amino)-1,4-dioxo-1,4-dihydronaphthalen-2-yl)acetamide (43). Following general synthetic method A, ethylaminomorpholine (0.30 mL, 2.2 mmol) and Et3N (0.31 mL, 2.2 mmol) were added to amide 35 (0.55 g, 2.2 mmol) in diethyl ether (20 mL). The reaction mixture was refluxed for 18 h. Product eluted from gradient column (0–15% MeOH in DCM) as a red solid (0.25 g, 34%); mp: 147.0–147.1 °C; 1H NMR (400 MHz, CDCl3) δ 8.12–8.01 (m, 2H), 7.72 (t, J = 7.5 Hz, 1H), 7.66–7.60 (m, 1H), 7.24 (s, 1H), 6.86 (s, 1H), 3.77 (t, J = 4.7 Hz, 4H), 3.60 (d, J = 7.8 Hz, 2H), 2.63 (s, 2H), 2.52 (s, 4H), 2.27 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 182.23, 170.67, 142.37, 134.63, 132.38, 130.55, 126.46, 126.14, 66.95, 56.87, 53.08, 39.11, 23.57; IR (cm−1) 3463, 3166, 2821, 1658, 1604, 1569; HRMS (+ESI): found m/z 344.1603, [M + H]+; C18H22N3O4 required 344.1605.
2-Methyl-1-(3-(piperidin-1-yl)propyl)-1H-naphtho[2,3-d]imidazole-4,9-dione (44). Following general procedure E, aqueous NaOH (2 M, 2 drops) was added to a solution of amide 39 (100 mg, 0.3 mmol) in EtOH (2 mL). The reaction mixture was heated at 50 °C for 1 h. The solvent was removed in vacuo yielding a yellowy-green solid (50 mg, 53%); mp: 125.6–126.0 °C; 1H NMR (400 MHz, CDCl3) δ 8.27–8.10 (m, 2H), 7.78–7.66 (m, 2H), 4.44 (t, J = 7.1 Hz, 2H), 2.63 (s, 3H), 2.35 (t, J = 6.5 Hz, 6H), 2.08–2.00 (m, 2H), 1.56 (q, J = 5.6 Hz, 4H), 1.45 (d, J = 5.7 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 179.06, 176.14, 153.48, 143.41, 133.65, 133.37, 133.20, 132.83, 132.21, 126.98, 126.44, 55.41, 54.49, 44.05, 27.22, 25.92, 24.40, 13.30; IR (cm−1) 2934, 2765, 1656, 1583; HRMS (+ESI): found m/z 338.1860, [M + H]+; C20H24N3O2 required 338.1863.
2-Ethyl-1-(3-(piperidin-1-yl)propyl)-1H-naphtho[2,3-d]imidazole-4,9-dione (45). Following general synthetic method E, aqueous NaOH (2 M, 6 drops) was added to a solution of amide 40 (130 mg, 0.4 mmol) in EtOH (12 mL). The reaction mixture was heated at 50 °C for 2 h. The solvent was removed in vacuo yielding a yellowy-green solid (67 mg, 54%). 1H NMR (300 MHz, CDCl3) δ 8.31–8.19 (m, 1H), 8.19–8.08 (m, 1H), 7.79–7.65 (m, 2H), 4.50–4.36 (m, 2H), 2.93 (q, J = 7.5 Hz, 2H), 2.36 (t, J = 6.6 Hz, 6H), 2.02 (p, J = 6.9 Hz, 3H), 1.56 (q, J = 5.5 Hz, 4H), 1.47 (t, J = 7.5 Hz, 5H), 1.34–1.20 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 179.10, 176.21, 157.84, 143.46, 133.59, 133.31, 133.18, 132.83, 132.15, 126.95, 126.37, 55.48, 54.51, 43.72, 27.54, 25.95, 24.40, 20.24, 11.83; IR (cm−1) 2935, 2768, 1653, 1584; HRMS (+ESI): found m/z 352.2016, [M + H]+; C21H26N3O2 required 352.2020.
1-(3-(Piperidin-1-yl)propyl)-2-propyl-1H-naphtho[2,3-d]imidazole-4,9-dione (46). Following general synthetic method E, aqueous NaOH (2 M, 4 drops) was added to a solution of amide 41 (31 mg, 0.08 mmol) in EtOH (5 mL). The reaction mixture was heated at 50 °C for 2 h. The solvent was removed in vacuo yielding a dark green solid (39 mg, 100%). 1H NMR (400 MHz, CDCl3) δ 8.28–8.17 (m, 1H), 8.17–8.05 (m, 1H), 7.77–7.67 (m, 2H), 4.48–4.40 (m, 1H), 3.62 (q, J = 7.1 Hz, 1H), 2.92–2.86 (m, 1H), 2.38 (d, J = 5.5 Hz, 4H), 2.07–1.96 (m, 2H), 1.89–1.80 (m, 2H), 1.55 (s, 3H), 1.55–1.40 (m, 4H), 1.03–0.90 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 176.21, 157.11, 133.61, 133.35, 133.20, 132.84, 126.97, 126.40, 77.24, 58.23, 55.46, 54.49, 43.75, 29.87, 27.54, 26.56, 25.85, 24.34, 22.65, 20.18, 18.34, 13.86, 13.77; IR (cm−1) 2829, 2869, 1655, 1587; HRMS (+ESI): found m/z 366.2173, [M + H]+; C22H28N3O2 required 366.2176.
2-Butyl-1-(3-(piperidin-1-yl)propyl)-1H-naphtho[2,3-d]imidazole-4,9-dione (47). Following general synthetic method E, aqueous NaOH (2 M, 4 drops) was added to a solution of amide 42 (64 mg, 0.16 mmol) in EtOH (5 mL). The reaction mixture was heated at 50 °C for 2 h. The product was purified by flash column chromatography using gradient elution (0–5% MeOH in DCM) to give a yellow sticky solid (29 mg, 47%). 1H NMR (400 MHz, CDCl3) δ 8.22 (m, 1H), 8.15–8.07 (m, 1H), 7.75–7.65 (m, 2H), 4.43 (t, J = 7.4 Hz, 2H), 2.87 (m, 2H), 2.46 (s, 4H), 2.08–2.00 (m, 2H), 1.84 (td, J = 8.1, 1.7 Hz, 2H), 1.62 (dd, J = 8.2, 3.3 Hz, 4H), 1.51–1.39 (m, 4H), 0.97 (td, J = 7.4, 1.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 206.98, 179.09, 176.25, 157.14, 143.59, 133.63, 133.33, 133.19, 132.86, 131.99, 127.00, 126.38, 55.27, 54.35, 53.43, 50.83, 43.61, 30.93, 29.87, 27.27, 26.57, 25.49, 24.09, 22.65, 13.79; IR (cm−1) 2928, 2853, 1620, 1589; HRMS (+ESI): found m/z 380.2331, [M + H]+; C23H30N3O2 required 380.2333.
2-Methyl-1-(2-morpholinoethyl)-1H-naphtho[2,3-d]imidazole-4,9-dione (48). Following general synthetic method E, aqueous NaOH (2 M, 3 drops) was added to a solution of amide 43 (65 mg, 0.2 mmol) in EtOH (4 mL). The reaction mixture was heated at 50 °C for 1 h. The solvent was removed in vacuo yielding a black-green solid (68 mg, 100%); mp: 133.3–133.8 °C; 1H NMR (400 MHz, CDCl3) δ 8.30–8.21 (m, 1H), 8.18–8.09 (m, 1H), 7.79–7.68 (m, 2H), 4.50 (t, J = 6.4 Hz, 2H), 3.68–3.64 (m, 4H), 2.76 (t, J = 6.4 Hz, 2H), 2.63 (s, 3H), 2.58–2.51 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 178.99, 176.33, 153.55, 143.24, 133.77, 133.45, 133.10, 132.81, 132.15, 127.04, 126.42, 66.94, 58.40, 54.03, 43.40, 13.52; IR (cm−1) 2948, 2931, 1656, 1589; HRMS (+ESI): found m/z 348.1313, [M + H]+; C18H19N3O3Na required 348.1319.
N-(1,4-Dioxo-3-((3-(piperidin-1-yl)propyl)amino)-1,4-dihydronaphthalen-2-yl)-2-phenylacetamide (50). Following general synthetic method A, aminopropylpiperidine (0.36 mL, 2.4 mmol) and Et3N (0.34 mL, 2.4 mmol) were added to a solution of amide 49 (0.78 g, 2.4 mmol) in THF (20 mL). The reaction mixture was refluxed for 18 h. The product was purified by flash column chromatography using gradient elution (0–10% MeOH in DCM) to give a red solid (0.71 g, 69%). 1H NMR (400 MHz, CDCl3) δ 8.08–7.99 (m, 2H), 7.69 (td, J = 7.5, 1.4 Hz, 1H), 7.59 (td, J = 7.6, 1.3 Hz, 1H), 7.48–7.38 (m, 4H), 7.36–7.30 (m, 1H), 7.22 (s, 1H), 3.83 (s, 2H), 3.42 (s, 2H), 2.49 (s, 6H), 1.76 (s, 6H), 1.49 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 182.32, 179.32, 171.58, 143.23, 134.93, 134.72, 132.64, 132.31, 130.75, 129.84, 129.23, 127.65, 126.61, 126.28, 54.73, 44.33, 43.10, 25.66, 24.24; IR (cm−1) 3239, 2931, 2771, 1604, 1568; HRMS (+ESI): found m/z 432.2280, [M + H]+; C26H30N3O3 required 432.2282.
1,4-Dioxo-3-((3-(piperidin-1-yl)propyl)amino)-1,4-dihydronaphthalen-2-ammonium chloride (53). HCl·dioxane (4 M, 1.1 mL, 4.4 mmol) was added to a solution of 52 (0.42 g, 0.81 mmol) in diethyl ether (10 mL) under nitrogen on ice. The reaction mixture was stirred at r.t. for 18 h. The resulting brown precipitate was collected (0.30 g, 100%); mp: 228.4–228.7 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.85 (dd, J = 7.1, 1.7 Hz, 2H), 7.73–7.63 (m, 2H), 3.38 (d, J = 12.2 Hz, 2H), 3.24 (t, J = 6.9 Hz, 2H), 3.05 (dd, J = 9.4, 6.6 Hz, 2H), 2.81 (t, J = 12.3 Hz, 2H), 1.82 (m, 4H), 1.60 (m, 4H), 1.38 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 180.14, 178.49, 135.21, 133.78, 131.49, 130.46, 126.42, 126.10, 83.49, 79.25, 78.92, 53.90, 52.83, 52.72, 44.18, 31.18, 31.03, 27.82, 22.87, 22.34, 21.44; IR (cm−1) 3369, 3080, 2936, 2482, 1622, 1561; HRMS (+ESI): found m/z 314.1860, [M]+; C18H24N3O2 required 314.1863.
8.2. Cytotoxicity assays
Reagents and Equipment. Phosphate buffered saline (Sigma Aldrich), trypsin–EDTA Solution 10× (Sigma Aldrich), Dulbecco's modified Eagle's medium with 10% foetal bovine serum (Sigma Aldrich), minimum essential medium (ThermoFisher Scientific), MTT Reagent (5 mg mL−1 MTT in PBS), MTT Solvent (4 mM HCl, 0.1% Nondet P-40, in isopropanol), dimethyl sulfoxide (DMSO; Sigma Aldrich), N-acetylcysteine (NAC, Sigma Aldrich) and reduced glutathione (GSH, Sigma Aldrich).
Methods. Cytotoxicity assays were performed as previously described.7 Briefly, HEC1A (5000/well) and MAD11 (2500/well) cells were seeded in 50 μL of media in 96-well tissue culture plates and incubated at 37 °C overnight prior to drug treatment. All compounds, except NAC and GSH, were reconstituted in dimethyl sulfoxide (DMSO), serially diluted in cell culture medium (to final concentrations of 160 μM, 80 μM, 40 μM, 20 μM, 10 μM, 5 μM, 2.5 μM, 1.25 μM) and 50 μL of each was added to wells in triplicate. For the thiol rescue experiments, NAC and GSH were prepared in cell culture media (neutralised to pH 7.4) and the cells were treated with 2 mM of each for 40 minutes prior to the addition of the compounds (BH10, 21, and 44). In order to eliminate variations caused by the presence of DMSO (vehicle), the drug dilutions and control wells were treated with the same final concentration of DMSO. The maximum concentration of DMSO added to the cells did not exceed 0.5% v/v. Cell viability was then determined after 48 h by the addition of 20 μL per well of 5 mg mL−1 thiazolyl blue tetrazolium bromide (MTT) reagent (3-hour incubation). Formazan crystals were solubilized in solvent (4 mM HCl, 0.1% NP-40 in isopropanol) and absorbance read at 590/620 nm using an EnSight Multimode plate reader (PerkinElmer). Viability of drug-treated cells is displayed as a percentage of control cells i.e. cells with equivalent concentrations of the appropriate vehicle (DMSO). The same experimental protocol used for HEC-1-A cells was applied to the CALU-1 and Mia-Pa-Ca-2 cancer cell lines, with the following modifications: CALU-1 cells were cultured in minimum essential medium (MEM) instead of Dulbecco's modified Eagle medium (DMEM).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) P. The half maximal inhibitory concentration (IC50), that is, the amount of compound required to inhibit cell viability by 50% relative to vehicle controls, was calculated using GraphPad Prism software. The selectivity ratio is the IC50 value of the non-cancerous cells (MAD11) divided by the IC50 value of the cancer cells (HEC1A), for each compound.
P. The half maximal inhibitory concentration (IC50), that is, the amount of compound required to inhibit cell viability by 50% relative to vehicle controls, was calculated using GraphPad Prism software. The selectivity ratio is the IC50 value of the non-cancerous cells (MAD11) divided by the IC50 value of the cancer cells (HEC1A), for each compound.8.3. Oxygen consumption rate assay
000 cells per well in complete media (DMEM supplemented with 10% (v/v) FBS, and 0.1% penicillin (10000 IU mL−1)/streptomycin (10 mg mL−1). Cells were then allowed to adhere for 24 hours in a 37 °C humidified incubator with 5% CO2. Prior to the commencement of the assay, cell culture media was replaced with Seahorse media (unbuffered phenol-red free DMEM supplemented with 25 mM glucose, 4 mM L-glutamine, and 1 mM sodium pyruvate, pH 7.4) and cells were incubated at 37 °C in a non-CO2 incubator for 40 minutes to allow for de-gassing. The cartridge plate was then equilibrated in the Seahorse XF analyser for 20 min, prior to adding the cell culture plate. Baseline measurements were recorded 3 times prior to the injection of the compounds. Oxygen Consumption Rate (OCR) was measured subsequently for 11 cycles, each consisting of a 3-minute waiting, 3-minute mixing, and 2-minute measurement period. Data from Seahorse assays was acquired using the Agilent Seahorse Wave Desktop (V2.6) software. The OCR was normalised to the third baseline reading.8.4. Pharmacokinetic profiling in vivo
8.5. In silico molecular docking study
The Keap1 protein (PDB code: 4XMB) was obtained from the Protein Data Bank. Molecular docking simulations were performed using Maestro software (Schrödinger, New York, NY, USA) in standard precision mode. All workflows, including protein preparation, LigPrep, and receptor grid generation, were executed with default parameter settings. The predicted protein-ligand complexes were further analyzed in Maestro and visualized using PyMOL (Schrödinger, New York, NY, USA).Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We thank the BMSF and NMR facilities at UNSW Mark Wainwright Analytical Centre for the structural determination of the synthesized compounds. Yao Cheng is thankful to UNSW Sydney for the University International Postgraduate Award. This research was supported by fellowships from the Hope Funds for Cancer Research (HFCR-14-06-04) and Cancer Institute NSW fellowships (Early Career 2018/ECF003 and Career Development 2021/CDF1120) to Frances Byrne.References
- K. Nurgali, R. T. Jagoe and R. Abalo, Editorial: Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae?, Front. Pharmacol., 2018, 9, 245, DOI:10.3389/fphar.2018.00245/full.
- G. Housman, S. Byler, S. Heerboth, K. Lapinska, M. Longacre, N. Snyder and S. Sarkar, Drug resistance in cancer: an overview, Cancers, 2014, 6, 1769–1792 CrossRef , Available from: https://www.mdpi.com/2072-6694/6/3/1769.
- D. Hanahan and R. A. Weinberg, Hallmarks of cancer: the next generation, Cell, 2011, 144, 646–674 CrossRef CAS PubMed , Available from: https://www.sciencedirect.com/science/article/pii/S0092867411001279?via%3Dihub.
- M. G. V. Heiden, L. C. Cantley and C. B. Thompson, Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation, Science, 2009, 324, 1029–1033, DOI:10.1126/science.1160809.
- O. Warburg, F. Wind and E. Negelein, The metabolism of tumors in the body, J. Gen. Physiol., 1927, 8, 519–530 CrossRef CAS PubMed , Available from: https://rupress.org/jgp/article/8/6/519/12408/THE-METABOLISM-OF-TUMORS-IN-THE-BODY.
- A. Chiarugi, C. Dolle, R. Felici and M. Ziegler, The NAD metabolome--a key determinant of cancer cell biology, Nat. Rev. Cancer, 2012, 12, 741–752 CrossRef CAS PubMed , Available from: https://www.nature.com/articles/nrc3340.
- F. L. Byrne, E. M. Olzomer, G. R. Marriott, L. E. Quek, A. Katen, J. Su, N. Kumar and K. L. Hoehn, Phenotypic screen for oxygen consumption rate identifies an anti-cancer naphthoquinone that induces mitochondrial oxidative stress, Redox Biol., 2020, 28, 101374 CrossRef CAS PubMed , Available from: https://www.sciencedirect.com/science/article/pii/S2213231719308845?via%3Dihub.
- Y. Cheng, J. P. Jones, T. T. Yu, E. M. Olzomer, J. Su, A. Katen, D. S. Black, G. Hart-Smith, E. S. Childress, M. R. Wilkins, I. A. Mateos, W. L. Santos, K. L. Hoehn, F. L. Byrne and N. Kumar, Design, synthesis and biological evaluation of glucose metabolism inhibitors as anticancer agents, Bioorg. Chem., 2024, 151, 107665 CrossRef CAS PubMed , Available from: https://www.sciencedirect.com/science/article/pii/S0045206824005704.
- T. Suzuki, A. Muramatsu, R. Saito, T. Iso, T. Shibata, K. Kuwata, S. I. Kawaguchi, T. Iwawaki, S. Adachi, H. Suda, M. Morita, K. Uchida, L. Baird and M. Yamamoto, Molecular Mechanism of Cellular Oxidative Stress Sensing by Keap1, Cell Rep., 2019, 28, 746–758 CrossRef CAS PubMed , Available from: https://www.sciencedirect.com/science/article/pii/S2211124719308204?via%3Dihub.
- V. Prachayasittikul, R. Pingaew, A. Worachartcheewan, C. Nantasenamat, S. Prachayasittikul, S. Ruchirawat and V. Prachayasittikul, Synthesis, anticancer activity and QSAR study of 1,4-naphthoquinone derivatives, Eur. J. Med. Chem., 2014, 84, 247–263 CrossRef CAS PubMed , Available from: https://www.sciencedirect.com/science/article/pii/S0223523414006308?via%3Dihub.
- K. W. Wellington, Understanding cancer and the anticancer activities of naphthoquinones – a review, RSC Adv., 2015, 5, 20309–20338 RSC , Available from: https://rsc.66557.net/en/content/articlelanding/2015/ra/c4ra13547d.
- J. Yuan, Z. Liu, Z. Zhang, D. Yan and W. Zhang, Synthesis and biological evaluation of naphthoquinone phenacylimidazolium derivatives, Bioorg. Med. Chem. Lett., 2021, 41, 127977 CrossRef CAS PubMed , Available from: https://www.sciencedirect.com/science/article/pii/S0960894X21002031?via%3Dihub.
- K. T. Fridianto, M. Li, K. Hards, D. A. Negatu, G. M. Cook, T. Dick, Y. Lam and M. L. Go, Functionalized Dioxonaphthoimidazoliums: A Redox Cycling Chemotype with Potent Bactericidal Activities against Mycobacterium tuberculosis, J. Med. Chem., 2021, 64, 15991–16007, DOI:10.1021/acs.jmedchem.1c01383.
- S. Ahenkorah, D. Coertzen, J. X. Tong, K. Fridianto, S. Wittlin, L. M. Birkholtz, K. S. W. Tan, Y. Lam, M. L. Go and R. K. Haynes, Antimalarial N (1),N (3)-Dialkyldioxonaphthoimidazoliums: Synthesis, Biological Activity, and Structure-activity Relationships, ACS Med. Chem. Lett., 2020, 11, 49–55, DOI:10.1021/acsmedchemlett.9b00457.
- S. H. Ho, M. Y. Sim, W. L. Yee, T. Yang, S. P. Yuen and M. L. Go, Antiproliferative, DNA intercalation and redox cycling activities of dioxonaphtho[2,3-d]imidazolium analogs of YM155: A structure-activity relationship study, Eur. J. Med. Chem., 2015, 104, 42–56 CrossRef CAS PubMed , Available from: https://www.sciencedirect.com/science/article/pii/S0223523415302701?via%3Dihub.
- J. E. Egleton, C. C. Thinnes, P. T. Seden, N. Laurieri, S. P. Lee, K. S. Hadavizadeh, A. R. Measures, A. M. Jones, S. Thompson, A. Varney, G. M. Wynne, A. Ryan, E. Sim and A. J. Russell, Structure-activity relationships and colorimetric properties of specific probes for the putative cancer biomarker human arylamine N-acetyltransferase 1, Bioorg. Med. Chem., 2014, 22, 3030–3054 CrossRef CAS PubMed , Available from: https://www.sciencedirect.com/science/article/pii/S0968089614001886?via%3Dihub.
- M. P. Brun, E. Braud, D. Angotti, O. Mondesert, M. Quaranta, M. Montes, M. Miteva, N. Gresh, B. Ducommun and C. Garbay, Design, synthesis, and biological evaluation of novel naphthoquinone derivatives with CDC25 phosphatase inhibitory activity, Bioorg. Med. Chem., 2005, 13, 4871–4879 CrossRef CAS PubMed , Available from: https://www.sciencedirect.com/science/article/pii/S0968089605003962.
- D. J. Huggins, W. Sherman and B. Tidor, Rational approaches to improving selectivity in drug design, J. Med. Chem., 2012, 55, 1424–1444, DOI:10.1021/jm2010332.
- T. Kayashima, M. Mori, H. Yoshida, Y. Mizushina and K. Matsubara, 1,4-Naphthoquinone is a potent inhibitor of human cancer cell growth and angiogenesis, Cancer Lett., 2009, 278, 34–40 CrossRef CAS PubMed , Available from: https://www.sciencedirect.com/science/article/pii/S0304383508009683.
- J. C. Lien, L. J. Huang, J. P. Wang, C. M. Teng, K. H. Lee and S. C. Kuo, Synthesis and antiplatelet, antiinflammatory, and antiallergic activities of 2-substituted 3-chloro-1,4-naphthoquinone derivatives, Bioorg. Med. Chem., 1997, 5, 2111–2120 CrossRef CAS PubMed , Available from: https://www.sciencedirect.com/science/article/pii/S0968089697001338.
- U. Sharma, D. Katoch, S. Sood, N. Kumar and B. Singh, Synthesis, antibacterial and antifungal activity of 2-amino-1,4-naphthoquinones using silica-supported perchloric acid (HClO4-SiO2) as a mild, recyclable and highly efficient heterogeneous catalyst, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2013, 52, 1431–1440 Search PubMed.
- M. Campora, C. Canale, E. Gatta, B. Tasso, E. Laurini, A. Relini, S. Pricl, M. Catto and M. Tonelli, Multitarget Biological Profiling of New Naphthoquinone and Anthraquinone-Based Derivatives for the Treatment of Alzheimer's Disease, ACS Chem. Neurosci., 2021, 12, 447–461, DOI:10.1021/acschemneuro.0c00624?ref=pdf.
- I. A. Schepetkin, A. S. Karpenko, A. I. Khlebnikov, M. O. Shibinska, I. A. Levandovskiy, L. N. Kirpotina, N. V. Danilenko and M. T. Quinn, Synthesis, anticancer activity, and molecular modeling of 1,4-naphthoquinones that inhibit MKK7 and Cdc25, Eur. J. Med. Chem., 2019, 183, 111719 CrossRef PubMed , Available from: https://www.sciencedirect.com/science/article/pii/S0223523419308712?via%3Dihub.
- E. Mezeiova, J. Janockova, R. Andrys, O. Soukup, T. Kobrlova, L. Muckova, J. Pejchal, M. Simunkova, J. Handl, P. Micankova, J. Capek, T. Rousar, M. Hrabinova, E. Nepovimova, J. L. Marco-Contelles, M. Valko and J. Korabecny, 2-Propargylamino-naphthoquinone derivatives as multipotent agents for the treatment of Alzheimer's disease, Eur. J. Med. Chem., 2021, 211, 113112 CrossRef CAS PubMed , Available from: https://www.sciencedirect.com/science/article/pii/S0223523420310849?via%3Dihub.
- V. K. Tandon, D. B. Yadav, H. K. Maurya, A. K. Chaturvedi and P. K. Shukla, Design, synthesis, and biological evaluation of 1,2,3-trisubstituted-1,4-dihydrobenzo[g]quinoxaline-5,10-diones and related compounds as antifungal and antibacterial agents, Bioorg. Med. Chem., 2006, 14, 6120–6126 CrossRef CAS PubMed , Available from: https://www.sciencedirect.com/science/article/pii/S0968089606003105.
- F. L. Byrne, I. K. Poon, S. C. Modesitt, J. L. Tomsig, J. D. Chow, M. E. Healy, W. D. Baker, K. A. Atkins, J. M. Lancaster, D. C. Marchion, K. H. Moley, K. S. Ravichandran, J. K. Slack-Davis and K. L. Hoehn, Metabolic vulnerabilities in endometrial cancer, Cancer Res., 2014, 74, 5832–5845 CrossRef CAS PubMed , Available from: https://aacrjournals.org/cancerres/article/74/20/5832/599112/Metabolic-Vulnerabilities-in-Endometrial.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00987h |
| This journal is © The Royal Society of Chemistry 2025 |