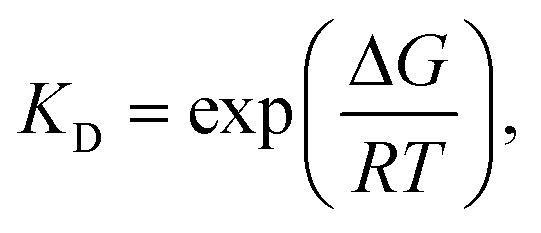DOI:
10.1039/D4MD00992D
(Research Article)
RSC Med. Chem., 2025,
16, 1758-1765
Transmembrane protease serine 2 (TMPRSS2) inhibitors screened from an Fv-antibody library for preventing SARS-CoV-2 infection
Received
17th December 2024
, Accepted 11th February 2025
First published on 13th February 2025
Abstract
Fv-antibodies targeting the transmembrane protease serine 2 (TMPRSS2) were screened from an Fv-antibody library for inhibiting SARS-CoV-2 infection. Fv-antibodies were derived from the variable region of heavy-chain immunoglobulin G (IgG), which consisted of three complementarity-determining regions (CDRs) and frame regions (FRs). The Fv-antibody library was prepared through site-directed mutagenesis of CDR3 region. The proteolytic cleavage site (S2′ site) of TMPRSS2 on the spike protein (SP) of SARS-CoV-2 was used as a screening probe for the library. Two Fv-antibodies were screened and subsequently expressed as soluble recombinant proteins. The binding affinities of the expressed Fv-antibodies were estimated using a surface plasmon resonance (SPR) biosensor. The two expressed Fv-antibodies specifically bound to the active site of TMPRSS2 which interacts with S2′ site in the proprotein convertase (PPC) region. The neutralizing activities of the two expressed Fv-antibodies were demonstrated using a cell-based infection assay with pseudo-viruses that expressed the SP of four types of SARS-CoV-2 variants: Wu-1 (D614), Delta (B.1.617.2), Omicron (BA.2), and Omicron (BA.4/5). Additionally, a docking simulation was performed to analyze the interaction between the screened Fv-antibodies and the active sites of TMPRSS2.
1. Introduction
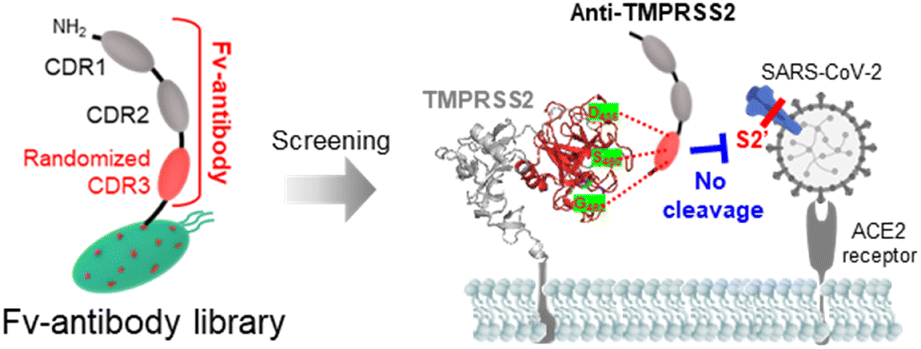
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) initiates infection when its spike protein (SP) binds to the angiotensin-converting enzyme 2 (ACE2) receptors on host cells.1–3 Specifically, the receptor-binding domain (RBD) of SP is known to directly interact with the ACE2 receptor.4,5 Several strategies have been reported to inhibit this interaction, encompassing approaches targeting different stages of SARS-CoV-2 infection, from attachment to viral entry into host cells.6–9 However, these strategies face several challenges owing to frequent mutations in the RBD of SP, which can alter its structure and affect the efficacy of medical interventions.10–12 Consequently, alternative methods have emerged that focus on inhibiting transmembrane protease serine 2 (TMPRSS2) on host cells, a critical enzyme responsible for cleaving SP during viral entry.13,14 The coronavirus HKU1 is one of the four endemic seasonal human coronaviruses (HKU1, 229E, NL63, and OC43) which cause the common cold worldwide, and the major receptor for these strains remain unidentified. Recent studies report that TMPRSS2 has a key role for the infection of HKU1. In these studies, the SP of HKU1 is reported to interact with TMPRSS2 and the nanobodies (VHH) targeting TMPRSS2 effectively inhibits the HKU1 entry into host cells.15,16 Following the initial binding of SP to the ACE2 receptor, membrane fusion between the virus and host cells is facilitated by the cleavage of SP. SP contains cleavage sites in the proprotein convertase (PPC) region, which includes the S1/S2 (residue: 682–685) and S2′ sites (residue: 809–815), targeted by the proteolytic enzyme furin and TMPRSS2, respectively.17,18 This cleavage process is crucial for SP activation, allowing the virus to entry host cells and initiate infection. Additionally, the virus can also enter cells through the endocytosis pathway, where cathepsin L cleaves the SP instead of TMPRSS2.19 Since SP activation plays a key role in viral entry, TMPRSS2 has gained significant attention as a potential therapeutic target for inhibiting SARS-CoV-2 infection.20,21
In this study, Fv-antibodies targeting TMPRSS2 were screened from an Fv-antibody library to inhibit SARS-CoV-2 infection. As shown in Scheme 1, Fv-antibodies were derived from the variable heavy (VH) region of immunoglobulin G (IgG). They consisted of three complementarity-determining regions (CDRs) and four framework regions (FRs).22,23 The CDR3 regions of the Fv-antibodies were randomized using site-directed mutagenesis. The prepared Fv-antibody library was expressed on the surface of E. coli using the autodisplay technology, which utilized the AIDA-1 system to transport target proteins to the bacterial surface.24 The library exhibited a high display density of 105 Fv-antibodies per E. coli and a diversity of 106 different Fv-antibodies per library.9,10,25–30 To screen the Fv-antibodies, the extracellular domain of TMPRSS2 (residue: 106–492), including the active sites (residue: 256–489), substrate-binding sites (residues: D435, S460, and G462), and catalytically active sites (residues: H296, D345, and S441),14,31 was used as a screening probe. This region was located in the pocket where the S2′ site of SP was known to specifically bind, facilitating the proteolytic cleavage necessary for viral entry. The screened Fv-antibodies were expressed as soluble recombinant proteins,32 and their specific binding activity to TMPRSS2 was analyzed using a SPR biosensor. To demonstrate the inhibition of SARS-CoV-2 infection using the screened Fv-antibodies, the neutralizing activity of the Fv-antibodies was assessed using a cell-based infection assay with pseudo-virus particles expressing the SPs of different SARS-CoV-2 variants, namely Wu-1 (D614), Delta (B.1.617.2), Omicron (BA.2 and BA.4/5).9,33–35 Additionally, a docking simulation was performed to analyze the interaction between the screened Fv-antibodies and TMPRSS2.
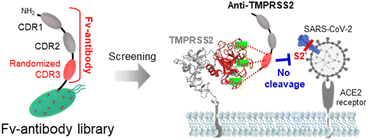 |
| | Scheme 1 TMPRSS2 inhibitors used to prevent SARS-CoV-2 infection, which were screened from an Fv-antibody library. | |
2. Experimental section
2.1 Materials
The expression vectors for extracellular TMPRSS2 (residue: 106–492, 69.7 kDa) and SARS-CoV-2 PPC region (residue: 661–900, 53.6 kDa) were synthesized by Cosmo Genetech (Seoul, South Korea). The materials necessary for producing SARS-CoV-2 variant pseudo-viruses particles have been described previously.9 HEK-Blue™ 293 cell line, which overexpress hACE2 and TMPRSS2 was purchased from InvivoGen Inc. (San Diego, CA, USA).
2.2 Screening of anti-TMPRSS2 Fv-antibodies
The Fv-antibody library was prepared as described in previous studies.9,10,25–30,34–38 Clones that exhibited binding activity to the TMPRSS2 probe (extracellular residue: 106–492, labeled with GFP) were screened using the following protocol. (1) The randomized CDR3 region of the Fv-antibodies was displayed on the surface of E. coli. (2) The TMPRSS2 probe (1 μM, 100 μL) was incubated with the Fv-antibody library (100 μL) for 1 h at 37 °C. (3) After washing with 0.01% PBST (containing 0.01% Tween 20), the clones with binding activity were sorted (n = 500) using a flow cytometer (FACSCalibur™, NJ, USA). (4) The selected clones were identified by sequencing the CDR3 region using DNA oligonucleotide sequencing.
2.3 Determination of binding affinity (KD) of anti-TMPRSS2 Fv
The KD of the anti-TMPRSS2 Fv was determined using an SPR biosensor (i-Cluebio, Seongnam, South Korea) as follows.39 (1) SPR chips made of BK-7 glass were initially coated with a 2 nm titanium layer, followed by a 48 nm gold layer. (2) The anti-TMPRSS2 Fv (20 μg mL−1, 100 μL) was immobilized onto the gold surface of the SPR chips by incubating for 16 h at 4 °C. (3) The chips were blocked with BSA (1 mg mL−1) for 1 h at 37 °C. (4) Following a wash with 0.01% PBST, TMPRSS2 antigen at varying concentrations (12.5 to 100.0 nM) was delivered at a flow rate of 25 μL min−1 for 10 min to facilitate binding. (5) dissociation was carried out by introducing PBS as a washing buffer at the same flow rate for 10 min.
2.4 Binding analysis of anti-TMPRSS2 Fv to TMPRSS2
The binding interactions between the anti-TMPRSS2 Fv and TMPRSS2 were investigated using the SPR biosensor, as described above. SARS-CoV-2 PPC (20 μg mL−1, 100 μL) was immobilized onto the gold surface of the SPR chips for 16 h at 4 °C. After washing with PBS, the binding of TMPRSS2 was monitored before and after treatment with the anti-TMPRSS2 Fv-antibodies at various concentrations (11.1–300.0 nM).
2.5 Production of pseudo-virus particles
SARS-CoV-2 SP variant pseudo-viruses, namely Wu-1 (D614), Delta (B.1.617.2), Omicron (BA.2 and BA.4/5), were produced using Lenti-X HEK293 cells. The production process comprised the following steps: (1) Lenti-X™ HEK293 cells (1 × 105 cells) were cultured in 15 mL of DMEM supplemented with 10% FBS for 1 d. (2) A mixture containing 10 μL of the transfection reagent FuGENE and 5 μg of each plasmid (SP pseudotyping vectors, pLVXS-ZsGreen1-Puro, and psPAX2) was prepared in 1 mL of Opti-MEM and incubated at 37 °C for 10 min. (3) Subsequently, the cell culture medium was replaced with 10 mL of fresh DMEM containing 10% FBS, and the transfection mixture obtained from Step 2 was added to the cells. (4) After three days of incubation, the supernatant was harvested by centrifugation at 500g for 10 min. (5) A Lenti-X™ concentrator was added to the supernatant and incubated for 3 h at 4 °C. (6) Pseudo-virus particles were pelleted by centrifugation at 1500g for 45 min and resuspended in DMEM. (7) The concentration of the pseudo-virus particles was quantified using Lenti-X qRT-PCR.
2.6 Evaluation of neutralizing effectiveness of anti-TMPRSS2 Fv
The neutralizing capability of the anti-TMPRSS2 Fv was assessed using an in vitro cell-based infection assay as follows: (1) HEK-Blue™ cells overexpressing hACE2-TMPRSS2 (5.0 × 104 cells per well) were seeded onto 96-well microplates pre-coated with poly L-lysine and incubated for 1 d. (2) The anti-TMPRSS2 Fv was added to the HEK-Blue™ cells and incubated for 30 min in DMEM (100 μL) containing 2% FBS at 37 °C. (3) Subsequently, the pseudo-virus particles were introduced to the HEK-Blue™ cells, followed by a 48 h incubation period. (4) Finally, the cells were viewed using a fluorescence microscope (Eclipse Ts2) to evaluate the neutralization capacity of the anti-TMPRSS2 Fv.
3. Result and discussion
3.1 Screening of Fv-antibodies against TMPRSS2
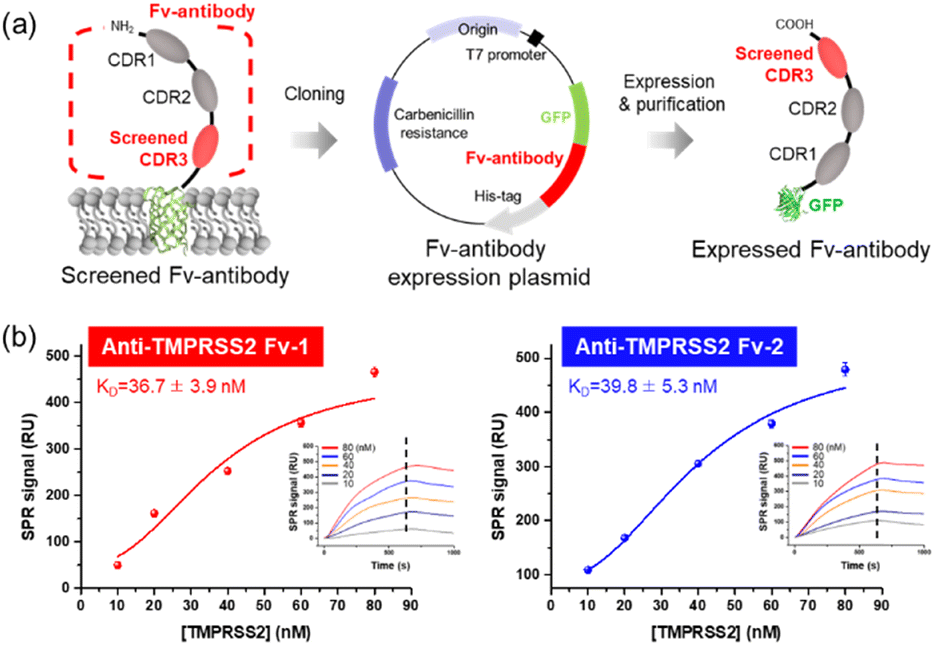
Fv-antibodies with binding affinities for the active sites of TMPRSS2 were screened to prevent SARS-CoV-2 from infecting host cells. As shown in Fig. 1(a), the Fv-antibodies were derived from the VH domain of IgG, which consisted of three CDRs and four FRs.25,40 The Fv-antibody library was prepared by randomizing eleven amino acid residues of CDR3 via site-directed mutagenesis.34,35 The Fv-antibody library was expressed on the outer membrane of E. coli using the autodisplay technology.9,10,25–30,34–38 The Fv-antibody library expressed on the outer membrane of E. coli exhibited a high surface density of up to 105 Fv-antibodies per E. coli and a high diversity of 106 Fv-antibodies per library.24,41 Using the Fv-antibody library, TMPRSS2 inhibitors were screened to prevent SARS-CoV-2 infection. As shown in Fig. 1(b), TMPRSS2 is a transmembrane protein with a molecular weight of 53.8 kDa, which is crucial for the viral fusion of SARS-CoV-2 with host cells. The extracellular domain of TMPRSS2 (residue: 106–492, PDB no.: 7MEQ) was expressed as a screening probe, which included active sites (residue: 256–489), a substrate-binding site (residues: D435, S460, and G462), and catalytically active sites (residues: H296, D345, and S441).14,31 This region was located in the pocket where the S2′ site of SP was known to specifically bind with, facilitating the proteolytic cleavage necessary for viral entry. As shown in Fig. 1(c), the fluorescence-labeled TMPRSS2 probe reacted with the Fv-antibody library, and positive clones were screened using flow cytometry. As shown in Fig. 1(d), a highly fluorescent region was observed in the flow cytometry dot plot, indicating potential binding interactions. In contrast, the control strain containing only CDR1 and CDR2 exhibited no significant fluorescence in the same region. These results indicated that the Fv-antibody library contained clones capable of specifically binding to the fluorescent TMPRSS2 probe through the amino acid sequence of CDR3 in the fluorescent clones. Clones with fluorescent signals were isolated via flow cytometry and grown on agar plates. Randomly selected clones were treated with the fluorescent TMPRSS probe, and their binding affinity was analyzed using flow cytometry. Oligonucleotide sequencing was performed for the highly fluorescent clones, and two final clones containing suitable oligonucleotide sequences in the CDR3 region were selected, as shown in Fig. 1(e). The oligonucleotide sequences and corresponding amino acid sequences of the screened CDR3 region of the Fv-antibodies are listed in Table 1.
 |
| | Fig. 1 Screening Fv-antibodies for specific binding affinity to TMPRSS2 using an Fv-antibody library. (a) Construction of an Fv-antibody library on the surface of E. coli. (b) Analysis of TMPRSS2 and expression of the TMPRSS2 probe (residue: 106–492) as a GFP fusion protein. (c) Scheme of screening process. (d) Screening of positive clones with binding affinity for the TMPRSS2 probe using a screening gate. (e) Flow cytometric analysis of TMPRSS2 probe activity in screened clones. | |
Table 1 Oligonucleotide and amino acid sequences of Fv-antibodies and the binding affinity (KD) of TMPRSS2
| Screened clone |
Sequences of CDR3 region |
Binding constant (KD) |
| Oligonucleotide (33 bp) |
Amino acid (11 mer) |
SPR (nM) |
| 1 |
5′-GAC CCT CCT CCT CCT GCC GTA GCT GCA GAT GTC-3′ |
1DPPPP 5AVAAD 11V |
36.7 |
| 2 |
5′-TGC CGC GAT CTA CTG GGT GTG GTC CGC GAT TTT-3′ |
1CRDLL 5GVVRD 11F |
39.8 |
3.2 Binding properties of screened Fv-antibodies
The screened Fv-antibodies were expressed as soluble proteins, along with GFP, as shown in Fig. 2(a). The co-expression of GFP offers several advantages: (1) it enhances the solubility of the Fv-antibodies, which have limited solubility,32,42 and (2) it facilitates effective immobilization of the Fv-antibodies onto the metal surface of biosensors.25 The binding affinities of the expressed Fv-antibodies were estimated using an SPR biosensor. After the Fv-antibodies were immobilized on the Au chip of the SPR biosensor, different concentrations of the TMPRSS2 probe were added, and the SPR signal was measured. Anti-TMPRSS2 Fv-1 and Fv-2 exhibited binding affinities (KD) of 36.7 and 39.8, respectively, as shown in Fig. 2(b). These calculated values were comparable to the known binding affinity between TMPRSS2 and SARS-CoV-2 SP (53 nM),14 highlighting the potential effectiveness of these Fv-antibodies as inhibitors. The binding affinity (KD) of SARS-CoV-1 SP for the host cell ACE2 receptor is reported to range from 31 to 100 nM. SARS-CoV-2 is also reported to have a binding affinity in the range of 4.7–10.0 nM.2,8,25,43,44
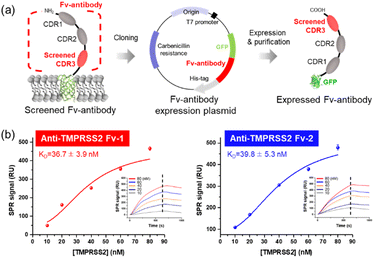 |
| | Fig. 2 Expression of Fv-antibodies (anti-TMPRSS2 Fv) and analysis of binding affinity. (a) Scheme for the expression of screened Fv-antibodies fused with GFP. (b) Evaluation of the binding affinity (KD) of the anti-TMPRSS2 Fv using an SPR biosensor (insets: SPR sensorgrams for TMPRSS2 in the concentrations range of 10–80 nM). | |
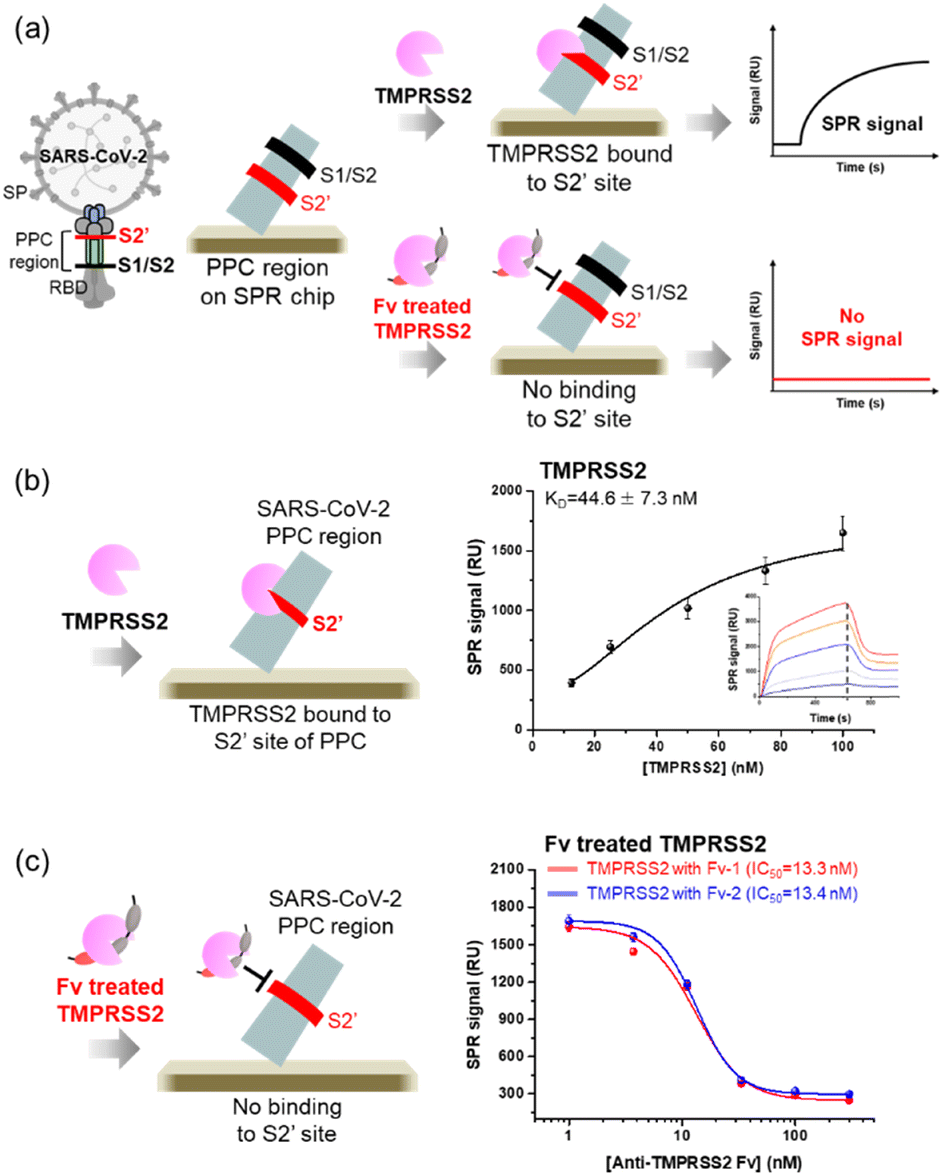
The specific binding of two Fv-antibodies to the active site of TMPRSS2 was confirmed by analyzing the interactions between the TMPRSS2 and the PPC region of SARS-CoV-2 SP, which includes the S2′ site. The SARS-CoV-2 SP contains cleavage sites in the PPC region, which includes the S1/S2 site (residue: 682–685) targeted by the proteolytic enzyme furin and the S2′ site (residue: 809–815) targeted by TMPRSS2.17,18 As shown in Fig. 3(a), the PPC region was immobilized on the Au chip of the SPR biosensor, and the binding of TMPRSS2 was monitored before and after treatment with Fv-antibodies. As shown in Fig. 3(b), when the TMPRSS2 probe was introduced to the SPR biosensor, a significant SPR signal was observed, indicating the binding of TMPRSS2 to the S2′ site.
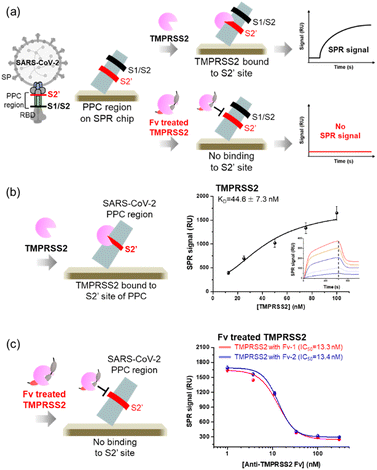 |
| | Fig. 3 TMPRSS2 inhibition assay using anti-TMPRSS2 Fv. (a) Schematic representation of the TMPRSS2 inhibition assay using an SPR biosensor. (b) Analysis of TMPRSS2 binding to the S2′ site of the SARS-CoV-2 PPC region (inset: SPR sensorgram for TMPRSS2 in the concentrations range of 12.5–100.0 nM). (c) Estimation of the IC50 of anti-TMPRSS2 Fv against TMPRSS2. | |
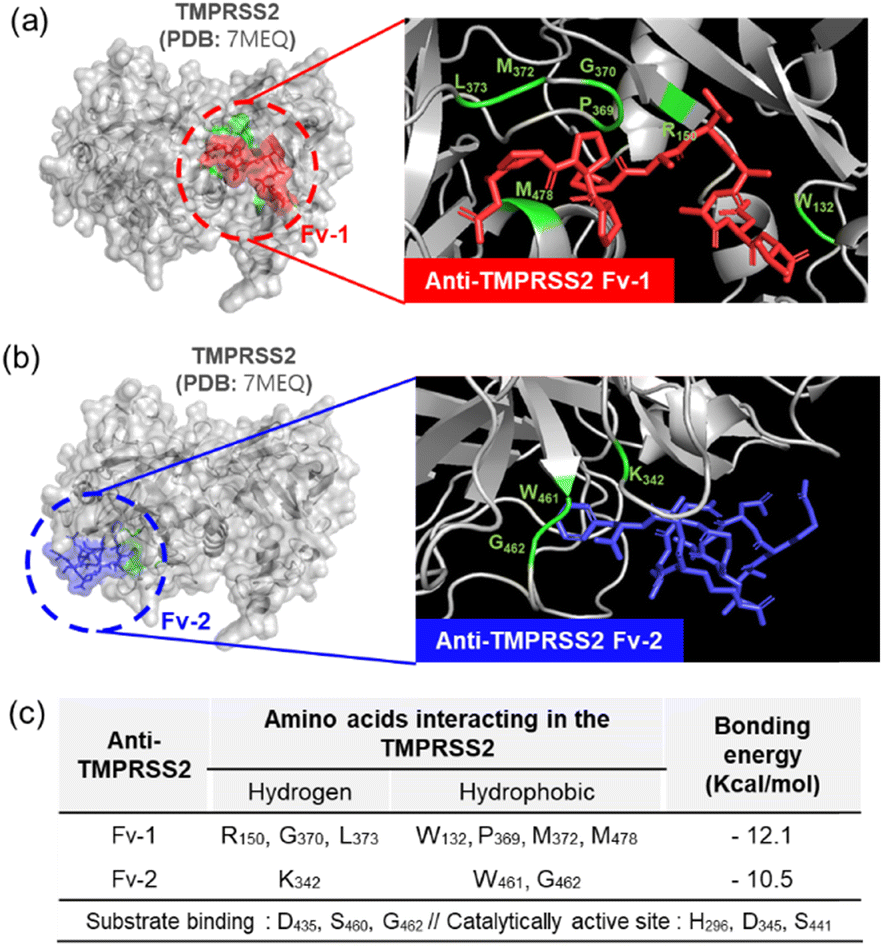
As shown in Fig. 3(c), when TMPRSS2 was pre-incubated with anti-TMPRSS2 Fv-1, a significantly decreased SPR signal was observed, indicating the blocking of the binding site of anti-TMPRSS2 Fv-1. Similar results were obtained after treatment with anti-TMPRSS2 Fv-2. Based on these results, the IC50 of anti-TMPRSS2 Fv-1 and Fv-2 was determined to be 13.3 nM and 13.4 nM, respectively. Both Fv-antibodies specifically bound to the active site of TMPRSS2 and effectively inhibited its proteolytic activity. To investigate the binding affinity between TMPRSS2 and the screened Fv-antibodies, docking simulations were performed using AutoDock Vina developed by Scripps Research (La Jolla, CA, USA).45 As shown in Fig. 4(a), anti-TMPRSS2 Fv-1 bound to the known active site of TMPRSS2, with a binding energy of −12.1 kcal mol−1, indicating spontaneous binding between TMPRSS2 and anti-TMPRSS2 Fv-1. This interaction involved hydrogen bonding with R150, G370, and L373 and hydrophobic interactions with W132, P369, M372, and M478 in anti-TMPRSS2 Fv-1. As shown in Fig. 4(b), anti-TMPRSS2 Fv-2 also bound to the active site of TMPRSS2, with a binding energy of −10.5 kcal mol−1, indicating spontaneous binding. This interaction involved hydrogen bonding with K342 and hydrophobic interactions with W461 and G462 in anti-TMPRSS2 Fv-2.
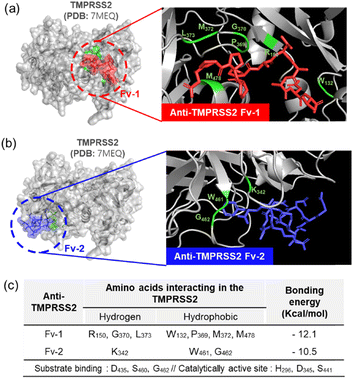 |
| | Fig. 4 Docking simulation analysis of anti-TMPRSS2 Fv to the TMPRSS2. (a) Binding sites of anti-TMPRSS2 Fv-1 and (b) anti-TMPRSS2 Fv-2. (c) Analysis of the binding sites between anti-TMPRSS2 Fv and TMPRSS2. | |
The interaction model with the lowest Gibbs free energy change (ΔG) value was selected as the optimal one. Additionally, from the measurement with SPR biosensor, the KD values of screened Fv-antibodies were estimated to be 36.7 nM for anti-TMPRSS2 Fv-1 and 39.8 nM for Fv-2 (Fig. 2(b)). From the docking simulation, ΔG values were calculated to be −12.1 kcal mol−1 for anti-TMPRSS2 Fv-1 and −10.5 kcal mol−1 for Fv-2, respectively. The KD value is related to ΔG by the following equation:
where Δ
G denoted the Gibbs free energy change (kcal mol
−1),
R was the gas constant (0.001987 kcal mol
−1 ×
K), and
T referred to the absolute temperature (Kelvin).
34,46 Considering the above equation, the
KD values of anti-TMPRSS2 Fv-1 and Fv-2 are as followers:
KD = 2.9 nM for anti-TMPRSS2 Fv-1 and 75.0 nM for Fv-2. In comparison with
KD values obtained from the SPR measurements, the calculated
KD values from the docking simulation were considered to be comparable levels. The results are presented in
Fig. 4(c), indicating that both anti-TMPRSS2 Fv-1 and Fv-2, which have high binding affinities, can spontaneously bind to the active site (residue: 256–489) of TMPRSS2.
3.3 Neutralizing activity of screened Fv-antibodies
The neutralizing activity of the Fv-antibodies against SARS-CoV-2 was evaluated using a cell-based infection assay.34,35In vitro cell-based assays employing pseudo-virus particles are known to correlate strongly with assays using live SARS-CoV-2 virus.47–49
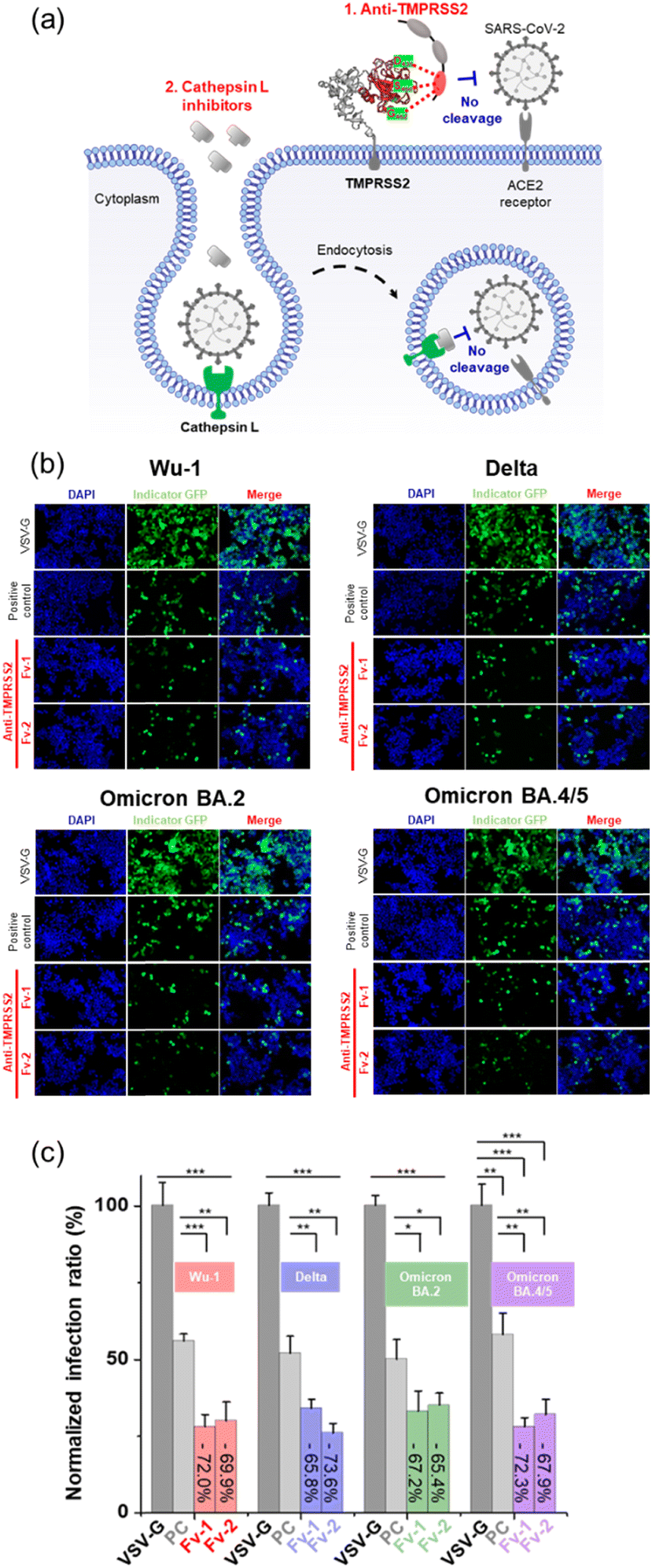
The HEK293 cell line stably expressing TMPRSS2 and the ACE2 receptor (HEK-Blue™ hACE-TMPRSS2) was used as the host cell for SARS-CoV-2 infection. Pseudo-viruses based on lentiviruses were prepared by expressing the SP of SARS-CoV-2 corresponding to the Wu-1 (D614), Delta (B.1.617.2), Omicron (BA.2), and Omicron (BA.4/5) variants.34 The pseudo-viruses included a GFP expression vector, which serves as a marker to indicate the successful infection of a host cell. After infection, the viruses introduce GFP-encoding genetic material into the host cell, leading to GFP expression and the generation of a fluorescence signal, as shown in Fig. 5(a). No fluorescence signals were observed when pseudoviral infection was inhibited by the Fv-antibodies. Therefore, the preventive activity of Fv-antibodies against viral infection can be estimated by measuring the fluorescence signal. First, the binding activity of the Fv-antibodies (labeled with GFP) to TMPRSS2 on host cells was observed, as shown in Fig. 5(b). In the case of anti-RBD Fv-antibody (labeled with GFP) which was used as a negative control was observed to produce no fluorescence signal because this Fv-antibody did not exhibit binding activity to TMPRSS2 on the host cell. This result represented that the binding of the two screened Fv-antibodies was specific to TMPRSS2 on the host cell. The neutralizing activity of the Fv-antibodies against SARS-CoV-2 infection was assessed using a pseudo-virus. The host cells were infected by pseudo-viruses corresponding to the Wu-1 (D614), Delta (B.1.617.2), Omicron (BA.2), and Omicron (BA.4/5) variants. As shown in Fig. 5(c), the assay was performed to compare the neutralizing activity of these Fv-antibodies at the concentration corresponding to their binding affinity (KD). The KD values of these Fv-antibodies were measured to be 36.7 nM for anti-TMPRSS2 Fv-1, 39.8 M for Fv-2 using SPR measurement (Fig. 2(b)). As shown in the fluorescence images, for all four variant strains of pseudo-virus particles, treatment with Fv-antibodies significantly reduced the fluorescence signals, indicating the inhibition of virus infection by the Fv-antibodies. For the statistical calculation of the neutralizing activity of the Fv-antibodies, the fluorescence signal was analyzed using flow cytometry. As shown in Fig. 5(d), the neutralizing activity of the Fv-antibodies was estimated to be in the range of 59.5–70.4% for four types of pseudo-virus particles in comparison with the infection of VSV-G. The SARS-CoV-2 cellular entry has been also reported to be possible through the endocytosis pathway where the cathepsin L plays a role in cleaving the SP. In order to estimate the contribution of endocytosis pathway to the SARS-CoV-2 infection, the cathepsin L inhibitor (#SCP0110, Sigma-Aldrich, MO, USA) was added at the same time as the treatment of the Fv-antibodies, as shown in Fig. 6(a). And then, the neutralizing activity of the Fv-antibodies was measured and compared with the assay results without cathepsin L inhibitor treatment. The infection inhibition ratio for anti-TMPRSS2 Fv-1 with (without) the treatment of cathepsin L inhibitor was estimated to be −72.0% (−63.5%) for Wu-1, −65.8% (−59.5%) for Delta, −67.2% (−64.4%) for Omicron BA.2, and −72.3% (−62.8%) for Omicron BA.4/5. Similarly, the infection inhibition ratio for anti-TMPRSS2 Fv-2 with (without) the treatment of cathepsin L inhibitor was estimated to be −69.9% (−62.9%) for Wu-1, −73.6% (−70.4%) for Delta, −65.4% (−60.3%) for Omicron BA.2, and −67.9% (−62.8%) for Omicron BA.4/5 (Fig. 6(b) and (c)). From these results the contribution of endocytosis pathway for the infection of SARS-CoV-2 was estimated to be approximately 5–10%, and there made not so significant difference in neutralizing activity of the Fv-antibodies. And, these trends were observed across the viral variants. These results represented the TMPRSS2 played major role for the infection of SARS-CoV-2 in comparison with the endocytosis pathway across different SARS-CoV-2 variants. These results indicated that (1) Fv-antibodies could effectively prevent the infection of SARS-CoV-2 by inhibiting the protease activity of TMPRSS2, and (2) similar levels of neutralizing activity could be achieved against different variant strains of SARS-CoV-2.
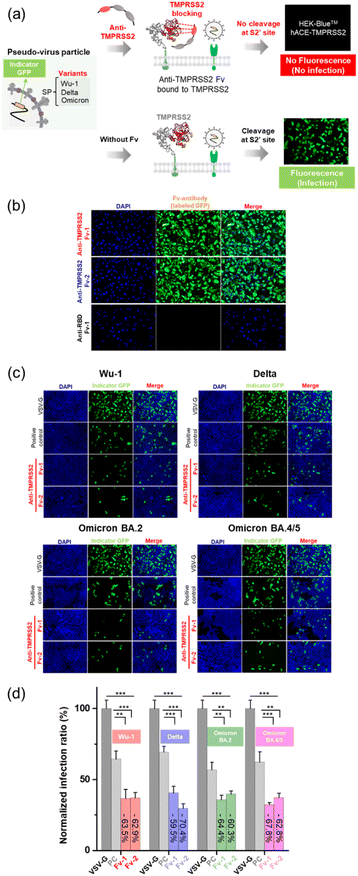 |
| | Fig. 5
In vitro infection assay using pseudo-virus particles (four different SARS-CoV-2 variants: Wu-1 D614, Delta B.1.617.2, Omicron BA.2, and Omicron BA.4/5) and HEK-Blue™ cells overexpressing hACE2-TMPRSS2. (a) Scheme of the in vitro infection assay using pseudo-virus particles. (b) Binding analysis of anti-TMPRSS2 with HEK-blue™ cells. (c) Evaluation of infection inhibition using fluorescence imaging with anti-TMPRSS2 Fv and pseudo-virus particles. (d) Inhibition ratio of SARS-CoV-2 infection. | |
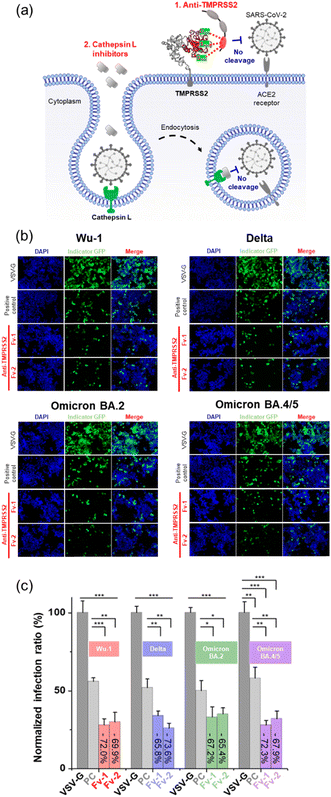 |
| | Fig. 6
In vitro infection assay of anti-TMPRSS2 Fv with the treatment of cathepsin L inhibitor. (a) Schematic representation of the process involving Fv-antibodies and cathepsin L inhibitor. (b) Fluorescence images after treatment of anti-TMPRSS2 Fv and cathepsin L inhibitor. (c) Inhibition ratio of SARS-CoV-2 infection for variant strains. | |
4. Conclusions
In this study, Fv-antibodies against TMPRSS2 were screened from an Fv-antibody library. The Fv-antibody library was constructed by site-directed mutagenesis of the CDR3 region and expressed on the outer membrane of E. coli using the autodisplay technology. The library exhibited an expression level exceeding 105 Fv-antibodies/E. coli and a diversity of over 106 Fv-antibodies/library. After screening, the selected Fv-antibodies were expressed as soluble recombinant proteins. Using an SPR biosensor, the binding affinities (KD) of anti-TMPRSS2 Fv-1 and Fv-2 were measured to be 36.7 nM and 39.8 nM, respectively. Additionally, anti-TMPRSS2 Fv-1 and anti-TMPRSS2 Fv-2 exhibited an IC50 of 13.3 nM and 13.4 nM, respectively. Docking simulations demonstrated that these Fv-antibodies specifically bind to the active sites of TMPRSS2, effectively blocking these regions of the protease. The neutralizing activity of anti-TMPRSS2 Fv-antibodies was measured using an in vitro cell-based infection assay. In this assay, we used pseudo-virus particles expressing the SP of SARS-CoV-2 variants, such as Wu-1 (D614), Delta (B.1.617.2), Omicron (BA.2), and Omicron (BA.4/5). The infection inhibition ratio for anti-TMPRSS2 Fv-1 with (without) the treatment of cathepsin L inhibitor was estimated to be −72.0% (−63.5%) for Wu-1, −65.8% (−59.5%) for Delta, −67.2% (−64.4%) for Omicron BA.2, and −72.3% (−62.8%) for Omicron BA.4/5. Similarly, the infection inhibition ratio for anti-TMPRSS2 Fv-2 with (without) the treatment of cathepsin L inhibitor was estimated to be −69.9% (−62.9%) for Wu-1, −73.6% (−70.4%) for Delta, −65.4% (−60.3%) for Omicron BA.2, and −67.9% (−62.8%) for Omicron BA.4/5. From these results the contribution of endocytosis pathway for the infection of SARS-CoV-2 was estimated to be approximately 5–10%, and there made not so significant difference in neutralizing activity of Fv-antibodies. And, these trends were observed across the viral variants. The neutralizing activity of the Fv-antibodies against multiple SARS-CoV-2 variants including Wu-1, Delta and Omicrons indicated their potential for cross-protection against future viral variants. This indicates that the targeted epitopes on TMPRSS2 can be a promising candidate for the development of pan-corona therapeutics.
Data availability
The data used in this article can be available upon request to the corresponding author.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Research Foundation of Korea [RS-2023-00209053, 2020R1A5A101913111, RS-2024-00401422, and NRF-2022K1A3A1A25081295] and the Ministry of Health & Welfare, Republic of Korea [RS-2022-KH128357(HV22C0131)].
References
- S. H. Hristova and A. M. Zhivkov, Viruses, 2023, 15, 1752 CrossRef CAS.
- J. Lan, J. Ge, J. Yu, S. Shan, H. Zhou, S. Fan, Q. Zhang, X. Shi, Q. Wang, L. Zhang and X. Wang, Nature, 2020, 581, 215–220 CrossRef CAS.
- D. E. Oh, J. S. Kim, I. K. Hwang, H.-S. Park, C.-S. Lee and T. H. Kim, BioChip J., 2023, 17, 393–401 CrossRef CAS.
- B. Jawad, P. Adhikari, R. Podgornik and W.-Y. Ching, J. Chem. Inf. Model., 2021, 61, 4425–4441 CrossRef CAS PubMed.
- K. H. Kim, Y. K. Yoo, N. E. Lee, J. Lee, C. Kim, S. Lee, J. Kim, S. J. Park, D. Lee and S. W. Lee, BioChip J., 2023, 17, 340–348 CrossRef CAS.
- B. Liu, Y. Shi, W. Zhang, R. Li, Z. He, X. Yang, Y. Pan, X. Deng, M. Tan and L. Zhao, Cell. Mol. Immunol., 2020, 17, 1098–1100 CrossRef CAS PubMed.
- X. Qi, B. Ke, Q. Feng, D. Yang, Q. Lian, Z. Li, L. Lu, C. Ke, Z. Liu and G. Liao, Chem. Commun., 2020, 56, 8683–8686 RSC.
- J. Zahradník, S. Marciano, M. Shemesh, E. Zoler, D. Harari, J. Chiaravalli, B. Meyer, Y. Rudich, C. Li and I. Marton, Nat. Microbiol., 2021, 6, 1188–1198 CrossRef PubMed.
- J. Jung, J. S. Sung, J.-H. Bong, T.-H. Kim, S. Kwon, H. E. Bae, M.-J. Kang, J. Jose, M. Lee and H.-J. Shin, Biosens. Bioelectron., 2023, 245, 115834 CrossRef.
- J. Jung, J. S. Sung, T.-H. Kim, M.-J. Kang, J. Jose, H.-J. Shin and J.-C. Pyun, BioChip J., 2024, 18, 318–329 CrossRef CAS.
- E. Pérez-Then, C. Lucas, V. S. Monteiro, M. Miric, V. Brache, L. Cochon, C. B. Vogels, A. A. Malik, E. De la Cruz and A. Jorge, Nat. Med., 2022, 28, 481–485 CrossRef PubMed.
- V. Servellita, A. M. Syed, M. K. Morris, N. Brazer, P. Saldhi, M. Garcia-Knight, B. Sreekumar, M. M. Khalid, A. Ciling and P.-Y. Chen, Cell, 2022, 185(1539–1548), e1535 Search PubMed.
- D. Bestle, M. R. Heindl, H. Limburg, O. Pilgram, H. Moulton, D. A. Stein, K. Hardes, M. Eickmann, O. Dolnik and C. Rohde, Life Sci. Alliance, 2020, 3, e202000786 CrossRef.
- M. Z. Salleh and Z. Z. Deris, Life, 2022, 12, 231 CrossRef CAS PubMed.
- N. Saunders, I. Fernandez, C. Planchais, V. Michel, M. M. Rajah, E. Baquero Salazar, J. Postal, F. Porrot, F. Guivel-Benhassine and C. Blanc, Nature, 2023, 624, 207–214 CrossRef CAS.
- M. McCallum, Y.-J. Park, C. Stewart, K. R. Sprouse, A. Addetia, J. Brown, M. A. Tortorici, C. Gibson, E. Wong and M. Ieven, Cell, 2024, 187(4231–4245), e4213 Search PubMed.
- J. Shang, Y. Wan, C. Luo, G. Ye, Q. Geng, A. Auerbach and F. Li, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 11727–11734 CrossRef CAS PubMed.
- Y. K. Lee and H. S. Song, BioChip J., 2024, 18, 171–185 CrossRef CAS.
- M.-M. Zhao, W.-L. Yang, F.-Y. Yang, L. Zhang, W.-J. Huang, W. Hou, C.-F. Fan, R.-H. Jin, Y.-M. Feng and Y.-C. Wang, Signal Transduction Targeted Ther., 2021, 6, 134 CrossRef CAS.
- M. Hoffmann, H. Kleine-Weber, S. Schroeder, N. Krüger, T. Herrler, S. Erichsen, T. S. Schiergens, G. Herrler, N.-H. Wu and A. Nitsche, Cell, 2020, 181(271–280), e278 Search PubMed.
- B. J. Fraser, S. Beldar, A. Seitova, A. Hutchinson, D. Mannar, Y. Li, D. Kwon, R. Tan, R. P. Wilson and K. Leopold, Nat. Chem. Biol., 2022, 18, 963–971 CrossRef CAS PubMed.
- J. Lu, T. Panavas, K. Thys, J. Aerssens, M. Naso, J. Fisher, M. Rycyzyn and R. W. Sweet, Mol. Immunol., 2014, 57, 274–283 CrossRef CAS.
- S. D'Angelo, F. Ferrara, L. Naranjo, M. F. Erasmus, P. Hraber and A. R. Bradbury, Front. Immunol., 2018, 9, 395 CrossRef.
- J. Jose and T. F. Meyer, Microbiol. Mol. Biol. Rev., 2007, 71, 600–619 CrossRef CAS.
- J. Jung, J.-H. Bong, J. S. Sung, J.-H. Park, T.-H. Kim, S. Kwon, M.-J. Kang, J. Jose and J.-C. Pyun, Biosens. Bioelectron., 2023, 237, 115439 CrossRef CAS PubMed.
- J. S. Sung, Y. Han, T. G. Yun, J. Jung, T.-H. Kim, F. Piccinini, M.-J. Kang, J. Jose, M. Lee and J.-C. Pyun, Int. J. Biol. Macromol., 2024, 265, 130854 CrossRef CAS PubMed.
- J. S. Sung, J. Jung, H. Hwang, J.-H. Bong, M.-J. Kang, J. Jose, M. Lee and J.-C. Pyun, ACS Pharmacol. Transl. Sci., 2023, 6, 1945–1957 CrossRef CAS PubMed.
- J. S. Sung, S. Kim, J. Jung, T.-H. Kim, S. Kwon, H. E. Bae, M.-J. Kang, J. Jose, M. Lee and J.-C. Pyun, ACS Pharmacol. Transl. Sci., 2023, 7, 150–160 CrossRef PubMed.
- J. S. Sung, J.-H. Bong, S. J. Lee, J. Jung, M.-J. Kang, M. Lee, W.-B. Shim, J. Jose and J.-C. Pyun, Biosens. Bioelectron., 2022, 202, 113976 CrossRef CAS PubMed.
- T.-H. Kim, J.-Y. Park, J. Jung, J. S. Sung, S. Kwon, H. E. Bae, H.-J. Shin, M.-J. Kang, J. Jose and J.-C. Pyun, J. Mater. Chem. B, 2024, 12, 3751–3763 RSC.
- M. Hussain, N. Jabeen, A. Amanullah, A. A. Baig, B. Aziz, S. Shabbir, F. Raza and N. Uddin, AIMS Microbiol., 2020, 6, 350 CAS.
- J.-D. Pédelacq, S. Cabantous, T. Tran, T. C. Terwilliger and G. S. Waldo, Nat. Biotechnol., 2006, 24, 79–88 CrossRef.
- M. Lee, B. Kang, J. Lee, J. Lee, S. T. Jung, C. Y. Son and S. S. Oh, Sci. Adv., 2022, 8, eabq6207 CrossRef CAS PubMed.
- J. Jung, J. S. Sung, S. Kwon, H. E. Bae, M.-J. Kang, J. Jose, M. Lee and J.-C. Pyun, RSC Med. Chem., 2024, 15, 3704–3710 RSC.
- J. Jung, S. Kwon, J. S. Sung, H. E. Bae, M.-J. Kang, J. Jose, M. Lee and J.-C. Pyun, ACS Pharmacol. Transl. Sci., 2024, 7, 3914–3920 CrossRef CAS PubMed.
- J. S. Sung, J. Jung, T.-H. Kim, S. Kwon, H. E. Bae, M.-J. Kang, J. Jose, M. Lee and J.-C. Pyun, Bioconjugate Chem., 2024, 35, 1324–1334 CrossRef CAS PubMed.
- J. Jung, J.-H. Bong, J. S. Sung, S. J. Lee, M. Lee, M.-J. Kang, J. Jose and J.-C. Pyun, Bioconjugate Chem., 2021, 32, 2213–2223 CrossRef CAS PubMed.
- J. Jung, J.-H. Bong, S. J. Lee, M.-J. Kim, J. S. Sung, M. Lee, M.-J. Kang, J. Song, J. Jose and J.-C. Pyun, ACS Appl. Bio Mater., 2021, 4, 3388–3397 CrossRef CAS PubMed.
- J.-H. Bong, S. J. Lee, J. Jung, J. S. Sung, M.-J. Kang, M. Lee, J. Jose and J.-C. Pyun, BioChip J., 2024, 18, 146–159 CrossRef CAS.
- J. L. Xu and M. M. Davis, Immunity, 2000, 13, 37–45 CrossRef CAS PubMed.
- G. Yoo, T. Saenger, J.-H. Bong, J. Jose, M.-J. Kang and J.-C. Pyun, Biochim. Biophys. Acta, 1848, 2015, 3126–3133 Search PubMed.
- M. Liu, B. Wang, F. Wang, Z. Yang, D. Gao, C. Zhang, L. Ma and X. Yu, Appl. Microbiol. Biotechnol., 2019, 103, 6071–6079 CrossRef CAS PubMed.
- D. Wrapp, N. Wang, K. S. Corbett, J. A. Goldsmith, C.-L. Hsieh, O. Abiona, B. S. Graham and J. S. McLellan, Science, 2020, 367, 1260–1263 CrossRef CAS PubMed.
- A. C. Walls, Y.-J. Park, M. A. Tortorici, A. Wall, A. T. McGuire and D. Veesler, Cell, 2020, 181(281–292), e286 Search PubMed.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS PubMed.
- S. Kumar, D. Bose, H. Suryawanshi, H. Sabharwal, K. Mapa and S. Maiti, PLoS One, 2011, 6, e23300 CrossRef CAS PubMed.
- I. Hyseni, E. Molesti, L. Benincasa, P. Piu, E. Casa, N. J. Temperton, A. Manenti and E. Montomoli, Viruses, 2020, 12, 1011 CrossRef PubMed.
- J. Nie, Q. Li, J. Wu, C. Zhao, H. Hao, H. Liu, L. Zhang, L. Nie, H. Qin and M. Wang, Emerging Microbes Infect., 2020, 9, 680–686 CrossRef CAS PubMed.
- M. Chen and X.-E. Zhang, Int. J. Biol. Sci., 2021, 17, 1574 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Min-Jung
Kang
a,
Min-Jung
Kang