
Structure-guided design of a truncated heterobivalent chemical probe degrader of IRE1α†
Breanna L.
Zerfas
*ab,
Yingpeng
Liu
ab,
Jianwei
Che
ab,
Katherine A.
Donovan
bc,
John M.
Hatcher
bc,
Fidel
Huerta
a,
Rebecca J.
Metivier
c,
Radosław P.
Nowak
ab,
Leah
Ragosta
a,
Tiffany
Tsang
a,
Eric S.
Fischer
 abc and
Lyn H.
Jones
*ab
abc and
Lyn H.
Jones
*ab
aCenter for Protein Degradation, Dana-Farber Cancer Institute, Boston, MA, USA. E-mail: breannal_zerfas@dfci.harvard.edu; lyn_jones@dfci.harvard.edu
bDepartment of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA, USA
cDepartment of Cancer Biology, Dana-Farber Cancer Institute, Boston, MA, USA
First published on 18th March 2025
Abstract
IRE1α is an ER protein involved in the unfolded protein response (UPR) and dysregulation of the ER stress pathway has been implicated in several diseases. Inhibitors of the cytoplasmic endonuclease or kinase domains of the enzyme have limited utility and targeted degradation would address additional scaffolding functions of the protein. Here, we describe the design and development of IRE1α proteolysis targeting chimeras (PROTACs) based on a lysine-reactive salicylaldehyde RNase inhibitor, and present the structure–activity relationships (SARs) that delivered the first highly selective degraders of a native ER-membrane associated protein. Medicinal chemistry optimization exploited ternary complex computational modelling to inform design, HiBiT-SpyTag IRE1α degradation and NanoBRET cereblon occupancy cell-based assays to generate SARs, and mass spectrometry-based proteomics to assess broad selectivity in an unbiased manner. Merging IRE1α and CRBN ligand chemotypes provided the truncated chimera CPD-2828 with physicochemical properties more akin to an oral molecular glue degrader than a traditional PROTAC.
Introduction
Inositol requiring enzyme alpha (IRE1α) is an ER-resident transmembrane protein that possesses an N-terminal domain within the ER lumen, and C-terminal cytosolic kinase and RNase domains. During ER stress, misfolded proteins accumulate in the ER which triggers IRE1α dimerization, causing autophosphorylation and activation of the RNase domain that cleaves X-box binding protein 1 (XBP1) mRNA, facilitating its subsequent splicing to yield XBP1s.1 The IRE1α interactome2 also regulates signaling through catalytic-independent mechanisms.3 The IRE1α-XBP1s axis of the unfolded protein response (UPR) is linked to tumor progression and immunosuppressive effects in a number of cancers and inhibitors of the kinase and RNase domains have been developed due to their therapeutic potential.4 However, potent and selective inhibitors of the ATP-site have been challenging to develop,5–7 which has been complicated by their potential to induce IRE1α oligomerization, leading to allosteric activation of the RNase domain. Additionally, the druggability of the RNase site is low due to its shallow and polar nature. A high-throughput screen identified reactive salicylaldehyde inhibitors that covalently engaged Lys907 in the binding site through the formation of a Schiff base (Fig. 1).8 MKC8866 was the most advanced compound from this family of inhibitors and had been in clinical trials for the treatment of solid tumors.9,10 Despite their covalent mode of action, these compounds possess moderate potency in cells due to difficulties in optimizing equilibrium binding interactions within the RNase pocket.11 | ||
| Fig. 1 Chemical structures of IRE1α RNase covalent inhibitors and cereblon molecular glue degraders. | ||
Targeted protein degradation (TPD) has emerged as an exciting new small molecule therapeutic modality that pharmacologically depletes proteins-of-interest (POIs).12,13 TPD often delivers enhanced potency and selectivity over traditional inhibitors. Molecular glue degraders such as the immunomodulatory imide drugs (IMiDs) thalidomide, lenalidomide and pomalidomide (Fig. 1) bind cereblon (CRBN), an adaptor for the CRL4CRBN E3 ubiquitin ligase complex, and remodel its surface to induce proximity with neosubstrates, triggering their polyubiquitination and subsequent proteasomal degradation.14,15 IMiDs have been incorporated into heterobifunctional proteolysis targeting chimeras (PROTACs)16 that also induce proximity between a POI and CRL4CRBN thus mediating TPD of the target.17,18 We reasoned that optimization of a PROTAC-mediated CRBN/IRE1α complex may address the low druggability of the RNase pocket and deliver modulators with improved efficacy over traditional inhibitors. Here we describe the use of computational modelling, engineered cell-based assays and mass spectrometry (MS) proteomics to design and develop the first degraders of IRE1α.19
Results and discussion
In order to develop PROTAC degraders we considered both kinase and RNase site inhibitors as POI ligands for IRE1α, and thalidomide derivatives for the CUL4-RBX1-DDB1-CRBN (CRL4CRBN) complex. To understand the likelihood of success for PROTACs based on either RNase or kinase domain ligands we performed computational modelling of the protein complex using Rosetta (version 3.9)20 employing the existing crystal structures of CRBN (PDB 4TZ4) with either RNase (PDB 4PL3) or kinase inhibitors (PDB 6URC) (Fig. 2). The model showed that the distance between the IMiD and RNase ligands was shorter than that for the kinase ligands, suggesting that design of a cell-permeable bivalent compound might be facilitated when employing an RNase ligand for the POI. | ||
| Fig. 2 Computational modelling of the IRE1α/CRBN complexes using kinase or RNase domains. The CRBN-lenalidomide crystal structure (PDB 4TZ4) was modelled using Rosetta 3.9 with a) IRE1α-Kira8 (PDB 6URC) or b) IRE1α-MKC9989 (PDB 4PL3) structures. The RNase ligand MKC9989 was modified to a reversible binding mimic that hydrogen bonds to Lys907 to enable Rosetta modeling. Distances shown are in angstroms. | ||
Moreover, computational modelling using multiple crystal structures of the IRE1α kinase domain showed extensive flexibility at the potential interface with CRBN, implying potential challenges in forming a stable ternary complex. We performed molecular dynamics (MD) simulations for representative complexes with respective ligands (without linkers) to investigate their stability. In the kinase/CRBN complex, due to the kinase domain flexibility, we observed a significant change in the distance between the ligands (extending to over 20 Å). Conversely, the RNase/CRBN complex was significantly more stable and the distance between the ligands remained relatively short.
Therefore, we focussed our design strategy on chemically linking RNase site inhibitors to CRBN modulators to create heterobifunctional degraders. To assess binding of new molecules to the RNase site, an activity-based biochemical screen was developed based on a previous method,11 which monitored the cleavage of a single hairpin RNA substrate that was labeled with AlexaFluor-647 at the 5′ end and a corresponding black hole quencher at the 3′ end, and measuring the change in fluorescence. Our previously reported NanoBRET assay was used to measure CRBN cell-based occupancy.21 IRE1α degradation in cells utilized a HiBiT-SpyTag fusion system, a chemical biology technology that we had developed previously to establish the feasibility of pharmacologically degrading the ER protein using the chemical probe dTAG13.19 This system allowed us to transiently label IRE1α with SpyCatcher-dTAG, to prevent any potential interference from a large protein tag when testing our targeted PROTACs.
Based on our computational model, it was apparent that the 3-position of the coumarin ring would provide a suitable attachment point for the PROTAC linker (Fig. 3). 4μ8C is a structurally simpler covalent inhibitor than MKC8866 and MKC9989 that lacks the 6-methoxy substituent (Fig. 1) and we noticed in the patent literature a related IRE1α inhibitor (Cpd II, Fig. 1) possessed a benzamide functionality that may facilitate the preparation of heterobifunctional molecules.22 Therefore, to aid chemical synthesis, the synthetic precursor acid 1 (Fig. 3) was used as the RNase binding element for the attachment of linkers that enabled conjugation to pomalidomide as the CRBN binding motif using simple amidation chemistry (details provided in the ESI†). Polar and apolar flexible linkers of various lengths were probed, as well as tethers with increased rigidity that may reduce entropic penalties upon binding (compounds 2–8). The piperazine-linked thalidomide derivative 8 was prepared based on the successful use of this substructure in PROTACs previously.23–26 These derivatives retained CRBN binding activity in cells, but weak or negligible RNase binding resulted in little or no IRE1α degradation (Fig. 3). To probe the linker exit vector from the IRE1α pocket we prepared the meta-isomers 10–16 from the precursor 9, but these derivatives also lacked significant degradation activity (Fig. 3).
 | ||
| Fig. 3 Biological profiles of IRE1α PROTACs 2–8, 10–16 and 18–24. Potency values are shown for CRBN cell-based occupancy (NanoBRET), inhibition of RNase activity and IRE1α HiBiT Dmax (maximum percentage degraded, 24 h treatment, maximum concentration 30 μM). HiBiT dose response curves are shown in Fig. S1 (ESI†). | ||
We reasoned that a design closer in structure to MKC8866, which provides a methylene tether to the amide linking function, may enhance IRE1α binding and allow for productive ternary complex formation without incorporating excessive flexibility. Pleasingly, several PROTACs prepared from precursor 17 possessed RNase inhibitory activity, and the piperazine amides (cf. Cpd II) were the most potent (23 and 24, Fig. 3). Compound 23 (CPD-2522) retained slightly higher RNase and CRBN occupancy and degraded IRE1α most potently in this set. Structure–activity relationships (SARs) were also explored by making small modifications to the structure (Fig. 4, 25–27), but these compounds were significantly less potent than CPD-2522, although extending the tether from the coumarin by a single –CH2– unit yielded 28 with similar IRE1α degradation activity. Surprisingly, when the methoxy substituent present in MKC8866 was incorporated into CPD-2522 to yield 29, the compound was a weaker degrader of IRE1α which may be due to lower RNase potency (Fig. 4). We suspected that the apparent difficulty in improving the degradation activity of 23 (CPD-2522) may be due to suboptimal ternary complex formation. We, and others, have described the significant impact CRBN ligand choice has on PROTAC degradation activity,27–31 and so we decided to explore a differentiated non-IMiD binding moiety to potentially enhance the induced proximity of IRE1α and CRBN. We turned to the N-aryl glutarimide motif that has been used extensively in the design of CRBN-based PROTACs,32 the substructure being reported in 159 patent applications at the time of writing (CAS SciFinder). Although few literature publications have described N-aryl glutarimide containing PROTACs,26,33,34 a recent report exemplified its use in the development of a potent AR degrader.35 The direct attachment of acid 17 (Fig. 3) to a piperidine substituted N-aryl glutarimide (the latter also used in 38 PROTAC patent applications), yielded design 30 (CPD-2828) where the piperidine amide satisfies both RNase and CRBN binding SARs (Fig. 5a and b and ESI†).19 Importantly, the short rigid piperidine ‘linker’ between CRBN and RNase pharmacophores allowed for the formation of a stable ternary complex following 200 ns MD simulations. During the simulation, we observed relatively large movements in the distal N-terminus of IRE1α while the C-terminus CRBN contact interface was stable. This was expected because the N-terminal kinase domain has no significant constraint given its distal location to the CRBN contacts and may even be beneficial to allow optimal engagement with the E2 enzyme for ubiquitin transfer. Even though 30 possessed similar CRBN and RNase binding affinity to 23 (CPD-2522), the PROTAC more effectively degraded IRE1α in cells. Increasing the linker length (to yield 31 and 32, Fig. 5c) was detrimental to degradation, as expected based on the computational model.
 | ||
| Fig. 4 Biological profiles of IRE1α PROTACs 25–29. Potency values are shown for CRBN cell-based occupancy (NanoBRET), inhibition of RNase activity and IRE1α HiBiT Dmax (maximum percentage degraded, 24 h treatment, maximum concentration 30 μM). | ||
 | ||
| Fig. 5 a) Computational model of the CRBN/IRE1α/CPD-2828 ternary complex. b) Structure and properties of the optimized IRE1α degrader CPD-2828 (CRBN and IRE1α binding regions of the molecule are highlighted). c) Structures and biological profiles of the PROTACs 30–33 and the negative control 34 (CPD-3121). | ||
Incorporation of the 6-methoxy substituent on the coumarin ring of 30 (cf. MKC8866), to give compound 33, reduced cellular degradation activity and CRBN occupancy, while the biochemical CRBN TR-FRET and RNase potencies were similar to 30 (Fig. 5c and S2†). These data suggest that PROTAC 33 possesses lower unbound levels in cells, which may be due to lower cell membrane permeability or higher non-specific binding.
Due to its significant IRE1α activity in the HiBiT line, 30 (CPD-2828) was profiled more broadly. 30 was shown to degrade endogenous IRE1α protein in HEK293T cells more potently than 23, consistent with the HiBiT data (Fig. 6a). In fact, 30 appeared to degrade more endogenous protein in HEK293T than the engineered HiBiT IRE1α protein at 10 μM and 24 hours treatment (Fig. 6a and S3†). 30 also degraded IRE1α more rapidly than 23, likely reflecting an improved rate of ternary complex formation, and possessed a hook effect only at the shorter timepoint of 6 hours incubation (Fig. S3†). The degradation of IRE1α by 30 was unaffected by the induction of ER stress using thapsigargin (Fig. S4†). From a drug development perspective, 30 had respectable in vitro metabolic stability in human liver microsomes (HLM) and hepatocytes (HHeps) and good solubility in biorelevant media, i.e. fed and fasted state simulated intestinal fluids (FeSSIF and FaSSIF, Fig. 5b).
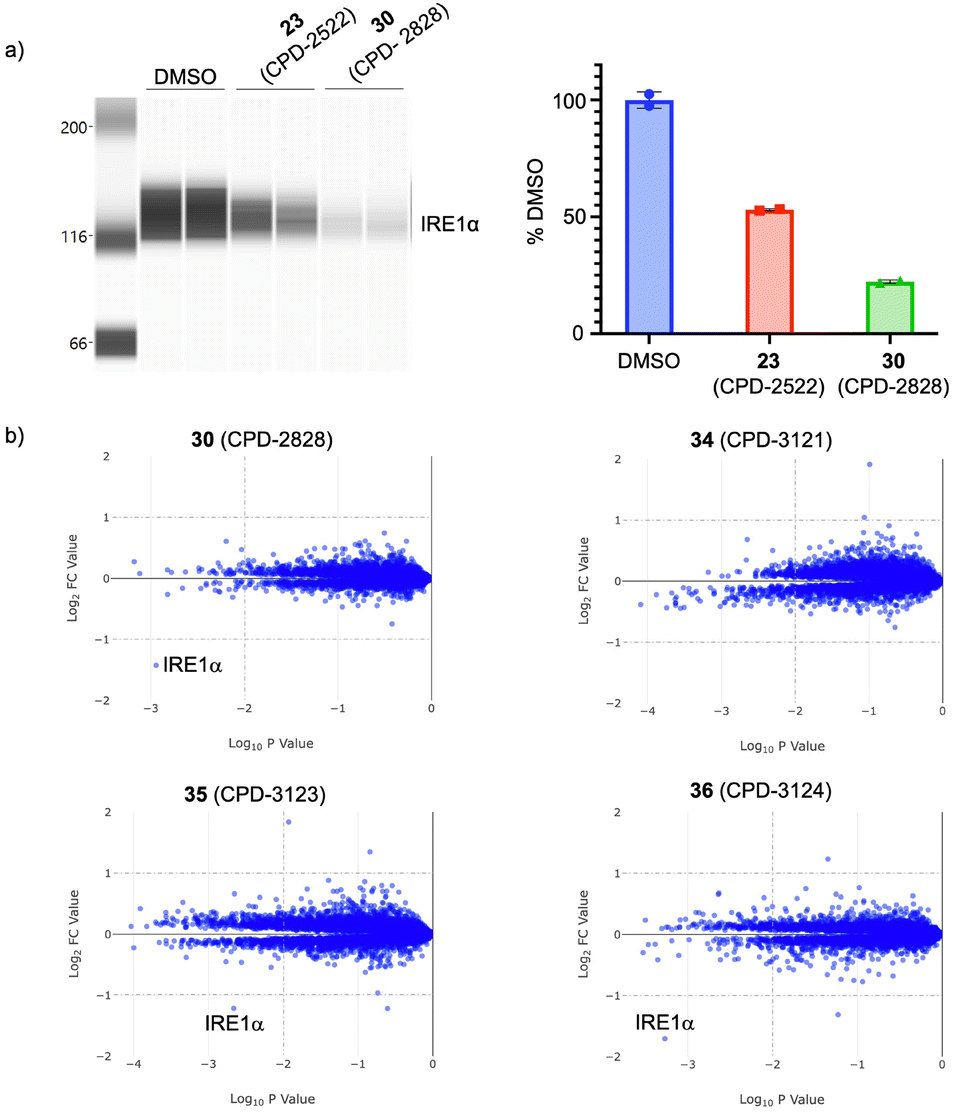 | ||
| Fig. 6 a) Effects of PROTAC degraders 23 (CPD-2522) and 30 (CPD-2828) on endogenous IRE1α levels in HEK293T cells (10 μM, 24 hours), analyzed using automated capillary electrophoresis (data were quantified by determining the ratio of the integration of the peaks for vinculin and IRE1α, and a percentage protein reduction calculated relative to DMSO). b) Quantitative MS proteomics analysis of the effects of 30, its separate enantiomers (35, CPD-3123 and 36, CPD-3124), and the negative control compound 34 (CPD-3121), on protein levels in cells (log2-fold change over DMSO; HEK293T, 10 μM, 5 hours). | ||
N-Methylation of glutarimide degraders has been shown to establish a steric clash within the tri-tryptophan cage of the thalidomide binding domain, and such compounds are useful negative controls in cell-based assays because they retain similar physicochemistry to the parent PROTAC but are unable to bind CRBN.17,36 The N-methyl derivative of 30 (CPD-3121, 34, Fig. 5) as expected, lacked any IRE1α degradation activity, thus confirming CRBN-mediated degradation.
Compound 30 is a racemate, and so the enantiomers 35 (CPD-3123) and 36 (CPD-3124) were prepared and screened separately (stereochemistry arbitrarily assigned, see ESI†). 35 and 36 possessed similar RNase inhibition (IC50 17 ± 2.4, 19.1 ± 0.9 μM) and CRBN occupancy (IC50 1.2 ± 0.15, 1.2 ± 0.08 μM) as expected,35 translating to comparable levels of IRE1α degradation activity (DC50 0.47 ± 0.08 μM, 0.40 ± 0.08 μM; Dmax 57.9 ± 1.4%, 65.3 ± 3.1%). Both enantiomers possessed excellent degradation selectivity, assessed using a MS proteomics platform we have previously described (Fig. 6b).19,3730 and its separate enantiomers do not degrade the canonical CRBN neosubstrates likely due to the lack of the hydrogen bonding acceptor carbonyl group present within the IMiD scaffold that interacts with the G-loop degron via the CRBN sensor loop Asn351 residue.38–40 As predicted, the negative control 34 (CPD-3121) was completely clean in the proteomics assay (Fig. 6b).
Conclusions
Degradation of IRE1α will inhibit both catalytic and non-catalytic functions of this key protein involved in the UPR, and selective chemical probes are needed to explore the therapeutic potential of perturbing the ER stress pathway. PROTACs allow for the modular design of targeted degraders, which is structurally-enabled through ternary complex computational modelling, as illustrated in this work and other research from our laboratory.27 The merging of the IRE1α and CRBN ligand chemotypes enabled delivery of the truncated heterobifunctional degrader 30 (CPD-2828) and its enantiomers with physicochemical properties closer to a molecular glue than a traditional PROTAC (30: MW 531, 6 rotatable bonds, 6 hydrogen bond acceptors, 3 hydrogen bond donors, clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P 3.7).41 For example, golcadomide (MW 535, 7 rotatable bonds, 6 H-bond acceptors, 3 H-bond donors, clogP 1.4) and mezigdomide (MW 567, 7 rotatable bonds, 5 H-bond acceptors, 2 H-bond donors, clogP 3.0), IKZF1/3 molecular glue degraders in clinical trials, have similar physicochemistry.42 This approach mirrors our previous studies integrating β-2 agonist and M3 muscarinic antagonist pharmacophores to create dual pharmacology bronchodilators with reduced flexibility.43,44 Compound 30 thus reflects the physicochemical continuum that can exist between formal heterobifunctional degraders and molecular glues that possess the same induced-proximity mode-of-action.45 These results suggest that the development of highly truncated or ‘linkerless’ PROTACs are feasible in certain instances. Our work challenges the dogma of categorizing degrader modalities based on their physicochemical properties. Our approach also exemplifies the utility of structure-guided design and E3 ligase scaffold hopping as viable strategies to advance selective degraders with oral drug-like properties for challenging targets such as IRE1α.
P 3.7).41 For example, golcadomide (MW 535, 7 rotatable bonds, 6 H-bond acceptors, 3 H-bond donors, clogP 1.4) and mezigdomide (MW 567, 7 rotatable bonds, 5 H-bond acceptors, 2 H-bond donors, clogP 3.0), IKZF1/3 molecular glue degraders in clinical trials, have similar physicochemistry.42 This approach mirrors our previous studies integrating β-2 agonist and M3 muscarinic antagonist pharmacophores to create dual pharmacology bronchodilators with reduced flexibility.43,44 Compound 30 thus reflects the physicochemical continuum that can exist between formal heterobifunctional degraders and molecular glues that possess the same induced-proximity mode-of-action.45 These results suggest that the development of highly truncated or ‘linkerless’ PROTACs are feasible in certain instances. Our work challenges the dogma of categorizing degrader modalities based on their physicochemical properties. Our approach also exemplifies the utility of structure-guided design and E3 ligase scaffold hopping as viable strategies to advance selective degraders with oral drug-like properties for challenging targets such as IRE1α.
The addition of a 6-methoxy substituent to the coumarin scaffold, which is present in the IRE1α inhibitor MKC8866 that was advanced to clinical trials, surprisingly reduced CRBN occupancy and decreased degradation potency, which we attribute to lower unbound levels in cells caused by an increase in lipophilicity and non-specific binding or possibly lower permeability. Our results demonstrate the importance of developing cellular and biochemical assays to help interpret SARs, which is especially important for PROTACs.46
These medicinal chemistry studies are preliminary and further work is needed to optimize degradation potency and efficacy. Clearly, lysine covalent engagement by the salicylaldehyde warhead would be expected to reduce catalytic efficiency of the resulting PROTAC.47 This may explain the initial challenges in developing a highly potent degrader, although further optimization of linker chemistry or scaffold hopping to alternative CRBN binding motifs could further enhance ternary complex formation and, as a consequence, IRE1α degradation efficacy. The recruitment of alternative E3 ubiquitin ligases, particularly those localized to the ER,3,48 may also improve degrader potency. Nevertheless, our results illustrate the remarkable substrate versatility of CRBN-based PROTACs and expands the nature of addressable targets using the modality.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
B. L. Z., F. H., R. P. N, L. R. and T. T. developed the assays and screened compounds; K. A. D., R. J. M. and E. S. F. developed the proteomics profiling methods and screened compounds; Y. L., J. C., J. M. H. and L. H. J. designed the compounds; J. C. performed the computational modelling; L. H. J. conceived and directed the project; L. H. J. and B. L. Z. wrote the manuscript with contributions from all authors.Conflicts of interest
L. H. J. is an advisor to ONO Pharma, Merck KGaA, Rapafusyn Pharmaceuticals, Matchpoint Therapeutics, Belharra Therapeutics, and Third Rock Ventures, and holds equity in Hyku Biosciences and Rapafusyn Pharmaceuticals. The Center for Protein Degradation at DFCI receives research funding from Deerfield. E. S. F. is an advisor and equity holder of Civetta Therapeutics, Proximity Therapeutics, Ajax Therapeutics, Neomorph Inc (also board of directors), Avilar Therapeutics, Stelexis Biosciences and Photys Therapeutics, equity holder in Lighthorse Therapeutics and a consultant to Astellas, Sanofi, Novartis, Deerfield, and EcoR1 capital. The Fischer laboratory receives or has received research funding from Novartis, Deerfield, Ajax, Interline, and Astellas. J. C. is a cofounder of Matchpoint therapeutics and consultant for Soltego, Allorion and Matchpoint Therapeutics, and holds equity in Soltego, Allorion, Matchpoint and M3 Bioinformatics & Technology Inc. K. A. D. is a consultant for Kronos Bio and Neomorph Inc.Acknowledgements
We thank J. Cubillos-Ruiz, L. H. Glimcher, and members of the CPD past and present, for useful discussions. We also thank Wuxi for compound synthesis, and for determining metabolic stability, solubility, membrane permeability, and CRBN occupancy of the compounds. We recognize Deerfield for funding this work.References
- L. H. Glimcher, A. H. Lee and N. N. Iwakoshi, Nat. Immunol., 2020, 21, 963–965 CrossRef CAS.
- C. Hetz and L. H. Glimcher, Mol. Cell, 2009, 35, 551–561 CrossRef CAS PubMed.
- S. Le Goupil, H. Laprade, M. Aubry and E. Chevet, J. Biol. Chem., 2024, 300, 107169 CrossRef CAS PubMed.
- J. R. Cubillos-Ruiz, S. E. Bettigole and L. H. Glimcher, Cell, 2017, 168, 692–706 CrossRef CAS PubMed.
- D. Korovesis, N. Rufo, R. Derua, P. Agostinis and S. H. L. Verhelst, ACS Chem. Biol., 2020, 15, 3106–3111 CrossRef CAS PubMed.
- E. Ferri, A. Le Thomas, H. A. Wallweber, E. S. Day, B. T. Walters, S. E. Kaufman, M. G. Braun, K. R. Clark, M. H. Beresini, K. Mortara, Y. A. Chen, B. Canter, W. Phung, P. S. Liu, A. Lammens, A. Ashkenazi, J. Rudolph and W. Wang, Nat. Commun., 2020, 11, 6387 CrossRef CAS PubMed.
- H. C. Feldman, R. Ghosh, V. C. Auyeung, J. L. Mueller, J. H. Kim, Z. E. Potter, V. N. Vidadala, B. G. K. Perera, A. Olivier, B. J. Backes, J. Zikherman, F. R. Papa and D. J. Maly, Nat. Chem. Biol., 2021, 17, 1148–1156 CrossRef CAS PubMed.
- K. Volkmann, J. L. Lucas, D. Vuga, X. Wang, D. Brumm, C. Stiles, D. Kriebel, A. Der-Sarkissian, K. Krishnan, C. Schweitzer, Z. Liu, U. M. Malyankar, D. Chiovitti, M. Canny, D. Durocher, F. Sicheri and J. B. Patterson, J. Biol. Chem., 2011, 286, 12743–12755 CrossRef CAS PubMed.
- S. E. Logue, E. P. McGrath, P. Cleary, S. Greene, K. Mnich, A. Almanza, E. Chevet, R. M. Dwyer, A. Oommen, P. Legembre, F. Godey, E. C. Madden, B. Leuzzi, J. Obacz, Q. Zeng, J. B. Patterson, R. Jäger, A. M. Gorman and A. Samali, Nat. Commun., 2018, 9, 3267 CrossRef PubMed.
- X. Sheng, H. Z. Nenseth, S. Qu, O. F. Kuzu, T. Frahnow, L. Simon, S. Greene, Q. Zeng, L. Fazli, P. S. Rennie, I. G. Mills, H. Danielsen, F. Theis, J. B. Patterson, Y. Jin and F. Saatcioglu, Nat. Commun., 2019, 10, 323 CrossRef CAS PubMed.
- M. Sanches, N. M. Duffy, M. Talukdar, N. Thevakumaran, D. Chiovitti, M. D. Canny, K. Lee, I. Kurinov, D. Uehling, R. Al-awar, G. Poda, M. Prakesch, B. Wilson, V. Tam, C. Schweitzer, A. Toro, J. L. Lucas, D. Vuga, L. Lehmann, D. Durocher, Q. Zeng, J. B. Patterson and F. Sicheri, Nat. Commun., 2014, 5, 4202 CrossRef CAS PubMed.
- P. P. Chamberlain and L. G. Hamann, Nat. Chem. Biol., 2019, 15, 937–944 CrossRef CAS PubMed.
- N. R. Kong and L. H. Jones, Clin. Pharmacol. Ther., 2023, 114, 558–568 CrossRef CAS PubMed.
- J. Krönke, N. D. Udeshi, A. Narla, P. Grauman, S. N. Hurst, M. McConkey, T. Svinkina, D. Heckl, E. Comer, X. Li, C. Ciarlo, E. Hartman, N. Munshi, M. Schenone, S. L. Schreiber, S. A. Carr and B. L. Ebert, Science, 2014, 343, 301–305 CrossRef PubMed.
- Q. L. Sievers, G. Petzold, R. D. Bunker, A. Renneville, M. Słabicki, B. J. Liddicoat, W. Abdulrahman, T. Mikkelsen, B. L. Ebert and N. H. Thomä, Science, 2018, 362, eaat0572 CrossRef PubMed.
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef CAS PubMed.
- J. Lu, Y. Qian, M. Altieri, H. Dong, J. Wang, K. Raina, J. Hines, J. D. Winkler, A. P. Crew, K. Coleman and C. M. Crews, Chem. Biol., 2015, 22, 755–763 CrossRef CAS PubMed.
- G. E. Winter, D. L. Buckley, J. Paulk, J. M. Roberts, A. Souza, S. Dhe-Paganon and J. E. Bradner, Science, 2015, 348, 1376–1381 CrossRef CAS PubMed.
- T. Tsang, F. Huerta, Y. Liu, J. Che, R. J. Metivier, S. Ferrao, K. A. Donovan, L. H. Jones, B. L. Zerfas and R. P. Nowak, ACS Chem. Biol., 2023, 18, 933–941 CrossRef CAS PubMed.
- R. P. Nowak, S. L. DeAngelo, D. Buckley, Z. He, K. A. Donovan, J. An, N. Safaee, M. P. Jedrychowski, C. M. Ponthier, M. Ishoey, T. Zhang, J. D. Mancias, N. S. Gray, J. E. Bradner and E. S. Fischer, Nat. Chem. Biol., 2018, 14, 706–714 CrossRef CAS PubMed.
- B. L. Zerfas, F. Huerta, H. Liu, G. Du, N. S. Gray, L. H. Jones and R. P. Nowak, Methods Enzymol., 2023, 681, 169–188 CAS.
- Q. Zeng, A. Toro, J. B. Patterson, W. S. Wade, Z. Zubovics, Y. Yang and Z. Wu, WO2011127070, 2011.
- X. Han, L. Zhao, W. Xiang, C. Qin, B. Miao, D. McEachern, Y. Wang, H. Metwally, L. Wang, A. Matvekas, B. Wen, D. Sun and S. Wang, J. Med. Chem., 2021, 64, 12831–12854 CrossRef CAS PubMed.
- Y. Sun, Z. Yang, Z. Zhang, Z. Li, L. Guo, H. Pan, X. Luo, D. Liu and Y. Rao, RSC Med. Chem., 2023, 14, 1562–1566 RSC.
- Z. Chen, M. Wang, D. Wu, L. Zhao, H. Metwally, W. Jiang, Y. Wang, L. Bai, D. McEachern, J. Luo, Q. Li, A. Matvekas, B. Wen, D. Sun, A. M. Chinnaiyan and S. Wang, J. Med. Chem., 2024, 67, 5351–5372 CrossRef CAS PubMed.
- M. N. O'Brien Laramy, S. Luthra, M. F. Brown and D. W. Bartlett, Nat. Rev. Drug Discovery, 2023, 22, 410–427 CrossRef PubMed.
- Y. Gao, B. Jiang, H. Kim, M. J. Berberich, J. Che, K. A. Donovan, J. M. Hatcher, F. Huerta, N. P. Kwiatkowski, Y. Liu, P. P. Liuni, R. J. Metivier, V. K. Murali, R. P. Nowak, T. Zhang, E. S. Fischer, N. S. Gray and L. H. Jones, J. Med. Chem., 2023, 66, 5524–5535 CrossRef CAS PubMed.
- D. Remillard, D. L. Buckley, J. Paulk, G. L. Brien, M. Sonnett, H. S. Seo, S. Dastjerdi, M. Wühr, S. Dhe-Paganon, S. A. Armstrong and J. E. Bradner, Angew. Chem., Int. Ed., 2017, 56, 5738–5743 CrossRef CAS PubMed.
- M. de Wispelaere, G. Du, K. A. Donovan, T. Zhang, N. A. Eleuteri, J. C. Yuan, J. Kalabathula, R. P. Nowak, E. S. Fischer, N. S. Gray and P. L. Yang, Nat. Commun., 2019, 10, 3468 CrossRef PubMed.
- X. Nie, Y. Zhao, H. Tang, Z. Zhang, J. Liao, C. M. Almodovar-Rivera, R. Sundaresan, H. Xie, L. Guo, B. Wang, H. Guan, Y. Xing and W. Tang, ChemBioChem, 2024, 25, e202300685 CrossRef CAS PubMed.
- J. Hu, J. Jarusiewicz, G. Du, G. Nishiguchi, S. Yoshimura, J. C. Panetta, Z. Li, J. Min, L. Yang, D. Chepyala, M. Actis, N. Reyes, B. Smart, C. H. Pui, D. T. Teachey, Z. Rankovic and J. J. Yang, Sci. Transl. Med., 2022, 14, eabo5228 CrossRef CAS PubMed.
- A. Kazantsev and M. Krasavin, Expert Opin. Ther. Pat., 2022, 32, 171–190 CrossRef CAS PubMed.
- O. H. Rathje, L. Perryman, R. J. Payne and D. W. Hamprecht, J. Med. Chem., 2023, 66, 11216–11236 CrossRef CAS PubMed.
- A. Murgai, I. Sosič, M. Gobec, P. Lemnitzer, M. Proj, S. Wittenburg, R. Voget, M. Gütschow, J. Krönke and C. Steinebach, Chem. Commun., 2022, 58, 8858–8861 RSC.
- S. Norris, X. Ba, J. Rhodes, D. Huang, G. Khambatta, J. Buenviaje, S. Nayak, J. Meiring, S. Reiss, S. Xu, L. Shi, B. Whitefield, M. Alexander, E. J. Horn, M. Correa, L. Tehrani, J. D. Hansen, P. Papa and D. S. Mortensen, J. Med. Chem., 2023, 66, 16388–16409 CrossRef CAS PubMed.
- I. V. Hartung, J. Rudolph, M. M. Mader, M. P. C. Mulder and P. Workman, J. Med. Chem., 2023, 66, 9297–9312 CrossRef CAS PubMed.
- K. A. Donovan, F. M. Ferguson, J. W. Bushman, N. A. Eleuteri, D. Bhunia, S. Ryu, L. Tan, K. Shi, H. Yue, X. Liu, D. Dobrovolsky, B. Jiang, J. Wang, M. Hao, I. You, M. Teng, Y. Liang, J. Hatcher, Z. Li, T. D. Manz, B. Groendyke, W. Hu, Y. Nam, S. Sengupta, H. Cho, I. Shin, M. P. Agius, I. M. Ghobrial, M. W. Ma, J. Che, S. J. Buhrlage, T. Sim, N. S. Gray and E. S. Fischer, Cell, 2020, 183, 1714–1731 CrossRef CAS PubMed , e1710.
- M. E. Matyskiela, T. Clayton, X. Zheng, C. Mayne, E. Tran, A. Carpenter, B. Pagarigan, J. McDonald, M. Rolfe, L. G. Hamann, G. Lu and P. P. Chamberlain, Nat. Struct. Mol. Biol., 2020, 27, 319–322 CrossRef CAS PubMed.
- C. Steinebach, A. Bricelj, A. Murgai, I. Sosič, L. Bischof, Y. L. D. Ng, C. Heim, S. Maiwald, M. Proj, R. Voget, F. Feller, J. Košmrlj, V. Sapozhnikova, A. Schmidt, M. R. Zuleeg, P. Lemnitzer, P. Mertins, F. K. Hansen, M. Gütschow, J. Krönke and M. D. Hartmann, J. Med. Chem., 2023, 66, 14513–14543 CrossRef CAS PubMed.
- H. Furihata, S. Yamanaka, T. Honda, Y. Miyauchi, A. Asano, N. Shibata, M. Tanokura, T. Sawasaki and T. Miyakawa, Nat. Commun., 2020, 11, 4578 CrossRef CAS PubMed.
- K. R. Hornberger and E. M. V. Araujo, J. Med. Chem., 2023, 66, 8281–8287 CrossRef CAS PubMed.
- J. D. Hansen, M. Correa, M. A. Nagy, M. Alexander, V. Plantevin, V. Grant, B. Whitefield, D. Huang, T. Kercher, R. Harris, R. K. Narla, J. Leisten, Y. Tang, M. Moghaddam, K. Ebinger, J. Piccotti, C. G. Havens, B. Cathers, J. Carmichael, T. Daniel, R. Vessey, L. G. Hamann, K. Leftheris, D. Mendy, F. Baculi, L. A. LeBrun, G. Khambatta and A. Lopez-Girona, J. Med. Chem., 2020, 63, 6648–6676 CrossRef CAS PubMed.
- L. H. Jones, J. Burrows, N. Feeder, P. Glossop, K. James, R. M. Jones, A. S. Kenyon, S. Patel, D. F. Roberts, M. D. Selby, R. S. Strang, E. F. Stuart, M. A. Trevethick, J. Watson, K. N. Wright and N. Clarke, Bioorg. Med. Chem. Lett., 2015, 25, 5121–5126 CrossRef CAS PubMed.
- R. Osborne, N. Clarke, P. Glossop, A. Kenyon, H. Liu, S. Patel, S. Summerhill and L. H. Jones, J. Med. Chem., 2011, 54, 6998–7002 CrossRef CAS PubMed.
- J. M. Tsai, R. P. Nowak, B. L. Ebert and E. S. Fischer, Nat. Rev. Mol. Cell Biol., 2024, 25, 740–757 CrossRef CAS PubMed.
- M. E. Bunnage, E. L. Chekler and L. H. Jones, Nat. Chem. Biol., 2013, 9, 195–199 CrossRef CAS PubMed.
- N. P. Grimster, RSC Med. Chem., 2021, 12, 1452–1458 RSC.
- E. J. Fenech, F. Lari, P. D. Charles, R. Fischer, M. Laétitia-Thézénas, K. Bagola, A. W. Paton, J. C. Paton, M. Gyrd-Hansen, B. M. Kessler and J. C. Christianson, eLife, 2020, 9, e57306 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5md00028a |
| This journal is © The Royal Society of Chemistry 2025 |