
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and anti-mycobacterial activity of novel medium-chain β-lactone derivatives: a multi-target strategy to combat Mycobacterium abscessus†
Thomas Francis‡
a,
Christina Dedaki‡b,
Phoebe Ananida-Dasenakib,
Dimitra Bolkab,
Kanellos Albanisb,
Filippos Foteinakisb,
Julie Mezquidaa,
Marie Hancea,
Alexandros Athanasoulisb,
Anna-Krinio Papagiorgoub,
Ioanna-Foteini Karampoulab,
George Georgitsisb,
Celia Jardinc,
Stéphane Audebertc,
Luc Camoin c,
Céline Crausted,
Stéphane Canaana,
Victoria Magrioti*b and
Jean-François Cavalier*a
c,
Céline Crausted,
Stéphane Canaana,
Victoria Magrioti*b and
Jean-François Cavalier*a
aAix-Marseille Univ., CNRS, LISM, Institut de Microbiologie de la Méditerranée FR3479, Marseille, France. E-mail: jfcavalier@imm.cnrs.fr
bDepartment of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis, Athens, 15771, Greece. E-mail: vmagriot@chem.uoa.gr
cAix-Marseille Univ., INSERM, CNRS, Institut Paoli-Calmettes, CRCM, Marseille Protéomique, Marseille, France
dIBMM, Univ Montpellier, CNRS, ENSCM, Montpellier, France
First published on 25th April 2025
Abstract
The constant emergence of drug-resistant mycobacteria, together with the lack of new antibiotics entering the market, has become a global public health problem that threatens the effective treatment of infectious diseases. The development of single molecules targeting different proteins should significantly reduce the emergence of resistant strains, and therefore represent a promising strategy to overcome such an issue. In this challenging context, a new series of 30 lipophilic compounds based on the β-lactone-core has been synthesized by varying the nature of the substituents on the lactone ring. The evaluation of their antibacterial activity against M. tuberculosis and M. abscessus, two major pathogenic mycobacteria, highlighted potential candidates. The VM038, VM040 and VM045 were active only against M. tuberculosis, while VM025, VM026 and VM043 inhibited the growth of both M. tuberculosis and the S and R variants of M. abscessus. Competitive click chemistry activity-based protein profiling revealed several potential M. abscessus target enzymes of VM043, the best extracellular growth inhibitor. Finally, when tested against intracellular bacteria, although VM043 was found inactive, VM025 & VM026 proved to be potent and promising inhibitors of intramacrophagic M. abscessus growth with minimal inhibitory concentrations (MIC50Raw) comparable to the standard antibiotic imipenem. Overall, these results strengthen the added value of our VM β-lactone derivatives not only in the fight against pathogenic mycobacteria, leading to the arrest of M. abscessus and/or M. tuberculosis growth through multitarget enzyme inhibition, but also as efficient probes to identify novel potential therapeutic targets using chemoproteomics approaches.
1. Introduction
The Mycobacterium genus consists of more than 200 species, which are mainly classified according to their pathogenicity and growth rate.1,2 In addition to Mycobacterium tuberculosis, the causative agent of tuberculosis (TB),3,4 nontuberculous mycobacteria (NTM) are opportunistic pathogens responsible for clinical syndromes ranging from skin to pulmonary infections (e.g., Mycobacterium abscessus5–7) especially in immunocompromised individuals.8–10 Furthermore, the emergence of multidrug-resistant isolates of M. tuberculosis or M. abscessus has significantly reduced treatment success rates, leading to higher incidence of treatment failure and mortality.6,11–13 Known as the “antibiotic and clinical nightmare”,6,14 M. abscessus is indeed considered one of the most drug-resistant mycobacterial species.15 This mycobacterium exists as two distinct colony morphotypes: smooth (S) and rough (R), which can adapt and develop differently in response to the host immune system therefore leading to different outcomes for the mycobacteria within its host.7,16,17 The R variant is particularly associated with severe lung infections5,12 and can persist for years, especially in patients with cystic fibrosis (CF).18A key factor limiting the treatment of mycobacterial infections is the presence of a complex, waxy, lipid-rich cell wall containing unique lipids such as mycolic acids.19,20 These lipids play a critical role in maintaining the structural integrity of the bacterial cell envelope and in modulating interactions with the host immune system.20,21 Moreover, this general composition and architecture is shared by all mycobacterial species contributing to their low permeability to many antibiotics thus limiting therapeutic options.19,20 Therefore, targeting the enzymes involved in mycobacterial lipid metabolism;22,23 which are mainly serine and cysteine enzymes (i.e., (Ser/Cys)-based enzymes);24–26 has emerged as a promising strategy to combat not only M. tuberculosis,27–31 but also other chronic mycobacterial infections like those caused by M. abscessus.24,32
β-Lactones are a class of four-membered cyclic esters characterized by a highly strained ring structure that confers significant reactivity and makes them potent inhibitors of (Ser/Cys)-based enzymes. The unique reactivity of β-lactones allows them to form a covalent, but often reversible, long-lived acyl enzyme complex with (Ser/Cys)-based enzymes as a result of nucleophilic attack of the catalytic serine (or cysteine) residue on the β-lactone ring.33,34 This characteristic has driven interest in their development as pharmacological agents, particularly for inhibiting enzymes involved in lipid metabolism. The best example of this family is the FDA-approved anti-obesity drug Orlistat (Fig. 1),33,35 which is also known to inhibit microbial (Ser/Cys)-based enzymes.36 Acting as a versatile (Ser/Cys)-hydrolase inhibitor, Orlistat impairs the activity of key mycobacterial enzymes involved in critical processes related to lipid metabolism, particularly in the biosynthesis of mycolic acids which are essential for the integrity of the bacterium's cell wall.37 Among them are the enzymes belonging to the cutinase-like family proteins including the essential M. tuberculosis phospholipase/thioesterase Cut6 (Rv3802c);38–40 enzymes belonging to the hormone-sensitive lipase (HSL) family member proteins (i.e., Lip-HSL);41,42 the polyketide synthase-13 thioesterase domain (Pks13-TE) as well as the mycolyltransferase antigen 85C.43,44 Finally, Orlistat has been shown to inhibit the growth of various mycobacterial species with minimum inhibitory concentrations (MIC) of 10–60 μg mL−1 (ref. 45) and demonstrated strong synergistic effects with vancomycin against M. tuberculosis H37Rv, reducing its MIC (50 μg mL−1) by approximately 16-fold.46 Various structural modifications of the Orlistat pharmacophore have been explored to enhance the specificity and antibacterial potency of newly synthesized analogs.40,45,47,48 In particular, the β-lactone EZ120 (Fig. 1) displayed promising antitubercular activity (MIC ∼0.7 μg mL−1 = 1.6 μM) and was found to target the antigen 85 enzymes and Pks13-TE.47
 | ||
| Fig. 1 Chemical structures of Orlistat,42 and EZ120;47 former VM001 and VM008 (ref. 45) as well as new synthesized related analogues (see Table S1† for structure details). | ||
The multi-target nature of β-lactones makes them promising candidates for the development of new derivatives that may target lipid-processing enzymes critical for mycobacterial growth and survival.
A few years ago, we reported the synthesis and antimycobacterial activity of a first series of 16 long- and medium-chain mono- and disubstituted β-lactones, namely VM001–VM016.45 Although M. abscessus growth was barely affected, six β-lactones were active against M. tuberculosis (MIC50 ∼ 20–65 μg mL−1), with VM008 [trans-(Z)-3-(hexadec-7-en-1-yl)-4-propyloxetan-2-one = trans-VM001] (Fig. 1) being the best growth inhibitor (MIC50 ∼ 19.7 μg mL−1).45
Given the promising antibacterial activities of this best compound,45 VM008 chemical structure has been used as a template to synthesize a new set of 27 β-lactone and 3 β-lactam derivatives (VM compounds – Fig. 1) by varying the nature of the R1 and R2 alkyl chains to modulate their lipophilicity as a means of improving their activity. Their respective anti-mycobacterial activity was further assessed against M. tuberculosis and the two variants S & R of M. abscessus. Remarkably, and contrary to the first series of β-lactone derivatives,45 the determined MIC revealed that some VM β-lactones and β-lactams were able to inhibit M. abscessus growth in vitro in culture broth medium and/or inside infected macrophages. In addition, using a competitive activity-based protein profiling approach,28,29 the potential target enzymes of the newly synthesized VM043, identified as the most active inhibitor of extracellular bacterial growth, were further identified.
2. Experimental section
2.1. Chemistry
Compounds 1a–g, 3a–c, 8a–b, 16, 21, 27, 32a–b and 38 were commercially available. The synthetic methods and the characterization data of all synthesized new β-lactones as well as intermediates are included in the ESI† file.General procedure I. Aldol reaction for the synthesis of α,β-substituted β-hydroxy acids (see Schemes 1, 5 and 6, methods A, F, G, H and I). To a stirring solution of diisopropylamine (3 mmol) in dry THF (2 mL), under argon at 0 °C, a solution of 1.6 M n-BuLi in hexane (3 mmol) was slowly added via syringe and the solution of LDA was stirred at 0 °C for 10 min. The carboxylic acid (1 mmol) in dry THF (3 mL) was then added and the solution was stirred at 0 °C for 1 h. Then, the appropriate aldehyde (1.3 mmol) in dry THF (2 mL) was added and the solution was stirred at 0 °C for 1 h and at room temperature overnight. The solvent was removed under reduced pressure. The reaction mixture was acidified with 1 N HCl and extracted with Et2O (3 × 30 mL). The organic layers were combined, washed with brine (30 mL) and dried. The solvent was removed and the product was purified by column chromatography eluting with a gradient of CHCl3/MeOH 97
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 to 95:5 (v/v).
:3 to 95:5 (v/v).
 | ||
| Scheme 1 Synthesis of β-lactones form an aldol reaction (method A). Reagents and conditions: (a) i) LDA (prepared in situ from (i-Pr)2NH and n-BuLi), dry THF, 0 °C, 1 h; ii) R2CHO, dry THF, 0 °C, 1 h, then r.t. 16 h; iii) 1 N HCl, 33–93%; (b) i) p-TsCl, dry pyridine, 0 °C, 1 h, then 4 °C, 3 days, 36–70%; ii) chromatographic separation of the racemic mixtures of cis- and trans-β-lactones. | ||
General procedure II. β-Lactone cyclization using p-TsCl in pyridine (see Scheme 1, method A). To a stirring solution of the β-hydroxy acid (1 mmol) in dry pyridine (2 mL), under argon at 0 °C, p-toluenesulfonyl chloride (2 mmol) in dry pyridine (1 mL) was added slowly via a syringe. The solution was stirred at 0 °C for 1 h and kept at 4 °C for 3 days. Then, Et2O (30 mL) was added, and the organic layer was washed with 10% Na2CO3 (2 × 30 mL), 1 N HCl (2 × 30 mL) and brine (30 mL). The organic layer was dried, and the solvent was removed in vacuo. The product was purified by column chromatography eluting with a gradient of hexane/EtOAc.
General procedure III. β-Lactone cyclization using EDC·HCl and DMAP (see Schemes 2 and 3, methods B–C). In a flame-dried flask under argon, a solution of the β-hydroxy acid (1 mmol) in dry CH2Cl2 (18 mL) was added, followed by EDC·HCl (1.6 mmol) and DMAP (0.1 mmol) and the solution was stirred at r.t. for 3 days. CH2Cl2 (20 mL) and H2O (20 mL) were added and then the organic layer was washed with brine (20 mL). The organic layer was dried, and the solvent was removed in vacuo. The product was purified by column chromatography eluting with a gradient of hexane/EtOAc.
 | ||
| Scheme 2 Synthesis of β-lactones from appropriately substituted β-keto ester (method B). Reagents and conditions: (a) i) CDI, dry THF, r.t., 6 h; ii) MgCl2, EtOCOCH2COOK, r.t., 18 h; iii) aq. HCl 1 N, 95%; (b) K2CO3, R1I, acetone/DMF, reflux, 18 h, 50–78%; (c) NaBH4, EtOH, 0 °C 30 min, then r.t. 3 h, 70–86%; (d) NaOH 1 N, 1,4-dioxane, r.t., 16 h, 54–85%; (e) for VM028: i) p-TsCl, dry pyridine, 0 °C, 1 h, then 4 °C, 3 days, 60%; ii) chromatographic separation of the racemic mixtures of cis- and trans-β-lactones; or for VM047 and VM048: EDC·HCl, DMAP, dry DCM, r.t., 72 h, 39–45%. | ||
 | ||
| Scheme 3 Synthesis of fluorinated β-lactones (methods C & D). Reagents and conditions: (a) i) CDI, dry THF, r.t., 6 h; ii) MgCl2, EtOCOCH2COOK, r.t., 18 h; iii) aq. HCl 1 N, 95%; (b) K2CO3, R1I, acetone/DMF, reflux, 18 h, 67–70%; (c) i) NaH, dry THF, −20 °C, 1 h, ii) Selectfluor, dry MeCN, −20 °C, 2.5 h, 78–82%; (d) NaBH4, EtOH, 0 °C 30 min, then r.t. 3 h, 61–82%; (e) NaOH 1 N or LiOH 1 N, THF or 1,4-dioxane, r.t., 16 h, 51–75%; (f) EDC·HCl, DMAP, dry DCM, r.t., 72 h, 29–36%; (g) i) LDA (prepared in situ from (i-Pr)2NH and n-BuLi), dry THF, −78 °C, 30 min, ii) NFSI, dry THF, −78 °C to r.t., 2.5 h, 23–39%. | ||
General procedure IV. Late-stage fluorination of β-lactones (see Scheme 3, method D). In a flame-dried flask with a solution of diisopropylamine (1.6 mmol) in dry THF (1 mL), under argon at 0 °C, a solution of n-BuLi 1.6 μ in hexane (1.6 mmol, 1 mL) was slowly added via a syringe and the solution of LDA was stirred at 0 °C for 10 min and then cooled at −78 °C. At −78 °C a solution of the β-lactone (1 mmol) in dry THF (7 mL) was added via a syringe and the solution was left stirring for 30 min at −78 °C. Then a solution of NFSI (2 mmol) in dry THF (2 mL) was added and the reaction was left to warm up to −15 °C and left stirring for additional 1.5 h at −15 °C. Then THF was removed in vacuo, EtOAc (30 mL) and 10% NaHCO3 (30 mL) were added. The organic layer was washed with brine (30 mL), dried and the solvent was removed in vacuo. The product was purified by column chromatography eluting with a gradient of hexane/EtOAc.
General procedure V. Deprotection of TIPS-protected terminal alkyne β-lactones using TBAF (see Scheme 5, methods F–H). In a flame-dried flask with a solution of the TIPS-protected terminal alkyne β-lactone (1 mmol) in dry THF (5 mL) under argon, TBAF (2 mmol) in dry THF (2 mL) was added and the mixture was left stirring for 3 h at r.t. The progress of the reaction was monitored by TLC. Once the starting material was consumed, saturated NH4Cl was added (20 mL) and the crude product was extracted with Et2O (3 × 20 mL). The organic layer was washed with brine (20 mL), dried and the solvent was removed in vacuo. The product was purified by column chromatography eluting with a gradient of hexane/EtOAc or hexane/Et2O.
3-Decyl-4-propyloxetan-2-one (VM025). Prepared according to general procedure II using 2f; purified by column chromatography eluting with a gradient of hexane/EtOAc starting 97
:3 to 95:5 (v/v). VM025 gives rise to VM026 and VM027.Isolated mixture of diastereomers after column chromatography (dr 1:1). Yield 70%; 1H NMR (200 MHz, CDCl3) δ 4.59–4.46 (m, 0.5H), 4.26–4.15 (m, 0.5H), 3.65–3.50 (m, 0.5H), 3.21–3.08 (m, 0.5H), 1.91–1.08 (m, 24H), 0.96 (t, J = 7 Hz, 3H), 0.86 (t, J = 7 Hz, 3H). 13C NMR (50 MHz, CDCl3) δ 172.5, 171.8, 78.1, 75.6, 56.3, 52.8, 36.7, 32.4, 32.1, 29.8, 29.7, 29.6, 29.5, 28.1, 27.8, 27.2, 24.1, 22.8, 19.1, 18.6, 14.3, 14.0, 13.9. HRMS (ESI) [M + Na]+: calcd for C16H30NaO2+ 277.2138, found 277.2135.
(±)-trans 3-Decyl-4-propyloxetan-2-one (VM026). Purified by 2nd column chromatography of VM025 eluting with a gradient of hexane/EtOAc 98
:2 to 95:5 (v/v). 1H NMR (200 MHz, CDCl3) δ 4.26–4.15 (m, 1H), 3.21–3.08 (m, 1H), 1.91–1.56 (m, 4H), 1.55–1.08 (m, 18H), 1.00–0.65 (m, 6H). 13C NMR (50 MHz, CDCl3) δ 172.2, 78.2, 56.4, 36.7, 32.1, 29.7, 29.5, 28.1, 27.2, 22.8, 18.6, 14.3, 14.0. HRMS (ESI) [M + Na]+: calcd for C16H30NaO2+ 277.2138, found 277.2137.
(±)-cis 3-Decyl-4-propyloxetan-2-one (VM027). Purified by 2nd column chromatography of VM025 eluting with a gradient of hexane/EtOAc 98
:2 to 95:5 (v/v). 1H NMR (200 MHz, CDCl3) δ 4.59–4.46 (m, 1H), 3.65–3.50 (m, 1H), 1.90–1.40 (m, 8H), 1.40–1.11 (m, 14H), 0.98 (t, J = 7 Hz, 3H), 0.88 (t, J = 7 Hz, 3H). 13C NMR (50 MHz, CDCl3) δ 171.8, 75.7, 52.8, 32.4, 32.0, 29.6, 29.5, 29.4, 29.3, 27.8, 24.1, 22.8, 19.0, 14.3, 14.0. HRMS (ESI) [M + Na]+: calcd for C16H30NaO2+ 277.2138, found 277.2138.
3-Fluoro-3-octyl-4-propyloxetan-2-one (VM043). Prepared according to general procedure III using 11a; purified by column chromatography eluting with hexane/EtOAc 95
:5 (v/v). Isolated mixture of diastereomers after column chromatography (dr 65:35). Yield 36%; yellowish oil; 1H NMR (400 MHz, CDCl3) δ 4.71–4.61 (m, 0.35H), 4.50–4.42 (m, 0.65H), 2.07–1.65 (m, 4H), 1.65–1.25 (m, 14H), 1.02 (t, J = 7.4 Hz, 3H), 0.90 (t, J = 6.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.66 (d, J = 25.3 Hz), 166.93 (d, J = 24.2 Hz), 102.90 (d, J = 217.2 Hz), 102.00 (d, J = 224.2 Hz), 83.44 (d, J = 25.3 Hz), 82.26 (d, J = 22.2 Hz), 32.36 (d, J = 23.2 Hz), 31.80, 31.77, 31.46 (d, J = 3.0 Hz), 30.95 (d, J = 5.1 Hz), 29.65, 29.39, 29.21, 29.11, 29.08, 28.72 (d, J = 23.2 Hz, CH2CF), 22.63, 22.62, 22.58, 22.54, 22.16, 22.12, 18.76, 17.95, 14.07, 13.79, 13.66. 19F NMR (376 MHz, CDCl3) δ −159.76, −173.43. HRMS (ESI) [M + Na]+: calcd for C14H25FNaO2+ 267.1731, found 267.1732.
3-(8-(Octyloxy)octyl)-4-propyloxetan-2-one (VM045). Prepared according to general procedure II using 2j; purified by column chromatography eluting with a gradient of hexane/EtOAc starting 97
:3 to 95:5 (v/v). Isolated mixture of diastereomers after column chromatography (dr 3:7). Yield 60%; colorless oil; 1H NMR (200 MHz, CDCl3) δ 4.59–4.50 (m, 0.7H), 4.26–4.20 (m, 0.3H), 3.65–3.55 (m, 0.7H), 3.32 (t, J = 7.0 Hz, 4H), 3.20–3.14 (m, 0.3H), 1.91–1.50 (m, 10H), 1.50–1.22 (m, 20H), 1.00 (t, J = 7 Hz, 3H), 0.86 (t, J = 7 Hz, 3H). 13C NMR (50 MHz, CDCl3) δ 172.28, 171.59, 77.92, 75.45, 70.98, 70.88, 56.17, 52.67, 36.50, 32.21, 31.83, 29.78, 29.75, 29.46, 29.36, 29.33, 29.27, 29.25, 29.22, 27.87, 27.58, 26.97, 26.20, 26.15, 23.92, 22.65, 18.89, 18.43, 14.08, 13.79, 13.75. HRMS (ESI) [M + Na]+: calcd for C22H42NaO3+ 377.3026, found 377.3029.
3-Fluoro-3-(8-(octyloxy)octyl)-4-propyloxetan-2-one (VM046). Prepared according to general procedure IV using VM045; purified by column chromatography eluting with hexane/Et2O 95
:5 (v/v). Isolated mixture of diastereomers after column chromatography (dr 55:45). Yield 23%; 1H NMR (400 MHz, CDCl3) δ 4.61–4.53 (m, 0.55H), 4.40–4.34 (m, 0.45H), 3.32 (t, J = 6.8 Hz, 4H), 1.98–1.14 (m, 30H), 0.93 (t, J = 7.3 Hz, 3H), 0.81 (t, J = 6.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 166.63 (d, J = 25.2 Hz), 165.91 (d, J = 23.90 Hz), 101.91 (d, J = 215.5 Hz), 101.00 (d, J = 224.3 Hz), 82.40 (d, J = 25.2 Hz), 81.24 (d, J = 21.4 Hz), 69.98, 69.86, 69.84, 31.31 (d, J = 2.5 Hz), 30.82, 30.43 (d, J = 2.5 Hz), 29.92 (d, J = 3.8 Hz), 28.75, 28.71, 28.56, 28.45, 28.30, 28.27, 28.24, 28.19, 28.18, 27.67 (d, J = 22.7), 25.18, 25.11, 21.65, 21.52 (d, J = 3.8 Hz), 21.10 (d, J = 3.8 Hz), 17.73, 16.92, 13.09, 12.78, 12.66. 19F NMR (376 MHz, CDCl3) δ −159.71, −173.39. HRMS (ESI) [M + Na]+: calcd for C22H41FNaO3+ 395.2932, found 395.2935.
(±)-trans 3-(Oct-7-yn-1-yl)-4-propyloxetan-2-one (VM053p). Prepared according to general procedure V using 35b; purified by column chromatography eluting with a gradient of hexane/EtOAc starting 97
:3 to 92:8 (v/v). Only (±)-trans diastereomers were isolated after column chromatography. Yield 98%; colorless oil; 1H NMR (400 MHz, CDCl3) δ 4.27–4.23 (m, 1H), 3.21–3.17 (m, 1H), 2.21 (t, J = 7 Hz, 2H), 1.96 (s, 1H), 1.91–1.80 (m, 2H), 1.79–1.69 (m, 2H), 1.57–1.42 (m, 10H), 1.01 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 171.57, 84.47, 77.93, 68.29, 56.14, 36.51, 28.77, 28.35, 28.26, 27.82, 26.86, 18.45, 18.33, 13.78. HRMS (ESI) [M + Na]+: calcd for C14H22NaO2+ 245.1512, found 245.1510.
(±)-trans 3-(8-Azidooctyl)-4-propyloxetan-2-one (VM055p). Prepared according to general procedure I using 39, followed by general procedure II; purified by column chromatography eluting with hexane/EtOAc 95
:5 (v/v). Only (±)-trans diastereomers were isolated after column chromatography. Yield 12%; colorless oil; 1H NMR (400 MHz, CDCl3) δ 4.27–4.23 (m, 1H), 3.28 (t, J = 7 Hz, 2H), 3.22–3.17 (m, 1H), 1.92–1.80 (m, 2H), 1.78–1.69 (m, 2H), 1.52–1.34 (m, 14H), 1.01 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 171.58, 77.91, 56.16, 51.45, 36.50, 29.18, 29.15, 29.00, 28.80, 27.87, 26.93, 26.65, 18.44, 13.77. HRMS (ESI) [M + Na]+: calcd for C14H25NaN3O2+ 290.1839, found 290.1835.
2.2. Biological evaluation
:1 (w/v) ratio and then lysed by mechanical disruption on a BioSpec Beadbeater. Each sample (300 μL – 0.3 mg total proteins) was further subjected to copper-free azide-alkyne cycloaddition and enrichment with DBCO-agarose bead 50% slurry (Click Chemistry Tools, ref. 1034) according to the manufacturer's instructions. The beads containing bound, biotinylated proteins were resuspended in 30 μL PBS buffer pH 7.4, snap frozen in liquid nitrogen and stored at −80 °C before mass spectrometry experiments. See ESI† for detailed protocol.The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (https://www.proteomexchange.org)63 via the PRIDE partner repository64 (https://www.ebi.ac.uk/pride/login) with the dataset identifiers PXD057836.
3. Results and discussion
3.1. Chemistry
Based on our previous results on the activity of long and medium chain substituted β-lactones,45 we decided to synthesize a new set of β-lactone derivatives bearing saturated aliphatic chains located at the α-position such as C6, C8, C10, C12, and an ether analogue of our most potent β-lactone VM001; while at the β-position a propyl chain that had proved to be the optimal substituent45 was used. We also synthesized β-lactone VM036, which has an oleyl chain at the β-position and a propyl chain at the α-position, i.e. in opposite positions compared to β-lactone VM001. Finally, we resynthesized β-lactones VM009, VM010, VM011 and VM012 to separate the racemic mixtures of cis- and trans-β-lactones to be tested independently. For the synthesis of these β-lactones, method A was followed (Scheme 1). The first step is an aldol reaction between the appropriate carboxylic acid and aldehyde using LDA as the base which deprotonates the proton in the α-position of the acid function and yields the corresponding β-hydroxy acids 2 after treatment with aqueous HCl. The β-hydroxy acids 2 were then cyclized using para-toluenesulfonyl chloride (p-TsCl) in dry pyridine, and the resulting β-lactones were further purified by column chromatography (Scheme 1). In all cases, the mixture of all 4 diastereomers was obtained. In the cases of VM019 and VM045 the cis- and trans-diastereomeric mixture were inseparable, while in the cases of VM009–VM012, VM025, VM036, VM038 and VM040 both cis- and trans-pure racemic mixtures were obtained after a second chromatography of the initial mixture of all 4 diastereomers. The cis- and trans-isomers were assigned by comparison of the corresponding 1H NMR and 13C NMR chemical shifts of α-CH and β-CH to our previous work for both β-hydroxy acids and β-lactones.45For the synthesis of β-lactones VM028, VM047 and VM048, a different synthetic route (method B – Scheme 2) was chosen either to insert the desired substituent from a commercially available starting material or to avoid any side reactions that would be caused by the use of LDA in the aldol reaction mentioned above. In this procedure, butyric acid 3 reacted with 1,1′-carbonyl diimidazole (CDI) and monoethyl malonic acid magnesium salt using Masamune's method65 to yield the corresponding β-keto esters 4. Deprotonation of the more acidic proton in α-position of the β-keto ester carbonyl function using less reactive K2CO3 base, followed by substitution reaction with an alkyl iodide bearing the desired R1 chain as substituent yielded the substituted β-keto esters 5 which were subsequently reduced using NaBH4. Hydrolysis of the resulting β-hydroxy esters 6 to the corresponding β-hydroxy acids 7 and a final cyclization reaction as described above using p-TsCl/pyridine afforded the final β-lactone VM028. For β-lactones VM047 and VM048, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl) with a catalytic amount of 4-dimethylaminopyridine (DMAP) was used. In the case of VM028 both racemic mixtures of cis- and trans-β-lactones were isolated by column chromatography, whereas the diastereomers of VM047 and VM048 were inseparable by column chromatography.
We also decided to incorporate a fluorine atom as second substituent at the α-position of the β-lactone ring. Fluorine atoms are widely used in compounds of medicinal interest as they enhance physicochemical properties such as solubility and lipophilicity, but also often increase the affinity of bioactive compounds for enzymes and receptors. To that end, two different synthetic procedures, methods C & D, were established as depicted in Scheme 3. Method C is similar to the previously described method B (Scheme 2) with the addition of an extra synthetic fluorination step (Scheme 3 – method C, step c) after the synthesis of the monosubstituted β-keto esters 8 and before the cyclisation step. The fluorination step that we introduced here for the first time included the use of sodium hydride for the deprotonation of the monosubstituted β-keto ester 8, followed by the use of Selectfluor to insert the fluorine atom. The β-lactones VM043 and VM044 were successfully prepared according to this procedure.
Furthermore, in order to incorporate the fluorine atom directly on the β-lactone ring, we further developed a more efficient fluorination method where the fluorination step would be performed at the last step of the synthesis, after the cyclisation. This proposed late-stage fluorination method would be much more efficient since it could be applied to already synthesized β-lactone molecules, or to new β-lactones such as VM045. Indeed, this new method D (Scheme 3) includes the use of LDA on the β-lactone and treatment with N-fluorobenzenesulfonimide (NFSI) as fluorinating electrophile. VM042 and VM046 which are the fluorinated analogues of VM001 and VM045, respectively, have been prepared via this late-stage fluorination method D.
Finally, three β-lactam analogues were synthesized. For this purpose, a synthetic method (Scheme 4 – method E) starting from the corresponding β-lactone was established. After opening the β-lactone ring by the nucleophilic attack of NaN3 to give the corresponding β-azido acids 12, the azide group was reduced by catalytic hydrogenation using 10% Pd/C, and finally the β-lactam ring of 13 was closed using Mukaiyama's reagent66 in a very dilute solution to avoid the intermolecular reaction (Scheme 4).
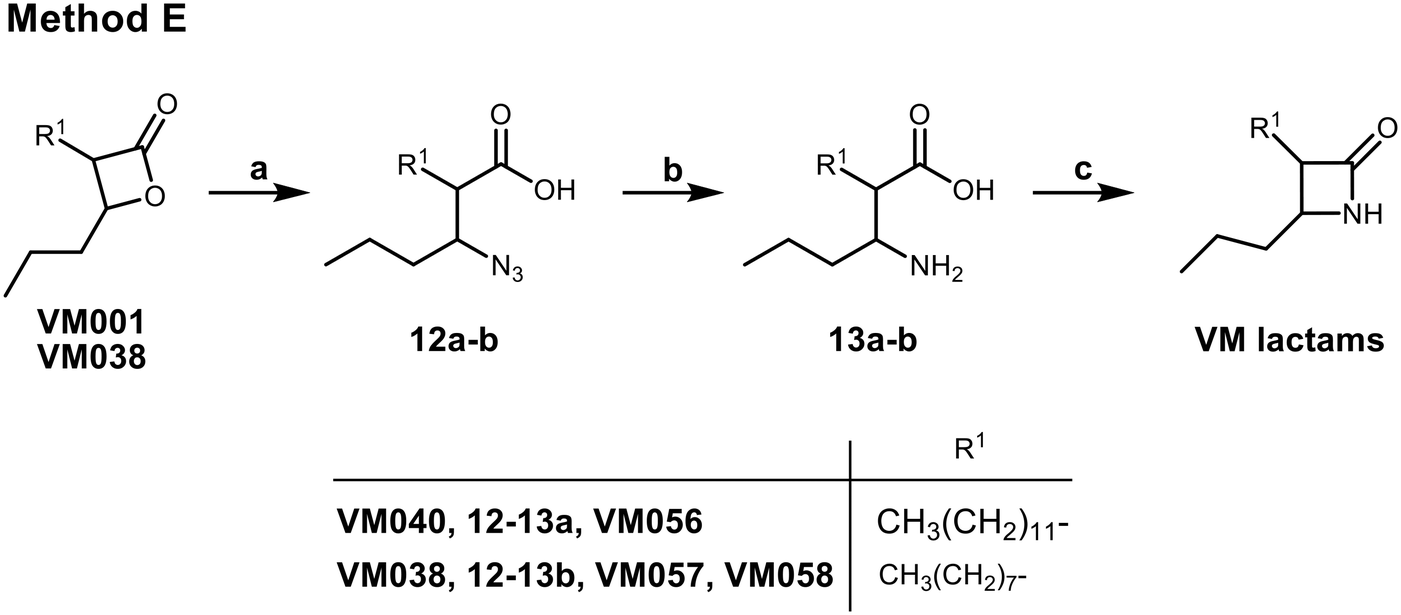 | ||
| Scheme 4 Synthesis of β-lactams VM056 and VM057 (method E). Reagents and conditions: (a) NaN3, DMF, 60 °C, 16 h, 65–77%; (b) H2, 10% Pd/C, MeOH, r.t., 2 h, 36–44%; (c) i) Mukaiyama's reagent, Et3N, CH3CN, reflux, 4 h, then r.t., 16 h, 65–71%; ii) chromatographic separation of the racemic mixture of the trans-β-lactone VM058. | ||
3.2. Antimycobacterial activity of the new β-lactone compounds
Drug susceptibility testing of the 27 β-lactone and 3 β-lactam derivatives was assessed against two pathogenic mycobacterial species: M. tuberculosis mc26230,49 and the two S & R variants of M. abscessus. The corresponding MIC50/MIC90 values for each molecule, as determined by the resazurin microtiter assay (REMA),30,45,53,54 are reported in Table 1. Amikacin (AMK) which is a core drug used in the treatment of M. abscessus, with MIC50/MIC90 of 4/32 and 8/32 μg mL−1 against the S & R variant,67 respectively; as well as a second-line injectable drug for the treatment of multidrug resistant TB, with MIC50/MIC90 ∼1 μg mL−1 against M. tuberculosis H37Rv,68 was used as reference drug.| Compounds | MIC50/MIC90a (μg mL−1) | ||
|---|---|---|---|
| M. abscessus CIP 104536T | M. tuberculosis mc26230 | ||
| Smooth | Rough | ||
| a Minimum inhibitory concentration leading to 50% or 90% growth inhibition (MIC50/MIC90) as determined by the resazurin microtiter assay (REMA).b Data from our previous study.45 AMK: amikacin. All reported values are expressed as mean ± SD of three independent assays. | |||
| Orlistatb | 44.4 ± 0.68/45.6 ± 0.56 | 57.0 ± 0.11/73.5 ± 0.50 | 11.0 ± 0.50/13.5 ± 1.0 |
| VM001 (C16:1 ω9)b | >100 | >100 | 31.8 ± 1.5/>100 |
| VM008 = trans-(R,R)-VM001b | >100 | >100 | 19.7 ± 1.3/31.9 ± 1.4 |
| VM009b | >100 | >100 | >100 |
| VM013 = trans-VM009b | >100 | >100 | 33.6 ± 0.21/42.9 ± 1.3 |
| VM020 = cis-VM009 | >100 | >100 | >100 |
| VM010b | >100 | >100 | 33.5 ± 0.26/69.9 ± 0.94 |
| VM017 = trans-VM010 | >100 | >100 | 45.8 ± 1.0/47.8 ± 1.8 |
| VM018 = cis-VM010 | >100 | >100 | 47.2 ± 1.2/53.1 ± 2.0 |
| VM011b | >100 | >100 | >100 |
| VM022 = trans-VM011 | >100 | 59.9 ± 2.1/68.9 ± 2.7 | >100 |
| VM024 = cis-VM011 | >100 | 41.9 ± 2.9/64.7 ± 0.66 | >100 |
| VM012b | >100 | >100 | >100 |
| VM021 = trans-VM012 | >100 | >100 | >100 |
| VM023 = cis-VM012 | >100 | 31.0 ± 2.8/>100 | >100 |
| VM019 (trans) | >100 | >100 | 65.2 ± 0.93/67.2 ± 0.61 |
| VM025 | 49.9 ± 1.3/52.4 ± 2.5 | 27.1 ± 0.80/>100 | 63.9 ± 1.6/65.1 ± 1.5 |
| VM026 = trans-VM025 | 47.2 ± 1.5/56.7 ± 2.2 | 25.6 ± 1.4/>100 | 59.2 ± 2.8/>100 |
| VM027 = cis-VM025 | >100 | >100 | >100 |
| VM028 (C18:1 ω9) | >100 | >100 | >100 |
| VM029 = trans-VM028 | >100 | >100 | >100 |
| VM030 = cis-VM028 | >100 | >100 | >100 |
| VM037 | >100 | >100 | >100 |
| VM036 = trans-VM037 | >100 | >100 | >100 |
| VM039 | >100 | >100 | 24.7 ± 1.7/51.3 ± 1.3 |
| VM038 = trans-VM039 | >100 | >100 | 17.7 ± 0.22/46.5 ± 1.6 |
| VM040 (trans) | >100 | >100 | 9.6 ± 0.45/29.1 ± 1.6 |
| VM041 (cis) | >100 | >100 | 17.4 ± 1.6/44.2 ± 1.7 |
| VM042 | >100 | >100 | 10.2 ± 3.6/40.1 ± 7.5 |
| VM043 = fluorinated VM038–039 | 33.8 ± 1.2/49.2 ± 3.4 | 59.8 ± 5.6/88.9 ± 0.3 | 30.2 ± 1.3/45.9 ± 2.5 |
| VM044 = fluorinated VM040–041 | >100 | >100 | 20.8 ± 0.2/24.1 ± 0.1 |
| VM045 | >100 | >100 | >100 |
| VM046 = fluorinated VM045 | >100 | >100 | 32.2 ± 4.2/45.0 ± 3.3 |
| VM047 | >100 | >100 | 46.2 ± 4.0/>100 |
| VM048 | >100 | >100 | 46.8 ± 8.7/>100 |
| VM056 = VM040 β-lactam | 47.3 ± 6.0/74.1 ± 3.9 | >100 | 15.2 ± 0.6/21.8 ± 0.7 |
| VM057 = trans-VM058 β-lactam | 41.9 ± 7.4/44.4 ± 6.2 | >100 | 35.5 ± 1.2/44.2 ± 1.1 |
| VM058 = β-lactam | 41.8 ± 6.3/46.6 ± 4.0 | >100 | 23.7 ± 0.2/44.2 ± 2.4 |
| AMK | 2.3 ± 0.12/3.4 ± 0.11 | 4.3 ± 0.15/5.9 ± 0.26 | 0.24 ± 0.01/0.37 ± 0.02 |
Nearly all β-lactone derivatives were active against M. tuberculosis mc26230 (i.e., 18 compounds) growth (Table 1). With M. tuberculosis, MIC50 values were indicative of either a poor (MIC50 > 100 μg mL−1 for VM020 to VM024, VM027 to VM030, VM036, VM037, and VM045), a weak (MIC50 ∼45–66 μg mL−1 for VM017 to VM019, VM025, VM026, VM047, and VM048), a moderate (MIC50 ∼20–35 μg mL−1 for VM039, VM043, VM044, VM046, VM057, and VM058), or a quite good (∼9.6–17.7 μg mL−1 for VM038, VM040 to VM042, and VM056) antibacterial activity comparable to that of Orlistat (11.0 μg mL−1) (Table 1). Of note, 15 of the 18 active β-lactone derivatives exhibited interesting MIC90 values in the range of 21–67 μg mL−1. Remarkably, most of these latter β-lactones exhibited MIC50 and MIC90 values in the same range. Collectively, the best growth inhibitors of M. tuberculosis mc26230 were VM040, VM044 and VM056 with MIC50/MIC90 in the range 9.6–20.8/21.8–29.1 μg mL−1, as compared to 0.24/0.37 μg mL−1 for amikacin (Table 1).
In the case of M. abscessus, only 6 β-lactones were found to block the growth of the S variant. These included VM025 (MIC50 = 49.9 ± 1.3 μg mL−1/MIC90 = 52.4 ± 2.5 μg mL−1), VM026 (MIC50 = 47.2 ± 1.5 μg mL−1/MIC90 = 56.7 ± 2.2 μg mL−1), VM043 (MIC50 = 33.8 ± 1.2 μg mL−1/MIC90 = 49.2 ± 3.4 μg mL−1), VM056 (MIC50 = 47.3 ± 6.0 μg mL−1/MIC90 = 74.1 ± 3.9 μg mL−1), VM057 (MIC50 = 41.9 ± 7.4 μg mL−1/MIC90 = 44.4 ± 6.2 μg mL−1), and VM058 (MIC50 = 41.8 ± 6.3 μg mL−1/MIC90 = 46.6 ± 4.0 μg mL−1) for which both MIC50 & MIC90 values were in the same order of magnitude (Table 1). Among these, three molecules were also active against M. abscessus R: VM025 (MIC50 = 27.1 ± 0.80 μg mL−1) and VM026 (MIC50 = 49.9 ± 1.3 μg mL−1) but with MIC90 values higher than 100 μg mL−1; and VM043 (MIC50 = 59.8 ± 5.6 μg mL−1 and a MIC90 = 88.9 ± 0.3 μg mL−1). Interestingly, although VM022 and VM024 were inactive against M. abscessus S (MIC > 100 μg mL−1), these two molecules were however found to impair the growth of the R morphotype with interesting, albeit moderate, MIC50/MIC90 values (MIC50 = 59.9 ± 2.1 μg mL−1/MIC90 = 68.9 ± 2.7 μg mL−1, and MIC50 = 41.9 ± 2.9 μg mL−1/MIC90 = 64.7 ± 0.66 μg mL−1, respectively). In addition, although VM023, which is also inactive against the S variant, has an interesting MIC50 value (31.0 ± 2.8 μg mL−1), its MIC90 was however higher than 100 μg mL−1. Overall, the best inhibitors of M. abscessus R growth were VM022, VM024, and VM043 which exhibited similar MIC50/MIC90 values, respectively (Table 1).
From all these results, some structure–activity relationships (SAR) tendencies can however be set up. First, and as mentioned in our previous report,45 trans-β-lactones were almost >2 times more active than the corresponding cis isomers, and nearly as active as the cis/trans isomeric mixtures. This is best illustrated by the antibacterial activity of VM026 (trans-VM025) on M. tuberculosis mc26230 and M. abscessus compared to the cis isomer VM027 (cis-VM025) and the cis/trans isomeric mixture VM025 (Table 1).
Remarkably, the three β-lactones VM025, VM026 (trans-VM025), and VM043 which were able to efficiently inhibit the growth of all mycobacteria were bearing a medium R1 octyl or decyl chain, and a short R2 propyl chain (Fig. 1). In terms of lipophilicity this translates into a narrow range of calculated logPo/w, determined using the iLOGP69 method from the SwissADME web tool,70 between 4.3 and 4.8 for these latter three compounds (Table S1†). Indeed, the insertion of a fluorine atom on the β-lactone ring of VM038 and VM039 (logPo/w ∼ 4.12) slightly increased the lipophilicity of the fluorinated derivative VM043 (logPo/w ∼ 4.31). This fluorine will also affect the electrophilic nature of the carbonyl, making it even more reactive and sensitive to nucleophilic attack by enzymes with nucleophilic residues, thus resulting in a significantly improved antibacterial activity for VM043 as compared to the parent molecules. Finally, switching from a β-lactone core (VM038 and VM039, and VM040 and VM041) to a β-lactam core (VM057 and VM058, and VM056, respectively) led to a partial improvement in antibacterial properties against M. abscessus S.
In summary, while VM025, VM026 and VM043 are active against all three mycobacterial strains, VM043 is the best growth inhibitor with MIC50/MIC90 values very similar to those of Orlistat.45,46 As already discussed in the case of others families of mycobacterial growth inhibitors,28,54 the reason for such differences in β-lactones activity against M. tuberculosis compared to M. abscessus S and R variants still remains unclear and would require further study. However, differences in membrane composition and permeability between these three mycobacterial species are likely to play a key role in this phenotype.
3.3. Synthesis of β-lactone activity-based probes
Having established the promising antibacterial activity of VM025, VM026 and VM043 against M. tuberculosis mc26230 and more especially M. abscessus, we further aimed at identifying their target enzymes using click-chemistry activity-based protein profiling (CC-ABPP) approach.26,71–74 Considering the covalent mechanism of action attributed to Orlistat34,42 and related β-lactone derivatives,72 a series of eight new molecules were synthesized by substituting the R1 or R2 alkyl chain with a terminal alkyne or azide group, to provide a panel of new activity-based probes (ABP), i.e., VM035p, and VM049p to VM055p probes, that would be capable of retaining the antibacterial activity of their respective parent molecules (Schemes 5 and 6 and Table 2). | ||
| Scheme 5 Synthesis of β-lactone probes bearing a terminal alkyne. Reagents and conditions: method B. (a) i) CDI, dry THF, r.t., 6 h; ii) MgCl2, EtOCOCH2COOK, r.t., 18 h; iii) aq. HCl 1 N, 82%; (b) K2CO3, R1I, acetone/DMF, reflux, 18 h, 76–83%; (c) NaBH4, EtOH, 0 °C 30 min, then r.t. 3 h, 69–75%; (d) NaOH 1 N, 1,4-dioxane, r.t., 16 h, 30–87%; (e) p-TsCl, dry pyridine, 0 °C, 1 h, then 4 °C, 3 days, 40–71%. Method F. (f) t-BuOH, DMAP, dry DCM, DCC, 75%; (g) i) n-BuLi, dry THF; ii) TIPS-Cl, 77%; (h) 15% TFA in DCM, 75%; (i) i) LDA (prepared in situ from (i-Pr)2NH and n-BuLi), dry THF; ii) butyraldehyde, dry THF, 0 °C to r.t., 66–67%; (j) TBAF, dry THF, 65–100%. Method G. (k) NaI, acetone, 95%; (l) n-BuLi, TIPS-acetylene, dry THF, −78 °C, 62%. Method H. (m) Propargyl alcohol, n-BuLi, THF/HMPA, −78 °C to r.t., 83–84%; (n) NaH, 1,2-ethylenediamine, 0 °C to 70 °C, 65–66%; (o) Jones reagent, acetone, 0 °C, 81%; (p) i) n-BuLi, dry THF; ii) TIPS-Cl, iii) K2CO3, THF/MeOH/H2O 3:1:1 (v/v/v), 38–64%. | ||
 | ||
| Scheme 6 Synthesis of β-lactone probes bearing a terminal azide (method I). Reagents and conditions: (a) conc. HBr, toluene, reflux, 68%; (b) TBAB, NaN3, benzene, reflux, 95%; (c) Jones reagent, acetone, 0 °C, 71%; (d) i) LDA (prepared in situ from (i-Pr)2NH and n-BuLi), dry THF; ii) butyraldehyde, dry THF, 0 °C to r.t.; iii) column chromatography; iv) p-TsCl, dry pyridine, 0 °C to 4 °C, 12%. | ||
| Compounds | MIC50/MIC90a (μg mL−1) | |||
|---|---|---|---|---|
| M. abscessus CIP 104536T | M. tuberculosis mc26230 | |||
| Smooth | Rough | |||
| a Minimum inhibitory concentration leading to 50% or 90% growth inhibition (MIC50/MIC90) as determined by the resazurin microtiter assay (REMA). All values are expressed as mean ± SD of three independent assays. The MIC values of the parent molecules derived from Table 1 are shown in parentheses. | ||||
| VM035p |  |
>100 | >100 | >100 |
| (= VM028 probe) | (>100) | (>100) | (>100) | |
| VM049p |  |
>100 | >100 | >100 |
| (= VM019 probe) | (>100) | (>100) | (65.2 ± 0.93/67.2 ± 0.61) | |
| VM050p |  |
>100 | >100 | >100 |
| (= VM019 probe) | (>100) | (>100) | (65.2 ± 0.93/67.2 ± 0.61) | |
| VM051p |  |
35.0 ± 7.1/>100 | 64.6 ± 0.1/>100 | 18.0 ± 0.2/23.9 ± 1.0 |
| (= VM025 probe) | (49.9 ± 1.3/52.4 ± 2.5) | (27.1 ± 0.80/>100) | (63.9 ± 1.6/65.1 ± 1.5) | |
| VM052p |  |
52.1 ± 5.5/>100 | >100 | 35.2 ± 0.1/45.7 ± 0.2 |
| (= VM038–VM039 probe) | (>100) | (>100) | (17.7–24.7/46.5–51.3) | |
| (= VM043 probe) | (33.8 ± 1.2/49.2 ± 3.4) | (59.8 ± 5.6/88.9 ± 0.3) | (10.2 ± 3.6/40.1 ± 7.5) | |
| VM053p |  |
63.4 ± 1.3/>100 | >100 | 35.1 ± 3.6/39.4 ± 6.4 |
| (= VM038–VM039 probe) | (>100) | (>100) | (17.7–24.7/46.5–51.3) | |
| (= VM043 probe) | (33.8 ± 1.2/49.2 ± 3.4) | (59.8 ± 5.6/88.9 ± 0.3) | (30.2 ± 1.3/45.9 ± 2.5) | |
| VM054p |  |
>100 | >100 | 18.5 ± 0.8/24.5 ± 1.0 |
| (= VM040–VM041 probe) | (>100) | (>100) | (9.6–17.4/29.1–44.2) | |
| VM055p |  |
39.0 ± 4.5/>100 | 55.7 ± 3.3/>100 | 34.2 ± 1.5/52.7 ± 1.4 |
| (= VM038–VM039 probe) | (>100) | (>100) | (17.7–24.7/46.5–51.3) | |
| (= VM043 probe) | (33.8 ± 1.2/49.2 ± 3.4) | (59.8 ± 5.6/88.9 ± 0.3) | (30.2 ± 1.3/45.9 ± 2.5) | |
For the synthesis of the β-lactones ABP that would be tested in CC-ABPP, several different synthetic routes were established (Scheme 5). The previous Method B was first applied on 4-pentynoic acid 14 as starting material following the steps previously presented in Scheme 2. Reaction of 14 with CDI and monoethyl malonic acid magnesium salt afforded the β-keto ester 15. Then, substitution with K2CO3 as base and the appropriate alkyl iodide followed by NaBH4 reduction and hydrolysis, led to the β-hydroxy acids 18. Finally, β-lactone ring formation using p-TsCl in dry pyridine produced the final VM035p and VM054p probes that bear a terminal alkyne at the β-position of the ring. Methods F–H were further designed to synthesize β-lactones that bear the terminal alkyne at the α-position of the ring. Method F utilizes commercially available 6-heptynoic acid 19 that was protected as a tert-butyl ester at the carboxyl end using DCC, catalytic DMAP and tert-butanol. Then, a TIPS protecting group was added with n-BuLi and TIPS-Cl at the terminal alkyne for protection, followed by TFA deprotection of the ester to yield the free acid 22. With the terminal alkyne group protected, the acid undergoes an aldol reaction using LDA and butyraldehyde, as previously described in Scheme 1, followed by cyclization with p-TsCl in dry pyridine. The final step of this synthesis is the deprotection of the terminal alkyne using TBAF in dry THF for the production of the diastereomeric mixture of β-lactone VM049p and the racemic trans-mixture of VM050p.
In method G, an ω-bromo carboxylic acid may be used. Here, 11-bromoundecanoic acid 25 is transformed into the corresponding 11-iodoundecanoic acid 26 using NaI in acetone, that is then used to a substitution reaction to TIPS-acetylene using n-BuLi in dry THF and HMPA, to yield the corresponding TIPS protected tridec-12-ynoic acid 27. This acid is then used in an aldol reaction, cyclization and final deprotection, as described in the above-mentioned method B to yield β-lactone VM051p.
All of the above-mentioned methods used commercially ω-alkynoic acids or ω-bromo acids as starting materials that are expensive and only few are commercially available. To that end, we designed a synthetic procedure (method H) starting from any bromoalkane and propargyl alcohol that would lead to any ω-alkynoic acid using inexpensive starting materials. Indeed, starting from 1-bromooctane or 1-bromononane 30 using propargyl alcohol and n-BuLi in dry THF and HMPA, the corresponding internal alkynyl alcohols 31 were prepared. A Zipper reaction75 using NaH and ethylenediamine moved the triple bond to the ω-end of the alcohol 32 and subsequent oxidation using Jones reagent in acetone yielded the desired terminal alkyne carboxylic acids 33. Protection of the alkyne with TIPS group, aldol reaction with butyraldehyde, cyclization and finally TIPS deprotection produced the β-lactones VM052p and VM053p.
For the terminal azide β-lactone probes, we started from 1,10-decanediol 36 which was monobrominated using HBr in toluene under reflux (Scheme 6, method I). Then, the bromine was substituted by the azide group using NaN3 and TBAB in benzene, followed by Jones oxidation, aldol reaction and cyclization to yield the terminal azide β-lactone VM055p.
3.4. Activity-based protein profiling (ABPP) approach for targets identification
The antimicrobial potency of the synthesized β-lactone probes [VM035p, VM049p to VM055p] was first evaluated against M. tuberculosis mc26230 and M. abscessus S and R variants (Table 2). Partial loss of activity was observed for VM049p and VM050p (i.e., pentynyl version of VM019), and even no activity for VM035p (i.e., butynyl version of VM028) as compared to unmarked parent molecules (Table 2). On the other hand, VM054p (i.e., pentynyl version of VM040 and VM041) bearing the alkyne on the alkyl chain in β-position, and VM051p (i.e., undecynyl version of VM025) retained comparable activity to their parent molecules against M. tuberculosis mc26230. The same trend was observed with VM052p and VM053p, the nonyl and octyl derivatives, respectively, of VM038 and VM039, which gained some antibacterial activity against M. abscessus S while displaying similar activity to the parent β-lactones against M. tuberculosis mc26230. Remarkably, the azide derivative VM055p not only retained good antibacterial activities as the unmarked parent VM038 and VM039 molecules against M. tuberculosis mc26230, but also showed same moderate antibacterial activity as VM043 against M. abscessus S and R variants despite the absence of the fluorine atom, with fold changes in MIC50/MIC90 of around ×0.9–1.2/>2.0.Overall, these results indicate that the choice of the alkyl chain length for the incorporation of a terminal alkyne bond, as well as its location in either the R1 or R2 position of the lactone cycle, is of great importance to avoid major changes in the antibacterial activity of the resulting β-lactone ABPs.
According to these results, VM055p proved to be the best inhibitor of M. abscessus S & R and M. tuberculosis mc26230 growth among the eight β-lactone probes tested, with MICs very similar to those of its parent molecules. This probe was therefore a logical choice for target enzyme identification through CC-ABPP workflow.26
To do so, exponential growth phase M. abscessus S cells were incubated with dimethyl sulfoxide (DMSO) and then treated with VM055p ABP (Fig. 2A). Bacteria were lysed, and the probe-labeled proteins was subjected to strain-promoted azide–alkyne cycloaddition with DBCO-agarose beads.58 Indeed, such strained cyclooctynes can exclusively react with azide-tagged biomolecules to form a stable 1,2,3-triazole linkage without the need for copper catalyst.76 Here, the use of crosslinked agarose resin activated with DBCO functional groups, allows affinity enrichment of VM055p-labeled proteins. Following on-bead tryptic digestion, the resulting peptides were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) and quantified by label-free quantitative analysis59 (Fig. 2). The comparative proteomic analysis between a control sample (i.e. DMSO-treated M. abscessus cells for non-specific binding to DBCO-agarose beads) and VM055p-treated samples led to the identification of 12 target enzymes, when applying p-value ≤ 0.05 and fold change ≥ 1.0 thresholds on the proteomics analysis results (Fig. 2A, Tables 3 and S2†).
 | ||
| Fig. 2 Click chemistry activity-based protein profiling on living M. abscessus S culture. CC-ABPP typical workflow for the identification of proteins covalently bound to (A) VM055p or (B) significantly outcompeted by VM043. A mid-log phase culture of M. abscessus S was pre-treated with either DMSO or VM043, and then labelled with VM055p probe. After cell lysis and strain-promoted azide–alkyne cycloaddition with DBCO-agarose beads, enriched samples were subjected to tryptic digestion. Tandem mass spectrometry analyses and subsequent differential peptides analysis allowed the identification of the target enzymes captured by VM055p and those outcompeted by addition of VM043. (C) Volcano plot of the differential analysis of the labeled proteome of samples pre-incubated with VM043 inhibitor prior to VM055p probe labeling vs. VM055p probe-labeled samples only, and showing the significance of two-sample t-test (−log(p-value)) versus fold change (log2(LFQ normalized intensity in [VM043 + VM055p] vs. VM055p)) on the y and x axes, respectively (n = 3 biological independent experiments per group). The dashed lines indicate the threshold of p-value ≤ 0.1 and a fold change ≥ 1.0. Blue dots indicate VM055p targets proteins that are outcompeted by addition of VM043; while red dots represent VM055p targets proteins that were not inhibited by VM043. See also Tables S2–S6.† | ||
| Gene name | Protein name | M. tuberculosis orthologs | ||||
|---|---|---|---|---|---|---|
| Rv number | Essentialityb | Locationc | Activity/function | Functional categoryd | ||
| a See also Tables S2 and S4.†b From ref. 82 and 83.c CF: culture filtrate; CW: cell wall; M: membrane fraction; WCL: whole cell lysate.d CW/CP: cell wall/cell processes; IM/R: intermediary metabolism/respiration; IP: information pathways; LM: lipid metabolism; Pe/PPE: PE-PGRS family protein; RP: regulatory protein. | ||||||
| MAB_3436 | Non-specific serine/threonine protein kinase | Rv0014c | In vitro growth | CF; WCL | Serine/threonine-protein kinase PknB | RP |
| MAB_4551c | Possible acylglycerol lipase | Rv0183 | CM; CW; WCL | Monoacylglycerol lipase | LM | |
| MAB_1026c | Hypothetical protein | Rv1926c | CF; CM; WCL | Immunogenic protein Mpt63 | CW/CP | |
| MAB_0302 | Putative quinolone synthase | Rv1260 | WCL | Probable oxidoreductase | IM/R | |
| MAB_4284c | Hypothetical protein | Rv3514 | PE-PGRS family protein PE_PGRS57 | PE/PPE | ||
| MAB_1895c | Hypothetical protein | Rv0278c | PE-PGRS family protein PE_PGRS3 | PE/PPE | ||
| MAB_0974 | Hypothetical protein | Rv3876 | CW; M; WCL | ESX-1 secretion-associated protein EspI | CW/CP | |
| MAB_3501 | Putative hydrolase | Rv3195 | Conserved hypotheticals | — | ||
| MAB_3566c | Putative cyclase | — | — | |||
| MAB_4924 | Hypothetical protein | Rv0040c | CL; WCL | Secreted proline rich protein Mtc28 | CW/CP | |
| MAB_2394 | Hypothetical protein | Rv0910 | Conserved hypotheticals | — | ||
| MAB_2912c | Probable aldolase | Rv0727c | M | Possible L-fuculose phosphate aldolase FucA | IM/R | |
Even though at this stage no genes have been reported to be essential for M. abscessus growth by saturated transposon mutagenesis;77 their corresponding orthologs in M. tuberculosis H37Rv were nevertheless reported using the KEGG database78,79 and then cross-referenced with the Mycobrowser database80 to provide further information on their essentiality, activity and predicted location.81 Although three of the captured proteins are only conserved hypotheticals, the remaining nine ranged in their functional category from intermediary metabolism/respiration (2 proteins), lipid metabolism (1 protein), regulatory pathways (1 protein), cell wall/cell processes (3 proteins), and PE/PPE family proteins (2 proteins). Interestingly, these include the probable non-specific serine/threonine protein kinase MAB_3436 whose ortholog Rv0014c is annotated as essential for the in vitro growth of M. tuberculosis.82,83
Interestingly, several hydrolases were detected, including a putative hydrolase (MAB_3501/Rv3195), a putative cyclase (MAB_3566c), a probable aldolase (MAB_2912c/Rv0727c), a putative quinolone synthase (MAB_0302/Rv1260), and the monoacylglycerol lipase (MAB_4551c/Rv0183) which has previously been reported to be inhibited by Orlistat.84
Since all β-lactones share a similar mechanism of enzyme inhibition, resulting from the formation of a covalent bond between the β-lactone ring and the catalytic site of the enzyme,33,34 and since VM055p showed comparable antibacterial activities to VM043 against both M. abscessus and M. tuberculosis mc26230, we decided to also use this latter probe in an in situ competitive ABPP approach.26 The M. abscessus S culture was then pre-incubated with VM043 prior to treatment with the VM055p probe and subsequent CC-ABPP experiment (Fig. 2B). To gain in statistically relevant results, a differential comparative proteomic analysis between the [VM043 + VM055p] captured proteome and that of the VM055p probe alone was then performed. As shown in the resulting volcano plot (Fig. 2C), pre-incubation with VM043 significantly and selectively impaired (≥2-fold inhibition) the capture of four proteins by VM055p (see blue dots in Fig. 2C), thereby suggesting that these latter enzymes are preferential targets of VM043 vs. VM055p (Table S3†). These included the probable aldolase MAB_2912c (Rv0727c), the putative quinolone synthase MAB_0302 (Rv1260), the uncharacterized protein MAB_2394 (Rv0910), and the non-specific serine/threonine protein kinase MAB_3436 (Rv0014c) (Tables 4 and S3†).
| # | Protein IDs | Protein names | Gene names | [VM043 + VM055p] vs. VM055p | |
|---|---|---|---|---|---|
| −log(p-value) | Fold change (log2) | ||||
| a See also Tables S2–S6.† The two lists of proteins are deduced from the differential volcano plot depicted in Fig. 2C. | |||||
| 1 | B1MCL9 | Probable aldolase | MAB_2912c | 1.319 | −1.810 |
| 2 | B1MFK1 | 2-Heptyl-3-hydroxy-4(1H)-quinolone synthase | MAB_0302 | 1.411 | −1.189 |
| 3 | B1MEQ6 | Non-specific serine/threonine protein kinase | MAB_3436 | 1.169 | −1.080 |
| 4 | B1MB54 | Uncharacterized protein | MAB_2394 | 1.201 | −1.073 |
| 5 | B1MF33 | Hypothetical dipeptidyl aminopeptidase | MAB_3564c | 3.543 | 2.606 |
| 6 | B1MDI7 | SnoaL-like domain-containing protein | MAB_3230c | 1.098 | 1.712 |
| 7 | B1MBL9 | Peptidyl-prolyl cis–trans isomerase | MAB_2559c | 1.897 | 1.431 |
| 8 | B1MEW6 | Uncharacterized protein | MAB_3496 | 3.839 | 1.030 |
Remarkably, this pre-incubation with VM043 also resulted in a statistically significant number of four enzymes being captured by the VM055p probe (see red dots in Fig. 2C); namely a hypothetical dipeptidyl aminopeptidase MAB_3564c, a snoaL-like domain-containing protein MAB_3230c (Rv2910c), an uncharacterized protein MAB_3496, and the peptidyl-prolyl cis–trans isomerase MAB_2559c whose ortholog Rv3909 is annotated as essential for M. tuberculosis H37Rv in vitro growth (Fig. 2C, Tables 4 and S3†). Of particular interest, although these latter four enzymes were also captured by the VM055p probe alone, they were however found to be below our p-value ≤ 0.05 and fold change ≥ 1.0 thresholds when compared to DMSO non-specific conditions (Table S3†).
These later findings thus suggest that the antibacterial activity of VM043 and VM055p against M. abscessus growth should be due to the inhibition of a comparable spectrum of target enzymes by these two molecules, resulting in a similar anti-mycobacterial activity. Therefore, a posteriori, it is not surprising that pre-incubation of M. abscessus S cells with VM043 modifies the availability/accessibility/selectivity of these target enzymes with respect to VM055p, since proteins that were covalently inhibited by VM043 can no longer react and be captured by VM055p.
Moreover, these proteomic data also suggest that, as previously reported for two other families of multi-target inhibitors,24,28,29,54,85 VM β-lactones would impair the growth of M. abscessus and M. tuberculosis by inhibiting of the activity of several enzymes involved in various important physiological processes. As a direct consequence, the likelihood of a strain developing resistance to such inhibitors would be very low, because resistant mutants should acquire multiple mutations in the same bacterial genome; making it difficult or impossible for the bacteria to adapt and survive.
3.5. Cell toxicity of beta-lactone derivatives
One of the keys to the success of M. tuberculosis86,87 as well as M. abscessus7,16,88 as lung pathogens is their ability to survive and replicate inside infected macrophages. As a result, new drugs must be able to inhibit only the intracellular growth of the pathogen and not be cytotoxic to host cells. In this regard, we determined for each β-lactone analog the concentration required to induce a 50% decrease in Raw264.7 murine macrophage cells viability,89 i.e. CC50 (ref. 55) (Table S7†). Among the 38 tested derivatives, VM049p and VM050p bearing a short pentynyl R2 chain and the three β-lactam analogs VM056 to VM058 exhibited high toxicity towards Raw264.7 cells with CC50 values in the range of 30–70 μg mL−1, close to their MIC90. Otherwise, except VM039 which is slightly toxic (CC50 = 119 ± 4 μg mL−1), the other molecules had no cytotoxic effect against Raw264.7 macrophages at the highest concentration tested (125 μg mL−1).3.6. Intramacrophagic susceptibility of M. abscessus to selected β-lactone derivatives
Given these latter findings, we further investigated the ability of the β-lactones to inhibit the intra-macrophagic growth of M. abscessus S.30,54,85,90,91 To achieve this goal, Raw264.7 cells were infected with M. abscessus S at a multiplicity of infection (MOI) of 10, and then incubated for 24 h with selected β-lactone compounds or with imipenem (IMP; 80 μg mL−1 ∼ 9.5 × MIC50Raw) used as positive drug control for this intracellular killing assay.54,85 The selected inhibitors tested were VM025, VM026 (= trans-VM025), and VM043 which are the best inhibitors of M. abscessus S & R growth in broth medium. The inactive growth inhibitor VM027 (= cis-VM025), as well as the two probes of VM043, VM053p and VM055p which differ in their “clickable” group (alkyne vs. azide), were also included (Table 5 and Fig. 3).| Compounds | CC50 (μg mL−1) | MIC50Raw (μg mL−1) | SI | |
|---|---|---|---|---|
| a Experiments were performed as described in the Experimental section. CC50: compound concentration leading to 50% Raw264.7 macrophages toxicity. IC50Raw: minimal compound concentration leading to a 50% decrease in RLU count as compared to untreated cells. Raw264.7 macrophages were infected by M. abscessus S-LuxG13 at a MOI of 10, and further treated with each β-lactone or IMP for 24 h. The viable mycobacteria were quantified by measurement of luminescence from luciferase-expressing M. abscessus S-LuxG13 within Raw264.7 macrophages. Untreated infected macrophages were used as control representing 100% of bacterial viability. MIC50Raw were calculated from curve fitting of RLU% as a function of the inhibitor concentration and are expressed as mean values of three independent assays.b Data from ref. 54. IMP, imipenem. ND: not determined. SI, selectivity index, SI = CC50/MIC50Raw. | ||||
| VM025 |  |
>125 | 13.6 ± 1.1 | >9.2 |
| VM026 = trans-VM025 |  |
>125 | 12.9 ± 1.2 | >9.7 |
| VM027 = cis-VM025 |  |
>125 | 27.9 ± 1.4 | >4.5 |
| VM043 | 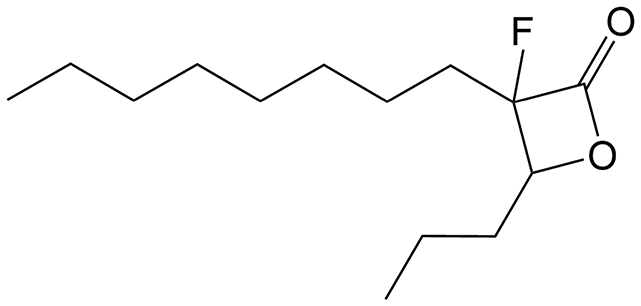 |
>125 | >100 | — |
| VM045 |  |
>125 | 18.9 ± 1.3 | >6.5 |
| VM046 = fluorinated-VM045 |  |
>125 | >100 | — |
| VM053p = non-fluorinated VM043 probe | 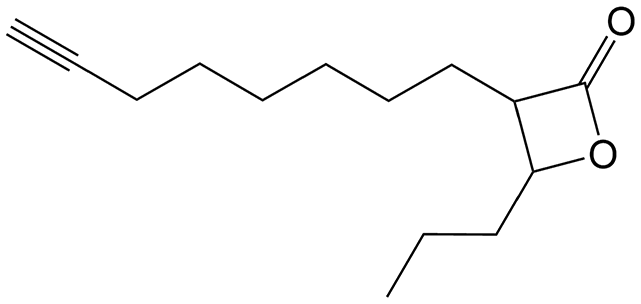 |
>125 | 48.6 ± 3.8 | >2.6 |
| VM055p = non-fluorinated VM043 probe |  |
>125 | 24.9 ± 3.3 | >5.0 |
| IMPb | 8.5 ± 1.4 | — | ||
 | ||
| Fig. 3 Intracellular activity of VM025, VM026, VM027, and VM043 as compared to imipenem (IMP). The activity of selected β-lactones on intracellular M. abscessus growth was tested in Raw264.7 murine macrophages. Cells were infected at a multiplicity of infection (MOI) of 10 with M. abscessus S-LuxG13 and treated with various concentrations of each inhibitor or IMP for 24 h. The viable mycobacteria were quantified by measurement of luminescence from luciferase-expressing M. abscessus S-LuxG13 within Raw264.7 macrophages. Untreated infected macrophages were used as control representing 100% of bacterial viability. Untreated infected macrophages were used as control representing 100% of bacterial viability. Results are shown as mean ± standard error of the mean (SEM) of three independent assays performed in triplicate. SC, solvent control (DMSO). ***, p-value <0.001. *, p-value <0.05. Statistical analysis was done using a Student's t-test. | ||
Among the compounds tested, VM025 and its trans-isomer VM026 exhibited promising antibacterial activity against intracellular M. abscessus growth, with an approximated MIC50Raw of around 13 μg mL−1 which is only 1.5 times higher than that of IMP (MIC50Raw = 8.5 μg mL−1) (Fig. 3 and Table 5). Interestingly, the cis-isomer VM027 which was not active against extracellular mycobacteria (Table 1), was however able to significantly decrease the intramacrophagic M. abscessus present 24 h post-infection, with a MIC50Raw of around 28 μg mL−1. On the other hand, VM043 bearing a fluorine atom at the C-3 position of the β-lactone ring had no activity against the intracellular growth of M. abscessus (Fig. 3, Table 5 and Fig. S1†). The potential “negative” effect of the fluorine atom was further confirmed by testing VM046 and its non-fluorinated analog, VM045 (Table 5 and Fig. S1†). Indeed, although VM046 was inactive, VM045 was able to significantly inhibit the intracellular growth of M. abscessus with a MIC50Raw of around 19 μg mL−1. Finally, the two non-fluorinated probes displayed quite good (MIC50Raw = 24.9 μg mL−1 for VM055p) to moderate (MIC50Raw = 48.6 μg mL−1 for VM053p) antibacterial activities against intracellular M. abscessus S growth (Table 5 and Fig. S1†).
The fact that MIC values determined in broth medium do not always correlate with the activity of the compounds against intracellular bacteria, is not new. In 2015, Vanderven et al. demonstrated that among 1359 hits tested in a screening assay against M. tuberculosis, only 141 compounds were able to inhibit both the extracellular and intracellular growth of the bacillus, and 132 inhibited bacterial replication inside macrophages with little or no inhibitory activity in broth medium.92 Another best example is the first-line anti-TB drug pyrazinamide93 which is inactive in vitro at neutral pH in conventional culture media, but displays potent activity against M. tuberculosis in an acidic environment (pH 5.5 or below) mimicking the endolysosomal pH of macrophages. Regarding multi-target inhibitors, such a discrepancy between extracellular and intracellular activities has also been reported and discussed in previous works with two other families of growth inhibitors of M. tuberculosis and M. abscessus; namely the oxadiazolone (OX) derivatives28,85 and the cyclipostins & cyclophostin (CyC) analogs.29,30,32,54 In particular, we have recently demonstrated that the CyCs which were active against intracellular M. abscessus growth accumulated in acidic compartments inside macrophage cells, and that this accumulation was essential for their delivery to mycobacteria-containing phagosomes and thus for their antimicrobial activity against intramacrophagic M. abscessus.30
Here, the fact that fluorine derivatives were fully inactive against intramacrophagic M. abscessus, as in the case of the best extracellular growth inhibitor VM043, may suggest a specific behavior or mode of action of these molecules inside infected macrophages. On the other hand, VM025 and VM026 which had a promising extracellular activity (MIC90 ∼54 μg mL−1) were 4 times more active against M. abscessus intracellular growth within macrophages (MIC50Raw ∼13 μg mL−1). One hypothesis to explain this clear preference for intracellularly replicating mycobacteria could be that the corresponding target enzyme(s) of these inhibitors would be more accessible and/or vulnerable during the intracellular lifestyle of M. abscessus compared to its extracellular replication. However, a specific response of the infected macrophages resulting from the stress effect of these compounds and leading to bacterial clearance cannot be excluded.
Given the previously determined very low toxicity of the β-lactones toward Raw264.7 cells with CC50 > 125 μg mL−1, the selectivity index (SI = CC50/MIC50Raw) of the intracellular inhibitors on M. abscessus vs. Raw264.7 cells was calculated and found to be in a promising range from >2.6 and up to >9.0 (Table 4).
From these findings, it can be assumed that the observed inhibitory potency of the β-lactone derivatives i) might result from the inhibition of specific but most likely distinct mycobacterial target enzymes between intramacrophagic- vs. extracellularly-replicating mycobacteria; or ii) might reflect differences in the uptake and accumulation of the different compounds inside the macrophage. Overall, these results suggest that the non-fluorinated derivatives would be able to enter the macrophages and arrest bacterial replication without exhibiting significant toxicity to the host cell, with comparable antibacterial activity to imipenem.
4. Conclusion
In the present work, a new series of lipophilic compounds based on β-lactone-core were synthesized by varying the nature of the substituents on the lactone ring. Evaluation of their antibacterial activity first highlighted VM038, VM040, VM043 and VM045 as potential candidates against M. tuberculosis. With respect to M. abscessus, the MIC determination of this set of 30 derivatives provided VM025 and VM026, in addition to the latter VM043, as efficient inhibitors of both S and R variants. A competitive click-chemistry ABPP approach and comparative chemical proteomics with a newly synthesized customized activity-based probe VM055p revealed several M. abscessus target enzymes of VM043, the best inhibitor of extracellular growth, compared to VM055p, thus confirming the multi-target nature of this family of molecules. When tested against intracellular bacteria, while VM043 was found inactive, VM025 & VM026 emerged as potent and promising inhibitors of intramacrophagic M. abscessus growth with MIC50Raw values comparable to the standard antibiotic imipenem. This dual activity is of major importance as it may affect the different stages of the infection process.Hence, thanks to their multitargeted covalent mechanism of action, our results underscore the added value of β-lactone probes. In particular, we anticipate that these probes would represent attractive tools against mycobacterial infections, and provide interesting insights into the different stages of the infection process that may lead to the arrest of two main mycobacterial pathogens, M. tuberculosis and/or M. abscessus. Identifying the proteins inactivated by our antibacterial activity-based probes would indeed reveal new potential targets for treating mycobacterial-related diseases, and contribute to background information for the development of new therapeutic strategies for elimination of either actively replicating or latent mycobacteria from infected individuals.
Further work to better understand the behavior of our β-lactone derivatives inside macrophage cells and consequently to elucidate their mode of action against intracellular M. abscessus is currently in progress.
Abbreviations
| ABP | Activity-based probe |
| ABPP | Activity-based protein profiling |
| AMK | Amikacin |
| CC50 | Compound concentration leading to 50% of cell cytotoxicity |
| CC-ABPP | Click-chemistry activity-based protein profiling |
| CF | Cystic fibrosis |
| IMP | Imipenem |
| MIC50/MIC90 | Minimal inhibitory concentration leading to 50% and 90% of bacterial growth inhibition, respectively |
| MIC50Raw | Minimal inhibitory concentration leading to 50% of bacterial growth inhibition inside infected macrophages |
| NTM | Nontuberculous mycobacterial |
| REMA | Resazurin microtiter assay |
| RLU | Relative luminescence unit |
| SI | Selectivity index |
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files. The mass spectrometry proteomics data are available online through the ProteomeXchange Consortium (http://www.proteomexchange.org) with the dataset identifiers PXD057836.Author contributions
Thomas Francis: formal analysis – investigation – visualization – writing-review & editing. Christina Dedaki: formal analysis – investigations – resources – writing-review & editing. Phoebe Ananida-Dasenaki: investigations – resources. Dimitra Bolka: investigations – resources. Kanellos Albanis: investigations – resources. Filippos Foteinakis: investigations – resources. Julie Mezquida: formal analysis – investigation. Marie Hance: formal analysis – investigation. Alexandros Athanasoulis: investigations – resources. Anna-Krinio Papagiorgou: investigations – resources. Ioanna-Foteini Karampoula: investigations – resources. George Georgitsis: investigations – resources. Celia Jardin: data curation – formal analysis – investigation. Stéphane Audebert: data curation – formal analysis – investigation – writing-review & editing. Luc Camoin: data curation – writing-review & editing. Céline Crauste: writing-review & editing. Stéphane Canaan: writing-review & editing. Victoria Magrioti: conceptualization – formal analysis – methodology – project administration – resources – supervision – validation – visualization – writing-original draft – writing-review & editing. Jean-François Cavalier: conceptualization – data curation – formal analysis – investigation – methodology – project administration – supervision – validation – visualization – writing-original draft – writing-review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the Centre National de la Recherche Scientifique (CNRS) and Aix-Marseille Université (AMU) and by the Special Account for Research Grants of the National and Kapodistrian University of Athens (SARG/NKUA). Proteomics analyses were done using the mass spectrometry facility of Marseille Proteomics (http://marseille-proteomique.univ-amu.fr) supported by IBISA, the Cancéropôle PACA, the Provence-Alpes-Côte d'Azur Region, the Institut Paoli-Calmettes, and Fonds Européen de Développement Regional (FEDER). T. F. PhD fellowship was supported by the Ministère de l'Enseignement Supérieur et de la Recherche Française.References
- E. Tortoli, Microbiological features and clinical relevance of new species of the genus Mycobacterium, Clin. Microbiol. Rev., 2014, 27, 727–752 CrossRef PubMed.
- E. Tortoli, T. Fedrizzi, C. J. Meehan, A. Trovato, A. Grottola, E. Giacobazzi, G. F. Serpini, S. Tagliazucchi, A. Fabio, C. Bettua, R. Bertorelli, F. Frascaro, V. De Sanctis, M. Pecorari, O. Jousson, N. Segata and D. M. Cirillo, The new phylogeny of the genus Mycobacterium: The old and the news, Infect., Genet. Evol., 2017, 56, 19–25 CrossRef PubMed.
- WHO, Global tuberculosis report, https://www.who.int/teams/global-tuberculosis-programme/data, (accessed accessed 20 November 2024).
- D. G. Russell, Mycobacterium tuberculosis: here today, and here tomorrow, Nat. Rev. Mol. Cell Biol., 2001, 2, 569–577 CrossRef CAS PubMed.
- E. Catherinot, J. Clarissou, G. Etienne, F. Ripoll, J. F. Emile, M. Daffe, C. Perronne, C. Soudais, J. L. Gaillard and M. Rottman, Hypervirulence of a rough variant of the Mycobacterium abscessus type strain, Infect. Immun., 2007, 75, 1055–1058 CrossRef CAS PubMed.
- R. Nessar, E. Cambau, J. M. Reyrat, A. Murray and B. Gicquel, Mycobacterium abscessus: a new antibiotic nightmare, J. Antimicrob. Chemother., 2012, 67, 810–818 CrossRef CAS PubMed.
- M. D. Johansen, J. L. Herrmann and L. Kremer, Non-tuberculous mycobacteria and the rise of Mycobacterium abscessus, Nat. Rev. Microbiol., 2020, 18, 392–407 CrossRef CAS PubMed.
- J. E. Stout, W. J. Koh and W. W. Yew, Update on pulmonary disease due to non-tuberculous mycobacteria, Int. J. Infect. Dis., 2016, 45, 123–134 CrossRef PubMed.
- J. M. Leung and K. N. Olivier, Nontuberculous mycobacteria in patients with cystic fibrosis, Semin. Respir. Crit. Care Med., 2013, 34, 124–134 CrossRef PubMed.
- A. L. Roux, E. Catherinot, F. Ripoll, N. Soismier, E. Macheras, S. Ravilly, G. Bellis, M. A. Vibet, E. Le Roux, L. Lemonnier, C. Gutierrez, V. Vincent, B. Fauroux, M. Rottman, D. Guillemot, J. L. Gaillard and Jean-Louis Herrmann for the OMA Group, Multicenter study of prevalence of nontuberculous mycobacteria in patients with cystic fibrosis in france, J. Clin. Microbiol., 2009, 47, 4124–4128 CrossRef PubMed.
- P. H. Candido, S. Nunes Lde, E. A. Marques, T. W. Folescu, F. S. Coelho, V. C. de Moura, M. G. da Silva, K. M. Gomes, M. C. Lourenco, F. S. Aguiar, F. Chitolina, D. T. Armstrong, S. C. Leao, F. P. Neves, F. C. Mello and R. S. Duarte, Multidrug-resistant nontuberculous mycobacteria isolated from cystic fibrosis patients, J. Clin. Microbiol., 2014, 52, 2990–2997 CrossRef PubMed.
- E. Catherinot, A. L. Roux, E. Macheras, D. Hubert, M. Matmar, L. Dannhoffer, T. Chinet, P. Morand, C. Poyart, B. Heym, M. Rottman, J. L. Gaillard and J. L. Herrmann, Acute respiratory failure involving an R variant of Mycobacterium abscessu, J. Clin. Microbiol., 2009, 47, 271–274 CrossRef PubMed.
- K. Dheda, T. Gumbo, G. Maartens, K. E. Dooley, R. McNerney, M. Murray, J. Furin, E. A. Nardell, L. London, E. Lessem, G. Theron, P. van Helden, S. Niemann, M. Merker, D. Dowdy, A. Van Rie, G. K. H. Siu, J. G. Pasipanodya, C. Rodrigues, T. G. Clark, F. A. Sirgel, A. Esmail, H.-H. Lin, S. R. Atre, H. S. Schaaf, K. C. Chang, C. Lange, P. Nahid, Z. F. Udwadia, C. R. Horsburgh, G. J. Churchyard, D. Menzies, A. C. Hesseling, E. Nuermberger, H. McIlleron, K. P. Fennelly, E. Goemaere, E. Jaramillo, M. Low, C. M. Jara, N. Padayatchi and R. M. Warren, The epidemiology, pathogenesis, transmission, diagnosis, and management of multidrug-resistant, extensively drug-resistant, and incurable tuberculosis, Lancet, 2017, 5, 291–360 Search PubMed.
- R. C. Lopeman, J. Harrison, M. Desai and J. A. G. Cox, Mycobacterium abscessus: Environmental Bacterium Turned Clinical Nightmare, Microorganisms, 2019, 7(3), 90 CrossRef CAS PubMed.
- B. J. Seaworth and D. E. Griffith, Therapy of Multidrug-Resistant and Extensively Drug-Resistant Tuberculosis, Microbiol. Spectrum, 2017, 5 DOI:10.1128/microbiolspec.tnmi7-0042-2017.
- A. L. Roux, A. Viljoen, A. Bah, R. Simeone, A. Bernut, L. Laencina, T. Deramaudt, M. Rottman, J. L. Gaillard, L. Majlessi, R. Brosch, F. Girard-Misguich, I. Vergne, C. de Chastellier, L. Kremer and J. L. Herrmann, The distinct fate of smooth and rough Mycobacterium abscessus variants inside macrophages, Open Biol., 2016, 6, 160185 CrossRef PubMed.
- Y. M. Boudehen and L. Kremer, Mycobacterium abscessus, Trends Microbiol., 2021, 29, 951–952 CrossRef CAS PubMed.
- B. E. Jönsson, M. Gilljam, A. Lindblad, M. Ridell, A. E. Wold and C. Welinder-Olsson, Molecular epidemiology of Mycobacterium abscessus, with focus on cystic fibrosis, J. Clin. Microbiol., 2007, 45, 1497–1504 CrossRef PubMed.
- P. J. Brennan and H. Nikaido, The envelope of mycobacteria, Annu. Rev. Biochem., 1995, 64, 29–63 CrossRef CAS PubMed.
- M. Jackson, The mycobacterial cell envelope-lipids, Cold Spring Harbor Perspect. Med., 2014, 4, a021105 CrossRef PubMed.
- S. M. Batt, D. E. Minnikin and G. S. Besra, The thick waxy coat of mycobacteria, a protective layer against antibiotics and the host's immune system, Biochem. J., 2020, 477, 1983–2006 CrossRef PubMed.
- G. Gago, L. Diacovich and H. Gramajo, Lipid metabolism and its implication in mycobacteria-host interaction, Curr. Opin. Microbiol., 2018, 41, 36–42 CrossRef CAS PubMed.
- T. van der Klugt, R. van den Biggelaar and A. Saris, Host and bacterial lipid metabolism during tuberculosis infections: possibilities to synergise host- and bacteria-directed therapies, Crit. Rev. Microbiol., 2024, 1–21 Search PubMed.
- J. F. Cavalier, C. D. Spilling, T. Durand, L. Camoin and S. Canaan, Lipolytic enzymes inhibitors: A new way for antibacterial drugs discovery, Eur. J. Med. Chem., 2021, 209, 112908 CrossRef CAS PubMed.
- G. Johnson, The alpha/beta Hydrolase Fold Proteins of Mycobacterium tuberculosis, With Reference to their Contribution to Virulence, Curr. Protein Pept. Sci., 2017, 18, 190–210 Search PubMed.
- R. Avellan, M. Sarrazin, C. D. Spilling, C. Crauste, S. Canaan and J.-F. Cavalier, Chapter 11 - Deciphering the physiological role of serine enzymes involved in mycobacterial lipid metabolism using activity-based protein profiling, in Biology of Mycobacterial Lipids, ed. Z. Fatima and S. Canaan, Academic Press, 2022, pp. 235–251, DOI:10.1016/B978-0-323-91948-7.00001-4.
- M. Li, H. V. Patel, A. B. Cognetta, 3rd, T. C. Smith 2nd, I. Mallick, J. F. Cavalier, M. L. Previti, S. Canaan, B. B. Aldridge, B. F. Cravatt and J. C. Seeliger, Identification of cell wall synthesis inhibitors active against Mycobacterium tuberculosis by competitive activity-based protein profiling, Cell Chem. Biol., 2022, 29, 883–896 CrossRef PubMed , e885.
- P. C. Nguyen, V. Delorme, A. Benarouche, A. Guy, V. Landry, S. Audebert, M. Pophillat, L. Camoin, C. Crauste, J. M. Galano, T. Durand, P. Brodin, S. Canaan and J.-F. Cavalier, Oxadiazolone derivatives, new promising multi-target inhibitors against M. tuberculosis, Bioorg. Chem., 2018, 81, 414–424 Search PubMed.
- P. C. Nguyen, V. Delorme, A. Benarouche, B. P. Martin, R. Paudel, G. R. Gnawali, A. Madani, R. Puppo, V. Landry, L. Kremer, P. Brodin, C. D. Spilling, J.-F. Cavalier and S. Canaan, Cyclipostins and Cyclophostin analogs as promising compounds in the fight against tuberculosis, Sci. Rep., 2017, 7, 11751 Search PubMed.
- M. Sarrazin, B. P. Martin, R. Avellan, G. R. Gnawali, I. Poncin, H. Le Guenno, C. D. Spilling, J. F. Cavalier and S. Canaan, Synthesis and Biological Characterization of Fluorescent Cyclipostins and Cyclophostin Analogues: New Insights for the Diagnosis of Mycobacterial-Related Diseases, ACS Infect. Dis., 2022, 8, 2564–2578 CrossRef CAS PubMed.
- K. R. Tallman, S. R. Levine and K. E. Beatty, Small Molecule Probes Reveal Esterases with Persistent Activity in Dormant and Reactivating Mycobacterium tuberculosis, ACS Infect. Dis., 2016, 2, 936–944 CrossRef CAS PubMed.
- P. C. Nguyen, A. Madani, P. Santucci, B. P. Martin, R. R. Paudel, S. Delattre, J. L. Herrmann, C. D. Spilling, L. Kremer, S. Canaan and J.-F. Cavalier, Cyclophostin and Cyclipostins analogues, new promising molecules to treat mycobacterial-related diseases, Int. J. Antimicrob. Agents, 2018, 51, 651–654 CrossRef CAS PubMed.
- P. Hadvary, W. Sidler, W. Meister, W. Vetter and H. Wolfer, The lipase inhibitor tetrahydrolipstatin binds covalently to the putative active site serine of pancreatic lipase, J. Biol. Chem., 1991, 266, 2021–2027 CrossRef CAS PubMed.
- A. Benarouche, V. Point, F. Carriere and J. F. Cavalier, Using the reversible inhibition of gastric lipase by Orlistat for investigating simultaneously lipase adsorption and substrate hydrolysis at the lipid-water interface, Biochimie, 2014, 101, 221–231 CrossRef CAS PubMed.
- A. M. Heck, J. A. Yanovski and K. A. Calis, Orlistat, a new lipase inhibitor for the management of obesity, Pharmacotherapy, 2000, 20, 270–279 CrossRef CAS PubMed.
- L. Haalck and F. Spener, On the inhibition of microbial lipases by tetrahydrolipstatin, Methods Enzymol., 1997, 286, 252–263 CAS.
- L. Kremer, C. De Chastellier, G. Dobson, K. J. C. Gibson, P. Bifani, S. Balor, J.-P. Gorvel, C. Locht, D. E. Minnikin and G. S. Besra, Identification and structural characterization of an unusual mycobacterial monomeromycolyl-diacylglycerol, Mol. Microbiol., 2005, 57, 1113–1126 CrossRef CAS PubMed.
- S. K. Parker, R. M. Barkley, J. G. Rino and M. L. Vasil, Mycobacterium tuberculosis Rv3802c encodes a phospholipase/thioesterase and is inhibited by the antimycobacterial agent tetrahydrolipstatin, PLoS One, 2009, 4, e4281 CrossRef PubMed.
- P. K. Crellin, J. P. Vivian, J. Scoble, F. M. Chow, N. P. West, R. Brammananth, N. I. Proellocks, A. Shahine, J. Le Nours, M. C. J. Wilce, W. J. Britton, R. L. Coppel, J. Rossjohn and T. Beddoe, Tetrahydrolipstatin inhibition, functional analyses, and three-dimensional structure of a lipase essential for mycobacterial viability, J. Biol. Chem., 2010, 285, 30050–30060 CrossRef PubMed.
- N. P. West, K. M. Cergol, M. Xue, E. J. Randall, W. J. Britton and R. J. Payne, Inhibitors of an essential mycobacterial cell wall lipase (Rv3802c) as tuberculosis drug leads, Chem. Commun., 2011, 47, 5166–5168 RSC.
- V. Delorme, S. V. Diomande, L. Dedieu, J.-F. Cavalier, F. Carriere, L. Kremer, J. Leclaire, F. Fotiadu and S. Canaan, MmPPOX inhibits Mycobacterium tuberculosis lipolytic enzymes belonging to the hormone-sensitive lipase family and alters mycobacterial growth, PLoS One, 2012, 7, e46493 CrossRef CAS PubMed.
- M. S. Ravindran, S. P. Rao, X. Cheng, A. Shukla, A. Cazenave-Gassiot, S. Q. Yao and M. R. Wenk, Targeting lipid esterases in mycobacteria grown under different physiological conditions using activity-based profiling with tetrahydrolipstatin (THL), Mol. Cell. Proteomics, 2014, 13, 435–448 CrossRef CAS PubMed.
- C. M. Goins, T. D. Sudasinghe, X. Liu, Y. Wang, G. A. O'Doherty and D. R. Ronning, Characterization of Tetrahydrolipstatin and Stereoderivatives on the Inhibition of Essential Mycobacterium tuberculosis Lipid Esterases, Biochemistry, 2018, 57, 2383–2393 CrossRef CAS PubMed.
- C. M. Goins, S. Dajnowicz, M. D. Smith, J. M. Parks and D. R. Ronning, Mycolyltransferase from Mycobacterium tuberculosis in covalent complex with tetrahydrolipstatin provides insights into antigen 85 catalysis, J. Biol. Chem., 2018, 293, 3651–3662 CrossRef CAS PubMed.
- P. Santucci, C. Dedaki, A. Athanasoulis, L. Gallorini, A. Munoz, S. Canaan, J.-F. Cavalier and V. Magrioti, Synthesis of long chain β-lactones and their antibacterial activities against pathogenic mycobacteria, ChemMedChem, 2019, 14, 349–358 CrossRef CAS PubMed.
- C. Rens, F. Laval, M. Daffe, O. Denis, R. Frita, A. Baulard, R. Wattiez, P. Lefevre and V. Fontaine, Effects of lipid-lowering drugs on vancomycin susceptibility of mycobacteria, Antimicrob. Agents Chemother., 2016, 60, 6193–6199 CrossRef CAS PubMed.
- J. Lehmann, T. Y. Cheng, A. Aggarwal, A. S. Park, E. Zeiler, R. M. Raju, T. Akopian, O. Kandror, J. C. Sacchettini, D. B. Moody, E. J. Rubin and S. A. Sieber, An Antibacterial beta-Lactone Kills Mycobacterium tuberculosis by Disrupting Mycolic Acid Biosynthesis, Angew. Chem., Int. Ed., 2018, 57, 348–353 CrossRef CAS PubMed.
- S. S. Khan, T. D. Sudasinghe, A. D. Landgraf, D. R. Ronning and S. J. Sucheck, Total Synthesis of Tetrahydrolipstatin, Its Derivatives, and Evaluation of Their Ability to Potentiate Multiple Antibiotic Classes against Mycobacterium Species, ACS Infect. Dis., 2021, 7, 2876–2888 CrossRef CAS PubMed.
- V. K. Sambandamurthy, S. C. Derrick, T. Hsu, B. Chen, M. H. Larsen, K. V. Jalapathy, M. Chen, J. Kim, S. A. Porcelli, J. Chan, S. L. Morris and W. R. Jacobs, Jr., Mycobacterium tuberculosis DeltaRD1 DeltapanCD: a safe and limited replicating mutant strain that protects immunocompetent and immunocompromised mice against experimental tuberculosis, Vaccine, 2006, 24, 6309–6320 CrossRef CAS PubMed.
- N. Andreu, A. Zelmer, T. Fletcher, P. T. Elkington, T. H. Ward, J. Ripoll, T. Parish, G. J. Bancroft, U. Schaible, B. D. Robertson and S. Wiles, Optimisation of bioluminescent reporters for use with mycobacteria, PLoS One, 2010, 5, e10777 CrossRef PubMed.
- B. S. Simcox, B. R. Tomlinson, L. N. Shaw and K. H. Rohde, Mycobacterium abscessus DosRS two-component system controls a species-specific regulon required for adaptation to hypoxia, Front. Cell. Infect. Microbiol., 2023, 13, 1144210 CrossRef CAS PubMed.
- D. G. Lee, Y. H. Hwang, E. J. Park, J. H. Kim and S. W. Ryoo, Clomiphene Citrate Shows Effective and Sustained Antimicrobial Activity against Mycobacterium abscessus, Int. J. Mol. Sci., 2021, 22, 11029 CrossRef CAS PubMed.
- J. C. Palomino, A. Martin, M. Camacho, H. Guerra, J. Swings and F. Portaels, Resazurin microtiter assay plate: simple and inexpensive method for detection of drug resistance in Mycobacterium tuberculosis, Antimicrob. Agents Chemother., 2002, 46, 2720–2722 CrossRef CAS PubMed.
- A. Madani, J. N. Ridenour, B. P. Martin, R. R. Paudel, A. Abdul Basir, V. Le Moigne, J. L. Herrmann, S. Audebert, L. Camoin, L. Kremer, C. D. Spilling, S. Canaan and J.-F. Cavalier, Cyclipostins and Cyclophostin Analogues as Multitarget Inhibitors That Impair Growth of Mycobacterium abscessus, ACS Infect. Dis., 2019, 5, 1597–1608 CrossRef CAS PubMed.
- C. Rodrigues Felix, R. Gupta, S. Geden, J. Roberts, P. Winder, A. Pomponi Shirley, C. Diaz Maria, K. Reed John, E. Wright Amy and H. Rohde Kyle, Selective Killing of Dormant Mycobacterium tuberculosis by Marine Natural Products, Antimicrob. Agents Chemother., 2017, 61 DOI:10.1128/aac.00743-17.
- T. Christophe, M. Jackson, H. K. Jeon, D. Fenistein, M. Contreras-Dominguez, J. Kim, A. Genovesio, J. P. Carralot, F. Ewann, E. H. Kim, S. Y. Lee, S. Kang, M. J. Seo, E. J. Park, H. Skovierova, H. Pham, G. Riccardi, J. Y. Nam, L. Marsollier, M. Kempf, M. L. Joly-Guillou, T. Oh, W. K. Shin, Z. No, U. Nehrbass, R. Brosch, S. T. Cole and P. Brodin, High content screening identifies decaprenyl-phosphoribose 2′ epimerase as a target for intracellular antimycobacterial inhibitors, PLoS Pathog., 2009, 5, e1000645 CrossRef PubMed.
- J. C. Jewett and C. R. Bertozzi, Cu-free click cycloaddition reactions in chemical biology, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- J. C. Jewett, E. M. Sletten and C. R. Bertozzi, Rapid Cu-free click chemistry with readily synthesized biarylazacyclooctynones, J. Am. Chem. Soc., 2010, 132, 3688–3690 CrossRef CAS PubMed.
- B. M. Babin, L. Atangcho, M. B. van Eldijk, M. J. Sweredoski, A. Moradian, S. Hess, T. Tolker-Nielsen, D. K. Newman and D. A. Tirrell, Selective Proteomic Analysis of Antibiotic-Tolerant Cellular Subpopulations in Pseudomonas aeruginosa Biofilms, mBio, 2017, 8 DOI:10.1128/mbio.01593-17.
- A. M. Frankenfield, J. Ni, M. Ahmed and L. Hao, Protein Contaminants Matter: Building Universal Protein Contaminant Libraries for DDA and DIA Proteomics, J. Proteome Res., 2022, 21, 2104–2113 CrossRef CAS PubMed.
- S. Tyanova and J. Cox, Perseus: A Bioinformatics Platform for Integrative Analysis of Proteomics Data in Cancer Research, in Cancer Systems Biology: Methods and Protocols, ed. L. von Stechow, Springer New York, New York, NY, 2018, pp. 133–148. DOI:10.1007/978-1-4939-7493-1_7.
- V. G. Tusher, R. Tibshirani and G. Chu, Significance analysis of microarrays applied to the ionizing radiation response, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5116–5121 CrossRef CAS PubMed.
- E. W. Deutsch, N. Bandeira, V. Sharma, Y. Perez-Riverol, J. J. Carver, D. J. Kundu, D. Garcia-Seisdedos, A. F. Jarnuczak, S. Hewapathirana, B. S. Pullman, J. Wertz, Z. Sun, S. Kawano, S. Okuda, Y. Watanabe, H. Hermjakob, B. MacLean, M. J. MacCoss, Y. Zhu, Y. Ishihama and J. A. Vizcaino, The ProteomeXchange consortium in 2020: enabling 'big data' approaches in proteomics, Nucleic Acids Res., 2020, 48, D1145–D1152 CAS.
- Y. Perez-Riverol, J. Bai, C. Bandla, D. Garcia-Seisdedos, S. Hewapathirana, S. Kamatchinathan, D. J. Kundu, A. Prakash, A. Frericks-Zipper, M. Eisenacher, M. Walzer, S. Wang, A. Brazma and J. A. Vizcaino, The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences, Nucleic Acids Res., 2022, 50, D543–D552 CrossRef CAS PubMed.
- D. W. Brooks, L. D. L. Lu and S. Masamune, C-Acylation under Virtually Neutral Conditions, Angew. Chem., Int. Ed., 2003, 18, 72–74 CrossRef.
- T. Mukaiyama, Reactions based on the onium salts of azaaromatics, Pure Appl. Chem., 1979, 51, 1337–1346 CAS.
- S. Singh, N. Bouzinbi, V. Chaturvedi, S. Godreuil and L. Kremer, In vitro evaluation of a new drug combination against clinical isolates belonging to the Mycobacterium abscessus complex, Clin. Microbiol. Infect., 2014, 20, O1124–1127 CrossRef CAS PubMed.
- Y. I. Ho, C. Y. Chan and A. F. Cheng, In-vitro activities of aminoglycoside-aminocyclitols against mycobacteria, J. Antimicrob. Chemother., 1997, 40, 27–32 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, iLOGP: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach, J. Chem. Inf. Model., 2014, 54, 3284–3301 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- B. F. Cravatt, A. T. Wright and J. W. Kozarich, Activity-based protein profiling: from enzyme chemistry to proteomic chemistry, Annu. Rev. Biochem., 2008, 77, 383–414 CrossRef CAS PubMed.
- T. Bottcher and S. A. Sieber, Beta-lactones as privileged structures for the active-site labeling of versatile bacterial enzyme classes, Angew. Chem., Int. Ed., 2008, 47, 4600–4603 CrossRef PubMed.
- G. M. Simon and B. F. Cravatt, Activity-based proteomics of enzyme superfamilies: serine hydrolases as a case study, J. Biol. Chem., 2010, 285, 11051–11055 CrossRef CAS PubMed.
- H. K. Lembke and E. E. Carlson, Activity-based probes in pathogenic bacteria: Investigating drug targets and molecule specificity, Curr. Opin. Chem. Biol., 2023, 76, 102359 CrossRef CAS PubMed.
- C. A. Brown and A. Yamashita, Saline hydrides and superbases in organic reactions. IX. Acetylene zipper. Exceptionally facile contrathermodynamic multipositional isomeriazation of alkynes with potassium 3-aminopropylamide, J. Am. Chem. Soc., 1975, 97, 891–892 CrossRef CAS.
- K. R. Venrooij, L. de Bondt and K. M. Bonger, Mutually Orthogonal Bioorthogonal Reactions: Selective Chemistries for Labeling Multiple Biomolecules Simultaneously, Top. Curr. Chem., 2024, 382, 24 CrossRef CAS PubMed.
- D. Rifat, L. Chen, B. N. Kreiswirth and E. L. Nuermberger, Genome-Wide Essentiality Analysis of Mycobacterium abscessus by Saturated Transposon Mutagenesis and Deep Sequencing, mBio, 2021, 12, e0104921 CrossRef PubMed.
- M. Kanehisa, Y. Sato, M. Kawashima, M. Furumichi and M. Tanabe, KEGG as a reference resource for gene and protein annotation, Nucleic Acids Res., 2016, 44, D457–D462 CrossRef CAS PubMed.
- H. Ogata, S. Goto, K. Sato, W. Fujibuchi, H. Bono and M. Kanehisa, KEGG: Kyoto Encyclopedia of Genes and Genomes, Nucleic Acids Res., 1999, 27, 29–34 CrossRef CAS PubMed.
- A. Kapopoulou, J. M. Lew and S. T. Cole, The MycoBrowser portal: a comprehensive and manually annotated resource for mycobacterial genomes, Tuberculosis, 2011, 91, 8–13 CrossRef CAS PubMed.
- E. V. Koonin, Orthologs, Paralogs, and Evolutionary Genomics, Annu. Rev. Genet., 2005, 39, 309–338 CrossRef CAS PubMed.
- J. E. Griffin, J. D. Gawronski, M. A. DeJesus, T. R. Ioerger, B. J. Akerley and C. M. Sassetti, High-Resolution Phenotypic Profiling Defines Genes Essential for Mycobacterial Growth and Cholesterol Catabolism, PLoS Pathog., 2011, 7, e1002251 CrossRef CAS PubMed.
- C. M. Sassetti, D. H. Boyd and E. J. Rubin, Genes required for mycobacterial growth defined by high density mutagenesis, Mol. Microbiol., 2003, 48, 77–84 CrossRef CAS PubMed.
- K. Cotes, R. Dhouib, I. Douchet, H. Chahinian, A. de Caro, F. Carriere and S. Canaan, Characterization of an exported monoglyceride lipase from Mycobacterium tuberculosis possibly involved in the metabolism of host cell membrane lipids, Biochem. J., 2007, 408, 417–427 CrossRef CAS PubMed.
- A. Madani, I. Mallick, A. Guy, C. Crauste, T. Durand, P. Fourquet, S. Audebert, L. Camoin, S. Canaan and J. F. Cavalier, Dissecting the antibacterial activity of oxadiazolone-core derivatives against Mycobacterium abscessus, PLoS One, 2020, 15, e0238178 CrossRef CAS PubMed.
- S. B. Cohen, B. H. Gern, J. L. Delahaye, K. N. Adams, C. R. Plumlee, J. K. Winkler, D. R. Sherman, M. Y. Gerner and K. B. Urdahl, Alveolar Macrophages Provide an Early Mycobacterium tuberculosis Niche and Initiate Dissemination, Cell Host Microbe, 2018, 24, 439–446 CrossRef CAS PubMed , e434.
- L. Huang, E. V. Nazarova and D. G. Russell, Mycobacterium tuberculosis: Bacterial Fitness within the Host Macrophage, Microbiol. Spectrum, 2019, 7, 7.2.04 CrossRef PubMed.
- T. F. Byrd and C. R. Lyons, Preliminary characterization of a Mycobacterium abscessus mutant in human and murine models of infection, Infect. Immun., 1999, 67, 4700–4707 CrossRef CAS PubMed.
- T. Parish, In vitro drug discovery models for Mycobacterium tuberculosis relevant for host infection, Expert Opin. Drug Discovery, 2020, 15, 349–358 CrossRef CAS PubMed.
- E. Le Run, M. Arthur and J. L. Mainardi, In Vitro and Intracellular Activity of Imipenem Combined with Tedizolid, Rifabutin, and Avibactam against Mycobacterium abscessus, Antimicrob. Agents Chemother., 2019, 63 DOI:10.1128/aac.01915-18.
- A. L. Lefebvre, V. Le Moigne, A. Bernut, C. Veckerle, F. Compain, J. L. Herrmann, L. Kremer, M. Arthur and J. L. Mainardi, Inhibition of the beta-Lactamase BlaMab by Avibactam Improves the In Vitro and In Vivo Efficacy of Imipenem against Mycobacterium abscessus, Antimicrob. Agents Chemother., 2017, 61 DOI:10.1128/aac.02440-16.
- B. C. VanderVen, R. J. Fahey, W. Lee, Y. Liu, R. B. Abramovitch, C. Memmott, A. M. Crowe, L. D. Eltis, E. Perola, D. D. Deininger, T. Wang, C. P. Locher and D. G. Russell, Novel inhibitors of cholesterol degradation in Mycobacterium tuberculosis reveal how the bacterium's metabolism is constrained by the intracellular environment, PLoS Pathog., 2015, 11, e1004679 CrossRef PubMed.
- P. Santucci, D. J. Greenwood, A. Fearns, K. Chen, H. Jiang and M. G. Gutierrez, Intracellular localisation of Mycobacterium tuberculosis affects efficacy of the antibiotic pyrazinamide, Nat. Commun., 2021, 12, 3816 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary information for this article is available online. Additional file 1: detailed protocols related to biological evaluation and to the synthesis of intermediates as well as new β-lactone derivatives; Table S1, code numbers, structures and logPO/W of all β-lactone compounds; Fig. S1, intracellular activity of VM045, VM046, as well as the two probes, VM053p and VM055p, against M. abscessus S infected macrophages; NMR spectra of the new compounds synthesized (PDF). Additional file 2: Tables S2–S6, target proteins identified from M. abscessus S culture, through CC-ABPP by LC-ESI-MS/MS analysis, using either VM055p probe alone, or following VM043 pre-incubation and labeling with VM055p probe; Table S7, cytotoxic activities of the β-lactone derivatives towards Raw264.7 murine macrophage cells (pdf). See DOI: https://doi.org/10.1039/d5md00102a |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |