
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding the HIV-CA protein and the ligands that bind at the N-terminal domain (NTD) - C-terminal domain (CTD) interface†
Stuart
Lang
 *
*
New Cambridge House, Bassingbourn Road, Litlington, Cambridgeshire, SG8 0SS, UK. E-mail: stuart.lang@cresset-group.com
First published on 17th April 2025
Abstract
Treatment and prevention of HIV/AIDS infections represents a significant global challenge, with this being the cause of a substantial number of deaths each year. HIV-CA, the protein responsible for protecting the viral RNA and facilitating reverse transcription, has emerged as an important target in drug discovery. This review applies various computer drug discovery tools for the analysis and understanding of not only the HIV-CA protein, but also the ligands reported to bind to the site at the NTD-CTD interface between two capsid monomer units. Combining this evaluation with reported experimental data, highlights the effects that changes to the ligands make to the binding affinity. This analysis, including identifying areas of the ligand that have not been adequately explored, allows for the generation of guidelines that can be applied to the design of novel ligands that bind to HIV-CA.
 Stuart Lang | Stuart Lang obtained his PhD at the University of Strathclyde and following a fellowship at the University if York has worked on drug discovery projects at a variety of stages. Through this he has developed a knowledge and understanding of synthetic medicinal chemistry and computer aided drug design both in the pharmaceutical industry (AstraZeneca) and in academic drug discovery (University of Strathclyde and Drug Discovery Unit at the University of Dundee). Currently working as the project manager in the Discovery team at Cresset, he works with a skilled and talented team of scientists to apply molecular modelling solutions for the discovery of novel bioactive molecules. |
1. Analysis of HIV-capsid (CA) bind site and its ligands
Human immunodeficiency viruses (HIV) are lentiviruses that contain two RNA genomes that infect hosts through reverse transcription into double stranded DNA, followed by integration into the hosts genome to ensure a productive infection.1 This initial infection can be subsequently followed by infection of CD4+ T cells and macrophages leading to acquired immune deficiency syndrome (AIDS).2 This review will focus specifically on the capsid (CA) protein, which has the role of protecting the viral RNA from the host's natural immune system and providing optimal conditions for reverse transcription. Focus will be on the ligands that interact with this protein at the N-terminal domain (NTD) and C-terminal domain (CTD) interface, specifically interaction with amino acids Gln50, Leu56, Asn57, Gln63, Met66, Leu69, Lys70, Ile73, Asn74, and Thr107 in the NTD and Ile37, Ser41, Tyr169, and Arg173 in the CTD.The HIV-CA protein, consisting of an of assembly roughly 1200–1500 monomer units of repeated hexamers (approximately 250) and pentamers (exactly 12) producing a fullerene cone geometry,3–5 provides an opportunity for therapeutic small molecule intervention to inhibit infection of the HIV virus. There are two strategies that have been proposed for small molecules interacting with HIV-CA, the first is premature uncoating of the protective HIV-CA protein before it has the opportunity to infect the cell, the second is stabilisation of the HIV-CA protein so that it is unable to release the viral RNA into the host cell.6,7
A key interaction that has been exploited in the design of molecules that bind to HIV-CA is one also demonstrated by two of the co-factors (CPSF6 and Nup153),8,9 which are both involved in capsid nuclear entry. This site, at the NTD-CTD, is predicted as being druggable using a pocket detection tool10 (Fig. 1A and B). The highlighted phenylalanine in both CPSF6 (Fig. 1C) and Nup153 (Fig. 1D) interact, through the phenyl ring, with Leu56, Met66, and the sidechain of Lys70 and, through the N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, with Asn57.11 An interesting point of note is the interaction with the sidechain of Lys70. While, due to the amine head group, lysine can be considered a polar amino acid, it also contains a lipophilic chain that links this amine group to the protein backbone. This sidechain, as is seen here, can make interactions with lipophilic groups.
O, with Asn57.11 An interesting point of note is the interaction with the sidechain of Lys70. While, due to the amine head group, lysine can be considered a polar amino acid, it also contains a lipophilic chain that links this amine group to the protein backbone. This sidechain, as is seen here, can make interactions with lipophilic groups.
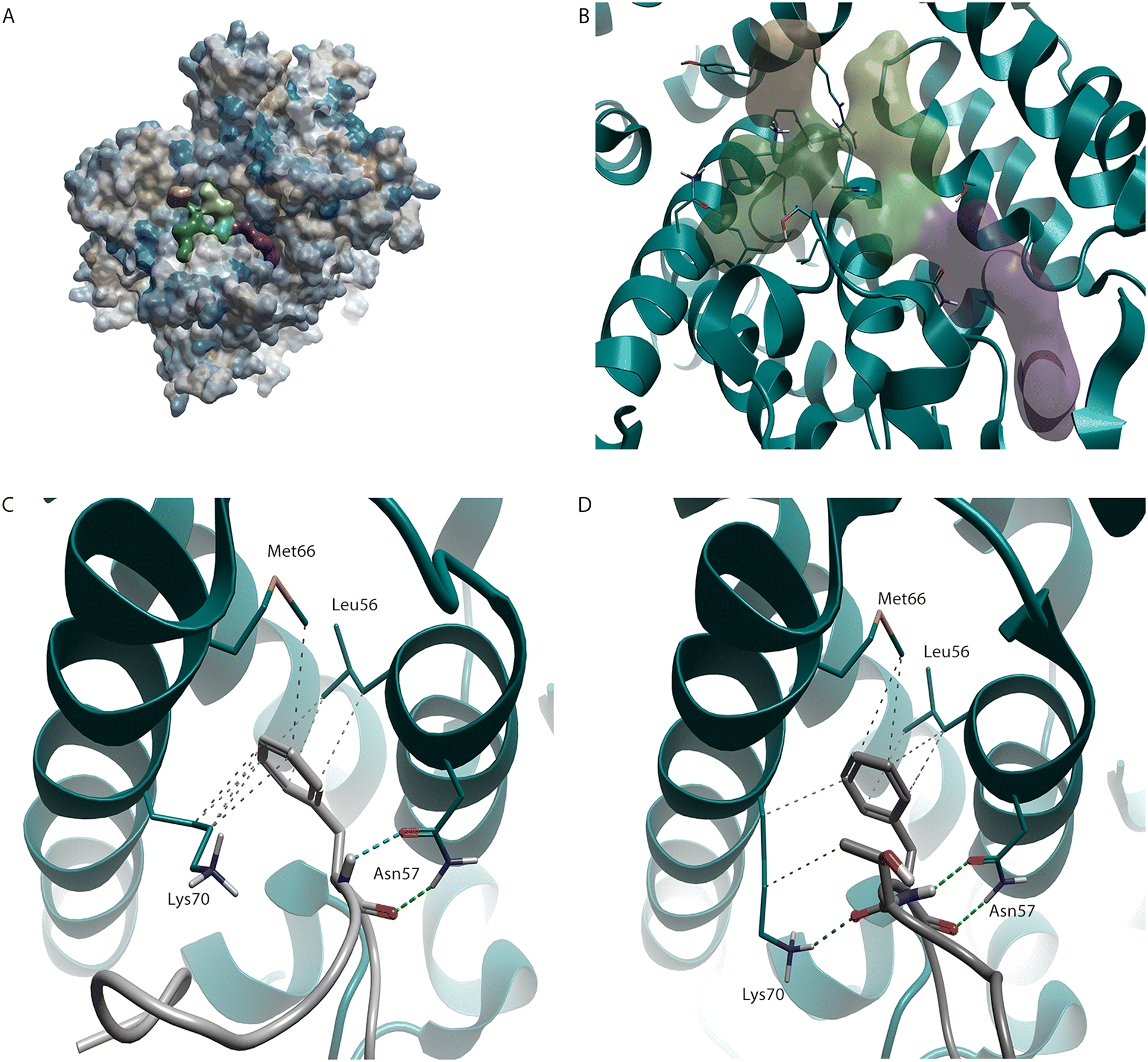 | ||
| Fig. 1 HIV-CA binding site. (A) HIV-CA protein (PDB: 5HGN) with hydrophobicity surface added. Areas coloured yellow represent areas of high lipophilicity while areas coloured blue represent areas of high hydrophilicity with grey coloured areas falling in an intermediate range. The green, orange, and purple regions (B) were detected using a pocket detection tool and represent the NTD-CTD binding site covered in this review. This pocket has been shown to bind to both co-factors CPSF6 (C, PDB: 8CL1) and Nup153 (D, PDB: 8CKY). | ||
These interactions are key recognition motifs present in all the small molecules that will be discussed in this review. This phenylalanine-glycine (FG) binding site12 is mainly lipophilic and is situated predominately in the NTD, however ligands that bind in this site can also, by accessing a suitable vector, extend into the interface between the NTD and the CTD. Lipophilic amino acids, particularly aromatic amino acids like phenylalanine, tyrosine, and tryptophan often make key interactions in protein–protein/peptide interactions.13 This is because groups of this type are not happy being exposed to the polar aqueous environment that can exist outside the protein, instead wanting to bury their lipophilicity in more suitable surroundings.14
One small molecule that takes advantage of this phenylalanine-based interaction with HIV-CA is PF-3450074 (1), herein referred to as PF-74 (Fig. 2A).15 This molecule contains a central phenylalanine unit that conserves the interactions, through the phenyl ring, with Leu56, Met66, and the sidechain of Lys70 along with the interactions, through the N–H and CO, with Asn57 (Fig. 2B).16 PF-74 (1) also contains a tertiary amide which, in the binding site, adopts a cis geometry allowing the phenyl ring to interact with the methyl group of Thr107.
 | ||
| Fig. 2 PF-74 (1) bound to HIV-CA (PDB: 4U0E). (A) PF-74 (1) 2D structure. (B) PF-74 (1) inside binding site of HIV-CA. (C) Hydrophobicity surface added to HIV-CA. 3D-RISM water analysis of PF-74 (1) bound to HIV-CA, with predicted thermodynamically favourable (green) and unfavourable (red) water molecules shown as spheres. (D) PF-74 (1) shown with positive (red), negative (blue), hydrophobic (gold), and van der Waals (yellow) field surfaces. (E) PF-74 (1) shown with positive (red), negative (blue), hydrophobic (gold), and van der Waals (yellow) field points. (F) PF-74 (1) shown with Electrostatic Complementarity™ (EC) to HIV-CA surface of with favourable (green) and unfavourable (red) regions highlighted. | ||
PF-74 (1) makes a series of interactions with the amino group on the head of Lys 70, with the CO making a hydrogen bonding interaction and indole making two cation–π interactions (one with each ring). The pyrrole component of the indole also makes a cation–π interaction with Arg173, which is situated in CTD. The N-H of the indole also makes a hydrogen bonding interaction with the CO in the backbone of Gln63.
A key analysis of this system is mapping the regions that predict if water molecules will be energetically favoured or unfavoured.17 Using a 3D-RISM calculation18 the positions that water is predicted to be located are shown as spheres, with the colours highlighting the water molecules that can be easily displaced (red) and those that will suffer a binding energy penalty if a ligand is displaced (green). It is clear from this analysis that there is space to grow the ligand in the region of the phenyl ring next to the cis amide (Fig. 2C) with the water molecules in this area being predicted to be energetically disfavoured.
The water molecules in the area around the methyl group of the cis amide (Fig. 2C), toward the NTD-CTD, are not as energetically unfavourable as those around the phenyl ring. However, as the majority of these predicted water molecules are coloured white, there is still an opportunity to explore these regions of the protein when developing molecules that bind to this site in the HIV-CA protein, a strategy adopted in the discovery of lenacapavir (also known as GS-6207).
Lenacapavir (2)19 binds to the same site as PF-74 (1) and there are similarities in the binding mode adopted for the two molecules. One of the major problems with PF-74 (1) is its poor metabolic stability, resulting in a short half-life.20,21 The peptidyl nature of PF-74 (1) makes it susceptible to enzymatic degradation of the amide bond. This metabolism issue was a key consideration in the development of lenacapavir (2). Not only has the phenyl ring present in the phenylalanine component of the molecule been replaced with a difluoroaryl ring, but the labile tertiary cis amide has been locked in the desired bioactive conformation by introduction of a pyridine ring (Fig. 3B).22 The nitrogen atom of this pyridine makes the same interaction with the N–H of Asn57 that was seen with the amide CO that it replaced in PF-74 (1), representing an example stabilising the molecule with use of a bioisostere replacement.23,24
 | ||
| Fig. 3 Lenacapavir (2) bound to HIV-CA (PDB: 6VKV). (A) Lenacapavir (2) 2D structure. (B) Lenacapavir (2) in binding site of HIV-CA. (C) Lenacapavir (2) shown with positive (red), negative (blue), hydrophobic (gold), and van der Waals (yellow) field surfaces. (D) Lenacapavir (2) shown with positive (red), negative (blue), hydrophobic (gold), and van der Waals (yellow) field points. (E) Lenacapavir (2) shown with Electrostatic Complementarity (EC) to HIV-CA surface of with favourable (green) and unfavourable (red) regions highlighted. | ||
The phenyl ring that interacts with Thr107 has been replaced with a chloro-indazole. The chlorine atom makes a halogen bond with the CO of Asn74. There is an additional interaction between the N–H of Asn74 and a SO, along with the sulfonamide N anion, of the sulfonamide substituent on the indazole ring. This indazole ring can also make a π–cation interaction with the amine on Lys70, which although also possible with the phenyl ring of PF-74 (1) was not displayed in the crystal structure (Fig. 2B). An additional factor that may give an improvement in binding affinity is that the highly substituted indazole is considerably larger than the phenyl ring that it replaced. This increase in volume will have resulted in displacement of unhappy water molecules, predicted to be present in the PF-74 (1) system (Fig. 2C).
A key difference with lenacapavir (2) and PF-74 (1) is in the pyrazole derivative that has replaced the indole at the NTD-CTD interface. There are two particularly noticeable variations, the first is that the pyrazole no longer contains a N-H therefore losing the H-bonding interaction with Gln63, although this is replaced with a hydrophobic interaction with the chain of the Gln63, the second is that the carbocyclic ring fills a different portion in the NTD-CTD interface than the indole arene system of PF-74 (1). This new binding mode causes movement of Met66 to bind to the group at the NTD-CTD interface and is now not interacting with the difluoroaryl ring. In addition to the previously discussed interaction the cyclopropane makes with Gln63, it also makes an additional hydrophobic interaction with the chain of Tyr169 in the CTD, along with the pyrazole ring conserving the CTD π–cation interaction with the Arg173.
In addition to optimisation within the silhouette of PF-74 (1), lenacapavir (2) also grows into a previously unexplored pocket of the protein. This region was previously identified as a druggable pocket using the pocket detection analysis (Fig. 1A and B, highlighted in purple). Through use of an alkyne linker, lenacapavir (2) can make interactions through the SO groups of a sulfone with a N–H on Asn57 and the O–H of Ser43. An additional hydrophobic interaction is made between the Me group attached to the sulfone and the sidechain of Gln50.
PF-74 (1) and lenacapavir (2) can also be expressed in terms of their field surface (Fig. 2D and 3C respectively),25 which can be used to calculate a molecular interaction potential for the respective ligand. This molecular field surface can be simplified to a field point (Fig. 2E and 3D), these field points can be used as a 3D molecular descriptor for ligand-based design and virtual screening.26 It is not only for the ligand that a field surface can be calculated, this can also be done with the protein. Generation of the electrostatic field surface for both ligand and protein allow the Electrostatic Complementarity™ (EC) surface to be calculated (Fig. 2F and 3E).27 Areas that are a match, or complement each other, are shown in green, with electrostatically mismatched regions shown in red. From a visual perspective the EC surface can be represented on either the protein or the ligand (it is shown on the ligand in Fig. 2F and 3E). This EC surface can be used to assess the areas of the ligand that are electrostatically compatible with the protein and the areas where a clash is observed.
A striking observation made from the analysis of the EC surface for PF-74 (1) is, while there is a definite interaction between the π-systems of the indole group with the NH3 cation on Lys70, this interaction is not electrostatically favourable (Fig. 2F). This may be due to the indole ring being a suboptimal position with respect to Lys70, because its N-H making an electrostatically favourable interaction with the amide CO. Although not governed by any interaction with the amine group in Lys70 the phenyl ring in the phenylalanine component of PF-74 (1) is also not electrostatically compatible with its protein environment. While there is slight improvement with the EC in lenacapavir (2), perhaps resulting from the introduction of electronegative fluorine atoms, there are still areas that have poor EC, most notably in the hydrophobic groups in the vicinity of the sulfone.
Lenacapavir (2) inspired the design of GSK878 (3),28 which maintains the difluoroaryl ring and the pyrazole that binds at the NTD-CTD interface (Fig. 4). It also contains the indazole ring, however because of the replacement of the pyridine with a quinazolinone, the CH2CF3 group can be trimmed back to a methyl group and still lock its axial chirality, specifically atropisomerism.29 Another interesting observation when comparing the 6VKV crystal structure with lenacapavir (2) bound and the 8FIU structure containing GSK878 (3) is that the Thr107 residue has rotated, with the Me group making a lipophilic contact with the indazole of lenacapavir (2), while the OH makes an H-bonding interaction with the CO in the quinazolinones group of GSK878 (3). Furthermore, the replacement of the sulfone in lenacapavir (2) with a morpholine in GSK878 (3) shows an improved EC profile.
 | ||
| Fig. 4 GSK878 (3) bound to HIV-CA (PDB: 8FIU). (A) GSK878 (3) 2D structure. (B) GSK878 (3) in binding site of HIV-CA. (C) GSK878 (3) shown with Electrostatic Complementarity (EC) to HIV-CA surface of with favourable (green) and unfavourable (red) regions highlighted. | ||
While the evolution of PF-74 (1) to lenacapavir (2) and subsequent design of GSK878 (3) represents a tour de force in rational structure-based drug design, an alternative approach to finding compounds that bind in this pocket is using high throughput screening (HTS).30 An HTS of ∼60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds was used as part of the process to identify BI-2 (4) (Fig. 5A), along with its structural analogue BI-1 (5).31 Crystallography of BI-2 (4) (Fig. 5B) shows, while it binds to similar pockets in the NTD to PF-74 (1), it does not make any interactions at NTD-CTD interface observed with the indole in PF-74 (1).16
000 compounds was used as part of the process to identify BI-2 (4) (Fig. 5A), along with its structural analogue BI-1 (5).31 Crystallography of BI-2 (4) (Fig. 5B) shows, while it binds to similar pockets in the NTD to PF-74 (1), it does not make any interactions at NTD-CTD interface observed with the indole in PF-74 (1).16
 | ||
| Fig. 5 BI-2 (4) bound to HIV-CA (PDB: 4U0F). (A) BI-2 (4) 2D structure. (B) BI-2 (4) in binding site of HIV-CA. (C) BI-2 (3) shown with Electrostatic Complementarity (EC) to HIV-CA surface with favourable (green) and unfavourable (red) regions highlighted. | ||
The unsubstituted phenyl ring of BI-2 (4) makes hydrophobic interactions with Leu56 and the sidechain of Lys70, like PF-74 (1). However, the position of the phenyl ring in BI-2 (4) is slightly different, allowing it to interact with Leu69. As was the case with lenacapavir (2), the interaction between this aryl system and Met66 is lost. However, as BI-2 (4) does not have a group at the NTD-CTD interface, there is no opportunity to regain this contact. An interesting observation when analysing the binding mode of BI-2 (4) (Fig. 5B) is that the amide group on the sidechain of Asn57 has rotated (compared to Fig. 2B, 3B, and 4B) to satisfy the alternative hydrogen bond donor/acceptor requirements with this scaffold.
The phenol ring of BI-2 (4) while located in the same pocket at the phenyl ring connected to the cis amide in PF-74 (1), does not interact with the same residues. In fact, the Thr107, which made a hydrophobic interaction with the phenyl ring in PF-74 (1) has rotated, as seen with GSK838 (3), in this system allowing the alcohol oxygen to make an H-bond with the N-H of the lactam in BI-2 (4). This places the phenol ring deeper in this pocket, shown by the water analysis (Fig. 2C) of PF-74 (1) to contain energetically unfavoured water molecules. The aryl system of this phenol makes hydrophobic interactions with the side chain of Lys70 and Ile73. An additional interaction on this system, which would not be possible with BI-1 (5), is the phenol OH which can make an H-bond with the CO of the Asn74, a residue that was also targeted with lenacapavir (2) and GSK878 (3).
While the phenyl and phenol rings of BI-2 (4) map well with their equivalent rings systems in compounds 1–3, the alternative orientation of the Asn57 primary amide makes growth toward Tyr169 and Arg171 in the CTD difficult with this scaffold. The vector exploited by compounds 1–3 is unavailable in BI-2 (4), with the pyrazole N acting as a hydrogen bond acceptor for the N-H of Asn57. Furthermore, the requirement of the pyrazole N–H to bind to the CO of Asn57 means that the vector is also not available for growing into the purple pocket (Fig. 1A and B). The presence of the lactam CO in BI-2 (4) also makes growth into this pocket from that position of the scaffold challenging.
From this analysis, it is possible to map the key residues that are interacting with the ligand and are likely to be responsible for activity. The residues that interact with the phenylalanine unit (and its bioisosteres) in the FG binding site are essential for the function of the capsid and therefore unlikely to be mutated. The main residue responsible for H-bonding is Asn57, which functions as both an H-bond donor and acceptor. The Leu56 and Lys70 are also key residues in this area that all the ligands described in this analysis bind to, with Met66 and Leu69 also providing binding opportunities.
Thr107 is an interesting amino acid residue, in both PF-74 (1) and lenacapavir (2) in that it makes a hydrophobic interaction with an aryl ring. However, if this group is rotated, exposing the alcoholic O–H group, it can make polar interactions such as H-bonding with the CO of the quinazolinones GSK878 (3) and the lactam N-H in BI-2 (4). This highlights that, while in molecules such as lenacapavir (2), it is possible to take advantage of the highly lipophilic nature of the protein to improve potency, there is an opportunity to interact with the polar groups that are presented within the pocket.
The Asn74 residue offers another opportunity within this pocket. Both lenacapavir (2) and GSK878 (3) make interactions with this residue via both a halogen bond with the chloro group and an H-bond with the SO and N anion of the sulfonamide that are both substituents on the indazole system. This residue also interacts with the phenolic OH of BI-2 (4), although interaction with Asn74 is not possible with either PF-74 (1) or BI-1 (5).
Interaction with amino acids that are in the CTD side of the NTD-CTD interface offers an attractive strategy, with both PF-74 (1) and lenacapavir (2) taking advantage of this. While many of the interactions made with groups at this interface are with amino acids in the NTD, such as Gln63 and Lys70, the π–cation interaction of PF-74 (1) with Arg173 provides an opportunity to also bind to the CTD, as does the lipophilic interaction with the cyclopropyl ring of lenacapavir (2) and GSK878 (3) to Tyr169.
This binding pose analysis also highlights the transient nature of the specific positions of the amino acids within the pocket, particularly those with flexible side chains. Along with the Thr107 rotation, we have also observed a rotation of the key Asn57 residue when binding to BI-2 (4) and BI-1 (5) compared to the other ligands, along with significant movement of the Met66 and Lys70 side chains. Similarly, the H-bonding interaction that the indole N–H of PF-74 (1) makes with CO in the amide side chain of Gln63 is completely different to the hydrophobic interaction that the cyclopropane of lenacapavir (2) and GSK878 (3) make with the lipophilic chain of Gln63. This means that care must be taken when designing new ligands as modifications made at one part of the molecule could have an impact on the binding of another group.
2. SAR analysis: Efficiency metrics and 3D-QSAR analysis
While docking32 and EC27 scored binding poses can give an indication of a ligand's probability of binding, the docking or EC scores that are associated with the ligand binding pose cannot be used as an absolute measurement of binding affinity. There are several reasons for this, but a key one is that the docking or EC score is only generated for the single ligand binding pose33 and protein conformation being analysed.34 The experimentally measured binding energy will be due to the contributions from many of the ligand and protein conformations that exist.35 Therefore, effort is needed when selecting a binding conformation to ensure that it is representative of the true bioactive conformation. It is also worth noting that a high energy conformation can still give a good docking score if it makes favourable interactions with the protein, however there will be an energy penalty associated with the ligand adopting this conformation that will result in the molecule being less active than the docking score would suggest.36Water molecules are used to fill the unoccupied pockets in a protein,37 as demonstrated with the 3D-RISM analysis (Fig. 2C), these can be energetically stable within the protein. Displacement of these water molecules may cause a lower binding affinity. With water molecules that are energetically unstable, displacement of these water molecules could lead to an increased binding affinity.38
As not all the parameters responsible for the binding affinity are captured in the protein-ligand binding pose, with the ligand's behaviour outside of the protein also being critical. A ligand that has a high degree of flexibility will exist in multiple low energy conformations while in solution.39 There will be an energy penalty associated with reorientating this ligand from this solution conformation(s) to the bioactive conformation, which may be higher in energy. Another factor that will affect a ligand's activity is its lipophilicity.40,41 Compounds that are highly lipophilic will prefer to bind to lipophilic areas of a protein target rather than the more hydrophilic aqueous environment that predominately exists outside the protein. This means that lipophilic molecules will appear to be more potent, although because they prefer to bind to proteins in general, as opposed to being in solution, there will be little specificity for the target protein compared to other proteins.42 This can lead to selectivity issues and can result in problems associated with toxicity43 and metabolism.44,45
To understand the contribution to binding energy for each part of a molecule it is necessary to look for patterns, or structure activity relationships (SAR).46 This SAR is not based on in silico analysis, but rather in finding patterns in the experimentally measured activity data that is associated with a series of compounds that share a binding site. As discussed, PF-74 (1), lenacapavir (2), GSK878 (3) and BI-2 (4) share a binding site, with shared interactions being made with common amino acid residues in the protein. This means that groups in similar positions can be compared. By analysis of the compounds that are structurally related (Fig. 6) it is possible to track the evolution of PF-74 (1) to lenacapavir (2) and its next generation analogues, including GSK878 (3).
 | ||
| Fig. 6 Key molecule that bind to same HIV-CA binding site – evolution from PF74 (1) to more advanced molecules. | ||
The introduction of the pyridine ring to replace the cis amide of PF-74 (1), while eliminating the possibility of rotation around the amide bond, introduces an aspect of axial chirality to the molecule. By placing larger groups at the ortho positions to the biaryl bond, such as the CH2CF3 on lenacapavir (2) or the dual effect of equivalent Me group coupled with the CO bond in molecules like 12 and GSK878 (3), it is possible to increase the energy needed to move from one form to another (Table 1).29
| Compound | pIC50 | clogP |
TPSA | LLE | LE | pIC50 (QSAR) |
|---|---|---|---|---|---|---|
| PF-74 (1) | 6.2 | 4.4 | 65 | 1.8 | 0.27 | 6.4 |
| Lenacapavir (2) | 9.7 | 8.2 | 158 | 1.4 | 0.21 | 9.7 |
| GSK878 (3) | 10.4 | 7.5 | 156 | 2.9 | 0.23 | 10.4 |
| BI-2 (4) | 5.7 | 2.6 | 78 | 3.1 | 0.36 | 5.8 |
| BI-1 (5) | 5.1 | 2.3 | 71 | 2.8 | 0.33 | 5.3 |
| 6 | 6.3 | 5.1 | 65 | 1.2 | 0.27 | 6.5 |
| 7 | 5.6 | 6.2 | 58 | −0.6 | 0.23 | 5.3 |
| 8 | 6.1 | 6.6 | 58 | −0.5 | 0.25 | 5.8 |
| 9 | 5.3 | 4.0 | 60 | 1.3 | 0.26 | 5.2 |
| GS-CA1 (10) | 9.4 | 8.4 | 158 | 1.2 | 0.21 | 9.5 |
| KFA-012 (11) | 10.1 | 7.9 | 158 | 2.2 | 0.23 | 10.1 |
| 12 | 10.3 | 6.9 | 144 | 3.4 | 0.26 | 9.9 |
In fact, it was shown that the binding energy for compound 12 in its more active axially chiral form is more than 1600 time more active than its less active partner. This represents an example of the benefits to activity, of locking a molecule in a bioactive conformation by restricting its conformational flexibility.28
From analysis of the LLE plot47 (Fig. 7A), the increase in activity in moving from PF-74 (1) to lenacapavir (2) has been driven by an increase in logP. Despite lenacapavir (2) being 3.8 log units more active than PF-74 (1) it has a lower LLE (1.4 compared to 1.8). The addition of the –Cl atom on the aromatic ring in compound 6, while allowing for an additional interaction with Asn74, gives a modest increase in activity, but because of the increase in logP of around 0.7 results in a drop in LLE with no effect in LE being observed for this change. The introduction of the –OH in BI-2 (4) to give an equivalent contact with Asn74 gives a larger jump in activity, coupled with an improvement in LLE when compared to BI-1 (5) that is unable to make this interaction.31
 | ||
| Fig. 7 (A) LLE plot of key molecule that bind to same HIV-CA binding site. (B) LE vs. LLE plot of key molecule that bind to same HIV-CA binding site. | ||
From analysis of the LE vs. LLE plot47 (Fig. 7B), lenacapavir (2) has a lower LE than PF-74 (1). One reason for this is that the addition of the pyridine ring. While this is required to reduce the metabolism of the molecule, comparing compound 7 with PF-74 (1) saw both a drop of potency and increase in logP.48 With the design of compound 7, there have been no steps taken to lock this biaryl system in the correct conformation in preference to the alternative conformations that exist as a result of the axial chirality that has been introduced, which is reflected with this drop in activity.29 While this modification only resulted in a moderate drop in LE, when comparing compound 7 with PF-74 (1), the LLE for this compound is now negative. The replacement of the indole in compound 7 with the pyrazole in compound 9, while leading to a slight drop in potency, reduced the logP significantly meaning that the LLE has now increased to 1.3 with the LE being at a similar level as observed in compound 7.
Compound 9 represents the minimum pharmacophore that has been elaborated upon to build the more advanced key ligands covered in this analysis. The aryl system attached to the pyridine can be replaced with an indazole that has been engineered to control the axial chirality and interact with the Asn74.49,50 The pyrazole has also been extended with hydrophobic groups that while displacing water molecules at the NTD-CTD interface also significantly increase the logP of the molecule. These more advanced molecules also explore the previously identified druggable pocket (Fig. 1A and B, highlighted in purple) using either a sulfone tethered by an alkyne (as shown in compounds 2, 10, and 11)51 or by replacement of the pyridine ring with a quinazolinone (as shown in compounds 3 and 12).28 The move from the pyridine to the quinazolinone, while only improving the activity slightly, reduces the logP considerably. This brings the LLE values of compounds 12 and GSK878 (3) in line with that observed for BI-1 (5) and BI-2 (4), but with significantly higher activity.31
QSAR, is a ligand-based technique that does not require any protein information in the calculation.52,53 However, as in this analysis, the protein structural information can be used to generate the ligand alignments, ensuring that the ligands are in the correct conformation to interact with the protein.54 Using Activity Atlas™, a qualitative QSAR tool, it is possible to generate an activity cliffs analysis of the system, which can allow a visual representation.55,56 Using a set of 147 ligands reported in the literature15,22,28,31,48,51,57–69 it is possible to map, when a relevant crystallographic ligand16,22,28 is added as a reference, the areas in the molecule that benefit from positive or negative electrostatics (Fig. 8A) and those that favour and disfavour hydrophobic groups (Fig. 8B). This analysis highlights the preference of a negative electrostatic field in the indazole ring of GSK878 (3). It is also evident that much of the activity of these molecules benefits from the addition of hydrophobic groups in various locations. However, this strategy has resulted in molecules with a high logP. Another aspect that is highlighted by this analysis it the lack of diversity that exists in the phenyl/difluoro motif of the molecule that interacts with the FG binding site, meaning that it is not possible to use this method to predict the changes in this region that will improve the activity of the molecule.
 | ||
| Fig. 8 Activity Atlas™ analysis with GSK878 (12) as a reference. (A) Activity cliff summary showing areas of favoured negative (blue) and positive (red) electrostatics. (B) Activity cliff summary showing areas of favourable (green) and unfavourable (purple) hydrophobics. | ||
Quantitative QSAR analysis53 can also lead to a better understanding of the system. Using the same ligand set that was used in the Activity Atlas analysis, it is possible to generate a QSAR model that can be used to predict the activity of compounds that are within the activity range of the set used to create the QSAR model (Table 1). Due to the significant crystallographic structural information that is available for this system, meaning the ligands can be aligned with a high degree of certainty, the model generated for this analysis has an R2 = 0.92 for the test set.
3. Increasing diversity through crystallisation of fragments
One of the limitations with the structural information that has been generated for ligands binding to HIV-CA is the lack of diversity that has been demonstrated to bind in the FG binding site. This limited variation in explored groups in this region is highlighted with the lack of predictability in the activity atlas analysis (Fig. 8) in both the electrostatic and hydrophobic parameters.In a recent study70 molecules with low molecular weight, or fragments,71,72 were shown to bind to HIV-CA. Fragments, due to their size are often identified as low binding affinity molecules, however the binding displayed can often be more efficient than that of molecules identified by HTS.73 While all the fragments reported have an aryl group in the FG binding site as expected (Fig. 9), it was demonstrated that an aromatic ring is not required in the pocket with Asn74 (Fig. 9B). In compound 13, the aromatic ring present in all previous examples, is replaced with a lactam that H-bonds to the amide sidechain of Asn74.
 | ||
| Fig. 9 Selected fragments known to bind to HIV-CA. (A) Fragments 13–19 2D structure. (B) Fragment 13 in binding site of HIV-CA (PDB: 8QUK). (C) Fragment 14 in binding site of HIV-CA (PDB: 8QUL). (D) Fragment 15 in binding site of HIV-CA (PDB: 8QUW). (E) Fragment 16 in binding site of HIV-CA (PDB: 8QUX). (F) Fragment 17 in binding site of HIV-CA (PDB: 8QUY). (G) Fragment 18 in binding site of HIV-CA (PDB: 8QV9). (H) Fragment 19 in binding site of HIV-CA (PDB: 8QVA). | ||
The crystallographic evidence presented demonstrates a preference for aromatic groups to be present in the phenylalanine region of PF-74 (1). These interactions are predominately lipophilicity, with the π-system of these ligands only occasionally making a π–cation interaction with Lys70 (Fig. 9B–D, and F). One example, compound 14 (Fig. 9C), showed that a pyridyl ring was tolerated in this position, as opposed to the regular phenyl group. The pyridyl nitrogen was able to make an interaction with the sulfur atom on the flexible sidechain of Met66 with the NH2 group making additional H-bonds with Asn57 and a water molecule that that is predicted by 3D-RISM to be part of a stable water network, also binding to Gln63.
Care is needed when introducing polarity to the phenylalanine pocket, introducing a phenolic OH, compound 15 (Fig. 9D), caused the entire fragment to adopt an alternative binding pose. While this adjustment is possible with fragments in a more complicated or optimised ligand, where adopting a new binding mode will not be possible, this type of change would result in a drop or complete loss of activity. However, in this case the new binding mode allows the phenolic OH to make a similar interaction with Asn74 as was seen with BI-2 (4) (Fig. 5B). Interaction with Asn74 provides an opportunity to introduce polarity to a molecule, in a region of the protein shown by 3D-RISM (Fig. 9B–H) to contain several energetically disfavoured waters.74 Maximising the occupancy of this pocket, by displacement of these water molecules,75 was a tactic used to optimise ligands such as lenacapavir (2)22 and GSK878 (3).28
It is also noteworthy that in some examples (Fig. 9D–H) an ethylene glycol molecule has been crystalised in this region. Ethylene glycol is commonly used as a co-solvent to obtain protein crystal structures, its presence in the obtained crystal structure can be used to identify areas of easily displaceable water.76 The water molecule is so easy to displace, even ethylene glycol can displace it.
The absence of the phenolic OH allows compound 16 to adopt the more expected binding pose (Fig. 9E), with the lactam making H-bonds with Asn57. Introduction of an additional carbonyl, as seen in compound 17 (Fig. 9F), induces a rotation of Thr107 to allow an H-bond to be made between this CO and the OH of Thr107. This orientation of Thr107 has also been observed in the crystal structures of GSK878 (3) (Fig. 4B) and BI-2 (4) (Fig. 5B).
With the addition of a Br group at the 7-position of the benximidazol-2-one in compound 18 (Fig. 9G), it was possible to increase the binding affinity with HIV-CA to a pKi of 5.3, with the Br group interacting with a water molecule77,78 that is shown to be stable in a 3D-RISM calculation. Taking advantage of the interactions seen with the NH2 group in compound 14 (Fig. 9C), it was possible to replace the Br with an NH2 to give compound 19 (Fig. 9H). Not only does compound 19 maintain the interaction with the stable water molecule, but it is also able to make an additional interaction with Asn57, with a pKi of 5.3 also being seen with this compound.
4. Guidelines for HIV-CA ligand design
Computational chemistry can be used to aid in the design and prioritisation of new ligands. A recent study described the use of Free Energy Perturbation (FEP) to rank molecules that bind to HIV-CA.79 Once the bioactive conformation of a ligand, or pseudo-ligand, is validated, virtual screening can be applied to identify potential binding ligands from a curated library of available ligands,80,81 but this approach is out of the scope of this review. From the analysis described in this review, it is possible to generate a series of guidelines to aid in the design of new HIV-CA ligands:• All active ligands have an aryl ring in the FG binding site, equivalent to the position of the phenylalanine in CPSF6 and Nup153. PF-74 (1) also contains a phenylalanine unit, with a lipophilic aryl replacement introduced to lenacapavir (2) and GSK878 (3) along with BI-2 (4). This phenyl system interacts with a pocket generated from Leu56, Met66, Leu69, Lys70, Ile73, with the absence of predicted water molecules in 3D-RISM showing that this pocket is well occupied.
It has been shown that a pyridyl N can be introduced, as seen with compound 14, with an interaction being made with Met66. But care needs to be added when adding more polar groups as introducing a phenolic OH, with compound 15, resulted in a change in binding mode. While there is potential to make a π–cation interaction with Lys70, the interactions are primarily lipophilic, it is unclear (based on the evidence presented) if saturated lipophilic groups are also tolerated.
• H-bonding with sidechain Asn57 is beneficial. This interaction occurs with amide groups CPSF6, Nup153, and PF-74 (1). The pyridine heterocycle used to replace an amide in lenacapavir (2) and the quinazolinones used in GSK878 (3) function as bioisosteres to mimic interaction with the NH of Asn57. This Asn57 sidechain can rotate, as seen with BI-2 (4). This orientation of the Asn57 sidechain has not been exploited as extensively. This may be because of limitations in vectors to grow toward the CTD with ligands that induce this Asn57 orientation.
• The pocket generated from Lys70, Ile73, Asn74, and Thr107 was mainly filled using lipophilic groups with PF-74 (1), lenacapavir (2), and GSK878 (3), with the latter two compounds making an interaction with the NH of Asn74 using a Cl. BI-2 (4) showed that it is possible to interact with the CO of Asn74 using a more polar OH group, an interaction also replicated with compound 15. Compound 13 showed that aromaticity is not needed in this pocket, at least providing an interaction is made with Asn74. Like Asn57, Thr107 has demonstrated two different orientations, one with the Me group making lipophilic interactions [e.g. PF-74 (1) and lenacapavir (2)] and another with the OH making polar interactions [e.g. GSK878 (3) and BI-2 (4)]. 3D-RISM shows that there is a network of unstable water molecules in this pocket, this could be exploited for increasing binding.
• The channel between the NTD and CTD is made primarily from Gln63, Met66, Lys70 in the NTD and Tyr169 and Arg173 in the CTD. While not all classes of molecule have exploited this interface between two monomers of the HIV-CA hexamer, it has been a strategy employed by the more advanced ligands. The binding mode of the indole in PF-74 (1) is predominately driven by the formation of π–cation sandwich between Lys70, the indole of PF-74 (1) and Arg173. An additional H-bond exists between the indole NH and the CO in the sidechain of Gln63.
However, the system developed for lenacapavir (2) and used in GSK878 (3), while maintaining a single π-system to facilitate a π–cation sandwich, builds in a different direction to make more lipophilic interactions with Gln66, Met66 and Tyr169. While this strategy has, at least in part, allowed a 3–4 log unit increase in potency, it has done so at the expense of the physical chemical profile of the molecule.
• There is a network of stable water molecules that need to be displaced to access the NTD-CTD interface, as seen with 3D-RISM analysis in Fig. 9. This means that a significant increase in binding affinity must be achieved in accessing this channel to counteract the penalty associated with displacing these water molecules.
• Access to additional residues Ser41, Gln50, and an additional interaction with the other face of Asn57 is achieved with lenacapavir (2) and interaction with Ile37 in the CTD is seen with GSK878 (3) by growing through a channel between Asn57 and Thr107. It is unclear, due to the limitation in published data, the benefit to binding of these additional interactions. However, like with building into the NTD-CTD interface used by the indole of PF-74 (1), 3D-RISM has shown that there are water molecules that need to be displaced to access these residues. While these were coloured white in the 3D-RISM (Fig. 2C), the increase in size of the ligand required to reach these residues, means there may be limited benefit to growing in this channel.
5. Conclusion
Analysis of the HIV-CA system using computational drug discovery tools relies on the initial generation of experimental data, such as x-ray crystallographic data for exploring the protein ligand interactions and initial proposal of a bioactive binding conformations, or activity data that can be used to build and validate a predictive QSAR model. The data analysed in this review has focused on the binding of a ligand to the primary protein of interest, however there are additional factors that require attention when developing ligands that bind to HIV-CA, such as metabolic stability, cytotoxicity, permeability, and solubility.82–84Visualisation of a ligand's binding to HIV-CA in 3D provide opportunities in generating designs, in effect to rationally design new ligands based on the binding conformation of an existing ligand. Modelling the binding conformation of the ligand in the active site, and the interactions made with the protein, allow alternative scaffolds to be explored that provide access to novel vectors, with these designs prioritised using techniques like EC and QSAR.
Further information is obtained by conducting an analysis of the water molecules in the binding site of HIV-CA. Water molecules fill the volume of the protein that is otherwise unoccupied. Through understanding of the binding energy of these water molecules, these water molecules can be classified as thermodynamically favourable and unfavourable. This information is particularly useful when growing ligands into additional pockets, for example in the optimisation of fragments.
Computational drug discovery tools will never replace the requirement to experimentally synthesize and test molecules in suitable assays. However, it should be used to provide insights that may otherwise go unnoticed. This reduces the number of molecules that require preparation and allows focus on synthesising the molecules that will most efficiently progress the project.
Data availability
The crystal structures analysed in this review have been previously published, with their PDB codes given throughout. In addition to this, all of these protein structures, along with their ligands have been aligned and superposed to the same reference frame, with this material being supplied as ESI† in .flr format. In addition to this overview of all the structures, the 3D-RISM water analysis files have also been shared as .flr files. This file format can be viewed using Flare (https://www.cresset-group.com/software/licensing-flare/), which includes a free visualizer license. The QSAR models are also shared in .flr format, although this is not compatible with the visualizer license. However, for qualifying researchers free academic license options are also available that will allow access to these QSAR files (https://www.cresset-group.com/software/academiclicensing/). Finally, all ligands used for the QSAR models has been shared in the same reference frame in a universally recognised 3D.sdf format, allowing this data set to be used with any CADD software.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Special thanks to Alana Thompson for her assistance in the generation of the images reported in this review.Notes and references
- N. M. Johnson, A. Francesca Alvarado, T. N. Moffatt, J. M. Edavettal, T. A. Swaminathan and S. E. Braun, Mol. ther., Methods Clin. Dev., 2021, 21, 451–465 CrossRef CAS PubMed.
- J. N. Blankson, Discov. Med., 2010, 9, 261–266 Search PubMed.
- G. Zhao, J. R. Perilla, E. L. Yufenyuy, X. Meng, B. Chen, J. Ning, J. Ahn, A. M. Gronenborn, K. Schulten, C. Aiken and P. Zhang, Nature, 2013, 497, 643–646 CrossRef CAS PubMed.
- J. Zhou, A. J. Price, U. D. Halambage, L. C. James, C. Aiken and W. I. Sundquist, J. Virol., 2015, 89, 270–282 Search PubMed.
- X. Zhang, L. Sun, S. Xu, X. Shao, Z. Li, D. Ding, X. Jiang, S. Zhao, S. Cocklin, E. de Clercq, C. Pannecouque, A. J. Dick, X. Liu and P. Zhan, Molecules, 2022, 27, 7640 CrossRef CAS PubMed.
- N.-Y. Chen, L. Zhou, P. J. Gane, S. Opp, N. J. Ball, G. Nicastro, M. Zufferey, C. Buffone, J. Luban, D. Selwood, F. Diaz-Griffero, I. Taylor and A. Fassati, Retrovirology, 2016, 13–28 Search PubMed.
- S. Thenin-Houssier and S. T. Valente, Curr. HIV Res., 2016, 14, 270–282 CrossRef CAS PubMed.
- A. Bhattacharya, S. L. Alam, T. Fricke, K. Zadrozny, J. Sedzicki, A. B. Taylor, B. Demeler, O. Pornillos, B. K. Granser-Pornillos, F. Diaz-Griffero, D. N. Ivanov and M. Yeager, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 18625–18630 CrossRef CAS PubMed.
- S. Zhuang and B. E. Torbett, Viruses, 2021, 13, 417 CrossRef CAS PubMed.
- V. Le Guilloux, P. Schmidtke and P. Tuffery, BMC Bioinformatics, 2009, 10, 168 CrossRef PubMed.
- J. C. V. Stacey, A. Tan, J. M. Lu, L. C. James, R. A. Dick and J. A. G. Briggs, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2220557120 CrossRef CAS PubMed.
- C. F. Dickson, S. Hertel, A. J. Tuckwell, N. Li, J. Ruan, S. C. Al-Izzi, N. Ariotti, E. Sierecki, Y. Gambin, R. G. Moris, G. J. Towers, T. Bocking and D. A. Jacques, Nature, 2024, 626, 836–842 CrossRef CAS PubMed.
- E. Lanzarotti, L. A. Defelipe, M. A. Marti and A. G. Turjanski, Aust. J. Chem., 2020, 12, 30 CAS.
- H. Gong, H. Zhang, J. Zhu, C. Wang, S. Sun, W.-M. Zheng and D. Bu, BMC Bioinformatics, 2017, 18, 70 CrossRef PubMed.
- W. S. Blair, C. Pickford, S. L. Irving, D. G. Brown, M. Anderson, R. Bazin, J. Cao, G. Ciaramella, J. Isaacson, L. Jackson, R. Hunt, A. Kjerrstrom, J. A. Nieman, A. K. Patrick, M. Perros, A. D. Scott, K. Whitby, H. Wu and S. L. Butler, PLoS Pathog., 2010, 6, e1001220 CrossRef PubMed.
- A. J. Price, D. A. Jacques, W. A. McEwan, A. J. Fletcher, S. Essig, J. W. Chin, U. D. Halambage, C. Aiken and L. C. James, PLoS Pathog., 2014, 10, e1004459 CrossRef PubMed.
- R. E. Skyner, J. L. McDonagh, C. R. Groom, T. van Mourik and J. B. O. Mitchell, Phys. Chem. Chem. Phys., 2015, 17, 6174–6191 RSC.
- E. L. Ratkova and M. V. Fedorov, J. Chem. Theory Comput., 2011, 7, 934–941 CrossRef PubMed.
- J. O. Link, M. S. Rhee, W. C. Tse, J. Zheng, J. R. Somoza, W. Rowe, R. Begley, A. Chiu, A. Mulato, D. Hansen, E. Singer, L. K. Tsai, R. A. Bam, C.-H. Chou, E. Canales, G. Brizgys, J. R. Zhang, J. Li, M. Graupe, P. Morganelli, Q. Liu, Q. Wu, R. L. Halcomb, R. D. Saito, S. D. Schroeder, S. E. Lazerwith, S. Bondy, D. Jin, M. Hung, N. Novikov, X. Liu, A. G. Villasenor, C. E. Cannizzaro, E. Y. Hu, R. L. Anderson, T. C. Appleby, B. Lu, J. Mwangi, A. Liclican, A. Niedziela-Majka, G. A. Papalia, M. H. Wong, S. A. Leavitt, Y. Xu, D. Koditek, G. J. Stepan, H. Yu, N. Pagratis, S. Clancy, S. Ahmadyar, T. Z. Cai, S. Sellers, S. A. Wolckenhauer, J. Ling, C. Callebaut, N. Margot, R. R. Ram, Y.-P. Liu, R. Hyland, G. I. Sinclair, P. J. Ruane, G. E. Crofoot, C. K. McDonald, D. M. Brainard, L. Lad, S. Swaminathan, W. I. Sundquist, R. Sakowicz, A. E. Chester, W. E. Lee, E. S. Daar, S. R. Yant and T. Cihlar, Nature, 2020, 584, 614–618 CrossRef CAS PubMed.
- M. Werle and A. Bernkop-Schnurch, Amino Acids, 2006, 30, 351–367 CrossRef CAS PubMed.
- P. F. Fitzpatrick, Biochemistry, 2003, 42, 14083–14091 CrossRef CAS PubMed.
- S. M. Bester, G. Wei, H. Zhao, D. Adu-Ampratwum, N. Iqbal, V. V. Courouble, A. C. Francis, A. S. Annamalai, P. K. Singh, N. Shkriabai, P. van Blerkom, J. Morrison, E. M. Poeschla, A. N. Engelman, G. B. Melikyan, P. R. Griffin, J. R. Fuchs, F. J. Asturias and M. Kvaratskhelia, Science, 2020, 370, 360–364 CrossRef CAS PubMed.
- S. Kumari, A. V. Carmona, A. K. Tiwari and P. C. Trippier, J. Med. Chem., 2020, 63, 12290–12358 CrossRef CAS PubMed.
- J. Tsien, C. Ju, R. R. Merchant and T. Qin, Nat. Rev. Chem., 2024, 8, 605–627 CrossRef CAS PubMed.
- J. G. Vinter, J. Comput.-Aided Mol. Des., 1994, 8, 653–668 CrossRef CAS PubMed.
- T. Cheeseright, M. Mackey, S. Rose and A. Vinter, J. Chem. Inf. Model., 2006, 42, 665–676 CrossRef PubMed.
- M. R. Bauer and M. D. Mackey, J. Med. Chem., 2019, 62, 3036–3050 CrossRef CAS PubMed.
- E. P. Gillis, K. Parcella, M. Bowsher, J. H. Cook, C. Iwuagwu, B. N. Naidu, M. Patel, K. Peese, H. Huang, L. Valera, C. Wang, K. Kieltyka, D. D. Parker, J. Simmermacher, E. Arnoult, R. T. Nolte, L. Wang, J. A. Bender, D. B. Frennesson, M. Saulnier, A. X. Wang, N. A. Meanwell, M. Belema, U. Hanumegowda, S. Jenkins, M. Krystal, J. F. Kadow, M. Cockett and R. Fridell, J. Med. Chem., 2023, 66, 1941–1954 CrossRef CAS PubMed.
- M. Basilaia, M. H. Chen, J. Secka and J. L. Gustafson, Acc. Chem. Res., 2022, 55, 2904–2919 CrossRef CAS PubMed.
- V. Blay, B. Tolani, S. P. Ho and M. R. Arkin, Drug Discovery Today, 2020, 25, 1807–1821 CrossRef CAS PubMed.
- L. Lamorte, S. Titolo, C. T. Lemke, N. Goudreau, J. F. Mercier, E. Wardrop, V. B. Shah, U. K. von Schwedler, C. Langelier, S. S. Banik and C. Aiken, Antimicrob. Agents Chemother., 2013, 57, 4622–4631 CrossRef CAS PubMed.
- B. J. Bender, S. Gahbauer, A. Luttens, J. Lyu, C. M. Webb, R. M. Stein, E. A. Fink, T. E. Balius, J. Carlsson, J. J. Irwin and B. J. Shoichet, Nat. Protoc., 2021, 16, 4799–4832 CrossRef CAS PubMed.
- L. Pinzi and G. Rastelli, Int. J. Mol. Sci., 2019, 20, 4331 CrossRef CAS PubMed.
- M. Ameral, D. B. Kokh, J. Bomke, A. Wegener, H. P. Buchstaller, H. M. Eggenweiler, P. Matias, C. Sirrenberg, R. C. Wade and M. Frech, Nat. Commun., 2017, 8, 2267 CrossRef PubMed.
- P. C. Agu, C. A. Afiukwa, O. U. Orji, E. M. Ezeh, I. H. Ofoke, C. O. Ogbu, E. I. Ugwuja and P. M. Aja, Sci. Rep., 2023, 13, 13398 CrossRef CAS PubMed.
- C. A. Chang, W. Chen and M. K. Gilson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1534–1539 CrossRef CAS PubMed.
- M. Maurer and C. Oostenbrink, J. Mol. Recognit., 2019, 32, e2810 CrossRef CAS PubMed.
- L. Wang, B. J. Berne and R. A. Friesner, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1326–1330 CrossRef CAS PubMed.
- S. J. Teague, Nat. Rev. Drug Discovery, 2003, 2, 527–541 CrossRef CAS PubMed.
- T. W. Johnson, R. A. Gallego and M. P. Edwards, J. Med. Chem., 2018, 61, 6401–6420 CrossRef CAS PubMed.
- M. L. Landry and J. J. Crawford, ACS Med. Chem. Lett., 2019, 11, 72–76 CrossRef PubMed.
- D. J. Huggins, W. Sherman and B. Tidor, J. Med. Chem., 2012, 55, 1424–1444 CrossRef CAS PubMed.
- F. Pognan, M. Beilmann, H. C. M. Boonen, A. Czich, G. Dear, P. Hewitt, T. Mow, T. Oinonen, A. Roth, T. Steger-Hartmann, J.-P. Valentin, F. van Goethem, R. J. Weaver and P. Newham, Nat. Rev. Drug Discovery, 2023, 22, 317–335 CrossRef CAS PubMed.
- A. F. Stepan, V. Mascitti, K. Beaumont and A. S. Kalgutkar, Med. Chem. Commun., 2013, 4, 631–652 RSC.
- J. Shanu-Wilson, L. Evans, S. Wrigley, J. Steele, J. Atherton and J. Boer, ACS Med. Chem. Lett., 2020, 11, 2087–2107 CrossRef CAS PubMed.
- R. Guha, Methods Mol. Biol., 2013, 993, 81–94 CrossRef CAS PubMed.
- R. J. Young and P. D. Leeson, J. Med. Chem., 2018, 61, 6421–6467 CrossRef CAS PubMed.
- R. L. Sahani, R. Diana-Rivero, S. K. V. Vernekar, L. Wangm, H. Du, H. Zhang, A. E. Castaner, M. C. Casey, K. A. Kirby, P. R. Tedbury, J. Xie, S. G. Sarafianos and Z. Wang, Viruses, 2021, 13, 479 CrossRef CAS PubMed.
- K. Singh, F. Gallazzi, K. J. Hill, D. H. Burke, M. J. Lange, T. P. Quinn, U. Neogi and A. Sonnerborg, Front. Microbiol., 2019, 10, 1227 CrossRef PubMed.
- S. Yant, A. Mulato, D. Hansen, W. C. Tse, A. Niedziela-Majka, J. R. Zhang, G. J. Stepan, D. Jin, M. H. Wong, J. M. Perreira, E. Singer, G. A. Papalia, E. Y. Hu, J. Zheng, B. Lu, S. D. Schroeder, K. Chou, S. Ahmadyar, A. Liclican, H. Yu, N. Novikov, E. Paoli, D. Gonik, R. R. Ram, M. Hung, W. H. McDougall, A. L. Brass, W. I. Sundquist, T. Cihlar and L. O. Link, Nat. Med., 2019, 25, 1377–1384 CrossRef CAS PubMed.
- S. M. Baster, D. Adu-Ampratwum, A. S. Annamalai, G. Wei, L. Briganti, B. C. Murphy, R. Haney, J. R. Fuchs and M. Kvaratskhelia, MBio, 2022, 13, e01804–e01822 Search PubMed.
- A. A. Lagunin, M. A. Romanova, A. D. Zadorozhny, N. S. Kurilenko, B. V. Shilov, P. V. Pogodin, S. M. Ivanov, D. A. Filimonov and V. V. Poroikov, Front. Pharmacol., 2018, 9, 1136 CrossRef CAS PubMed.
- E. N. Muratov, J. Bajorath, R. P. Sheridan, I. V. Tetko, D. Filimonov, V. Poroikov, T. I. Oprea, I. I. Baskin, A. Varnek, A. Roitberg, O. Isayev, S. Curtalolo, D. Fourches, Y. Cohen, A. Aspuru-Guzik, D. A. Winkler, D. Agrafiotis, A. Cherkazov and A. Tropsha, Chem. Soc. Rev., 2020, 49, 3525–3564 RSC.
- H.-H. Hsu, Y.-C. Hsu, L.-J. Chang and J.-M. Yang, BMC Genomics, 2017, 18, 104 CrossRef PubMed.
- M. Cruz-Monteagudo, J. L. Medina-Franco, Y. Perez-Castillo, O. Nicolotti, M. N. D. S. Cordeiro and F. Borges, Drug Discovery Today, 2014, 19, 1069–1080 CrossRef CAS PubMed.
- D. Stumpfe, H. Hu and J. Bajorath, ACS Omega, 2019, 4, 14360–14368 CrossRef CAS PubMed.
- J. P. Xu, J. D. Branson, R. Lawrence and S. Cocklin, Bioorg. Med. Chem. Lett., 2016, 26, 824–828 CrossRef CAS PubMed.
- G. Wu, W. A. Zalloum, M. E. Meuser, L. Jing, D. Kang, C.-H. Chen, Y. Tian, F. Zhang, S. Cocklin, K.-H. Lee, X. Liu and P. Zhan, Eur. J. Med. Chem., 2018, 158, 478–492 CrossRef CAS PubMed.
- M. Graupe, S. J. Henry, J. O. Link, C. W. Rowe, R. D. Saito, S. D. Schroeder, D. Stefanidis, W. C. Tse and J. R. Zhang, WIPO, WO2018035359A1, 2018 Search PubMed.
- S. K. V. Vernekar, R. L. Sahani, M. C. Casey, J. Kankanala, L. Wang, K. A. Kirby, H. Du, H. Zhang, P. R. Tedbury, J. Xie, S. G. Sarafianos and Z. Wang, Viruses, 2020, 12, 452 CrossRef CAS PubMed.
- L. Wang, M. C. Casey, S. K. V. Vernekar, H. T. Do, R. L. Sahani, K. A. Kirby, H. Du, A. Hachiya, H. Zhang, P. R. Tedbury, J. Xie, S. G. Sarafianos and Z. Wang, Eur. J. Med. Chem., 2020, 200, 112427 CrossRef CAS PubMed.
- L. Sun, A. Dick, M. E. Meuser, T. Huang, W. A. Zalloum, C.-H. Chen, S. Cherukupalli, S. Xu, X. Ding, P. Gao, D. Kang, E. De Clercq, C. Pannecouque, S. Cocklin, K.-H. Lee, X. Liu and P. Zhan, J. Med. Chem., 2020, 63, 4790–4810 CrossRef CAS PubMed.
- L. Wang, M. C. Casey, S. K. V. Vernekar, R. L. Sahani, J. Kankanala, K. A. Kirby, H. Du, A. Hachiya, H. Zhang, P. R. Tedbury, J. Xie, S. G. Sarafianos and Z. Wang, Eur. J. Med. Chem., 2020, 204, 112626 CrossRef CAS PubMed.
- X. Zhang, W. Hu, F. He and C. Ye, WIPO, WO2021104413A1, 2021 Search PubMed.
- L. Wang, M. C. Casey, S. K. V. Vernekar, R. L. Sahani, K. A. Kirby, H. Du, H. Zhang, P. R. Tedbury, J. Xie, S. G. Sarafianos and Z. Wang, Acta Pharm. Sin. B, 2021, 11, 810–822 CrossRef CAS PubMed.
- W. M. McFadden, A. A. Snyder, K. A. Kirby, P. R. Tedbury, M. Raj, Z. Wang and S. G. Sarafianos, Retrovirology, 2021, 18, 41 CrossRef CAS PubMed.
- J. Li, X. Jiang, A. Dick, P. P. Sharma, C.-H. Chem, B. Rathi, D. Kang, Z. Wang, X. Ji, K.-H. Lee, S. Cocklin, X. Liu and P. Zhan, Bioorg. Med. Chem., 2021, 48, 116414 CrossRef CAS PubMed.
- X. Zhang, L. Sun, S. Xu, X. Shao, Z. Li, D. Ding, X. Jiang, S. Zhao, S. Cocklin, E. De Clercq, C. Pannecouque, A. Dick, X. Liu and P. Zhan, Molecules, 2022, 27, 7640 CrossRef CAS PubMed.
- S. Xu, L. Sun, W. A. Zalloum, T. Huang, X. Zhang, D. Ding, X. Shao, X. Jiang, F. Zhao, S. Cocklin, E. De Clercq, C. Pannecouque, A. Dick, X. Liu and P. Zhan, Molecules, 2022, 27, 8415 CrossRef CAS PubMed.
- S. Lang, D. A. Fletcher, A.-P. Petit, N. Luise, P. Fyfe, F. Zuccotto, D. Porter, A. Hope, F. Bellany, C. Kerr, C. J. Mackenzie, P. G. Wyatt and D. W. Gray, ChemMedChem, 2024, 19, e202400025 CrossRef CAS PubMed.
- D. E. Scott, A. G. Coyne, S. A. Hudson and C. Abell, Biochemistry, 2012, 51, 4990–5003 CrossRef CAS PubMed.
- D. A. Erlanson, S. W. Fesik, R. E. Hubbard, W. Jahnke and H. Jhote, Nat. Rev. Drug Discovery, 2016, 15, 605–619 CrossRef CAS PubMed.
- J. D. St. Denis, R. J. Hall, C. W. Murray, T. D. Heightman and D. C. Rees, RSC Med. Chem., 2021, 12, 321–329 RSC.
- J. Michel, J. Tirado-Rives and W. L. Jorgensen, J. Am. Chem. Soc., 2009, 131, 15403–15411 CrossRef CAS PubMed.
- P. Matricon, R. R. Sureshm, Z.-G. Gao, N. Panel, K. A. Jacobson and J. Carlsson, Chem. Sci., 2021, 12, 960–968 RSC.
- H. Bansia, P. Mahanta, N. H. Yennawar and S. Ramakumar, J. Chem. Inf. Model., 2021, 61, 1322–1333 CrossRef CAS PubMed.
- A. Rudling, A. Orro and J. Carlsson, J. Chem. Inf. Model., 2018, 58, 350–361 CrossRef CAS PubMed.
- B. Z. Zsido and C. Hetenyi, Curr. Opin. Struct. Biol., 2021, 67, 1–8 CrossRef CAS PubMed.
- S. Xu, S. Wang, Y. Zhou, N. Foley, L. Sun, L. Walsham, K. Tang, D. Shi, X. Shi, Z. Zhang, X. Jiang, S. Gao, X. Liu, C. Pannecouque, D. C. Goldstone, A. Dick and P. Zhang, J. Med. Chem., 2024, 67, 19057–19076 CrossRef CAS PubMed.
- W. P. Walters and R. Wang, J. Chem. Inf. Model., 2020, 60, 4109–4111 CrossRef CAS PubMed.
- S. Lang and M. J. Slater, J. Med. Chem., 2024, 67, 6897–6898 CrossRef CAS PubMed.
- G. Brizgys, E. Canales, C.-H. Chou, M. Graupe, Y. E. Hu, J. O. Link, Y. Yu, R. D. Saito, S. D. Schroeder, J. R. Somoza, W. C. Tse and J. R. Zhang, WIPO, WO2014134566A2, 2014 Search PubMed.
- G. Brizgys, E. Canales, C.-H. Chou, M. Graupe, R. D. Saito, S. D. Schroeder, W. C. Tse, J. R. Zhang and J. Li, WIPO, WO2019161017A1, 2019 Search PubMed.
- J. Farand, M. Graupe, T. Guney, D. Kato, J. Li, J. O. Link, J. B. C. Mack, D. M. Mun, R. D. Saito, W. J. Watkins and J. R. Zhang, WIPO, WO2023102239A1, 2023 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5md00111k |
| This journal is © The Royal Society of Chemistry 2025 |