DOI:
10.1039/D5MD00161G
(Research Article)
RSC Med. Chem., 2025, Advance Article
Improved anti-breast cancer activity through structural modification of fused pyran derivatives and mechanistic investigations†
Received
19th February 2025
, Accepted 3rd May 2025
First published on 7th May 2025
Abstract
Novel fused pyran derivatives were synthesized and evaluated for anticancer activity against MCF7 (ATCC – HTB-22, breast), A549 (ATCC – CCL-185, lung), and HCT116 (ATCC – CCL-247, colorectal) cancer cell lines. Most compounds exhibited broad-spectrum activity. Among them, imidazole-containing derivatives 8a and 8b demonstrated potent anti-breast cancer activity with IC50 values of 8.24 ± 0.19 μM and 4.22 ± 0.81 μM, respectively. Both compounds were nontoxic to MCR5 (ATCC – CCL-171, human diploid fibroblast) cell line. In vitro studies using MCF7 cells showed that 8a and 8b significantly reduced colony formation and inhibited spheroid progression. Cell cycle analysis revealed G1 arrest, a mechanism distinct from the standard drug etoposide. Furthermore, these compounds induced persistent DNA damage and both early and late apoptosis. In addition, ex ovo CAM assay revealed a significant reduction in new blood vessel formation over time compared to untreated controls. Network pharmacology identified EGFR, SRC, and GSK3B as potential targets, and molecular docking studies confirmed strong binding affinities to these proteins. This interaction is proposed to inhibit uncontrolled breast cancer cell growth. Additionally, the compounds exhibited favorable pharmacokinetic properties in silico. These findings suggest that novel fused pyran derivatives, particularly 8a and 8b, are promising candidates for further preclinical development as breast cancer therapeutics.
1. Introduction
Cancer continues to be a major global health concern and a leading cause of mortality worldwide.1 Among various cancer types, female breast cancer is the most prevalent and remains the primary cause of cancer-related deaths.2 According to the GLOBOCAN 2020 report, approximately 2.3 million new breast cancer cases were diagnosed, with around 685![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths attributed to the disease as of 2020.1–3 Despite significant advancements in treatment options, there remains an urgent need for novel therapeutic strategies, particularly for aggressive subtypes.4–7
000 deaths attributed to the disease as of 2020.1–3 Despite significant advancements in treatment options, there remains an urgent need for novel therapeutic strategies, particularly for aggressive subtypes.4–7
Recent advancements in drug development have highlighted fused pyran/chromene derivatives as promising antitumor agents.8–10 These compounds, found in various natural sources, exhibit significant anticancer activity against various malignancies.8–10 For instance, Tephrosin has shown efficacy against lung cancer through the autophagy pathway and against pancreatic cancer by generating reactive oxygen species.11,12 Calanone has demonstrated activity against cervical carcinoma by upregulating the p53 tumor suppressor gene,13 while Acronycine has been effective against ovarian, lung, and colon cancers by inducing DNA damage.14 Similarly, Seselin has the potential to fight skin cancer by inhibiting tumour-promoting mechanisms.15 Further studies on fused pyran/chromene derivatives have shown their ability to inhibit tubulin polymerization by binding to or near the colchicine-binding site, triggering apoptosis through caspase activation and inducing G2/M cell cycle arrest, ultimately leading to cancer cell death.16 Additionally, these molecules disrupt tumor vascular endothelial cell capillary tubules, indicating their potential as vascular-disrupting agents.17 EPC2407 (Crolibulin™), a potent microtubule-targeting agent, has progressed to phase I/II clinical trials for anaplastic thyroid cancer.8,18,19 Another lead compound, MX-58151, is a robust tubulin destabilizer, triggering cell apoptosis through caspse3/7 activation and nuclear fragmentation.20,21 Furthermore, recent reports indicate that fused pyran/chromene derivatives selectively inhibit formyl peptide receptor-1 (FPR-1), thereby blocking calcium ion (Ca2+) flux and suppressing chemotaxis in human neutrophils, ultimately preventing their migration to inflammatory sites.22 These compounds have also been shown to specifically target triple-negative breast cancer cells, significantly inhibiting their proliferation while exerting minimal effects on hormone-responsive cells.23 All these findings highlight the therapeutic potential of fused pyran/chromene derivatives in developing novel anticancer therapies.
A comprehensive literature review on fused pyran/chromene derivatives highlights their ability to inhibit cancer cells through diverse mechanisms of action. For instance, 3-amino-9-methoxy-1-aryl-1H-benzo[f]chromene-2-carbonitrile (A) demonstrated significant potency against MCF7/ADR cells.24 It effectively inhibited P-gp expression, induced apoptosis, caused cell cycle arrest at various phases, disrupted mitochondrial transmembrane potential, and activated caspase 3/7, collectively promoting apoptotic cell death.24 Similarly, 3-amino/3,5-diamino-1-aryl-1H-benzo[f]chromene-2-carbonitrile (B) showed a significant effect on multiple mechanisms, including induction of apoptosis, flow cytometry analysis, and regulation of apoptosis-related genes.25,26 Moreover, 3-amino-8-bromo/methoxy-1-aryl-1H-benzo[f]chromene-2-carbonitrile (C) displayed potent and selective cytotoxic activity against MCF7, HCT-116, and HepG-2 cell lines, targeting the c-Src kinase enzyme and exhibiting anti-invasion effects on MDA-MB-231 cells.27 Furthermore, 2-amino-4-(3-nitrophenyl)-5-oxo-4,5-dihydropyrano[3,2-c]chromene-3-carbonitrile (D) showed a strong antiproliferative effect mediated through DNA interaction.28 The structures of compounds A–D are presented in Fig. 1. In addition to their anticancer properties, fused pyran derivatives have been reported as potential anticonvulsant agents,29 antidiabetic agents,30 antitubercular agents,31 antimicrobial agents,32 and inhibitors of estrogen biosynthesis,33 and monoamine oxidase (MAO).34
 |
| | Fig. 1 Examples of fused pyran/chromene derivatives (A–D) as potential anticancer agents with multiple mechanisms of action. | |
Nitrogen-containing heterocycles are common drug fragments with immense importance in medicine, as they are abundant in nature and serve as key components in numerous biologically active compounds, such as vitamins, hormones, antibiotics, etc.35 Analysis of U.S. FDA-approved drugs reveals that around 60% of approved small-molecule drugs contain N-based heterocycles, highlighting their structural significance in drug design.36 The prevalence of these compounds in pharmacology is primarily attributed to their stability and efficiency in the human body and their ability to form hydrogen bonds with DNA to enhance their therapeutic potential, particularly in cancer treatment.37 Recognizing the therapeutic potential of fused pyran derivatives and as a part of our endeavour to investigate the anticancer potentials of heterocyclic small molecules,38–42 we recently reported the synthesis and anticancer evaluation of various fused pyran derivatives featuring N-based heterocycles.43,44 Notably, some compounds demonstrated significant inhibitory activity against lung, breast, and colon cancer cell lines.43,44 In addition, the potent compounds were identified to inhibit colony formation and various phases of the cell cycle, as well as induce DNA double-strand breaks and apoptosis in the tested cancer cells.44 Encouraged by these results and to enhance the efficacy of the compounds, we have designed several new fused pyran derivatives, as illustrated in Fig. 2. All the designed molecules were synthesized and screened for their in vitro anticancer activity against MCF7, A549, and HCT116 cell lines, and the potent compounds were evaluated for their mechanism of action.
 |
| | Fig. 2 The design strategy of new fused pyran derivatives from the reported potent anticancer agents. | |
2. Results and discussion
2.1 Chemical synthesis
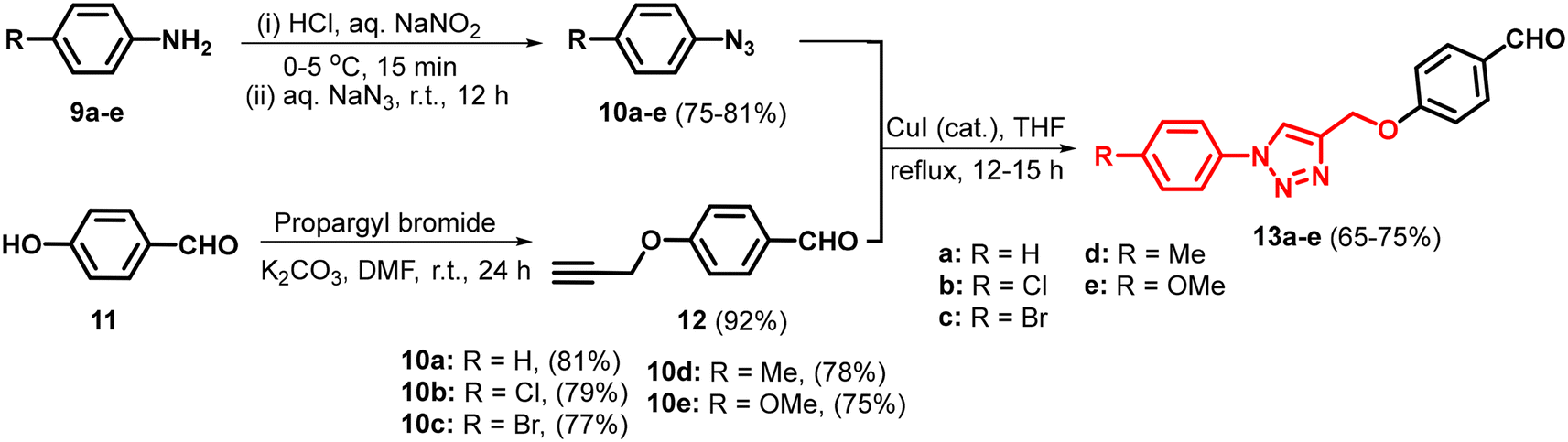
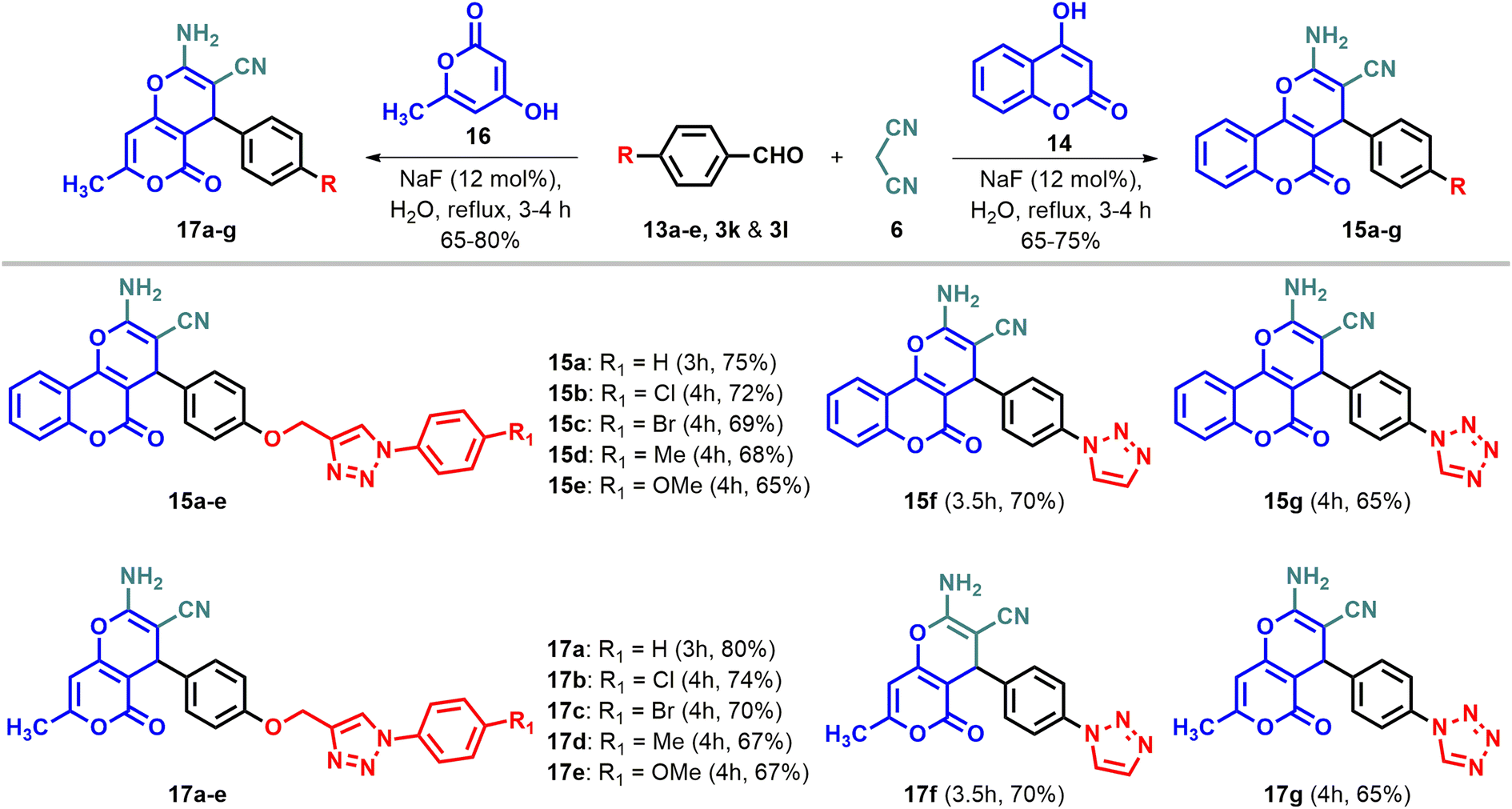
The three-component condensation reaction of 4-hydroxycoumarin/4-hydroxy-6-methyl-2H-pyran-2-one/5-hydroxy-2-(hydroxymethyl)-4H-pyran-4-one/cyclohexane-1,3-dione/5,5-dimethylcyclohexane-1,3-dione with N-based heterocycles substituted arylaldehydes and malononitrile in water, catalyzed by sodium fluoride (NaF), produced diverse fused pyran derivatives.44 In vitro anticancer evaluation of all the compounds led to identifying potential anti-lung, anti-breast and anti-colon cancer agents (I–IV, Fig. 2). To enhance the anticancer efficacy, various substituted pyrazoles, imidazoles, triazoles and tetrazoles were introduced as outlined in Schemes 2 and 4. The intermediates, N-heterocycle-substituted arylaldehydes (3a–l) were synthesized through SNAr reactions of 4-fluorobenzaldehyde (1) with substituted secondary amines, including pyrazole (2a–e), imidazole (2f–j), 1,2,3-triazole (2k), and 1,2,3,4-tetrazole (2l), in the presence of potassium carbonate in DMF at 130 °C (Scheme 1). Substituted 1,2,3-triazole-based aldehydes (13a–e) were prepared by following the synthetic route depicted in Scheme 3.45 The intermediates required, i.e. azide (10) was obtained through the classical diazotization–azidation method. Whereas, o-propargylation of commercially available 4-hydroxybenzaldehyde (11) was achieved by treating it with propargyl bromide in the presence of anhydrous K2CO3 in DMF. Subsequently, a copper-catalyzed azide–alkyne cycloaddition (CuAAC) between azide (10) and terminal alkyne (12) resulted in the formation of 13a–e in 65–75% yield.45 Finally, the desired products (7a–e, 8a–e, 15a–g, & 17a–g) were synthesized by reacting cyclohexane-1,3-dione (4), 5,5-dimethylcyclohexane-1,3-dione (5), 4-hydroxycoumarin (14), and 4-hydroxy-6-methyl-2H-pyran-2-one (16) individually with N-heterocycle-substituted benzaldehydes (3a–l)/1,2,3-triazole substituted arylaldehydes (13a–e) and malononitrile in H2O, using 12 mol% sodium fluoride (NaF) as a catalyst. The reactions were performed at reflux temperature, yielding 65–80% of the products within 3–4 hours.
 |
| | Scheme 1 Synthesis of N-based heterocycles substituted arylaldehydes (3a–l). | |
 |
| | Scheme 2 Synthesis of novel N-based heterocycles embedded fused pyran derivatives (7a–e & 8a–e). | |
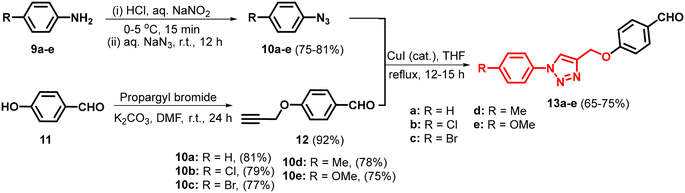 |
| | Scheme 3 Synthesis of 1,2,3-triazole substituted arylaldehydes (13a–e). | |
 |
| | Scheme 4 Synthesis of novel fused pyran derivatives (15a–g & 17a–g). | |
Structures of all the target compounds were confirmed by spectral studies such as FT-IR, 1H NMR, 13C NMR, and LC-HRMS. From the 1H NMR spectra, the appearance of a singlet peak at 4.10–4.60 ppm confirms the presence of the pyran-C4 proton. Moreover, the presence of a peak at 34–38 ppm in the 13C NMR spectra corresponding to the pyran-C4 carbon provided additional evidence of the formation of the pyran ring. Furthermore, the molecular ion peak from the mass spectra also confirmed the product formation. The purity of all tested compounds was greater than 99.5% as determined by LC-HRMS.
2.2 Effects of fused pyran derivatives on cancer cell viability
The cytotoxic effects of all the synthesized compounds were systematically evaluated using the MTT assay across multiple cancer cell lines, such as A549 (ATCC – CCL-185, lung carcinoma), HCT116 (ATCC – CCL-247, colon carcinoma), and MCF7 (ATCC – HTB-22, breast carcinoma), and a noncancerous human diploid fibroblast cell line, MCR5 (ATCC – CCL-171). As summarized in Table 1, the most tested compounds demonstrated dose-dependent cytotoxicity across all selected cancer cell lines, suggesting their potential as effective anti-cancer agents (Fig. 3). Determining half maximal inhibitory concentration (IC50) values revealed varying degrees of potency among the compounds. Specifically, compounds 8a and 8b exhibited the most significant cytotoxic activity against the MCF7 cell line, with IC50 values of 8.24 ± 0.19 μM and 4.22 ± 0.81 μM, respectively. Notably, compound 8a demonstrated a 1.5-fold higher potency, while compound 8b exhibited a 3-fold increase in potency compared to the previously reported fused pyran derivative (III, Fig. 2).44 The structure–activity relationship (SAR) study against the MCF-7 cell lines is presented in Fig. 4. These low IC50 values suggest a strong inhibitory effect on MCF7 cell viability, highlighting the superior efficacy of these compounds in targeting breast cancer cells. In addition, both compounds were found to be nontoxic to MRC5 cells. The enhanced and selective cytotoxicity observed with these compounds (8a and 8b) underscores their potential therapeutic relevance and justifies their selection for further mechanistic studies in MCF7 cell lines. The MTT assay confirmed the synthesized compounds' cytotoxic capabilities and provided crucial quantitative insights into their relative effectiveness across different cancer types. These findings contribute to the ongoing efforts to develop potent anti-cancer agents and support the continued investigation of compounds 8a and 8b in subsequent evaluations.
Table 1 IC50 values of fused pyran derivatives against MCF7, A549, HCT116 and MRC5. Data are shown as mean ± SD, n = 3
| Compound |
Cell lines |
| MCF7 |
A549 |
HCT116 |
MRC5 |
| IC50 (μM) |
| 7a |
190.47 ± 5.34 |
89.68 ± 4.38 |
139.13 ± 3.89 |
>200 |
| 7b |
23.98 ± 2.27 |
147.08 ± 3.61 |
163.48 ± 3.14 |
>200 |
| 7c |
22.92 ± 1.31 |
>200 |
193.79 ± 6.01 |
>200 |
| 7d |
56.13 ± 3.79 |
23.56 ± 1.29 |
45.70 ± 0.67 |
>200 |
| 7e |
15.03 ± 0.78 |
17.96 ± 0.45 |
72.21 ± 0.96 |
>200 |
| 8a |
8.24 ± 0.19 |
111.46 ± 4.91 |
142.96 ± 6.35 |
>200 |
| 8b |
4.22 ± 0.81 |
109.56 ± 4.94 |
129.16 ± 5.79 |
>200 |
| 8c |
83.25 ± 2.11 |
133.38 ± 5.81 |
161.97 ± 6.91 |
>200 |
| 8d |
>200 |
19.82 ± 0.94 |
136.80 ± 5.82 |
>200 |
| 8e |
74.87 ± 0.17 |
30.60 ± 1.85 |
77.57 ± 1.58 |
>200 |
| 15a |
>200 |
>200 |
>200 |
146.5 ± 0.53 |
| 15b |
>200 |
>200 |
>200 |
>200 |
| 15c |
>200 |
>200 |
>200 |
66.9 ± 1.6 |
| 15d |
>200 |
>200 |
>200 |
>200 |
| 15e |
>200 |
>200 |
>200 |
>200 |
| 15f |
144.92 ± 2.75 |
72.86 ± 2.26 |
163.25 ± 5.45 |
156 ± 0.48 |
| 15g |
>200 |
>200 |
>200 |
>200 |
| 17a |
>200 |
80.51 ± 4.68 |
>200 |
>200 |
| 17b |
36.06 ± 0.31 |
20.44 ± 1.87 |
47.44 ± 4.13 |
>200 |
| 17c |
>200 |
>200 |
>200 |
>200 |
| 17d |
>200 |
>200 |
>200 |
>200 |
| 17e |
52.25 ± 1.34 |
>200 |
>200 |
>200 |
| 17f |
30.32 ± 1.73 |
>200 |
>200 |
>200 |
| 17g |
39.21 ± 2.71 |
164.10 ± 3.26 |
>200 |
>200 |
| I44 |
— |
2.25 ± 0.67 |
— |
— |
| II44 |
— |
0.23 ± 0.12 |
— |
— |
| III44 |
12.46 ± 2.72 |
— |
— |
— |
| IV44 |
— |
— |
7.58 ± 1.01 |
— |
 |
| | Fig. 3 IC50 comparison graph of top selected compounds [A] and the dose-dependent inhibition of the viability of cancer cells under compound treatment at various concentrations (100–1.56 μM) [B]. The statistical analysis was conducted using one-way ANOVA and post hoc Dunnett's test. The data represent mean SD (n = 3), *P ≤ 0.01, **P ≤ 0.001, ***P ≤ 0.0001, and ****P ≤ 0.00001. | |
 |
| | Fig. 4 Structure–activity relationship (SAR) study of fused pyran derivatives against MCF-7 cell lines. | |
2.3 Inhibitory effects of fused pyran derivatives on colony formation and spheroid formation
The data from the colony formation assays for MCF-7 cells treated with compounds 8a and 8b were systematically analyzed to determine the percentage of colony formation at various inhibitory concentrations (IC10, IC30, IC50, IC70, and IC90). For compound 8a, the percentage of colony formation progressively decreased from 94.3 ± 4.04% at IC10 to 5.67 ± 4.04% at IC90, demonstrating a clear dose-dependent response. Similarly, for compound 8b, the percentage of colony formation declined from 97.7 ± 1.53% at IC10 to 15 ± 5% at IC90. Intermediate inhibitory concentrations also showed marked reductions, with IC50 values of 74.3 ± 2.08% for compound 8a and 76 ± 2.65% for compound 8b, reflecting their potential to impair cellular reproductive viability Fig. 5. Colony formation assays serve as a critical tool in cancer research, providing insights into cancer cells long-term survival and proliferative potential under various treatment conditions. This assay uniquely measures the ability of single cells to grow into colonies over time, making it a gold standard for assessing the cytotoxic and anti-proliferative effects of anticancer agents. By evaluating the percentage of colony formation across a range of concentrations, the assay provides a robust quantitative assessment of a compound's efficacy in impairing clonogenic survival, a hallmark of tumorigenicity. The sharp reductions in colony formation observed with compounds 8a and 8b underscore their potential therapeutic relevance, as they effectively inhibit the colony-forming ability of MCF-7 cells, a widely used model for breast cancer research. The results indicate that both compounds exhibit significant anti-proliferative activity, with compound 8a demonstrating a steeper colony formation decline at higher concentrations compared to compound 8b. These findings are particularly significant as the ability to inhibit colony formation reflects a compound's potential to prevent tumor regrowth and metastasis, critical factors in improving cancer treatment outcomes.
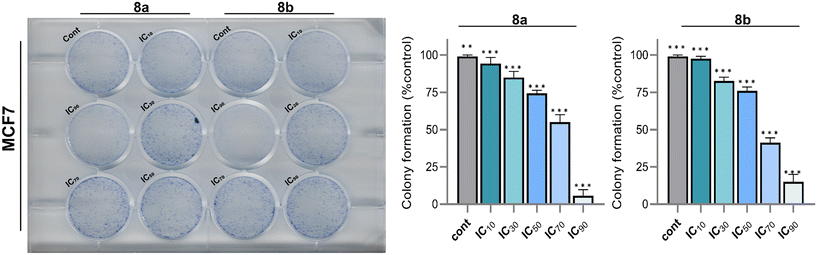 |
| | Fig. 5 Compounds 8a and 8b colony formation ability on MCF7. The statistical analysis was performed using a two-way ANOVA and Tukey's test. The data represent mean ± SD (n = 3), ns P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. | |
The spheroid formation analysis has emerged as a critical in vitro model for cancer drug discovery due to its ability to closely mimic the three-dimensional (3D) architecture and the microenvironment of solid tumors. Unlike traditional two-dimensional (2D) monolayer cultures, spheroids replicate key tumor characteristics such as cell–cell and cell–matrix interactions, oxygen and nutrient gradients, and resistance to chemotherapeutic agents. This makes spheroid models highly suitable for evaluating the efficacy of candidate compounds, as they provide a more physiologically relevant environment to assess tumor progression, cellular proliferation, and drug penetration. By monitoring spheroid dynamics, researchers can obtain valuable insights into the anti-tumor potential of new compounds and better predict their in vivo efficacy. In this study, compounds 8a and 8b were evaluated for their anti-tumor activity using MCF7 spheroids over a treatment period of 72 hours. MCF7 cells, a well-established breast cancer cell line, readily form compact spheroids that allow for robust monitoring of growth and progression under treatment conditions. The results demonstrated a significant inhibitory effect of compounds 8a and 8b on spheroid progression. In untreated control spheroids, a steady and measurable increase in spheroid size was observed over time, which indicates continuous cellular proliferation and tumor-like growth. Conversely, spheroids treated with compounds 8a and 8b showed a marked reduction in size throughout the experimental duration, signifying an inhibition of cellular growth and potential cytotoxic effects Fig. 6. Specifically, after 72 hours of treatment, spheroids exposed to compound 8a exhibited a pronounced reduction in size compared to untreated controls, indicating a robust suppression of cellular proliferation and spheroid expansion. Compound 8b produced a similar effect, demonstrating its ability to impede spheroid growth over the same period significantly. The consistent reduction in spheroid size under both treatment conditions highlights the efficacy of these compounds in targeting tumor-like structures and inhibiting growth in a 3D environment. Furthermore, the observed inhibition suggests that compounds 8a and 8b may disrupt key processes required for tumor progression, such as cell division, nutrient uptake, or survival signaling within the spheroid.
 |
| | Fig. 6 Effects of 8a and 8b on spheroid formation and growth in MCF7 cell lines over 72 hours. Data are presented as mean ± standard deviation from three independent experiments. | |
2.4 Impact of the fused pyran derivatives on the cancer cell cycle
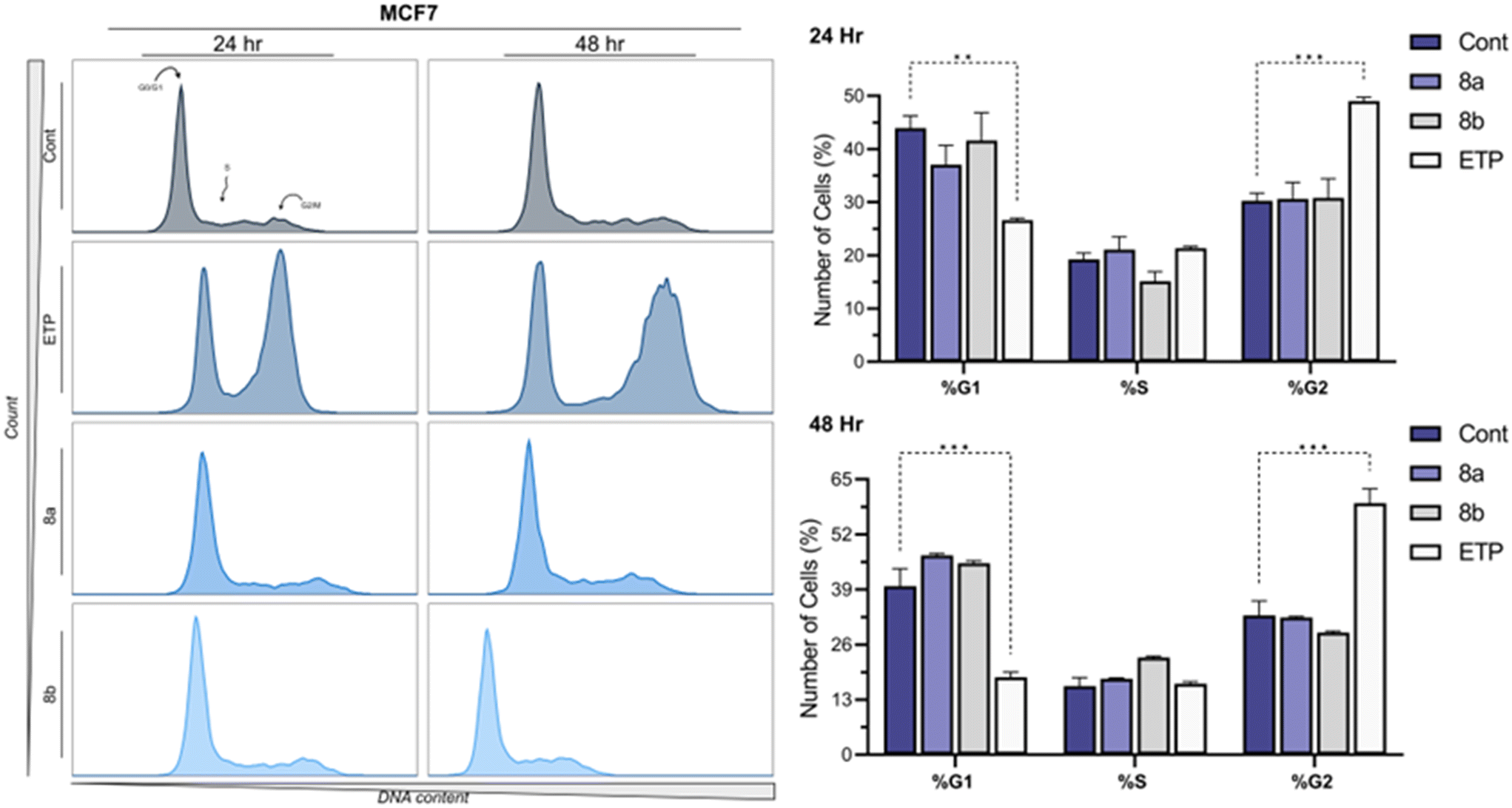
The cell cycle distribution of MCF7 cells treated with compounds 8a and 8b at their IC50 concentrations was analyzed using flow cytometry following propidium iodide staining, with Etoposide (ETP) at 10 μM as a positive control and 0.1% DMSO as the vehicle control. The results revealed significant alterations in the cell cycle profile at both 24 and 48 hours of incubation. After 24 hours, compound 8a induced a modest decrease in the percentage of cells in the G1 phase (37.08 ± 5.07%) compared to the control (43.93 ± 3.26%), coupled with an increase in the S phase population (21.11 ± 3.39%) and a slight shift in the G2 phase (30.63 ± 4.35%). Similarly, compound 8b caused a reduction in G1 phase cells (41.59 ± 7.44%) with a concurrent decrease in the S phase population (15.11 ± 2.59%) and a marginal increase in G2 phase cells (30.79 ± 5.11%). In contrast, ETP treatment resulted in a pronounced G2/M phase arrest (49.03 ± 0.99%), with a significant reduction in the G1 phase population (26.61 ± 0.54%), consistent with its known mechanism as a topoisomerase II inhibitor. After 48 hours, the impact of both compounds became more distinct. Compound 8a prominently increased the G1 phase population (47.08 ± 0.6%) compared to the control (39.82 ± 5.78%), suggesting a potential G1 phase arrest. Meanwhile, 8b showed a similar trend with an elevated G1 phase population (45.22 ± 0.82%) and a reduced G2 phase population (28.83 ± 0.52%). These observations contrasted sharply with ETP, which induced a robust G2/M phase arrest (59.36 ± 4.79%) and a significant reduction in the G1 phase population (18.3 ± 1.67%). The control cells maintained relatively consistent distribution across all phases at both time points, reflecting the typical cell cycle dynamics of untreated MCF7 cells Fig. 7. Studying the cell cycle is essential in cancer drug research because abnormalities in its regulation are a key feature of cancer. Targeting specific cell cycle phases allows new drugs to stop cancer cells from dividing while minimizing effects on normal cells. In this study, compounds 8a and 8b showed their ability to alter the cell cycle of MCF7 cells, mainly by causing G1 arrest, suggesting their potential as effective cancer treatments. Comparing these effects to those of Etoposide highlights the different ways anticancer drugs work. These results set the stage for more research to understand the molecular pathways behind these changes.
 |
| | Fig. 7 Flow cytometry analysis and quantitative evaluation of cell cycle distribution after 24 and 48 hours of treatment with the compounds 8a and 8b in the MCF7 cell line. The statistical analysis was performed using a two-way ANOVA and Tukey's test. The data represent mean ± SD (n = 3), ns P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. | |
2.5 Ability of the fused pyran derivatives to induce DNA double-strand breaks (DSBs)
γH2AX labelling, a widely recognized marker for double-strand breaks (DSBs), pivotal role in evaluating DNA damage in cancer cells treated with anticancer compounds. In the present study, γH2AX labelling was utilized to assess DNA damage in the MCF7 cell line following treatment with compounds 8a and 8b at their IC50 concentrations for 24 and 48 hours, alongside etoposide (ETP, a positive control at 10 μM) and 0.1% DMSO as a vehicle control. Flow cytometry analysis revealed distinct patterns of γH2AX expression that correlated with treatment duration and the nature of the compounds, providing insights into the potential of these compounds as DNA damage-inducing agents. At 24 hours, the control group showed minimal γH2AX labelling (0.56 ± 0.66), indicating negligible baseline DNA damage. In contrast, etoposide treatment markedly increased γH2AX levels (8.8 ± 0.62), consistent with its well-documented role in inducing DSBs via topoisomerase II inhibition. Both compounds 8a and 8b exhibited significant DNA damage induction compared to the control, with γH2AX levels of 2.89 ± 0.99 and 4.71 ± 0.75, respectively. Compound 8b elicited a stronger γH2AX response than compound 8a, suggesting higher potency in inducing DSBs within the same incubation period. A more pronounced difference was observed at 48 hours, highlighting the time-dependent effects of the treatments. γH2AX levels in the control group were slightly elevated (1.44 ± 0.68), likely due to natural cellular processes over extended incubation. Etoposide demonstrated a dramatically increase in γH2AX expression (39.44 ± 1.69), consistent with sustained and severe DNA damage. Similarly, Compounds 8a and 8b showed substantial γH2AX labelling (20.3 ± 2.74 and 21.62 ± 3.38, respectively), further indicating the ability of these compounds to induce persistent DNA damage. Compound 8b again exhibited slightly higher activity than compound 8a, though the difference was not statistically significant within the margin of experimental variability Fig. 8. The observed γH2AX responses for compounds 8a and 8b suggest their potential mechanisms of action may involve the induction of DNA damage, a hallmark of effective chemotherapeutics. Furthermore, the time-dependent increase in γH2AX expression highlights the sustained activity of these compounds, which may contribute to their cytotoxic effects against cancer cells. These results warrant further investigation into the detailed molecular pathways and potential therapeutic applications of compounds 8a and 8b.
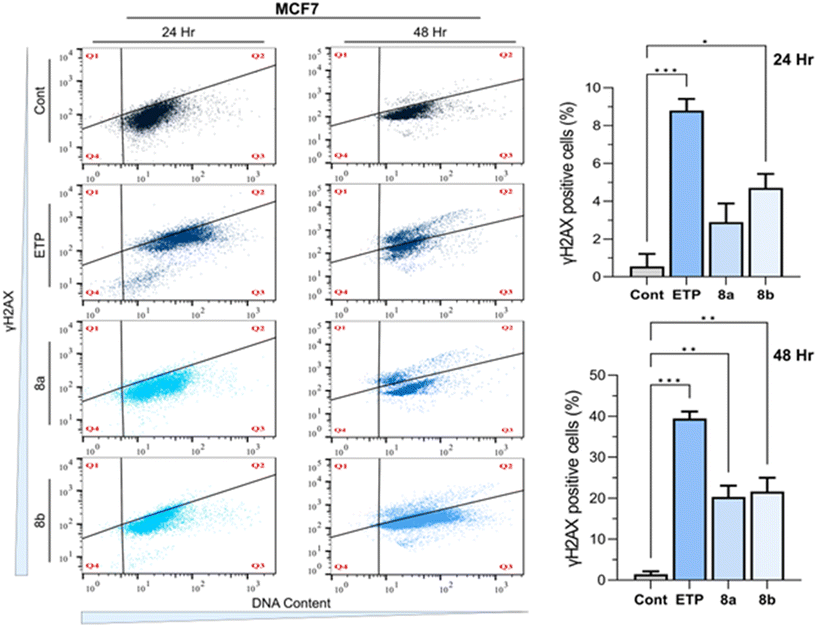 |
| | Fig. 8 Flow cytometry analysis and quantitative assessment of γH2AX expression following 24 and 48 hours of treatment with the respective compounds in MCF7 cell lines. The statistical analysis was performed using a two-way ANOVA and Tukey's test. The data represent mean ± SD (n = 3), ns P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. | |
2.6 Efficacy of fused pyran derivatives in inducing apoptosis in MCF7 cell lines
Apoptosis, a programmed cell death mechanism, pivotal in cancer drug discovery. It is often dysregulated in cancer cells, allowing them to evade cell death and proliferate uncontrollably. Compounds that can restore or induce apoptosis selectively in cancer cells are of significant interest in developing of anticancer therapies. This study assessed the apoptotic effects of compounds 8a and 8b in MCF7 breast cancer cells, using annexin V staining and flow cytometry at their IC50 concentrations over 24 and 48 hour incubation periods. The results were compared to etoposide (10 μM) as a positive control and 0.1% DMSO as the untreated control. At 24 hours, etoposide significantly increased late apoptotic cells to 12.25 ± 0.08%, compared to 7.58 ± 0.03% in the control group, reflecting its potent pro-apoptotic effect. Early apoptotic cells were also elevated in the etoposide group (6.42 ± 0.27%) relative to the control (3.15 ± 0.12%). Compound 8a showed a modest increase in early apoptosis (4.27 ± 0.20%) and late apoptosis (7.6 ± 0.01%) compared to the control, while compound 8b exhibited a slight increase in late apoptosis (7.87 ± 0.02%) but a decrease in early apoptosis (2.66 ± 0.12%). Both compounds preserved a high percentage of live cells, with 86.4 ± 0.15% and 87.55 ± 0.08% for 8a and 8b, respectively, compared to 87.7 ± 1.03% in the control group. After 48 hours, the apoptotic effects became more pronounced. Etoposide treatment led to a substantial increase in late apoptotic cells (11.8 ± 0.29%) and a modest rise in early apoptotic cells (3.28 ± 0.16%), with live cells reduced to 83.15 ± 0.50%. Compound 8a markedly increased early apoptosis (5.89 ± 0.20%) and late apoptosis (6.17 ± 0.13%), indicating a time-dependent enhancement of its pro-apoptotic activity. Similarly, compound 8b showed an increase in both early apoptosis (5.27 ± 0.13%) and late apoptosis (9.46 ± 0.22%), with fewer live cells (84.45 ± 0.64%) compared to the 24 hour time point. Interestingly, compound 8b also reduced debris/dead cells (0.82 ± 0.30%), suggesting it may limit necrosis while inducing apoptosis more effectively over time Fig. 9. These results highlight the potential of compounds 8a and 8b to modulate apoptotic pathways in MCF7 cells. The apoptotic response elicited by both compounds was time-dependent, with increased efficacy observed at 48 hours. While etoposide demonstrated the highest apoptotic activity, the observed effects of 8a and 8b, particularly in increasing early apoptotic populations, underscore their therapeutic potential. Mechanistically, the ability of these compounds to induce apoptosis while maintaining a significant proportion of live cells may indicate selective targeting of cancer cells without widespread cytotoxicity.
 |
| | Fig. 9 Flow cytogram and quantitative assessment of apoptotic cells following 24 and 48 hours of treatment with compounds 8a and 8b in the MCF7 cell line. The statistical analysis was performed using a two-way ANOVA and Tukey's test. The data represent mean ± SD (n = 3), ns P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. | |
2.7 Effect of fused pyran derivatives on angiogenesis
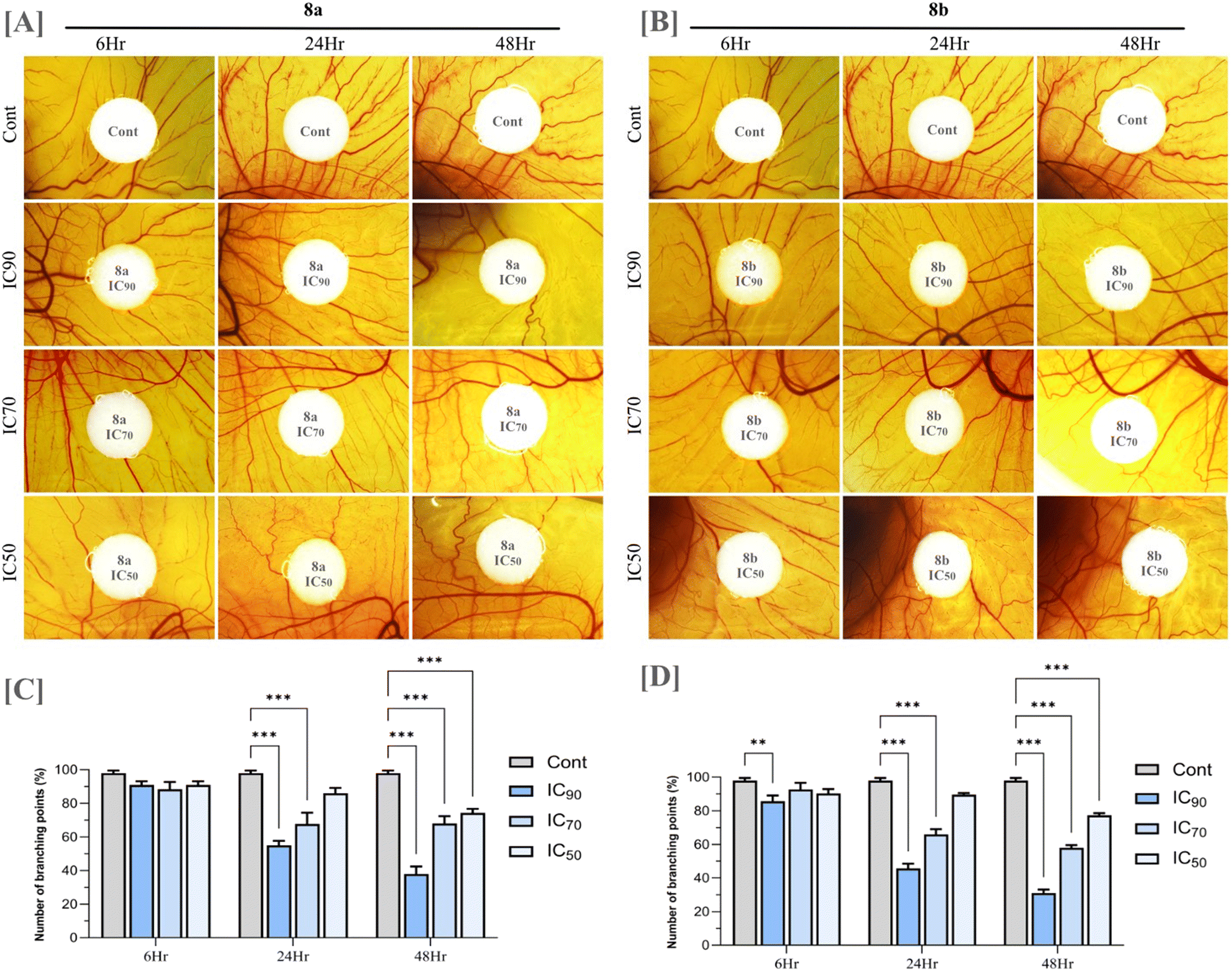
To evaluate the anti-angiogenic potential of compounds 8a and 8b, an ex ovo chorioallantoic membrane (CAM) assay was performed. Angiogenesis, the formation of new blood vessels from pre-existing vasculature, is a fundamental process in both physiological and pathological conditions. In the context of cancer, angiogenesis plays a critical role in tumor development and progression by supplying oxygen and essential nutrients to rapidly growing tumor cells. It also facilitates metastatic spread by providing a conduit for tumor cells to enter the circulation. The CAM assay is a widely used in vivo model for studying angiogenesis due to its highly vascularized nature and ease of manipulation. The ex ovo version allows for direct visualization and quantification of blood vessel growth in response to experimental treatments, making it an ideal platform to assess the impact of potential anti-angiogenic compounds. Upon treatment with compounds 8a and 8b, a significant reduction in new blood vessel formation was observed over time compared to untreated controls. This inhibitory effect was found to be concentration-dependent, with higher concentrations resulting in a more pronounced suppression of vascular growth (Fig. 10). The treated CAMs exhibited thin, underdeveloped vasculature, suggesting that both compounds effectively hinder angiogenic activity. While anti-angiogenic effects are not a universal characteristic of all anticancer agents and may in some cases be attributed to off-target actions, the ability to disrupt angiogenesis can be a valuable therapeutic asset. By limiting tumor vascularization, such compounds may contribute to reduced tumor growth and metastatic potential. The observed anti-angiogenic properties of compounds 8a and 8b enhance their overall anticancer profile and support further investigation into their mechanisms of action and therapeutic potential.
 |
| | Fig. 10 Photomicrograph of CAM vascular area around the drug applied site (A) compound 8a (B) compound 8b (C and D) quantification of angiogenesis based on the number of vascular branching points. The statistical analysis was performed using a two-way ANOVA and Tukey's test. The data represent mean ± SD (n = 5), ns P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. | |
2.8 Network pharmacological analysis of fused pyran derivatives: identifying targets and biological mechanisms
Over the past decade, drug discovery has followed a “one drug, one disease” paradigm, where each drug is designed to target a specific disease. The action of drugs can be understood through the “lock and key” model, in which a drug acts like a selective key that binds to its target, or “lock”. However, drug effectiveness can improve when a single key fits multiple locks, allowing it to impact multiple targets. Viewing drug action through the lens of network biology may provide insights into how drugs bind to different targets. In 2017, Hopkins introduced a novel concept of network pharmacology to identify various drug targets using the available biomedical data in the postgenomic era, systems biology, and polypharmacology. The emergence of network pharmacology helps to examine the multi-pathway interactions in cancer (how different pathways interact and influence each other) and helps the researchers to understand drug-response signature datasets, which contain information on how cancer cells respond to various drugs and thereby decode more complex mechanism of drug action.46–49 Therefore, we applied a network pharmacology approach to identify the targets of fused pyran derivatives against lung, breast, and colorectal cancer. Subsequently, molecular docking was performed on these identified targets followed by in vitro analysis to determine whether the predicted mechanisms from the network pharmacology and molecular docking studies are accurate in real biological systems. The study is framed as a three-phase workflow stated below: (i) identification of the potential targets of compound 8a and 8b based on its association with breast cancer through retrieval from databases; (ii) establishment of the core targets based on the interaction relationships by network analysis; and (iii) validation of the potential targets by molecular docking verification and in vitro assessment.
2.8.1 Identification of potential targets. Swiss target prediction was used to identify potential targets for compounds 8a and 8b. For compound 8a, approximately 100 likely protein targets were predicted, with 60% being kinases, 20% family A G protein receptors, 6.7% nuclear receptors, and 6.7% electrochemical transporters Fig. 11A. For compound 8b, around 101 potential protein targets were identified, with 60% being kinases, 13% phosphodiesterases, 6.7% electrochemical transporters, 6.7% family B G protein receptors, 6.7% family A G protein receptors, and another 6.7% kinases Fig. 11B.
 |
| | Fig. 11 A and B represent the overview of the Swiss target prediction of compounds 8a and 8b. C and D represent the venn diagram of common targets in each set. | |
A comprehensive database search was conducted for breast cancer across multiple resources. For breast cancer, data from Malacards (18253 records), Disginet (16200 records), and Omim (250 records) were merged, yielding 18276 unique gene targets after removing duplicates. To identify co-relative targets specific to each compound, a Venn diagram was constructed using the Venny website.50 This approach helped pinpoint common targets across each compound's dataset, revealing their specificity for breast cancer. Notably, compound 8a was found to have 97 common targets for breast cancer, while compound 8b shared 91 common targets for breast cancer. These identified targets, considered potential anticancer candidates, were selected for further analysis (Fig. 11C and D).
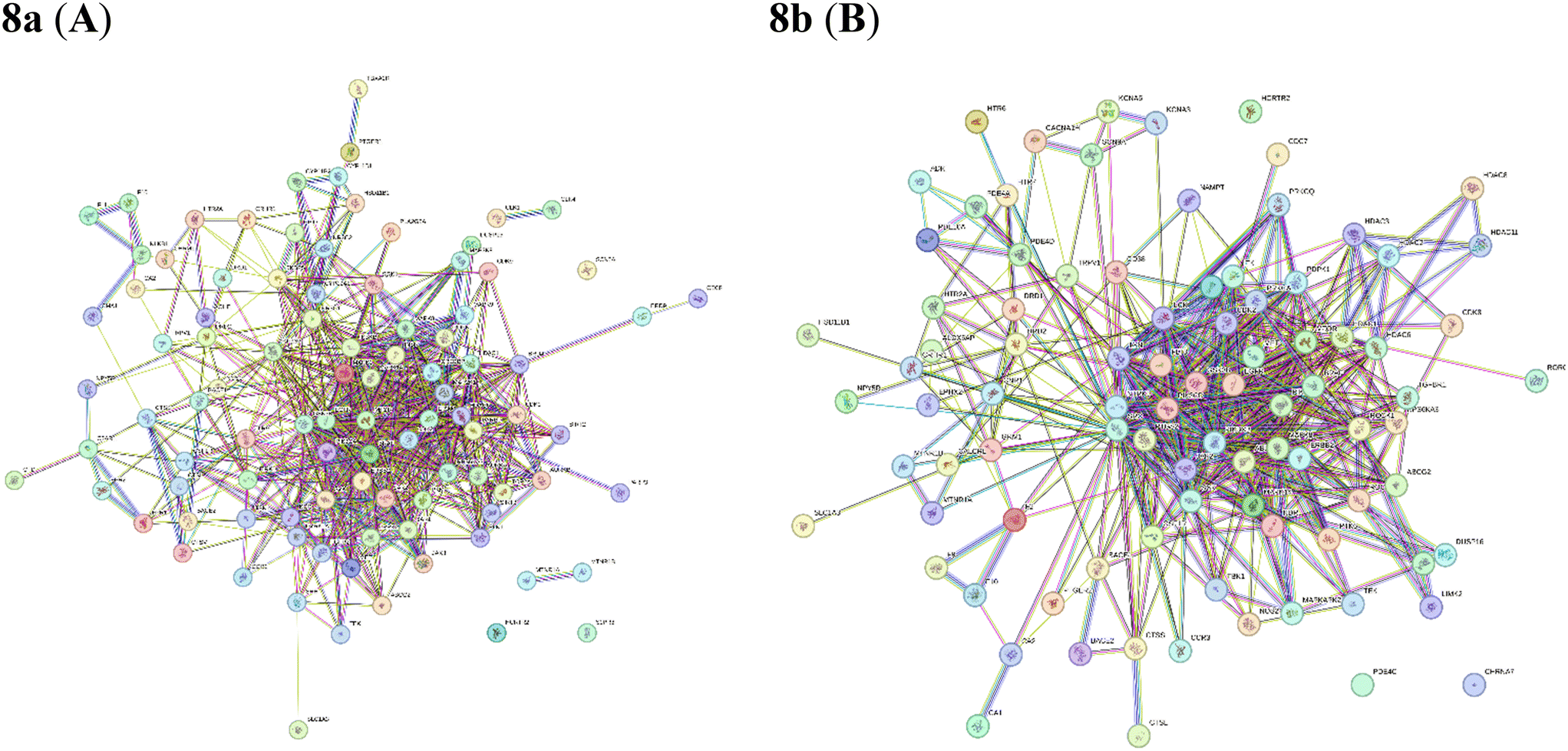
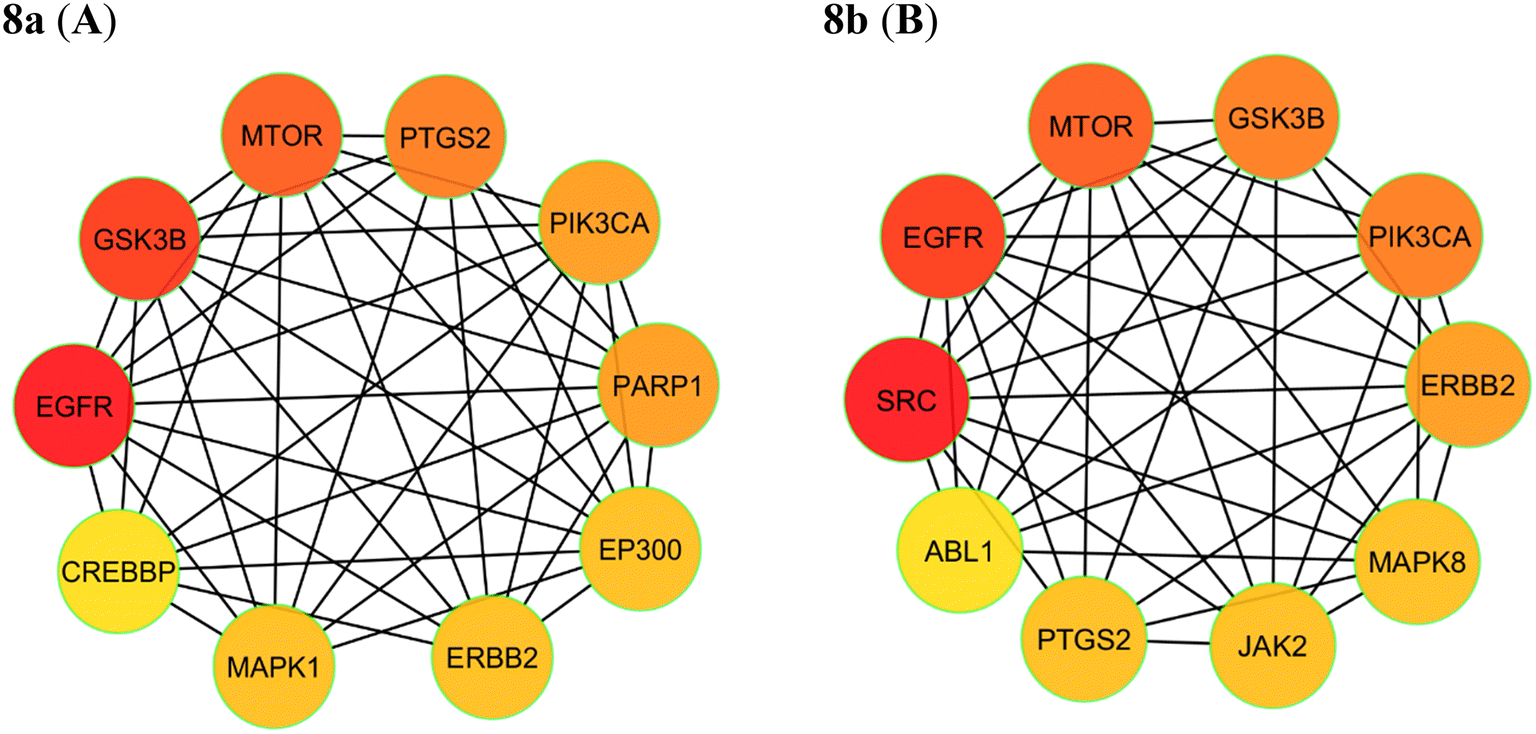
2.8.2 Network interaction and its ranking. The identifiable targets of compounds associated with the respective cancer were introduced into the STRING web server to obtain the functional protein-association network. The protein–protein interaction (PPI) network of these anti-cancer targets was constructed. The identified 97 targets of compound 8a and 91 targets of compound 8b associated with breast cancer were introduced to obtain the functional protein-association network. For 8a, STRING database identified 97 nodes and 578 edges, representing a network of interconnected genes (Fig. 12A). The top ten hub genes, ranked by the degree method through the CytoHubba plug-in in Cytoscape, were EGFR, GSK3B, MTOR, PTGS2, PIK3CA, PARP1, EP300, ERBB2, MAPK1, CREBBP (Fig. 13A). These genes, with high degree values, highlight their potential significance in the development of breast cancer. For 8b string database identified 91 nodes and 500 edges, representing a network of interconnected genes (Fig. 12B). The top ten hub genes, ranked by the degree method through the CytoHubba plug-in in Cytoscape, were SRC, EGFR, MTOR, GSK3B, PIK3CA, ERBB2, MAPK8, JAK2, PTGS2, ABL1 (Fig. 12B).
 |
| | Fig. 12 STRING database analysis of compounds 8a and 8b. | |
 |
| | Fig. 13 Hub genes are coloured and sized according to their degree from red to yellow (A) 8a and (B) 8b. | |
2.8.3 Molecular docking studies. The top two hub genes identified for compound 8a were EGFR and GSK3B, while for compound 8b, they were SRC and EGFR. Both compounds were imported into Schrodinger Maestro and prepared to generate all possible 3D conformations. The 3D protein structures of EGFR (PDB ID: 4I23), GSK3B (PDB ID: 4ACC), and SRC (PDB ID: 3QLF) were retrieved from the PDB database and processed using the Protein Preparation Wizard. XP docking was then performed for these targets with both compounds.EGFR is a receptor tyrosine kinase, and its crystal structure includes a co-crystallized ligand, dacomitinib. The coordinates (−0.26, −52.67, −22.5) of dacomitinib were used as the center of the grid for docking the compounds. For GSK3B, a serine/threonine protein kinase, the crystal structure is a homodimer, and chain A was selected for the study. The structure features a co-crystallized inhibitor, 7YG, with coordinates (17.46, 18.83, 10.69) used to define the grid center. Similarly, SRC, a protein tyrosine kinase, also forms a homodimer, and chain A was chosen for the analysis. The crystal structure of SRC is co-crystallized with the inhibitor pyrazolopyrimidine 5, and its coordinates (6.22, −33.92, −6.7) were used for grid generation.
The docking analysis of compounds 8a and 8b against various target proteins revealed differences in their binding affinities. For compound 8a, docking with EGFR yielded a docking score of −5.42 kcal mol−1 and a binding energy of −53.69 kcal mol−1 (Fig. 14A), with interactions predominantly through hydrophobic contacts. When docked with GSK3B, 8a showed a docking score of −5.99 kcal mol−1 and a binding energy of −40.05 kcal mol−1, forming hydrogen bonds with Val135 (bond length: 2.35 Å; D–H⋯A angle: 177.1°) (Fig. 14B). In contrast, compound 8b exhibited a docking score of −5.51 kcal mol−1 and a binding energy of −64.77 kcal mol−1 against SRC, forming one hydrogen bond (bond length: 2.07 Å; D–H⋯A angle: 141.9°) and a salt bridge (distance: 2.94 Å) with Asp348, along with a second hydrogen bond (bond length: 2.07 Å; D–H⋯A angle: 140.4°) with Glu339 (Fig. 15A). When docked with EGFR, 8b showed a docking score of −4.66 kcal mol−1 and a binding energy of −41.54 kcal mol−1, forming a hydrogen bond (bond length: 2.07 Å; D–H⋯A angle: 129.7°) with Leu718 (Fig. 15B). The docking scores, binding energies, interacting residues and free energy (ΔG) parameters for compounds 8a and 8b are summarized in Tables 2 and 3.
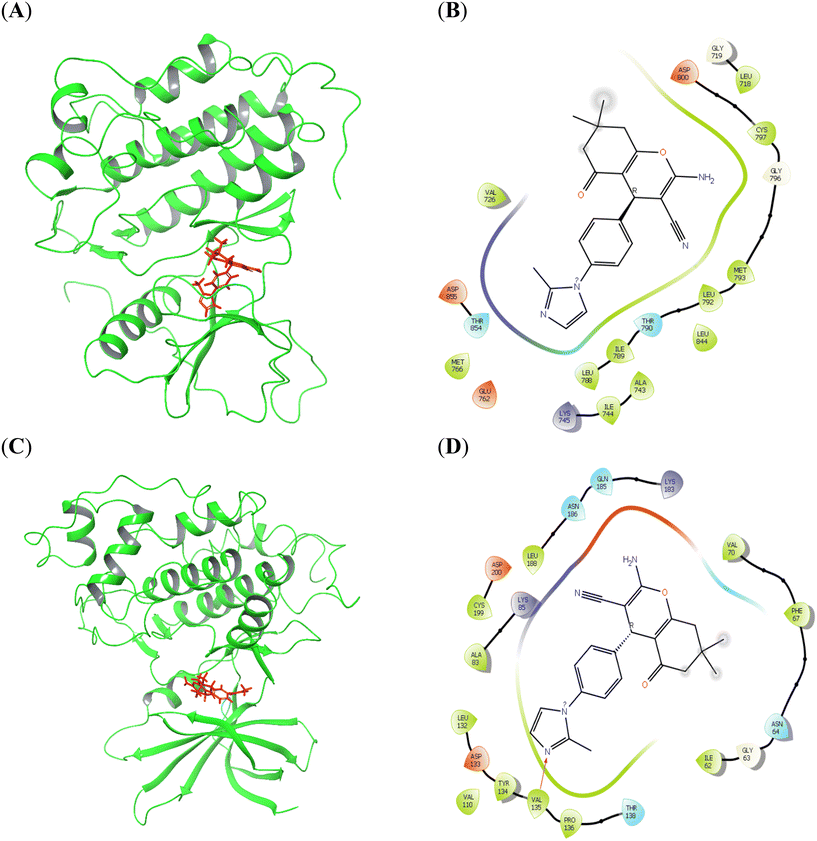 |
| | Fig. 14 Molecular docking analysis of compound 8a against EGFR (A and B) and GSK3B (C and D) in 3D and 2D. | |
 |
| | Fig. 15 Molecular docking analysis of compound 8b against SRC (A and B) and EGFR (C and D) in 3D and 2D. | |
Table 2 The docking scores and binding energies of 8a and 8b against the target proteins
| Compound |
Target protein |
Dock score (kcal mol−1) |
Interacting residues |
MMGBSA binding energy (kcal mol−1) |
| 8a |
EGFR |
−5.42 |
— |
−53.69 |
| GSK3B |
−5.99 |
Val135 (H-bond) |
−40.05 |
| 8b |
SRC |
−5.517 |
Asp348 (H-bond and salt bridge) Glu339 (H-bond) |
−64.77 |
| EGFR |
−4.66 |
Leu718 (H-bond) |
−41.54 |
Table 3 The free energy (ΔG) parameters and the binding score of compounds 8a and 8b with its respective targets. All the energies are reported in kcal mol−1
| |
MMGBSA, ΔG, bind |
MMGBSA, ΔG, bind, Coulomb |
MMGBSA, ΔG, bind, covalent |
MMGBSA, ΔG, bind, H-bond |
MMGBSA, ΔG, bind, lipo |
MMGBSA, ΔG, bind, packing |
MMGBSA, ΔG, bind, SelfCont. |
MMGBSA, ΔG, bind, Solv. GB |
MMGBSA, ΔG, bind, vdW |
| 8a-EGFR |
−53.69 |
−8.88 |
7.56 |
−0.32 |
−37.35 |
0.00 |
0.00 |
21.77 |
−36.48 |
| 8a-GSK3B |
−40.05 |
−14.44 |
4.16 |
−1.12 |
−29.44 |
−0.00 |
0.00 |
31.90 |
−31.11 |
| 8b-SRC |
−64.77 |
27.00 |
2.44 |
−1.05 |
−43.32 |
0.00 |
0.00 |
−15.70 |
−34.13 |
| 8b-EGFR |
−41.54 |
−5.72 |
4.68 |
−0.26 |
−31.15 |
0.00 |
0.00 |
27.68 |
−36.76 |
Breast cancer is a heterogeneous disease characterized by diverse pathological features and clinical implications.51 The subtypes of breast cancer, including luminal A, luminal B, HER2-enriched, and triple-negative, exhibit distinct gene expression profiles and treatment responses.52 Network pharmacology was employed to identify key target proteins in breast cancer, followed by molecular docking studies to assess the binding affinity of the synthesized compounds to these targets.46,47 This approach resulted in the identification of epidermal growth factor receptor (EGFR), proto-oncogene tyrosine-protein kinase Src (SRC), and glycogen synthase kinase 3 beta (GSK3B) as targets for docking studies. These proteins play critical roles in breast cancer development and progression. Molecular docking simulations conducted on EGFR, SRC, and GSK3B demonstrated favorable binding affinity with fused pyran derivatives. This observation suggests that the compounds may interact with these proteins at their active sites, potentially resulting in the inhibition of their oncogenic functions.
EGFR is a receptor tyrosine kinase that initiates intracellular signaling cascades upon binding to its ligands.53,54 This process involves pathways including PI3K/Akt and MAPK, which regulate cell proliferation, survival, and differentiation.55 In breast cancer, EGFR is frequently overexpressed or exhibits activating mutations, resulting in the activation of these pathways and subsequent uncontrolled cell growth.56 EGFR signaling promotes cell cycle progression and DNA repair mechanisms. Disruption of these processes by EGFR inhibition can lead to DNA damage accumulation and trigger apoptosis.57 SRC is a non-receptor tyrosine kinase involved in various cellular processes, including cell growth, adhesion, migration, angiogenesis, and survival.58 It is involved in cell survival signaling and DNA repair pathways.58 Elevated SRC activity is observed in breast cancer, contributing to tumor progression, invasion, and metastasis.58,59 SRC can interact with and phosphorylate EGFR, enhancing EGFR signaling and promoting tumor growth.60 Its inhibition by fused pyran derivatives can disrupt these processes, resulting in increased DNA damage and apoptosis. GSK3B is a serine/threonine kinase with diverse functions in cellular processes, including cell cycle regulation,61 apoptosis,62 and DNA repair.63 Its role in cancer is complex and context-dependent. In breast cancer, GSK3B can be involved in both tumor-promoting and tumor-suppressing functions, contingent upon the specific context and signaling pathways involved.64,65 Targeting these proteins with drugs can disrupt oncogenic signaling, leading to apoptosis and DNA breaks.
2.9 ADME prediction
The Swiss ADME webserver was utilized to calculate the physicochemical properties of the compounds. Several parameters, including lipophilicity, number of hydrogen bond donors, number of hydrogen bond acceptors, water solubility, and drug-likeness, were calculated based on pre-trained models. A radar plot was generated for each molecule, wherein the colored zone represented a suitable physicochemical space for oral bioavailability (Fig. 16). Compounds 8a and 8b exhibited similar physicochemical properties, rendering them promising candidates for further investigation (Table 4). Both compounds possessed a molecular weight of 374.44 g mol−1 and demonstrated moderate water solubility. Each compound contained four hydrogen bond acceptors and one donor, indicating their capacity to interact with biological molecules. Both compounds exhibited high gastrointestinal (GI) absorption, indicating good oral bioavailability, although neither demonstrates permeability of the blood–brain barrier (BBB). Furthermore, both 8a and 8b are substrates of P-glycoprotein (P-gp) and inhibitors of several cytochrome P450 enzymes (CYP1A2, CYP2C19, CYP2C9, and CYP3A4), which may have implications for drug metabolism and potential drug–drug interactions. They do not inhibit CYP2D6, which suggests a lower risk of interfering with the metabolism of other drugs that rely on this enzyme. Both compounds exhibited a logKp value of −6.47 cm s−1, indicating low skin permeation. In terms of drug-likeness, both compounds pass Lipinski's rule of five with no violations and meet the criteria for Ghose, Veber, Egan, and Muegge rules, suggesting that they possess favorable characteristics for oral drug development. Their bioavailability score was 0.56, indicating moderate potential for effective absorption.
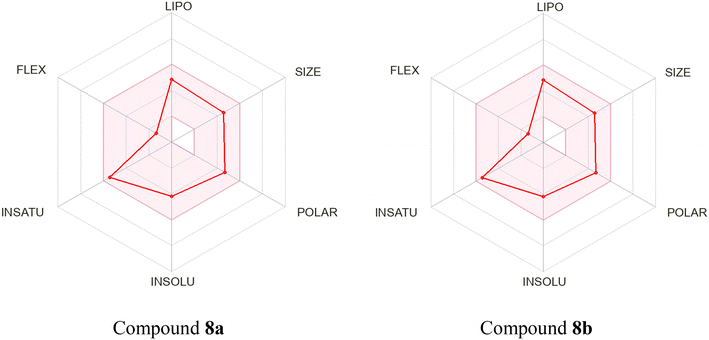 |
| | Fig. 16 Bioavailability radar and score prediction of compounds 8a and 8b were generated using SwissADME. | |
Table 4 The physicochemical properties of compounds 8a and 8b
| Properties |
8a |
8b |
| Water solubility |
Moderately soluble |
Moderately soluble |
| Number of H-bond acceptors |
4 |
4 |
| Number of H-bond donors |
1 |
1 |
| GI absorption |
High |
High |
| BBB permeant |
No |
No |
| P-gp substrate |
Yes |
Yes |
| CYP1A2 inhibitor |
Yes |
Yes |
| CYP2C19 inhibitor |
Yes |
Yes |
| CYP2C9 inhibitor |
Yes |
Yes |
| CYP2D6 inhibitor |
No |
No |
| CYP3A4 inhibitor |
Yes |
Yes |
| LogKp (skin permeation) |
−6.47 cm s−1 |
−6.47 cm s−1 |
| Druglikeness |
|
|
| Lipinski |
Yes; 0 violation |
Yes; 0 violation |
| Ghose |
Yes |
Yes |
| Veber |
Yes |
Yes |
| Egan |
Yes |
Yes |
| Muegge |
Yes |
Yes |
| Bioavailability score |
0.56 |
0.56 |
3. Conclusion
In this study, we successfully designed and synthesized a series of N-heterocyclic substituted fused pyran derivatives using NaF catalyst in an aqueous solvent. All the fused pyran derivatives were evaluated for their anti-proliferative activity through in vitro assay on MCF7, A549 and HCT116 cell lines, and several compounds exhibited promising anticancer properties. Notably, compounds 8a and 8b demonstrated significant inhibitory effects on the MCF7 cell line, with IC50 values of 8.24 ± 0.19 and 4.22 ± 0.81 μM, respectively. These compounds were nontoxic to the human diploid fibroblast cell line, MCR5. Colony formation assays revealed a marked reduction in the colony-forming capacity of MCF7 cells treated with compounds 8a and 8b, suggesting their potential to inhibit cellular proliferation and tumor progression. Spheroid formation assays in three-dimensional cultures further demonstrated consistent decreases in spheroid size, highlighting the efficacy of these compounds in targeting tumor-like structures in a 3D environment. Compounds 8a and 8b were shown to induce G1 phase cell cycle arrest in MCF7 cells, which is a key mechanism in limiting cancer cell proliferation. Comparisons with the standard drug, etoposide, further elucidated their therapeutic potential. Additionally, DNA damage analysis revealed high-intensity γH2AX responses for compounds 8a and 8b, indicating that their mechanisms of action may involve the induction of DNA damage, a characteristic of effective chemotherapeutics. Furthermore, apoptosis assays demonstrated the capacity of these compounds to modulate apoptotic pathways, particularly by increasing the early apoptotic populations in MCF7 cells. In addition, ex ovo CAM assay (angiogenesis analysis) revealed a significant reduction in new blood vessel formation over time compared to untreated controls. The docking studies of compounds 8a and 8b with targets revealed favorable binding affinities with EGFR, SRC, and GSK3β. This interaction of compounds with targets potentially disrupts critical signaling pathways that drive breast cancer development and progression. In silico ADME analysis indicates favorable pharmacokinetic properties. These findings collectively elucidate the therapeutic potential of compounds 8a and 8b as promising candidates for further development as effective anticancer agents.
4. Experimental section
4.1 Chemistry
All the solvents and reagents were purchased from Sigma Aldrich, TCI, Thermo Fischer, SRL, or Spectrochem and used without further purification unless otherwise mentioned. The melting point of all the compounds was determined using the Stuart-SMP20 capillary melting point apparatus and are uncorrected. FTIR spectra were recorded on a JASCO FT-IR 4700 spectrometer by dispersing the compounds in potassium bromide pellets. Thin layer chromatography (TLC) was performed on silica gel plates (60 F254, 0.25 mm, Merck) using a 4:1 (v/v) mixture of hexanes and ethyl acetate and components were visualized under UV light or with iodine. Chromatographic separation was carried out on 100–200 mesh silica gel. 1H NMR and 13C NMR spectra were measured using a 500 MHz JEOL spectrometer in CDCl3/DMSO-d6 solvent using TMS as an internal standard. High-resolution mass spectra (HRMS) data were obtained using an Agilent 6530 ESI-QTOF mass spectrometer (mass accuracy of 5 ppm) in positive ion mode with a reverse-phase C18 column.
Synthesis of 4-(3-methyl-1H-pyrazol-1-yl)benzaldehyde (3a). 3-Methyl-1H-pyrazole (2a, 164.2 mg, 2 mmol) and anhydrous potassium carbonate (829.23 mg, 6 mmol) were added to a solution of 4-fluorobenzaldehyde (1, 248.22 mg, 2 mmol) in dimethylformamide (5 mL). The obtained reaction mixture was heated at 130 °C for 19 h. The mixture was cooled to room temperature, poured into water (50 mL) and extracted the crude product with ethyl acetate (20 mL) thrice. The combined organic layer was dried over Na2SO4, concentrated under reduced pressure, and purified by column chromatography using 20% ethyl acetate in hexanes to obtain the pure product. Yield: 70%. Pale yellow solid. Melting point: 95–96 °C. IR (KBr, νmax, cm−1): 3136 (C–H), 2861, 2753 (C–H, aldehyde), 1703 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1613 (CC), 1518 (CN). 1H NMR (500 MHz, CDCl3): δ 9.97 (s, 1H), 7.94–7.90 (m, 3H), 7.82–7.80 (m, 2H), 6.30 (d, J = 2.3 Hz, 1H), 2.37 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 191.1, 152.1, 144.4, 133.6, 131.4, 127.7, 118.3, 109.1, 13.9. MS (ESI-QTOF) for C11H10N2O [M + H]+ calculated 187.0866, found 187.0871.
O), 1613 (CC), 1518 (CN). 1H NMR (500 MHz, CDCl3): δ 9.97 (s, 1H), 7.94–7.90 (m, 3H), 7.82–7.80 (m, 2H), 6.30 (d, J = 2.3 Hz, 1H), 2.37 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 191.1, 152.1, 144.4, 133.6, 131.4, 127.7, 118.3, 109.1, 13.9. MS (ESI-QTOF) for C11H10N2O [M + H]+ calculated 187.0866, found 187.0871.Compounds 3b–l were synthesized following the same procedure described for compound 3a.
4-(4-Methyl-1H-pyrazol-1-yl)benzaldehyde (3b). 4-Methyl-1H-pyrazole (2b, 164.2 mg, 2 mmol) was used as a reagent. Yield: 68%. White solid. Melting point: 88–90 °C. IR (KBr, νmax, cm−1): 3100 (C–H), 2812, 2754 (C–H, aldehyde), 1704 (CO), 1612 (CC), 1512 (CN). 1H NMR (500 MHz, CDCl3): δ 9.97 (s, 1H), 7.94–7.92 (m, 2H), 7.82–7.78 (m, 3H), 7.57 (s, 1H), 2.15 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 191.0, 144.4, 143.3, 133.8, 131.4, 125.4, 119.7, 118.3, 9.0. MS (ESI-QTOF) for C11H10N2O [M + H]+ calculated 187.0866, found 187.0867.
4-(3,5-Dimethyl-1H-pyrazol-1-yl)benzaldehyde (3c). 3,5-Dimethyl-1H-pyrazole (2c, 192.26 mg, 2 mmol) was used as a reagent. Yield: 56%. Yellow liquid. IR (KBr, νmax, cm−1): 3136 (C–H), 2831, 2721 (C–H, aldehyde), 1694 (CO), 1604 (CC), 1511 (CN). 1H NMR (500 MHz, CDCl3): δ 9.86 (s, 1H), 7.82–7.76 (m, 2H), 7.51 (d, J = 8.4 Hz, 1H), 7.02 (d, J = 8.4 Hz, 1H), 5.91 (s, 1H), 2.25 (s, 3H), 2.14 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 191.2, 150.2, 144.7, 139.8, 134.2, 132.1, 130.6, 123.8, 119.3, 108.8, 13.5, 13.0. MS (ESI-QTOF) for C12H12N2O [M + H]+ calculated 201.1022, found 201.1021.
4-(4-Bromo-1H-pyrazol-1-yl)benzaldehyde (3d). 4-Bromo-1H-pyrazole (2d, 293.94 mg, 2 mmol) was used as a reagent. Yield: 60%. White solid. Melting point: 120–124 °C. IR (KBr, νmax, cm−1): 3120 (C–H), 2812, 2730 (C–H, aldehyde), 1694 (CO), 1612 (CC), 1502 (CN). 1H NMR (500 MHz, CDCl3): δ 10.00 (s, 1H), 8.03 (s, 1H), 7.98–7.96 (m, 2H), 7.83–7.81 (m, 2H), 7.71 (s, 1H).13C NMR (126 MHz, CDCl3): δ 190.9, 143.6, 142.8, 134.6, 131.5, 127.2, 118.7, 97.2. MS (ESI-QTOF) for C10H7BrN2O [M + H]+ calculated 250.9815, found 250.9819.
4-(3-(Trifluoromethyl)-1H-pyrazol-1-yl)benzaldehyde (3e). 3-(Trifluoromethyl)-1H-pyrazole (2e, 272.15 mg, 2 mmol) was used as a reagent. Yield: 56%. Pale yellow liquid. IR (KBr, νmax, cm−1): 3138 (C–H), 2821, 2739 (C–H, aldehyde), 1690 (CO), 1617 (CC), 1511 (CN). 1H NMR (500 MHz, CDCl3): δ 10.01 (s, 1H), 8.06 (d, J = 1.5 Hz, 1H), 7.99–7.97 (m, 2H), 7.90–7.88 (m, 2H), 6.76 (d, J = 2.3 Hz, 1H). 13C NMR (126 MHz, CDCl3): δ 190.9, 145.10 (q, J = 38.8 Hz), 143.4, 135.2, 131.4, 128.6, 119.80 (m), 119.7, 107.0. MS (ESI-QTOF) for C11H7F3N2O [M + H]+ calculated 241.0583, found 241.0581.
4-(2-Methyl-1H-imidazol-1-yl)benzaldehyde (3f). 2-Methyl-1H-imidazole (2f, 164.2 mg, 2 mmol) was used as a reagent. Yield: 70%. Yellow solid. Melting point: 84–86 °C. IR (KBr, νmax, cm−1): 3164 (C–H), 2839, 2750 (C–H, aldehyde), 1684 (CO), 1600 (CC), 1511 (CN). 1H NMR (500 MHz, CDCl3): δ 10.04 (s, 1H), 8.00–7.98 (m, 2H), 7.46 (d, J = 8.4 Hz, 2H), 7.03 (s, 2H), 2.40 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 190.9, 144.6, 142.9, 135.6, 131.1, 128.5, 125.8, 120.3, 14.1. MS (ESI-QTOF) for C11H10N2O [M + H]+ calculated 187.0866, found 187.0872.
4-(4-Methyl-1H-imidazol-1-yl)benzaldehyde (3g). 4-Methyl-1H-imidazole (2g, 164.2 mg, 2 mmol) was used as a reagent. Yield: 75%. Yellow liquid. IR (KBr, νmax, cm−1): 3124 (C–H), 2831, 2748 (C–H, aldehyde), 1694 (CO), 1603 (CC), 1511 (CN). 1H NMR (500 MHz, CDCl3): δ 10.06 (s, 1H), 8.02–8.01 (m, 2H), 7.60 (s, 1H), 7.46 (d, J = 8.4 Hz, 2H), 6.92 (s, 1H), 2.22 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 190.7, 141.9, 140.6, 134.7, 134.5, 131.7, 120.6, 114.0, 13.7. MS (ESI-QTOF) for C11H10N2O [M + H]+ calculated 187.0866, found 187.0867.
4-(5-Methyl-1H-imidazol-1-yl)benzaldehyde (3h). 5-Methyl-1H-imidazole (2h, 164.2 mg, 2 mmol) was used as a reagent. Yield: 64%. Yellow solid. Melting point: 80–83 °C. IR (KBr, νmax, cm−1): 3156 (C–H), 2835, 2748 (C–H, aldehyde), 1703 (CO), 1607 (CC), 1520 (CN). 1H NMR (500 MHz, CDCl3): δ 10.00 (s, 1H), 7.97 (d, J = 8.4 Hz, 2H), 7.86 (d, J = 1.5 Hz, 1H), 7.51 (d, J = 8.4 Hz, 2H), 7.07 (s, 1H), 2.28 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 190.8, 141.9, 140.6, 134.7, 134.5, 131.7, 120.6, 114.0, 13.7. MS (ESI-QTOF) for C11H10N2O [M + H]+ calculated 187.0866, found 187.0873.
4-(2-Ethyl-1H-imidazol-1-yl)benzaldehyde (3i). 2-Ethyl-1H-imidazole (2i, 192.26 mg, 2 mmol) was used as a reagent. Yield: 57%. Yellow liquid. IR (KBr, νmax, cm−1): 3149 (C–H), 2852, 2762 (C–H, aldehyde), 1690 (CO), 1600 (CC), 1514 (CN). 1H NMR (500 MHz, CDCl3): δ 10.03 (s, 1H), 7.99–7.97 (m, 2H), 7.45–7.43 (m, 2H), 7.04 (d, J = 1.5 Hz, 1H), 6.99 (d, J = 1.5 Hz, 1H), 2.67 (q, J = 7.4 Hz, 2H), 1.22 (t, J = 7.6 Hz, 3H). 13C NMR (126 MHz, CDCl3): δ 190.9, 149.5, 142.9, 135.7, 131.0, 128.4, 126.1, 120.3, 20.8, 12.3. MS (ESI-QTOF) for C12H12N2O [M + H]+ calculated 201.1022, found 201.1031.
4-(4-Bromo-1H-imidazol-1-yl)benzaldehyde (3j). 4-Bromo-1H-imidazole (2j, 293.94 mg, 2 mmol) was used as a reagent. Yield: 62%. Yellow solid. Melting point: 159–162 °C. IR (KBr, νmax, cm−1): 3101 (C–H), 2808, 2735 (C–H, aldehyde), 1690 (CO), 1612 (CC), 1511 (CN). 1H NMR (500 MHz, CDCl3): δ 10.03 (s, 1H), 8.03–8.00 (m, 2H), 7.83 (d, J = 1.5 Hz, 1H), 7.55–7.53 (m, 2H), 7.34 (d, J = 1.5 Hz, 1H). 13C NMR (126 MHz, CDCl3): δ 190.5, 140.9, 135.6, 135.0, 131.8, 121.3, 118.0, 117.2. MS (ESI-QTOF) for C10H7BrN2O [M + H]+ calculated 250.9815, found 250.9811.
4-(1H-1,2,3-Triazol-1-yl)benzaldehyde (3k). 1H-1,2,3-Triazole (2k, 138.12 mg, 2 mmol) was used as a reagent. Yield: 58%. White solid. Melting point: 106–108 °C. IR (KBr, νmax, cm−1): 3101 (C–H), 2854, 2744 (C–H, aldehyde), 1699 (CO), 1617 (CC), 1507 (CN). 1H NMR (500 MHz, CDCl3): δ 10.03 (s, 1H), 8.26–8.24 (m, 2H), 8.01–7.98 (m, 2H), 7.86 (s, 2H). 13C NMR (126 MHz, CDCl3): δ 191.1, 143.7, 136.7, 135.1, 131.2, 119.2. MS (ESI-QTOF) for C9H7N3O [M + H]+ calculated 174.0662, found 174.0664.
4-(1H-Tetrazol-1-yl)benzaldehyde (3l). 1H-Tetrazole(2l, 140.11 mg, 2 mmol) was used as a reagent. Yield: 50%. Pale grey solid. Melting point: 98–99 °C. IR (KBr, νmax, cm−1): 3100 (C–H), 2862, 2733 (C–H, aldehyde), 1691 (CO), 1604 (CC), 1504 (CN). 1H NMR (500 MHz, CDCl3): δ 10.10 (s, 1H), 8.71 (s, 1H), 8.36 (d, J = 9.2 Hz, 2H), 8.10 (d, J = 10.7 Hz, 2H). 13C NMR (126 MHz, CDCl3): δ 190.7, 153.5, 140.4, 137.0, 131.3, 120.5. MS (ESI-QTOF) for C8H6N4O [M + H]+calculated 175.0614, found 175.0623.
Synthesis of 4-((1-phenyl-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde (13a). To a mixture of azidobenzene 10a (238.24 mg, 5 mmol) and 4-(prop-2-yn-1-yloxy)benzaldehyde 12 (800.26 mg, 5 mmol) in anhydrous THF (10 mL) was added catalytic amount of copper iodide. Then, the reaction mixture was refluxed for 12 h. After completion of the reaction (monitored by TLC), the mixture was filtered and washed with cold THF, and the crude product present in the filtrate was extracted with ethyl acetate (3 × 20 mL) three times. The combined organic layer was washed with brine solution, dried over Na2SO4, and concentrated using a rotary evaporator. The product obtained was recrystallized using a mixture of chloroform and hexanes (8:2 v/v) to afford the pure product. Yield: 74%. Off-white solid. Melting point: 100–103 °C. IR (KBr, νmax, cm−1): 3124 (C–H), 2808, 2735 (C–H, aldehyde), 1690 (CO), 1617 (CC), 1507 (CN). 1H NMR (500 MHz, CDCl3): δ 9.83 (s, 1H), 8.02 (s, 1H), 7.81–7.77 (m, 2H), 7.68–7.65 (m, 2H), 7.48–7.44 (m, 2H), 7.40–7.37 (m, 1H), 7.09–7.06 (m, 2H), 5.31 (s, 2H). 13C NMR (126 MHz, CDCl3): δ 190.8, 163.1, 144.0, 136.8, 132.0, 130.4, 129.8, 129.0, 121.1, 120.6, 115.1, 62.1. MS (ESI-QTOF) for C16H13N3O2 [M + H]+ calculated 280.1081, found 280.1082.Compounds 13b–e were synthesized by following the same procedure as described for compound 13a.
4-((1-(4-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde (13b). 1-Azido-4-chlorobenzene (10b, 307.12 mg, 2 mmol) was used as a reagent. Yield: 75%. Pale brown solid. Melting point: 138–140 °C, IR (KBr, νmax, cm−1): 3082 (C–H), 2837, 2762 (C–H, aldehyde), 1709 (CO), 1606 (CC), 1504 (CN). 1H NMR (500 MHz, CDCl3): δ 9.87 (s, 1H), 8.06 (s, 1H), 7.84–7.82 (m, 2H), 7.69–7.67 (m, 2H), 7.50–7.47 (m, 2H), 7.13–7.11 (m, 2H), 5.35 (s, 2H). 13C NMR (126 MHz, CDCl3): δ 190.8, 163.1, 144.3, 135.4, 135.0, 132.1, 130.6, 130.1, 121.9, 121.2, 115.2, 62.1. MS (ESI-QTOF) for C16H12ClN3O2 [M + H]+ calculated 314.0691, found 314.0690.
4-((1-(4-Bromophenyl)-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde (13c). 1-Azido-4-bromobenzene (10c, 396.04 mg, 2 mmol) was used as a reagent. Yield: 70%. Pale yellow solid. Melting point: 159–161 °C. IR (KBr, νmax, cm−1): 3065 (C–H), 2835, 2753 (C–H, aldehyde), 1685 (CO), 1612 (CC), 1516 (CN). 1H NMR (500 MHz, CDCl3): δ 9.88 (s, 1H), 8.07 (s, 1H), 7.86–7.83 (m, 2H), 7.66–7.61 (m, 4H), 7.14–7.11 (m, 2H), 5.36 (s, 2H). 13C NMR (126 MHz, CDCl3): δ 190.9, 163.1, 135.9, 133.1, 132.1, 130.6, 122.8, 122.1, 121.2, 121.0, 115.2, 62.1. MS (ESI-QTOF) for C16H12BrN3O2 [M + H]+ calculated 358.0186, found 358.0189.
4-((1-(p-Tolyl)-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde (13d). 1-Azido-4-methylbenzene (10d, 266 mg, 2 mmol) was used as a reagent. Yield: 69%. Off-white solid. Melting point: 134–136 °C. IR (KBr, νmax, cm−1): 3133 (C–H), 2812, 2733 (C–H, aldehyde), 1699 (CO), 1612 (CC), 1525 (CN). 1H NMR (500 MHz, CDCl3): δ 9.88 (s, 1H), 8.03 (s, 1H), 7.85–7.83 (m, 2H), 7.60–7.58 (m, 2H), 7.30 (d, J = 7.6 Hz, 2H), 7.14–7.11 (m, 2H), 5.36 (s, 2H), 2.41 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 190.9, 163.2, 143.9, 139.3, 134.7, 132.1, 130.5, 130.4, 121.3, 120.6, 115.2, 62.2, 21.2. MS (ESI-QTOF) for C17H15N3O2 [M + H]+calculated 294.1237, found 294.1233.
4-((1-(4-Methoxyphenyl)-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde (13e). 1-Azido-4-methoxybenzene (10e, 298 mg, 2 mmol) was used as a reagent. Yield: 65%. Pale orange solid. Melting point: 130–133 °C. IR (KBr, νmax, cm−1): 3051 (C–H), 2803, 2721 (C–H, aldehyde), 1694 (CO), 1612 (CC), 1520 (CN). 1H NMR (500 MHz, CDCl3): δ 9.87 (s, 1H), 7.99 (s, 1H), 7.84–7.82 (m, 2H), 7.62–7.59 (m, 2H), 7.14–7.11 (m, 2H), 7.02–6.98 (m, 2H), 5.35 (s, 2H), 3.84 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 190.9, 163.2, 160.1, 143.8, 132.1, 130.5, 130.4, 122.4, 121.4, 115.2, 114.9, 62.2, 55.7. MS (ESI-QTOF) for C17H15N3O3 [M + H]+ calculated 310.1186, found 310.1182.
Synthesis of 2-amino-4-(4-(3-methyl-1H-pyrazol-1-yl)phenyl)-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (7a). To a solution of 4-(3-methyl-1H-pyrazol-1-yl)benzaldehyde (3a, 186.07 mg, 1 mmol) in water (2 mL) was added cyclohexane-1,3-dione (4, 112.05 mg, 1 mmol), malononitrile (5, 66.06 mg, 1 mmol), and sodium fluoride (5 mg, 12 mol%). The obtained reaction mixture was refluxed for 3 h. After completion of the reaction, the mixture was cooled to room temperature, and the solid product obtained was filtered and washed with water (10 mL) three times to remove the NaF. The crude product obtained was purified by column chromatography using 30% ethyl acetate in hexanes to get the pure product. Yield: 80%. Off-white solid. Melting point: 224–226 °C. IR (νmax, cm−1): 3381, 3216 (–NH2), 3134 (C–H), 2186 (CN), 1676 (CO), 1617 (CC), 1222 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.25 (d, J = 2.3 Hz, 1H), 7.64–7.61 (m, 2H), 7.20–7.19 (m, 2H), 7.01 (s, 2H), 6.27 (d, J = 2.3 Hz, 1H), 4.19 (s, 1H), 2.60–2.54 (m, 2H), 2.29–2.22 (m, 5H), 1.97–1.83 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 196.4, 165.0, 159.0, 149.9, 142.7, 138.9, 128.8, 128.7, 120.3, 118.6, 114.1, 108.1, 58.4, 36.9, 35.5, 27.0, 20.3, 14.0. MS (ESI-QTOF) for C20H18N4O2 [M + H]+ calculated 347.1503, found 347.1505.Compounds 7b–e, 8a–e, 15a–g and 17a–g were synthesized using the same procedure outlined for compound 7a.
2-Amino-4-(4-(4-methyl-1H-pyrazol-1-yl)phenyl)-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (7b). 4-(4-Methyl-1H-pyrazol-1-yl)benzaldehyde (3b, 186.21 mg, 1 mmol) was used as a reagent. Yield: 77%. Off-white solid. Melting point: 255–258 °C. IR (KBr, νmax, cm−1): 3344, 3179 (–NH2), 3032 (C–H), 2194 (CN), 1686 (CO), 1630 (CC), 1236 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.15 (s, 1H), 7.62 (d, J = 8.4 Hz, 2H), 7.49 (s, 1H), 7.20 (d, J = 8.4 Hz, 2H), 7.00 (s, 2H), 4.19 (s, 1H), 2.60–2.56 (m, 2H), 2.27–2.20 (m, 2H), 2.00–2.09 (m, 3H), 1.91–1.83 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 196.4, 165.1, 159.0, 142.9, 142.0, 138.9, 128.7, 126.5, 120.3, 118.6, 118.0, 114.1, 58.4, 36.9, 35.5, 27.0, 20.3, 9.3. MS (ESI-QTOF) for C20H18N4O2 [M + H]+ calculated 347.1503, found 347.1501.
2-Amino-4-(4-(3,5-dimethyl-1H-pyrazol-1-yl)phenyl)-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (7c). 4-(3,5-Dimethyl-1H-pyrazol-1-yl)benzaldehyde (3c, 200.24 mg, 1 mmol) was used as a reagent. Yield: 71%. Mustard yellow solid. Melting point: 220–224 °C. IR (KBr, νmax, cm−1): 3335, 3183 (–NH2), 2941 (C–H), 2189 (CN), 1680 (CO), 1635 (CC), 1255 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 7.35 (d, J = 8.4 Hz, 2H), 7.22 (d, J = 8.4 Hz, 2H), 7.02 (s, 2H), 6.00 (s, 1H), 4.22 (s, 1H), 2.61–2.57 (m, 2H), 2.28–2.23 (m, 5H), 2.12 (s, 3H), 1.98–1.87 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 196.5, 165.2, 159.1, 148.2, 143.9, 139.5, 138.7, 128.3, 124.5, 120.3, 114.1, 107.6, 58.4, 36.9, 35.6, 27.0, 20.3, 13.9, 12.7. MS (ESI-QTOF) for C21H20N4O2 [M + H]+ calculated 361.1659, found 361.1660.
2-Amino-4-(4-(4-bromo-1H-pyrazol-1-yl)phenyl)-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (7d). 4-(4-Bromo-1H-pyrazol-1-yl)benzaldehyde (3d, 251.08 mg, 1 mmol) was used as a reagent. Yield: 72%. Grey solid. Melting point: 220–224 °C. IR (KBr, νmax, cm−1): 3310, 3188 (–NH2), 3120 (C–H), 2182 (CN), 1683 (CO), 1642 (CC), 1243 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.91 (s, 1H), 8.67 (s, 1H), 8.48 (s, 1H), 8.04 (d, J = 1.5 Hz, 1H), 7.81 (s, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.24 (d, J = 8.4 Hz, 1H), 7.02 (s, 1H), 4.20 (s, 1H), 2.63–2.56 (m, 2H), 2.29–2.20 (m, 2H), 1.94–1.85 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 196.4, 165.1, 158.9, 145.0, 142.9, 142.6, 137.9, 131.1, 128.9, 120.2, 113.9, 106.7, 58.2, 36.8, 35.7, 27.0, 20.3. MS (ESI-QTOF) for C19H15BrN4O2 [M + H]+ calculated 411.0451, found 411.0448.
2-Amino-5-oxo-4-(4-(3-(trifluoromethyl)-1H-pyrazol-1-yl)phenyl)-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (7e). 4-(3-(Trifluoromethyl)-1H-pyrazol-1-yl)benzaldehyde (3e, 240.18 mg, 1 mmol) was used as a reagent. Yield: 70%. Yellow solid. Melting point: 200–204 °C. IR (KBr, νmax, cm−1): 3386, 3306 (–NH2), 2964 (C–H), 2195 (CN), 1693 (CO), 1640 (CC), 1249 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.61 (s, 1H), 7.72 (d, J = 8.4 Hz, 2H), 7.30 (d, J = 8.4 Hz, 2H), 7.03 (s, 2H), 6.98 (d, J = 2.3 Hz, 1H), 4.24 (s, 1H), 2.62–2.57 (m, 2H), 2.30–2.20 (m, 2H), 1.96–1.84 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 196.4, 165.2, 159.0, 145.0, 142.9, 142.7, 137.9, 131.1, 129.0, 120.8, 120.2, 113.9, 106.7, 58.2, 36.8, 35.7, 27.0, 20. MS (ESI-QTOF) for C20H15F3N4O2 [M + H]+ calculated 401.1220, found 401.1222.
2-Amino-7,7-dimethyl-4-(4-(2-methyl-1H-imidazol-1-yl)phenyl)-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (8a). 4-(2-Methyl-1H-imidazol-1-yl)benzaldehyde (3f, 186.21 mg, 1 mmol) was used as a reagent. Yield: 79%. Pale yellow solid. Melting point: 229–232 °C. IR (KBr, νmax, cm−1): 3381, 3312 (–NH2), 2991 (C–H), 2194 (CN), 1699 (CO), 1630 (CC), 1255 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 7.33 (d, J = 8.4 Hz, 2H), 7.25 (d, J = 8.4 Hz, 3H), 7.05 (s, 2H), 6.85 (s, 1H), 4.24 (s, 1H), 2.50 (s, 2H), 2.25–2.10 (m, 5H), 1.01 (s, 3H), 0.95 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 196.3, 163.4, 159.1, 144.9, 144.0, 136.6, 128.7, 127.6, 125.6, 121.5, 120.2, 112.9, 58.4, 50.5, 35.7, 32.4, 28.8, 27.5, 14.2. MS (ESI-QTOF) for C22H22N4O2 [M + H]+ calculated 375.1816, found 375.1810.
2-Amino-7,7-dimethyl-4-(4-(4-methyl-1H-imidazol-1-yl)phenyl)-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (8b). 4-(4-Methyl-1H-imidazol-1-yl)benzaldehyde (3g, 186.21 mg, 1 mmol) was used as a reagent. Yield: 77%. Off-white solid. Melting point: 230–234 °C. IR (KBr, νmax, cm−1): 3326, 3183 (–NH2), 2941 (C–H), 2194 (CN), 1690 (CO), 1625 (CC), 1236 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.03 (s, 1H), 7.46 (d, J = 8.4 Hz, 2H), 7.34 (s, 1H), 7.21 (d, J = 8.4 Hz, 2H), 7.02 (s, 2H), 4.20 (s, 1H), 2.49 (s, 2H), 2.24–2.06 (m, 5H), 1.00 (s, 3H), 0.93 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 196.3, 163.2, 159.0, 143.7, 138.7, 136.1, 135.2, 129.1, 120.6, 120.2, 114.8, 112.9, 58.5, 50.5, 35.6, 32.4, 28.9, 27.4, 14.1. MS (ESI-QTOF) for C22H22N4O2 [M + H]+ calculated 375.1816, found 375.1815.
2-Amino-7,7-dimethyl-4-(4-(5-methyl-1H-imidazol-1-yl)phenyl)-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (8c). 4-(5-Methyl-1H-imidazol-1-yl)benzaldehyde (3h, 272.15 mg, 1 mmol) was used as a reagent. Yield: 73%. Pale yellow solid. Melting point: 225–228 °C. IR (KBr, νmax, cm−1): 3312, 3188 (–NH2), 2959 (C–H), 2194 (CN), 1685 (CO), 1621 (CC), 1245 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.05 (s, 1H), 7.46 (d, J = 8.4 Hz, 2H), 7.35 (s, 1H), 7.21 (d, J = 9.2 Hz, 2H), 7.02 (s, 2H), 4.19 (s, 1H), 2.49 (s, 2H), 2.24–2.06 (m, 5H), 1.00 (s, 3H), 0.93 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 196.3, 163.2, 159.0, 143.7, 138.6, 136.0, 135.2, 129.1, 120.7, 120.2, 114.8, 112.9, 58.5, 50.5, 35.6, 32.4, 28.9, 27.4, 14.1. MS (ESI-QTOF) for C22H22N4O2 [M + H]+ calculated 375.1816, found 375.1811.
2-Amino-4-(4-(2-ethyl-1H-imidazol-1-yl)phenyl)-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (8d). 4-(2-Ethyl-1H-imidazol-1-yl)benzaldehyde (3i, 200.24 mg, 1 mmol) was used as a reagent. Yield: 70%. Yellow solid. Melting point: 230–233 °C. IR (KBr, νmax, cm−1): 3358, 3321 (–NH2), 3138 (C–H), 2203 (CN), 1690 (CO), 1653 (CC), 1245 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 7.31 (d, J = 8.4 Hz, 2H), 7.26–7.21 (m, 3H), 7.06 (s, 2H), 6.88 (s, 1H), 4.24 (s, 1H), 2.56–2.50 (m, 4H), 2.25–2.10 (m, 2H), 1.07 (t, J = 7.6 Hz, 3H), 1.01 (s, 3H), 0.94 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 196.3, 163.4, 159.1, 148.8, 145.1, 136.4, 128.7, 127.5, 126.0, 121.5, 120.2, 112.9, 58.4, 50.5, 35.7, 32.4, 28.8, 27.5, 20.6, 12.7. MS (ESI-QTOF) for C23H24N4O2 [M + H]+ calculated 389.1972, found 389.1978.
2-Amino-4-(4-(4-bromo-1H-imidazol-1-yl)phenyl)-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (8e). 4-(4-Bromo-1H-imidazol-1-yl)benzaldehyde (3j, 251.08 mg, 1 mmol) was used as a reagent. Yield: 73%. Yellow solid. Melting point: 197–200 °C. IR (KBr, νmax, cm−1): 3358, 3307 (–NH2), 3147 (C–H), 2185 (CN), 1691 (CO), 1653 (CC), 1250 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.17 (s, 1H), 7.87 (d, J = 1.5 Hz, 1H), 7.52 (d, J = 8.4 Hz, 2H), 7.24 (d, J = 9.2 Hz, 2H), 7.04 (s, 2H), 4.22 (s, 1H), 2.50 (s, 2H), 2.24–2.07 (m, 2H), 1.01 (s, 3H), 0.93 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 196.2, 163.3, 159.1, 144.8, 136.4, 135.3, 129.1, 121.2, 120.1, 118.1, 116.0, 112.9, 58.4, 50.5, 35.7, 32.4, 28.8, 27.5. MS (ESI-QTOF) for C21H19BrN4O2 [M + H]+ calculated 439.0764, found 439.0768.
2-Amino-5-oxo-4-(4-((1-phenyl-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-4H,5H-pyrano[3,2-c]chromene-3-carbonitrile (15a). 4-((1-Phenyl-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde (13a, 279.29 mg, 1 mmol) was used as a reagent. Yield: 75%. White solid. Melting point: 220–223 °C. IR (KBr, νmax, cm−1): 3436, 3248 (–NH2), 3178 (C–H), 2181 (CN), 1708 (CO), 1662 (CC), 1259 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.90 (s, 1H), 7.86 (t, J = 7.2 Hz, 3H), 7.68–7.65 (m, 1H), 7.56 (t, J = 8.0 Hz, 2H), 7.47–7.41 (m, 3H), 7.35 (s, 2H), 7.17 (d, J = 9.2 Hz, 2H), 6.99 (d, J = 8.0 Hz, 2H), 5.17 (s, 2H), 4.38 (s, 1H). 13C NMR (126 MHz, DMSO-d6): δ 160.1, 158.5, 157.7, 153.7, 152.6, 144.4, 137.1, 136.4, 133.4, 130.4, 129.4, 129.3, 125.2, 123.4, 123.0, 120.7, 119.9, 117.1, 115.2, 113.5, 104.8, 61.6, 58.7, 36.7. MS (ESI-QTOF) for C28H19N5O4 [M + H]+ calculated 490.1510, found 490.1511.
2-Amino-4-(4-((1-(4-chlorophenyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-5-oxo-4H,5H-pyrano[3,2-c]chromene-3-carbonitrile (15b). 4-((1-(4-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde (13b, 313.74 mg, 1 mmol) was used as a reagent. Yield: 72%. White solid. Melting point: 255–258 °C. IR (KBr, νmax, cm−1): 3468, 3261 (–NH2), 3101 (C–H), 2185 (CN), 1717 (CO), 1653 (CC), 1250 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.93 (s, 1H), 7.92 (d, J = 9.2 Hz, 2H), 7.85 (d, J = 6.9 Hz, 1H), 7.68–7.62 (m, 3H), 7.46–7.40 (m, 2H), 7.35 (s, 2H), 7.17 (d, J = 8.4 Hz, 2H), 6.98 (d, J = 8.4 Hz, 2H), 5.17 (s, 2H), 4.38 (s, 1H). 13C NMR (126 MHz, DMSO-d6): δ 160.1, 158.5, 157.6, 153.7, 152.6, 144.6, 136.5, 135.9, 133.5, 133.4, 130.4, 129.3, 125.2, 123.4, 123.0, 122.4, 119.9, 117.1, 115.2, 113.5, 104.8, 61.6, 58.6, 36.7. MS (ESI-QTOF) for C28H18ClN5O4 [M + H]+ calculated 524.1120, found 524.1115.
2-Amino-4-(4-((1-(4-bromophenyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-5-oxo-4H,5H-pyrano[3,2-c]chromene-3-carbonitrile (15c). 4-((1-(4-Bromophenyl)-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde (13c, 358.19 mg, 1 mmol) was used as a reagent. Yield: 69%. Off-white solid. Melting point: 223–225 °C. IR (KBr, νmax, cm−1): 3458, 3243 (–NH2), 3096 (C–H), 2189 (CN), 1703 (CO), 1653 (CC), 1255 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.93 (s, 1H), 7.85 (d, J = 8.4 Hz, 3H), 7.76 (d, J = 9.2 Hz, 2H), 7.66 (t, J = 7.6 Hz, 1H), 7.46–7.40 (m, 2H), 7.35 (s, 2H), 7.17 (d, J = 9.2 Hz, 2H), 6.98 (d, J = 9.2 Hz, 2H), 5.17 (s, 2H), 4.38 (s, 1H). 13C NMR (126 MHz, DMSO-d6): δ 160.1, 158.4, 157.6, 153.7, 152.6, 144.6, 136.5, 136.2, 133.4, 133.3, 129.4, 125.2, 123.4, 123.0, 122.6, 121.9, 119.8, 117.1, 115.2, 113.4, 104.7, 61.6, 58.6, 36.7. MS (ESI-QTOF) for C28H18BrN5O4 [M + H]+ calculated 568.0615, found 568.0613.
2-Amino-5-oxo-4-(4-((1-(p-tolyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-4H,5H-pyrano[3,2-c]chromene-3-carbonitrile (15d). 4-((1-(p-Tolyl)-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde (13d, 293.32 mg, 1 mmol) was used as a reagent. Yield: 68%. White solid. Melting point: 245–247 °C. IR (KBr, νmax, cm−1): 3453, 3233 (–NH2), 3022 (C–H), 2184 (CN), 1716 (CO), 1621 (CC), 1236 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.84 (s, 1H), 7.85 (d, J = 7.6 Hz, 1H), 7.74 (d, J = 9.2 Hz, 2H), 7.68–7.64 (m, 1H), 7.46–7.40 (m, 2H), 7.35 (t, J = 3.8 Hz, 4H), 7.17 (d, J = 9.2 Hz, 2H), 6.98 (d, J = 9.2 Hz, 2H), 5.16 (s, 2H), 4.38 (s, 1H), 2.33 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 160.1, 158.5, 157.7, 153.7, 152.6, 144.3, 138.9, 136.4, 134.9, 133.4, 130.7, 129.2, 125.2, 123.2, 123.0, 120.6, 119.9, 117.1, 115.2, 113.5, 104.8, 61.6, 58.7, 36.7, 21.1. MS (ESI-QTOF) for C29H21N5O4 [M + H]+ calculated 504.1666, found 504.1668.
2-Amino-4-(4-((1-(4-methoxyphenyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-5-oxo-4H,5H-pyrano[3,2-c]chromene-3-carbonitrile (15e). 4-((1-(4-Methoxyphenyl)-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde (13e, 309.32 mg, 1 mmol) was used as a reagent. Yield: 65%. Off-white solid. Melting point: 253–255 °C. IR (KBr, νmax, cm−1): 3408, 3275 (–NH2), 3101 (C–H), 2185 (CN), 1703 (CO), 1667 (CC), 1245 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.78 (s, 1H), 7.86–7.65 (m, 4H), 7.46–7.40 (m, 2H), 7.35 (s, 2H), 7.17 (d, J = 8.4 Hz, 2H), 7.09 (d, J = 9.2 Hz, 2H), 6.98 (d, J = 8.4 Hz, 2H), 5.15 (s, 2H), 4.38 (s, 1H), 3.78 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 160.1, 159.8, 158.5, 157.7, 153.7, 152.6, 144.2, 136.4, 133.4, 130.5, 129.4, 125.2, 123.3, 123.0, 122.4, 119.9, 117.1, 115.4, 115.2, 113.5, 104.8, 61.6, 58.7, 56.1, 36.7. MS (ESI-QTOF) for C29H21N5O4 [M + H]+ calculated 520.1615, found 520.1614.
4-(4-(1H-1,2,3-Triazol-1-yl)phenyl)-2-amino-5-oxo-4H,5H-pyrano[3,2-c]chromene-3-carbonitrile (15f). 4-(1H-1,2,3-Triazol-1-yl)benzaldehyde (3k, 173.17 mg, 1 mmol) was used as a reagent. Yield: 70%. White solid. Melting point: 257–259 °C. IR (KBr, νmax, cm−1): 3345, 3212 (–NH2), 3042 (C–H), 2201 (CN), 1721 (CO), 1630 (CC), 1236 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.23–8.21 (m, 1H), 8.12–8.06 (m, 2H), 7.93 (d, J = 8.4 Hz, 2H), 7.88 (d, J = 8.4 Hz, 1H), 7.69 (t, J = 6.9 Hz, 1H), 7.48–7.42 (m, 5H), 4.52 (s, 1H). 13C NMR (126 MHz, DMSO-d6): δ 160.1, 158.5, 154.1, 152.7, 143.4, 138.7, 138.4, 137.0, 133.6, 133.0, 129.6, 125.2, 123.1, 119.7, 119.5, 119.2, 117.2, 113.5, 104.1, 58.1, 37.1. MS (ESI-QTOF) for C21H13N5O3 [M + H]+ calculated 384.1091, found 384.1086.
4-(4-(1H-Tetrazol-1-yl)phenyl)-2-amino-5-oxo-4H,5H-pyrano[3,2-c]chromene-3-carbonitrile (15g). 4-(1H-Tetrazol-1-yl)benzaldehyde (3l, 174.16 mg, 1 mmol) was used as a reagent. Yield: 65%. Yellow solid. Melting point: 240–243 °C. IR (KBr, νmax, cm−1): 3371, 3261 (–NH2), 3178 (C–H), 2208 (CN), 1708 (CO), 1648 (CC), 1250 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 9.19 (s, 1H), 8.01 (d, J = 8.4 Hz, 2H), 7.88 (d, J = 8.0 Hz, 1H), 7.69 (t, J = 7.6 Hz, 1H), 7.56 (d, J = 8.4 Hz, 2H), 7.48–7.43 (m, 4H), 4.59 (s, 1H). 13C NMR (126 MHz, DMSO-d6): δ 160.1, 158.5, 154.4, 154.3, 152.8, 146.0, 135.6, 133.6, 130.1, 125.3, 123.1, 120.8, 119.6, 117.2, 113.5, 103.8, 57.8, 37.2. MS (ESI-QTOF) for C20H12N6O3 [M + H]+ calculated 385.1044, found 385.1046.
2-Amino-7-methyl-5-oxo-4-(4-((1-phenyl-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-4H,5H-pyrano[4,3-b]pyran-3-carbonitrile (17a). 4-((1-Phenyl-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde (13a, 279.29 mg, 1 mmol) was used as a reagent. Yield: 80%. Brownish red solid. Melting point: 167–170 °C. IR (KBr, νmax, cm−1): 3382, 3310 (–NH2), 3178 (C–H), 2190 (CN), 1699 (CO), 1650 (CC), 1239 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.93 (s, 1H), 7.90 (d, J = 7.6 Hz, 2H), 7.59 (t, J = 7.9 Hz, 2H), 7.49 (t, J = 7.4 Hz, 1H), 7.14–7.11 (m, 4H), 7.01 (d, J = 8.6 Hz, 2H), 6.25 (s, 1H), 5.20 (s, 2H), 4.23 (s, 1H), 2.20 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 162.7, 161.3, 158.0, 157.9, 157.0, 143.9, 136.6, 136.2, 129.9, 128.7, 128.6, 122.8, 120.2, 119.4, 114.6, 101.0, 97.9, 61.1, 58.1, 35.5, 19.3. MS (ESI-QTOF) for C25H19N5O4 [M + H]+ calculated 454.1510, found 454.1515.
2-Amino-4-(4-((1-(4-chlorophenyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-7-methyl-5-oxo-4H,5H-pyrano[4,3-b]pyran-3-carbonitrile (17b). 4-((1-(4-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde (13b, 313.74 mg, 1 mmol) was used as a reagent. Yield: 74%. Pale orange solid. Melting point: 200–203 °C. IR (KBr, νmax, cm−1): 3408, 3340 (–NH2), 3120 (C–H), 2194 (CN), 1708 (CO), 1644 (CC), 1245 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.93 (s, 1H), 7.93–7.91 (m, 2H), 7.65–7.62 (m, 2H), 7.13 (s, 2H), 7.09 (d, J = 8.4 Hz, 2H), 6.97 (d, J = 8.4 Hz, 2H), 6.22 (s, 1H), 5.17 (s, 2H), 4.20 (s, 1H), 2.17 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 163.3, 161.9, 158.6, 158.5, 157.5, 144.6, 136.7, 135.9, 133.6, 130.4, 129.2, 123.4, 122.4, 119.9, 115.1, 101.5, 98.5, 61.5, 58.6, 35.9, 19.8. MS (ESI-QTOF) for C25H18ClN5O4 [M + H]+ calculated 488.1120, found 488.1122.
2-Amino-4-(4-((1-(4-bromophenyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-7-methyl-5-oxo-4H,5H-pyrano[4,3-b]pyran-3-carbonitrile (17c). 4-((1-(4-Bromophenyl)-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde (13c, 358.19 mg, 1 mmol) was used as a reagent. Yield: 70%. Off-white solid. Melting point: 206–209 °C. IR (KBr, νmax, cm−1): 3403, 3321 (–NH2), 3106 (C–H), 2198 (CN), 1703 (CO), 1648 (CC), 1236 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.93 (s, 1H), 7.86 (d, J = 8.4 Hz, 2H), 7.76 (d, J = 9.2 Hz, 2H), 7.13–7.08 (m, 4H), 6.97 (d, J = 9.2 Hz, 2H), 6.22 (s, 1H), 5.17 (s, 2H), 4.20 (s, 1H), 2.17 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 163.3, 161.9, 158.6, 158.5, 157.5, 144.7, 136.7, 136.3, 133.3, 129.2, 123.4, 122.6, 121.9, 119.9, 115.1, 101.5, 98.5, 61.6, 58.6, 36.0, 19.8. MS (ESI-QTOF) for C25H18BrN5O4 [M + H]+ calculated 532.0615, found 532.0610.
2-Amino-7-methyl-5-oxo-4-(4-((1-(p-tolyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-4H,5H-pyrano[4,3-b]pyran-3-carbonitrile (17d). 4-((1-(p-Tolyl)-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde (13d, 293.32 mg, 1 mmol) was used as a reagent. Yield: 67%. Pale yellow solid. Melting point: 208–211 °C. IR (KBr, νmax, cm−1): 3408, 3307 (–NH2), 3179 (C–H), 2189 (CN), 1708 (CO), 1658 (CC), 1255 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.85 (s, 1H), 7.75 (d, J = 8.4 Hz, 2H), 7.36 (d, J = 8.4 Hz, 2H), 7.13 (s, 2H), 7.09 (d, J = 9.2 Hz, 2H), 6.98 (d, J = 8.4 Hz, 2H), 6.22 (s, 1H), 5.16 (s, 2H), 4.20 (s, 1H), 2.34 (s, 3H), 2.17 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 163.3, 161.9, 158.6, 158.5, 157.6, 144.3, 138.9, 136.7, 134.9, 130.8, 129.2, 123.2, 120.6, 119.9, 115.1, 101.5, 98.5, 61.6, 58.6, 36.0, 21.1, 19.8. MS (ESI-QTOF) for C26H21N5O4 [M + H]+ calculated 468.1666, found 468.1668.
2-Amino-4-(4-((1-(4-methoxyphenyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-7-methyl-5-oxo-4H,5H-pyrano[4,3-b]pyran-3-carbonitrile (17e). 4-((1-(4-Methoxyphenyl)-1H-1,2,3-triazol-4-yl)methoxy)benzaldehyde (13e, 309.32 mg, 2 mmol) was used as a reagent. Yield: 67%. Pale pink solid. Melting point: 202–205 °C. IR (KBr, νmax, cm−1): 3330, 3211 (–NH2), 3106 (C–H), 2194 (CN), 1722 (CO), 1648 (CC), 1245 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.99 (s, 1H), 8.93 (s, 1H), 8.38 (s, 1H), 7.97–7.91 (m, 2H), 7.64 (t, J = 7.6 Hz, 2H), 7.30 (d, J = 8.4 Hz, 1H), 7.13–7.08 (m, 2H), 6.97 (d, J = 8.4 Hz, 1H), 6.22 (s, 1H), 5.37 (s, 2H), 5.17 (s, 3H), 4.20 (s, 1H), 2.17 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 163.3, 161.9, 159.8, 158.6, 158.5, 157.6, 144.2, 136.7, 130.5, 129.2, 123.3, 122.4, 119.9, 115.4, 115.1, 101.5, 98.5, 61.6, 58.6, 56.1, 36.0, 19.8. MS (ESI-QTOF) for C26H21N5O5 [M + H]+ calculated 484.1615, found 484.1615.
4-(4-(1H-1,2,3-Triazol-1-yl)phenyl)-2-amino-7-methyl-5-oxo-4H,5H-pyrano[4,3-b]pyran-3-carbonitrile (17f). 4-(1H-1,2,3-Triazol-1-yl)benzaldehyde (3k, 173.17 mg, 1 mmol) was used as a reagent. Yield: 70%. Off-white solid. Melting point: 232–234 °C. IR (KBr, νmax, cm−1): 3372, 3309 (–NH2), 3178 (C–H), 2203 (CN), 1700 (CO), 1658 (CC), 1236 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 8.06 (s, 2H), 7.93 (d, J = 8.4 Hz, 2H), 7.36 (d, J = 9.2 Hz, 2H), 7.23 (s, 2H), 6.26 (s, 1H), 4.35 (s, 1H), 2.19 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 163.6, 161.9, 158.8, 158.6, 143.7, 138.7, 136.9, 129.5, 119.8, 119.2, 100.8, 98.5, 58.0, 36.4, 19.9. MS (ESI-QTOF) for C18H13N5O3 [M + H]+calculated 348.1091, found 348.1091.
4-(4-(1H-Tetrazol-1-yl)phenyl)-2-amino-7-methyl-5-oxo-4H,5H-pyrano[4,3-b]pyran-3-carbonitrile (17g). 4-(1H-Tetrazol-1-yl)benzaldehyde (3l, 174.16 mg, 1 mmol) (2e, 272.15 mg, 2 mmol) was used as a reagent. Yield: 65%. Brown solid. Melting point: 227–230 °C. IR (KBr, νmax, cm−1): 3385, 3280 (–NH2), 3106 (C–H), 2185 (CN), 1690 (CO), 1644 (CC), 1245 (C–O–C). 1H NMR (500 MHz, DMSO-d6): δ 9.20 (s, 1H), 8.01 (d, J = 8.3 Hz, 2H), 7.47 (d, J = 8.3 Hz, 2H), 7.27 (s, 2H), 6.27 (s, 1H), 4.42 (s, 1H), 2.20 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 163.8, 161.9, 159.0, 158.7, 154.4, 146.3, 135.5, 129.9, 120.8, 119.7, 100.6, 98.5, 57.7, 36.6, 19.9. MS (ESI-QTOF) for C17H12N6O3 [M + H]+ calculated 349.1044, found 349.1047.
4.2 Anticancer activity
Cell viability assessment, colony formation assay, cell cycle analysis, DNA double-strand break evaluation, and apoptosis detection were performed following our previously reported protocol.44 Etoposide (1 μM) served as the reference compound for cell cycle analysis and DNA double-strand break assessment. Data are expressed as mean ± standard error of the mean. Statistical differences between control and treated groups were assessed using a one-way and two-way analysis of variance (ANOVA) followed by Tukey and Dunnett post hoc test using GraphPad Prism 9 software. FACS data were quantified by FlowJo software.
Spheroid formation assay. Spheroid formation and growth were assessd using agar molds prepared with the 3D Petri Dish® system (Microtissues®). Agar was liquefied in normal saline, sterilized, and cast into the molds. Once solidification, the molds were rinsed with the appropriate culture medium and placed into 24-well plates. Cells were seeded onto the molds and allowed to adhere for 20 minutes. Subsequently, an additional medium containing various concentrations of the test drugs was added. Live-cell imaging was conducted using an Olympus CellVivo Inverted Microscope, and spheroid dimensions were analyzed with Olympus CellSens software.
Ex ovo CAM assay. Fertilized chicken eggs were sourced from a local supplier and incubated at 37.5 °C with a humidity of 65% and periodic rotation for three days. On the third day, the eggs were carefully opened into sterile weighing boats, each covered with a sterile lid, and placed in a cell culture-grade CO2 incubator maintained at 37 °C and 5% CO2 concentration. To assess the angiogenic potential of various compounds, on the seventh day, glass filter paper discs, previously dipped in designated drug concentrations and dried, were applied to the chorioallantoic membrane CAM vascular zones. The development of the vascular networks was monitored, and images were captured periodically. These images were subsequently analysed using AngioTool software to quantify vascular growth. The CAM experiments are not considered to be animal experimentation from a regulatory point of view in Europe (EU directive 2010/63/EU), European Commission (2021/2784(RSP)). Therefore, they are exempt from Institutional Animal Ethics Committee (IAEC) approval in EU countries.
4.3 Network interaction and its ranking
4.3.1 Data collection and preparation. Fused pyrazole derivatives 8a and 8b demonstrated good activity in vitro against a breast cancer cell line MCF7. Based on this key information, various databases were utilized to identify targets related to breast cancer. In this study, the Swiss Target Prediction database66 was employed to screen for potential targets of compounds 8a and 8b. Breast cancer hits were obtained from the Disgenet,67 Malacard68 and OMIM69 (accessed on October 15, 2024) using the keyword searches “breast cancer”. The overlapping targets identified from both database searches for each compound set will provide a relevant anticancer target, which is taken for further analysis.
4.3.2 Establishment of STRING network and module formation. Protein–protein interaction (PPI) data for the identified targets were generated using the STRING Database (https://stringdb.org/, accessed on 15 October 2024).70 A list of target genes was provided as input to STRING, which offers valuable insights into protein interactions and correlations, assigning confidence scores to evaluate their reliability. The overlapping targets for each cancer type were entered into the database, with the Homo sapiens category selected for network visualization. A medium confidence threshold ranging from 0.4 to 0.7 was applied to construct the interaction network. The resulting network was then imported into Cytoscape,71 and the top 10 hub genes were identified and ranked using the CytoHubba app.72
4.4 Molecular docking
Molecular docking is a protocol used to simulate the binding of a ligand to target proteins. Docking was performed using Schrödinger Maestro.73 The respective protein targets, selected after ranking using CytoHubba, were retrieved from the PDB database. The proteins were prepared using a standard protocol, which involved removing crystallized water molecules, adding missing loops and side chains, and performing a short minimization step. Grid generation was performed by selecting the center of the co-crystallized ligand as the reference. This was followed by extra precision docking using the Glide module. Binding free energy calculation was done for each of the docked complexes using a prime module.74
4.5 ADME analysis
The physicochemical properties of the compounds were obtained from the SwissADME webserver.75 SwissADME is a freely accessible online tool that evaluates the pharmacokinetics, drug-likeness, and medicinal chemistry properties of compounds. The SMILES notation of the respective compounds 8a and 8b was input, and an analysis was conducted to calculate various parameters, including compliance with Lipinski's rule of five, lipophilicity, and water solubility.
Data availability
The primary data supporting this work are included in the ESI.†
Author contributions
Fabitha K. synthesized all the target compounds and performed data analysis and validation. Hiba Haneefa P. T. prepared and characterized the intermediates. Anoop Kallingal and Natalia Maciejewska conducted the biological assays. Varun Thachan Kundil conducted in silico studies. Fabitha K. drafted the manuscript with assistance from Anoop Kallingal and Varun Thachan Kundil. Dr. Janardhan Banothu designed the project, provided support, supervised the entire study, and reviewed, and edited the manuscript for final submission. Dr. Majed Alharbi reviewed the manuscript. All the authors approved the final version of the manuscript.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
JB sincerely thanks the Science and Engineering Research Board (SERB) for awarding the research grant (DST No: EEQ/2020/000303) and the Centre for Innovation, Entrepreneurship & Incubation (CIEI), NIT Calicut for the financial support under Students Innovative Project 2023–24. FK sincerely acknowledges the Ministry of Education and the National Institute of Technology Calicut for the fellowship. All the authors thanks to Varun Thachan Kundil for conducting in silico studeis. All the authors are grateful to the CMC, NIT Calicut for NMR characterization and the Director, NIT Calicut for providing research facilities. We sincerely thank DST-FIST for providing the HRMS facility (SR/FIST/CS-I/2019/120) to the Department of Chemistry, NIT Calicut.
References
- F. Bray, M. Laversanne, H. Sung, J. Ferlay, R. L. Siegel, I. Soerjomataram and A. Jemal, Ca-Cancer J. Clin., 2024, 74, 229–263, DOI:10.3322/caac.21834.
- L. Wilkinson and T. Gathani, Br. J. Radiol., 2021, 95, 20211033, DOI:10.1259/bjr.20211033.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249, DOI:10.3322/caac.21660.
- N. Kerru, P. Singh, N. Koorbanally, R. Raj and V. Kumar, Eur. J. Med. Chem., 2017, 142, 179–212, DOI:10.1016/j.ejmech.2017.07.033.
- S. Sharmin, M. M. Rahaman, M. Martorell, J. Sastre-Serra, J. Sharif-Rad, M. Butnariu, I. C. Bagiu, R. V. Bagiu and M. T. Islam, Cancer Cell Int., 2021, 21, 1–19, DOI:10.1186/s12935-021-02309-9.
- F. Ye, S. Dewanjee, Y. Li, N. K. Jha, Z.-S. Chen, A. Kumar, Vishakha, T. Behl, S. K. Jha and H. Tang, Mol. Cancer, 2023, 22, 105, DOI:10.1186/s12943-023-01805-y.
- J. Wang, K. Lei and F. Han, Riv. Eur. Sci. Med. Farmacol., 2018, 22, 3855–3864, DOI:10.26355/eurrev_201806_15270.
- S. A. Patil, R. Patil, L. M. Pfeffer and D. D. Miller, Future Med. Chem., 2013, 5, 1647–1660, DOI:10.4155/fmc.13.126.
- A. Chaudhary, K. Singh, N. Verma, S. Kumar, D. Kumar and P. P. Sharma, Mini-Rev. Med. Chem., 2022, 22, 2736–2751, DOI:10.2174/1389557522666220331161636.
- M. Costa, T. A. Dias, A. Brito and F. Proença, Eur. J. Med. Chem., 2016, 123, 487–507, DOI:10.1016/j.ejmech.2016.07.057.
- J. Li, X. Wang, Y. Fang and C. Wang, J. Asian Nat. Prod. Res., 2010, 12, 992–1000, DOI:10.1080/10286020.2010.513034.
- J. Du, F. Jiang, S. Xu, Z. Huang, L. L. Chen and L. Li, J. Cancer, 2021, 12, 270–280, DOI:10.7150/jca.50360.
- H. Ekowati, I. Astuti and Mustofa, Indones. J. Chem., 2010, 10, 247–251, DOI:10.22146/ijc.21467.
- N. Guilbaud, L. Kraus-Berthier, F. Meyer-Losic, V. Malivet, C. Chacun, M. Jan, F. Tillequin, S. Michel, M. Koch, B. Pfeiffer, G. Atassi, J. Hickman and A. Pierré, Clin. Cancer Res., 2001, 7, 2573–2580 CAS.
- H. Nishino, T. Okuyama, M. Takata, S. Shibata, H. Tokuda, J. Takayasu, T. Hasegawa, A. Nishino, H. Ueyama and A. Iwashima, Carcinogenesis, 1990, 11, 1557–1561, DOI:10.1093/carcin/11.9.1557.
- V. Raj and J. Lee, Front. Chem., 2020, 8, 1–23, DOI:10.3389/fchem.2020.00623.
- T. Kottha and V. Gnanasambandam, ChemistrySelect, 2018, 3, 8381–8386, DOI:10.1002/slct.201801210.
- W. Kemnitzer, J. Drewe, S. Jiang, H. Zhang, Y. Wang, J. Zhao, S. Jia, J. Herich, D. Labreque, R. Storer, K. Meerovitch, D. Bouffard, R. Rej, R. Denis, C. Blais, S. Lamothe, G. Attardo, H. Gourdeau, B. Tseng, S. Kasibhatla and S. X. Caiet, J. Med. Chem., 2004, 47, 6299–6310, DOI:10.1021/jm049640t.
- Y. Liu, D. Yang, Z. Hong, S. Guo, M. Liu, D. Zuo, D. Ge, M. Qin and D. Sun, Eur. J. Med. Chem., 2018, 146, 185–193, DOI:10.1016/j.ejmech.2018.01.052.
- S. Kasibhatla, H. Gourdeau, K. Meerovitch, J. Drewe, S. Reddy, L. Qiu, H. Zhang, F. Bergeron, D. Bouffard, Q. Yang, J. Herich, S. Lamothe, S. X. Cai and B. Tseng, Mol. Cancer Ther., 2004, 3, 1365–1374, DOI:10.1158/1535-7163.1365.3.11.
- H. Gourdeau, L. Leblond, B. Hamelin, C. Desputeau, K. Dong, I. Kianicka, D. Custeau, C. Boudreau, L. Geerts, S.-X. Cai, J. Drewe, D. Labrecque, S. Kasibhatla and B. Tseng, Mol. Cancer Ther., 2004, 3, 1375–1383, DOI:10.1158/1535-7163.1375.3.11.
- I. A. Schepetkin, L. N. Kirpotina, A. I. Khlebnikov, N. Cheng, R. D. Ye and M. T. Quinn, Biochem. Pharmacol., 2014, 92, 627–641, DOI:10.1016/j.bcp.2014.09.027.
- A. Alneyadi, Z. N. Nizami, H. E. Aburawi, S. Hisaindee, M. Nawaz, S. Attoub, G. Ramadan, N. Benhalilou, M. Al Azzani, Y. Elmahi, A. Almeqbali, K. Muhammad, A. H. Eid, R. Vijayan and R. Iratni, Cancers, 2023, 15, 2682, DOI:10.3390/cancers15102682.
- L. M. Al-harbi, E. A. Al-Harbi, R. M. Okasha, R. A. El-Eisawy, M. A. A. El-nassag, H. M. Mohamed, A. M. Fouda, A. A. Elhenawy, A. Mora, A. M. El-Agrody and H. K. A. El-Mawgoud, J. Enzyme Inhib. Med. Chem., 2023, 38, 2155814, DOI:10.1080/14756366.2022.2155814.
- T. H. Afifi, R. M. Okasha, H. E. A. Ahmed, J. Ilaš, T. Saleh and A. S. Abd-El-Aziz, EXCLI J., 2017, 16, 868–902, DOI:10.17179/excli2017-356.
- M. H. Ahagh, G. Dehghan, M. Mehdipour, R. Teimuri-Mofrad, E. Payami, N. Sheibani, M. Ghaffari and M. Asadi, Bioorg. Chem., 2019, 93, 103329, DOI:10.1016/j.bioorg.2019.103329.
- H. E. A. Ahmed, M. A. A. El-Nassag, A. H. Hassan, R. M. Okasha, S. Ihmaid, A. M. Fouda, T. H. Afifi, A. Aljuhani and A. M. El-Agrody, J. Enzyme Inhib. Med. Chem., 2018, 33, 1074–1088, DOI:10.1080/14756366.2018.1476503.
- T. C. Braga, M. M. Silva, E. O. O. Nascimento, E. C. D. da Silva, Y. F. Rego, M. Mandal, Z. A. de Souza, A. L. T. G. Ruiz, J. E. de Carvalho, F. T. Martins, I. M. Figueiredo, T. M. de Aquino, C. M. da Silva, B. Mandal, G. Brahmachari, J. C. C. Santos and A. de Fatima, Eur. J. Med. Chem. Rep., 2022, 4, 100030, DOI:10.1016/j.ejmcr.2022.100030.
- P. Rawat and S. M. Verma, Drug Des., Dev. Ther., 2016, 10, 2779–2788, DOI:10.2147/DDDT.S111266.
- R. Soni, S. D. Durgapal, S. S. Soman and J. J. Georrge, Arabian J. Chem., 2019, 12, 701–708, DOI:10.1016/j.arabjc.2016.11.011.
- W. Zhao, B. Wang, Y. Liu, L. Fu, L. Sheng, H. Zhao, Y. Lu and D. Zhang, Eur. J. Med. Chem., 2020, 189, 112075, DOI:10.1016/j.ejmech.2020.112075.
- M. Mashhadinezhad, M. Mamaghani, M. Rassa and F. Shirini, ChemistrySelect, 2019, 4, 4920–4932, DOI:10.1002/slct.201900405.
- Y. Kanbe, M. Kim, M. Nishimoto, Y. Ohtake, N. Kato, T. Tsunenari, K. Taniguchi, I. Ohizumi, S. Kaiho, K. Morikawa, J. Jo, H. Limb and H. Kimb, Bioorg. Med. Chem., 2006, 14, 4803–4819, DOI:10.1016/j.bmc.2006.03.020.
- K. Takao, U. Shiori, H. Kamauchi and Y. Sugita, Bioorg. Chem., 2019, 87, 594–600, DOI:10.1016/j.bioorg.2019.03.042.
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, Molecules, 2020, 25, 1909, DOI:10.3390/molecules25081909.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274, DOI:10.1021/jm501100b.
- H. B. Broughton and I. A. Watson, J. Mol. Graphics Modell., 2004, 23, 51–58, DOI:10.1016/j.jmgm.2004.03.016.
- P. Telukuntla, M. Chandrakanth, P. G. Amrutha, N. M. Thomas, R. Gondru, K. R. Valluru and J. Banothu, Tetrahedron Lett., 2024, 146, 155180, DOI:10.1016/j.tetlet.2024.155180.
- R. Deshineni, C. G. Arya, J. Banothu, R. Velpula and G. Chellamella, Chem. Data Collect., 2020, 30, 100546, DOI:10.1016/j.cdc.2020.100546.
- J. Banothu, R. Gondru, R. Bavantula, Y. Velivela and P. A. Crooks, Bioorg. Med. Chem. Lett., 2015, 25, 106–112, DOI:10.1016/j.bmcl.2014.10.100.
- R. Gali, J. Banothu, M. Porika, R. Velpula, R. Bavantula and S. Abbagani, RSC Adv., 2014, 4, 53812–53819, 10.1039/C4RA11428K.
- R. Gali, J. Banothu, M. Porika, R. Velpula, S. Hnamte, R. Bavantula, S. Abbagani and S. Busi, Bioorg. Med. Chem. Lett., 2014, 24, 4239–4242, DOI:10.1016/j.bmcl.2014.07.030.
- K. Fabitha, C. G. Arya, M. Chandrakanth and J. Banothu, Res. Chem. Intermed., 2023, 49, 997–1014, DOI:10.1007/s11164-022-04929-w.
- K. Fabitha, A. Kallingal, N. Maciejewska, C. G. Arya, M. Chandrakanth, N. M. Thomas, Y. Li, R. Gondru, M. Munikumar and J. Banothu, New J. Chem., 2024, 48, 8038–8054, 10.1039/d4nj00824c.
- K. Fabitha, C. G. Arya, P. Shyam, R. Gondru and J. Banothu, Polycyclic Aromat. Compd., 2022, 43, 7093–7109, DOI:10.1080/10406638.2022.2130369.
- S. Wang, Y. Tong, T. Ng, L. Lao, J. K. W. Lam, K. Y. Zhang, Z. Zhang and S. C. W. Sze, Chin. Med., 2015, 10, 1–12, DOI:10.1186/s13020-015-0051-z.
- A. L. Hopkins, Nat. Biotechnol., 2007, 25, 1110–1111, DOI:10.1038/nbt1007-1110.
- A. S. Azmi, Future Med. Chem., 2012, 4, 939–941, DOI:10.4155/fmc.12.44.
- X. Liang, H. Li and S. Li, Mol. BioSyst., 2014, 10, 1014–1022, 10.1039/c3mb70507b.
- J. C. Oliveros, (2007–2015) Venny, An Interactive Tool for Comparing Lists with Venn's Diagrams, https://bioinfogp.cnb.csic.es/tools/venny/index.html.
- A. Prat and C. M. Perou, Mol. Oncol., 2010, 5, 5–23, DOI:10.1016/j.molonc.2010.11.003.
- X. Dai, T. Li, Z. Bai, Y. Yang, X. Liu, J. Zhan and B. Shi, Am. J. Cancer Res., 2015, 5, 2929–2943 CAS.
- P. Wee and Z. Wang, Cancers, 2017, 9, 52, DOI:10.3390/cancers9050052.
- H. Masuda, D. Zhang, C. Bartholomeusz, H. Doihara, G. N. Hortobagyi and N. T. Ueno, Breast Cancer Res. Treat., 2012, 136, 331–345, DOI:10.1007/s10549-012-2289-9.
- S. Jahani-Sherafat, M. Azimirad, H. Raeisi, M. A. Looha, S. Tavakkoli, H. A. Amoli, S. Moghim, M. Rostami-Nejad, A. Yadegar and M. R. Zali, Mol. Biol. Rep., 2024, 51, 265, DOI:10.1007/s11033-024-09273-3.
- A. E. Maennling, M. K. Tur, M. Niebert, T. Klockenbring, F. Zeppernick, S. Gattenlöhner, I. Meinhold-Heerlein and A. F. Hussain, Cancers, 2019, 11, 1826, DOI:10.3390/cancers11121826.
- T. Fang, Y. Zhang, V. Y. Chang, M. Roos, C. M. Termini, L. Signaevskaia, M. Quarmyne, P. K. Lin, A. Pang, J. Kan, X. Yan, A. Javier, K. Pohl, L. Zhao, P. Scott, H. A. Himburg and J. P. Chute, Blood, 2020, 136, 441–454, DOI:10.1182/blood.2020005895.
- D. L. Wheeler, M. Iida and E. F. Dunn, Oncologist, 2009, 14, 667–678, DOI:10.1634/theoncologist.2009-0009.
- Y. Fukumoto, M. Morii, T. Miura, S. Kubota, K. Ishibashi, T. Honda, A. Okamoto, N. Yamaguchi, A. Iwama, Y. Nakayama and N. Yamaguchi, J. Biol. Chem., 2014, 289, 12313–12329, DOI:10.1074/jbc.M113.533752.
- W. Wu, L. M. Graves, G. N. Gill, S. J. Parsons and J. M. Samet, J. Biol. Chem., 2002, 277, 24252–24257, DOI:10.1074/jbc.m200437200.
- K. Park, H. Yang, S. Youn, H. Yang, I. Chae, B. Oh, M. Lee, Y. Park, Y. Choi, H. Kim and K. Walsh, Arterioscler., Thromb., Vasc. Biol., 2003, 23, 1364–1369, DOI:10.1161/01.ATV.0000081633.53390.B4.
- E. Beurel and R. S. Jope, Prog. Neurobiol., 2006, 79, 173–189, DOI:10.1016/j.pneurobio.2006.07.006.
- Y. Yang, T. Lei, S. Du, R. Tong, H. Wang, J. Yang, J. Huang, M. Sun, Y. Wang and Z. Dong, Int. J. Oncol., 2018, 52, 709–720, DOI:10.3892/ijo.2018.4237.
- R. He, S. Du, T. Lei, X. Xie and Y. Wang, Oncol. Rep., 2020, 44, 2373–2385, DOI:10.3892/or.2020.7817.
- J. Luo, Cancer Lett., 2009, 273, 194–200, DOI:10.1016/j.canlet.2008.05.045.
- A. Daina, O. Michielin and V. Zoete, Nucleic Acids Res., 2019, 47, W357–W364, DOI:10.1093/nar/gkz382.
- J. Piñero, J. M. Ramírez-Anguita, J. Saüch-Pitarch, F. Ronzano, E. Centeno, F. Sanz and L. I. Furlong, Nucleic Acids Res., 2020, 48, D845–D855, DOI:10.1093/nar/gkz1021.
- N. Rappaport, N. Nativ, G. Stelzer, M. Twik, Y. Guan-Golan, T. I. Stein, I. Bahir, F. Belinky, C. P. Morrey, M. Safran and D. Lancet, Database, 2013, 2013, bat018, DOI:10.1093/database/bat018.
- A. Hamosh, A. F. Scott, J. S. Amberger, C. A. Bocchini and V. A. McKusick, Nucleic Acids Res., 2005, 33, D514–D517 CrossRef CAS , https://pmc.ncbi.nlm.nih.gov/articles/PMC539987/.
- D. Szklarczyk, R. Kirsch, M. Koutrouli, K. Nastou, F. Mehryary, R. Hachilif, A. L. Gable, T. Fang, N. T. Doncheva, S. Pyysalo, P. Bork, L. J. Jensen and C. V. Mering, Nucleic Acids Res., 2023, 51, D638–D646 CrossRef CAS PubMed , https://pubmed.ncbi.nlm.nih.gov/36370105/.
- P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, N. Amin, B. Schwikowski and T. Ideker, Genome Res., 2003, 13, 2498–2504 CrossRef CAS , https://pmc.ncbi.nlm.nih.gov/articles/PMC403769/.
- C. Chin, S. Chen, H. Wu, C. Ho, M. Ko and C. Lin, BMC Syst. Biol., 2014, 8, 1–7 CrossRef PubMed , https://pubmed.ncbi.nlm.nih.gov/25521941/.
- Y. Yang, K. Yao, M. P. Repasky, K. Leswing, R. Abel, B. K. Shoichet and S. V. Jerome, J. Chem. Theory Comput., 2021, 17, 7106–7119, DOI:10.1021/acs.jctc.1c00810.
- M. P. Jacobson, D. L. Pincus, C. S. Rapp, T. J. F. Day, B. Honig, D. E. Shaw and R. A. Friesner, Proteins: Struct., Funct., Bioinf., 2004, 55, 351–367, DOI:10.1002/prot.10613.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717, DOI:10.1038/srep42717.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Anoop Kallingal
a,
Anoop Kallingal