
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, molecular modelling investigation and biological studies of novel human carbonic anhydrase inhibitors based on coumalic acid†
Virginia Pontecorviae,
Andrea Angelib,
Luigi Cutarellac,
Mattia Mori c,
Daniela Seccia,
Michele Colucciaa,
Simone Carradorid,
Anna Troiania,
Federico Pepia,
Chiara Salvittia,
Alessia Di Noia,
Mattia Spanoa,
Francesca Arrighia,
Elena De Falcoef,
Antonella Bordine,
Emanuela Berrinoag,
Arianna Granesea,
Paola Chimenti*a,
Paolo Guglielmi*a and
Claudiu T. Supuranb
c,
Daniela Seccia,
Michele Colucciaa,
Simone Carradorid,
Anna Troiania,
Federico Pepia,
Chiara Salvittia,
Alessia Di Noia,
Mattia Spanoa,
Francesca Arrighia,
Elena De Falcoef,
Antonella Bordine,
Emanuela Berrinoag,
Arianna Granesea,
Paola Chimenti*a,
Paolo Guglielmi*a and
Claudiu T. Supuranb
aDepartment of Drug Chemistry and Technologies, Sapienza University of Rome, P.le A. Moro 5, 00185 Rome, Italy. E-mail: paola.chimenti@uniroma1.it; paolo.guglielmi@uniroma1.it
bNEUROFARBA Department, Pharmaceutical and Nutraceutical Section, University of Florence, Via Ugo Schiff 6, 50019 Sesto Fiorentino, FI, Italy
cDepartment of Biotechnology, Chemistry and Pharmacy, University of Siena, Via Aldo Moro 2, 53100 Siena, Italy
dDepartment of Pharmacy, “G. d'Annunzio” University of Chieti-Pescara, Via dei Vestini 31, 66100 Chieti, Italy
eDepartment of Medico-Surgical Sciences and Biotechnologies, Sapienza University of Rome, Corso della Repubblica 79, 04100, Latina, Italy
fMaria Cecilia Hospital, Gruppo Villa Maria Care & Research, Via Corriera, 1, 48033 Cotignola, Italy
gDepartment of Life Science, Health, and Health Professions, Link Campus University, Via del Casale di San Pio V, 44 – 00165, Rome, Italy
First published on 30th May 2025
Abstract
The present study provides the design, synthesis, molecular modelling investigation and biological evaluation of a series of coumalic acid-based selective inhibitors of hCA IX and XII. Based on the previously obtained results with carboxamide analogues of coumalic acid, here we explored some modifications of the original scaffold with the aim to expand our knowledge about these compounds and to promote structure–activity relationship (SAR) studies. Structural modifications as lactone-to-lactam conversion, repositioning of the carboxylic acid moiety, and the introduction of ester or amidic linkers, as well as triazole-based elongation via click chemistry have been performed. The synthesised compounds were tested for their inhibitory activities against hCA I, II, IX, and XII showing the lactone moiety to be crucial for inhibitory potency. Particularly, ester derivatives demonstrated selectivity for hCA IX and XII, with certain compounds exhibiting micromolar affinities. Notably, compound 8, featuring a bromine substitution, displayed the highest selectivity and potency against hCA IX and XII. Molecular docking studies further elucidated the binding mechanisms, revealing that the lactone ring hydrolysis plays a significant role in the inhibition process. These results offer valuable insights into the structure–activity relationships (SAR) of coumalic acid analogues and support their further biological investigation for cancer therapy by targeting hCA IX and XII.
1. Introduction
Homeostasis is the crucial state required for the optimal physical, chemical, and internal functioning of all living systems.1 Consequently, different variables are constantly controlled such as fluid balance, ion concentration, and pH equilibrium between intra- and extra-cellular environments.2 Among the different homeostatic mechanisms, carbonic anhydrases (CAs, EC 4.2.1.1) play an important role as pH regulators.3,4 These ubiquitous metalloenzymes catalyse the reversible hydration of CO2 in HCO3−, an important reaction involved in a plethora of fundamental processes.5,6 To date, eight distinct genetic families have been defined α-, β-, γ-, δ-, η-, ζ-, θ-, and ι-, with the latter having been very recently discovered.7–11 The α-CAs are the best-characterised enzymes as they are expressed in vertebrates, fungi, protozoa, plants, algae, diatoms, archaea, and bacteria.12 The fifteen human carbonic anhydrase isoforms (hCAs) belong to the α-family and cover a pivotal role in a multitude of physiological functions such as respiration, transport of carbon dioxide between metabolising tissues and lungs, pH homeostasis, electrolyte secretion in various tissues/organs, as well as biosynthetic reactions.1,6 However, these enzymes may also contribute to pathological processes when their dysregulated expression and/or abnormal activity occurs (e.g., in glaucoma, oedema, epilepsy, altitude sickness, and cancer).13–16 For this reason, different libraries of compounds have been explored as CA inhibitors (CAIs) and activators.17–22The transmembrane tumour-related isoforms CA IX and XII are overexpressed in solid hypoxic tumours due to their activation by the HIF pathway.23–25 Their mechanism of action is associated with intra- and extra-cellular pH regulation which contributes to tumour proliferation and invasiveness.26,27 Accordingly, their inhibition could be considered as a prognostic tool and a therapeutic target for tumour treatment.28–30 Among the CAIs developed so far, the coumarin-based ones occupy a prominent place due to their selective inhibition mechanism against hCA isoforms IX and XII.21,31–33 The lactone portion of the coumarin core undergoes hydrolysis by the Zn(II) bound hydroxide ion, leading to the cinnamic acid product, or the open form, considered the real inhibitor of the enzyme.34 The ability of the latter to bind the entrance of the active site cavity, where the differences among the isoforms are higher, results in increased selectivity. In 2019 Cornelio and colleagues performed the molecular simplification of the saccharin scaffold to obtain potent hCAs inhibitors based on the 5-arylisothiazol-3(2H)-one core (as mono or dioxides at the sulphur atom).35 Recently, we employed a similar approach on coumarin nucleus to design a series of potent and selective hCA IX and XII inhibitors provided of the 2H-pyran-2-one core (Fig. 1).36 These compounds, obtained from the commercially available coumalic acid, retained the lactone moiety that should represent the molecular attribute responsible for the inhibition of hCAs, through (i) its putative hydrolysis provoked by the enzyme and successive binding in the entrance of the active site, or (ii) anchoring the zinc-bound water in its hydrolysed/not-hydrolysed form. Position 5 of the 2H-pyran-2-one core was provided with an amide linker group bearing different (un)substituted aromatic or (hetero)(cyclo)aliphatic residues.36 With the aim to expand our knowledge about these compounds and to promote structure–activity relationship (SAR) studies, we explored some modifications of the original scaffold (Fig. 1).
 | ||
| Fig. 1 Design of the novel coumalic acid derivatives. | ||
The reasons behind these structural changes can be resumed as follows:
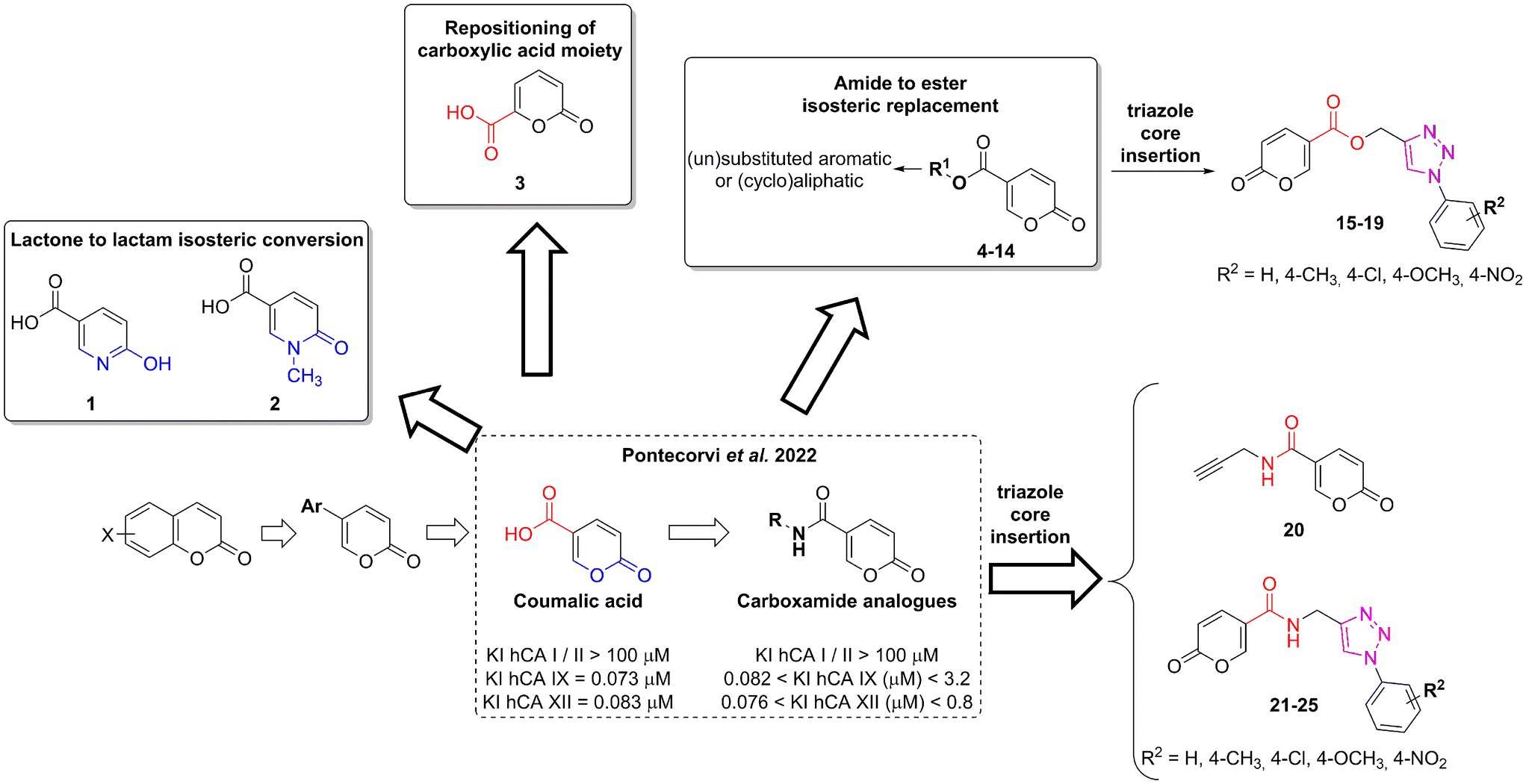
• The docking studies performed in the previous work underlined the importance of the lactone moiety in obtaining potent hCA IX and XII inhibitors.36 To challenge this claim, we evaluated the effects of lactone to lactam replacement on hCA inhibition (compounds 1–2).
• With the aim of investigating the importance of the carboxylic acid moiety position, we tried to move this fragment from position 5 of the original coumalic acid (2-oxo-2H-pyran-5-carboxylic acid) to position 6, as in the compound 3 (2-oxo-2H-pyran-6-carboxylic acid, Fig. 1).
• Novel coumalic acid analogues provided with ester moiety in spite of amidic of the formers already published, have been designed to investigate the influence of nitrogen to oxygen isosteric substitution at the linker moiety (compounds 4–14).
• The insertion of the triazole ring on the coumarin core has been extensively investigated to obtain potent and selective hCA IX and XII inhibitors.37–39 Taking into account this evidence, we also evaluated the effect of molecular “elongation” obtained by exploiting click chemistry to insert triazole core between the amide (or ester) linker, and the (un)substituted aromatic residues (compounds 16–24).40
All the compounds have been evaluated against four hCA isoforms, the two cancer-related ones (hCA IX and XII) and the two off-targets hCA I and II. Moreover, to provide a structural elucidation of the detected KI values for coumalic acid derivatives towards hCA IX and hCA XII, molecular docking simulations were performed. The further aim of this study is to understand the relationships between the coumalic acid isomers and their different inhibitory activities. The mechanism of action of coumalic acid derivatives in carbonic anhydrases inhibition is based on literature reports, showing that the coumalic acid ring is hydrolysed by the Zn(II)-coordinated hydroxide ion or water molecule within the hCAs active site, generating the corresponding E/Z cinnamic acid derivatives.36,38,40
2. Chemistry
Compounds 1–3, corresponding to the two lactam derivatives (molecules 1 and 2) and the coumalic acid analogue endowed with carboxylic acid moiety at position 6 (compound 3), have been commercially obtained. On the contrary, the derivatives 4–25 have been achieved by employing different synthetic procedures (Scheme 1I and II). | ||
| Scheme 1 Synthetic approaches employed for the synthesis of compounds 4–25. | ||
The synthesis of the compounds provided with ester linker (4–14) has been performed through a multi-step synthetic approach. In the first step, coumalic acid was converted into the corresponding acyl chloride A (Scheme 1I) by refluxing in thionyl chloride (SOCl2). The acyl chloride A was employed in the subsequent reaction with (un)substituted aromatic or cycloaliphatic alcohol, in absence of solvent, at a temperature of 75 °C for 3 h or 12 h depending on the reagents, to achieve the corresponding ester derivatives (Scheme 1I).41
Compound 20, the amidic analogue of derivative 14, has been synthesised by means of a slightly modified procedure, by reacting the acyl chloride A with propargyl amine in the presence of diisopropylethylamine (DIPEA) as base. The reaction was performed in diethyl ether (Et2O) at reduced temperature (0–4 °C). Compounds 14 and 20 correspond to the alkyne intermediates employed in the “click” reactions exploited to obtain the triazole derivatives provided with the ester (15–19) or amidic (21–25) linker, respectively.39 Particularly, these compounds (14 and 20) were reacted with azides (that in turn have been obtained from corresponding amines with NaN3 and NaNO2 in 6 M HCl), in the presence of CH3COOH, CuI, and DIPEA in dichloromethane through a copper(I)-catalysed click reaction.39
Structural characterization of the final compounds 4–25 was obtained by means of 1H-NMR and 13C-NMR spectroscopic analysis. Elemental analyses for C, H, and N were recorded on a Perkin-Elmer 240 B microanalyzer and the analytical results were within ±0.4% of the theoretical values for all compounds (data not shown).
3. Inhibition assay
Compounds 1–25 were evaluated for their ability to inhibit four different isoforms of hCA, the two off-targets hCA I and II and the two tumour-related isoforms hCA IX and XII. The inhibitory activity data (KI) obtained for compounds 1–25 are reported in Table 1 along with the KI values of coumalic acid and the related carboxamide analogues which exhibited the best outcomes in our previous work (8A, 9A and 11A, structures in blue in Table 1).36 The inhibitory activity data of these compounds (coumalic acid, 8A, 9A and 11A) have been added with the aim of performing an exhaustive structure–activity relationship (SAR) analysis. One of the first purposes of this study was to demonstrate the importance that the lactone moiety plays in the inhibition of hCA as suggested by our previously accomplished molecular modelling studies.36 To confirm this aspect, the replacement of the lactone fragment with the lactam one has been performed (derivatives 1–2) showing the loss of inhibitory activity, these compounds being ineffective against all the evaluated isoforms (KI > 100 μM). The other structural modification attempted concerns the change of position of the carboxylic acid group of the coumalic acid from position 5 to 6, to obtain the isomer 3. Surprisingly, compound 3 resulted to be devoid of inhibitory activity with KI > 100 μM against all the evaluated isoforms (see section 4, molecular modelling).| Compound | Structure | KIa (μM) | |||
|---|---|---|---|---|---|
| hCA I | hCA II | hCA IX | hCA XII | ||
| a Mean from 3 different assays, by a stopped-flow technique (errors were in the range of ±5–10% of the reported values). | |||||
| Coumalic acid36 |  |
>100 | >100 | 0.073 | 0.083 |
| 1 | 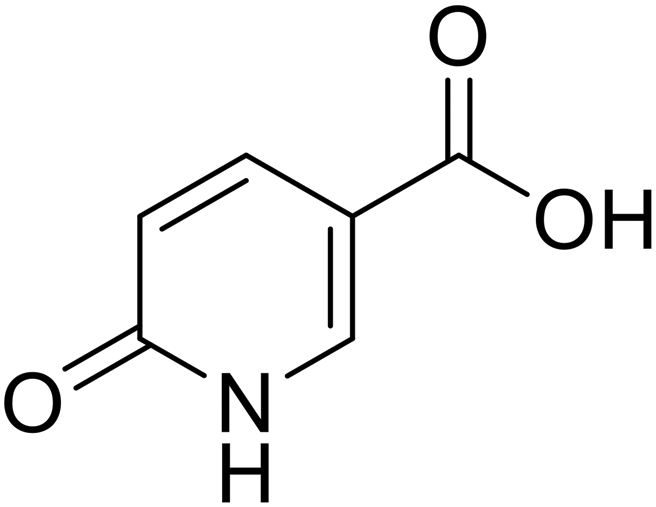 |
>100 | >100 | >100 | >100 |
| 2 |  |
>100 | >100 | >100 | >100 |
| 3 | 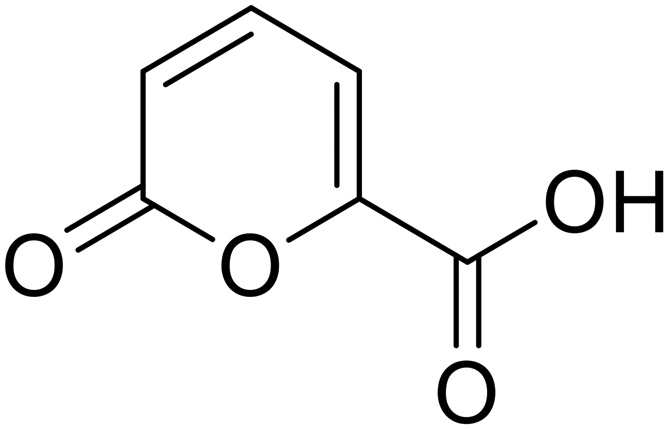 |
>100 | >100 | >100 | >100 |
| 4 | 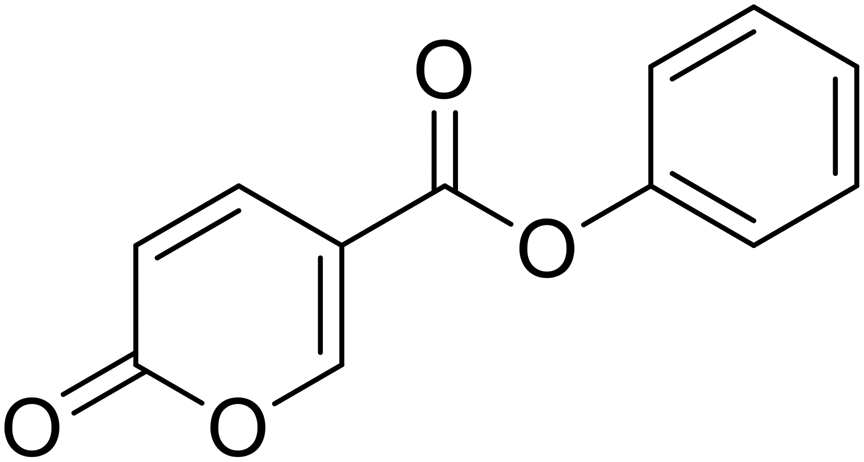 |
>100 | >100 | 21.4 | 65.7 |
| 5 | 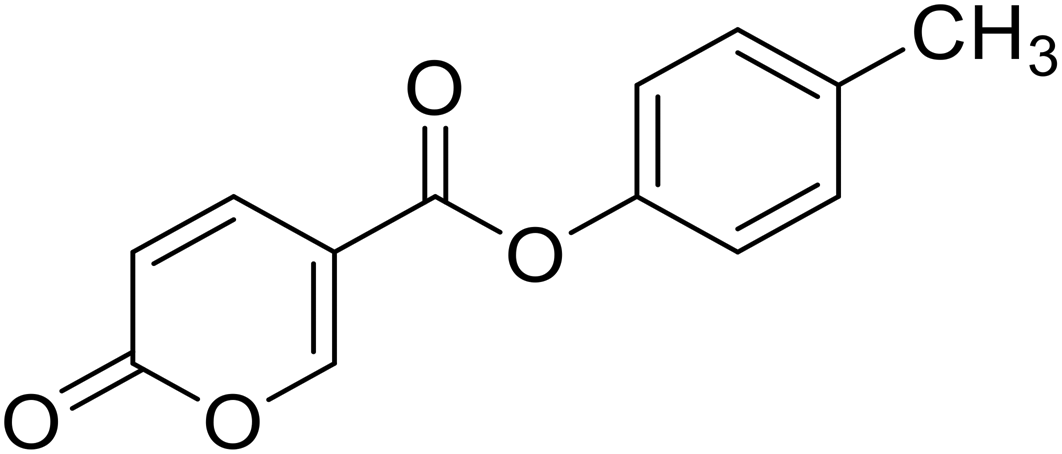 |
>100 | >100 | 12.21 | 1.83 |
| 6 | 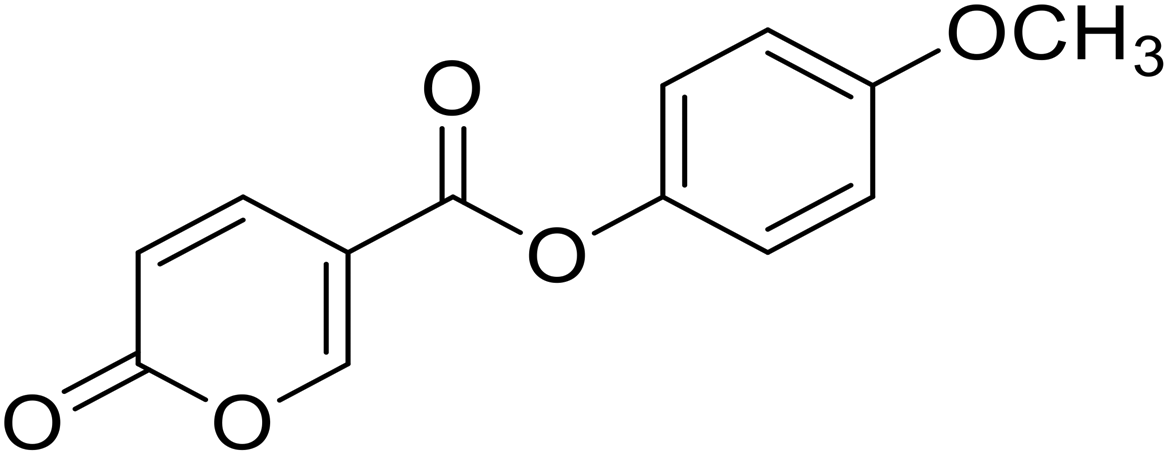 |
>100 | >100 | 10.17 | 1.65 |
| 7 | 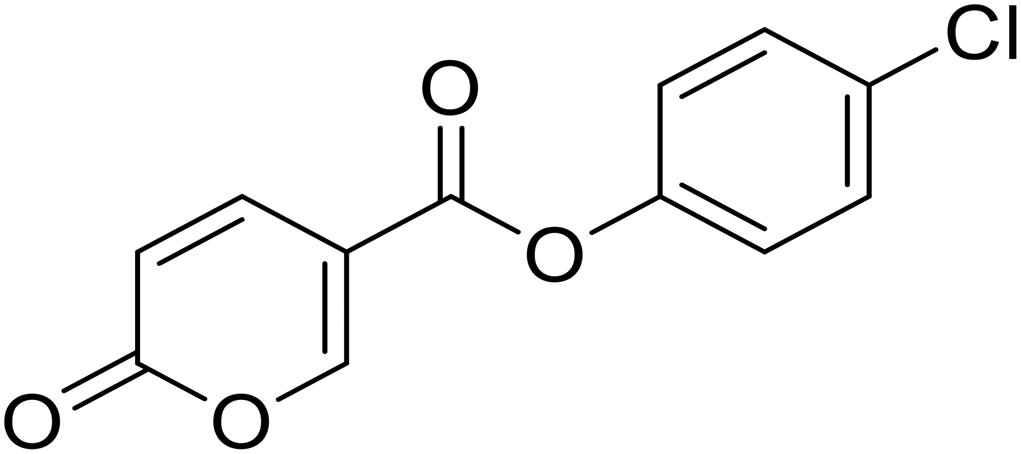 |
>100 | >100 | 15.1 | 76.3 |
| 8 | 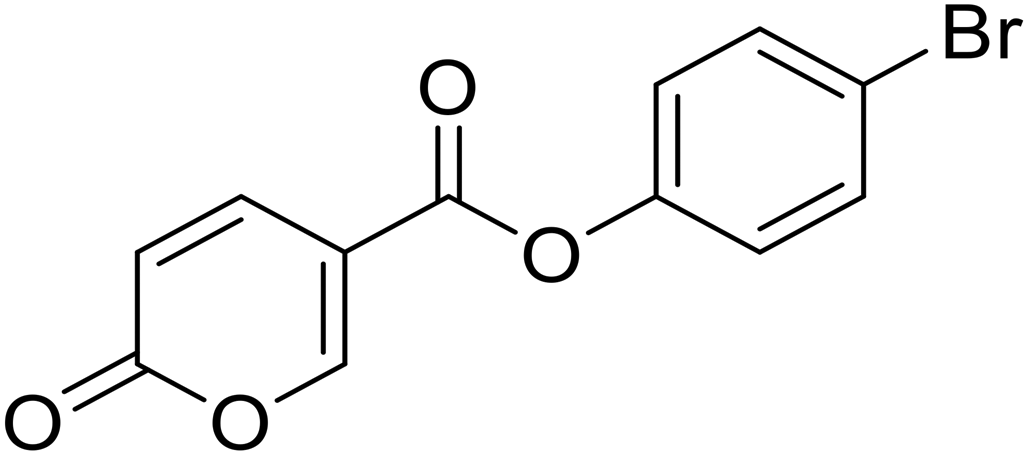 |
>100 | >100 | 3.14 | 0.91 |
| 8A36 | 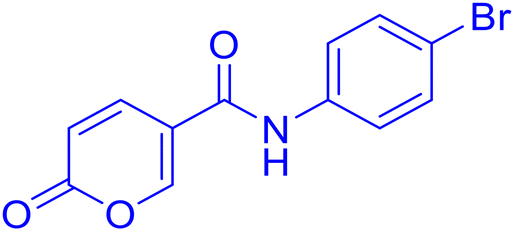 |
>100 | >100 | 0.082 | 0.089 |
| 9 | 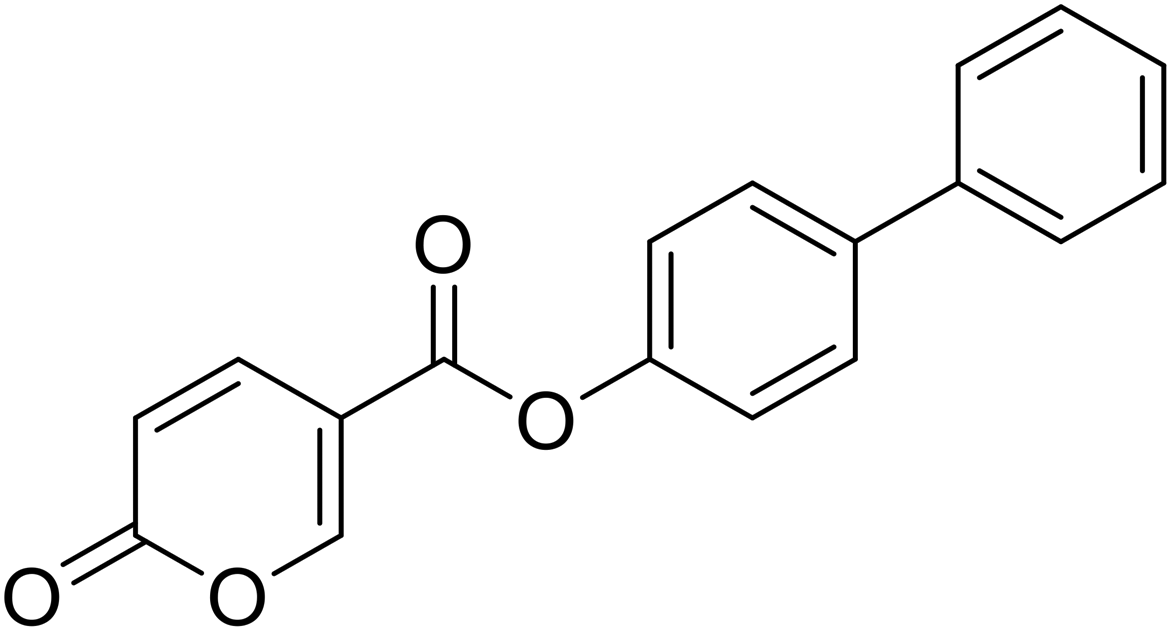 |
>100 | >100 | 11.43 | >100 |
| 9A36 | 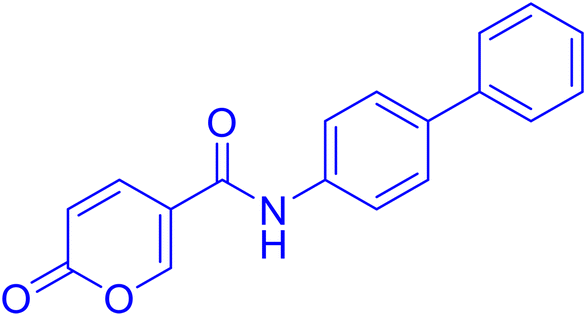 |
>100 | >100 | 0.083 | 0.076 |
| 10 | 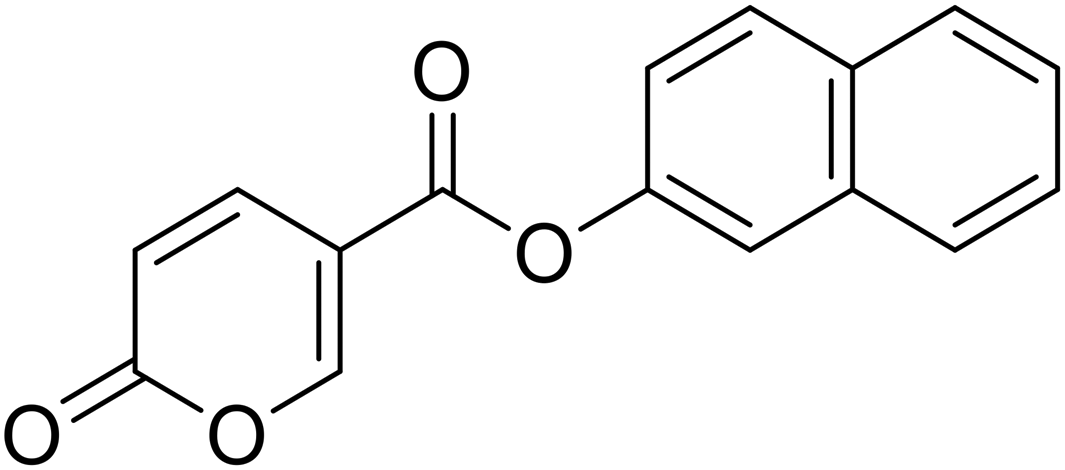 |
>100 | >100 | 14.8 | 68.9 |
| 11 |  |
>100 | >100 | 19.91 | 3.12 |
| 11A36 | 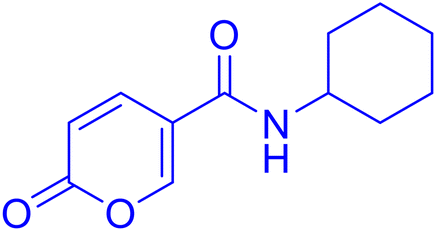 |
>100 | >100 | 0.098 | 0.086 |
| 12 | 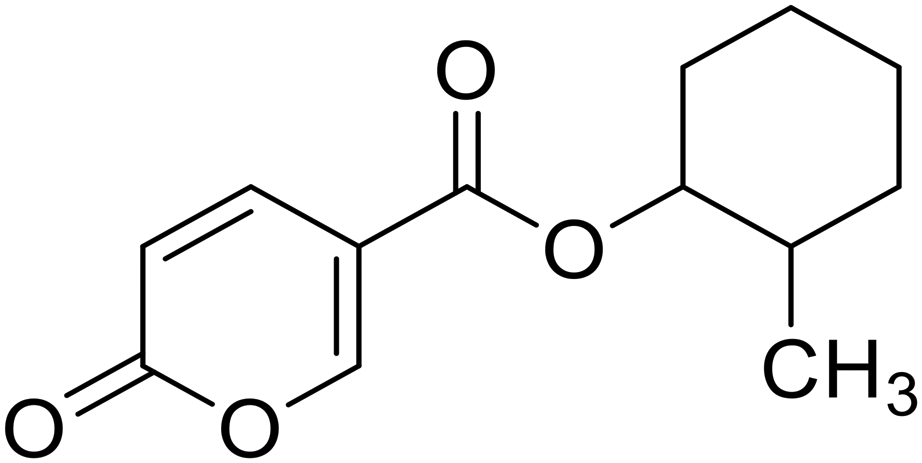 |
>100 | >100 | 12.3 | 82.8 |
| 13 | 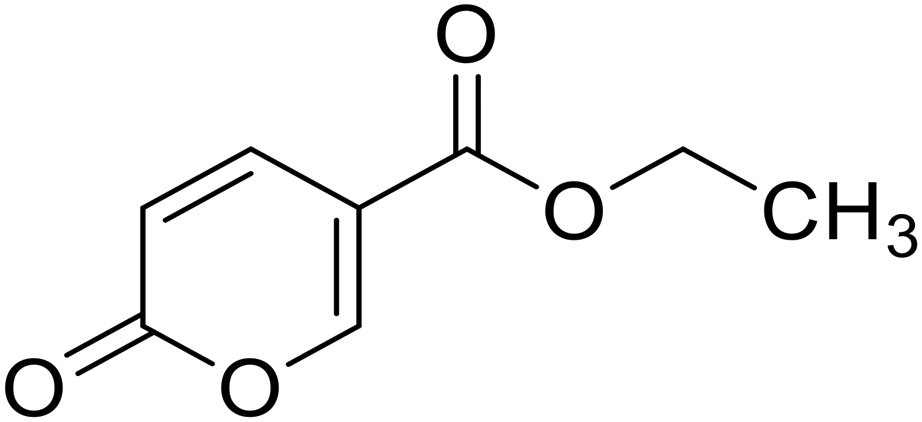 |
>100 | >100 | 14.4 | 92.6 |
| 14 | 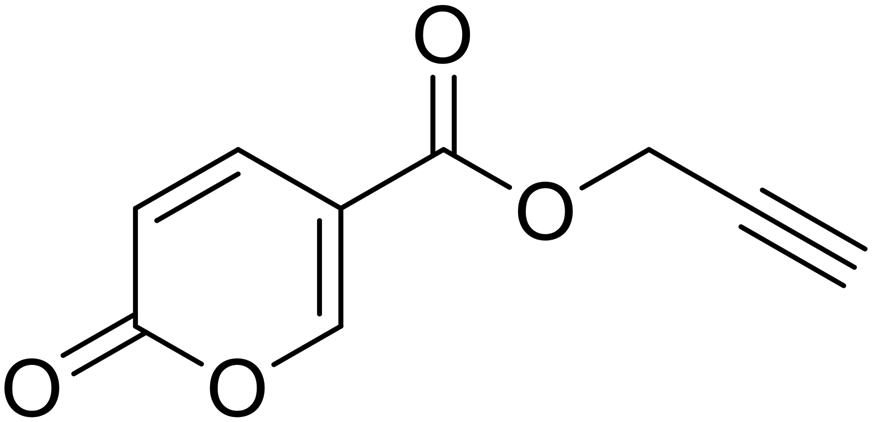 |
>100 | >100 | 17.9 | 76.3 |
| 15 | 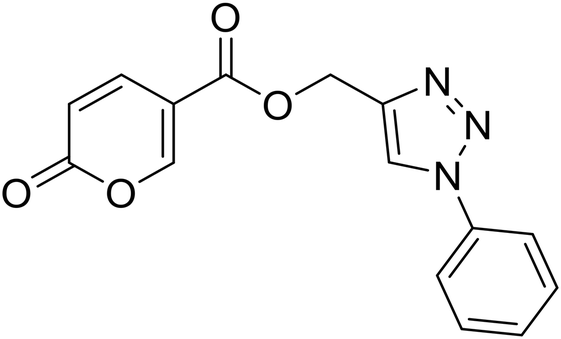 |
>100 | >100 | 18.6 | 55.8 |
| 16 | 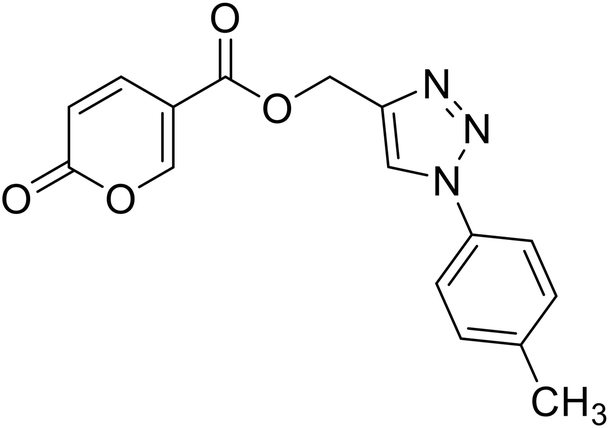 |
>100 | >100 | 9.8 | 62.6 |
| 17 | 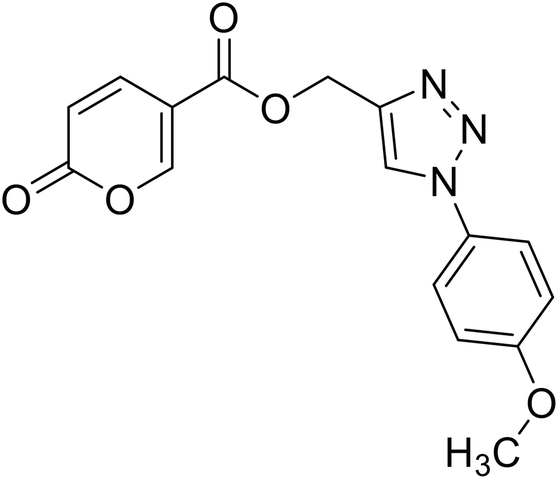 |
>100 | >100 | 11.4 | 54.8 |
| 18 |  |
>100 | >100 | 21.9 | 61.3 |
| 19 | 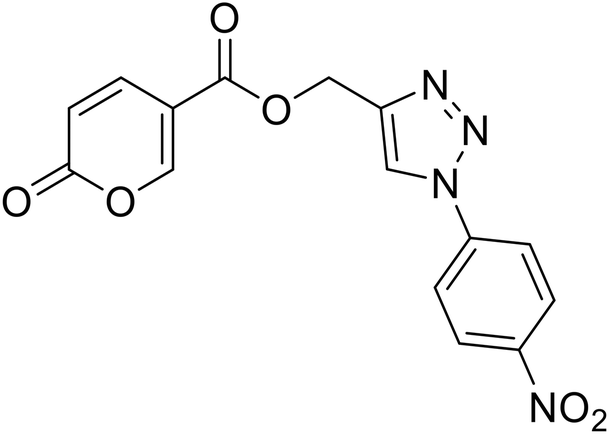 |
>100 | >100 | 20.0 | 62.8 |
| 20 | 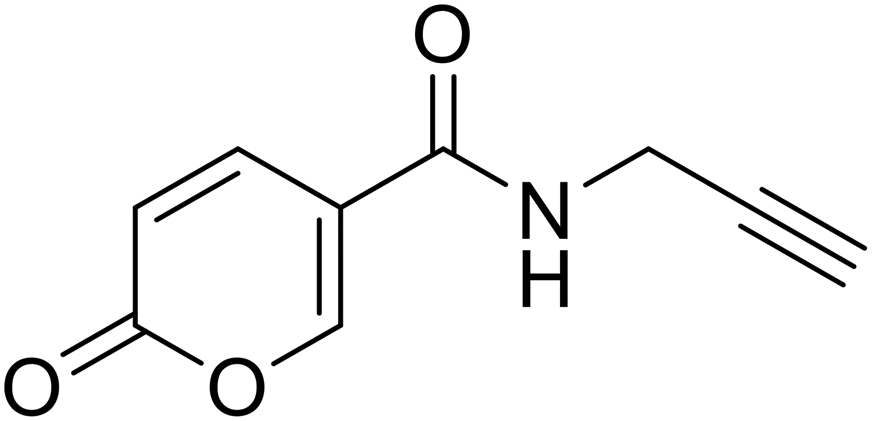 |
>100 | >100 | 22.8 | 68.1 |
| 21 | 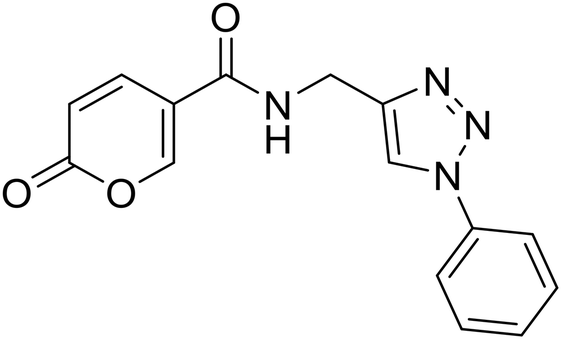 |
>100 | >100 | 21.3 | 62.0 |
| 22 | 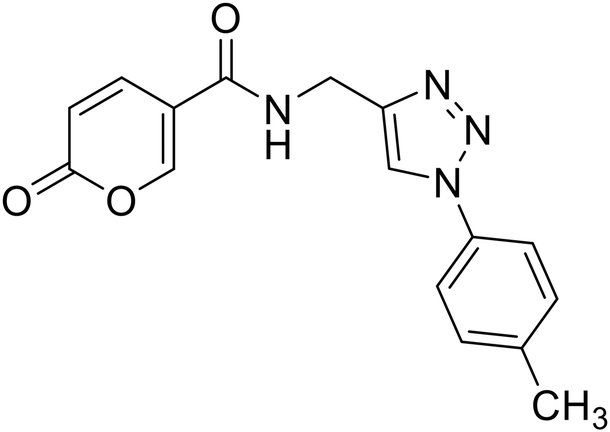 |
>100 | >100 | 14.5 | 57.8 |
| 23 | 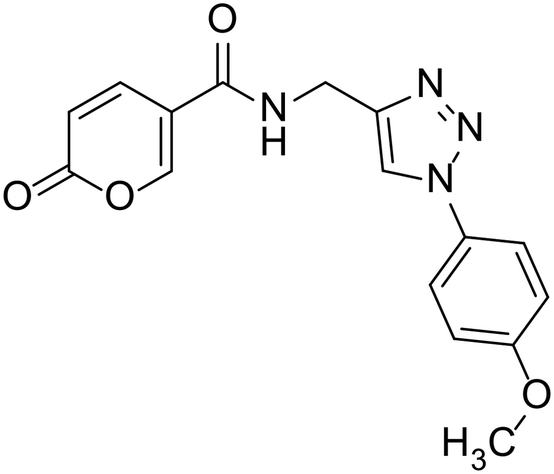 |
>100 | >100 | 23.5 | 58.7 |
| 24 | 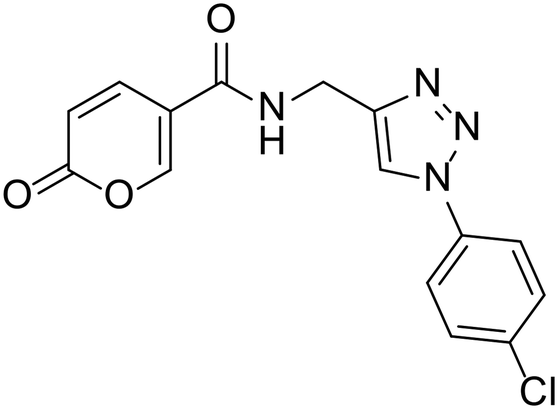 |
>100 | >100 | 18.2 | 61.2 |
| 25 | 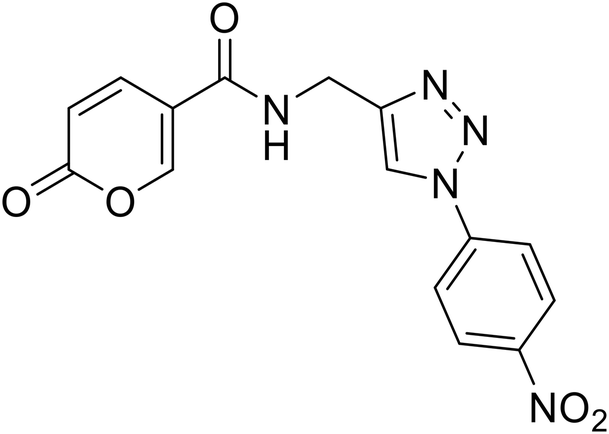 |
>100 | >100 | 7.5 | 63.4 |
| AAZ | 0.25 | 0.012 | 0.026 | 0.006 | |
The compounds obtained by the amide to ester replacement of the linker moiety (4–14, Table 1) preserved the selectivity against the hCA IX and XII isoforms, these compounds being ineffective against the two off-targets (KI hCA I/II > 100 μM). However, these derivatives showed a diminished affinity against the two cancer-related isoforms with KI values falling in the micromolar range (Table 1). These results demonstrate that the NH/O isosteric replacement at the linker moiety had detrimental effects on potency. Besides this general observation, some SAR considerations can be performed. In particular, it is possible to note that the insertion of substituent groups (compounds 5–10) on the phenyl ring generally led to the increase of inhibitory activity, especially against hCA IX. As for hCA XII, the inhibition values were found to be highly dependent on the substituents considered. The best results were obtained for compound 8, endowed with the bromine atom at the para position of the phenyl ring. Indeed, concerning the unsubstituted parent compound 4, derivative 8 exhibited a 7-fold and a 71-fold increase of hCA IX and XII inhibition, respectively, resulting as the most potent hCA XII inhibitor of the entire series (KI hCA IX = 3.14 μM; KI hCA XII = 0.91 μM, Table 1). As for compounds 7 (4-Cl), 9 (4-Ph) and 10 (1-naphthyl), comparable inhibition values against hCA IX were recorded when compared to the unsubstituted analogues 4. On the other hand, reduced (7–10) or absent (9) affinity against hCA XII isoform was observed. Considering the (cyclo)aliphatic derivatives (11–14), only compound 11 exhibited a single-digit micromolar inhibition of hCA XII (KI hCA XII = 3.12 μM) the others being endowed with KIs values greater than 10 μM against both the isoforms. Interestingly, the carboxamide analogue of compound 11 (Table 1, 11A) was one of the most active compounds of the previously reported series, thus further confirming the unfavourable effects of the N/O isosteric substitution at the linker moiety. Keeping into account the beneficial effects that the insertion of triazole linker exerts on the coumarin core,37–39 we proposed similar modifications on this scaffold, by combining the triazole ring with the carboxamide or ester linker group (compounds 15–19 and 21–25). Although maintaining the selectivity against the two tumour-related isoforms, being these compounds ineffective against hCA I and II (KI > 100 μM), the inhibitory activity data still evidenced a detrimental effect of this molecular elongation with most of the compounds exhibiting poor inhibition of both the isoforms, regardless of linker type and/or substituent group. As a matter of fact, only two compounds endowed with the triazole ring (16 and 25), showed KI values below 10 μM against hCA IX; conversely, the affinity of most of the compounds was quite scarce with KI values greater than 20 μM.
4. Molecular modelling studies
Coumalic acid and compounds 3 and 11 were docked to the crystallographic structure of hCA IX (PDB-ID: 3IAI) and hCA XII (PDB-ID: 1JD0) by the GOLD program. Considering the inhibition mechanism of these derivatives through the hydrolysis of the lactone moiety, the resulting open enolic forms in both E and Z configurations of the double bond were also docked. The Zn(II)-bound water molecule was kept as a part of the receptor during molecular docking simulations.36,43 Docking results of the closed form of all the examined compounds showed that all of them are able to fit the hCA IX and hCA XII catalytic sites. Coumalic acid and 11 are able to anchor the Zn(II)-bound water molecule through their lactone moiety, while 3 is unable to form a coordination bond with it. Instead, 3 establishes only an H-bond interaction with the carboxyl moiety with the Zn(II)-bound water molecule, which likely fails to activate the lactone ring-opening mechanism, thus explaining its lack of activity against both hCAs (KI > 100). The binding mode of coumalic acid against hCA IX was reproduced such as in a previous study36 (Fig. 2A), while its binding to hCA XII was simulated herein, showing that the molecule establishes H-bond interaction with Asn62, Gln92 and Thr199 residues (Fig. 2D). Compound 3 in hCA IX establishes H-bond interactions with Gln92, Tyr7 and a π–π stacking interaction with His94 (Fig. 2B), whereas in hCA XII establishes H-bond interactions with Thr200, Asn62 and π–π stacking interaction with His94 (Fig. 2E). Focusing on the derivative 11, it demonstrates a higher inhibitory activity against hCA XII (KI = 3.12 μM) compared to hCA IX (KI = 19.91 μM). Analysis of docking results with the closed form of the molecule shows that in hCA IX, 11 establishes H-bond interaction with Gln92 and a π–π stacking interaction with His94, while the cyclohexyl moiety binds in proximity of a lipophilic Leu/Val-rich cluster composed of Leu91, Val121, Leu123, Val131, Leu135, Leu141, Val134 (Fig. 2C). Similarly, in hCA XII, 11 establishes the same interaction network that is reinforced by an additional H-bond interaction with Tyr7 (Fig. 2F) which is not established in hCA IX.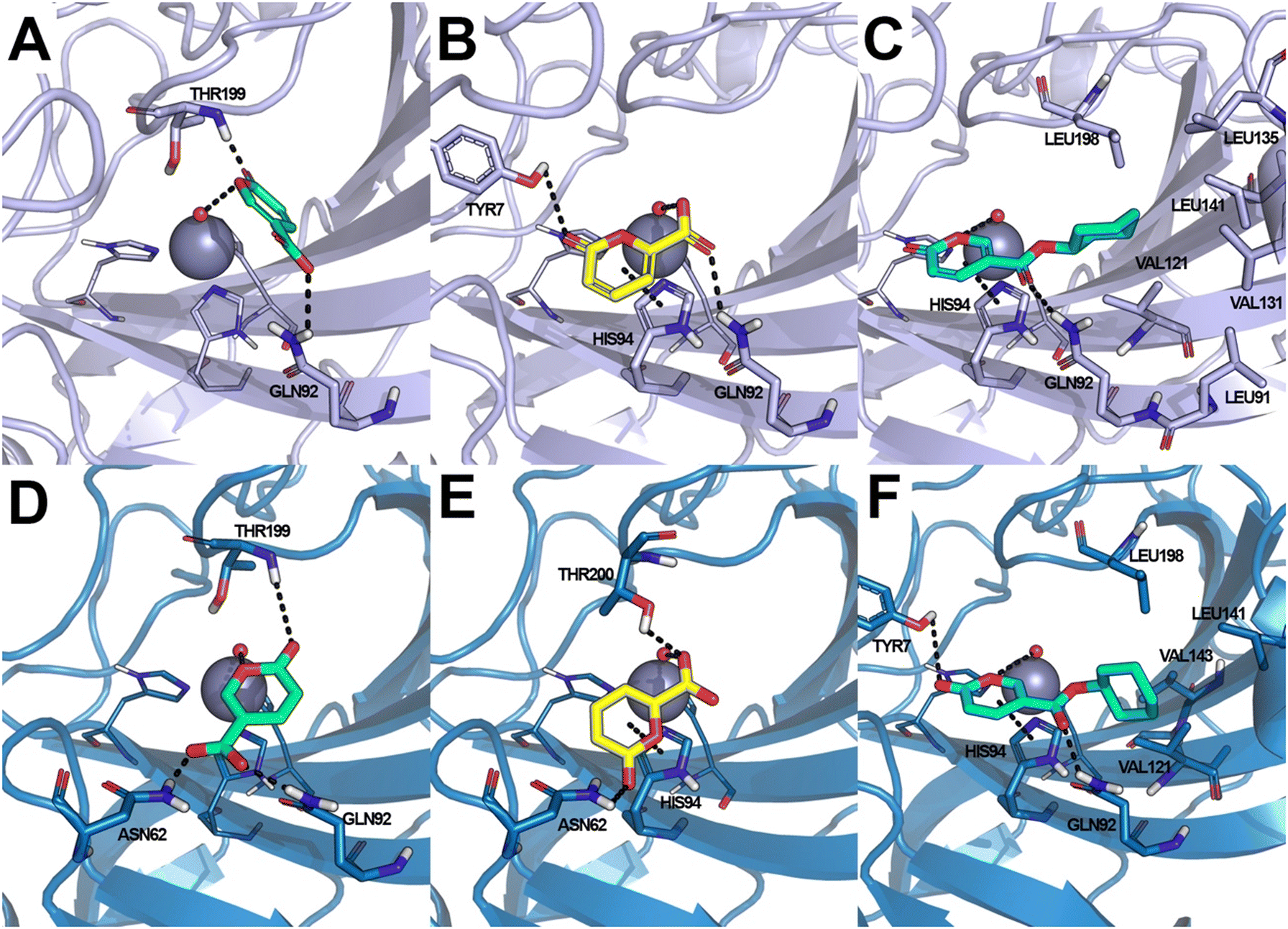 | ||
| Fig. 2 Docking-based binding mode of coumalic acid (A and D), 3 (B and E) and 11 (C and F) derivatives within the catalytic site of hCA IX (A–C) and hCA XII (D–F). The structure of hCA IX is shown as violet cartoon, whereas the structure of hCA XII is shown as blue cartoon. Residues not involved in active interaction with the inhibitors are omitted for clarity. Coumalic acid is shown as green sticks, 3 as yellow sticks and 11 as cyan sticks. Interactions are highlighted by black dashed lines, while residues involved in the interactions are represented as sticks. The Zn(II) ion is shown as a grey sphere, while the coordinated water molecule is represented as a small red sphere. | ||
Moreover, the analysis of the respective cinnamic acids in the open E and Z forms of 11 provides additional insight into the higher inhibitory activity of 11 against hCA XII compared to hCA IX. In fact, considering the Z conformation, which is reported to be more relevant than E in hCAs inhibition,36 11 establishes with hCA XII an additional H-bond to Asn62. Furthermore, the cyclohexyl moiety occupies the hydrophobic cluster of hCA XII, unlike the pose obtained in hCA IX, where the same portion is docked on the opposite side (Fig. 3A). The E conformation of 11 binds to both hCA IX and hCA XII through the same interaction network: H-bond interactions with Gln92, Thr199, Thr200 and with the Zn(II)-bound water molecule (Fig. 3B–D).
 | ||
| Fig. 3 Docking-based binding mode on hCA IX and hCA XII of the corresponding cinnamic acid in conformation E and Z of the double bond of 11. The structure of hCA IX is shown as violet cartoon, whereas the structure of hCA XII is shown as blue cartoon. Residues not involved in active interaction with the inhibitors are omitted for clarity. Compound 11 is shown as cyan sticks. Interactions are highlighted by black dashed lines, while residues involved in interaction are shown as sticks. The Zn(II) ion is shown as a grey sphere, while the coordinated water molecule is represented as a small red sphere. | ||
5. Materials and methods
5.1 Chemistry
All the reagents, coumalic acid, 1-methyl-6-oxo-1,6-dihydropyridine-3-carboxylic acid (compound 2) and other starting materials, employed in the synthetic procedures as well as HPLC analysis were obtained from commercial suppliers and were used without further purification. All melting points (m.p.) reported were uncorrected (temperatures are reported in °C) and measured on a Stuart® melting point apparatus SMP1. Nuclear magnetic resonance spectra (1H-NMR: 400 MHz and 13C-NMR: 101 MHz) were recorded in DMSO-d6 or CDCl3 using an Avance III 400 MHz spectrometer (Bruker, Milan, Italy). The processing and analyses of the NMR data were carried out with MestreNova software. Chemical shifts are reported in parts per million (ppm) and the coupling constants (J) are expressed in Hertz (Hz). Splitting patterns are designated as follows: s, singlet; d, doublet; t, triplet; q, quadruplet; m, multiplet; brs, broad singlet; dd, double of doublets.Accurate mass measurements were performed with an Orbitrap Exploris 120 mass spectrometer (Thermo-Fisher Scientific, Waltham, MA, USA) coupled to an AP-MALDI (ng) ultra-high resolution (UHR) ion source (MassTech Inc., Columbia, MD, USA). The samples were prepared by adapting a spotting procedure reported elsewhere.44 Briefly, 100 μl of acetonitrile/water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) were added to ca. 1 mg of each synthesized compound and 1 μl of the suspensions thus obtained was dropped on the wells of the MALDI plate (Opti-TOF 184 spot sample insert, SCIEX, Italy). The analytes were then covered with an equal volume of a 5 mg ml−1 solution of alpha-cyano-4-hydroxycinnamic acid (CHCA) dissolved in acetonitrile/ultrapure water/trifluoroacetic acid 50/50/0.1% (LaserBio Labs, Valbonne, France) used as a MALDI matrix.
:1 v/v) were added to ca. 1 mg of each synthesized compound and 1 μl of the suspensions thus obtained was dropped on the wells of the MALDI plate (Opti-TOF 184 spot sample insert, SCIEX, Italy). The analytes were then covered with an equal volume of a 5 mg ml−1 solution of alpha-cyano-4-hydroxycinnamic acid (CHCA) dissolved in acetonitrile/ultrapure water/trifluoroacetic acid 50/50/0.1% (LaserBio Labs, Valbonne, France) used as a MALDI matrix.
After solvent evaporation, the samples were irradiated with an UV laser at a wavelength of 355 nm operating in spiral motion mode at a repetition rate of 1000 Hz with 1% laser energy. Other parameters, i.e. pos ionization voltage (V) and ion transfer tube temperature (°C), were in turn optimized to maximize the ionic intensity of each compound. Mass spectra were acquired in positive ion mode over a mass range of 150–500 Da with a mass resolution of 120000@m/z 200 and a maximum injection time of 100 ms. The instrument was daily calibrated with Flexmix Calibration solution (Thermo-Fisher Scientific, Waltham, MA, USA) and fluorantrene was used as an internal lock mass by activating the EASY-IC source option allowing for a scan-to-scan mass scale recalibration in each analysis. Raw data were processed by using FreeStyle software v. 1.8 provided with the Xcalibur package. Δ values < 0.5 ppm between the theoretical and measured masses of the corresponding protonated species were obtained for all the compounds analysed. Due to the high mass accuracy achieved, measured masses were reported with five decimal places.
Thin layer chromatography (TLC) was performed on 0.2 mm thick silica gel–aluminium backed plates (60 F254, Merck) was employed to monitor all the reactions. Preparative flash column chromatography was carried out on silica gel (230–400 mesh, G60 Merck). The yields shown are not optimised. Drying of organic solution was accomplished with anhydrous sodium sulphate. Evaporation of the solvent after reaction was carried out on a rotary evaporator. Elemental analyses for C, H, and N were recorded on a Perkin-Elmer 240 B microanalyzer (PerkinElmer, Waltham, MA, USA) and the analytical results are within ±0.4% of the theoretical values for all compounds (data not shown).
General procedure for the synthesis of 2-oxo-2H-pyran-5-carbonyl chloride (A). To the powder of coumalic acid (1 eq.) placed in the round bottom flask, was dropwise added thionyl chloride (3.8 eq.). The synthesis was carried out under reflux conditions and stirred for 8 hours, after which a 1
:4 mixture of dichloromethane and cyclohexane was added. The obtained suspension was filtered and the solid crystallised in cyclohexane to afford the title compound A as a brown solid (yield 55%, mp 72–75 °C). 1H NMR (400 MHz, CDCl3-d): δ 6.38 (dd, J = 9.9, 1.1 Hz, 1H, CHPyr), 7.76 (dd, J = 9.9, 2.8 Hz, 1H, CHPyr), 8.57 (dd, J = 2.8, 1.1 Hz, 1H, CHPyr). 13C NMR (101 MHz, CDCl3-d): δ 115.3 (CPyr), 116.5 (CHPyr), 140.2 (CHPyr), 158.1 (CHPyr), 162.7 (CO), 162.8 (COCl).
General procedure B. A mixture of 2-oxo-2H-pyran-5-carbonyl chloride (A) (1 eq.) and the proper alcohol (1 eq.) was heated at T = 75 °C under inert atmosphere (N2) in absence of solvents and stirred for 3 to 12 h (TLC monitoring), depending on alcohol reactivity. The reaction was quenched with slush and extracted with dichloromethane (3 × 40 mL). The organics were reunited, washed with NaOH 2N and dried over sodium sulfate. The drying agent was removed by filtration and the organic phase was evaporated in vacuo. The obtained crude product was purified by silica gel column chromatography eluting with 33% ethyl acetate in petroleum ether to afford the intended product. When necessary, the solids obtained were further crystallised from petroleum ether to afford the desired compounds.
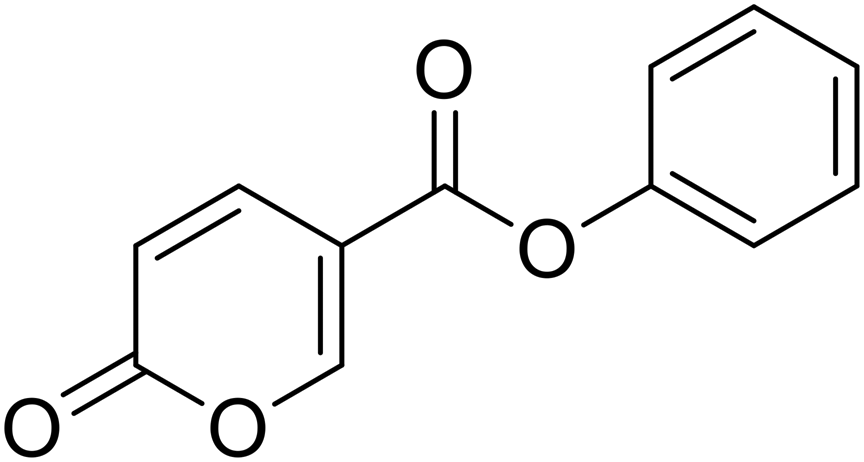
Phenyl 2-oxo-2H-pyran-5-carboxylate (4). Synthesised according to the general procedure B using phenol as starting material to afford the titled compound 4 as a white solid (51% yield). 1H NMR (400 MHz, CDCl3-d): δ 6.41 (dd, J = 9.8, 1.1 Hz, 1H, CHPyr), 7.16 (dd, J = 7.9, 1.7 Hz, 2H, Ar), 7.29 (t, J = 7.5 Hz, 1H, Ar), 7.43 (t, J = 7.9 Hz, 2H, Ar), 7.89 (dd, J = 9.8, 2.7 Hz, 1H, CHPyr), 8.51 (dd, J = 2.7, 1.1 Hz, 1H, CHPyr). 13C NMR (101 MHz, CDCl3-d): δ 111.6 (CHPyr), 115.5 (CPyr), 121.4 (2 × Ar), 126.4 (Ar), 129.6 (2 × Ar), 141.5 (Ar), 150.0 (CHPyr), 159.1 (CHPyr), 159.5 (CO), 161.6 (COOR). Measured monoisotopic mass [C12H8O4 + H]+: m/z 217.04954 (Δ: 0.00 ppm) – theoretical monoisotopic mass: m/z 217.04954.

p-Tolyl 2-oxo-2H-pyran-5-carboxylate (5). Synthesised according to the general procedure B using p-cresol as starting material to afford the titled compound 5 as a white solid (25% yield). 1H NMR (400 MHz, DMSO-d6): δ 2.33 (s, 3H, CH3), 6.50 (dd, J = 9.8, 1.1 Hz, 1H, CHPyr), 7.08–7.18 (m, 2H, Ar), 7.26 (d, J = 8.2 Hz, 2H, Ar), 7.93 (dd, J = 9.8, 2.7 Hz, 1H, CHPyr), 8.85 (d, J = 2.3 Hz, 1H, CHPyr). 13C NMR (101 MHz, DMSO-d6): δ 20.8 (CH3), 111.5 (CPyr), 115.3 (CHPyr), 121.9 (2 × Ar), 130.4 (2 × Ar), 135.9 (Ar), 142.5 (Ar), 148.3 (CHPyr), 160.0 (CHPyr), 160.8 (CO), 162.3 (COOR). Measured monoisotopic mass [C13H10O4 + H]+: m/z 231.06511 (Δ: −0.35 ppm) – theoretical monoisotopic mass: m/z 231.06519.

4-Methoxyphenyl 2-oxo-2H-pyran-5-carboxylate (6). Synthesized according to the general procedure B using 4-methoxyphenol as starting material to afford the titled compound 6 as a yellow solid (36% yield). 1H NMR (400 MHz, DMSO-d6): δ 3.78 (s, 3H, CH3), 6.50 (d, J = 9.8 Hz, 1H, CHPyr), 7.00 (d, J = 8.8 Hz, 2H, Ar), 7.18 (d, J = 8.7 Hz, 2H, Ar), 7.93 (dd, J = 9.9, 2.7 Hz, 1H, CHPyr), 8.85 (d, J = 2.6 Hz, 1H, CHPyr). 13C NMR (101 MHz, DMSO-d6): δ 55.9 (CH3), 111.5 (CPyr), 114.9 (2 × Ar), 115.3 (CHPyr), 123.0 (2 × Ar), 142.6 (Ar), 143.8 (Ar), 157.6 (CHPyr), 160.0 (CHPyr), 160.8 (CO), 162.4 (COOR). Measured monoisotopic mass [C13H10O5 + H]+: m/z 247.06009 (Δ: −0.04 ppm) – theoretical monoisotopic mass: m/z 247.06010.
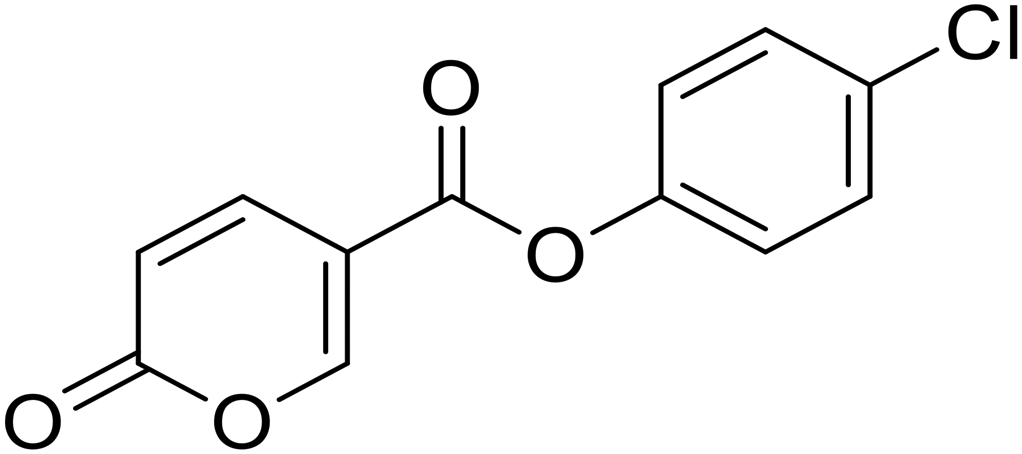
4-Chlorophenyl 2-oxo-2H-pyran-5-carboxylate (7). Synthesized according to the general procedure B using 4-chlorophenol as starting material to afford the titled compound 7 as a white solid (61% yield). 1H NMR (400 MHz, DMSO-d6): δ 6.51 (dd, J = 9.8, 1.1 Hz, 1H, CHPyr), 7.25–7.44 (m, 2H, Ar), 7.47–7.69 (m, 2H, Ar), 7.94 (dd, J = 9.8, 2.7 Hz, 1H, CHPyr), 8.88 (dd, J = 2.7, 1.1 Hz, 1H, CHPyr). 13C NMR (101 MHz, DMSO-d6): δ 111.3 (CHPyr), 115.3 (CPyr), 124.2 (2 × Ar), 130.0 (2 × Ar), 130.8 (Ar), 142.5 (Ar), 149.2 (CHPyr), 159.9 (CHPyr), 161.1 (CO), 162.0 (COOR). Measured monoisotopic mass [C12H7ClO4 + H]+: m/z 251.01050 (Δ: −0.24 ppm) – theoretical monoisotopic mass: m/z 251.01056.
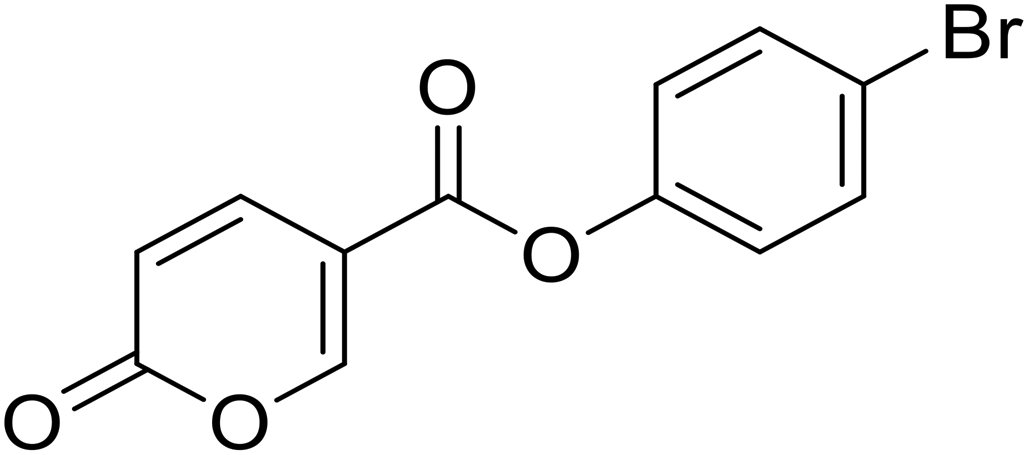
4-Bromophenyl 2-oxo-2H-pyran-5-carboxylate (8). Synthetized according to the general procedure B using 4-bromophenol as starting material to afford the titled compound 8 as a white solid (67% yield). 1H NMR (400 MHz, DMSO-d6): δ 6.51 (dd, J = 9.8, 1.1 Hz, 1H, CHPyr), 7.24–7.29 (m, 2H, Ar), 7.63–7.70 (m, 2H, Ar), 7.93 (dd, J = 9.8, 2.7 Hz, 1H, CHPyr), 8.87 (dd, J = 2.7, 1.1 Hz, 1H, CHPyr). 13C NMR (101 MHz, DMSO-d6): δ 111.3 (CHPyr), 115.3 (CPyr), 119.0 (Ar), 124.6 (2 × Ar), 132.9 (2 × Ar), 142.5 (Ar), 149.7 (CHPyr), 159.9 (CHPyr), 161.1 (CO), 161.9 (COOR). Measured monoisotopic mass [C12H7BrO4 + H]+: m/z 249.96006 (Δ: 0.03 ppm) – theoretical monoisotopic mass: m/z 294.96005.
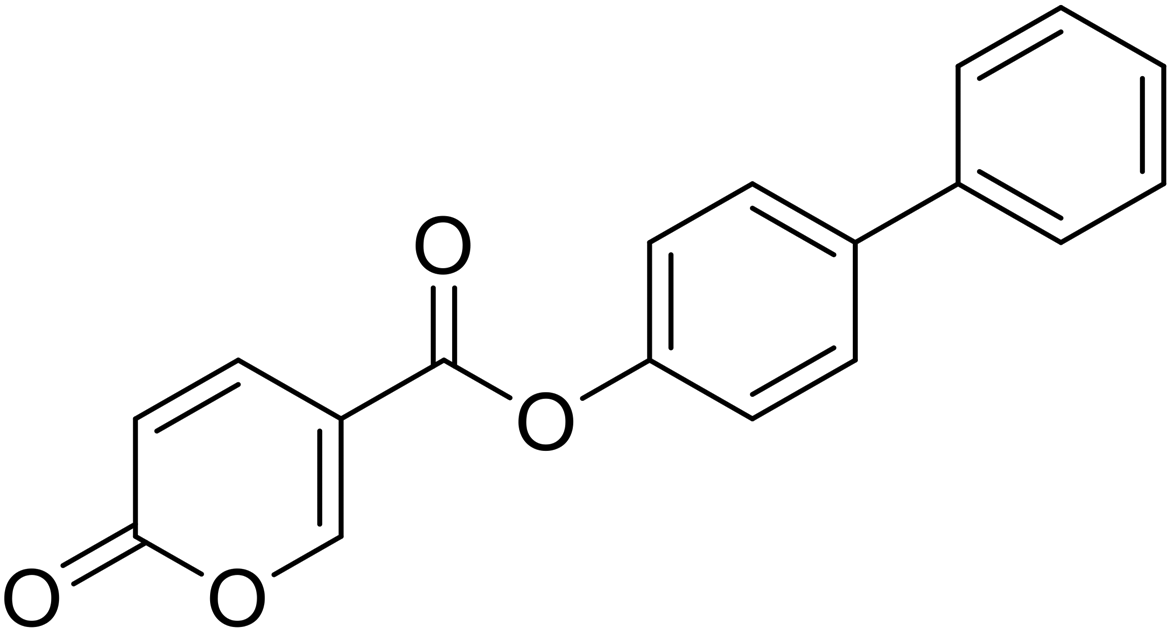
[1,1′-Biphenyl]-4-yl 2-oxo-2H-pyran-5-carboxylate (9). Synthetized according to the general procedure B using [1,1′-biphenyl]-4-ol as starting material to afford the titled compound 9 as a white solid (60% yield). δ 1H NMR (400 MHz, DMSO-d6): δ 6.55–6.48 (m, 1H, CHPyr), 7.43–7.32 (m, 3H, Ar), 7.48 (t, J = 7.5 Hz, 2H, Ar), 7.72–7.66 (m, 2H, Ar), 7.79–7.72 (m, 2H, Ar), 7.96 (dd, J = 9.8, 2.7 Hz, 1H, CHPyr), 8.91–8.85 (m, 1H, CHPyr). 13C NMR (101 MHz, DMSO-d6): δ 111.5 (CPyr), 115.3 (CHPyr), 122.7 (2 × Ar), 127.1 (2 × Ar), 128.1 (Ar), 128.3 (2 × Ar), 129.4 (2 × Ar), 138.7 (Ar), 139.7 (Ar) 142.6 (CHPyr), 150.0 (Ar), 160.1 (CHPyr), 161.0 (COOR), 162.2 (CO). Measured monoisotopic mass [C18H12O4 + H]+: m/z 293.08081 (Δ: −0.10 ppm) – theoretical monoisotopic mass: m/z 293.08084.
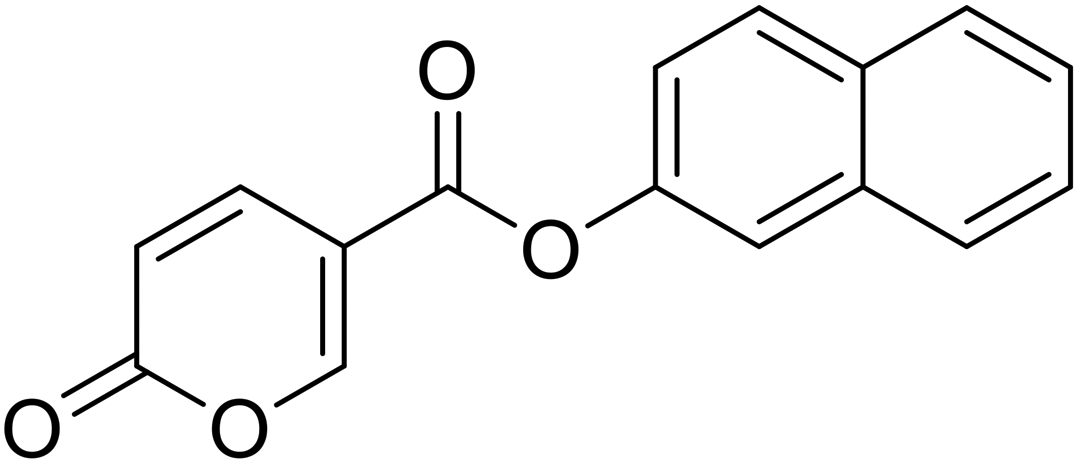
Naphthalen-2-yl 2-oxo-2H-pyran-5-carboxylate (10). Synthetized according to the general procedure B using naphthalen-2-ol as starting material to afford the titled compound 10 as a white solid (60% yield). 1H NMR (400 MHz, DMSO-d6): δ 6.53 (dd, J = 9.8, 1.1 Hz, 1H, CHPyr), 7.45 (dd, J = 8.9, 2.4 Hz, 1H, CHPyr), 7.57 (tt, J = 7.0, 5.2 Hz, 3H, Ar), 7.83 (d, J = 2.3 Hz, 1H, Ar), 7.91–8.08 (m, 3H, Ar), 8.86–9.01 (m, 1H, CHPyr). 13C NMR (101 MHz, DMSO-d6): δ 111.5 (CPyr), 115.3 (CHPyr), 119.1 (Ar), 121.8 (Ar), 126.5 (Ar), 127.3 (Ar), 128.0 (Ar), 128.2 (Ar), 129.9 (Ar), 131.6 (Ar), 133.7 (Ar), 142.5 (Ar), 148.1 (CHPyr), 160.0 (CHPyr), 161.0 (CO), 162.4 (COOR). Measured monoisotopic mass [C16H10O4 + H]+: m/z 267.06517 (Δ: −0.07 ppm) – theoretical monoisotopic mass: m/z 267.06519.
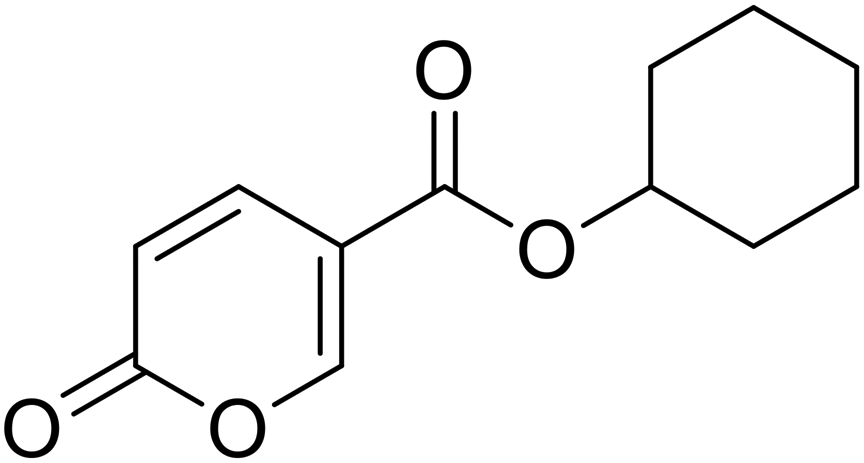
Cyclohexyl 2-oxo-2H-pyran-5-carboxylate (11). Synthetized according to the general procedure B using cyclohexanol as starting material to afford the titled compound 11 as a white solid (58% yield). 1H NMR (400 MHz, DMSO-d6): δ 1.24–1.43 (m, 3H, CH2Chx), 1.46–1.55 (m, 3H, CH2Chx), 1.67–1.87 (m, 4H, CH2Chx), 4.89 (tt, J = 8.3, 3.7 Hz, 1H, CHChx), 6.43 (dd, J = 9.8, 1.3 Hz, 1H, CHPyr), 7.82 (dd, J = 9.8, 2.6 Hz, 1H, CHPyr), 8.59 (dd, J = 2.7, 1.3 Hz, 1H, CHPyr). 13C NMR (101 MHz, DMSO-d6): δ 23.3 (2 × CH2Chx), 25.3 (CH2Chx), 31.3 (2 × CH2Chx), 73.5 (CH2Chx), 112.2 (CPyr), 115.2 (CHPyr), 142.5 (CHPyr), 159.5 (CHPyr), 160.2 (CO), 162.6 (COOR). Measured monoisotopic mass [C12H14O4 + H]+: m/z 233.09650 (Δ: 0.04 ppm) – theoretical monoisotopic mass: m/z 233.09649.
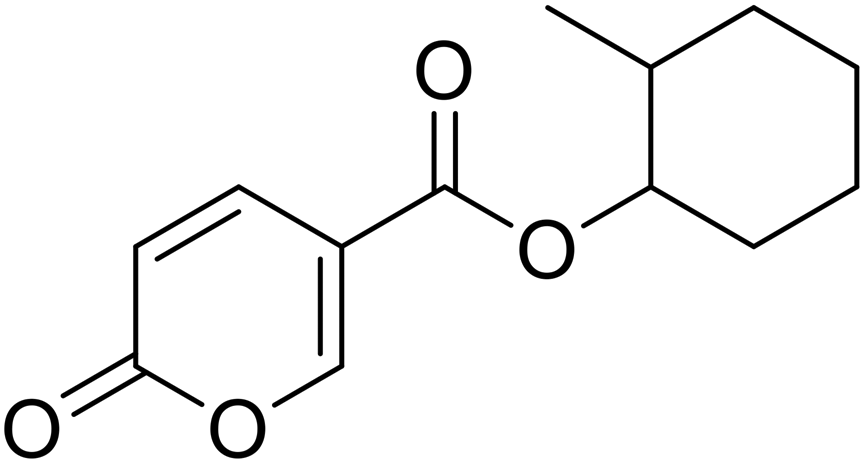
2-Methylcyclohexyl 2-oxo-2H-pyran-5-carboxylate (12). Synthetized according to the general procedure B using 2-methylcyclohexanol as starting material to afford the titled compound 12 as a white solid (22% yield). 1H NMR (400 MHz, DMSO-d6): δ 0.87 (dd, J = 9.8, 6.6 Hz, 3H, CH3), 1.04–1.16 (m, 1H, CH2Chx), 1.22 (dt, J = 12.5, 3.5 Hz, 1H, CH2Chx), 1.29–1.37 (m, 1H, CH2Chx), 1.47 (dq, J = 8.7, 4.4 Hz, 1H, CH2Chx), 1.57–1.68 (m, 2H, CH2Chx), 1.74 (tt, J = 9.1, 4.1 Hz, 2H, CH2Chx), 1.91–1.98 (m, 1H, CHChx), 4.52 (td, J = 9.9, 4.4 Hz, 1H, CHChx), 6.44 (dd, J = 9.8, 4.0 Hz, 1H, CHPyr), 7.82 (dd, J = 9.7, 2.7 Hz, 1H, CHPyr), 8.61 (dd, J = 11.1, 2.6 Hz, 1H, CHPyr). 13C NMR (101 MHz, DMSO-d6): δ 18.7 (CH3), 24.5 (2 × CH2Chx), 25.1 (2 × CH2Chx), 31.5 (2 × CH2Chx), 33.1 (2 × CH2Chx), 37.0 (CHChx), 79.4 (CHChx), 112.0 (CPyr), 115.2 (CHPyr), 142.5 (CHPyr), 159.5 (CHPyr), 160.1 (CO), 162.9 (COOR). Measured monoisotopic mass [C13H16O4 + H]+: m/z 237.11216 (Δ: 0.08 ppm) – theoretical monoisotopic mass: m/z 237.11214.
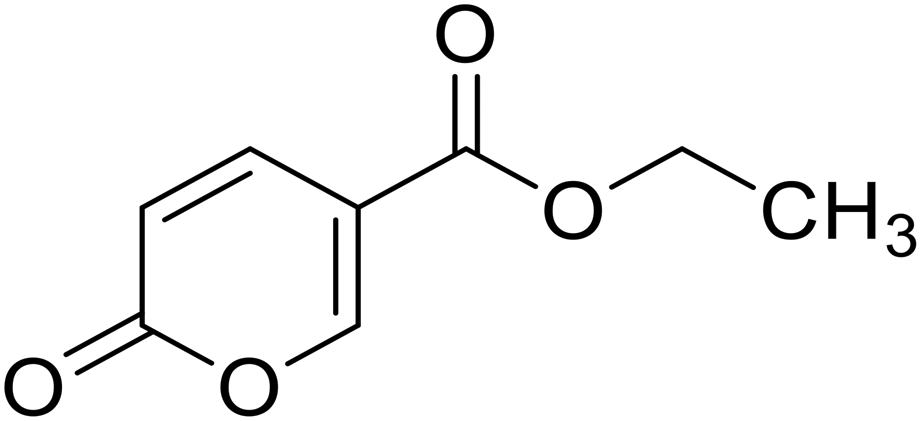
Ethyl 2-oxo-2H-pyran-5-carboxylate (13). Synthetized according to the general procedure B using ethanol as starting material to afford the titled compound 13 as a white solid (53% yield). 1H NMR (400 MHz, DMSO-d6): δ 1.27 (t, J = 7.1 Hz, 3H, CH3), 4.26 (q, J = 7.1 Hz, 2H, CH2), 6.42 (dd, J = 9.8, 1.1 Hz, 1H, CHPyr), 7.81 (dd, J = 9.8, 2.6 Hz, 1H, CHPyr), 8.57 (dd, J = 2.6, 1.2 Hz, 1H, CHPyr). 13C NMR (101 MHz, DMSO-d6): δ 14.4 (CH3), 61.5 (CH2), 112.0 (CPyr), 115.2 (CHPyr), 142.5 (CHPyr), 159.6 (CHPyr), 160.1 (CO), 163.2 (COOR). Measured monoisotopic mass [C8H8O4 + H]+: m/z 169.04955 (Δ: 0.06 ppm) – theoretical monoisotopic mass: m/z 169.04954.
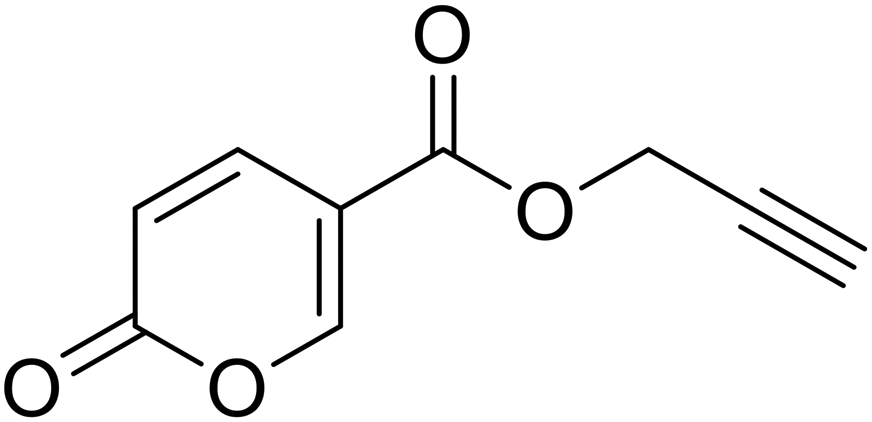
Prop-2-yn-1-yl 2-oxo-2H-pyran-5-carboxylate (14). To 1 eq. of intermediate A, were added 2 eq. of propargyl alcohol under inert atmosphere (N2) at T = 75 °C. The mixture was stirred for 12 h and quenched with slush. The mixture was extracted with dichloromethane (3 × 40 mL), the organics were reunited dried over sodium sulfate and filtered to remove the drying agent. The organic phase was evaporated in vacuo and the crude purified by silica gel column chromatography eluting with 33% ethyl acetate in n-hexane to afford the titled compound 14 as a white solid (yield 58%, mp 80–84 °C). 1H NMR (400 MHz, DMSO-d6): δ 3.64 (q, J = 2.2 Hz, 1H, CH), 4.91 (t, J = 1.8 Hz, 2H, CH2), 6.44 (d, J = 9.8 Hz, 1H, CHPyr), 7.82 (dt, J = 9.8, 1.9 Hz, 1H, CHPyr), 8.55–8.73 (m, 1H, CHPyr). 13C NMR (101 MHz, DMSO-d6): δ 53.2 (CH2), 78.4 (C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 78.8 (CCH), 111.3 (CPyr), 115.3 (CHPyr), 142.3 (CHPyr), 160.0 (CHPyr), 160.2 (CO), 162.6 (COOR). Measured monoisotopic mass [C9H6O4 + H]+: m/z 179.03392 (Δ: 0.17 ppm) – theoretical monoisotopic mass: m/z 179.03389.
CH), 78.8 (CCH), 111.3 (CPyr), 115.3 (CHPyr), 142.3 (CHPyr), 160.0 (CHPyr), 160.2 (CO), 162.6 (COOR). Measured monoisotopic mass [C9H6O4 + H]+: m/z 179.03392 (Δ: 0.17 ppm) – theoretical monoisotopic mass: m/z 179.03389.
General procedure C. To a mixture of CuI (0.01 eq.), DIPEA (0.1 eq.), and CH3COOH (0.1 eq.) in dichloromethane was added 2-oxo-N-(prop-2-yn-1-yl)-2H-pyran-5-carboxamide (14, 1 eq.) or prop-2-yn-1-yl 2-oxo-2H-pyran-5-carboxylate (20, 1 eq.) dissolved in dichloromethane, at room temperature. The solution was left to stir for 10 minutes and then the proper azide (1.05 eq.), was synthesized as previously reported.36 The resultant mixture was stirred for 12 h (TLC monitoring). Then depending on compound behavior, filtration or extraction was performed. The derivatives producing precipitate were filtered and purified by silica gel column chromatography eluting with 10% ethyl acetate in n-hexane to afford the final compound. The derivatives that did not precipitate were extracted with dichloromethane (3 × 40 mL), the organics were reunited, dried over sodium sulfate and filtered to remove the drying agent. The organic phase was evaporated in vacuo and the crude was purified with chromatographic procedures on silica gel employing 10% ethyl acetate in n-hexane as mobile phase.
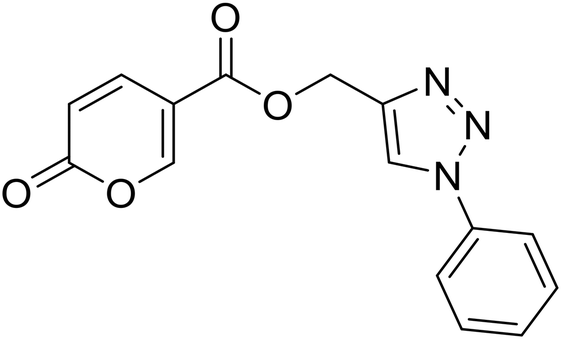
(1-Phenyl-1H-1,2,3-triazol-4-yl)methyl 2-oxo-2H-pyran-5-carboxylate (15). Synthetized according to the general procedure D to afford the titled compound 15 as a bright brown solid (37% yield). 1H NMR (400 MHz, CDCl3-d): δ 5.51 (s, 2H, CH2), 6.34 (dd, J = 9.8, 1.1 Hz, 1H, CHPyr), 7.45–7.50 (m, 1H, Ar), 7.51–7.58 (m, 2H, Ar), 7.72–7.76 (m, 2H, Ar), 7.79 (dd, J = 9.9, 2.6 Hz, 1H, CHPyr), 8.10 (s, 1H, CHTriaz), 8.34 (dd, J = 2.6, 1.1 Hz, 1H, CHPyr). 13C NMR (101 MHz, CDCl3-d): δ 58.3 (CH2), 111.7 (CPyr), 115.4 (CHPyr), 120.7 (2 × Ar), 122.5 (CHTriaz), 129.2 (Ar), 129.9 (2 × Ar), 136.8 (Ar), 141.5 (CHPyr), 142.7 (CTriaz), 158.7 (CHPyr), 159.6 (COPyr), 163.0 (COOR). Measured monoisotopic mass [C15H11N3O4 + H]+: m/z 298.08228 (Δ: 0.17 ppm) – theoretical monoisotopic mass: m/z 298.08223.
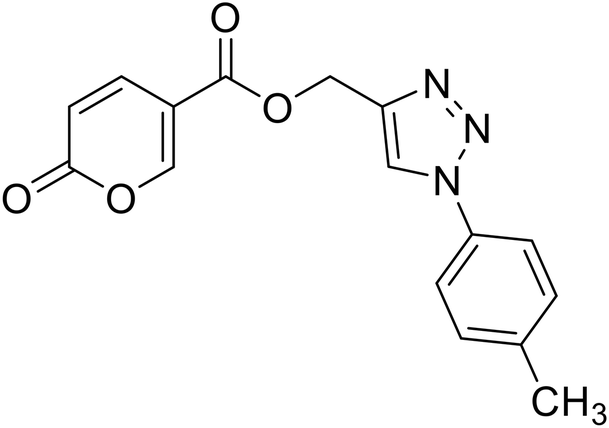
(1-(p-Tolyl)-1H-1,2,3-triazol-4-yl)methyl 2-oxo-2H-pyran-5-carboxylate (16). Synthetized according to the general procedure D to afford the titled compound 16 as a white solid (28% yield). 1H NMR (400 MHz, CDCl3-d): δ 2.42 (s, 3H, CH3), 5.49 (s, 2H, CH2), 6.33 (dd, J = 9.9, 1.1 Hz, 1H, CHPyr), 7.28–7.42 (m, 2H, Ar), 7.54–7.67 (m, 2H, Ar), 7.78 (dd, J = 9.9, 2.6 Hz, 1H, CHPyr), 8.06 (s, 1H, CHTriaz), 8.33 (dd, J = 2.6, 1.1 Hz, 1H, CHPyr). 13C NMR (101 MHz, CDCl3-d): δ 21.1 (CH3), 58.4 (CH2), 111.7 (CPyr), 115.4 (CHPyr), 120.6 (2 × Ar), 122.5 (CHTriaz), 130.4 (2 × Ar), 134.5 (Ar), 139.3 (Ar), 141.5 (CHPyr), 142.6 (CTriaz), 158.7 (CHPyr), 159.6 (COPyr), 163.0 (COOR). Measured monoisotopic mass [C16H13N3O4 + H]+: m/z 312.09794 (Δ: 0.19 ppm) – theoretical monoisotopic mass: m/z 312.09788.
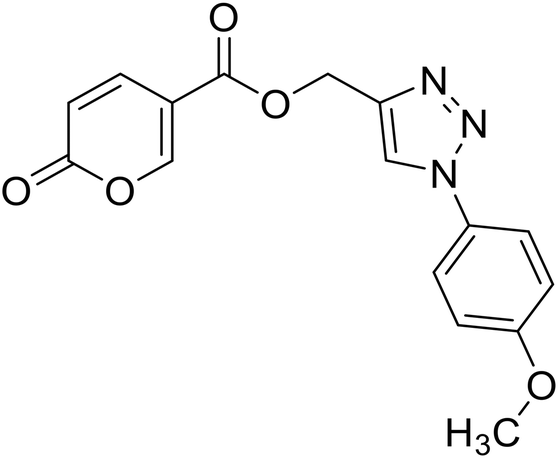
(1-(4-Methoxyphenyl)-1H-1,2,3-triazol-4-yl)methyl 2-oxo-2H-pyran-5-carboxylate (17). Synthetized according to the general procedure D to afford the titled compound 17 as an orange solid (26% yield). 1H NMR (400 MHz, CDCl3-d): δ 3.87 (s, 3H, CH3), 5.49 (s, 2H, CH2), 6.33 (dd, J = 9.9, 1.2 Hz, 1H, CHPyr), 6.97–7.09 (m, 2H, Ar), 7.58–7.67 (m, 2H, Ar), 7.78 (dd, J = 9.8, 2.7 Hz, 1H, CHPyr), 8.01 (s, 1H, CHTriaz), 8.33 (dd, J = 2.6, 1.1 Hz, 1H, CHPyr). 13C NMR (101 MHz, CDCl3-d): δ 55.7 (CH3), 58.4 (CH2), 111.7 (CPyr), 114.9 (2 × Ar), 115.4 (CHPyr), 122.3 (2 × Ar), 122.6 (CHTriaz), 127.7 (Ar), 130.2 (Ar), 141.5 (CHPyr), 142.5 (CTriaz), 158.7 (CHPyr), 159.6 (COPyr), 160.1 (Ar), 163.0 (COOR). Measured monoisotopic mass [C16H13N3O5 + H]+: m/z 328.09281 (Δ: 0.03 ppm) – theoretical monoisotopic mass: m/z 328.09280.
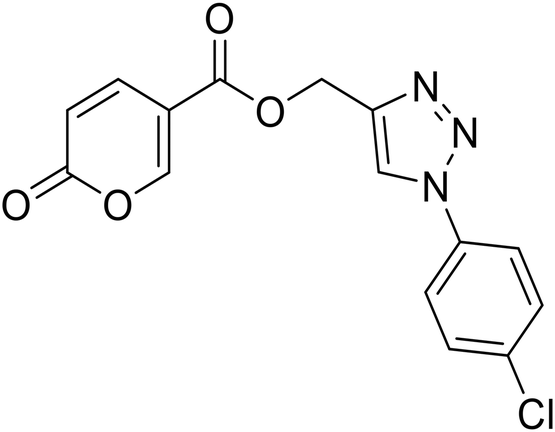
(1-(4-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl 2-oxo-2H-pyran-5-carboxylate (18). Synthetized according to the general procedure D to afford the titled compound 18 as a white solid (24% yield). 1H NMR (400 MHz, CDCl3-d): δ 5.50 (s, 2H, CH2), 6.34 (dd, J = 9.8, 1.1 Hz, 1H, CHPyr), 7.48–7.58 (m, 2H, Ar), 7.66–7.73 (m, 2H, Ar), 7.78 (dd, J = 9.8, 2.6 Hz, 1H, CHPyr), 8.08 (s, 1H, CHTriaz), 8.34 (dd, J = 2.6, 1.1 Hz, 1H, CHPyr). 13C NMR (101 MHz, CDCl3-d): δ 58.2 (CH2), 111.6 (CPyr), 115.4 (CHPyr), 121.8 (2 × Ar), 122.4 (CHTriaz), 130.1 (2 × Ar), 135.0 (Ar), 135.3 (Ar), 141.4 (CHPyr), 143.0 (CTriaz), 158.7 (CHPyr), 159.6 (COPyr), 163.0 (COOR). Measured monoisotopic mass [C15H10ClN3O4 + H]+: m/z 332.04328 (Δ: 0.06 ppm) – theoretical monoisotopic mass: m/z 332.04326.
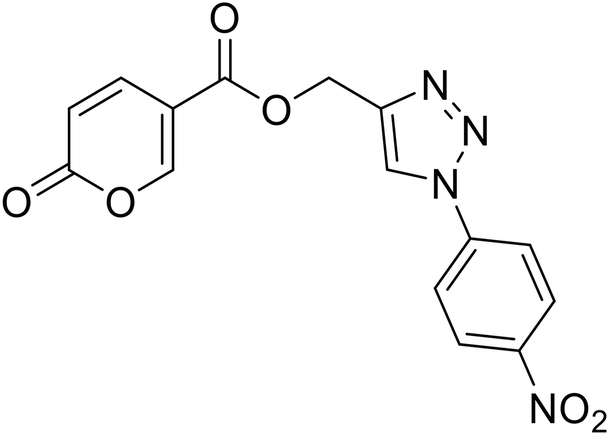
(1-(4-Nitrophenyl)-1H-1,2,3-triazol-4-yl)methyl 2-oxo-2H-pyran-5-carboxylate (19). Synthetized according to the general procedure D to afford the titled compound 19 as a white solid (15% yield). 1H NMR (400 MHz, CDCl3-d): δ 5.52 (s, 2H, CH2), 6.34 (dd, J = 9.9, 1.1 Hz, 1H, CHPyr), 7.78 (dd, J = 9.8, 2.6 Hz, 1H, CHPyr), 7.94–8.05 (m, 2H, Ar), 8.23 (s, 1H, CHTriaz), 8.34 (dd, J = 2.6, 1.1 Hz, 1H, CHPyr), 8.40–8.47 (m, 2H, Ar). 13C NMR (101 MHz, CDCl3-d): δ 58.0 (CH2), 111.5 (CPyr), 115.5 (CHPyr), 120.7 (2 × Ar), 122.4 (CHTriaz), 125.7 (2 × Ar), 140.9 (Ar), 141.3 (CHPyr), 143.7 (CTriaz), 147.5 (Ar), 158.8 (CHPyr), 159.5 (COPyr), 163.0 (COOR). Measured monoisotopic mass [C15H10N4O6 + H]+: m/z 343.06733 (Δ: 0.06 ppm) – theoretical monoisotopic mass: m/z 343.06731.
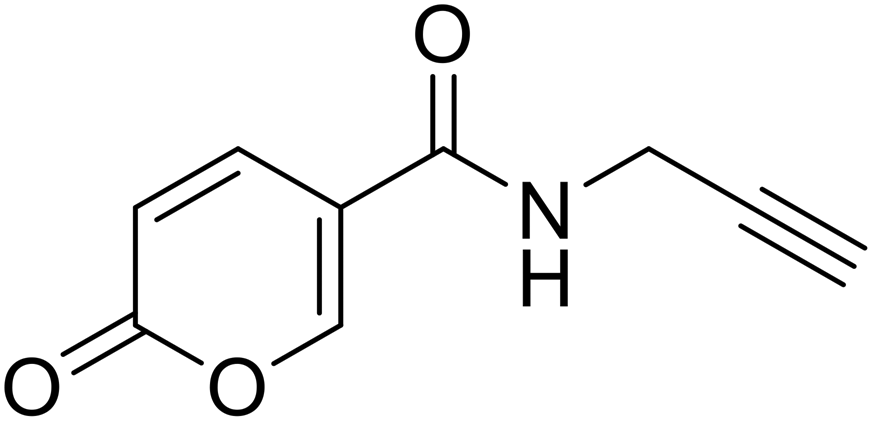
2-Oxo-N-(prop-2-yn-1-yl)-2H-pyran-5-carboxamide (20). To a solution of 2-oxo-2H-pyran-5-carbonyl chloride (A, 1 eq.) in diethyl ether, N,N-diisopropylethylamine (DIPEA, 1.5 eq.) and the propargyl amine (1.1 eq.) were added. The mixture was stirred at T = 4 °C (ice-bath) for 12 h (TLC monitoring). Then the reaction was added of diethyl ether and the organic phase extracted with HCl 2N (3 × 20 mL). The organic phase was dried over sodium sulfate, filtered to remove the drying agent and evaporated in vacuo. The crude has been purified by silica gel column chromatography eluting with 50% ethyl acetate in n-hexane, affording the titled compound 20 as a yellow powder (yield 51%, mp 131–135 °C).1H NMR (400 MHz, DMSO-d6): δ 3.14 (t, J = 2.5 Hz, 1H, CH), 4.01 (dd, J = 5.4, 2.6 Hz, 2H, CH2), 6.41 (dd, J = 9.8, 1.2 Hz, 1H, CHPyr), 7.86 (dd, J = 9.8, 2.8 Hz, 1H, CHPyr), 8.38 (dd, J = 2.7, 1.2 Hz, 1H, CHPyr), 8.86 (t, J = 5.5 Hz, 1H, NHCO). 13C NMR (101 MHz, DMSO-d6): δ 28.4 (CH2), 73.3 (C
CH), 80.7 (CCH), 114.4 (CPyr), 114.6 (CHPyr), 142.1 (CHPyr), 155.7 (CHPyr), 160.0 (CO), 161.9 (NHCO). Measured monoisotopic mass [C9H7NO3 + H]+: m/z 178.04989 (Δ: 0.11 ppm) – theoretical monoisotopic mass: m/z 178.04987.
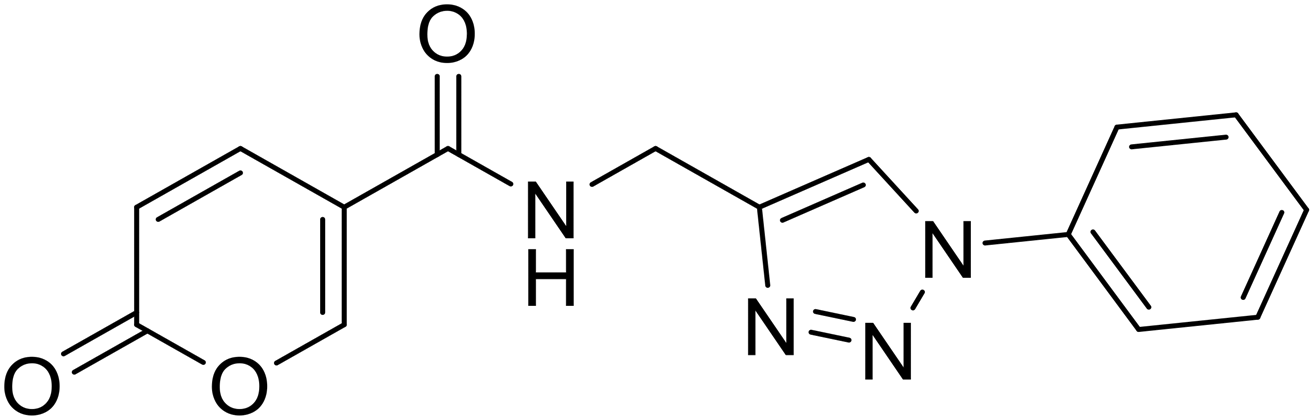
2-Oxo-N-((1-phenyl-1H-1,2,3-triazol-4-yl)methyl)-2H-pyran-5-carboxamide (21). Synthetized according to the general procedure C to afford the titled compound 21 as a white solid (32% yield). 1H NMR (400 MHz, DMSO-d6): δ 3.74 (d, J = 5.6 Hz, 2H, CH2), 5.51–5.66 (m, 1H, CHPyr), 6.60–6.67 (m, 1H, Ar), 6.69–6.83 (m, 2H, Ar), 7.01–7.07 (m, 2H, Ar), 7.09 (dd, J = 9.8, 2.6 Hz, 1H, CHPyr), 7.59 (dd, J = 9.8, 2.6 Hz, 1H, CHPyr), 7.77–7.89 (m, 1H, CHTriaz), 8.17 (t, J = 5.5 Hz, 1H, NHCO). 13C NMR (101 MHz, DMSO-d6): δ 35.1 (CH2), 114.9 (CPyr), 115.2 (CHPyr), 120.4 (2 × Ar), 121.5 (CHTriaz), 128.9 (Ar), 130.2 (2 × Ar), 137.2 (Ar), 142.8 (CHPyr), 146.1 (CTriaz), 155.9 (CHPyr), 160.5 (COPyr), 162.6 (NHCO). Measured monoisotopic mass [C15H12N4O3 + H]+: m/z 297.09827 (Δ: 0.17 ppm) – theoretical monoisotopic mass: m/z 297.09822.
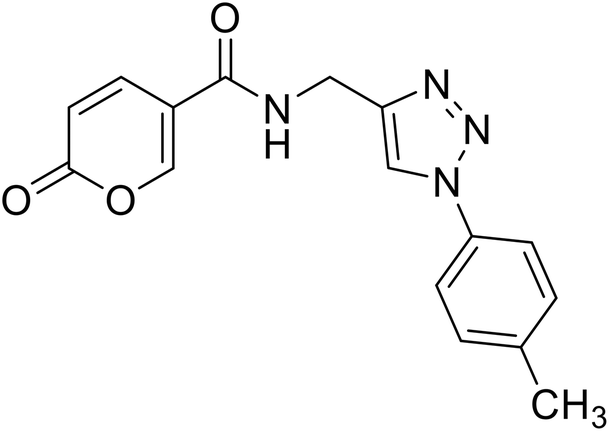
2-Oxo-N-((1-(p-tolyl)-1H-1,2,3-triazol-4-yl)methyl)-2H-pyran-5-carboxamide (22). Synthetized according to the general procedure C to afford the titled compound 22 as a white solid (26% yield). 1H NMR (400 MHz, DMSO-d6): δ 2.37 (s, 3H, CH3), 4.55 (d, J = 5.6 Hz, 2H, CH2), 6.36–6.46 (m, 1H, CHPyr), 7.38 (d, J = 8.1 Hz, 2H, Ar), 7.76 (d, J = 8.4 Hz, 2H, Ar), 7.91 (dd, J = 9.8, 2.7 Hz, 1H, CHPyr), 8.38–8.50 (m, 1H, CHPyr), 8.63 (s, 1H, CHTriaz), 9.00 (t, J = 5.6 Hz, 1H, NHCO). 13C NMR (101 MHz, DMSO-d6): δ 21.0 (CH3), 35.1 (CH2), 115.0 (CPyr), 115.2 (CHPyr), 120.3 (2 × Ar), 121.6 (CHTriaz), 130.7 (2 × Ar), 134.9 (Ar), 138.7 (Ar), 142.9 (CHPyr), 146.1 (CTriaz), 156.0 (CHPyr), 160.6 (COPyr), 162.7 (NHCO). Measured monoisotopic mass [C16H14N4O3 + H]+: m/z 311.11389 (Δ: 0.06 ppm) – theoretical monoisotopic mass: m/z 311.11387.
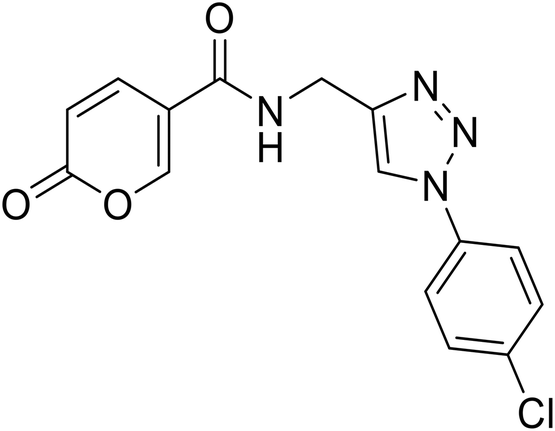
N-((1-(4-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl)-2-oxo-2H-pyran-5-carboxamide (24). Synthetized according to the general procedure C to afford the titled compound 24 as a white solid (34% yield). 1H NMR (400 MHz, CDCl3): δ 4.56 (d, J = 5.6 Hz, 2H, CH2), 6.42 (dd, J = 9.8, 1.1 Hz, 1H, CHPyr), 7.63–7.68 (m, 2H, Ar), 7.91 (dd, J = 9.8, 2.7 Hz, 1H, CHPyr), 7.93–7.97 (m, 2H, Ar), 8.42 (dd, J = 2.7, 1.1 Hz, 1H, CHPyr), 8.72 (s, 1H, CHTriaz), 9.02 (t, J = 5.6 Hz, 1H, NHCO). 13C NMR (101 MHz, CDCl3): δ 35.1 (CH2), 115.0 (CPyr), 115.2 (CHPyr), 121.8 (CHTriaz), 122.1 (2 × Ar), 130.3 (2 × Ar), 133.4 (Ar), 135.9 (Ar), 142.9 (CTriaz), 146.4 (CHPyr), 156.0 (CHPyr), 160.6 (COPyr), 162.7 (NHCO). Measured monoisotopic mass [C15H11ClN4O3 + H]+: m/z 331.05931 (Δ: 0.21 ppm) – theoretical monoisotopic mass: m/z 331.05924.
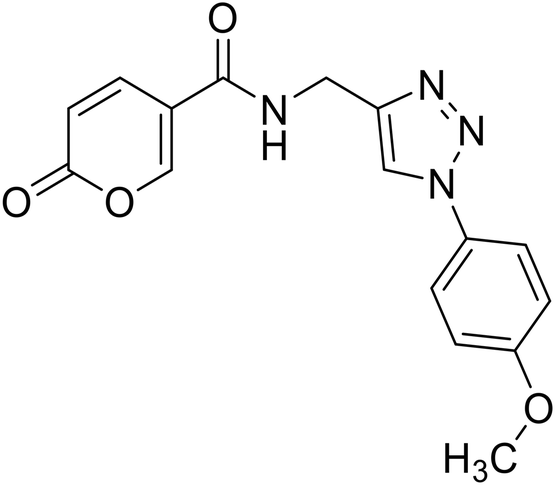
N-((1-(4-Methoxyphenyl)-1H-1,2,3-triazol-4-yl)methyl)-2-oxo-2H-pyran-5-carboxamide (23). Synthetized according to the general procedure C to afford the titled compound 23 as an orange solid (16% yield). 1H NMR (400 MHz, DMSO-d6): δ 3.82 (s, 3H, CH3), 4.54 (d, J = 5.6 Hz, 2H, CH2), 6.40 (dd, J = 9.8, 1.1 Hz, 1H, CHPyr), 7.04–7.18 (m, 2H, Ar), 7.73–7.85 (m, 2H, Ar), 7.90 (dd, J = 9.8, 2.7 Hz, 1H, CHPyr), 8.41 (dd, J = 2.7, 1.1 Hz, 1H, CHPyr), 8.55 (s, 1H, CHTriaz), 8.98 (t, J = 5.6 Hz, 1H, NHCO). 13C NMR (101 MHz, DMSO-d6): δ 35.1 (CH2), 56.0 (CH3), 114.9 (CPyr), 115.2 (CHPyr), 115.3 (2 × Ar), 121.7 (CHTriaz), 122.0 (2 × Ar), 130.6 (Ar), 142.8 (CHPyr), 145.9 (CTriaz), 156.0 (Ar), 159.7 (CHPyr), 160.5 (COPyr), 162.6 (NHCO). Measured monoisotopic mass [C16H14N4O4 + H]+: m/z 327.10890 (Δ: 0.37 ppm) – theoretical monoisotopic mass: m/z 327.10878.
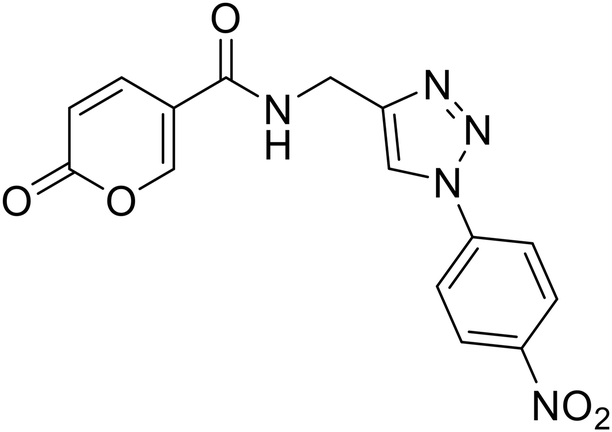
N-((1-(4-Nitrophenyl)-1H-1,2,3-triazol-4-yl)methyl)-2-oxo-2H-pyran-5-carboxamide (25). Synthetized according to the general procedure C to afford the titled compound 25 as a white solid (25% yield). 1H NMR (400 MHz, DMSO-d6): δ 4.58 (d, J = 5.6 Hz, 2H, CH2), 6.41 (dd, J = 9.8, 1.1 Hz, 1H, CHPyr), 7.91 (dd, J = 9.8, 2.7 Hz, 1H, CHPyr), 8.23 (d, J = 9.1 Hz, 2H, Ar), 8.28–8.51 (m, 3H, Ar + HTriaz), 8.89 (s, 1H, CHPyr), 9.05 (t, J = 5.6 Hz, 1H, NHCO). 13C NMR (101 MHz, DMSO-d6): δ 35.0 (CH2), 115.0 (CPyr), 115.2 (CHPyr), 120.9 (2 × Ar), 122.2 (Ar), 126.0 (2 × Ar), 141.4 (CTriaz), 142.8 (CHTriaz), 147.0 (Ar), 147.1 (CHPyr), 156.0 (CHPyr), 160.5 (CO), 162.7 (NHCO). Measured monoisotopic mass [C15H11N5O5 + H]+: m/z 342.08330 (Δ: 0.03 ppm) – theoretical monoisotopic mass: m/z 342.08329.
5.2 Inhibition assay
An Applied Photophysics stopped-flow instrument was used to assay the CA-catalyzed CO2 hydration activity.42 Phenol red (at a concentration of 0.2 mM) was used as an indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.4) as a buffer and 20 mM Na2SO4 (to maintain constant ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constant. Enzyme concentrations ranged between 5–12 nM. For each inhibitor, at least six traces of the initial 5–10% of the reaction were used to determine the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of the inhibitor (0.1 mM) were prepared in distilled–deionized water and dilutions up to 0.01 nM were conducted thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 6 h at room temperature prior to the assay, to allow for the formation of the E–I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 and the Cheng–Prusoff equation as reported earlier and represent the mean from at least three different determinations.5.3 Molecular modelling
The crystallographic structures of hCA IX at 2.2 Å resolution (PDB-ID: 3IAI, chain A)45 and hCA XII at 1.50 Å resolution (PDB-ID: 1JD0, chain A),46 both in complex with acetazolamide (AAZ), were used as rigid receptors in molecular docking simulations. In hCA IX, sequence alignment with human hCA IX deposited in the UniProt database (Q16790)47 revealed the C14S single point mutation compared to the hCA IX crystallized sequence. However, this mutation is at a distance higher than 20 Å from the catalytic site, thus it was not further accounted for in molecular docking simulations as in previous studies.36 The PDB sequence of PDB-ID: 1JD0 matches the UniProt sequence O43570, so no mutations are present. Small molecules were sketched in 2D with the Picto software (version 4.5.4.1, OpenEye Cadence Molecular Sciences, Santa Fe, NM) and converted into a 3D structure with OMEGA (version 4.2.0.1, OpenEye Cadence Molecular Sciences, Santa Fe, NM).48,49 Ligand protonation state was assigned by QUACPAC (version 2.2.0.1, OpenEye Cadence Molecular Sciences, Santa Fe, NM),50 while energy minimization was carried out by SZYBKI (version 2.5.0.1, OpenEye Cadence Molecular Sciences, Santa FE, NM)51 using the MMFF94S force field.52,53 Molecular docking was carried out with GOLD (The Cambridge Crystallographic Data Centre, Cambridge, UK) version 2023.1.54,55 In both structures, re-docking of AAZ was performed to validate the docking protocol, and the obtained pose overlaps with the co-crystalized AAZ (RMSD < 2.0 Å) in both crystals with the same docking settings. The ChemPLP fitness function for docking and scoring was used. Based on structural and literature evidence, the binding site for docking simulations was centred on the catalytic Zn(II) ion, and it was assigned a radius of 10 Å. The Zn(II)-coordinated water molecule was modelled as described previously,56,57 and it was kept as fixed in molecular docking (toggle “on” option). The receptor was prepared with the GOLD docking wizard implemented in Hermes visualizer, which is an integral part of the GOLD interface. Ten runs for each ligand were stored and submitted to visual inspection.6. Conclusion
In this study, a series of novel coumalic acid derivatives was synthesized and the compounds were evaluated for their inhibitory activity against human carbonic anhydrase isoforms, particularly the tumour-associated isoforms hCA IX and hCA XII. The results from this work contribute valuable insights into the structure–activity relationship (SAR) of coumalic acid-based inhibitors, with the goal of developing selective inhibitors for cancer therapy. The key findings from the SAR analysis highlight the critical role of the lactone moiety in the inhibition of hCA IX and XII. The replacement of the lactone with a lactam (compounds 1–2) resulted in a complete loss of activity, confirming the importance of the lactone ring and its open form (cinnamic acid) in enzyme inhibition. Furthermore, the position of the carboxylic acid group within the coumalic acid core was found to significantly influence inhibitory potency. Indeed, the moving this group from the position 5 of the coumalic acid to position 6 (compound 3) provoked the dramatic loss of inhibitory activity against isoforms IX and XII, emphasizing the structural precision required for effective binding to the enzyme active site. The modifications of the linker moiety, particularly the isosteric replacement of amide with ester (compounds 4–14), demonstrated a clear reduction in potency against hCA IX and XII, although selectivity for these isoforms was maintained. Notably, the introduction of substituents on the phenyl ring (compounds 5–10) enhanced inhibitory activity, with compound 8 emerging as the most potent inhibitor of hCA XII, showcasing its potential as a lead compound for further development. The incorporation of a triazole ring as part of a “click chemistry” approach (compounds 15–25) aimed at molecular elongation did not yield compounds with improved potency, suggesting that excessive structural complexity may hinder the desired inhibitory effect. However, compounds 16 and 25 did show some promise, indicating that further optimization of the triazole-linked derivatives could still provide valuable insights. Molecular docking studies supported the experimental results, showing that the coumalic acid derivatives bind effectively to the active sites of hCA IX and XII, with key interactions involving the Zn(II)-bound water molecule, critical for enzyme inhibition. In particular, compound 11 demonstrated a higher selectivity and potency for hCA XII, reinforcing the importance of specific structural features in achieving selective inhibition. In conclusion, the coumalic acid-based derivatives described in this study offer valuable starting points for the development of selective inhibitors targeting tumour-related carbonic anhydrase isoforms. These findings provide a deeper understanding of the molecular mechanisms underlying hCA inhibition and suggest potential avenues for further refinement of these compounds as therapeutic agents for cancer treatment. Future work should focus on optimizing the lead compounds identified in this study, exploring additional modifications to enhance their potency, selectivity, and pharmacological properties.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization, review and editing: P. G., V. P., C. T. S., D. S., and P. C. Supervision, P. G., D. S., C. T. S. Investigation: V. P., F. A., M. C., E. D. F., A. B., M. M., M. S., L. C., A. A., E. B., C. S., A. T., F. P., A. D. N. and A. G. Writing – original draft preparation: P. G., V. P., M. M., and L. C. Writing – review & editing: S. C., C. T. S., D. S., P. C., and A. A. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
M. M.–L. C.: We thank OpenEye Cadence Molecular Sciences for their free academic license.Virginia Pontecorvi was supported by Fondazione Umberto Veronesi.
References
- J. S. Torday, Homeostasis as the mechanism of evolution, Biology, 2015, 4(3), 573–590 CrossRef CAS PubMed.
- G. E. Billman, Homeostasis: The Underappreciated and Far Too Often Ignored Central Organizing Principle of Physiology, Front. Physiol., 2020, 11, 200 CrossRef PubMed.
- C. T. Supuran, A simple yet multifaceted 90 years old, evergreen enzyme: Carbonic anhydrase, its inhibition and activation, Bioorg. Med. Chem. Lett., 2023, 23, 129411 CrossRef PubMed.
- M. Y. Mboge, B. P. Mahon, R. McKenna and S. C. Frost, Carbonic anhydrases: Role in pH control and cancer, Metabolites, 2018, 8, 19 CrossRef PubMed.
- C. T. Supuran and A. Scozzafava, Carbonic anhydrases as targets for medicinal chemistry, Bioorg. Med. Chem., 2007, 4336–4350 CrossRef CAS PubMed.
- C. T. Supuran, Structure and function of carbonic anhydrases, Biochem. J., 2016, 473(14), 2023–2032 CrossRef CAS PubMed.
- C. T. Supuran and C. Capasso, An overview of the bacterial carbonic anhydrases, Metabolites, 2017, 7, 56 CrossRef PubMed.
- K. D'Ambrosio, C. T. Supuran and G. De Simone, Are Carbonic Anhydrases Suitable Targets to Fight Protozoan Parasitic Diseases?, Curr. Med. Chem., 2018, 25(39), 5266–5278 CrossRef PubMed.
- A. Nocentini, C. T. Supuran and C. Capasso, An overview on the recently discovered iota-carbonic anhydrases, J. Enzyme Inhib. Med. Chem., 2021, 1988–1995 CrossRef CAS PubMed.
- J. G. Ferry, The γ class of carbonic anhydrases, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 374–381 CrossRef CAS PubMed.
- Y. Hirakawa, M. Senda and K. Fukuda, et al., Characterization of a novel type of carbonic anhydrase that acts without metal cofactors, BMC Biol., 2021, 19, 105 CrossRef CAS PubMed.
- S. C. Frost, Physiological Functions of the Alpha Class of Carbonic Anhydrases, in Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications, ed. S. C. Frost and R. McKenna, Springer Netherlands, Dordrecht, 2014, pp. 9–30 Search PubMed.
- S. Kumar, S. Rulhania, S. Jaswal and V. Monga, Recent advances in the medicinal chemistry of carbonic anhydrase inhibitors, Eur. J. Med. Chem., 2021, 209, 112923 CrossRef CAS PubMed.
- E. Berrino and C. T. Supuran, Novel approaches for designing drugs that interfere with pH regulation, Expert Opin. Drug Discovery, 2019, 231–248 CrossRef CAS PubMed.
- P. Guglielmi, S. Carradori, C. Campestre and G. Poce, Novel therapies for glaucoma: a patent review (2013-2019), Expert Opin. Ther. Pat., 2019, 29(10), 769–780 CrossRef CAS PubMed.
- C. P. S. Potter and A. L. Harris, Diagnostic, prognostic and therapeutic implications of carbonic anhydrases in cancer, Br. J. Cancer, 2003, 2–7 CrossRef CAS PubMed.
- C. T. Supuran, How many carbonic anhydrase inhibition mechanisms exist?, J. Enzyme Inhib. Med. Chem., 2016, 345–360 CrossRef CAS PubMed.
- K. D'Ambrosio, A. Di Fiore and E. Langella, Dual targeting carbonic anhydrase inhibitors as promising therapeutic approach: a structural overview, Front. Mol. Biosci., 2025, 12, 1511281 CrossRef CAS PubMed.
- K. A. Mishra and K. K. Sethi, Unveiling tomorrow: Carbonic anhydrase activators and inhibitors pioneering new frontiers in Alzheimer’s disease, Arch. Pharm., 2024, 358, e2400748 CrossRef PubMed.
- P. Singh, M. Arifuddin, C. T. Supuran and S. G. Nerella, Carbonic anhydrase inhibitors: Structural insights and therapeutic potential, Bioorg. Chem., 2025, 156, 108224 CrossRef CAS PubMed.
- B. Zengin Kurt, G. Celebi and D. Ozturk Civelek, et al., Tail-Approach-Based Design and Synthesis of Coumarin-Monoterpenes as Carbonic Anhydrase Inhibitors and Anticancer Agents, ACS Omega, 2023, 8(6), 5787–5807 CrossRef CAS PubMed.
- C. T. Supuran, Carbonic anhydrase activators, Future Med. Chem., 2018, 561–573 CrossRef CAS PubMed.
- Y. Lou, P. C. McDonald and A. Oloumi, et al., Targeting tumor hypoxia: Suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors, Cancer Res., 2011, 71(9), 3364–3376 CrossRef CAS.
- E. Švastová, A. Hulíková and M. Rafajová, et al., Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH, FEBS Lett., 2004, 577(3), 439–445 CrossRef PubMed.
- S. Pastorekova, S. Parkkila, J. Pastorek and C. T. Supuran, Carbonic anhydrases: Current state of the art, therapeutic applications and future prospects, J. Enzyme Inhib. Med. Chem., 2004, 199–229 CrossRef CAS PubMed.
- C. B. Mishra, M. Tiwari and C. T. Supuran, Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: Where are we today?, Med. Res. Rev., 2020, 2485–2565 CrossRef CAS PubMed.
- D. Neri and C. T. Supuran, Interfering with pH regulation in tumours as a therapeutic strategy, Nat. Rev. Drug Discovery, 2011, 767–777 CrossRef CAS PubMed.
- C. T. Supuran, Structure-based drug discovery of carbonic anhydrase inhibitors, J. Enzyme Inhib. Med. Chem., 2012, 759–772 CrossRef CAS PubMed.
- J. Y. Winum, M. Rami, A. Scozzafava, J. L. Montero and C. Supuran, Carbonic anhydrase IX: A new druggable target for the design of antitumor agents, Med. Res. Rev., 2008, 445–463 CrossRef CAS PubMed.
- C. T. Supuran, V. Alterio and A. Di Fiore, et al., Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: Three for the price of one, Med. Res. Rev., 2018, 1799–1836 CrossRef PubMed.
- C. T. Supuran, Coumarin carbonic anhydrase inhibitors from natural sources, J. Enzyme Inhib. Med. Chem., 2020, 1462–1470 CrossRef PubMed.
- E. Berrino, S. Carradori and F. Carta, et al., A Multitarget Approach against Neuroinflammation: Alkyl Substituted Coumarins as Inhibitors of Enzymes Involved in Neurodegeneration, Antioxidants, 2023, 12, 2044 CrossRef CAS PubMed.
- S. Mahammad Ghouse, K. Bahatam and A. Angeli, et al., Synthesis and biological evaluation of new 3-substituted coumarin derivatives as selective inhibitors of human carbonic anhydrase IX and XII, J. Enzyme Inhib. Med. Chem., 2023, 38, 2185760 CrossRef PubMed.
- A. Maresca, C. Temperini and H. Vu, et al., Non-zinc mediated inhibition of carbonic anhydrases: Coumarins are a new class of suicide inhibitors, J. Am. Chem. Soc., 2009, 131(8), 3057–3062 CrossRef CAS PubMed.
- B. Cornelio, M. Laronze-Cochard and R. Miambo, et al., 5-Arylisothiazol-3(2H)-one-1,(1)-(di)oxides: A new class of selective tumor-associated carbonic anhydrases (hCA IX and XII) inhibitors, Eur. J. Med. Chem., 2019, 175, 40–48 CrossRef CAS PubMed.
- V. Pontecorvi, M. Mori and F. Picarazzi, et al., Novel Insights on Human Carbonic Anhydrase Inhibitors Based on Coumalic Acid: Design, Synthesis, Molecular Modeling Investigation, and Biological Studies, Int. J. Mol. Sci., 2022, 23, 7950 CrossRef CAS.
- Y. Aimene, R. Eychenne and F. Rodriguez, et al., Synthesis, crystal structure, inhibitory activity and molecular docking of coumarins/sulfonamides containing triazolyl pyridine moiety as potent selective carbonic anhydrase IX and XII inhibitors, Crystals, 2021, 11, 1076 CrossRef CAS.
- P. S. Thacker, P. L. Tiwari and A. Angeli, et al., Synthesis and biological evaluation of coumarin-linked 4-anilinomethyl-1,2,3-triazoles as potent inhibitors of carbonic anhydrases ix and xiii involved in tumorigenesis, Metabolites, 2021, 11, 225 CrossRef CAS PubMed.
- P. Guglielmi, G. Rotondi and D. Secci, et al., Novel insights on saccharin- and acesulfame-based carbonic anhydrase inhibitors: design, synthesis, modelling investigations and biological activity evaluation, J. Enzyme Inhib. Med. Chem., 2020, 35(1), 1891–1905 CrossRef CAS PubMed.
- A. Angeli and C. T. Supuran, Click chemistry approaches for developing carbonic anhydrase inhibitors and their applications, J. Enzyme Inhib. Med. Chem., 2023, 38, 2166503 CrossRef PubMed.
- R. H. Wiley and A. J. Hart, 2-Pyrones. IX. 2-Pyrone-6-carboxylic acid and its derivatives, J. Am. Chem. Soc., 1954, 76, 1942 CrossRef CAS.
- E. Lucarini, V. D'Antogiovanni and L. Antonioli, et al., Study of Chalcogen Aspirin Derivatives with Carbonic Anhydrase Inhibitory Properties for Treating Inflammatory Pain, ACS Med. Chem. Lett., 2024, 15, 1559–1565 CrossRef CAS PubMed.
- N. Dinh Thanh, N. Thi Kim Giang, D. Son Hai, V. Ngoc Toan, H. Thi Kim Van and N. Minh Tri, Sulfonyl thiourea derivatives from 2-aminodiarylpyrimidines: In vitro and in silico evaluation as potential carbonic anhydrase I, II, IX, and XII inhibitors, Chem. Biol. Drug Des., 2024, 103, e14494 CrossRef PubMed.
- A. Di Noi, A. Massaro, C. Salvitti, M. Managò, F. Cosentino, R. Piro, M. Suman, F. Pepi, A. Tata and A. Troiani, AP-MALDI-MS reveals adulteration of dried oregano leaves, J. Food Compos. Anal., 2025, 139, 107121 CrossRef CAS.
- V. Alterio, M. Hilvo and A. Di Fiore, Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16233–16238 CrossRef CAS PubMed.
- D. A. Whittington, A. Waheed and B. Ulmasov, Crystal structure of the dimeric extracellular domain of human carbonic anhydrase XII, a bitopic membrane protein overexpressed in certain cancer tumor cells, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 9545–9550 CrossRef CAS PubMed.
- A. Bateman, M. J. Martin and S. Orchard, et al., UniProt: the Universal Protein Knowledgebase in 2023, Nucleic Acids Res., 2023, 51(D1), D523–D531 CrossRef CAS PubMed.
- E. Berrino, S. Carradori and A. Angeli, et al., Dual carbonic anhydrase ix/xii inhibitors and carbon monoxide releasing molecules modulate LPS-mediated inflammation in mouse macrophages, Antioxidants, 2021, 10(1), 1–24 CrossRef PubMed.
- P. C. D. Hawkins, A. G. Skillman, G. L. Warren, B. A. Ellingson and M. T. Stahl, Conformer generation with OMEGA: Algorithm and validation using high quality structures from the protein databank and cambridge structural database, J. Chem. Inf. Model., 2010, 50(4), 572–584 CrossRef CAS PubMed.
- C. T. Supuran and W. A. Donald, Metalloenzymes, 2024 Search PubMed.
- M. D'Ascenzio, D. Secci and S. Carradori, et al., 1,3-Dipolar Cycloaddition, HPLC Enantioseparation, and Docking Studies of Saccharin/Isoxazole and Saccharin/Isoxazoline Derivatives as Selective Carbonic Anhydrase IX and XII Inhibitors, J. Med. Chem., 2020, 63(5), 2470–2488 CrossRef PubMed.
- T. A. Halgren, Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94, J. Comput. Chem., 1996, 17(5–6), 490–519 CrossRef CAS.
- T. A. Halgren, Merck molecular force field. III. Molecular geometries and vibrational frequencies for MMFF94, J. Comput. Chem., 1996, 17(5–6), 553–586 CrossRef CAS.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, Development and validation of a genetic algorithm for flexible docking11Edited by F. E. Cohen, J. Mol. Biol., 1997, 267(3), 727–748 CrossRef CAS PubMed.
- M. L. Verdonk, J. C. Cole, M. J. Hartshorn, C. W. Murray and R. D. Taylor, Improved protein–ligand docking using GOLD, Proteins: Struct., Funct., Bioinf., 2003, 52(4), 609–623 CrossRef CAS PubMed.
- A. Gumus, I. D'Agostino and V. Puca, et al., Cyclization of acyl thiosemicarbazides led to new Helicobacter pylori α-carbonic anhydrase inhibitors, Arch. Pharm., 2024, 357, e2400548 CrossRef CAS PubMed.
- Y. Cau, D. Vullo and M. Mori, et al., Potent and selective carboxylic acid inhibitors of tumor-associated carbonic anhydrases IX and XII, Molecules, 2018, 23, 17 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5md00208g |
| This journal is © The Royal Society of Chemistry 2025 |