
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design, synthesis and evaluation of pyrrolobenzodiazepine (PBD)-based PROTAC conjugates for the selective degradation of the NF-κB RelA/p65 subunit†
Peiqin Jin a,
Md. Mahbub Hasan‡
a,
Andrea G. S. Pepperb,
Simon Mitchellb,
Khondaker Miraz Rahman§
*a and
Chris Pepper§*ab
a,
Md. Mahbub Hasan‡
a,
Andrea G. S. Pepperb,
Simon Mitchellb,
Khondaker Miraz Rahman§
*a and
Chris Pepper§*ab
aInstitute of Pharmaceutical Science, School of Cancer and Pharmaceutical Sciences, King's College London, London SE1 9NH, UK. E-mail: k.miraz.rahman@kcl.ac.uk
bBrighton and Sussex Medical School, University of Brighton and University of Sussex, Brighton BN1 9PX, UK. E-mail: c.pepper@bsms.ac.uk
First published on 8th May 2025
Abstract
NF-κB signalling is frequently dysregulated in human cancers making it an attractive therapeutic target. Despite concerted efforts to generate NF-κB inhibitors, direct pharmacological inhibition of the kinases mediating canonical NF-κB has failed due to on-target toxicities in normal tissues. So, alternative strategies, designed to target specific components of the NF-κB signalling machinery, have the potential to selectively inhibit tumour cells whilst reducing the toxicities associated with broad inhibition of NF-κB in non-malignant cells. Here we present evidence that a C8-linked pyrrolobenzodiazepine (PBD) containing proteolysis-targeting chimera (PROTAC) selectively degrades the NF-κB subunit, RelA/p65, in a proteasome-dependent manner. Our lead PROTAC (JP-163-16, 15d) showed cytotoxicity with mean LC50 values of 2.9 μM in MDA-MB-231 cells, 0.14 μM in MEC-1 cells and 0.23 μM in primary chronic lymphocytic leukaemia cells. In contrast, 15d was two-logs less toxic in primary B- and T-lymphocytes (mean LD50 19.1 μM and 36.4 μM, respectively). Importantly, the development of 15d, by conjugating the C8-linked PBD with a cereblon-targeting ligand using a five-carbon linker, abolished the ability of the C8-linked PBD to bind to DNA, whilst demonstrating cytotoxicity in cancer cells associated with the degradation of RelA/p65. Mechanistically, 15d displayed PROTAC credentials through the selective degradation of NF-κB RelA/p65 in a proteasome-dependent manner and showed a five-fold reduction in potency in the cereblon deficient, lenalidomide resistant, myeloma cell line, RPMI-8226. To our knowledge, this work describes the first PROTAC capable of selective degradation of a single NF-κB subunit and highlights the therapeutic potential of our strategy for the treatment of RelA/p65-dependent tumours.
Introduction
The nuclear factor kappa-light chain-enhancer of activated B-cells (NF-κB) is a transcription factor that plays a pivotal role in inflammatory and immune responses, and its abnormal activation is associated with various pathogenic effects.1,2 For example, an over-expressed level of NF-κB signalling contributes to metastasis in triple-negative breast cancer (TNBC).3 In haematological malignancies, elevated nuclear expression of the NF-κB subunit RelA/p65 is commonly associated with more aggressive tumour cell growth, increased tumour burden and the emergence of drug resistance.4 Given the pivotal role that NF-κB plays in the development and progression of a range of human cancers, it seems logical to develop strategies to target abnormal NF-κB signalling for the treatment of these diseases.5However, direct targeting of transcription factors (TFs) has long been considered intractable due to the lack of well-defined enzymatic binding pockets and limited H-bond donors and acceptors, rendering them ‘undruggable’.6,7 To date, only a small number of molecules have been shown to be capable of interacting with NF-κB.8 One such molecule, (−)-DHMEQ, a synthetic compound derived from epoxyquinomicin C, can inhibit the nuclear transport of the transcriptional subunit RelA/p65 (Fig. 1a).9 Unfortunately, further development of this compound series was halted due to pharmacokinetic issues.10 Pyrrolobenzodiazepines (PBDs) (Fig. 1b) are highly cytotoxic agents derived from Streptomyces.11 They exert their primary anti-tumour effect by selectively binding into the minor groove of DNA in a sequence-selective manner. Subsequent studies showed that these compounds were also able to target DNA motifs related to TF signalling.11,12 Hu et al. synthesized a C8-conjugated PBD hybrid named IN6CPBD, and cellular studies revealed that this compound induced cellular apoptosis in A375 cells by repressing activation of NF-κB.13 Similarly, KMR-28-39 (also named ‘TSG-1301’) was shown to interfere with the binding of NF-κB protein to its cognate DNA motifs11,14 (Fig. 1c). This competitive inhibition resulted in nanomolar cytotoxicity in leukaemia cells.14 Further SAR studies showed that compound 13 (Fig. 1d) caused a high level of inhibition of RelA/p65-DNA binding in chronic lymphocytic leukaemia (CLL) cells.12,14 Other molecules, like CRL1101 and IT-901 (Fig. 1e and f), were also reported to block NF-κB subunits RelA/p65 and c-Rel8,15 but none of these compounds have been approved for use as anticancer agents.
 | ||
| Fig. 1 Chemical structure of reported NF-κB targeting compounds (a) (−)-DHMEQ, (b) PBD core, (c) KMR-28-39 (TSG-1301), (d) Cpd 13, (e) CRL1101, and (f) IT-901. | ||
Recently, proteolysis targeting chimeras (PROTACs) have emerged as a promising alternative strategy for selectively interfering with the activity of TFs like NF-κB. Unlike traditional small molecule inhibitors or antibodies, PROTACs recruit the ubiquitin-protease system to break down a specific protein of interest (POI), leading to target protein degradation (TPD). The PROTAC recognises the protein target via its POI ligand, while the E3 ligand located at the other end of the molecule will bring an E3 ligase proximate to the target substrate for consequent POI ubiquitination.16,17 This ubiquitinated protein is subsequently degraded in the proteasome.16,17 Due to their distinct mechanism of action, PROTACs can target non-druggable proteins that lack enzymatic activities, such as TFs, and they do not need to be maintained at high concentrations to induce their therapeutic effect,18–20 which can diminish on-target toxicity concerns.18–20 PROTACs also have the potential to re-sensitise cancer cells to chemotherapy due to their ability to degrade proteins associated with drug resistance.21 Considering these unique therapeutic characteristics, PROTAC technology has the potential to expand the library of drug candidates for clinical purposes. Indeed, PROTACs have already shown remarkable ability to promote anti-cancer activity.18
This project set out to develop PROTACs that can selectively degrade the NF-κB protein RelA/p65. The objective was to deplete this single NF-κB subunit, which is over-expressed in some cancer cells,4,5 whilst preserving the other components of the NF-κB signalling machinery. In so doing, we hoped to selectively target the tumour cells and diminish on-target toxicities in normal cells. Although other researchers have developed NF-κB targeting PROTACs,22 to date there are no reports of PROTACs that are able to selectively degrade a single NF-κB subunit. Although PBDs are well-characterised DNA interacting agents, more recently, C8-conjugated PBDs have been shown to also interact with proteins.23 We used PBD derivatives that were shown by in silico modelling to be able to bind to RelA/p65 (Fig. 2). The PBDs were then conjugated with cereblon (CRBN)-recruiting PROTAC building blocks via multi-step synthetic routes. These synthesised molecules were then biologically screened for their toxicity in cancer cells and non-malignant B- and T-lymphocytes. Subsequently, preliminary investigations were carried out to establish their mechanism of action.
 | ||
| Fig. 2 Docking of PBD-PROTAC with the RelA/p65-p50-DNA complex (PDB:1VKX). (a) Molecular docking of the designed PBD-PROTAC (15d); (b) zoomed in figure showing the 15d binding pose in the RelA/p65-DNA binding interface; (c) overlapped docking results of PBD controls within the RelA/p65-DNA interface; (d) the typical 2D ligand-interface binding projection of PBD ligands. Here fluorine-substituted PBD (JP-193-12) was selected as an example, while the dashed lines highlight the protein–ligand interactions. (e) Chemical structure of the docked PBD building blocks. | ||
Results and discussion
Design and synthesis of RelA/p65-targeting PROTACs
To design NF-κB targeting PROTACs, C8-conjugated PBD molecules were selected as RelA/p65-targeting ligands as these C8-phenyl linked short PBDs had been previously shown to inhibit the activity of the canonical RelA/p65 NF-κB subunit, implying their potential as NF-κB-targeting POI ligands.12 The initial concept was to produce RelA/p65-targeting PROTACs that were linked to CRBN-based building blocks. For a PROTAC to work, it's two functional components must be connected by a linker. One part of the molecule selectively binds the target protein, whilst the second part of the molecule recruits a cellular enzyme, an E3 ligase. This enables the PROTAC to recruit an E3 ligase enzyme to come into close contact with the target protein, enabling the protein to be marked for degradation in the cellular proteasome. Previous research on early-stage PROTAC design used a linear chain of alkyl units as a linker, which allowed the length of the linker to be modified (typically 5–11 carbon chain).17,22,24,25 Here, we selected a 5-carbon aliphatic linker for our preliminary study as molecular modelling suggested that this would allow the CRBN-ligand (lenalidomide) to be bioavailable once the POI had been captured. The designed structure and the docking simulation are shown in Fig. 2 (PBD: 1VKX). It was noted that the PBD moiety was stabilized at the interface between the DNA segment and RelA/p65 NF-κB subunit, while the CRBN ligand domain protruded outside the structure, suggesting that it would remain accessible for CRBN binding. Given the substantial interactions with RelA/p65, we hypothesized that this PBD molecule may have sufficient RelA/p65 binding to serve as a POI ligand.The expanded figure shows that the POI ligand binds in the RelA/p65 domain between Glu193 and Lys218 (Fig. 2b). Further investigation of the short C8-phenyl linked short PBD molecules suggested a similar binding pattern where the PBD core resides inside, and the aniline moiety is relatively solvent-exposed (Fig. 2c and d). To synthesize the molecules, the synthetic scheme of the PROTAC building block was based on the conditions reported by Qiu et al. (Scheme 1a).26 The PBD core was then synthesized as RelA/p65-targeting ligands as shown in Scheme 1b.12,14 PBD-based PROTACs were generated as shown in Scheme 1c. Di-tert-butyl decarbonate was used to install a Boc-protecting group towards the exposed amine of the starting material 9. The product 10 underwent nitro reduction catalysed by Pd–C, while the reduced amine was then attached to the PROTAC building block 2 to generate 12. In the next step, TFA was used to deprotect the Boc group and the product was thus coupled with the PBD core via the EDC/DMAP mediated amide coupling reaction. Finally, the THP and alloc protecting groups were simultaneously removed via pyrrolidine and tetrakis(triphenylphosphine)palladium(0), to obtain the final series 15a–15c. 15d was directly synthesized via the EDC/DMAP mediated amide coupling reaction. For the synthesis of PBD controls 20a–20d, the aromatic-substituted amine was initially conjugated to the PBD core via an amide coupling reaction, and the products were obtained by removing THP and alloc protecting groups (Scheme 2). The cytotoxicity of the synthesised PROTACs was compared with the individual constituent PBD controls.
 | ||
| Scheme 1 Synthesis of (a) lenalidomide-based CRBN building block JP-163-05; (b) PBD core; (c) PBD-based PROTACs.12,14 | ||
 | ||
| Scheme 2 Synthesis of PBD controls. | ||
15d exhibits cytotoxicity in MEC-1 cells
To evaluate the therapeutic potential of the PBD-PROTACs and their constituent PBD building blocks, all the samples were tested in the chronic lymphocytic leukaemia (CLL) cell line, MEC-1. Cells were exposed to PROTACs or their PBD building blocks for 48 h, and drug-induced apoptosis was quantified using annexin V and 7-AAD labelling using flow cytometry. Given the known DNA-binding characteristics of PBDs, we used a FRET-melting assay to compare the ability of the PROTAC molecules and their constituent PBD molecules to interact with DNA. All the compounds were serially diluted (10 μM) and then added to annealed FAM-TAMRA labelled AT-rich single stranded hairpin DNA. DNA Engine Opticom was used to determine the DNA melting characteristics. The sample was initially incubated at 30 °C for 3 h and then gradually increased to 100 °C. The FAM-TAMRA fluorescent signal was detected at 0.5 °C intervals and the mean melting point was determined using GraphPad Prism software. The melting point difference between each compound and naked ssDNA (ΔTm) was calculated for comparison.The screening results are presented in Table 1, and the dose–response curves are attached in Fig. S12.† Compound 15d showed the highest anti-tumour effects in MEC-1 cells. The result of the FRET-melting assay is shown in Fig. 3. 50 equivalents of PBD (10 μM) induced the shift of melting point of the DNA by ΔTm of 1.25 °C, whereas the same equivalence of PROTAC compound 15d showed almost no change in ΔTm, suggesting that these compounds do not interact with DNA. Consequently, the high toxicity of 15d in cancer cell lines and primary CLL cells is unlikely to be attributable to the ability of the PROTAC to bind to DNA. These data suggest a distinct mechanism of action of the PROTAC when compared to its PBD building block.
| PROTAC | MW | LC50 (μM) | ΔTm (°C) | PBD building blocks | MW | LC50 (μM) | ΔTm (°C) |
|---|---|---|---|---|---|---|---|
| 15d (JP-163-16) | 777.88 | 0.14 ± 0.02 | −0.82 | 20d (MMH-165-26) | 422.49 | 0.31 ± 0.14 | 1.25 |
| 15a | 795.87 | 0.81 ± 1.47 | 1.05 | 20a | 440.48 | 0.03 ± 0.29 | 2.17 |
| 15b | 791.91 | 0.84 ± 0.48 | 0.73 | 20b | 436.51 | 1.18 | −0.06 |
| 15c | 807.91 | >1000 | 1.27 | 20c | 452.51 | 1.31 ± 0.74 | 0.70 |
 | ||
| Fig. 3 FRET melting assay results reveal that the PROTAC (15d) does not significantly interact with DNA. Each compound was mixed with an AT-rich DNA sequence at 10 μM. 15d did not increase the melting temperature suggesting that it does not interact with DNA. In contrast, the PBD building block (20d) caused a marked increase in DNA melting temperature, confirming its ability to bind to and stabilise DNA. | ||
15d causes RelA/p65 degradation in a proteasome-dependent manner
To further explore the relative potency and mechanism of action of 15d, the lead PROTAC molecule was tested in MEC-1 cells, primary CLL cells derived from patients (n = 8) and normal B-and T-lymphocytes (n = 5). Cells were exposed to 15d, its PBD building block 20d, or lenalidomide for 48 h and drug-induced apoptosis was quantified using the same assay as described above. 15d and 20d showed potent anti-tumour effects in MEC-1 cells, while lenalidomide had a negligible impact on MEC-1 cell viability (Fig. 4a). Primary CLL cells were also sensitive to the apoptotic effects of 15d. In contrast, normal B- and T-lymphocytes were more than two logs less sensitive to the effects of the PROTAC (Fig. 4b). In parallel experiments, RelA/p65 expression was measured in MEC-1 cells treated with 15d or 20d. Fig. 4c–e show that both agents induced a marked reduction in RelA/p65 expression. However, the dose–response patterns of the two molecules were different. 20d induced a dose-dependent reduction in RelA/p65 expression. In contrast, 15d showed a similar reduction in RelA/p65 with all the concentrations tested. This supports the concept that 15d may have catalytic properties, which enable the recycling of PROTAC molecules after the target protein is degraded. In contrast, the PBD, 20d, demonstrated an occupancy-driven mechanism of action; a more obvious concentration-dependent reduction in RelA/p65 was observed in MEC-1 cells treated with 20d. Given its known DNA-binding activity, it seems likely that dose-dependent minor groove binding contributes to the cytotoxicity of 20d, which could, in turn, result in higher levels of competitive blockade of RelA/p65 DNA binding sites.12,27 In contrast, the PROTAC molecule, 15d, appeared to induce an event-driven pharmacology consistent with POI degradation. Interestingly, although the PROTAC induced similar levels of RelA/p65 reduction at both 0.25 μM and 1 μM, the higher concentration of 15d induced a stronger anti-tumour effect. This unique pattern could be caused by the ‘hook effect’ frequently observed with PROTAC compounds.20,27,28 | ||
| Fig. 4 (a) Overlaid dose–response curves for 15d, 20d, and lenalidomide in the MEC-1 cell line. Dose–response curves were generated using annexin V/7-AAD data following 48 h of exposure to each compound. The LD50 values were interpolated from each individual dose–response curve using GraphPad Prism 10, with all experiments performed in triplicate. (b) Shows the relative cytotoxic effect of 15d in MEC-1 cells, primary CLL cells and normal B- and T-lymphocytes. Normal lymphocytes were more than two logs less sensitive to the effects of 15d when compared with malignant B cells. RelA/p65 expression was significantly reduced in MEC-1 cells treated for 24 h with (c) 15d and (d) 20d. (e) In contrast to 20d, 15d did not show a dose-dependent reduction in RelA/p65, which suggests an event-driven mechanism of action consistent with other PROTACs. Furthermore, co-treatment with the proteasome inhibitor, MG-132, demonstrated a proteasome-dependent mechanism of action. (f) The cytotoxic and proteasome inhibitory effects of MG-132 were evaluated in MEC-1 cells and a concentration of 0.18 μM was chosen for the subsequent combination studies. (g) 15d shows a proteasome dependent mechanism of action. (h) In contrast, the PBD building block, 20d, showed a proteasome-independent mechanism of action. (i) The cytotoxic effects of 15d were shown to be dependent on CRBN expression by the five-fold reduction in potency in the CRBN deficient myeloma cell line, RPMI-8226. (j) In contrast, the PBD building block, 20d, showed similar potency in RPMI-8226 cells. All LC50 values were interpolated from individual dose–response curves using GraphPad Prism 10. Results are shown as the mean of five independent experiments carried out in duplicate. Statistical significance was determined using paired t-tests * p < 0.05. | ||
To confirm the proteasome dependency on the cytotoxic effects of 15d in MEC-1 cells, cells were co-treated with the proteasome inhibitor MG-132. We initially established the cytotoxic and proteasome inhibitory effects of MG-132 in MEC-1 cells (Fig. 4f). MEC-1 cells were treated with increasing concentrations of the proteasome inhibitor, MG-132 (0.1–2.5 μM) for 48 h. Aliquots of cells were first assessed for their apoptotic response to MG-132 using annexin V and 7-AAD labelling. In parallel, proteasome activity was evaluated using a proteasome activity assay kit (Abcam). The kit uses an AMC-tagged peptide substrate (proteasome substrate (Succ-LLVY-AMC in DMSO), which releases free, highly fluorescent AMC (Ex/Em 350/440 nm) in the presence of proteolytic activity. Due to the high sensitivity of MEC-1 cells to the effects of MG-132, a concentration of 0.18 μM MG-132 was used in combination with 15d or 20d.
This concentration of MG-132 reduced cellular proteasome activity by approximately 40–50% without inducing significant apoptosis. Each experiment was repeated five times in duplicate, and the mean LC50 values were calculated for each experiment. Subsequently the matched LC50 values (± the addition of MG-132) were plotted and the difference between the LC50 values was determined using the paired t-test (Fig. 4g and h). The results indicate that blocking the proteasome significantly repressed the cytotoxicity of 15d (p < 0.05), while there was no significant difference in LC50 values when MG-132 was co-administered with the PBD 20d. This provided further evidence that the anti-tumour effect of 15d is caused, at least in part, by proteasome-dependent degradation of RelA/p65. However, it should be noted that 15d was not able to entirely deplete the cellular expression of this molecule. Although molecular modelling showed excellent binding characteristics of the PBD PROTAC with RelA/p65, it may not result in the optimal degradation of RelA/p65.12 The scale of target protein degradation is reliant on the stability and positive cooperativity of the ternary complex, so a ligand bearing an inferior binding affinity could still induce effective protein degradation with considerable selectivity.19,20 Issues like charged repulsion between the E3 ligase and POI or steric clashes induced by unfavourable PROTAC conformation are more likely to be the cause of the incomplete RelA/p65 degradation.7,18,19 Although we did not directly explore ternary complex formation i.e., the binding of the PROTAC to both the protein of interest (RelA/p65) and the target E3 ligase (CRBN), we did examine the cytotoxic effects of 15d in the multiple myeloma cell line, RPMI-8226. These cells have very low CRBN protein expression and are consequently resistant to lenalidomide.29 Using these cells we were able to confirm that the mechanism of action of 15d is, at least in part, dependent on CRBN; the PROTAC showed a five-fold reduction in efficacy in RPMI-8226 cells (Fig. 4i). In contrast, the PBD building block 20d showed similar cytotoxicity in RPMI-8226 cells and MEC-1 cells (Fig. 4j) confirming that its cytotoxic effects were independent of CRBN.
Previous research indicated that PBD molecules fused with benzofuran and pyrrole terminal were selective for RelA/p65 inhibition. In contrast, benzene-fused short PBDs caused very limited RelA/p65 perturbance, but with significant effects on other NF-κB subunits.12 Consequently, the effect of 15d was evaluated on other NF-κB subunits (RelB and cRel) as described below.
15d selectively degrades the NF-κB RelA/p65 subunit
To ascertain the selectivity of 15d for the degradation of the RelA/p65 NF-κB subunit, MEC-1 cells were treated for 24 hours with a range of concentrations of 15d (0–1 μM). Cells were then harvested, fixed and permeabilised and labelled with fluorescence-labelled antibodies against the NF-κB subunits p65 (APC), RelB (Corallite 488) and cRel (PE). Protein expression was quantified using a CytoFLEX LX flow cytometer. Fig. 5 shows that 15d induced a marked reduction in RelA/p65 expression at 0.5 μM and 1 μM. In contrast, no significant change in RelB was observed at the same concentrations. Although a small but significant reduction in cRel was noted at 0.5 μM, this was not replicated at 1 μM. This suggests that 15d selectivity depletes RelA/p65, which adds to its promising characteristics as a lead PROTAC compound. To our knowledge, 15d represents the first example of a RelA/p65 selective PROTAC that does not substantially impact RelB or cRel.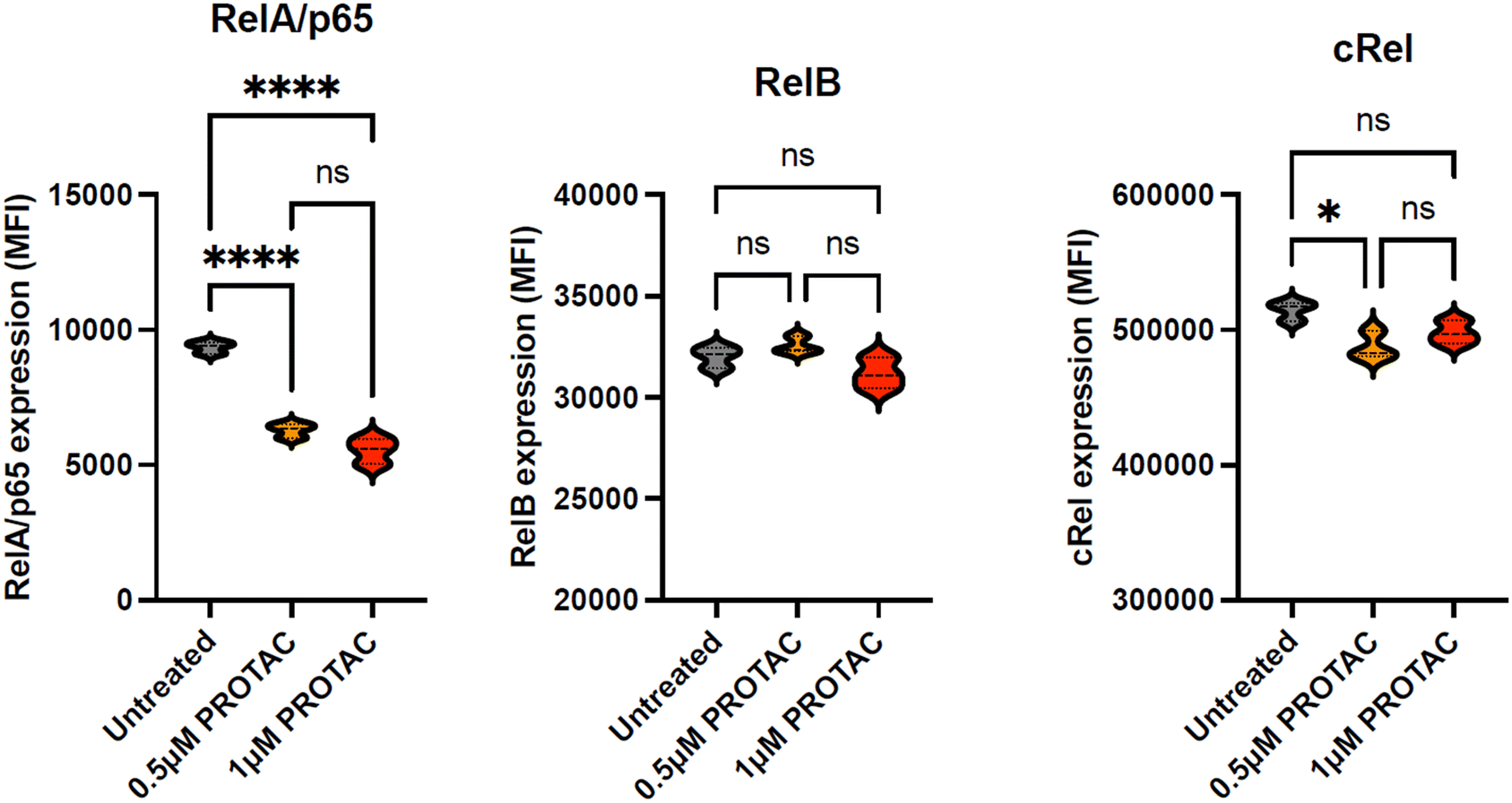 | ||
| Fig. 5 Comparison the effect of 15d (PROTAC) on the expression of three NF-κB subunits RelA/p65, RelB and cRel. Each subunit was quantified using fluorescence-labelled antibodies; all experiments were performed three times in duplicate and data are presented as violin plots. Statistical significance was determined using the Kruskal–Wallis test with Dunn's multiple comparison post hoc correction. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. | ||
Effects of PROTAC 15d on MDA-MB-231 cell viability
We next investigated the potency of 15d in the triple-negative breast cancer cell line (MDA-MB-231). This cell line was selected as it over expresses RelA/p65 and represents a cancer type with significant clinical unmet need.30,31 As shown in Fig. 6a, 15d was more potent than its constituent molecules, the POI targeting PBD, 20d and the CRBN E3 ligase ligand, lenalidomide. The cells were treated with serial dilutions of each compound and then incubated for 48 hours. FITC annexin V and 7-AAD labelling was used to evaluate cell viability using flow cytometry. In keeping with our findings in MEC-1 and primary CLL cells, 15d and MMH-165-26 both demonstrated high tumour suppressive effects in cancer cells with LC50 values in the low micromolar range, and 15d was significantly more potent than its PBD building block, 20d (Fig. 6c). The CRBN ligand, lenalidomide, showed low cytotoxicity even at the highest concentration tested. | ||
| Fig. 6 Evaluation of 15d in MDA-MB-231 cells. (a) Dose–response curves for 15d, 20d, and lenalidomide in MDA-MB-231 cells. Cells were treated with a range of concentrations of the PROTAC (15d) and the individual constituent molecules (20d and lenalidomide). Dose–response curves were generated using annexin V/7-AAD data following 48 h of exposure to each compound. The LC50 values were interpolated from each individual dose–response curve using GraphPad Prism 10. All experiments were performed in triplicate. (b) Shows an example of the gating strategy used to identify viable and apoptotic MDA-MB-231 cells. The percentage of viable cells was defined by cells being annexin-V and 7AAD negative. (c) The mean LC50 values (+SD) for 15d and 20d are shown for three independent experiments carried out in triplicate. 15d was significantly more cytotoxic than 20d, ** p < 0.001. | ||
15d promotes the degradation of RelA/p65 in MDA-MB-231 cells in a proteasome-dependent manner
Next, to confirm the PROTAC mechanism of action of 15d, MDA-MB-231 cells were co-treated with the proteasome inhibitor, MG-132, to evaluate whether this altered the tumour suppressive effects.32 MG-132 was much less cytotoxic in MDA-MB-231 cells when compared with MEC-1 cells, and again a concentration of MG-132 was selected that did not have significant cytotoxicity but inhibited proteasome activity by approximately 50% (Fig. 7a).33 Analysis of the cytotoxic dose–response curves and the impact of MG-132 on proteasomal activity in MDA-MB-231 cells revealed that 1 μM MG-132 caused >50% reduction in proteasome activity in MDA-MB-231 cells without inducing a significant reduction in cell viability when compared with the untreated controls (Fig. 7a and b). Based on the evaluation of the effects of MG-132 in MDA-MB-231 cells, cells were then treated with 15d, 20d and lenalidomide, with and without the addition of 1 μM MG-132. | ||
| Fig. 7 The effects of the proteasome inhibitor, MG-132, in the MDA-MB-231 cell line. (a) MG-132 induced dose-dependent cytotoxicity which was (b) associated with a dose-inhibition of proteasomal activity. A dose of 1 μM MG-132 did cause a significant increase in cytotoxicity but induced a >50% reduction in proteasome activity. All experiments were performed in triplicate and statistical significance was determined using the Kruskal–Wallis test with Dunn's multiple comparison post hoc. ** p < 0.01, *** p < 0.001, **** p < 0.0001. (c) Overlaid dose–response curves of MDA-MB-231 cells treated with 15d with and without the addition of the proteasome inhibitor, MG-132. (d) 20d and (e) lenalidomide. The cytotoxic effect of 15d was significantly reduced by co-treatment with 1 μM MG-132. This was not the case for 20d or lenalidomide. All experiments were performed in triplicate and statistical significance was determined using the Kruskal–Wallis test with Dunn's multiple comparison post hoc correction. *** p < 0.001. (f) 15d mediated depletion of RelA/p65 expression in MDA-MB-231 cells was dependent on proteasome activity. Experiments were performed in triplicate and statistical significance was determined using the Kruskal–Wallis test with Dunn's multiple comparison post hoc correction. ** p < 0.01, *** p < 0.001. | ||
Co-treatment with MG-132 significantly reduced the cytotoxic effect of 15d, which indicated a proteasome-dependent mechanism of action (Fig. 7c). In contrast, MG-132 did not significantly alter the cytotoxicity of 20d, implying again that the mechanism of action of this PBD compound was independent of proteasomal function (Fig. 7d). In the case of lenalidomide, blocking proteasome activity increased its cytotoxicity, but this was not statistically significant (Fig. 7e).33 This result indicated that the mechanism of the PBD was distinct from that of 15d as it was not dependent on proteasomal activity. As a C8-linked short PBD, 20d can bind into the minor groove of DNA and form a covalent bond with guanine molecules. This may contribute to its effect on NF-κB as it facilitates sequence-selective binding at promoter regions containing the guanine-rich NF-κB binding motifs, thereby disrupting NF-κB signal transduction.11,12,34 In this case, 20d may retain its anti-tumour activity after the blockade of proteasome function.
As for the E3 ligase ligand, lenalidomide, used to develop 15d, it did not significantly induce cell death as a single agent, but co-administration of MG-132 mildly improved tumour suppression. This could be caused by the additive impact of combination treatment as lenalidomide promotes immune cell activation that might slightly improve tumour sensitivity to other agents bearing distinct mechanisms, like MG-132.35–37 Subsequently, the ability of 15d to reduce RelA/p65 expression in MDA-MB-231 cells was evaluated using flow cytometry. Cells were treated with increased concentration of 15d for 24 h, and then they were harvested and permeabilised followed by labelling with an APC-labelled RelA/p65 antibody. A significant reduction in RelA/p65 expression was observed after treating with 0.5 μM and 1 μM 15d, which when co-administrated with 1 μM MG-132 reversed this effect, consistent with a proteasome-dependent mechanism of action (Fig. 4f). It was noted that 0.5 μM administration of 15d caused a similar depletion of RelA/p65 to that achieved with 1 μM. Due to the bifunctional characteristic of PROTAC molecules, high intracellular levels of PROTAC may lead to saturated binding of its relative binary complexes, which competitively restricts the formation of the effective ternary complex required for target protein degradation.20,27,28,38 It is possible that 15d saturated the binding sites of RelA/p65 and/or CRBN in MDA-MB-231 cells at 1 μM, which may restrict the formation of the POI–PROTAC–E3 complex thereby limiting the capacity for RelA/p65 degradation.20,28 In summary, 15d induced cytotoxicity in MDA-MB-231 cells and promoted the degradation of RelA/p65 in a proteasome-dependent manner.
Conclusions
Targeting the NF-κB signalling pathway has long been an appealing therapeutic strategy due to its role in regulating a range of cellular processes. The aberrant protein expression of one or more NF-κB subunits often results in increased NF-κB signalling, which is associated with pathogenic effects such as tumour proliferation, angiogenesis, and drug resistance. Here, we report the first prototype PROTAC capable of selectively degrading the NF-κB subunit RelA/p65. The POI targeting ligand was a C8-linked short PBD; simulated docking experiments showed strong binding to RelA/p65 protein in a region where the protein was predicted to interact with NF-κB DNA motifs. This supported its use as a selective ligand for targeting RelA/p65 protein if incorporated into a bifunctional PROTAC molecule. Consequently, PBD molecules were conjugated with a lenalidomide-based building block via an amide coupling reaction. A series of PBD PROTACs were synthesized, and all final products were purified either by flash column chromatography or preparative chromatography. Biological screening indicated that the lead compound, 15d, showed potency in the TNBC breast cancer cell line, MDA-MB-231 (LC50 = 2.9 μM), CLL cell line MEC-1 (LC50 = 0.14 μM) and primary CLL B cells derived from eight patients (LC50 = 0.23 μM). In all cases, this was associated with the selective depletion of RelA/p65 in a proteasome-dependent manner. It is noteworthy that the cytotoxicity of 15d was two logs lower in non-malignant B- and T-lymphocytes derived from healthy volunteers and was five-fold lower in RPMI-8226 cells, which possess very low levels of CRBN protein expression.The proteasome-recruitment mechanism of 15d was confirmed by the reversal of RelA/p65 depletion when cells were co-treated with the proteasome inhibitor, MG-132. Furthermore, a FRET-melting assay confirmed that this compound did not interact with DNA, which was in contrast with the strong DNA interaction shown by its constituent PBD, 20d. Hence, the cytotoxicity of 15d appeared to be predominately driven by proteasome-dependent RelA/p65 degradation. Further screening of the small library and comparison with simulated modelling results implied that PROTAC potency may be partially related to the binding affinity of the POI ligand, while it was also reliant on ternary complex formation as demonstrated by the reduced toxicity and RelA/p65 degradation in the presence of MG-132 and reduced cytotoxicity in RPMI-8226 cells which have low cereblon expression. It is worth noting that 15d was not able to abolish RelA/p65 expression, which suggests that further PROTAC optimisation may be possible. Despite the incomplete target degradation observed in our studies, the work presented here demonstrates, for the first time, that it is possible to produce a PROTAC with the ability to preferentially degrade a single NF-κB subunit, RelA/p65. As such, this may represent an important step towards unlocking the potential of NF-κB as a therapeutic target. In particular, the generation of a PROTAC with the ability to selectively degrade RelA/p65 may open the door to more effective and better tolerated treatments for human pathologies that are associated with RelA/p65 overexpression, including a range of cancers and autoimmune disorders.
Experimental section
Molecular docking
The protein data file was accessed from the PBD data bank (p65 protein data: 1VKX); all compound ligands were prepared and generated via either Chem 3D or Avogadro. All the compounds were calculated and performed with energy minimization via MMFF94. For p65-targeting ligands, molecular docking was performed by the Vina molecular docking programme using VEGA-ZZ modelling software. The parameters were set as X: −11.3, Y: 40.4, Z: 76.7; box: 30, 30, 30; exhaustiveness: 20; binding mode: 16. All docking results were visualised via Discovery Studio. The result was visualised via PyMOL.Chemistry materials and methods
All synthetic chemicals, building blocks, and solvents were purchased from Fluorochem, Sigma-Aldrich, and Thermo-Fisher Scientific. All reactions monitored via thin-layer chromatography (TLC) were performed by using Supelco TLC silica gel 60 F254 aluminium plates. The TLC plates were visualised using a UV lamp at 254 nm. Purification through flash column chromatography was performed in a glass column with silica gel as the stationary phase (230–400 mesh, 60 Å). Preparative HPLC was also used for purifying some products. An Agilent 1260 Preparative LC system was applied using H2O (solvent A) and acetonitrile (solvent B) as the mobile phase with a Monolithic C18 50 × 4.6 mm LC column (Phenomenex) as the stationary phase. Methods A, B, C, and D were used for purification (flow rate: 20 ml min−1). Formic acid was added (0.1%) into both solvent A and B to maintain an acidic mobile phase condition.Method A
The gradient was initially kept at 60% solvent A and 40% solvent B, while it was ramped up to 60% B over 2 min. Solvent B was then increased to 70% over 2 min, which was further ramped up to 80% over 2.5 min. The solvent reached 90% B over 0.5 min and kept for 2.5 min, which was then returned to 40% B over 1.5 min.Method B
The gradient was initially started from 90% A and 10% B and kept for 1 min, and then ramped up to 20% B over 1.5 min. B was then increased to 30% over 2 min, followed by ramping up to 40% B over 2 min. Subsequently, B was raised to 70% within 0.5 min, and then B was increased to 85% over 1 min, and then ramped up to 90% over 1 min. The solvent was finally returned to 10% B over 1 min.Method C
The gradient was initially started from 90% A with 10% B that is kept for 1 min, and solvent B was increased to 30% over 1.5 min. B was subsequently increased to 50% over 3.5 min, and then raised to 90% over 2 min and kept for 1 min. The gradient was finally reduced to 10% B over 1 min.Method D
The gradient was initially started from 90% A with 10% B that is kept for 1 min, and solvent B was increased to 50% over 5 min. The gradient was subsequently raised to 90% B over 2 min, and it was kept for 1 min. Solvent B was finally returned to 10% over 1 min.High-performance liquid chromatography-tandem mass spectrometry (LCMS) was applied for monitoring reaction and characterizing products. The product analysis was carried out using an Agilent 1260 separating system using H2O (solvent A) and acetonitrile (solvent B) as the mobile phase, while a monolithic C18 50 × 4.6 mm LC column (Phenomenex) worked as the stationary phase. Method E (10 min) and method F (5 min) were used for analysis (flow rate: 0.5 mL min−1; inject volume: 200 μL), while samples were split and passed through an Agilent 6120 quadrupole mass spectrometer. Formic acid was added (0.1%) into both solvent A and B to maintain an acid mobile phase condition.
Method E (10 min run)
Solvent A (95%) with solvent B (5%) was maintained for 2 min, and then ramped up to 50% solvent B in 3 min. The gradient was retained for 1 min and then solvent B was increased to 95% in 1.5 min. Solvent B was finally returned to 5% in 1.5 min and maintained for 1 min.Method F (5 min run)
Solvent A (95%) with solvent B (5%) was ramped up to 90% in 3 min, while solvent B was then ramped up to 95% within 0.5 min. The solvent gradient was kept for 1 min, and then solvent B was reduced to 5% within 0.5 min.Cell culture
CLL cells, the CLL cell line, MEC-1, the breast cancer cell line, MDA-MB-231 and the lenalidomide resistant myeloma cell line, RPMI-8226 were selected for testing the cytotoxicity of the PROTAC molecules. Primary CLL cells were collected from patients and normal B- and T-lymphocytes were obtained from healthy volunteers. All experiments were performed in accordance with the U.K. Human Tissue Authority guidelines, and experiments were approved by the local research ethics committee (17/SW/0263). Informed consent was obtained from all human participants in this study. Cell lines were acquired from DSMZ (MEC-1 and RPMI-8226) or ATCC (MDA-MB-231); cells were maintained in RPMI 1640 media (primary CLL cells, MEC-1 and RPMI-8226) or DMEM media (MDA-MB-231) with the addition of 10% FBS, 1% L-glutamine, and 1% penicillin streptomycin. The cells were seeded 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per mL at 37 °C in a humidified atmosphere containing 5% CO2. The cells were split every 48 hours; cell count and viability were measured each time.
000 cells per mL at 37 °C in a humidified atmosphere containing 5% CO2. The cells were split every 48 hours; cell count and viability were measured each time.
Cell counting
10 μL of cell suspension was mixed with 10 μL of trypan blue. Subsequently, 10 μL of the mixture was pipetted into a cell counting slide. The slide was inserted into a Countess 3 cell counter (ThermoFisher Scientific) to quantify the cell count and cell viability.Apoptotic assay for RelA/p65-targeting PROTACs
500000 cells per well were aliquoted into 24-well plates following resuspension with 1 mL of appropriate medium. All test compounds were dissolved in DMSO as 1 mM stock solution. Subsequently, working stocks of the PROTACs and their individual constituent molecules were produced by serial dilutions in a 96-well plate: RelA/p65-targeting PROTACs (1 μM, 0.5 μM and 0.25 μM, 0.125 μM, and 0.0625 μM). 10 μL of each dilution was then transferred to the cell suspensions in the 24-well plate. The plates were then incubated for 48 hours at 37 °C with 5% CO2. Samples were then harvested into 1.5 mL Eppendorf tubes and centrifuged at 500 × g for 5 min. The supernatant was then poured off and the cell pellets resuspended in 96 μL annexin V binding buffer and 4 μL FITC annexin V (both Biolegend) was added to each tube. The tubes were then incubated in the dark for 10 min prior to the addition of 4 μL 7-AAD. Finally, the cells were analysed using a CytoFLEX LX (Beckman Coulter) flow cytometer. In all cases, 10000 events were recorded. Apoptosis was quantified using CytExpert software, while the percentage of apoptotic cells was defined as annexin-V positive and 7-AAD positive, or annexin-V positive and 7-AAD negative. The data was further analysed using GraphPad prism to calculate the LC50 values for each compound using non-linear regression analysis.
Evaluation of NF-κB subunit expression following treatment with PROTAC
Aliquots of 0.5 × 106 MEC-1 cells were treated with increasing concentrations of 15d for 24 h. Cells were harvested by centrifugation, fixed using Cyto-Fast™ fix/perm buffer set (Biolegend) for 20 min at 37 °C. Cells were then washed in Cyto-Fast™ Perm Wash solution and centrifuged at 300 × g for 5 min before being resuspended in 100 μL Perm Wash solution followed by the addition of 5 μL APC-labelled RelA/p65 antibody (Biolegend), 5 μL PE-labelled cRel antibody (eBiosciences) and 5 μL corallite 488-labelled RelB antibody (ThermoFisher). Cells were incubated for 20 min prior to washing in cell staining buffer, centrifugation at 300 × g for 5 min and resuspension in 100 μL cell staining buffer prior to acquisition of the data on a CytoFLEX LX flow cytometer.MG-132 proteasome inhibition assay
To determine whether the toxicity of 15d was dependent on proteasome activity, MDA-MB-231 cells were treated with increasing concentration of the proteasome inhibitor, MG-132 (0.1–10 μM) for 48 h. Aliquots of cells were first assessed for their apoptotic response to MG-132 using annexin V and 7-AAD labelling (as described above). In parallel, proteasome activity was assessed using a proteasome activity assay kit (Abcam). The kit uses an AMC-tagged peptide substrate (proteasome substrate (Succ-LLVY-AMC in DMSO), which releases free, highly fluorescent AMC (Ex/Em 350/440 nm) in the presence of proteolytic activity. MEC-1 cells were very sensitive to the cytotoxic effects of MG-132. So, in this cell line, combination studies were carried out with 0.18 μM MG-132. Treatment of MEC-1 cells with 0.18 μM MG-132 caused >40% reduction in proteasome activity without significant effects on cellular viability. Subsequently, 10 μL MG-132 stock was added to the 500000 cells per mL MEC-1 cell with increasing concentration of 15d or the PBD building block, 20d. All treated cells were cultured for 48 h at 37 °C with 5% CO2. The cells were then harvested by centrifugation (300 × g for 5 min) and then incubated with annexin V and 7-AAD, prior to analysis by flow cytometry (as described above.).
FRET melting assay
The single-strand oligonucleotide FRET hairpin was purchased from Eurogentec Ltd, tagged with TAM at 5′ and TAMRA at 3′ terminal (sequence: 5′-FAM-TAT-AAG-ATA-TAT-ATA-TTT-TTT-TAT-ATA-TAT-CTT-ATA-TAMRA-3′). Nuclease-free water was added to prepare 20 μM ssDNA stock solution, and it was further diluted to 400 nM using 50 mM K cacodylate buffer (pH = 7.4). The prepared ssDNA sample was annealed at 85 °C for 5 min and then allowed to cool down to room temperature and then stored at −20 °C for completing the annealing process. PBD controls and PROTACs were prepared as 20 μM working solutions diluted with 50 mM K cacodylate buffer (pH = 7.4). 25 μL compound working solution was added to 25 μl DNA stock in the well of the Bio-Rad 96-well plate. DNA Engine Opticom was used for melting. The sample was initially incubated at 30 °C for 3 h and then the temperature was gradually increased to 100 °C. The fluorescence signal was detected at intervals of 0.5 °C. The mean of the melting point was analysed via GraphPad prism, and the melting point difference between the sample and naked ssDNA (ΔTm) was calculated for comparison.Data analysis
All biological data was calculated and plotted in GraphPad Prism. The standard deviations were presented as error bars in the plotted graph. The sigmoid dose–response curves were plotted using non-linear regression (4 parameters) to obtain LC50 values (the concentration of drug required to kill 50% of the cells in culture). As for significance testing, the mean values in the two groups were measured and compared. The data were initially subjected to normality testing using the Shapiro–Wilk and Kolmogorov–Smirnov test. If the data passed the normality test, it was then further evaluated using a paired t-test to assess whether there was a significance in the data before and after treatment. It was considered significantly different when the p value of the testing groups <0.05 with 95% confidence interval. If the data failed the normality test, they were subsequently evaluated using the Kruskal–Wallis test with Dunn's multiple comparison post hoc correction, if more than one set of pairs were analysed.Abbreviations
| 7-AAD | 7-Aminoactinomycin D |
| BAIB | Bis(acetoxy)iodobenzene |
| CRBN | Cereblon |
| DCM | Dichloromethane |
| dd | Double of doublets |
| DHP | Dihydropyran |
| DMAP | 4-Dimethylaminopyridine |
| DMF | Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| EA | Ethyl acetate |
| EDC | 1-Ethyl-3-(3-dimethyl aminopropyl)carbodiimide |
| FBS | Fetal bovine serum |
| FITC | Fluorescein isothiocyanate |
| FRET | Fluorescence resonance energy transfer |
| G | Guanine |
| HPLC | High performance liquid chromatography |
| HRMS | High resolution mass spectrometry |
| ImiDs | Immunomodulatory drugs |
| LC50 | Concentration of the toxic substance lethal to half of test cells |
| LC-MS | Liquid chromatography-mass spectrometry |
| Lys | Lysine |
| m | multiplet |
| M | Molar |
| NF-κB | Nuclear transcription kappa B |
| nM | Nanomolar |
| NMP | N-Methyl-2-pyrrolidone |
| NMR | Nuclear magnetic resonance |
| PBD | Pyrrolobenzodiazepine |
| PBS | Phosphate-buffered saline |
| PROTAC | Proteolysis targeting chimera |
| q | Quartet |
| RPMI | Roswell Park Memorial Institute |
| s | Singlet |
| SAR | Structure–activity relationships |
| ssDNA | Single-stranded DNA |
| T | Thymine |
| t | Triplet |
| TAMRA | Carboxytetramethylrhodamine |
| TEMPO | (2,2,6,6-Tetramethylpiperidin-1-yl)oxyl |
| TF | Transcription factor |
| TFA | Trifluoroacetic acid |
| THF | Tetrahydrofuran |
| TLC | Thin-layer chromatography |
| TPD | Targeted protein degradation |
| UV | Ultraviolet |
| ΔTm | Variation of the melting temperature |
| m/z | Mass-to-charge ratio |
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
K. M. R. and C. P. contributed equally. P. J., M. M. H., A. G. S. P., S. M., K. M. R., and C. P. designed the experiments and analysed the data. P. J., and M. M. H. designed, synthesized, and analysed the compounds. P. J., A. G. S. P., and C. P. performed the biological experiments. P. J., M. M. H., and K. M. R. performed the docking studies. P. J., K. M. R., and C. P. wrote the manuscript, with edits from M. M. H., A. G. S. P., and S. M.Conflicts of interest
The authors have no relevant conflicts of interest.Acknowledgements
This research was funded in part by a self-funded PhD studentship (P. J.) and by personal research funding held by C. P. The chemistry work was performed at King's College London, while the biology evaluations were performed at Brighton and Sussex Medical School, University of Sussex. 20d (MMH-165-26) and 3 (MMH-165-31) were obtained from Md. Mahbub Hassan.References
- T. Liu, L. Zhang, D. Joo and S.-C. Sun, NF-κB Signaling in Inflammation, Signal Transduction Targeted Ther., 2017, 2(1), 17023 CrossRef PubMed.
- K. Taniguchi and M. Karin, NF-KB, Inflammation, Immunity and Cancer: Coming of Age, Nat. Rev. Immunol., 2018, 18(5), 309–324 Search PubMed.
- C. L. Duran, G. S. Karagiannis, X. Chen, V. P. Sharma, D. Entenberg, J. S. Condeelis and M. H. Oktay, Cooperative NF-KB and Notch1 Signaling Promotes Macrophage-Mediated MenaINV Expression in Breast Cancer, Breast Cancer Res., 2023, 25(1), 37 CrossRef CAS PubMed.
- S. Hewamana, S. Alghazal, T. T. Lin, M. Clement, C. Jenkins, M. L. Guzman, C. T. Jordan, S. Neelakantan, P. A. Crooks, A. K. Burnett, G. Pratt, C. Fegan, C. Rowntree, P. Brennan and C. Pepper, The NF-KB Subunit Rel A Is Associated with in Vitro Survival and Clinical Disease Progression in Chronic Lymphocytic Leukemia and Represents a Promising Therapeutic Target, Blood, 2008, 111(9), 4681–4689 CrossRef CAS PubMed.
- D. Verzella, A. Pescatore, D. Capece, D. Vecchiotti, M. V. Ursini, G. Franzoso, E. Alesse and F. Zazzeroni, Life, Death, and Autophagy in Cancer: NF-KB Turns up Everywhere, Cell Death Dis., 2020, 11(3), 210 CrossRef PubMed.
- A. S. Bhagwat and C. R. Vakoc, Targeting Transcription Factors in Cancer, Trends Cancer, 2015, 1(1), 53–65 CrossRef PubMed.
- K. Li and C. M. Crews, PROTACs: Past, Present and Future, Chem. Soc. Rev., 2022, 51(12), 5214–5236 Search PubMed.
- J. Zhuang, Q. Liu, D. Wu and L. Tie, Current Strategies and Progress for Targeting the “Undruggable” Transcription Factors, Acta Pharmacol. Sin., 2022, 43(10), 2474–2481 Search PubMed.
- K. Horie, J. Ma and K. Umezawa, Inhibition of Canonical NF-KB Nuclear Localization by (−)-DHMEQ via Impairment of DNA Binding, Oncol. Res., 2015, 22(2), 105–115 CrossRef PubMed.
- Y. Shono, A. Z. Tuckett, S. Ouk, H.-C. Liou, G. Altan-Bonnet, J. J. Tsai, J. E. Oyler, O. M. Smith, M. L. West, N. V. Singer, E. Doubrovina, D. Pankov, C. V. Undhad, G. F. Murphy, C. Lezcano, C. Liu, R. J. O'Reilly, M. R. M. van den Brink and J. L. Zakrzewski, A Small-Molecule c-Rel Inhibitor Reduces Alloactivation of T Cells without Compromising Antitumor Activity, Cancer Discovery, 2014, 4(5), 578–591 Search PubMed.
- D. E. Thurston and I. Pysz, Chemistry and Pharmacology of Anticancer Drugs, CRC Press, Boca Raton, 2nd edn, 2021 Search PubMed.
- D. B. Corcoran, T. Lewis, K. S. Nahar, S. Jamshidi, C. Fegan, C. Pepper, D. E. Thurston and K. M. Rahman, Effects of Systematic Shortening of Noncovalent C8 Side Chain on the Cytotoxicity and NF-KB Inhibitory Capacity of Pyrrolobenzodiazepines (PBDs), J. Med. Chem., 2019, 62(4), 2127–2139 CrossRef CAS PubMed.
- W.-P. Hu, F.-Y. Tsai, H.-S. Yu, P.-J. Sung, L.-S. Chang and J.-J. Wang, Induction of Apoptosis by DC-81-Indole Conjugate Agent Through NF-KB and JNK/AP-1 Pathway, Chem. Res. Toxicol., 2008, 21(7), 1330–1336 Search PubMed.
- K. M. Rahman, P. J. M. Jackson, C. H. James, B. P. Basu, J. A. Hartley, M. de la Fuente, A. Schatzlein, M. Robson, R. B. Pedley, C. Pepper, K. R. Fox, P. W. Howard and D. E. Thurston, GC-Targeted C8-Linked Pyrrolobenzodiazepine–Biaryl Conjugates with Femtomolar in Vitro Cytotoxicity and in Vivo Antitumor Activity in Mouse Models, J. Med. Chem., 2013, 56(7), 2911–2935 CrossRef CAS PubMed.
- H. Kanzaki, A. Chatterjee, H. Hossein, X. Zhang, S. Chung, N. Deng, V. K. Ramanujan, X. Cui, M. I. Greene and R. Murali, Disabling the Nuclear Translocalization of RelA/NF-KB by a Small Molecule Inhibits Triple-Negative Breast Cancer Growth, Breast Cancer: Targets Ther., 2021, 13, 419–430 Search PubMed.
- Y. Zou, D. Ma and Y. Wang, The PROTAC Technology in Drug Development, Cell Biochem. Funct., 2019, 37(1), 21–30 CrossRef CAS PubMed.
- S.-L. Paiva and C. M. Crews, Targeted Protein Degradation: Elements of PROTAC Design, Curr. Opin. Chem. Biol., 2019, 50, 111–119 CrossRef CAS PubMed.
- M. Pettersson and C. M. Crews, PROteolysis TArgeting Chimeras (PROTACs) — Past, Present and Future, Drug Discovery Today: Technol., 2019, 31, 15–27 Search PubMed.
- D. P. Bondeson, B. E. Smith, G. M. Burslem, A. D. Buhimschi, J. Hines, S. Jaime-Figueroa, J. Wang, B. D. Hamman, A. Ishchenko and C. M. Crews, Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead, Cell Chem. Biol., 2018, 25(1), 78–87 CrossRef CAS PubMed.
- R. G. Guenette, S. W. Yang, J. Min, B. Pei and P. R. Potts, Target and Tissue Selectivity of PROTAC Degraders, Chem. Soc. Rev., 2022, 51(14), 5740–5756 RSC.
- J. Mannion, V. Gifford, B. Bellenie, W. Fernando, L. Ramos Garcia, R. Wilson, S. W. John, S. Udainiya, E. C. Patin, C. Tiu, A. Smith, M. Goicoechea, A. Craxton, N. Moraes de Vasconcelos, N. Guppy, K. M. J. Cheung, N. J. Cundy, O. Pierrat, A. Brennan, T. I. Roumeliotis, G. Benstead-Hume, J. Alexander, G. Muirhead, S. Layzell, W. Lyu, V. Roulstone, M. Allen, H. Baldock, A. Legrand, F. Gabel, N. Serrano-Aparicio, C. Starling, H. Guo, J. Upton, M. Gyrd-Hansen, M. MacFarlane, B. Seddon, F. Raynaud, I. Roxanis, K. Harrington, S. Haider, J. S. Choudhary, S. Hoelder, T. Tenev and P. Meier, A RIPK1-Specific PROTAC Degrader Achieves Potent Antitumor Activity by Enhancing Immunogenic Cell Death, Immunity, 2024, 57(7), 1514–1532 CrossRef CAS PubMed.
- J. Liu, H. Chen, H. Ü. Kaniskan, L. Xie, X. Chen, J. Jin and W. Wei, TF-PROTACs Enable Targeted Degradation of Transcription Factors, J. Am. Chem. Soc., 2021, 143(23), 8902–8910 CrossRef CAS PubMed.
- Y. Surani, M. Wand, P. Picconi, M. Di Palma, R. Zenezini Chiozzi, M. Hasan, M. Maynard-Smith, R. Steiner, K. Rahman, C. Hind and M. Sutton, Convergent evolution of antibiotic resistance mechanisms between synthetic pyrrolobenzodiazepines (PBDs) and the naturally occurring albicidin in multidrug resistant Klebsiella pneumoniae, Research Square, 2024, preprint, DOI:10.21203/rs.3.rs-4901630/v1.
- H. Zhou, L. Bai, R. Xu, D. McEachern, K. Chinnaswamy, R. Li, B. Wen, M. Wang, C.-Y. Yang, J. L. Meagher, D. Sun, J. A. Stuckey and S. Wang, SD-91 as A Potent and Selective STAT3 Degrader Capable of Achieving Complete and Long-Lasting Tumor Regression, ACS Med. Chem. Lett., 2021, 12(6), 996–1004 CrossRef CAS PubMed.
- U. Patel, J. P. Smalley and J. T. Hodgkinson, PROTAC Chemcal Probes for Histone Deacetylase Enzymes, RSC Chem. Biol., 2023, 4(9), 623–634 RSC.
- X. Qiu, N. Sun, Y. Kong, Y. Li, X. Yang and B. Jiang, Chemoselective Synthesis of Lenalidomide-Based PROTAC Library Using Alkylation Reaction, Org. Lett., 2019, 21(10), 3838–3841 CrossRef CAS PubMed.
- M. Pettersson and C. M. Crews, PROteolysis TArgeting Chimeras (PROTACs) — Past, Present and Future, Drug Discovery Today:Technol., 2019, 31, 15–27 CrossRef PubMed.
- K. M. Riching, E. A. Caine, M. Urh and D. L. Daniels, The Importance of Cellular Degradation Kinetics for Understanding Mechanisms in Targeted Protein Degradation, Chem. Soc. Rev., 2022, 51(14), 6210–6221 RSC.
- M. Heider, R. Eichner, J. Stroh, V. Morath, A. Kuisl, J. Zecha, J. Lawatscheck, K. Baek, A. K. Garz, M. Rudelius, F. C. Deuschle, U. Keller, S. Lemeer, M. Verbeek, K. S. Götze, A. Skerra, W. A. Weber, J. Buchner, B. A. Schulman, B. Kuster, V. Fernández-Sáiz and F. Bassermann, The IMiD target CRBN determines HSP90 activity toward transmembrane proteins essential in multiple myeloma, Mol. Cell, 2021, 81(6), 1170–1186.e10, DOI:10.1016/j.molcel.2020.12.046 , Epub 2021 Feb 10. PMID: 33571422; PMCID: PMC7980223.
- F. Kassam, K. Enright, R. Dent, G. Dranitsaris, J. Myers, C. Flynn, M. Fralick, R. Kumar and M. Clemons, Survival Outcomes for Patients with Metastatic Triple-Negative Breast Cancer: Implications for Clinical Practice and Trial Design, Clin. Breast Cancer, 2009, 9(1), 29–33 Search PubMed.
- N. A. Espinoza-Sánchez, J. Enciso, R. Pelayo and E. M. Fuentes-Pananá, An NFKB-Dependent Mechanism of Tumor Cell Plasticity and Lateral Transmission of Aggressive Features, 2018, vol. 9 Search PubMed.
- M. Winzker, A. Friese, U. Koch, P. Janning, S. Ziegler and H. Waldmann, Development of a PDEδ-Targeting PROTACs That Impair Lipid Metabolism, Angew. Chem., 2020, 132(14), 5644–5650 CrossRef.
- Y. Zhang, B. Yang, J. Zhao, X. Li, L. Zhang and Z. Zhai, Proteasome Inhibitor Carbobenzoxy-L-Leucyl-L-Leucyl-L-Leucinal (MG132) Enhances Therapeutic Effect of Paclitaxel on Breast Cancer by Inhibiting Nuclear Factor (NF)-KB Signaling, Med. Sci. Monit., 2018, 24, 294–304 Search PubMed.
- L. Ferguson, S. Bhakta, K. R. Fox, G. Wells and F. Brucoli, Synthesis and Biological Evaluation of a Novel C8-Pyrrolobenzodiazepine (PBD) Adenosine Conjugate. A Study on the Role of the PBD Ring in the Biological Activity of PBD-Conjugates, Molecules, 2020, 25(5), 1243 Search PubMed.
- C. Brosseau, K. Colston, A. G. Dalgleish and C. Galustian, The Immunomodulatory Drug Lenalidomide Restores a Vitamin D Sensitive Phenotype to the Vitamin D Resistant Breast Cancer Cell Line MDA-MB-231 through Inhibition of BCL-2: Potential for Breast Cancer Therapeutics, Apoptosis, 2012, 17(2), 164–173 Search PubMed.
- L. Yin, X. Wen, Q. Lai, J. Li and X. Wang, Lenalidomide Improvement of Cisplatin Antitumor Efficacy on Triple-Negative Breast Cancer Cells In�vitro, Oncol. Lett., 2018, 15(5), 6469–6474 Search PubMed.
- J. G. Gribben, N. Fowler and F. Morschhauser, Mechanisms of Action of Lenalidomide in B-Cell Non-Hodgkin Lymphoma, J. Clin. Oncol., 2015, 33(25), 2803–2811 CrossRef CAS PubMed.
- B. ab I. Thomas, H. L. Lewis, D. H. Jones and S. E. Ward, Central Nervous System Targeted Protein Degraders, Biomolecules, 2023, 13(8), 1164 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The ESI is available on the publications website. 1H NMR, 13C NMR, LC-MS spectra of the final products. 2D docking results of short benzene-fused PBDs (docx). HRMS spectra of the final products (PDF). See DOI: https://doi.org/10.1039/d5md00316d |
| ‡ Current address: Department of Genetic Engineering and Biotechnology, Faculty of Biological Sciences, University of Chittagong, Chattogram, 4331, Bangladesh. |
| § Joint senior authors. |
| This journal is © The Royal Society of Chemistry 2025 |