
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DORA-XGB: an improved enzymatic reaction feasibility classifier trained using a novel synthetic data approach†
Yash
Chainani‡
ab,
Zhuofu
Ni‡
ab,
Kevin M.
Shebek
ab,
Linda J.
Broadbelt
 ab and
Keith E. J.
Tyo
*ab
ab and
Keith E. J.
Tyo
*ab
aDepartment of Chemical and Biological Engineering, Northwestern University, Evanston, IL, USA. E-mail: k-tyo@northwestern.edu
bCenter for Synthetic Biology, Northwestern University, Evanston, IL, USA
First published on 2nd November 2024
Abstract
Retrobiosynthesis tools harness the inherent promiscuities of enzymes for the de novo design of novel biosynthetic pathways to key small molecules. Many existing pathway search algorithms rely on exhaustively enumerating the space of all possible enzymatic reactions using generalized rules, followed by an extensive analysis of the ensuing reaction network to extract candidate pathways for experimental validation. While this approach is comprehensive, many false positive reactions are often generated given the permissiveness of such reaction rules. Here, we have developed DORA-XGB, a enzymatic reaction feasibility classifier. DORA-XGB can be used within our DORAnet framework to assess whether newly enumerated enzymatic reactions and pathways would be feasible. To curate a training dataset for our model, we extracted enzymatic reactions from public databases and screened them for their general thermodynamic feasibility. We then considered alternate reaction centers on known substrates to strategically generate infeasible reactions with high confidence, thereby circumventing the lack of negative data in the literature. In training our model, we also experimented with various molecular fingerprinting techniques and configurations for assembling reaction fingerprints, taking into account not just primary substrate and primary product structures, but cofactor structures as well. Our model's utility is demonstrated through favorable benchmarking against a previously published classifier, the successful recovery of newly published reactions, and the ranking of previously predicted pathways for the biosynthesis of propionic acid from pyruvate.
Design, System, ApplicationRetrobiosynthesis tools aid in elucidating novel pathways for the sustainable biomanufacturing of small molecules. Such tools, however, may suggest many false positive reactions that are far too dissimilar from the canonical reaction/s that a given enzyme is known to catalyze, thereby demanding unrealistic extents of enzyme promiscuity. Here, we aim to reduce false positive predictions and enhance the accuracy of retrobiosynthesis tools by developing a machine learning model to reliably predict the feasibility of proposed enzymatic reactions. In designing this model, we innovated around the lack of infeasible reactions in the literature by introducing the concept of “alternate reaction centers”. These are functional groups that despite being identical to the catalyzed moiety on a substrate, remain uncatalyzed in a reported reaction. Our novel hypothesis enables us to strategically infer infeasible reactions from known positive reactions with higher confidence than previous approaches which assume any unseen reaction to be infeasible. After synthetically generating negative data from known reactions, we trained a supervised learning classifier and optimized it via a Bayesian hyperparameter optimization approach. Our model can be instantly dropped into pathway discovery workflows and even further improved upon in the future by incorporating additional features, such as enzyme sequence data. |
1. Introduction
Metabolic engineering is crucial in enabling the sustainable biomanufacturing of commodity chemicals, biofuels, and therapeutics.1–3 Enzymes with desirable activities that could lead to the synthesis of such key molecules have already been extensively documented in publicly available metabolic databases such as the Kyoto encyclopedia of genes and genomes (KEGG),4 BRENDA,5 and MetaCyc.6 Relying solely on known enzymatic reactions, however, is insufficient for exploring the entire space of possible molecules that could be manufactured through biosynthetic pathways. Synthesizing many valuable molecules may, in fact, require novel, non-native enzymatic reactions that have yet to be recorded in any database or the literature.7,8 The ability to predict and investigate such promiscuous, underground enzymatic activity is therefore necessary to expand the portfolio of chemicals that can be synthesized biologically.To this end, retrobiosynthesis tools can accelerate the de novo design of biosynthetic pathways to key products in an automated manner that circumvents costly trial-and-error based experiments.9–14 Such tools, including our in-house platform, DORAnet (formerly Pickaxe v2.0 (ref. 15)), typically use reaction rules or templates to recursively transform simple precursors, such as glucose or glycerol, into downstream metabolites of interest. These reaction rules, in turn, digitally encode for the potential promiscuities of enzymes by searching for substructure matches between native and non-native, predicted substrates.16,17 Despite their comprehensiveness, rule-based algorithms can generate many false positive reactions demanding unrealistic extents of enzyme promiscuity.
Throughout this work, we define positive reactions as those in which a moiety or reaction center, e.g., a carboxylic acid group, on a substrate successfully undergoes an enzyme-catalyzed reaction, such as decarboxylation. Consequently, false positives refer to predicted reactions within which a moiety that is known not to be transformed, thus representing a negative reaction, is incorrectly transformed by a reaction rule. Such false positives can occur if the substructure match required by a given rule only spans a small chemical neighborhood around a substrate's reaction center. For instance, our previously published JN1224MIN generalized rules16 predict enzyme promiscuity by considering only the reactive moieties present on a substrate and not its surrounding chemical groups, which may still influence catalysis due to their steric or electron donating/withdrawing effects. Our subsequently upgraded intermediate rules (available at https://github.com/tyo-nu/MINE-Database) incorporate some chemical context around reaction centers but still result in large metabolic in silico network expansions (MINEs).
Although such large networks ensure that the space of all possible reactions has been adequately explored, the high false positive rate arising from the permissible nature of reaction rules often results in far more pathways than can be thoroughly analyzed. This impedes the selection of promising pathways for experimental validation by users. DORAnet and other retrobiosynthesis tools would therefore benefit from the development of an automated reaction feasibility filter to elucidate only the most feasible and realistic of reactions suggested within a network expansion. While a variety of chemical similarity and molecular weight filters already exist within DORAnet to prune MINEs on-the-fly, these do not evaluate the feasibility of reactions generated and cannot be used to rank pathways once a network has been created.
To develop a filter or a model that can classify predicted reactions as feasible or infeasible, both positive and negative examples are needed. While thousands of observed reactions have already been recorded in BRENDA,5 KEGG,4 and MetaCyc,6 data for infeasible reactions are rare. A common approach in overcoming this lack of negative data is to assume unreported reactions as negative. This assumption can certainly aid in synthetically generating negative examples but has two key drawbacks. First, assuming that any unreported reaction must necessarily be infeasible can lead to generating negative reactions that are so strongly dissimilar to known, positive reactions (Fig. 1(a)) that the resulting classifier will suffer from a high degree of uncertainty in trying to predict the feasibilities of reactions with more intermediate degrees of similarity. The ideal distribution of positive and negative reactions should instead meaningfully delineate the boundary of reaction feasibility, allowing the sampling of negative data from more confident infeasible reactions only while positive data is sampled from known reactions. This would enable a classifier trained on such examples to also generalize well to real-world reactions wherein positive and negative examples are not so dissimilar. Moreover, the ‘unreported is negative’ assumption may lead to mislabelling understudied reactions that could very well be feasible, but because they have yet to be studied and/or published, are labeled as infeasible (Fig. 1(a)). Such mislabelling would introduce false negatives in a training set and also defeats the purpose of retrobiosynthesis tools, which will inevitably suggest new reactions. The explicit recovery of unreported profiles using methods such as collaborative filtering has been discussed in the literature,18,19 but the current space of substrates for any given enzyme is so limited that these methods may not apply to most classes of enzymatic reactions. Still, if negative reaction data can be reliably obtained, many artificial intelligence algorithms have been shown to be effective at demarcating complex decision boundaries in binary classification tasks across a diversity of domains, from predicting antimalarial bioactivity20 to the segmentation of coal mining faces.21
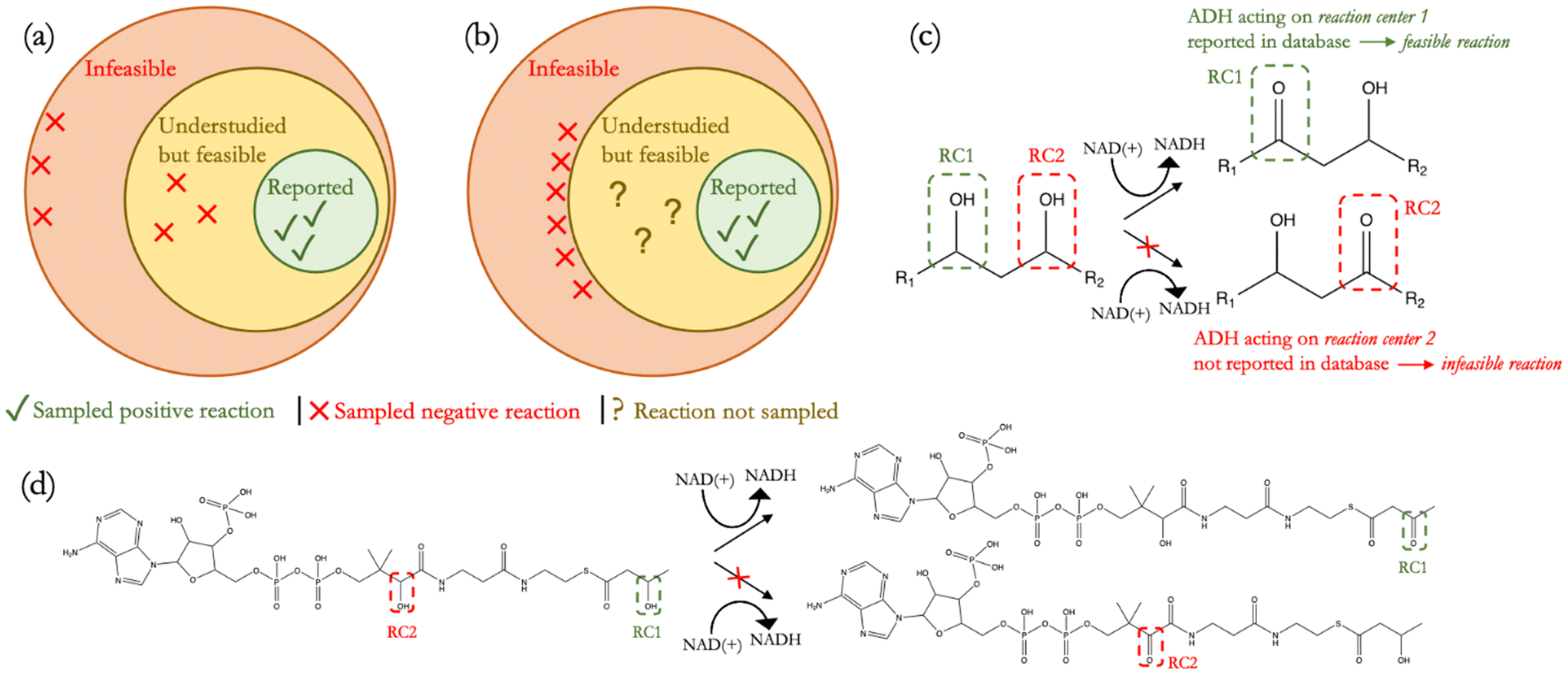 | ||
| Fig. 1 (a) It is common to assume that all reactions complementary to the set of reported reactions are infeasible. This assumption, however, may lead to sampling negative reactions that are too distant from positive reactions and/or mislabelling understudied reactions, which may very well may be feasible, as infeasible. Incorporating such false negatives and true negatives that are so strongly dissimilar to true positives can erode training data quality; (b) ideally, a high-quality training set should not comprise false negatives while true negatives should be as close to true positives as possible; (c) here, we propose the “alternate reaction center” assumption to more confidently infer infeasible reactions from reported reactions. Using the generalized alcohol dehydrogenase (ADH) transformation as an example, infeasible ADH reactions can be strategically inferred from a substrate with two or more alcohol reaction centers (RCs), wherein the oxidation of only one alcohol group (RC1) has been reported but not of the other (RC2). In this case, native or engineered ADH enzymes have been tested on the same substrate, but only one of the two possible reactions has been observed, allowing the other reaction to be inferred as being infeasible; (d) depicted here is an example of an infeasible ADH reaction with 3-hydroxybutyryl-coenzyme A (CoA) in which the generalized ADH transformation is not observed on the CoA group. | ||
Here, we address this lack of negative data in the literature by proposing the stricter “alternate reaction center” assumption, which enabled us to strategically and more confidently infer negative reactions that more closely resemble positive reactions (Fig. 1(b)) and used our dataset to train a supervised reaction feasibility classifier. We define a reaction center as the group of atoms in a substrate that directly participates in a reaction. Rather than treating all unreported reactions as negative datapoints, we posit that if an enzyme is observed to catalyze the transformation of a particular chemical moiety on a substrate but not that of other, identical moieties (alternate reaction centers) on the same substrate, then the transformation of those other, identical moieties represents products that could have also been formed in the same reaction but, since they were not observed, are infeasible products (Fig. 1(c) and (d)). In curating positive reactions from metabolic databases, we also screened such reactions for their thermodynamic feasibility across a range of metabolite concentrations.22 Since reported reactions are often a part of broader pathways that may only be specific to certain organisms, our thermodynamic screen elucidated which reactions would truly be most feasible in a diversity of contexts. The combination of this thermodynamic screen and our proposed “alternate reaction center” assumption created a dataset to train our classifier to evaluate reaction feasibility as a function of both reaction thermodynamics and enzyme specificity. To assess the applicability of our classifier, we tested DORA-XGB across various use-cases. Our model was found to achieve a high recall on newly discovered MetaCyc6 and EcoCyc23 reactions and also outperformed another published, deep-learning based feasibility classifier24 when benchmarked against these new reactions. Moreover, DORA-XGB was able to distinguish between feasible and infeasible reactions when tested on a high-throughput metabolomics dataset.25 Finally, we implemented our feasibility classifier as a filter in the design of propionic acid biosynthesis pathways26 and were able to achieve greater than 95% reduction of infeasible compounds and reactions while still preserving meaningful predictions of the most promising pathways towards propionic acid. Altogether, we demonstrate that our enzymatic reaction feasibility classifier is generalizable across different classes of reactions and enhances the computational prediction of enzyme promiscuity towards various applications.
2. Materials and methods
2.1 Curation of known enzymatic reactions from public metabolic databases
Processing of known enzymatic reaction data has been detailed in our previous publication.16 Briefly, experimentally validated reactions were curated from three publicly available metabolic databases: BRENDA,5 KEGG,4 and Metacyc.6 ChemAxon's structure checker (https://www.chemaxon.com) and RDKit's neutralization module (https://www.rdkit.org) were used for neutralization of molecules and removal of stereochemistry in preprocessing curated reactions. All transport and racemization reactions, along with reactions involving cofactors only, were not considered. Forward and reverse directions were considered as separate enzymatic reactions. Subsequently, all sanitized reactions were mapped to reaction rules within JN1224MIN,16 our previously published generalized enzymatic rule set, so as to categorize reactions by their minimal bond change pattern around the reaction center. A list of common cofactors (CoA, water, etc.) and cofactor pairs (ATP/ADP, NAD/NADH, etc.) can be found in our previous publication.16 After removing null entries and duplicates across the three databases as well as considering each reaction in both forward and reverse directions, our final dataset comprised 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 065 unique reactions that had been mapped to at least one rule in JN1224MIN.
065 unique reactions that had been mapped to at least one rule in JN1224MIN.
2.2 Screening curated reactions through calculation of free energies
In this study, we used eQuilibrator 3.0 (ref. 27) to calculate the change in Gibbs free energy due to a reaction, ΔrG′. The eQuilibrator 3.0 software platform uses the component contribution method for first estimating the standard Gibbs free energy change due to a reaction, ΔrG°, at reactant concentrations of 1 M. These ΔrG° values were then adjusted to our specified conditions of temperature 298 K, ionic strength 0.25 M, pH 7.4, and pMg 3.0 to calculate ΔrG′ under common cellular conditions. A ChemAxon license was used to compute pKa values of new compounds and add them to an eQuilibrator SQLite compound database that was maintained locally and initially downloaded through the Zenodo data repository (https://www.zenodo.org/records/4128543). In computing ΔrG′ values, instead of using a fixed concentration of 1 M for all metabolites, we allowed metabolite concentrations to vary within a predetermined range of 0.1 mM to 100 mM and optimized for the minimum ΔrG′ value,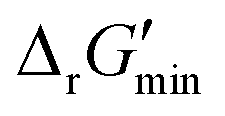 , that can be attained within this range. In computing
, that can be attained within this range. In computing 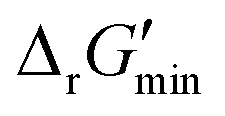 values for each reaction, the concentrations of cofactors are subject to ratios (e.g. [NADH]/[NAD+] = 0.1, [ATP]/[ADP] = 10) that have been empirically measured and reported in the literature.28 We considered NADH/NAD and NADPH/NADP as distinct cofactor pairs in this work since they are bound by different concentration ratios. The complete list of cofactor ratios used in computing
values for each reaction, the concentrations of cofactors are subject to ratios (e.g. [NADH]/[NAD+] = 0.1, [ATP]/[ADP] = 10) that have been empirically measured and reported in the literature.28 We considered NADH/NAD and NADPH/NADP as distinct cofactor pairs in this work since they are bound by different concentration ratios. The complete list of cofactor ratios used in computing 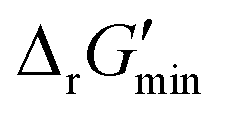 values is outlined in Section 3.1 of the ESI.† Our approach is a simplified and truncated version of solving the maximum/minimum driving force (MDF) problem, which seeks a set of metabolite concentrations that minimizes the ΔrG′ value of the most thermodynamically uphill or bottlenecked reaction within a multi-step pathway.22 ΔrG values of any kind were not computed for reactions within which at least one species was represented by an incomplete (as indicated by an asterisk) simplified molecular input linear entry system (SMILES) string and/or possessed uncommon atoms (i.e., atoms other than C, O, N, P, S, and H). The distributions of
values is outlined in Section 3.1 of the ESI.† Our approach is a simplified and truncated version of solving the maximum/minimum driving force (MDF) problem, which seeks a set of metabolite concentrations that minimizes the ΔrG′ value of the most thermodynamically uphill or bottlenecked reaction within a multi-step pathway.22 ΔrG values of any kind were not computed for reactions within which at least one species was represented by an incomplete (as indicated by an asterisk) simplified molecular input linear entry system (SMILES) string and/or possessed uncommon atoms (i.e., atoms other than C, O, N, P, S, and H). The distributions of 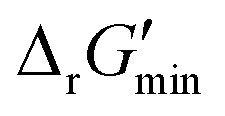 values for various classes of enzymatic reactions can be found in ESI† Section 3.2 and Fig. S2–S7.
values for various classes of enzymatic reactions can be found in ESI† Section 3.2 and Fig. S2–S7.
2.3 Assigning a threshold for thermodynamic feasibility
After computing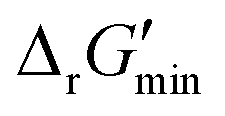 values for each reaction in our curated dataset, a threshold value was required to label reactions as feasible or infeasible. Instead of a classic threshold value of 0 kJ mol−1 to delineate thermodynamic feasibility, we set a threshold of −10 kJ mol−1, i.e., reactions with
values for each reaction in our curated dataset, a threshold value was required to label reactions as feasible or infeasible. Instead of a classic threshold value of 0 kJ mol−1 to delineate thermodynamic feasibility, we set a threshold of −10 kJ mol−1, i.e., reactions with 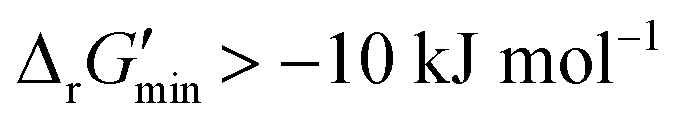 were labelled as infeasible (Fig. 2(a)) while reactions with
were labelled as infeasible (Fig. 2(a)) while reactions with 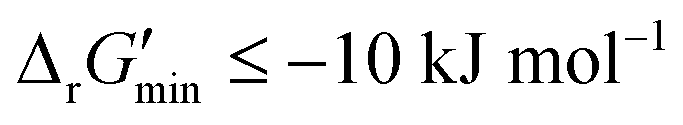 were labelled as feasible (Fig. 2(b)). Our decision in setting this threshold is informed by the flux-force efficacy relationship, which highlights that thermodynamic potentials are inextricably linked to reaction kinetics:22
were labelled as feasible (Fig. 2(b)). Our decision in setting this threshold is informed by the flux-force efficacy relationship, which highlights that thermodynamic potentials are inextricably linked to reaction kinetics:22Here, J+ is the flux carried in the forward direction of a given reaction at temperature T while J− is the flux carried in the reverse direction at the same temperature. According to this relationship, reactions that operate at ΔrG′ ∼ 0 kJ mol−1 may be thermodynamically efficient but are kinetically inefficient since the net flux in the forward direction, J+ − J−, would only comprise exactly 50% of the total flux, J+ + J−, carried throughout the reaction. By contrast, at our threshold of
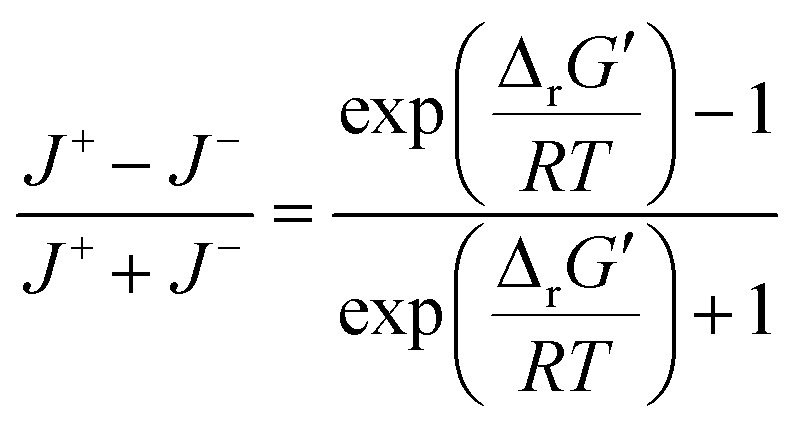
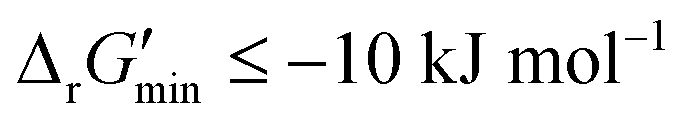 for thermodynamic feasibility, 96.5% of the total flux is in the forward direction only. Given that we consider forward and reverse directions of reported enzymatic reactions as distinct, our threshold ensures that each curated reaction can be treated independently. Reactions for which a ΔrG′ value could not be computed by eQuilibrator 3.0 or could be computed but with an excessively large uncertainty of 1.00 × 105 kJ mol−1 were not assigned a thermodynamic feasibility label. For such reactions, at least one of the species involved likely comprised at least one chemical group for which a thermodynamic contribution has yet to be reported or established with a high degree of certainty. Overall, we were able to confidently compute
for thermodynamic feasibility, 96.5% of the total flux is in the forward direction only. Given that we consider forward and reverse directions of reported enzymatic reactions as distinct, our threshold ensures that each curated reaction can be treated independently. Reactions for which a ΔrG′ value could not be computed by eQuilibrator 3.0 or could be computed but with an excessively large uncertainty of 1.00 × 105 kJ mol−1 were not assigned a thermodynamic feasibility label. For such reactions, at least one of the species involved likely comprised at least one chemical group for which a thermodynamic contribution has yet to be reported or established with a high degree of certainty. Overall, we were able to confidently compute 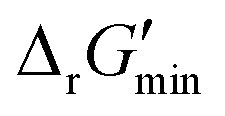 for 22803 reactions and label them accordingly for their thermodynamic feasibility.
for 22803 reactions and label them accordingly for their thermodynamic feasibility.
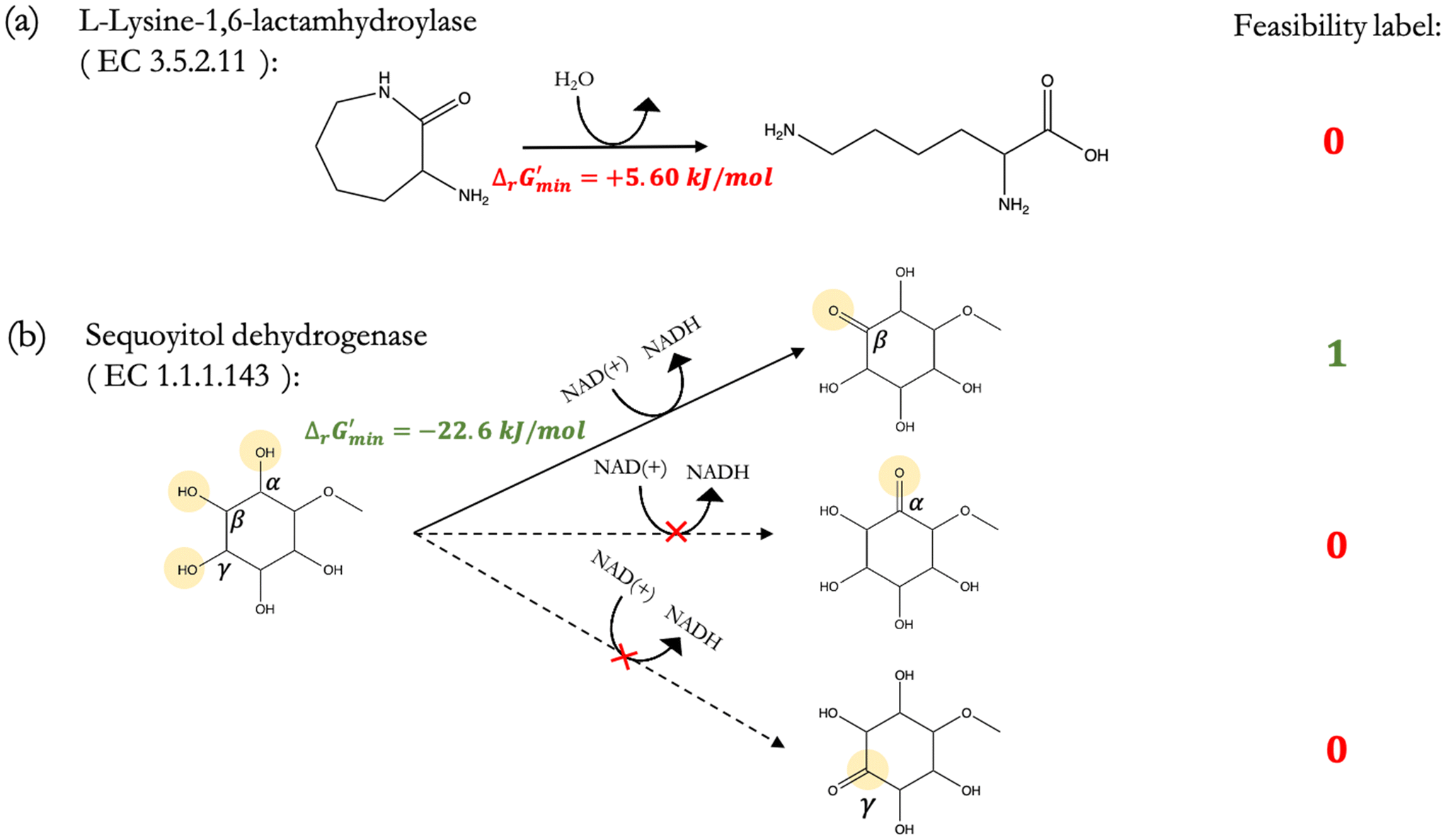 | ||
Fig. 2 (a) Curated reactions in our training dataset are labelled first using the minimum change in Gibbs free energy 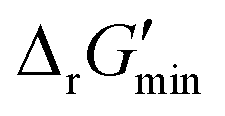 that can be released during the reaction across a predetermined range of metabolite concentrations. Reactions such as that catalyzed by the enzyme L-lysine-1,6-lactamhydrolase – with enzyme classification (EC) 3.5.2.11 – for which we find that that can be released during the reaction across a predetermined range of metabolite concentrations. Reactions such as that catalyzed by the enzyme L-lysine-1,6-lactamhydrolase – with enzyme classification (EC) 3.5.2.11 – for which we find that  are labelled as infeasible; (b) conversely, reactions for which are labelled as infeasible; (b) conversely, reactions for which 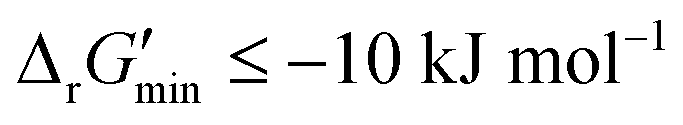 are labelled as feasible. For thermodynamically feasible reactions, such as that catalyzed by sequoyitol dehydrogenase (EC 1.1.1.143), in which only the hydroxyl group in the β position to the methoxy sequoyitol is oxidized but not the hydroxy groups in the α and γ positions to this methoxy group, our “alternate reaction center” assumption can be applied to confidently infer infeasible reactions. are labelled as feasible. For thermodynamically feasible reactions, such as that catalyzed by sequoyitol dehydrogenase (EC 1.1.1.143), in which only the hydroxyl group in the β position to the methoxy sequoyitol is oxidized but not the hydroxy groups in the α and γ positions to this methoxy group, our “alternate reaction center” assumption can be applied to confidently infer infeasible reactions. | ||
2.4 Synthetic generation of non-reacting substrates
In this study, we have proposed the “alternate reaction center” assumption, where a reactant must have two or more identical moieties, and an enzyme is known to act on only one of these moieties but not on the other (Fig. 1(c), (d) and 2(b)). Since the unreactive moiety was confronted with the enzyme in the original experiment, but a product resulting from the transformation of that moiety was not reported, we believe that this is a more rigorous assumption than the commonly used “unreported is negative” assumption wherein the entire space of reactions outside the space of published reactions is simply considered to be infeasible.Here, we utilized DORAnet to synthetically generate a total of 116412 unique infeasible reactions by considering alternate reaction centers (Fig. 2(b)). These synthetically generated infeasible reactions were then pooled together with reactions previously found to be thermodynamically infeasible to give a total of 122573 negative reactions and 16642 positive reactions (ESI† Section 3.3 and Fig. S8(a)–(c)). For curated and thermodynamically feasible monosubstrate reactions of the form A + cofactors → B + cofactors, all possible products beyond B are enumerated by expanding on A using only the general rule/s onto which this monosubstrate reaction had been mapped. For curated and thermodynamically feasible multisubstrate reactions of the form A + B + cofactors → C + D + cofactors, all possible products beyond (C, D) product pairs are iteratively enumerated first from A and then from B using only the general rule/s onto which this multisubstrate reaction has been mapped. Expanding on substrates only with mapped JN1224MIN rule/s follows from our “alternate reaction center” assumption.
By contrast, to generate the “unreported is negative” dataset, we utilized DORAnet and all 1224 reaction rules to expand upon each curated substrate/s and consequently enumerate the entire space of products. This resulted in a huge space of more than 11 million negative reactions. To directly compare models trained on datasets generated under each assumption, we randomly down-sampled 116412 negative reactions from these 11 million negative reactions. All tautomeric forms of compounds involved in reactions were comprehensively enumerated as part of our dataset.
2.5 Mitigating class imbalance between feasible and infeasible reactions for machine learning
Given that the number of negative reactions in our dataset of 139215 labelled reactions far outweighs the number of positive reactions by a ratio of nearly 8:1, any classifier trained on such imbalanced data would be biased towards making negative predictions by default.29 In order to counteract this class imbalance, we utilized the synthetic minority over-sampling technique30 (SMOTE) available through scikit-learn's imbalanced-learn Python package (imb-learn; https://www.imbalanced-learn.org). SMOTE creates synthetic reaction fingerprints from the positive reactions in our dataset by randomly picking a single positive reaction and the fingerprints of its k nearest neighbors (we set k = 5). The selected fingerprints are then added together to introduce over-sampled positive reactions into our dataset, which aids in balancing out negative reactions. SMOTE was performed only on our training dataset to prevent any leakage between training and testing sets. The class-weight hyperparameter was also tuned in training our machine learning models to further mitigate class imbalance.
2.6 Constructing reaction feature vectors using primary substrate, primary product, and cofactor structures with various molecular fingerprints and configurations
In constructing feature vectors to represent enzymatic reactions, we fingerprinted the chemical structures of all species involved in a given reaction instead of fingerprinting only primary reactant and primary product structures (Fig. 3). Chemical structures of all participating compounds were converted to molecular fingerprints by first removing any stereochemical information from their SMILES string and then taking the canonical form of the SMILES string using RDKit. We remove stereochemical information from all species because our reaction rule templates do not take stereochemistry into account. To decide on the optimal fingerprinting method, machine learning models were trained on a small prototyping set of 3013 alcohol dehydrogenase reactions featurized using five types of molecular fingerprints: (1) extended connectivity fingerprints with 2048 bits and a radius of fragmentation of 2 bonds (ECFP4),31,32 (2) atom pair fingerprints33 with 2048 bits, (3) MinHashed atom pair fingerprints34 with a radius of 2 and 2048 bits (MAP4), (4) Molecular ACCess System Keys35 (MACCS), and (5) Mordred.36 Of these five fingerprinting techniques, ECFP4, atom pair, and MAP4 are hashed fingerprints, while MACCS and Mordred are descriptor-based fingerprints.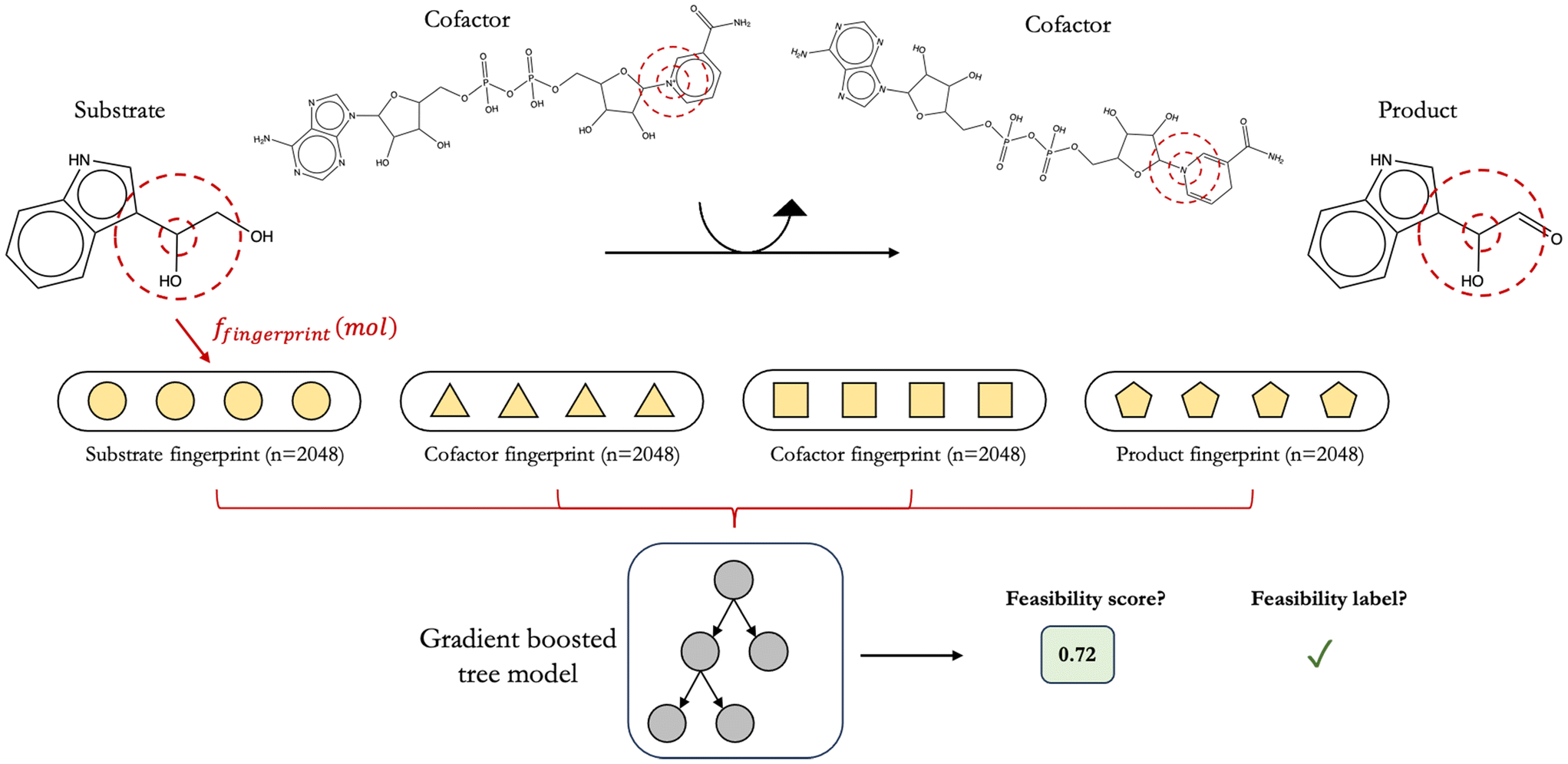 | ||
| Fig. 3 While it is common to ignore cofactor chemical structures and focus only on primary reactant and product structures in featurizing enzymatic reactions, in this work, we fingerprint all participating species to construct reaction feature vectors. Our decision is guided by the knowledge that cofactors also mechanistically participate in reactions alongside primary reactants and products. Including their structures can therefore lead to more interpretable models. As such, to predict feasibility, the input to DORA-XGB is a reaction string comprising all species, and the output is a feasibility score ranging from 0 to 1. This output score can also be converted to a predicted label based on our reported thresholds. We have experimented with various methods to arrange molecular fingerprints along a reaction feature vector and have provided the thresholds for each arrangement. | ||
Alongside attempting various molecular fingerprinting techniques, we experimented with four different methods to arrange these fingerprints for the assembly of reaction feature vectors. Briefly, we explored arranging reactant and product fingerprints in the order of ascending as well as descending molecular weights and through simple operations, such as the element-wise addition and concatenation of reactant and product fingerprints (“add then concatenate”) or the element-wise subtraction of the sum of product fingerprints from that of reactant fingerprints (“add then subtract”). We attempt such configurations to determine the pattern of fingerprints in our reaction vectors that would yield the highest predictive performance. Since different reactions involve different numbers of species, to ensure uniformity in reaction feature vector length, we zero-padded shorter vectors to the length of the longest reaction vector in our dataset for each fingerprinting technique and configuration.
2.7 Prototyping machine learning models with different architectures and fingerprints on a smaller set of alcohol dehydrogenase reactions
In training reaction feasibility classifiers, four different popular architectures are considered, namely logistic regression, random forests, support vector machines, and gradient boosted XGBoost models.37 Twenty models arising from a combination of these four architectures and the five molecular fingerprinting methods mentioned above are prototyped on a smaller dataset of 1254 feasible and 1759 infeasible alcohol dehydrogenase reactions to find the highest performing architecture-fingerprint pair (ESI† Section 4.1 and Fig. S9). A stratified 80/10/10 split ratio was used with scikit-learn in distributing these 3013 alcohol dehydrogenase reactions into train, validation, and test sets, respectively. Stratified splits ensure that the distribution of feasible to infeasible alcohol dehydrogenase reactions is largely maintained across each of the three sets.The hyperparameters of this alcohol dehydrogenase classifier were optimized on its validation set via a Bayesian optimization procedure38 with the objective of maximizing the classifier's area under its precision-recall curve (AUPRC). We opted for a Bayesian approach to tuning model hyperparameters because the large size of our final dataset necessitated an efficient and targeted search of hyperparameter space. Other approaches to hyperparameter tuning such as a grid-search or random-search would be far too exhaustive and also less effective given that in these searches, information from the model's previous performance is not used to inform the choice of hyperparameters in the next iteration.39 A Bayesian approach therefore allowed us to most efficiently balance exploration of new hyperparameter combinations with the exploitation of successful ones so as to reach optimal model hyperparameters in fewer iterations. We downloaded and used the algorithm that was freely available at https://www.github.com/bayesian-optimization/BayesianOptimization in order to perform our optimization. All regularization terms, where applicable, were included in our tuning procedure for each model so as to mitigate over-fitting. In training this classifier, reaction fingerprints are created by arranging molecular fingerprints in the order [substrate, NAD, product, NADH] for alcohol dehydrogenase reactions in the oxidation direction and [substrate, NADH, product, NAD] for reactions in the reduction direction. After this initial prototyping phase, ECFP4 fingerprints and an XGBoost model were chosen as the preferred fingerprint-architecture combination for training future models (Fig. 3).
2.8 Training a consolidated classifier and comparing performance against individual classifiers
In order to determine if training multiple individual feasibility classifiers, each specific to a single rule, would be the most accessible approach to predicting feasibility or if a single, consolidated classifier would be best, we trained 33 individual classifiers with the XGBoost architecture. These were trained on the top 33 classes of enzymatic reactions that had the most reactions mapped to them in our curated dataset. Reaction fingerprints for training these 33 classifiers were trained with the “add then concatenate” reaction fingerprint configuration. In order to directly compare performance between individual and consolidated models, our consolidated model was also trained using the “add then concatenate configuration”.For all classifiers, stratified train/validation/test sets were created from corresponding reaction data using an 80/10/10 split ratio. The hyperparameters of all classifiers were also optimized using the Bayesian approach described above for consistency. Moreover, for the final dataset of 139215 reactions, stratified train/validation/test splits were performed iteratively on a rule-by-rule basis for each family of reactions that had been mapped to at least one generalizable rule under JN1224MIN. This guarantees the presence of each generalized transformation across all three sets. In creating these splits, unit tests were performed to ensure that any duplicate reaction fingerprints are removed and consequently, that there is no leakage of reaction fingerprints between all three sets.
2.9 Mining benchmark datasets to test feasibility classifier
We mined three experimentally validated enzymatic reaction datasets to test whether our classifier could assess the feasibility of novel reactions. First, we extracted newly documented reactions in timesplit MetaCyc and EcoCyc datasets. Part of our training data was derived from EcoCyc V21 and MetaCyc V21, both published in 2017. Thus, 2810 and 270 reactions newly documented in MetaCyc V24 and EcoCyc V24, respectively, which were both published in 2021, were mined as benchmarking datasets. For these datasets, reactions were considered in both directions, and feasibility labels were assigned on the basis of thermodynamic feasibility as described in Materials and methods 2.2–2.3. Benchmarking studies were then performed with DeepRFC, an existing classification model already published in the literature. DeepRFC was downloaded and installed from the publicly available bitbucket code repository: https://bitbucket.org/kaistsystemsbiology/deeprfc.Since DeepRFC was trained on monosubstrate reactions only, even though our classifier is able to make predictions on multisubstrate reactions, only monosubstrate reactions were retained in our benchmarking set to enable a fair comparison between models. Overall, 1281 newly reported monosubstrate reactions could be confidently labelled for their thermodynamic feasibility and were used for benchmarking. For feasibility classification of a given reaction by DeepRFC, we used their stipulated threshold of 0.32 (calculated by subtracting half the predicted standard deviation from the predicted mean).
Finally, we mined an E. coli metabolomics dataset, which utilized a nontargeted approach to enable high-throughput identification of novel, underground metabolic reactions. 2799 accurate masses were identified in the metabolite cocktail of their experimental setup, and we were able to assign structures for 2578 masses that correspond to 737 unique metabolites by matching the reported metabolite names with those listed in the ModelSEED Biochemistry database (for which compounds are available at https://modelseed.org/biochem/compounds). In their work, 30 novel, unique enzymatic reactions had been experimentally discovered by 12 novel enzymes whose new functions were experimentally validated. Expanding from the cocktail of metabolites, we enumerated 16796 monosubstrate reactions that could have been catalyzed but were never observed or were not present in known EcoCyc reactions and thus could be plausibly treated as infeasible test reactions.
3. Results and discussion
3.1 Strategic generation of synthetic infeasible reactions enables exploration of enzymatic reaction feasibility boundaries more precisely
A common approach to generate synthetic negative data – given the lack of reported unsuccessful reactions – follows the “unreported is negative” assumption. This assumption considers all enumerated reactions that have not been observed as being infeasible. Such an assumption has been used previously for enzyme promiscuity prediction24,40 as well as reaction feasibility prediction within organic chemistry41 but can lead to sampling negative reactions that are too dissimilar from known, positive reactions as well as mislabelling potentially feasible reactions as infeasible.Here, we instead propose a novel and more strategic way of inferring negative reactions from known, positive reactions. We denote our approach to synthetically generating such negative data as the “alternate reaction center” assumption. While the “unreported is negative” assumption samples negative examples from the space of all reactions outside the corpus of reported reactions, our “alternate reaction center” assumption only samples negative examples from a smaller space of reactions for which infeasibility can be more confidently established (Fig. 1(b)). Our assumption is put into practice by considering metabolites with two or more identical molecular moieties on which an enzyme is known to transform only one of these moieties but not the other/s (Fig. 1(c) and (d)). This assumption is informed by the fact that many enzymes known to catalyze a given functional group transformation have already been validated on such metabolites. These enzymes – whether native or engineered – could have possessed a certain degree of promiscuity to catalyze the same transformation on other identical reaction centers within the same substrate. Given that no instances of reactions were observed on these alternate reaction centers, however, we can assume the conditions of such enzymatic transformations as less favorable and confidently categorize such reactions as infeasible. Further, examining alcohol dehydrogenase reactions through dimensionality reduction techniques reveals that putative infeasible products are evenly distributed amongst feasible products, thereby indicating a uniform sampling of chemical space, free of any biases (ESI† Fig. S8(a)).
3.2 Screening curated reactions for thermodynamic feasibility
Along with introducing our novel “alternate reaction center” assumption, we have screened curated reactions for their thermodynamic feasibility. This was done by optimizing for the minimum Gibbs free energy of reaction,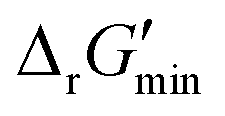 , that can be released under typical cellular conditions when the concentrations of all metabolites participating in a given reaction are constrained to a predetermined range with cofactor concentrations subject to empirically determined ratios28 (outlined in ESI† Section 1.1). Our optimization of
, that can be released under typical cellular conditions when the concentrations of all metabolites participating in a given reaction are constrained to a predetermined range with cofactor concentrations subject to empirically determined ratios28 (outlined in ESI† Section 1.1). Our optimization of 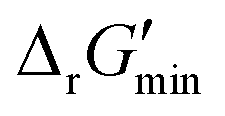 is a truncated version of solving the MDF problem22 (see Materials and methods 2.2–2.3) to eliminate thermodynamic bottlenecks along multi-step pathways. Reactions with
is a truncated version of solving the MDF problem22 (see Materials and methods 2.2–2.3) to eliminate thermodynamic bottlenecks along multi-step pathways. Reactions with 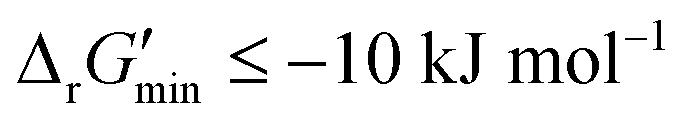 are labelled as feasible while reactions with
are labelled as feasible while reactions with 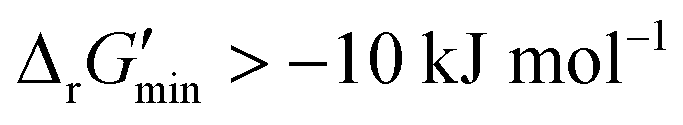 are labelled as infeasible (Fig. 2). This screen, along with our stringent threshold for thermodynamic feasibility, are essential components of our model development pipeline for the following reasons.
are labelled as infeasible (Fig. 2). This screen, along with our stringent threshold for thermodynamic feasibility, are essential components of our model development pipeline for the following reasons.
First, our curated reaction set comprises reactions in both directions. This follows from our previously published JN1224MIN rule set also comprising bidirectional operators, enabling retrobiosynthesis tools, such as DORAnet, the flexibility of being used in both the forward and reverse synthesis directions. The presence of such bidirectional rules is true for other rule sets published in the literature as well.17,42 While every enzymatic reaction is microscopically reversible, not all reactions are macroscopically reversible under typical cellular conditions. A thermodynamic screen can therefore quantitatively determine which set of functional group transformations and their associated reactions are most biochemically “realistic” and energetically favorable within general cellular contexts. For instance, alcohol dehydrogenase reactions that fall under the 1.1.1.x enzyme classification (EC) number are often favorable in both the oxidation and reduction directions (ESI† Fig. S3). Monooxygenation reactions (EC 1.14.13.x), by contrast, are only favorable in the direction of oxygen consumption (ESI† Fig. S2), given the extremely high energetic barrier that needs to be overcome to form NADH and oxygen as products in the reverse monooxygenation direction. Further, instead of the threshold of 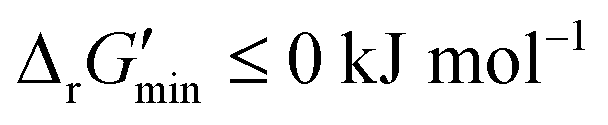 that is commonly invoked, our less permissive thermodynamic feasibility threshold of
that is commonly invoked, our less permissive thermodynamic feasibility threshold of 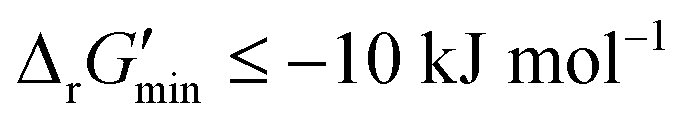 allows us to truly treat each reaction as independent of its reverse. This is because when
allows us to truly treat each reaction as independent of its reverse. This is because when 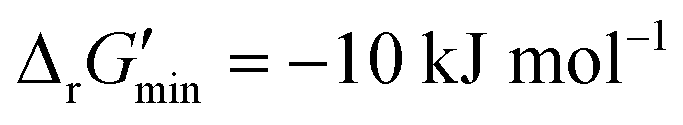 , 96.5% of the flux in a given reaction is carried in the forward direction only owing to the flux–force efficacy relationship (see Materials and methods 2.2–2.3).
, 96.5% of the flux in a given reaction is carried in the forward direction only owing to the flux–force efficacy relationship (see Materials and methods 2.2–2.3).
Finally, our thermodynamic screen is necessary since the presence of a reported reaction in a metabolic database does not simply guarantee its general feasibility. Often, databases report enzymatic reactions within the context of a broader pathway in a specific organism, wherein multiple factors, such as enzyme concentrations,22 cellular compartmentalization,43 energy coupling to other exothermic reactions,44,45 and metabolic channeling46–48 may help to drive a reaction forward. Given the difficulty of simultaneously accounting for all of these factors, our stricter bound of thermodynamic feasibility can help determine if a reported reaction would truly be feasible outside of the context in which it was reported. Consequently, our DORA-XGB classifier is able to evaluate the feasibility of novel reactions as a function of both enzyme specificity and reaction thermodynamics (Fig. 2).
3.3 Machine learning models with XGBoost architecture and hashed fingerprints provide best predictive performance
In order to determine which machine learning architecture and molecular fingerprinting method provided the best performance, we prototyped a smaller classifier trained only on alcohol dehydrogenase reactions. We trained four popular architectures (logistic regression, random forests, support vector machines, and XGBoost models) using five molecular fingerprints (ECFP4,31,32 MAP4,34 Atom Pair,33 MACCS,35 and Mordred36) for a total of 20 models.The AUPRC score is our metric of choice in this work given the considerable imbalance between positive and negative reactions in our dataset. With imbalanced data, accuracy is a misleading metric for model performance since any classifier that predicts negative by default would be largely correct anyway.49 Meanwhile, precision and recall are better at identifying the minority class but need to be evaluated at specified thresholds. AUPRC, by contrast, calculates the trade-off between precision and recall at all possible thresholds and does not overvalue negative datapoints, unlike the area under the receiver operating characteristic curve. AUPRC scores on both the validation data and the test data for alcohol dehydrogenase reactions showed similar trends (ESI† Section 4.1 and Fig. S9(a) and (b)) – hashed fingerprints (ECFP4, atom pair, MAP4) perform better than descriptor-based ones (MACCS, Mordred) for all machine learning architectures. This could be attributed to the automated and unbiased nature of hashed fingerprints accounting for diverse structural information. Considering hashed fingerprints only, all architectures are found to perform comparably well, but tree-based ensemble architectures were the fastest to train given their parallelizability. XGBoost in particular comes built in with both L1 and L2 regularization, enabling it to better mitigate overfitting and generalize well beyond the training data.37
As a result, we chose XGBoost and ECFP4 fingerprints as our final machine learning architecture-fingerprint combination. ECFP4 fingerprints are valuable because they highlight local molecular substructures within a set diameter. Atom pair fingerprints, meanwhile, are able to highlight more distant pairs of atoms. Both fingerprints were found to perform well when we prototyped the alcohol dehydrogenase classifier (ESI† Fig. S9(a) and (b)) and could have been used to provide equally important insights into the feasibility of novel reactions. Other studies have also used a combination of both fingerprints to represent chemical space in an unbiased manner.34 To create reaction feature vectors for this initial study, all participating molecular structures were first converted to ECFP4 fingerprints with 2048 bits. Then, molecular fingerprints are arranged in the order [substrate, NAD, product, NADH] for alcohol dehydrogenase reactions in the oxidation direction and [substrate, NADH, product, NAD] for reactions in the reduction direction.
3.4 Featurization of cofactor structures along with reactant and product structures to build a consolidated classifier for all enzymatic reactions
It is common practice to featurize only primary reactant and/or primary product structures when training machine learning models on enzymatic reactions.24,40 To build our classifier, however, we also included cofactor fingerprints in constructing reaction feature vectors (Fig. 3). Our decision is guided by the fact that cofactor molecules also participate mechanistically in enzymatic reactions but are often ignored. Further, it is theoretically possible to achieve the same generalized chemical transformation on a substrate through different chemistries of cofactors. For instance, the addition of a hydroxyl group to a substrate may be achieved either through a reductase (EC 1.17.1.x) or a monooxygenase (EC 1.14.13.x). If catalyzed by a monooxygenase, this reaction would be far more thermodynamically favorable than if it were catalyzed by a reductase given the significantly higher Gibbs free energy released when NADH and oxygen are utilized as cofactors on the reactants' side. A model trained without any cofactor information, however, would treat both reactions as identical and fail to assign a commensurately lower score to the reductase-catalyzed reaction. This inclusion of cofactors is widely underappreciated and not frequently attempted in current reaction prediction literature to our knowledge.In order to analyze reactions with multiple substrates or products, decisions have to be made on how to combine the feature vectors for each molecule on the left and right hand side of the reaction before they are fed to XGBoost. Four configurations in which to arrange molecular fingerprints along reaction feature vectors were used in this study (ESI† Section 4.2 and Fig. S10). Briefly, these involved arranging fingerprints in ascending and descending molecular weights of the corresponding species as well as through simple element-wise addition or subtraction of molecular fingerprints. The “add then concatenate” fingerprint approach described earlier (see Methods and materials 2.6) yielded nominally better results; therefore, we used the “add then concatenate” approach going forward.
With this reaction fingerprint configuration, we considered whether to build multiple individual classifiers specific to each class of enzymatic transformation or to build a single, consolidated model that would be generalizable to all reaction classes. Since 64.3% of our curated reactions are covered by 33 distinct types of enzymatic transformations, we built an individual XGBoost classifier for each of these 33 transformation types and then compared the performance of these models against a single, consolidated XGBoost model trained simultaneously on the data from all 33 transformation types. The overall AUPRC score of our consolidated model of 0.92 was found to exceed the average AUPRC of 0.86 across 33 individual models (ESI† Fig. S12). This indicates that the diversity in training data and its quantity are instrumental to the performance of our consolidated classifier. Further, given the overhead required in separately training and applying 33 different models based on an input reaction class, we chose to build a single, comprehensive classifier applicable to all reaction types.
Our consolidated model performs well with a high AUPRC score of 0.92, precision of 0.81, recall of 0.87, and F1 of 0.84 (Fig. 4) when these metrics were computed on our test set. The recall, precision, and F1 scores reported were calculated at an optimum threshold of 0.593. This threshold was in turn determined by considering 100 linearly spaced thresholds between 0 and 1 and then selecting the threshold that yielded the highest F1 score.
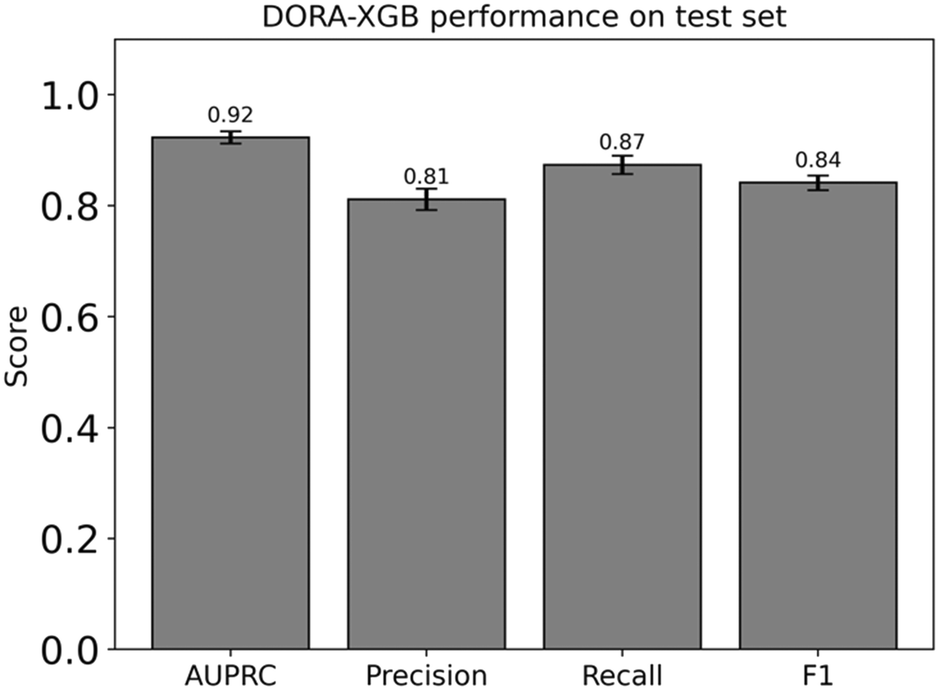 | ||
| Fig. 4 Our consolidated model performs well against our test set across all metrics of AUPRC, precision and recall. The metrics reported here are based on an XGBoost model tested on reactions that were featurized by concatenating the element-wise sum of all reactant fingerprints with that of all product fingerprints. The precision, recall, and F1 values reported here were calculated at a threshold value of 0.593. This threshold was in turn determined by considering 100 possible thresholds between 0 and 1 and then selecting the threshold at which the F1 value would reach a maximum. All models were trained using a train/validation/test split of 80/10/10, and hyperparameters were optimized using a Bayesian hyperparameter optimization procedure. Errors bars represent 95% confidence intervals calculated through a bootstrapped resampling of predicted labels and true labels over 1000 iterations. | ||
3.5 Enzymatic reaction feasibility classifier successfully retrieves newly discovered enzymatic reactions in publicly available metabolic databases
The generalizability of DORA-XGB outside of our training and test data was then explored by deploying our model to predict the feasibility of novel enzymatic reactions in three datasets that have recently been published but were not used as part of our training, validation, and test datasets. Part of our training data has been derived from MetaCyc v21.0 and EcoCyc v21.0, both published in 2017. As such, newly documented reactions in MetaCyc v24.0 (2021) and EcoCyc V24.0 (2021) could be used to further test model performance.Similar to the processing of our training set, these newly reported reactions were considered in both directions and screened for thermodynamic feasibility. On all 1281 newly-reported monosubstrate reactions for which thermodynamic feasibility could be confidently established, DORA-XGB outperforms another published deep learning method, DeepRFC (Fig. 5). Negative reactions for DeepRFC's training were generated with the “unreported is negative” assumption. The higher performance of DORA-XGB against this other classifier as well as our in-house “unreported is negative” dataset (ESI† Section 4.6 and Fig. S14) thus underscores the utility of our “alternate reaction center” hypothesis.
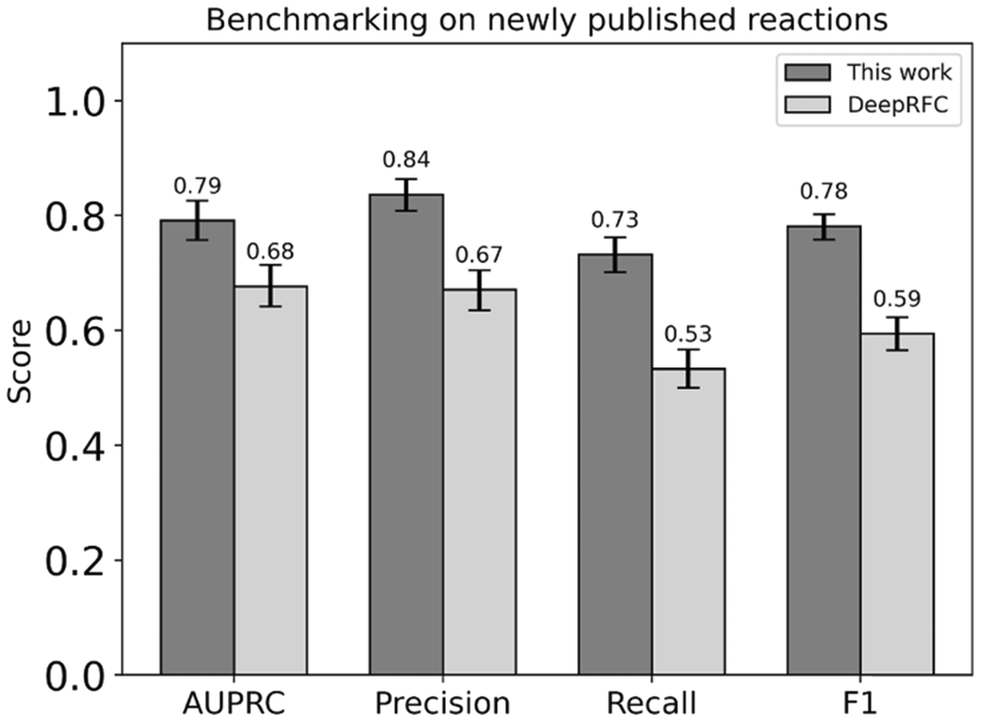 | ||
| Fig. 5 Our feasibility classifier outperforms another previously published model, DeepRFC, across all metrics of AUPRC, precision, recall, and F1 when both classifiers are tested against 1281 newly reported reactions with monosubstrates that neither classifier had been exposed to during training, testing or validation. | ||
Interestingly, the performance of DORA-XGB across all metrics drops between our test set and this external benchmarking set. This decline is due to two reasons. First, the distribution of enzymatic transformations in our benchmarking set is inherently different from that in the training, validation, and testing sets. This is expected given that the benchmarking set typically represents an out-of-distribution sample anyway. Crucially, since reactions in the benchmarking set were published later, there is a bias towards transformations that are rarely seen in the training, validation, and testing sets. Given that DORA-XGB had fewer opportunities to confront such rarer transformations during training, its performance understandably declines in benchmarking (ESI† Section 4.9, Fig. S16 and S17).
We then also used DORA-XGB to predict underground metabolism in an E. coli nontargeted metabolomics dataset.25 Promiscuous enzymatic activities often lead to underground metabolism, which are undocumented reactions or those without enzyme annotations, existing even in well-studied organisms. While current genome scale models fail to capture the entirety of such metabolism, computational resources such as our MINE database (publicly available at https://minedatabase.ci.northwestern.edu) have been developed to exhaustively enumerate possible reactions in E. coli. Distinguishing true positive novel reactions from the enormous space of computationally generated reactions, however, remains a challenge. In this E. coli metabolomics experimental setup, 30 newly discovered monosubstrate reactions were observed and validated as enzyme concentrations were tuned.25 Since these enzymes were extensively tested on native metabolites in E. coli, any undiscovered reactions that could have occurred on these metabolites that follow from the above transformations were labelled as plausibly infeasible reactions. Thus, we generated a dataset consisting of reactions that are 1) feasible and known, 2) feasible and novel, and 3) plausibly infeasible. We applied our feasibility classifier, and our model was able to recover 28 out of 30 newly discovered novel and feasible reactions (ESI† Table S1A), and on plausibly infeasible reactions, our model predicted 12372 out of 16796 reactions as infeasible. Our recovery of novel feasible reactions indicated that the model could successfully retrieve experimentally validated underground reactions. For prediction of infeasible reactions, despite lower performance than on the test data, the classifier could still assist efforts of filtering out numerous implausible reactions and prioritizing the most feasible underground reactions to be discovered with more extensive experiments.
3.6 Filtering out infeasible reactions greatly enhances efficiency of pathway design
Retrobiosynthesis tools rely on enzymatic reaction rules to explore novel biosynthetic reaction networks. Such rules can be overly permissive, which helps exhaustively enumerate all possible enzymatic reactions but may also generate many false positive predictions.Our updated DORAnet platform has implemented several on-the-fly filters to remove reactions in each generation that may lead farther away from the intended target molecule. One of these is a Tanimoto similarity filter with manually defined similarity cutoffs. With this, molecules that are too structurally dissimilar from the target are discarded after each generation. Such filters can improve pathway search efficiencies but do not speak to the feasibility of reactions that constitute a found pathway. Our reaction feasibility classifier could therefore be used to predict the feasibility of novel reactions and quickly filter out false positive reactions that demand unrealistic extents of enzyme promiscuity while still preserving molecules that can be feasibly reached after each generation. As such, our classifier enhances the confidence of newly predicted reactions while also improving the computational efficiency.
To test the utility of DORA-XGB in designing novel pathways, we deployed our classifier on our previously predicted biosynthetic pathways from pyruvate to propionic acid.26 We first reproduced the set of pathways that lead from pyruvate to acrylic acid in exactly three steps, where acrylic acid was an important precursor for propionic acid pathways and just one step away from the target (Fig. 6a). Starting from pyruvate, a network expansion using all 1224 generalized rules resulted in an enormous space of more than 750000 compounds and more than 1300000 reactions over three generations.
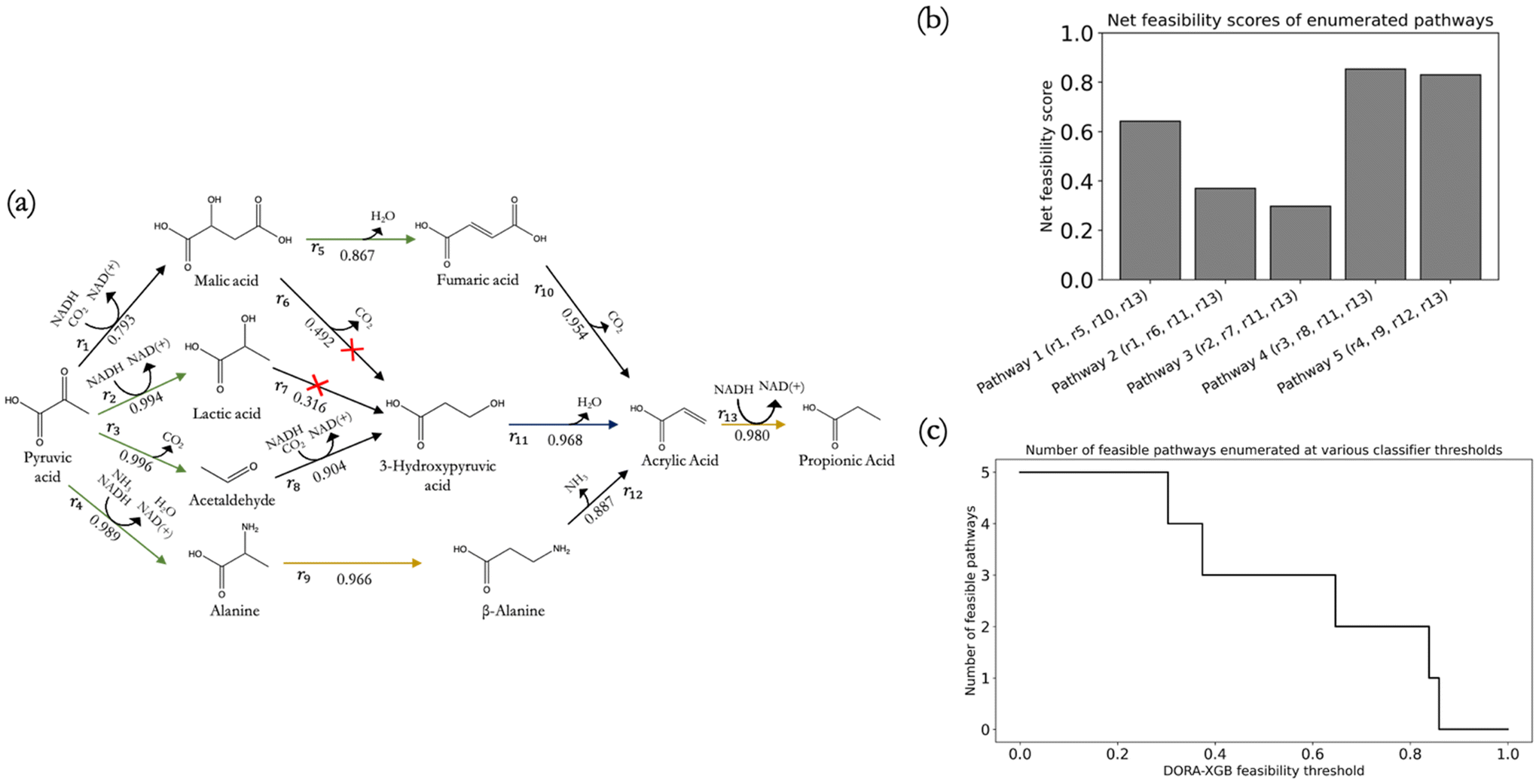 | ||
| Fig. 6 (a) Five candidate pathways comprising four reaction steps exactly from pyruvate to propionic acid are shown. These were proposed in our original propionic acid biosynthesis paper and are reproduced here by DORAnet. The feasibility score for each reaction is predicted by DORA-XGB and labelled under each arrow. Green arrows represent known reactions while yellow arrows represent reactions that may be catalyzed by enzymes that are known to be promiscuous; (b) the net feasibility score of each of the five pathways shown can be computed by taking the product of each constituent reaction's feasibility score, thereby enabling the ranking of pathways on the basis of their net feasibilities; (c) alternatively, pathways can also be pruned on-the-fly by tuning the feasibility threshold above which a reaction can be labelled as feasible. Users may specify their own feasibility thresholds or use the optimum thresholding values reported in this work. | ||
Using our classifier, pathways with any infeasible reactions below our set threshold were filtered out. As a result, 13 out of the 15 reactions in the original publication were predicted as feasible. Reassuringly, predicted reactions that had been reported in the literature and predicted reactions that were known from the literature to be catalyzed by promiscuous enzymes both received high feasibility scores. This resulted in three out of the five candidate pathways being predicted as feasible (Fig. 6(a)). In addition to discarding infeasible pathways, all pathways were also ranked with a net pathway feasibility score. This was computed by taking the product of all constituent reactions' feasibility scores (Fig. 6(b)) along a pathway. Users can also explore other ways to aggregate reaction feasibility scores, such as taking the average of reaction feasibility scores within a pathway.
As our classifier threshold was varied, an overall 96.3% reduction of new compounds and 96.9% reduction of new reactions was achieved after three generations (ESI† Fig. S15(a) and (b)), which in turn led to significant computational efficiency. This threshold can be customized by users to achieve their desired balance between precision and recall on novel reactions. A high classifier threshold would result in more reactions filtered out, thus improving precision at the expense of recall, and vice versa. We also tested the effect of varying the threshold by exploring the entire spectrum in increments of 0.01 of classifier thresholds from 0 to 1 and observed the resulting number of feasible reactions and compounds remaining as well as the number of pathways still predicted as feasible. At thresholds as low as 0.06, DORA-XGB can begin discarding infeasible pathways while at thresholds up until 0.84, DORA-XGB was able to retain the most feasible pathways within the network before all pathways were classified as infeasible. Ultimately, researchers could take advantage of the tunable nature of DORA-XGB to strike a balance between comprehensiveness and runtime and apply the feasibility classifier as an on-the-fly reaction filter and/or pathway ranking metric based on specific applications.
4. Conclusion
Here, we have developed DORA-XGB, a robust, generalizable, and user-friendly machine learning classification model based on the XGBoost architecture to evaluate the feasibility of novel enzymatic reactions. Our classifier augments retrobiosynthesis tools, such as DORAnet, and improves their accuracy by filtering out false positive reaction predictions that inevitably arise in using rule-based tools given the permissiveness of reaction rules.In order to train DORA-XGB, we required both positive and negative data. Although infeasible reactions are rarely published, we overcome this lack of negative data through our proposed “alternate reaction center” assumption to confidently infer infeasible reactions from known, positive ones. Our assumption involves examining reported substrates with multiple identical chemical moieties wherein only one of those centers is known to undergo enzymatic catalysis and not the others. With this insight, we first screened reported reactions for their thermodynamic feasibility and then, synthetically generated negative reactions from thermodynamically feasible ones. Our publicly available DORAnet platform and JN1224MIN rule set were used to generate such negative reactions, which were then pooled together with known ones to create a comprehensive, high-quality training dataset.
We subsequently demonstrated the effectiveness of our dataset and consequently, of DORA-XGB to predict the feasibility of reactions across various scenarios. Our model was able to identify novel feasible reactions when tested against a time split reaction dataset and a nontargeted metabolomics dataset. It could also be integrated within DORAnet as a custom reaction filter, where infeasible reactions after each generation would be filtered out to accelerate pathway design and preserve only the most promising pathways for experimental validation.
To train DORA-XGB, we also explored a range of molecular fingerprinting methods to construct reaction fingerprints from molecular fingerprints. In doing so, we recognized that cofactors also mechanistically play a role in enzymatic reactions and therefore included their fingerprints alongside those of primary substrates and primary products. This enabled us to fully capture the various chemistries involved in biochemical transformations. We note, however, that while DORA-XGB can predict the generalized feasibility of biochemical reactions, it currently cannot predict how feasible it would be for a given enzyme to catalyze a query reaction. This is because DORA-XGB does not yet take any enzyme information, such as sequence or structure, into account. Future work may involve incorporating such information through sequence embeddings50,51 as well as featurizing molecules with message-passing graph neural networks52–54 rather than with molecular fingerprints so as to better capture the long-range interactions between various functional groups within molecules.
Ultimately, our aim in this work was to provide a rigorous workflow for synthetically generating negative reaction data. Using the assumptions introduced in this work, we demonstrate that with reliable data, even relatively simple machine learning models can lead to good predictive performance. We have provided our classifier as an open-source tool on our lab Github page: https://github.com/tyo-nu/DORA_XGB.
Data availability
Data for this article can be found at the MetaCyc pathway database (https://www.metacyc.org) as well as the Kyoto Encyclopedia of Genes and Genomes (https://www.genome.jp/kegg/) and was obtained through an academic license. Data from the BRENDA enzyme database can be found at https://www.brenda-enzymes.org. DORA-XGB models are available at https://github.com/tyo-nu/DORA_XGB.Author contributions
Yash Chainani: methodology and software – developed the methodology as well as the pipeline to train, test, and validate DORA-XGB. Data curation – contributed to the extraction of metabolomics data. Writing – original draft and subsequent edits. Visualization – generated the figures in this manuscript. Zhuofu Ni: conceptualization – conceptualized the approach of this study and the idea of alternate reaction centers. Methodology – developed the methodology as well as early iterations of feasibility models. Data curation – curated raw data for this study. Writing – original draft. Visualization – contributed to figures in this manuscript. Kevin M. Shebek: software – contributed in set up of thermodynamics and eQuilibrator workflow. Linda J. Broadbelt: conceptualization – conceptualized the approach of this study. Investigation. Formal analysis. Writing – review & editing, were the principal investigators who directed this project, contributed to the data analysis and interpretation, as well as edited the manuscript. Keith E. J. Tyo: conceptualization – conceptualized the approach of this study. Investigation. Formal analysis. Writing – review & editing, were the principal investigators who directed this project, contributed to the data analysis and interpretation, as well as edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr. Christopher Henry, Dr. Danielle Tullman-Ercek, Dr. Jacob Martin, Dr. Tracey Dinh, Dr. Bapi Mandal, Dr. Sai Praneet Batchu, Shivani Kozarekar, Stefan Pate, Geoffrey Bonnanzio, Rawia Marafi, and Margaret Guilarte-Silva for their invaluable insights and constructive discussions. The funding for Yash Chainani for this study was partly provided for by the Northwestern University Graduate School Cluster Fellowship in Biotechnology, Systems, and Synthetic Biology, which is affiliated with the Biotechnology Training Program, and partly by the DOE Joint BioEnergy Institute (https://www.jbei.org) supported by the U.S. Department of Energy, Office of Science, Biological and Environmental Research Program under Award Number DE-AC02-05CH11231 with Lawrence Berkeley National Laboratory. The funding for Zhuofu Ni for this study was partly provided for by the U.S. Department of Energy (DOE), Office of Science, Office of Biological and Environmental Research under Award Number DE-SC0018249, and partly by an Institute of Sustainability and Energy at Northwestern (ISEN) Fellowship. This research project was supported in part through the computational resources and staff contributions provided by the Quest high performance computing facility at Northwestern University, which is jointly supported by the Office of the Provost, the Office of Research, and Northwestern University Information Technology. This research also used resources of the National Energy Research Scientific Computing Center (NERSC), a Department of Energy Office of Science User Facility using NERSC award ERCAP0028489.Notes and references
- J. D. Keasling, Science, 2010, 330, 1355–1358 CrossRef CAS.
- N. Fackler, B. D. Heijstra, B. J. Rasor, H. Brown, J. Martin, Z. Ni, K. M. Shebek, R. R. Rosin, S. D. Simpson, K. E. Tyo, R. J. Giannone, R. L. Hettich, T. J. Tschaplinski, C. Leang, S. D. Brown, M. C. Jewett and M. Köpke, Annu. Rev. Chem. Biomol. Eng., 2021, 12, 439–470 CrossRef.
- S. Y. Lee, H. U. Kim, T. U. Chae, J. S. Cho, J. W. Kim, J. H. Shin, D. I. Kim, Y.-S. Ko, W. D. Jang and Y.-S. Jang, Nat. Catal., 2019, 2, 18–33 CrossRef CAS.
- M. Kanehisa, M. Furumichi, M. Tanabe, Y. Sato and K. Morishima, Nucleic Acids Res., 2017, 45, 353–361 CrossRef.
- I. Schomburg, L. Jeske, M. Ulbrich, S. Placzek, A. Chang and D. Schomburg, J. Biotechnol., 2017, 261, 194–206 CrossRef CAS PubMed.
- R. Caspi, R. Billington, I. M. Keseler, A. Kothari, M. Krummenacker, P. E. Midford, W. K. Ong, S. Paley, P. Subhraveti and P. D. Karp, Nucleic Acids Res., 2020, 48, 445–453 CrossRef.
- M. A. Campodonico, B. A. Andrews, J. A. Asenjo, B. O. Palsson and A. M. Feist, Metab. Eng., 2014, 25, 140–158 CrossRef CAS PubMed.
- V. Hatzimanikatis, C. Li, J. A. Ionita and L. J. Broadbelt, Curr. Opin. Struct. Biol., 2004, 14, 300–306 CrossRef CAS PubMed.
- P. Carbonell, P. Parutto, C. Baudier, C. Junot and J.-L. Faulon, ACS Synth. Biol., 2014, 3, 565–577 CrossRef CAS PubMed.
- B. Delépine, T. Duigou, P. Carbonell and J.-L. Faulon, Metab. Eng., 2018, 45, 158–170 CrossRef.
- A. Kumar, L. Wang, C. Y. Ng and C. D. Maranas, Nat. Commun., 2018, 9, 184 CrossRef.
- P. A. Saa, M. P. Cortés, J. López, D. Bustos, A. Maass and E. Agosin, Biotechnol. J., 2019, 14, 1800734 CrossRef PubMed.
- N. Hadadi and V. Hatzimanikatis, Curr. Opin. Chem. Biol., 2015, 28, 99–104 CrossRef CAS PubMed.
- J. G. Jeffryes, R. L. Colastani, M. Elbadawi-Sidhu, T. Kind, T. D. Niehaus, L. J. Broadbelt, A. D. Hanson, O. Fiehn, K. E. J. Tyo and C. S. Henry, J. Cheminf., 2015, 7, 44 Search PubMed.
- K. M. Shebek, J. Strutz, L. J. Broadbelt and K. E. J. Tyo, BMC Bioinf., 2023, 24, 106 CrossRef PubMed.
- Z. Ni, A. E. Stine, K. E. J. Tyo and L. J. Broadbelt, Metab. Eng., 2021, 65, 79–87 CrossRef CAS PubMed.
- T. Duigou, M. du Lac, P. Carbonell and J.-L. Faulon, Nucleic Acids Res., 2019, 47, 1229–1235 CrossRef.
- C. Lan, S. N. Chandrasekaran and J. Huan, IEEE/ACM Trans. Comput. Biol. Bioinf., 2019, 1 CAS.
- R. Kurczab, S. Smusz and A. J. J. Bojarski, ChemInform, 2014, 6, 32 CrossRef.
- S. Egieyeh, J. Syce, S. F. Malan and A. Christoffels, PLoS One, 2018, 13, e0204644 CrossRef.
- Z. Xing, S. Zhao, W. Guo, F. Meng, X. Guo, S. Wang and H. He, Energy, 2023, 285, 128771 CrossRef.
- E. Noor, A. Bar-Even, A. Flamholz, E. Reznik, W. Liebermeister and R. Milo, PLoS Comput. Biol., 2014, 10, e1003483 CrossRef.
- P. D. Karp, W. K. Ong, S. Paley, R. Billington, R. Caspi, C. Fulcher, A. Kothari, M. Krummenacker, M. Latendresse, P. E. Midford, P. Subhraveti, S. Gama-Castro, L. Muñiz-Rascado, C. Bonavides-Martinez, A. Santos-Zavaleta, A. Mackie, J. Collado-Vides, I. M. Keseler and I. Paulsen, EcoSal Plus, 2018, 8, 10–1128 CrossRef.
- Y. Kim, J. Y. Ryu, H. U. Kim, W. D. Jang and S. Y. Lee, Biotechnol. J., 2021, 16, 2000605 CrossRef CAS.
- D. C. Sévin, T. Fuhrer, N. Zamboni and U. Sauer, Nat. Methods, 2017, 14, 187–194 CrossRef.
- A. Stine, M. Zhang, S. Ro, S. Clendennen, M. C. Shelton, K. E. J. Tyo and L. J. Broadbelt, Biotechnol. Prog., 2016, 32, 303–311 CrossRef CAS.
- M. E. Beber, M. G. Gollub, D. Mozaffari, K. M. Shebek, A. I. Flamholz, R. Milo and E. Noor, Nucleic Acids Res., 2022, 50, 603–609 Search PubMed.
- B. D. Bennett, E. H. Kimball, M. Gao, R. Osterhout, S. J. Van Dien and J. D. Rabinowitz, Nat. Chem. Biol., 2009, 5, 593–599 Search PubMed.
- J. Chakraborty, S. Majumder and T. Menzies, Proceedings of the 29th ACM Joint Meeting on European Software Engineering Conference and Symposium on the Foundations of Software Engineering, 2021, pp. 429–440 Search PubMed.
- N. V. Chawla, K. W. Bowyer, L. O. Hall and W. P. Kegelmeyer, J. Artif. Intell. Res., 2002, 16, 321–357 Search PubMed.
- H. L. Morgan, J. Chem. Doc., 1965, 5, 107–113 Search PubMed.
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742–754 Search PubMed.
- R. E. Carhart, D. H. Smith and R. Venkataraghavan, J. Chem. Inf. Comput. Sci., 1985, 25, 64–73 CAS.
- A. Capecchi, D. Probst and J.-L. Reymond, J. Cheminf., 2020, 12, 43 Search PubMed.
- J. L. Durant, B. A. Leland, D. R. Henry and J. G. Nourse, J. Chem. Inf. Comput. Sci., 2002, 42, 1273–1280 Search PubMed.
- H. Moriwaki, Y.-S. Tian, N. Kawashita and T. Takagi, J. Cheminf., 2018, 10, 4 Search PubMed.
- T. Chen and C. Guestrin, Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, 2016, pp. 785–794 Search PubMed.
- J. Wu, X.-Y. Chen, H. Zhang, L.-D. Xiong, H. Lei and S.-H. Deng, J. Electron. Sci. Technol., 2019, 17, 26–40 Search PubMed.
- A. H. Victoria and G. Maragatham, Evol. Syst., 2021, 12, 217–223 CrossRef.
- G. M. Visani, M. C. Hughes and S. Hassoun, Bioinformatics, 2021, 37, 2017–2024 CrossRef CAS.
- M. H. S. Segler and M. P. Waller, Neural-Symbolic Machine Learning for Retrosynthesis and Reaction Prediction, Chem. – Eur. J., 2017, 23, 5966–5971 CrossRef CAS.
- P. P. Plehiers, G. B. Marin, C. V. Stevens and K. M. J. Van Geem, ChemInform, 2018, 10, 11 CrossRef.
- L. Bar-Peled and N. Kory, Nat. Metab., 2022, 4, 1232–1244 CrossRef.
- S. Kok, B. U. Kozak, J. T. Pronk and A. J. A. Maris, FEMS Yeast Res., 2012, 12, 387–397 CrossRef.
- S. Nath, Theory Biosci., 2022, 141, 249–260 CrossRef CAS.
- M. H. Abernathy, L. He and Y. J. Tang, Biotechnol. Adv., 2017, 35, 805–814 CrossRef CAS.
- K. Jørgensen, A. V. Rasmussen, M. Morant, A. H. Nielsen, N. Bjarnholt, M. Zagrobelny, S. Bak and B. L. Møller, Curr. Opin. Plant Biol., 2005, 8, 280–291 CrossRef.
- V. Pareek, Z. Sha, J. He, N. S. Wingreen and S. J. Benkovic, Mol. Cell, 2021, 81, 3775–3785 CrossRef CAS PubMed.
- M. Kim and K.-B. Hwang, PLoS One, 2022, 17, e0271260 CrossRef CAS.
- Z. Lin, H. Akin, R. Rao, B. Hie, Z. Zhu, W. Lu, N. Smetanin, R. Verkuil, O. Kabeli, Y. Shmueli, M. Fazel-Zarandi, T. Sercu, S. Candido and A. Rives, Science, 2023, 379, 1123–1130 CrossRef CAS PubMed.
- T. Yu, H. Cui, J. C. Li, Y. Luo, Y. G. Jiang and H. Zhao, Science, 2023, 379, 1358–1363 CrossRef CAS PubMed.
- D. Jiang, Z. Wu, C. Y. Hsieh, G. Chen, B. Liao, Z. Wang, C. Shen, D. Cao, J. Wu and T. Hou, J. Cheminf., 2021, 13, 1–23 Search PubMed.
- Y. Wang, Z. Li and A. B. Farimani, Machine Learning in Molecular Sciences, 2023, pp. 21–66 Search PubMed.
- M. Tang, B. Li and H. Chen, Curr. Opin. Struct. Biol., 2023, 81, 102616 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4me00118d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |