
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding stable adsorption states in flexible soft porous coordination polymers through free energy profiles†
James E.
Carpenter
 a,
Jean Galliano Vega
Díaz
b,
Johnathan
Robinson
c and
Yamil J.
Colón
*a
a,
Jean Galliano Vega
Díaz
b,
Johnathan
Robinson
c and
Yamil J.
Colón
*a
aDepartment of Chemical and Biomolecular Engineering, University of Notre Dame, USA. E-mail: ycolon@nd.edu
bDepartment of Chemical Engineering, University of Puerto Rico – Mayagüez, USA
cMork Family Department of Chemical Engineering and Materials Science, University of Southern California, USA
First published on 11th December 2024
Abstract
Soft porous coordination polymers (SPCPs) are flexible porous materials comprised of metal–organic polyhedrons (MOPs) connected by organic linkers, with potential in adsorption applications. We performed molecular simulations of various SPCPs that vary in the length and flexibility of the organic linkers to address how the flexibility can result in various configurations and affects adsorption performance. We examined free energy profiles as a function of volume of different SPCPs while varying methane loading, resulting in different stable configurations. We found significant differences in the volume of the stable configurations and their number for the various structures, with more flexible linkers having more stable configurations in free energy. We also characterized the textural properties and methane adsorption isotherms of the stable configurations for the SPCPs and analyzed density profiles of the adsorption in the various configurations. Altogether, our examination can be used to predict the relevant configurations of the SPCPs at a given loading and provides molecular-level understanding of how the flexibility of the organic linkers affects the structure of the system and adsorption performance.
Design, System, ApplicationSoft porous coordination polymers (SPCPs) are an important class of flexible porous materials with applications for separations, sensing, catalysis, storage, etc. We examine herein the effect of methane adsorbate loading on free energy profiles to determine different stable configurations. It becomes clear in our examination that the choice of flexible linker has a strong impact on both the number of stable states and the range of volumes possible for an SPCP. For each of these stable configurations we also examined textural properties such as surface area, volume, and pore size distribution, as well as adsorption isotherms and specifics of pore filling behavior. From the examination of different SPCP configurations we are able to determine the stable state at any given adsorbate loading and gain an understanding of how the adsorbate affects the organic linker and thus the SPCP structure and adsorption. From this understanding we can better design SPCPs for specific applications. |
Introduction
Soft porous coordination polymers (SPCPs) are materials that are composed of metal–organic polyhedron (MOP)1–4 nodes interconnected by flexible organic linkers, either in a crystalline topology or an amorphous structure (Fig. 1).5,6 This composition allows flexibility and thus response to external stimulus.7,8 It also contributes to the sheer scale of design space that is available, similar to the space available to metal–organic frameworks (MOFs).9 SPCPs are inherently flexible, allowing high mechanical stability as gels and films comparable to polymers of intrinsic microporosity (PIMs),10 and being solution processable,11 but maintain permanent porosity through the MOPs. As a class of materials, SPCPs show potential for adsorption applications such as gas storage,12–16 separations,17–20 catalysis,21–23 sensing,24,25 and drug delivery.26–29 In addition, SPCPs have recently been shown by Wang et al. to be capable of simultaneous detection and removal of pharmaceuticals at trace-levels due in part to their processability and mechanical stability, as well as their tunable pore structure and composition.20 Given their potential in these applications and more, there arises a need to understand the molecular-level details that determine their properties, including adsorption characteristics, while accounting for the flexible nature of SPCPs.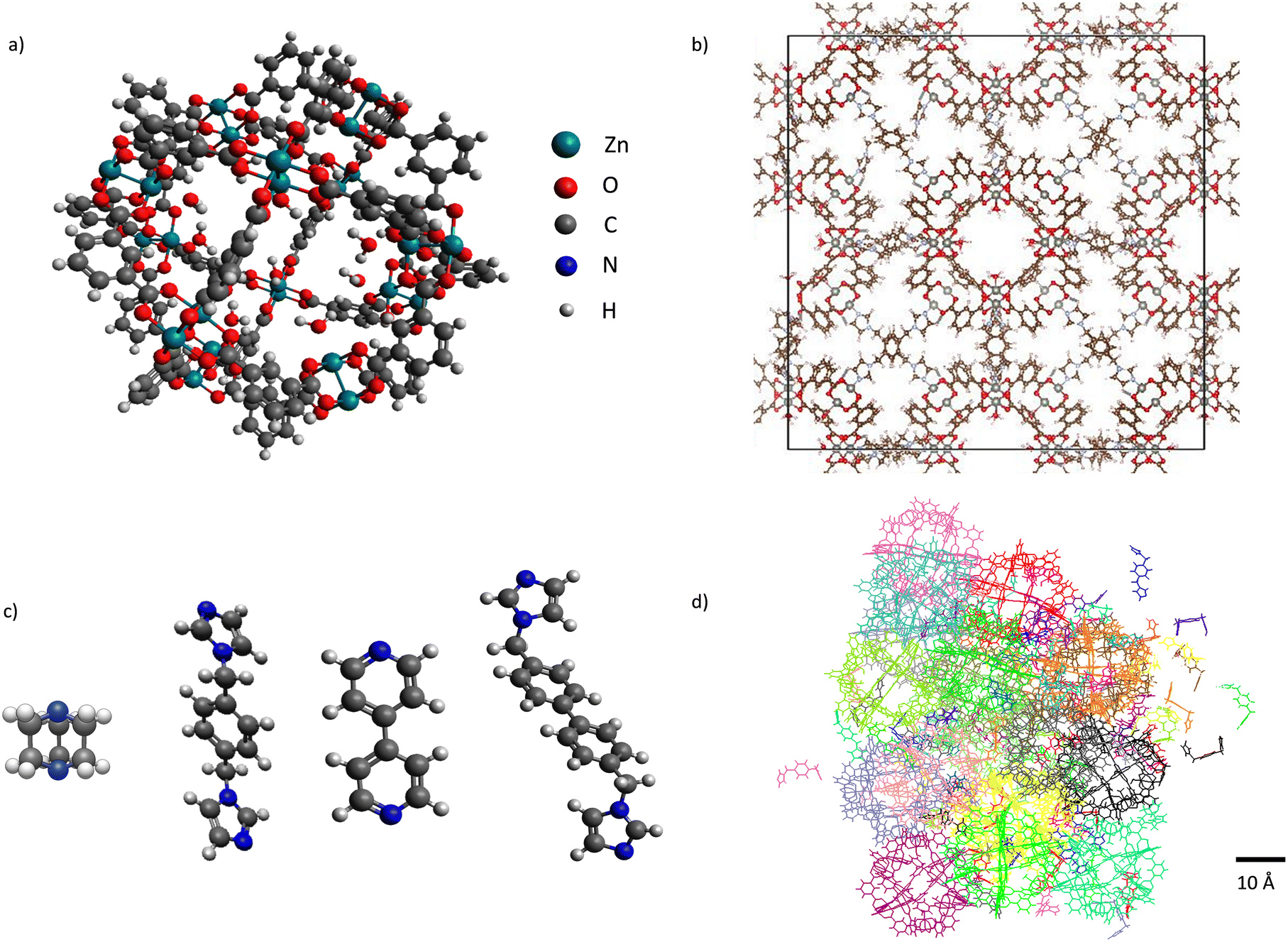 | ||
| Fig. 1 Left: SPCP building blocks – a) ZnMOP node and c) Dabco, bix, bipyridine, and bibPh linkers. Right: b) crystalline structure of ZnMOP–bix in the fcu topology, and d) an amorphous colloid construction of ZnMOP–bix. Reproduced from ref. 6, with the permission of AIP Publishing. | ||
Methods for simulating adsorption in flexible porous materials include flexible snapshot methods, the ellipsoid model, and hybrid Monte Carlo/molecular dynamics. Flexible snapshot methods successfully incorporate flexibility into simulations and predict loadings and deliverable capacities more accurately than rigid methods, but do not account for deformations induced by the adsorbate itself.30,31 This method has been applied to a subset of the CoRE MOF database32 where, for MOFs that do not show a significant volume change, the impact of flexibility on uptake was only notable for non-dilute conditions, whereas selectivity was significantly affected in both dilute and non-dilute conditions.30 It has also been shown that the flexible snapshot method can be applied to systems such as C8 aromatic adsorption in MIL-47 and MOF-48 where flexibility plays a significant role as long as adsorbed molecules do not strongly affect the ensemble of structures available to the porous material.33 The ellipsoid method allows prediction of the importance of framework flexibility for materials based on atomic positions and thermal parameters without implementing a force field, allowing insights on adsorption as long as an experimental crystal structure is available, but again does not account for adsorption induced deformation.31,34–36 This method has been applied to 15 randomly selected MOFs from the CoRE database where it saw some success in making qualitatively accurate predictions for loadings under non-dilute conditions and Henry's constants under dilute conditions.31 Hybrid Monte Carlo/molecular dynamics methods are carried out by iterating grand canonical Monte Carlo (GCMC) and molecular dynamics simulations, incorporating the flexibility of the porous material and effects of the adsorbate on structure deformation, allowing determination of the structure of the porous material and accurate prediction of adsorption behavior.37–39 Hybrid methods have been applied to the set of MOFs IRMOF-1, UiO-66, ZIF-8, and MIL-53 and compared to experimental results, and the qualitative behavior of the isotherms was captured, including the phase transition in MIL-53 from narrow-pore to large-pore.31 Systematic differences in the isotherms are attributed to inaccuracies arising from the force field UFF4MOF.40 Hybrid methods have also been applied to soft polymers such as PIMs41 and polyimides.42,43 In this application they have shown the ability to capture the swelling and plasticization behavior that significantly affect the structure and adsorption behavior of soft polymers, and allow an examination of how swelling affects specific materials and may improve performance in PIM-1.41 However, the high computational cost associated with these methods limits their application for high-throughput studies.31
One existing method for the analysis of the mechanics of and adsorption in flexible porous materials is in work by Evans and coworkers44 based on previous works examining both rigid and flexible MOFs.45,46 In their paper, they examine the structure and behavior of the flexible crystalline material DUT-49 through both a thermodynamic lens and the mechanics of the linker present in the system. Through their examination, they determined how the phenomenon of negative gas adsorption arises as a result of the shift in the stable configuration of DUT-49. Essentially, the interaction of adsorbing methane with the flexible framework causes a transition to the closed-pore phase due to a buckling of the linker that expels methane from the framework, resulting in the drop in loading with increasing pressure that defines negative gas adsorption. In this paper we implement this method to examine adsorption in SPCPs and build an understanding of the impact flexibility has on the structure of SPCPs and adsorption.
Herein we examine stable configurations of crystalline SPCPs at various adsorbate loadings through their free energy profiles, allowing us to account for flexibility, including adsorption induced deformation, and use these insights to select relevant SPCP configurations. For each relevant configuration we then apply grand canonical Monte Carlo (GCMC) methods to obtain methane adsorption isotherms at 298 K. In the process, we additionally explore the effect of linker flexibility on stable configurations of SPCPs and pore-filling behavior through GCMC density profiles.
Methods
All-atom molecular dynamics (MD) simulations are run in LAMMPS.47 SPCP .cif files are produced in ToBaCCo 3.0 (ref. 48) using the 1,4-bis(imidazole-1-ylmethyl)benzene (bix), bipyridine (bipy), 4,4′-bis(imidazole-1-ylmethyl)biphenyl (bibPh), and 1,4-diazabicyclo[2.2.2]octane (Dabco) linkers, MOP-1 (metal–organic polyhedron) analogs with Zn in the paddlewheel as nodes, and the fcu topology.49 These .cifs are converted to LAMMPS data files with UFF4MOF parameters50,51 in lammps-interface.52Free energy profiles as a function of unit cell volume are obtained using thermodynamic integration on pressure and volume data obtained from MD simulations.44Eqn (1) is used, where the change in free energy, denoted F, is found by numerically integrating average system pressures at a given volume V′ (〈Pi(V′)〉) from a reference volume (Vref) to a volume of interest V.
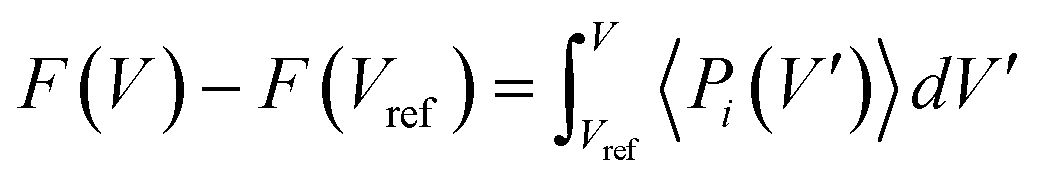 | (1) |
SPCP textural properties are calculated using Zeo++.53,54 LAMMPS data files are converted into cssr files and passed into Zeo++ to obtain surface area, occupiable volume, and pore size distributions. All properties are obtained using a probe size of 1.4 Å, equivalent to a helium atom. Surface area is obtained using 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 samples, while volumes and pore size distributions use 50000 samples.
000 samples, while volumes and pore size distributions use 50000 samples.
Adsorption simulations are run using RASPA.55 Isotherms and density profiles are obtained from GCMC simulations, using 100000 initialization cycles and 100000 production cycles with translation and swap (insertion/deletion) moves with relative probabilities of 0.5 and 1.0 respectively. The same forcefields are used as in MD simulations, UFF4MOF for the crystal structure and TraPPE for methane.
Visualizations of crystalline SPCP structures are done using VESTA,56 and visualization of gas density profiles is done with Paraview.57
Results and discussion
Free energies as a function of loading
From the free energy profiles obtained as described above, we can determine the stable state of a crystalline SPCP at a given loading as the point with minimum free energy. We focus first on the Dabco system (Fig. 2). The free energy plot examines the relationship between side length of a cubic unit cell and the free energy of the system, with one curve per methane loading. In the free energy profile we see only one distinct minimum for each loading, indicating a single stable phase for all loadings. Additionally, we see that this minimum is consistent across loadings, except for a slight increase in stable volume at high loading, which we attribute to a slight stretching of the Dabco linker due to the high methane loading. This is in line with the fact that Dabco is the stiffest linker we examine, and when we visualize the crystal structure we see a consistent configuration. Based on the single stable phase for Dabco, we can calculate the adsorption for this system using the crystal structure corresponding to the minimum free energy.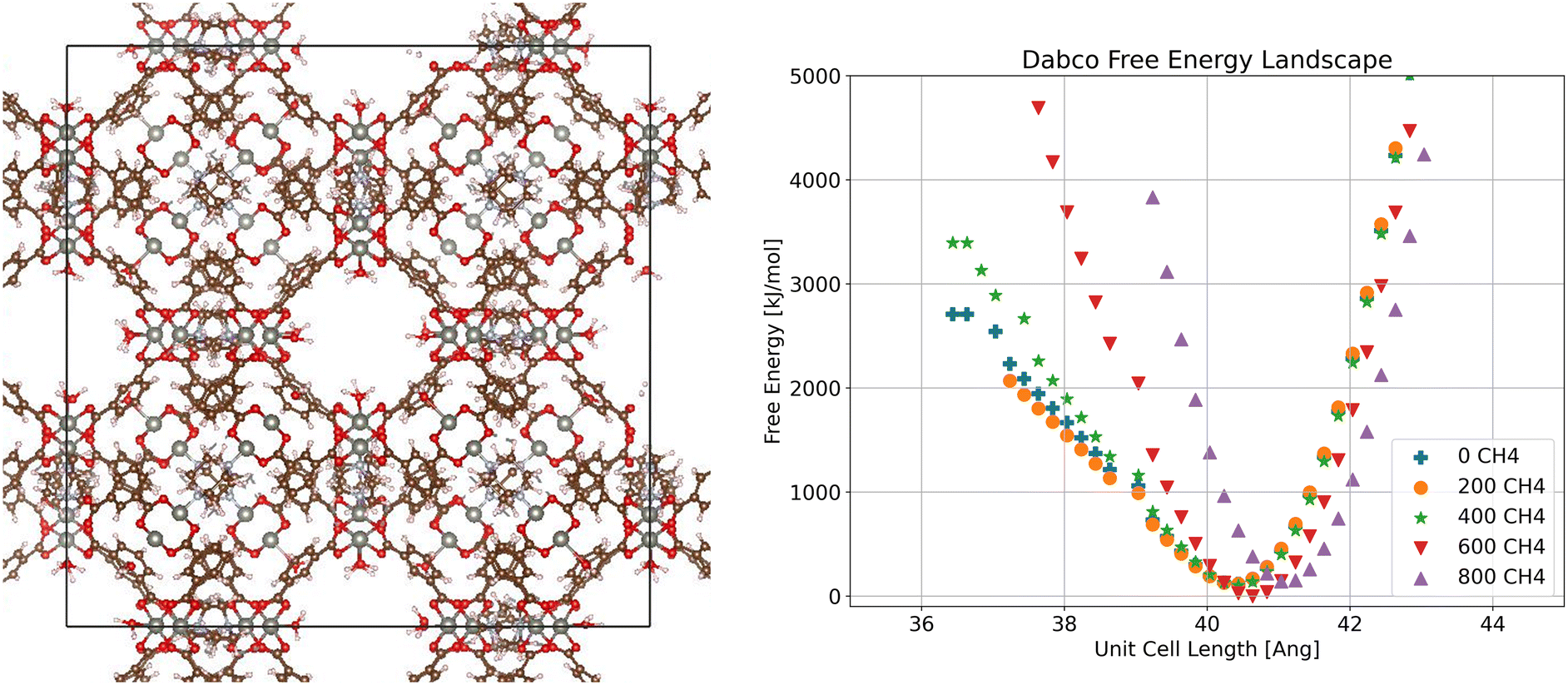 | ||
| Fig. 2 Visualization of crystalline ZnMOP–Dabco system and free energy landscape of the system as a function of unit cell side length and methane loading in molecules/unit cell. | ||
Moving now to the bipy system (Fig. 3), we see a constant global free energy minimum for all loadings. However, we also see another minimum at lower volume at low loadings (0 and 200 molecules per unit cell), corresponding to a metastable state. This indicates that it is possible for the bipy system to enter a closed pore state, though not favorable at 298 K with methane loading. This suggests that two different states may be observed at different temperatures, as temperature can dramatically change the free energy minima for these systems, as is the case with DUT-49,44,58 and as a result adsorption behavior.
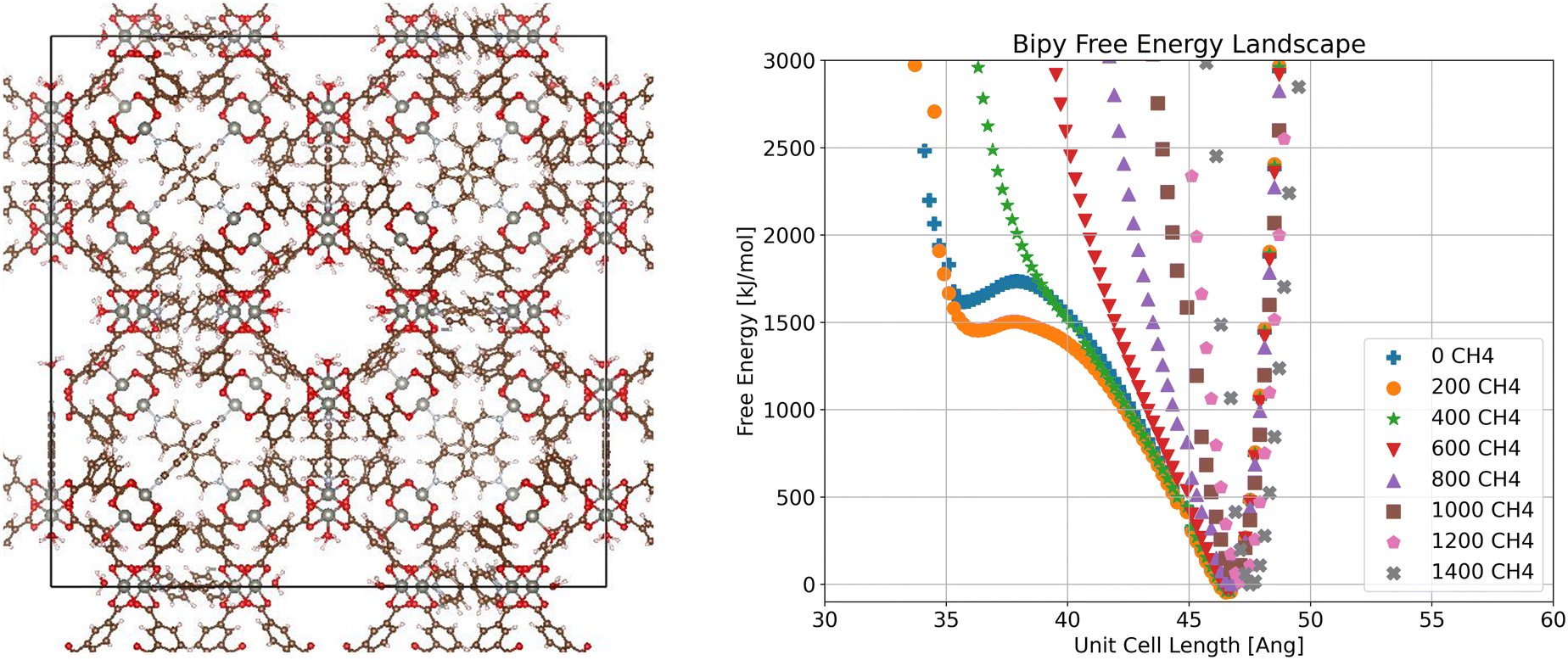 | ||
| Fig. 3 Visualization of crystalline ZnMOP–bipy system and free energy landscape of the system as a function of unit cell side length and methane loading in molecules per unit cell. | ||
Examining next the bix system (Fig. 4), we observe a shift in the global minimum as methane loading increases. At low loadings the low volume configuration is the most stable, a closed-pore configuration possible because of the flexibility of the bix linker allowing bix to fold. As methane loading increases, the stable volume increases slightly as seen at 400 molecules per unit cell. By 600 molecules per unit cell, the free energy minimum corresponding to the closed pore state has disappeared entirely, resulting in a single wide energy well between the closed pore and open pore states. At a loading of 1000 molecules per unit cell, the energy minimum corresponding to the open pore state starts to appear and becomes more pronounced as loading increases.
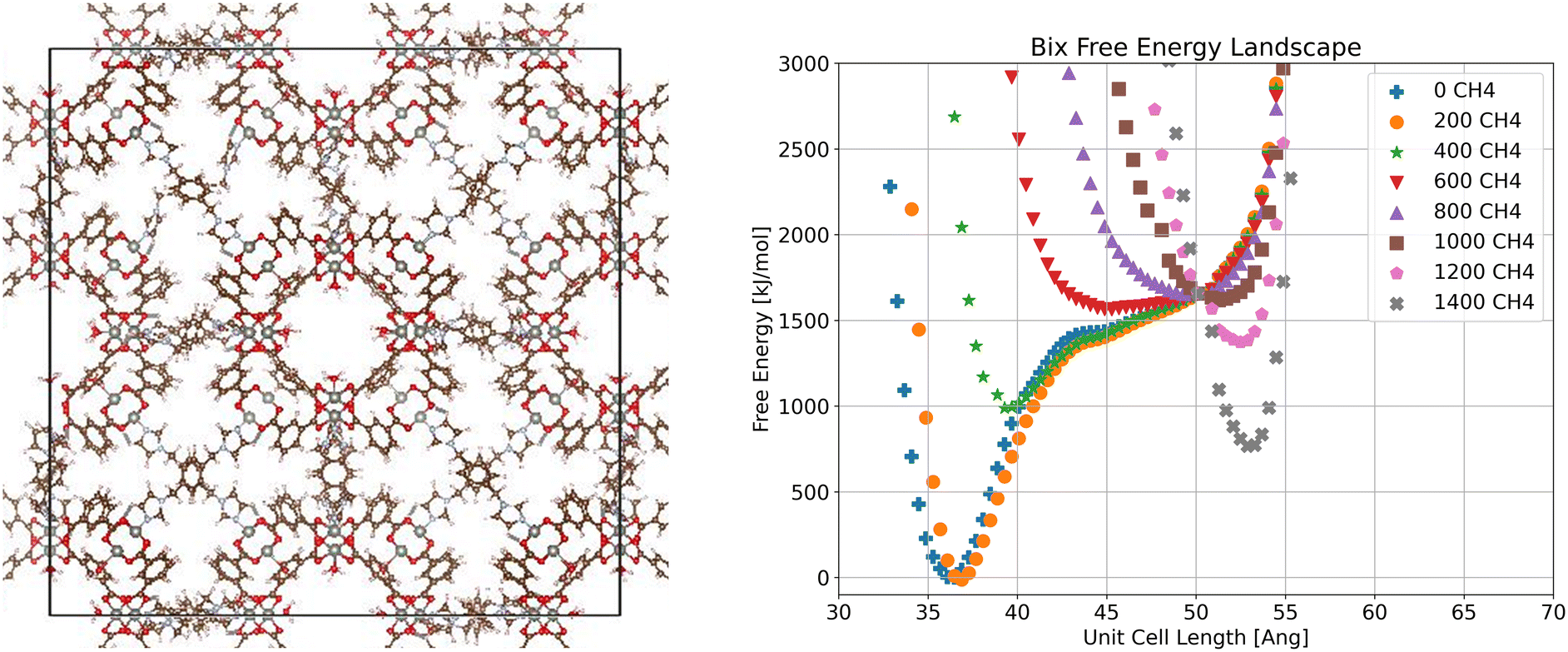 | ||
| Fig. 4 Visualization of crystalline ZnMOP–bix system and free energy landscape of the system as a function of unit cell side length and methane loading in molecules per unit cell. | ||
In the bibPh system (Fig. 5), we again see multiple global minima based on the loading of methane but observe a continuous change in volume instead of a switch between distinct configurations, as for bix. We do still see a fully closed pore state at low loading, but the energy minimum has shifted to a higher volume by 200 molecules per unit cell and only reaches the fully open pore state at the highest examined loading of 1300 molecules per unit cell. As bibPh is the most flexible linker examined, we attribute this to multiple configurations of the linker causing it to vary in length. It is also worth noting that the bibPh system has the widest free energy wells of any of the systems we examine, and thus has the least precisely set stable configuration.
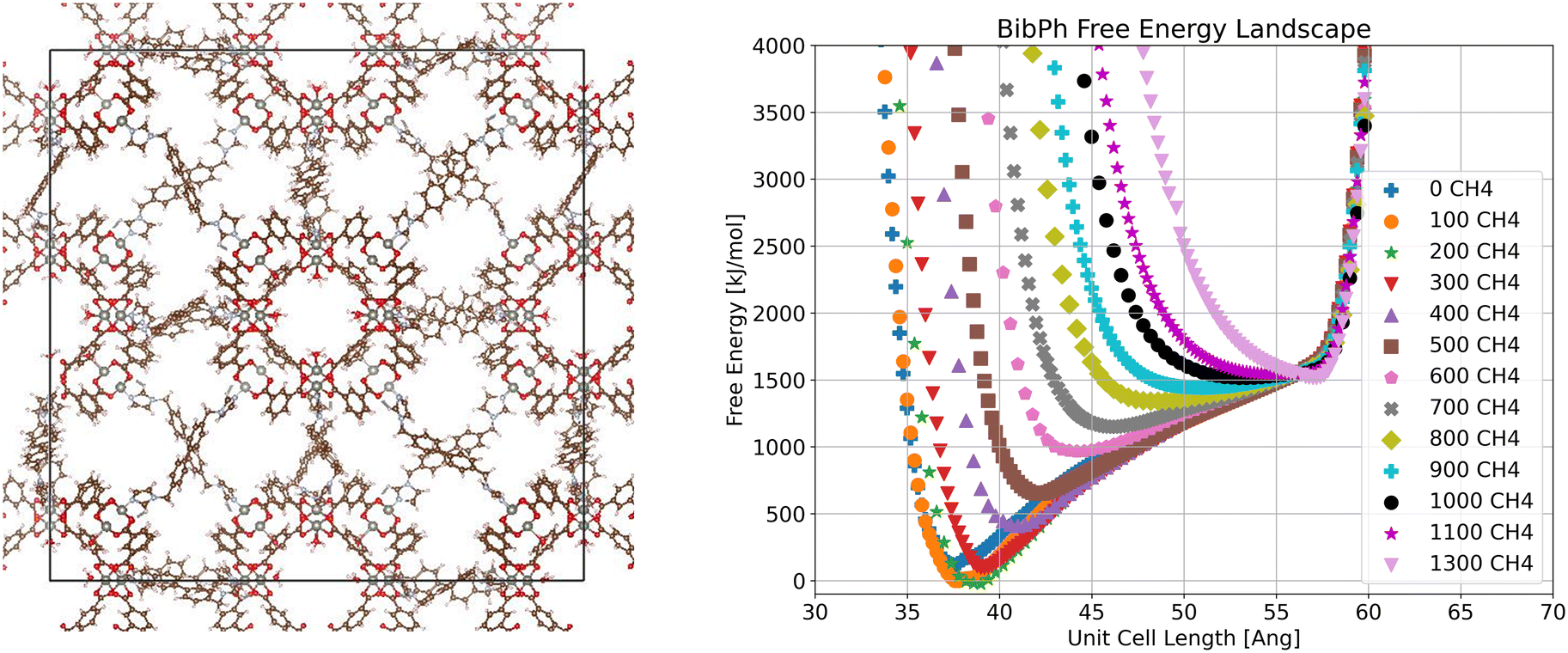 | ||
| Fig. 5 Visualization of crystalline ZnMOP–bibPh system and free energy landscape of the system as a function of unit cell side length and methane loading in molecules per unit cell. | ||
From the differences in free energy profiles between each system, we can see that a primary factor involved in different states of the SPCP is the linker configuration, whether it is in an extended or bent state. To corroborate this, we examine the ZnMOP–bix system visually (Fig. 6). We can see clearly that the low volume configurations (side lengths 36.4 Å and 39.2 Å) are contracted because of a collapsed bix linker. In the high volume configurations (side lengths 49.6 Å and 52.8 Å), the linkers are extended, opening the pore structure. We similarly analyze the ZnMOP–bipy system and find again that in the low volume configuration the linkers are folded, and in the high volume system they are extended (Fig. S1†).
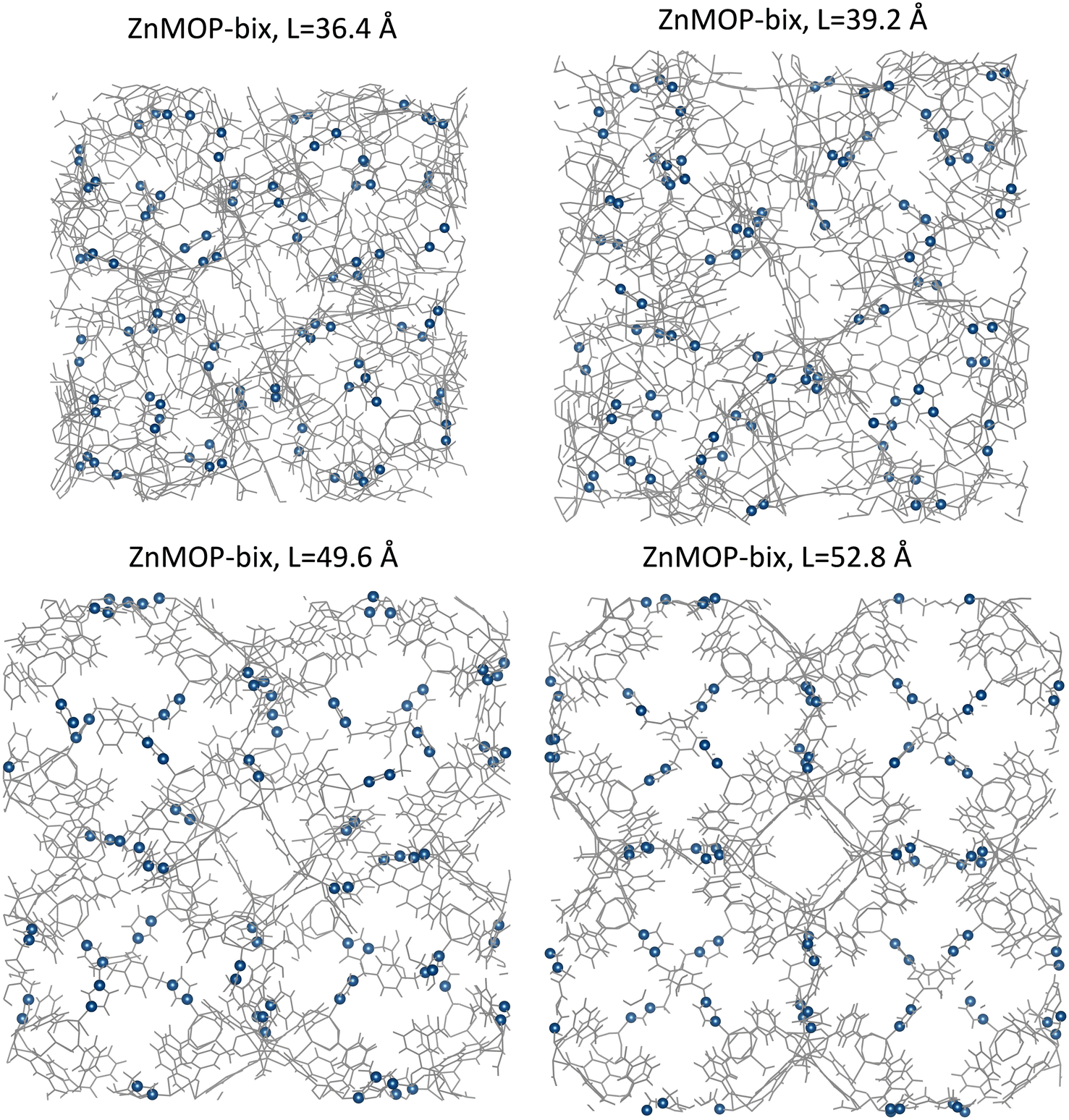 | ||
| Fig. 6 Visualization of crystalline ZnMOP–bix system at the various free energy minima that appear in Fig. 4. Highlighted in blue are the nitrogen atoms present in each bix linker. Upper panels: closed pore, low volume states. Lower panels: open pore, high volume states. | ||
Textural properties as a function of loading
We next examine the properties of each system at each volume, including surface area, accessible pore volume, accessible volume fraction, and pore size distribution.For the accessible volume and volume fraction (Table 1) we see the trends we expect: increasing loading pushes the more flexible SPCPs towards an open configuration, increasing the accessible volume and volume fraction. For surface area, we see a more nuanced trend, increasing surface area per volume to start but reaching a maximum and then decreasing as the SPCP approaches its fully expanded state at high loadings.
| Linker | Unit cell length [Å] | Surface area [m2 cm−3] | Accessible volume [cm3 g−1] | Accessible volume fraction |
|---|---|---|---|---|
| Dabco | 40.37 | 2266 | 0.620 | 0.398 |
| Bipy | 35.69 | 2129 | 0.141 | 0.142 |
| 46.49 | 1847 | 1.230 | 0.555 | |
| Bix | 36.4 | 2105 | 0.141 | 0.138 |
| 39.2 | 2543 | 0.299 | 0.235 | |
| 49.6 | 1739 | 1.537 | 0.596 | |
| 52.8 | 1474 | 2.066 | 0.664 | |
| BibPh | 37.58 | 2125 | 0.171 | 0.163 |
| 42.18 | 2463 | 0.505 | 0.340 | |
| 46.18 | 2171 | 0.933 | 0.479 | |
| 50.98 | 1731 | 1.581 | 0.603 | |
| 56.98 | 1277 | 2.617 | 0.715 |
For pore size distributions (Fig. 7), we see two distinct trends with increasing volume. First, increasing unit cell volume corresponds with increasing pore sizes, making clear the transition between closed pore and open pore states for the bipy, bix, and bibPh systems. We do see one consistent pore size between all linkers and unit cell volumes at 8 Å, corresponding to the ZnMOP interior pore. The second trend we see is a sharpening of the peaks, indicating more consistent pore sizes at larger volumes. These can be explained by the difference in the number of possible linker configurations available; at high volumes the linkers are fully extended and fixed in one configuration, resulting in more consistent pores. We also see a primary set of pores that appear in each material. A set of small pores between 5 and 10 Angstroms corresponding to the MOP interior pore, a medium grouping of pores, and a large set of pores. This is best visualized in Fig. S4.†
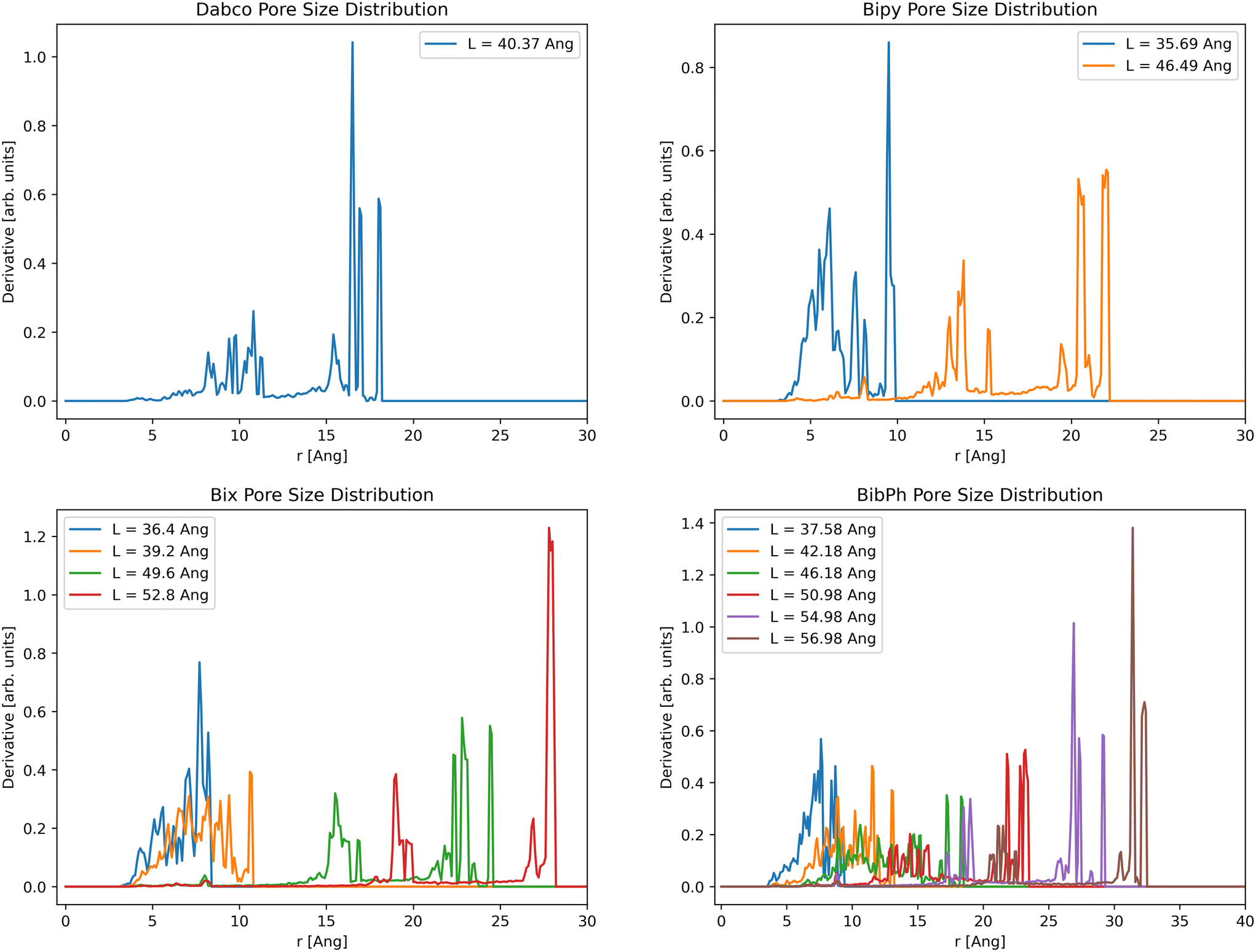 | ||
| Fig. 7 Pore size distributions for each system at various free energy minima, denoted in the legend by the side length of the unit cell. The x-axis for each plot is the pore size, and the y-axis is change in accessible volume with respect to probe size. | ||
Adsorption in different SPCP structures
For each stable SPCP state determined from free energy profiles we run an adsorption isotherm calculation in RASPA (Fig. 8). From there we can determine the loading and pressure for an isotherm based on the fixed volume of the stable state. For the Dabco and bipy systems we only have one state of interest, as seen from the free energy profiles which contain only one global minimum. In the bix system we have two main states: the open pore, and the closed pore, and a few intermediate states during the transition. Lastly, in the bibPh system, we have a continuous transition, so it becomes harder to determine where the isotherms for this system fit together. However, since we know the most stable configuration for each loading, and the corresponding pressures for each loading, we select the relevant isotherm and find the correct pressure for a given loading. As such, the overall isotherm for the fully flexible system can be obtained by stitching together the isotherms obtained from the separate rigid structures.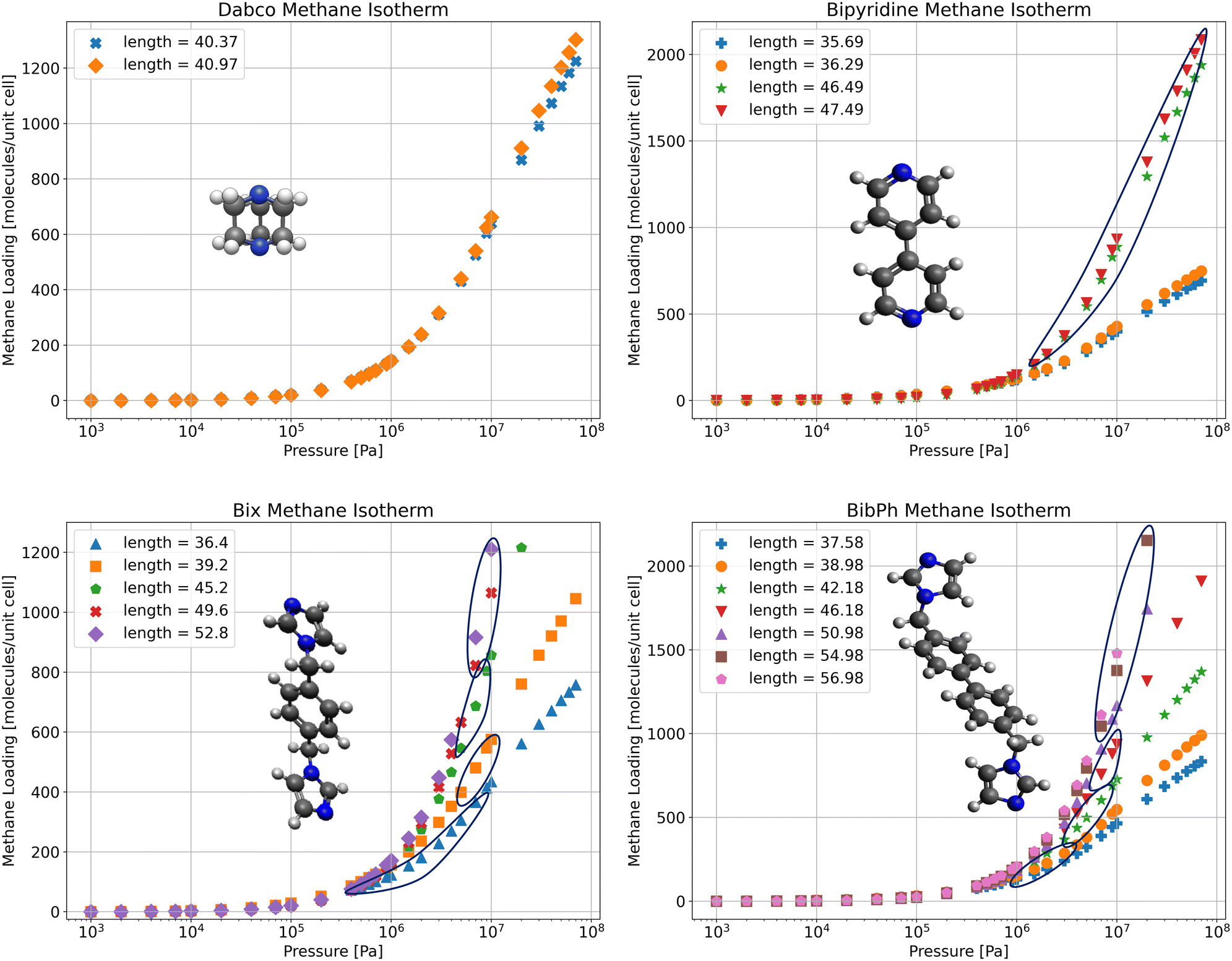 | ||
| Fig. 8 Methane adsorption isotherms of the stable configurations for each crystalline system – ZnMOP–Dabco, ZnMOP–bipy, ZnMOP–bix, ZnMOP–bibPh. Also included are circles around the stable region for a given loading for any system with distinct isotherms as a result of their flexibility. | ||
Density profiles
We move now to determine the locations at which adsorbate molecules accumulate within SPCP frameworks during adsorption. To do so, we examine density profiles and find which pores fill over the course of a full adsorption isotherm. In Fig. 9 we visualize methane density in ZnMOP–bipy, one of the systems with a single free energy minimum. We see that the regions of highest density are always the metal paddlewheel sites. Outside of that, the MOP interior pore sees localization of methane at low pressures, indicating that these are the first pores to begin filling during methane uptake. As pressure increases, methane density remains high at the metal paddlewheels, and otherwise becomes more diffuse around the system.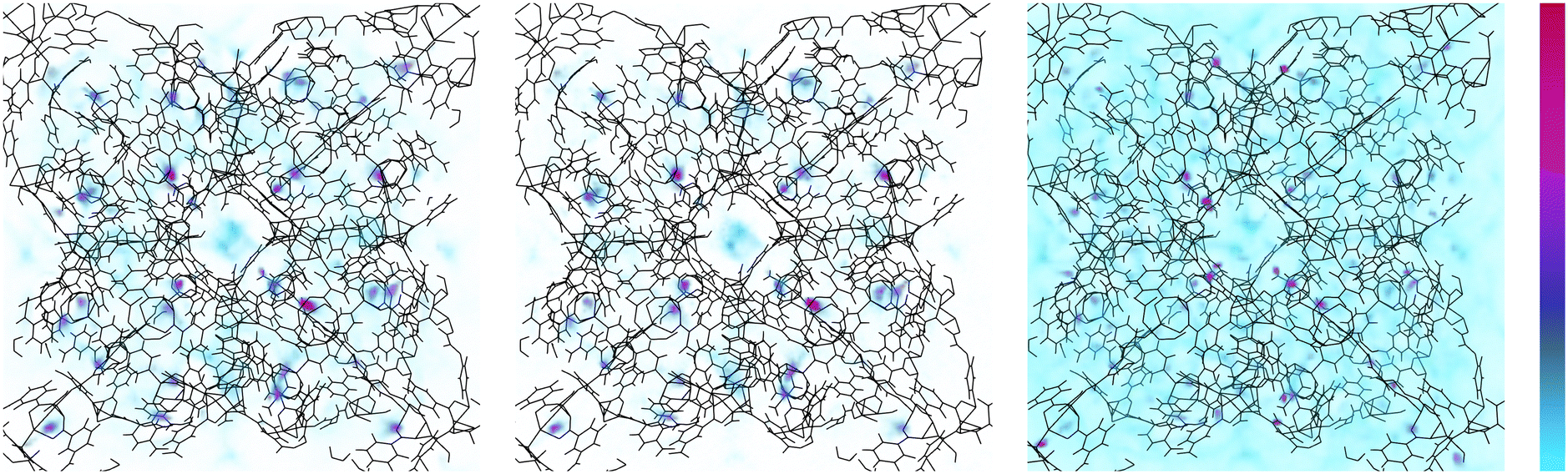 | ||
| Fig. 9 Density profiles of methane adsorption in ZnMOP–bipy with unit cell side length 46.49 Å. Increasing pressure from left to right (7 × 104, 7 × 105, 4 × 107 Pa). Scale is shown on the right, light blue is low density, red is high density. | ||
Examining now the pore-filling behavior in a system which exhibits a change in stable configuration during adsorption, we look at methane adsorption density in ZnMOP–bix (Fig. 10). Once again, we see high methane density at the metal paddlewheel sites at all pressures. However, at low pressures, in the closed pore configuration we see a high methane density in a ring-like formation around the MOP and lower density inside the MOP. This remains the case with increasing pressure until the structure transitions to the open pore configuration, at which point we see similar behavior as in ZnMOP–bipy, with methane becoming more diffuse throughout the system. Importantly, this indicates that the MOP interior pore plays a role in the transition between the closed pore and open pore states.
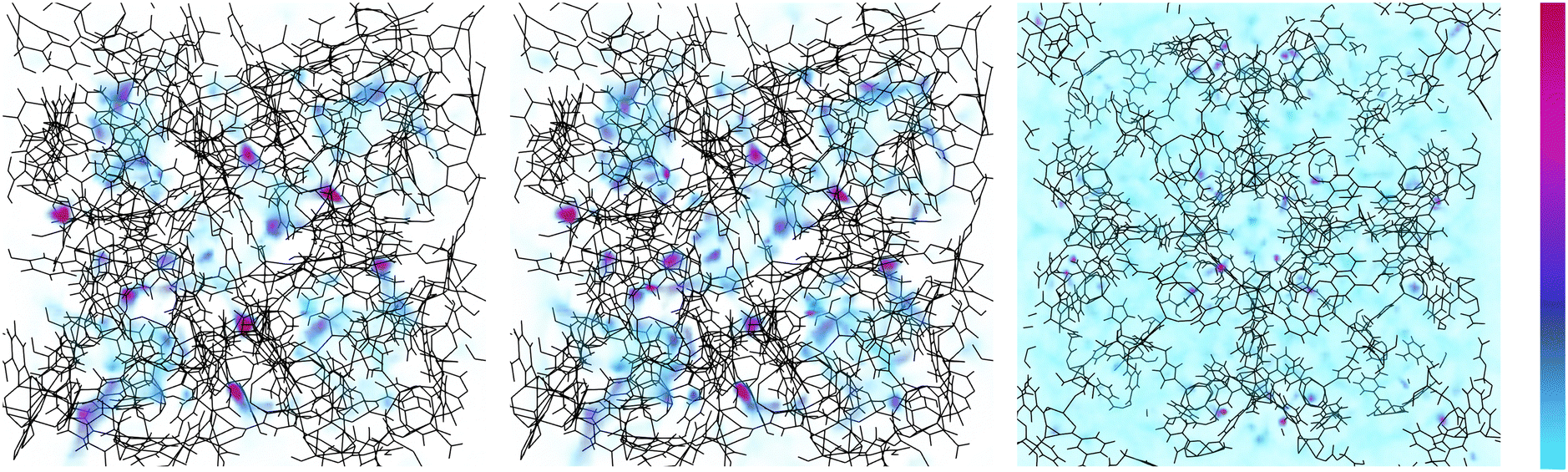 | ||
| Fig. 10 Density profiles of methane adsorption in ZnMOP–bix. Increasing pressure from left to right (7 × 104, 7 × 105, 2 × 107 Pa). States shown are the minimum free energy state; left and center are the closed pore state (unit cell side length 36.4 Å) far right is the open pore state (unit cell side length 52.8 Å). Scale is shown on the right, light blue is low density, red is high density. | ||
Conclusion
We have examined here the use of free energy profiles to determine stable states of four SPCPs with different linkers at different adsorbate loadings. Relative free energies obtained from molecular dynamics simulations offer a computationally inexpensive way to determine the relevant configuration of inherently flexible structures in cases where adsorption has a direct effect on the state of the porous material. We also examined the effect of linker choice on the number and volume of distinct configurations an SPCP has, and on how the methane is distributed through the system, and thus the relation between adsorbate and adsorbent. In our analysis we found that the stiffest linkers, Dabco and bipy, result in an SPCP with one primary global free energy minimum, and therefore one stable phase. The other linkers, bix and bibPh, are more flexible, and result in an SPCP with different stable phases depending on methane loading, with increased methane loading corresponding to more open pores. For textural properties, we see consistent increases in accessible volume as the SPCP transitions to an open pore configuration. For surface area, we see that the highest point for the bix and bibPh linkers is slightly above the fully closed-pore configuration, and for the bipy linker the closed pore configuration has the highest surface area. Finally, we used density profiles to observe the pore filling behavior of each SPCP. This analysis shows interesting behavior in the pore filling behavior that occurs in these SPCPs. For the stiff linker bipy, we see that there is a strong localization of methane around the Zn metal sites, and a weaker localization in the MOP interior pore. As the pressure increases, there remains a localization of methane at the metal sites and a more diffuse distribution through the rest of the SPCP. For the more flexible bix linker, initial localization of methane again occurs at the metal site, but not in the MOP interior pore, resulting in a ring structure around it. At high pressure, behavior is like the bipy SPCP, with localization around the metal sites and through the rest of the pore structure.This work showcases the impact of linker flexibility on the adsorption behavior of SPCPs, allowing us to see how the configuration of the SPCP is directly related to the loading of the adsorbing gas and its interaction with the SPCP structure. The use of free energy profiles allows us to determine the stable phase of the SPCP for any specific loading, and as a result the structure that should be used for a GCMC simulation to obtain a point on the isotherm of the fully flexible SPCP.
Future works will expand on the thermodynamic region explored for these materials. This includes exploring other temperatures, as well as different SPCPs, using active learning techniques59,60 to obtain as much information about the adsorption space as possible in as few simulations as possible.
Data availability
Input scripts are available in this project's Github repository: https://github.com/jec34/SPCP_FEP_Adsorption.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors thank American Chemical Society Petroleum Research Fund #66075-DNI5 for financial support. Authors also thank the University of Notre Dame Center for Research Computing for computational resources. This work was partially supported by the National Science Foundation (NSF) under award EEC-2244410, and we gratefully acknowledge this support.References
- V. Guillerm, D. Kim, J. F. Eubank, R. Luebke, X. Liu and K. Adil, et al. A supermolecular building approach for the design and construction of metal–organic frameworks, Chem. Soc. Rev., 2014, 43(16), 6141–6172 RSC.
- D. Kim, X. Liu and M. S. Lah, Topology analysis of metal–organic frameworks based on metal–organic polyhedra as secondary or tertiary building units, Inorg. Chem. Front., 2015, 2(4), 336–360 RSC.
- A. Khobotov-Bakishev, L. Hernández-López, C. von Baeckmann, J. Albalad, A. Carné-Sánchez and D. Maspoch, Metal–Organic Polyhedra as Building Blocks for Porous Extended Networks, Adv. Sci., 2022, 9(11), 2104753 CrossRef CAS.
- Z. Wang, C. V. Santos, A. Legrand, F. Haase, Y. Hara and K. Kanamori, et al. Multiscale structural control of linked metal–organic polyhedra gel by aging-induced linkage-reorganization, Chem. Sci., 2021, 12(38), 12556–12563 RSC.
- Z. Wang, C. V. Santos, A. Legrand, F. Haase, Y. Hara and K. Kanamori, et al. Multiscale structural control of linked metal–organic polyhedra gel by aging-induced linkage-reorganization, Chem. Sci., 2021, 12(38), 12556–12563 RSC.
- J. E. Carpenter and Y. J. Colón, Atomistic simulation of soft porous coordination polymers, J. Chem. Phys., 2023, 158(7), 074901 CrossRef CAS.
- N. Li, J. Pang, F. Lang and X. H. Bu, Flexible Metal–Organic Frameworks: From Local Structural Design to Functional Realization, Acc. Chem. Res., 2024, 57(16), 2279–2292 CrossRef CAS.
- Y. Li, Y. Wang, W. Fan and D. Sun, Flexible metal–organic frameworks for gas storage and separation, Dalton Trans., 2022, 51(12), 4608–4618 RSC.
- D. Kim, X. Liu and M. S. Lah, Topology analysis of metal–organic frameworks based on metal–organic polyhedra as secondary or tertiary building units, Inorg. Chem. Front., 2015, 2(4), 336–360 RSC.
- K. Polak-Kraśna, R. Dawson, L. T. Holyfield, C. R. Bowen, A. D. Burrows and T. J. Mays, Mechanical characterisation of polymer of intrinsic microporosity PIM-1 for hydrogen storage applications, J. Mater. Sci., 2017, 52(7), 3862–3875 CrossRef.
- A. Carné-Sánchez, G. A. Craig, P. Larpent, V. Guillerm, K. Urayama and D. Maspoch, et al. A Coordinative Solubilizer Method to Fabricate Soft Porous Materials from Insoluble Metal–Organic Polyhedra, Am. Ethnol., 2019, 131(19), 6413–6416 Search PubMed.
- A. C. Sudik, A. R. Millward, N. W. Ockwig, A. P. Côté, J. Kim and O. M. Yaghi, Design, Synthesis, Structure, and Gas (N2, Ar, CO2, CH4, and H2) Sorption Properties of Porous Metal-Organic Tetrahedral and Heterocuboidal Polyhedra, J. Am. Chem. Soc., 2005, 127(19), 7110–7118 CrossRef CAS PubMed.
- N. S. Bobbitt and R. Q. Snurr, Molecular modelling and machine learning for high-throughput screening of metal-organic frameworks for hydrogen storage, Mol. Simul., 2019, 45(14–15), 1069–1081 CrossRef CAS.
- Y. J. Colón, D. Fairen-Jimenez, C. E. Wilmer and R. Q. Snurr, High-Throughput Screening of Porous Crystalline Materials for Hydrogen Storage Capacity near Room Temperature, J. Phys. Chem. C, 2014, 118(10), 5383–5389 CrossRef.
- Y. Lin, C. Kong, Q. Zhang and L. Chen, Metal-Organic Frameworks for Carbon Dioxide Capture and Methane Storage, Adv. Energy Mater., 2017, 7(4), 1601296 CrossRef.
- A. Torrisi, C. Mellot-Draznieks and R. G. Bell, Impact of ligands on CO2 adsorption in metal-organic frameworks: First principles study of the interaction of CO2 with functionalized benzenes. I. Inductive effects on the aromatic ring, J. Chem. Phys., 2009, 130(19), 194703 CrossRef PubMed.
- J. Sang, F. Wei and X. Dong, Gas adsorption and separation in metal–organic frameworks by PC-SAFT based density functional theory, J. Chem. Phys., 2021, 155(12), 124113 CrossRef CAS PubMed.
- X. Zhao, Y. Wang, D. S. Li, X. Bu and P. Feng, Metal–Organic Frameworks for Separation, Adv. Mater., 2018, 30(37), 1705189 CrossRef.
- J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh and Y. J. Jeon, et al. A homochiral metal–organic porous material for enantioselective separation and catalysis, Nature, 2000, 404(6781), 982–986 CrossRef CAS.
- Z. Wang, I. Pacheco-Fernández, J. E. Carpenter, T. Aoyama, G. Huang and A. Pournaghshband Isfahani, et al. Pore-networked membrane using linked metal-organic polyhedra for trace-level pollutant removal and detection in environmental water, Commun. Mater., 2024, 5(1), 1–13 CrossRef.
- J. Liang, Z. Liang, R. Zou and Y. Zhao, Heterogeneous Catalysis in Zeolites, Mesoporous Silica, and Metal–Organic Frameworks, Adv. Mater., 2017, 29(30), 1701139 CrossRef PubMed.
- J. Jiao, C. Tan, Z. Li, Y. Liu, X. Han and Y. Cui, Design and Assembly of Chiral Coordination Cages for Asymmetric Sequential Reactions, J. Am. Chem. Soc., 2018, 140(6), 2251–2259 CrossRef CAS PubMed.
- A. Corma, H. García and F. X. Llabrés i Xamena, Engineering Metal Organic Frameworks for Heterogeneous Catalysis, Chem. Rev., 2010, 110(8), 4606–4655 CrossRef CAS.
- J. A. Greathouse, N. W. Ockwig, L. J. Criscenti, T. R. Guilinger, P. Pohl and M. D. Allendorf, Computational screening of metal–organic frameworks for large-molecule chemical sensing, Phys. Chem. Chem. Phys., 2010, 12(39), 12621–12629 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Metal–Organic Framework Materials as Chemical Sensors, Chem. Rev., 2012, 112(2), 1105–1125 CrossRef CAS PubMed.
- H. D. Lawson, S. P. Walton and C. Chan, Metal–Organic Frameworks for Drug Delivery: A Design Perspective, ACS Appl. Mater. Interfaces, 2021, 13(6), 7004–7020 CrossRef CAS.
- M. C. Bernini, D. Fairen-Jimenez, M. Pasinetti, A. J. Ramirez-Pastor and R. Q. Snurr, Screening of bio-compatible metal–organic frameworks as potential drug carriers using Monte Carlo simulations, J. Mater. Chem. B, 2014, 2(7), 766–774 RSC.
- C. Gaudin, D. Cunha, E. Ivanoff, P. Horcajada, G. Chevé and A. Yasri, et al. A quantitative structure activity relationship approach to probe the influence of the functionalization on the drug encapsulation of porous metal-organic frameworks, Microporous Mesoporous Mater., 2012, 157, 124–130 CrossRef CAS.
- P. Horcajada, C. Serre, M. Vallet-Regí, M. Sebban, F. Taulelle and G. Férey, Metal–Organic Frameworks as Efficient Materials for Drug Delivery, Am. Ethnol., 2006, 118(36), 6120–6124 Search PubMed.
- M. Agrawal and D. S. Sholl, Effects of Intrinsic Flexibility on Adsorption Properties of Metal–Organic Frameworks at Dilute and Nondilute Loadings, ACS Appl. Mater. Interfaces, 2019, 11(34), 31060–31068 CrossRef CAS PubMed.
- Z. Yu, D. M. Anstine, S. E. Boulfelfel, C. Gu, C. M. Colina and D. S. Sholl, Incorporating Flexibility Effects into Metal–Organic Framework Adsorption Simulations Using Different Models, ACS Appl. Mater. Interfaces, 2021, 13(51), 61305–61315 CrossRef CAS.
- Y. G. Chung, E. Haldoupis, B. J. Bucior, M. Haranczyk, S. Lee and H. Zhang, et al. Advances, Updates, and Analytics for the Computation-Ready, Experimental Metal–Organic Framework Database: CoRE MOF 2019, J. Chem. Eng. Data, 2019, 64(12), 5985–5998 CrossRef CAS.
- J. A. Gee and D. S. Sholl, Effect of Framework Flexibility on C8 Aromatic Adsorption at High Loadings in Metal–Organic Frameworks, J. Phys. Chem. C, 2016, 120(1), 370–376 CrossRef CAS.
- G. Y. Gor, P. Huber and N. Bernstein, Adsorption-induced deformation of nanoporous materials—A review, Appl. Phys. Rev., 2017, 4(1), 011303 Search PubMed.
- C. Balzer, R. T. Cimino, G. Y. Gor, A. V. Neimark and G. Reichenauer, Deformation of Microporous Carbons during N2, Ar, and CO2 Adsorption: Insight from the Density Functional Theory, Langmuir, 2016, 32(32), 8265–8274 CrossRef CAS.
- G. Y. Gor and A. V. Neimark, Adsorption-Induced Deformation of Mesoporous Solids, Langmuir, 2010, 26(16), 13021–13027 CrossRef CAS PubMed.
- D. M. Anstine, D. Tang, D. S. Sholl and C. M. Colina, Adsorption space for microporous polymers with diverse adsorbate species, npj Comput. Mater., 2021, 7(1), 1–9 CrossRef.
- S. M. J. Rogge, R. Goeminne, R. Demuynck, J. J. Gutiérrez-Sevillano, S. Vandenbrande and L. Vanduyfhuys, et al. Modeling Gas Adsorption in Flexible Metal–Organic Frameworks via Hybrid Monte Carlo/Molecular Dynamics Schemes, Adv. Theory Simul., 2019, 2(4), 1800177 CrossRef CAS.
- L. Zhang, Z. Hu and J. Jiang, Sorption-Induced Structural Transition of Zeolitic Imidazolate Framework-8: A Hybrid Molecular Simulation Study, J. Am. Chem. Soc., 2013, 135(9), 3722–3728 CrossRef CAS PubMed.
- M. A. Addicoat, N. Vankova, I. F. Akter and T. Heine, Extension of the Universal Force Field to Metal–Organic Frameworks, J. Chem. Theory Comput., 2014, 10(2), 880–891 CrossRef CAS.
- G. Kupgan, A. G. Demidov and C. M. Colina, Plasticization behavior in polymers of intrinsic microporosity (PIM-1): A simulation study from combined Monte Carlo and molecular dynamics, J. Membr. Sci., 2018, 565, 95–103 CrossRef CAS.
- S. Velioğlu, M. G. Ahunbay and S. B. Tantekin-Ersolmaz, Investigation of CO2-induced plasticization in fluorinated polyimide membranes via molecular simulation, J. Membr. Sci., 2012, 417–418, 217–227 CrossRef.
- M. Balçık and M. G. Ahunbay, Prediction of CO2-induced plasticization pressure in polyimides via atomistic simulations, J. Membr. Sci., 2018, 547, 146–155 CrossRef.
- J. D. Evans, L. Bocquet and F. X. Coudert, Origins of Negative Gas Adsorption, Chem, 2016, 1(6), 873–886 CAS.
- S. M. J. Rogge, L. Vanduyfhuys, A. Ghysels, M. Waroquier, T. Verstraelen and G. Maurin, et al. A Comparison of Barostats for the Mechanical Characterization of Metal–Organic Frameworks, J. Chem. Theory Comput., 2015, 11(12), 5583–5597 CrossRef CAS.
- J. Wieme, L. Vanduyfhuys, S. M. J. Rogge, M. Waroquier and V. Van Speybroeck, Exploring the Flexibility of MIL-47(V)-Type Materials Using Force Field Molecular Dynamics Simulations, J. Phys. Chem. C, 2016, 120(27), 14934–14947 CrossRef CAS.
- A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown and P. S. Crozier, et al. LAMMPS – a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales, Comput. Phys. Commun., 2022, 271, 108171 CrossRef CAS.
- Y. J. Colón, D. A. Gómez-Gualdrón and R. Q. Snurr, Topologically Guided, Automated Construction of Metal–Organic Frameworks and Their Evaluation for Energy-Related Applications, Cryst. Growth Des., 2017, 17(11), 5801–5810 CrossRef.
- Y. J. Colón and S. Furukawa, Understanding the role of linker flexibility in soft porous coordination polymers, Mol. Syst. Des. Eng., 2020, 5(1), 284–293 RSC.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations, J. Am. Chem. Soc., 1992, 114(25), 10024–10035 CrossRef CAS.
- M. A. Addicoat, N. Vankova, I. F. Akter and T. Heine, Extension of the Universal Force Field to Metal–Organic Frameworks, J. Chem. Theory Comput., 2014, 10(2), 880–891 CrossRef CAS PubMed.
- P. G. Boyd, S. M. Moosavi, M. Witman and B. Smit, Force-Field Prediction of Materials Properties in Metal-Organic Frameworks, J. Phys. Chem. Lett., 2017, 8(2), 357–363 CrossRef CAS PubMed.
- T. F. Willems, C. H. Rycroft, M. Kazi, J. C. Meza and M. Haranczyk, Algorithms and tools for high-throughput geometry-based analysis of crystalline porous materials, Microporous Mesoporous Mater., 2012, 149(1), 134–141 CrossRef CAS.
- M. Pinheiro, R. L. Martin, C. H. Rycroft, A. Jones, E. Iglesia and M. Haranczyk, Characterization and comparison of pore landscapes in crystalline porous materials, J. Mol. Graphics Modell., 2013, 44, 208–219 CrossRef CAS PubMed.
- D. Dubbeldam, S. Calero, D. E. Ellis and R. Q. Snurr, RASPA: molecular simulation software for adsorption and diffusion in flexible nanoporous materials, Mol. Simul., 2016, 42(2), 81–101 CrossRef CAS.
- K. Momma and F. Izumi, VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44(6), 1272–1276 CrossRef CAS.
- J. Ahrens, B. Geveci and C. Law, 36 – ParaView: An End-User Tool for Large-Data Visualization, in Visualization Handbook, Elsevier Inc, 2004, p. 717, LXX-731, LXXII Search PubMed.
- S. Krause, J. D. Evans, V. Bon, I. Senkovska, F. X. Coudert and D. M. Többens, et al. The role of temperature and adsorbate on negative gas adsorption transitions of the mesoporous metal–organic framework DUT-49, Faraday Discuss., 2021, 225, 168–183 RSC.
- E. Osaro, K. Mukherjee and Y. J. Colón, Active Learning for Adsorption Simulations: Evaluation, Criteria Analysis, and Recommendations for Metal–Organic Frameworks, Ind. Eng. Chem. Res., 2023, 62(33), 13009–13024 CrossRef CAS.
- K. Mukherjee, E. Osaro and Y. J. Colón, Active learning for efficient navigation of multi-component gas adsorption landscapes in a MOF, Digital Discovery, 2023, 2(5), 1506–1521 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4me00154k |
| This journal is © The Royal Society of Chemistry 2025 |