
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransfer learning accelerated discovery of conjugated oligomers for advanced organic photovoltaics†
Siyan
Deng
 ,
Jing Xiang
Ng
and
Shuzhou
Li
*
,
Jing Xiang
Ng
and
Shuzhou
Li
*
School of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798, Singapore. E-mail: lisz@ntu.edu.sg
First published on 31st March 2025
Abstract
Machine learning accelerates material discovery which includes selection of candidate small molecules and polymers for high-efficiency organic photovoltaic (OPV) materials. However, conventional machine learning models suffer from data scarcity for conjugated oligomers, crucial for OPV material production. To address this challenge, transfer learning within a graph neural network was introduced to reduce the data requirement while accurately predicting the electronic properties of the conjugated oligomers. By leveraging on transfer learning using original conjugated oligomer data and pre-trained models from the renowned PubChemQC dataset, the limitations posed by insufficient data were mitigated. The models in this study achieved a low mean absolute error, ranging from 0.46 to 0.74 eV, for the HOMO, LUMO, and HOMO–LUMO gap. An original candidate dataset of 3710 conjugated oligomers was constructed for materials discovery, and a high-throughput screening pipeline was developed by integrating the models with density functional theory. This pipeline effectively identified 46 promising conjugated oligomer candidates, showcasing its effectiveness in accelerating the discovery of advanced materials for organic photovoltaics. These results demonstrated the potential of the approach used in this study to overcome data scarcity while accelerating the discovery of new innovative materials in organic electronics.
Design, System, ApplicationConjugated oligomers are key components in organic photovoltaic (OPV) materials, yet their design and optimization are limited by the lack of sufficient training data for conventional machine learning models. To address this, we utilized a transfer learning approach within a graph neural network (GNN) framework, leveraging pre-trained models from the PubChemQC dataset. This strategy enabled accurate predictions of crucial electronic properties such as the HOMO, LUMO, and HOMO–LUMO gap, significantly mitigating the challenges of data scarcity. The developed system incorporates these GNN models into a high-throughput screening pipeline, combined with density functional theory (DFT) for validation, allowing efficient exploration of 3710 candidate oligomers. This led to the identification of 46 promising conjugated oligomer candidates for OPV applications. The integrated ML-DFT approach offers a robust solution to navigate large design spaces with reduced computational effort, facilitating the discovery of advanced OPV materials with enhanced performance. The methodology has immediate applications in accelerating the identification of novel organic semiconductors for energy conversion. Future applications could involve expanding this approach to other organic electronic materials, enhancing the efficiency of material discovery processes across diverse areas in organic electronics and renewable energy technologies. |
1. Introduction
Organic photovoltaic (OPV) devices are promising alternatives to conventional silicon-based photovoltaic devices, offering lightweight, flexible, and energy efficient solutions.1–5 Given this versatility, OPV becomes suitable for a wide range of applications, from small flexible electronics in healthcare5–7 to industrial solar panels in solar farms.8–10 The performance of OPV devices is critically dependent on the electronic properties of the active layer, which can be composed of small molecules,11–14 polymers,15–18 or conjugated oligomers.19–22 One major limitation in OPV material development is the difficulty of achieving both high electronic performance and good processability, as many high-performance materials suffer from poor solubility.23,24 Conjugated oligomers overcome this issue by offering tunable electronic properties to enhance performance and improved solubility to facilitate processability.25,26 Given these advantages, ongoing research is imperative to explore and identify novel conjugated oligomers that can further enhance OPV performance. However, discovering conjugated oligomers for OPV applications remains a challenge due to the complex relationship between molecular structure and electronic performance. Additionally, the vast chemical space further amplifies that complexity.In recent studies, machine learning (ML) has proven to be a powerful tool for accelerating the discovery and optimization of various materials, notably OPV materials.27–40 By leveraging large datasets and advanced algorithms, ML models performed well in predicting the properties of new materials, achieving high accuracy and reducing the time and cost associated with traditional experimental approaches. Despite this potential, the application of ML in OPV materials screening has predominantly focused on small molecules27,28 and polymers.29–32 More importantly, ML models struggle to predict the electronic properties of long conjugated oligomers due to the complex structure–property relationships.41 This complexity arises from the intermediate chain length of oligomers, which creates a balance of localized and delocalized electronic states that are difficult to model accurately with ML techniques. Possible solutions to this challenge include using calculated descriptors or the development of sophisticated deep learning models with very large datasets. However, obtaining calculated descriptors is both expensive and tedious, and large datasets on properties and structures needed to train deep learning models are unavailable for conjugated oligomers. These limitations underscore the need for more innovative approaches to leverage the power of ML for property–structure prediction, while addressing the constraints imposed by data scarcity and the complex structure–property relationship of conjugated oligomers.
Transfer learning, a subset of deep learning, involves using pre-trained models trained on large and related datasets to enhance the performance of models on smaller, specific datasets. Several studies have demonstrated the potential of transfer learning used in materials science research, particularly in fields that typically suffer from data scarcity.42–45 Some examples includes the prediction of the bulk modulus and dielectric constant of crystal structures,42 as well as the prediction of toxicity, yield, and odor of organic materials.43 Specific to conjugated oligomers, one study utilized a model pre-trained on a large dataset from the Clean Energy Project Database and then fine-tuned it using a dataset of 400 molecules to predict the power conversion efficiencies of oligomers based on the molecular structure.44 However, that study only considered oligomers with low degree of polymerisation (up to 5) and a small variety of only 20 different monomers. Similarly, in another study, transfer learning was introduced to predict the electronic properties of conjugated oligomers. However, that study only explored the effect of degrees of polymerization using a single monomer type, failing to account for the great variety of monomers present in oligomers.45 While these studies highlight the effectiveness of transfer learning, their chemical scope remains relatively narrow, limiting their generalizability to diverse oligomer compositions. Expanding the chemical space is crucial for improving model applicability in large-scale material screening, as it allows for the identification of novel candidates beyond previously studied structures. A broader chemical space enables more extensive candidate screening, which is essential when considering the transition from lab-scale discovery to practical fabrication. Other practical considerations such as viability of production, physical properties and cost of production exist in addition to electronic properties. With a larger pool of candidates, the probability of finding a material with both desirable electronic properties and practical feasibility is increased. To fulfil this, it is essential to develop models that can operate across a broader chemical space. This includes accommodating a wider range of polymerization degrees and a larger diversity of monomer compositions. Developing such models would significantly enhance the screening process and facilitate the discovery of high-efficiency OPV materials.
In this study, transfer learning within graph neural networks (GNNs) was introduced to address the issue of data scarcity during the screening of conjugated oligomers for candidates as OPV material's backbone. A pre-trained model was developed using the PubChemQC dataset.46 PubChemQC is one of the largest quantum chemistry databases, offering high computational accuracy with the B3LYP/6-31G* level of theory.47,48 By leveraging the extensive coverage of this dataset, the model's generalizability is significantly enhanced, allowing it to make more accurate predictions across a diverse range of chemical structures. Next, an original conjugated oligomer database was constructed using density functional theory (DFT). This dataset consists of 610 unique oligomers with polymerization degrees varying between 4 and 10 and the oligomers are made up of 131 distinct monomer units. Together, a new model was built using the pre-trained model and fine-tuned with the original oligomer dataset. The transfer learning technology successfully mitigated the limitations posed by data scarcity, enabling the construction of models that accurately predict the electronic properties of conjugated oligomers over a broader chemical space, encompassing a wider range of polymerization degrees and a greater diversity of monomer compositions. Due to its broader applicability and improved predictive power, the model is better suited for real-world material discovery. The developed models achieve a low mean absolute errors of 0.74 eV for the highest occupied molecular orbital (HOMO), 0.46 eV for the lowest unoccupied molecular orbital (LUMO), and 0.54 eV for the HOMO–LUMO gap. Furthermore, to balance computational efficiency and predictive accuracy, a high-throughput screening pipeline which combines these advanced models with DFT calculations was developed. This pipeline enabled the identification of 46 promising conjugated oligomer candidates for OPV materials, accelerating the discovery of possible innovative materials for organic electronics applications.
2. Methods
2.1 Data preparation
An original dataset, named CO-610, contains 610 conjugated oligomers and their corresponding electronic properties, including HOMO, LUMO, and gap. In this context, all subsequent mentions of “gap” refer to the HOMO–LUMO gap. Each oligomer is composed of a single type of monomer, with polymerization degrees ranging from 4 to 10. These oligomers encompass a total of 131 different monomers, collected from published literature.49 The oligomers with different polymerization degrees were generated through a custom tool using the RDKit library.50 The electronic properties of each oligomer were then calculated using DFT at the B3LYP/6-31G* level.47,48 Additionally, 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 pieces of data from PubChemQC were used for pre-training the models.46 This pre-training dataset is referred to as PubChemQC-100 K. PubChemQC-100 K includes organic small molecules and their corresponding electronic properties. The details of data selection are provided in the ESI.†
000 pieces of data from PubChemQC were used for pre-training the models.46 This pre-training dataset is referred to as PubChemQC-100 K. PubChemQC-100 K includes organic small molecules and their corresponding electronic properties. The details of data selection are provided in the ESI.†
2.2 Model development
A transfer learning framework was employed to develop predictive models, as illustrated in Fig. 1. SchNet was selected for this study due to its incorporation of continuous-filter convolution layers, which model atomic interactions as a function of interatomic distance, making it particularly well-suited for predicting quantum chemical properties.51 Since the electronic properties of conjugated oligomers depend strongly on molecular geometry, SchNet's ability to capture smooth variations in the electronic structure ensures more accurate predictions. Additionally, SchNet offers a balance between accuracy and computational efficiency, making it suitable for large-scale molecular screening. The SchNet architecture consists of embedding layers, interaction blocks, and property prediction layers. The embedding layers map atomic types to high-dimensional vectors, while the interaction blocks incorporate continuous-filter convolution operations, which update atomic representations based on neighboring atomic positions within a cut-off distance of 5 Å. The property prediction layers, consisting of fully connected neural networks, map these atomic embeddings to molecular electronic properties such as HOMO, LUMO, and the gap.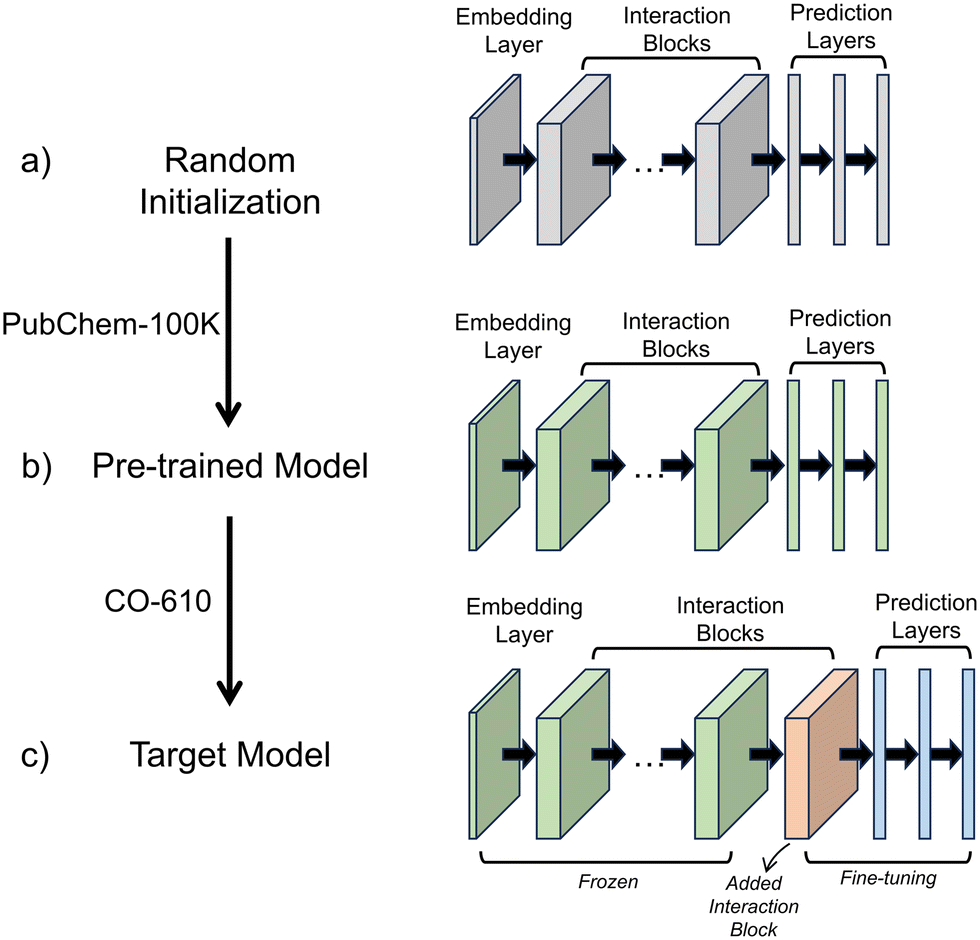 | ||
| Fig. 1 Schematic of the transfer learning protocol used in this work. The SchNet model, consisting of an embedding layer, interaction blocks, and prediction layers, is first randomly initialized. The model is then pre-trained on the PubChem-100 K dataset to learn general chemical representations. After pre-training, an additional interaction block is added, and the model is fine-tuned using the CO-610 dataset, which contains the target conjugated oligomers. | ||
This framework involves three key stages: random initialization, pre-training, and fine-tuning. Initially, the model is initialized with random parameters (Fig. 1a). The pre-training phase is crucial for learning generalizable chemical knowledge before fine-tuning on a smaller, specialized dataset. SchNet is first pre-trained on the PubChemQC-100 K dataset (Fig. 1b), a large-scale quantum chemistry dataset containing diverse molecular structures and electronic properties. This step allows the model to capture fundamental molecular interactions and structure–property relationships, creating a well-initialized parameter space rather than relying on random initialization. Pre-training mitigates overfitting, enhances model stability, and reduces the need for extensive hyperparameter tuning during fine-tuning. Following pre-training, the pre-trained layers remain frozen to preserve the general chemical knowledge acquired from a diverse set of molecular structures. This prevents catastrophic forgetting, ensuring that the small CO-610 dataset does not overwrite the broad chemical understanding learned during pre-training. While pre-training provides a strong foundation for molecular representation, predicting the HOMO, LUMO, and gap in conjugated oligomers requires further adaptation due to their unique electronic behavior. To address this, a new interaction block is introduced during fine-tuning (Fig. 1c). The interaction block plays a crucial role in refining the captured molecular interactions. Rather than modifying the embedding or property prediction layers, adding a new interaction block allows the model to capture atom-type-dependent and environment-dependent electronic effects, particularly those related to π-conjugation and electron delocalization. This refinement step ensures that the model effectively encodes conjugation-specific electronic interactions.
In this work, SchNet was configured with an embedding dimension of 128. The pre-trained model contained six interaction blocks, while the transfer learning model included seven interaction blocks, with one additional block added post-pre-training. The Adam optimizer was used with an initial learning rate of 0.0001 for pre-training, reduced to 0.00001 for fine-tuning. Pre-training was conducted for 1000 epochs, followed by 3000 epochs of fine-tuning, with early stopping applied to prevent overfitting. The dataset was divided into 400 training samples, 100 validation samples, and 110 test samples, with training conducted using a batch size of 64. The pre-training phase on the PubChemQC-100 K dataset was conducted on an NVIDIA A100-SXM4-40GB GPU with 16 physical CPU cores, requiring approximately 14 hours to complete for 1000 epochs. Fine-tuning on the CO-610 dataset utilized the same hardware and took approximately 1.8 hours for 3000 epochs, benefiting from pre-trained representations that accelerated convergence.
2.3 High-throughput screening
To identify potential conjugated oligomers with desirable electronic properties for photovoltaic applications, a candidate dataset was constructed. This dataset includes 3710 oligomers derived from 530 different monomers, with polymerization degrees ranging from 4 to 10. The oligomers were generated using the same method described in section 2.1, with monomers sourced from published literature.49Initially, optimized models were employed to predict the HOMO, LUMO, and gap of the oligomers in the candidate dataset. This initial screening step effectively reduced the dataset to a smaller subset of promising candidates. Subsequently, more accurate and computationally intensive DFT calculations were performed. These calculations provided a precise evaluation of the electronic properties, further refining the selection of potential conjugated oligomers for photovoltaic applications. This two-step screening process, which combines the efficiency of machine learning models with the precision of DFT calculations, significantly reduces computational costs while ensuring the identification of high-potential materials. The initial model-based screening allows for the rapid elimination of less promising candidates, while the follow-up DFT calculations ensure that the selected candidates meet the stringent electronic property requirements for photovoltaic applications.
3. Results
3.1 Dataset analysis
The CO-610 dataset covers a broad chemical space, providing a solid foundation for analyzing the electronic properties of conjugated oligomers. The CO-610 dataset includes 610 oligomers derived from 131 different types of monomers, with polymerization degrees varying from 4 to 10. To ensure relevance for OPV applications, monomers were selected based on their ability to form conjugated oligomers, with 89% containing aromatic rings and the remaining 11% being aliphatic monomers. Among the aromatic monomers, 56% contain a single aromatic ring, 28% contain two aromatic rings, and 5% contain three aromatic rings, contributing to an overall aromatic ring count ranging from 4 to 30 per oligomer. This diversity enriches the dataset, allowing the model to capture a wide range of structural variations. In addition to aromaticity, the dataset includes a significant portion of heteroatom-containing monomers, with 65% containing sulfur (S), 17% nitrogen (N), and 16% oxygen (O). Incorporating heteroatoms into conjugated polymers is a well-established strategy, as it facilitates fine-tuning of electronic properties, molecular geometry, solid-state packing, and processability—all critical for OPV performance. The dataset also maintains a balanced distribution of polymerization degrees, ensuring robust learning across different chain lengths. Specifically, lower polymerization degrees (n = 4–5) account for 30%, mid-range polymerization (n = 6–8) account for 49%, and higher polymerization degrees (n = 9–10) account for 21%. This even representation across polymerization degrees allows the model to learn from a broad range of molecular structures, improving its ability to generalize across different oligomer sizes. The extensive variety in monomer types and polymerization degrees ensures that the dataset encompasses a large chemical space, thereby increasing the likelihood that the model will learn from a broader spectrum of chemical knowledge. More importantly, the CO-610 dataset reveals the intricate and complex nature of structure–property relationships in conjugated oligomers.Firstly, Fig. 2 illustrates a comparative sensitivity analysis of oligomers with polymerization degrees ranging from n = 4 to n = 10 against a standardized heptameric baseline (n = 7), with each column representing oligomers derived from a unique monomer but at varying degrees of polymerization. The arrows in the figure indicate the direction of increasing polymerization degree. The deviations in electronic properties are evident across all three plots, demonstrating that the degree of polymerization significantly influences the electronic properties of the conjugated oligomers. Moreover, the sensitivity varies across properties, with the gap being the most sensitive, reflecting the cumulative effect of variations in both HOMO and LUMO levels. The deviations of the HOMO and LUMO are generally within ±0.5 eV, but the deviations in the gap are more pronounced, often exceeding ±0.5 eV. Specifically, in Fig. 2a, the points above the zero line are more densely clustered, indicating that as the polymerization degree increases, the increase in HOMO levels becomes smaller. Conversely, in Fig. 2b, the points below the zero line are more densely clustered, indicating that with an increase in polymerization degree, the decrease in LUMO levels becomes less pronounced. This observation aligns with the known behavior of conjugated oligomers, where electronic properties tend to stabilize beyond a certain conjugation length. Furthermore, different monomers exhibit varying sensitivities to changes in polymerization degree. For example, oligomers derived from monomer 0 exhibit a significant reduction in the gap, greater than 1.0 eV, as the polymerization degree increases from 4 to 10, indicating that longer chains enhance electron delocalization. In contrast, oligomers derived from monomer 23 show minimal changes in the gap, less than 0.1 eV, over the same range of polymerization degrees, suggesting a negligible impact of chain length on their electronic properties.
 | ||
| Fig. 2 Sensitivity analysis of conjugated oligomers by polymerization degree. This figure presents a comparative sensitivity analysis of oligomers with polymerization degrees ranging from n = 4 to n = 10 against a standardized heptameric baseline (n = 7). The scatter colors denote the values of the (a) HOMO, (b) LUMO, and (c) energy gap, respectively. The arrows in the figure indicate the direction of increasing polymerization degree. | ||
Secondly, as shown in Fig. S1,† the dataset exhibits significant variability in electronic properties, with HOMO levels ranging from −8.0 eV to −3.4 eV, LUMO levels between −5.4 eV and 0.4 eV, and gap levels spanning from 0.1 eV to 5.3 eV. This wide distribution underscores the diverse electronic environments and the complex interplay between molecular structure and electronic properties.
Lastly, investigating the electronic properties of specific oligomers revealed that simple linear correlations between structure and properties were insufficient to capture the intricacies of their electronic behavior. For instance, as shown in Fig. S3,† oligomers with identical backbone structures exhibited significantly different HOMO and LUMO levels. Both oligomers incorporate a thiophene ring with an ethynyl group attached. In the first oligomer, each thiophene ring features a methoxy group (–OCH3), while in the second oligomer, the methoxy group is substituted with a cyano group (–CN). This substitution results in the HOMO level shifting from −4.56 eV to −5.69 eV, the LUMO level shifting from −0.97 eV to −2.45 eV, and the gap changing from 3.59 eV to 3.20 eV. This suggests that subtle variations in substituents or conformations play a crucial role in determining electronic properties.
Overall, the analysis of the CO-610 dataset demonstrates that the structure–property relationships in conjugated oligomers are highly complex. This complexity necessitates advanced modeling approaches to accurately predict electronic properties and identify promising conjugated oligomers for OPV applications. Furthermore, the broad chemical space covered by the CO-610 dataset ensures that the models developed will have a comprehensive understanding of the potential electronic behaviors.
3.2 Model performance
To validate the effectiveness of the transfer learning approach, a comparison was made between the performance of the transfer learning models and models trained solely on the CO-610 dataset without transfer learning. Fig. 3a illustrates the comparative performance of direct learning and transfer learning models in predicting the HOMO, LUMO, and gap across various polymerization degrees. Fig. S5† shows a comparison between predicted and calculated values for different properties, further highlighting the performance differences between direct learning and transfer learning. The MAE indicates that direct learning models achieve acceptable accuracy within the mid-range of polymerization degrees (6 to 8), but their performance significantly declines at the higher and lower ends of the polymerization spectrum. For instance, the MAE for HOMO predictions by the direct learning model increases from 0.91 eV at polymerization degree 6 to 2.93 eV at degree 10. This MAE increase suggests a limitation of direct learning models in accurately modeling conjugated oligomers across a wide range of polymerization degrees. For oligomers with higher polymerization degrees (9 to 10), the extremely poor performance of direct learning models can be attributed to the low distribution of these oligomers within the CO-610 dataset, as shown in Fig. S2.† The scarcity of training samples for higher polymerization degrees results in larger prediction errors due to insufficient data to capture their electronic properties accurately. However, for oligomers with lower polymerization degrees (4 to 5), their distribution is comparable to those in the mid-range (6 to 8), yet the MAE remains high. One possible reason is that electronic sensitivity is higher at lower polymerization degrees as discussed previously in Fig. 2. This increased sensitivity makes it more challenging for the model to generalize and accurately predict the properties of shorter oligomers. However, transfer learning models demonstrate improved performance across the full range of polymerization degrees, evidenced by lower MAE values throughout. The improvement is primarily due to the ability of transfer learning to leverage knowledge from pre-trained models on larger datasets, which enhances the model's generalization and prediction accuracy across different polymerization degrees. At higher polymerization degrees, transfer learning mitigates prediction errors caused by the scarcity of training samples. For lower polymerization degrees, it effectively captures subtle variations in electronic properties despite increased sensitivity to small structural changes. This broader knowledge base allows the model to adapt to the specific characteristics of the oligomers, resulting in consistently lower MAE values and superior performance compared to direct learning models. | ||
| Fig. 3 a) Mean average errors (MAEs) for the HOMO, LUMO, and gap using direct (red) and transfer learning (blue) methods across different polymerization degrees (n). b) The distribution of HOMO, LUMO, and gap values in the PubChemQC-100 K and CO-610 datasets. | ||
Moreover, the impact of transfer learning on prediction accuracy varies across different molecular properties. As demonstrated in Table 1, the most substantial accuracy improvements are observed for HOMO predictions. Specifically, the MAE for HOMO predictions decreases from 1.34 eV with direct learning to 0.74 eV with transfer learning, reflecting a 44.8% reduction in error. Similarly, predictions of LUMO energy levels exhibit a significant enhancement, with the MAE decreasing from 0.68 eV to 0.46 eV, which corresponds to a 32.4% reduction. Although less pronounced, the improvements for the gap are also noteworthy, with the MAE reducing from 0.71 eV to 0.54 eV, indicating a 23.9% improvement. The variation in enhancement can be attributed to the similarity in property distributions between PubChemQC-100 K and CO-610, as depicted in Fig. 3b. The greater similarity in HOMO energy distributions results in more substantial improvements through transfer learning. For the LUMO and gap, despite the higher variance between PubChemQC-100 K and CO-610, transfer learning still achieves notable gains, demonstrating the model's robustness and adaptability.
| Properties | Direct learning (eV) | Transfer learning (eV) |
|---|---|---|
| HOMO | 1.34 | 0.74 |
| LUMO | 0.67 | 0.46 |
| Gap | 0.71 | 0.54 |
Overall, this comparative analysis underscores the improved generalization and predictive accuracy of transfer learning models. The robustness and adaptability of transfer learning are highlighted by its ability to effectively address the limitations of direct learning models, particularly in regions with sparse data and higher electronic sensitivity. This analysis demonstrates the significant advantages of transfer learning in modeling the electronic properties of conjugated oligomers across a wide range of polymerization degrees.
3.3 High-throughput screening
The high-throughput screening process employed in this study combines ML predictions and DFT calculations to identify potential conjugated oligomers for OPV application. This two-step approach efficiently narrows down the candidate pool to select conjugated oligomers with desirable electronic properties.The parallel coordinate plots in Fig. 4a and b provide a visual representation of how the candidate materials were progressively refined through HOMO, LUMO, and gap selection criteria. The funnel diagram in the center summarizes this sequential filtering approach. This workflow efficiently filters candidate materials while balancing computational efficiency and predictive accuracy. Initially, the transfer learning models were used to predict the HOMO, LUMO, and gap for the 3710 candidate oligomers in the candidate dataset. To account for the inherent prediction errors of ML models, we adjusted the selection criteria to include a margin of error equivalent to half the MAE of the ML models. The original criteria were set to HOMO levels between −6.5 and −4.9 eV, LUMO levels between −4.5 and −3.0 eV, and gap between 1.1 and 2.0 eV.52 Given that the MAEs for the HOMO, LUMO, and gap were 0.74 eV, 0.46 eV, and 0.54 eV, respectively, we expanded the selection range by adding half the MAE to each threshold. The adjusted criteria became HOMO levels between −6.87 and −4.53 eV, LUMO levels between −4.73 and −2.77 eV, and band gaps between 0.83 and 2.27 eV.52 This adjustment prevented the premature exclusion of potential candidates whose true properties might fall within the desired range but were slightly misestimated due to ML prediction errors. Fig. 4a presents a parallel coordinate plot visualizing these ML-predicted values. Each blue line represents a candidate oligomer, showing its ML-predicted HOMO, LUMO, and gap values. The green line highlights the subset of 256 oligomers that passed ML screening, meeting the adjusted selection criteria. This ML-based high-throughput screening step enabled a significant reduction in the number of candidates by filtering out those that did not meet the desired electronic criteria. By applying these adjusted criteria, the initial pool of 3710 oligomers was reduced to 256 candidates for further investigation.
 | ||
| Fig. 4 Screening process. a) ML screening: parallel coordinate plot of HOMO, LUMO, and gap predictions for the initial 3710 candidates. b) DFT screening: parallel coordinate plot showing the DFT-calculated HOMO, LUMO, and gap for the refined selection of 256 candidates, from which 46 final candidates were selected. | ||
The refined set of 256 candidates from the ML screening was then subjected to DFT calculations, which are more accurate but computationally intensive. Unlike ML predictions, DFT calculations were evaluated using the original selection criteria: HOMO levels between −6.5 and −4.9 eV, LUMO levels between −3.0 and −4.5 eV, and gaps between 1.1 and 2.0 eV.52Fig. 4b illustrates the parallel coordinate plot of the DFT-calculated HOMO, LUMO, and gap for the candidates that passed ML screening. Each green line represents a candidate refined through DFT calculations, providing a higher-accuracy evaluation of electronic properties. Orange lines highlight the 46 final candidates, which satisfy the original OPV-relevant electronic selection criteria under DFT assessment. This DFT refinement step ensured that the candidates identified by ML screening truly met the required electronic criteria when evaluated with a more accurate method. The distribution of HOMO, LUMO, and gap values in Fig. 4b indicates that while some ML-selected candidates fell outside the final DFT selection criteria, a substantial number remained within the desired range, confirming the effectiveness of the ML pre-screening step. The SMILES of these 46 final candidates is shown in Table S1.† The monomer structures of the final 46 OPV candidates are presented in Fig. 5. These candidates represent the most promising materials for further experimental validation and potential application in OPV devices.
 | ||
| Fig. 5 Monomer structures of the final 46 OPV candidates. The candidates with same monomer but different polymerization degree are not shown here. | ||
Notably, a significant number of these monomers are oligothiophene-based, which are among the most extensively studied and utilized conjugated oligomers in OPV materials.53–57 Among the oligothiophene-based candidates, monomer 187, thieno[3,2-b]pyrrole, stands out as it has been widely used as a building block in OPV materials.58–60 This monomer features fused thiophene and pyrrole rings and was first developed in 1957 by Matteson and Snyder.61 It possesses a low-lying HOMO level, which is beneficial for long-term stability.59 Monomer 295 is part of the benzothiophene-based family, while monomer 433 belongs to the 2,1,3-benzothiadiazole-based family. These two families are also widely studied and utilized in OPV materials.62–66 The presence of these well-known and widely studied monomers among our final candidates validates our screening methodology, demonstrating that our process is robust and capable of identifying promising materials that align with current OPV research trends. Furthermore, most of the monomers we identified are novel and not commonly used in OPV materials, providing new avenues for material development.
Overall, our combined ML and DFT screening approach effectively balances computational efficiency with predictive accuracy, resulting in a robust evaluation of potential OPV materials. The identification of monomers such as thieno[3,2-b]pyrrole, along with benzodithiophene- and benzothiadiazole-based families, which are already well-documented in OPV research, serves as validation for our methodology. This demonstrates that our screening process can reliably identify promising candidates with proven relevance. Moreover, our method has also highlighted several novel candidates offering new avenues for OPV material development. These newly identified candidates offer valuable insights and a solid foundation for subsequent experimental work, holding significant potential for future OPV applications.
4. Conclusion
This study successfully integrates ML predictions and DFT calculations to accelerate the discovery of high-performance conjugated oligomers for OPV applications. By introducing transfer learning within a GNN, we addressed the challenge of data scarcity, enabling accurate predictions of the electronic properties of conjugated oligomers across a wide range of polymerization degrees and various monomer types. The transfer learning models achieved a MAE of 0.74 eV for the HOMO, 0.46 eV for the LUMO, and 0.54 eV for the gap. A high-throughput screening pipeline was developed to seamlessly combine these ML models with DFT calculations, effectively narrowing down an initial set of 3710 oligomer candidates to 46 promising ones. This two-step screening process not only reduces computational costs but also ensures the selection of high-potential candidates. The methodology demonstrated here identifies conjugated oligomers with significant potential for future experimental validation, paving the way for the practical application of novel and efficient OPV materials.Data availability
The PubChemQC dataset is accessible at https://nakatamaho.riken.jp/pubchemqc.riken.jp/.The data selection criteria for PubChemQC-100 K can be found in the ESI† (SI.docx). Additionally, the CO-610 dataset is available in the ESI† (SI.csv).
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research/project is supported by the National Research Foundation, Singapore (NRF) under NRF's Medium Sized Centre: Singapore Hybrid-Integrated Next-Generation μ-Electronics (SHINE) Centre funding programme. Any opinions, findings and conclusions or recommendations expressed in this material are those of the author(s) and do not reflect the views of the National Research Foundation, Singapore. We acknowledge financial support from the Ministry of Education (MOE) of Singapore under Academic Research Fund Tier 2 (MOE-T2EP20221-0003) and Academic Research Fund Tier 1 (RG5/22). The computational work for this article was partially carried out at National Supercomputing Centre, Singapore (https://www.nscc.sg).References
- O. Inganäs, Organic photovoltaics over three decades, Adv. Mater., 2018, 30(35), 1800388 Search PubMed.
- J. Luke, E. J. Yang, C. Labanti, S. Y. Park and J.-S. Kim, Key molecular perspectives for high stability in organic photovoltaics, Nat. Rev. Mater., 2023, 8(12), 839–852 Search PubMed.
- H. Yao, J. Wang, Y. Xu, S. Zhang and J. Hou, Recent progress in chlorinated organic photovoltaic materials, Acc. Chem. Res., 2020, 53(4), 822–832 Search PubMed.
- J. Yi, G. Zhang, H. Yu and H. Yan, Advantages, challenges and molecular design of different material types used in organic solar cells, Nat. Rev. Mater., 2024, 9(1), 46–62 Search PubMed.
- T. P. Huynh and H. Haick, Autonomous flexible sensors for health monitoring, Adv. Mater., 2018, 30(50), 1802337 Search PubMed.
- F. C. Chen, Emerging organic and organic/inorganic hybrid photovoltaic devices for specialty applications: low-level-lighting energy conversion and biomedical treatment, Adv. Opt. Mater., 2019, 7(1), 1800662 Search PubMed.
- N. Cui, Y. Song, C.-H. Tan, K. Zhang, X. Yang, S. Dong, B. Xie and F. Huang, Stretchable transparent electrodes for conformable wearable organic photovoltaic devices, npj Flexible Electron., 2021, 5(1), 31 CAS.
- J. Yang, D. Vak, N. Clark, J. Subbiah, W. W. Wong, D. J. Jones, S. E. Watkins and G. Wilson, Organic photovoltaic modules fabricated by an industrial gravure printing proofer, Sol. Energy Mater. Sol. Cells, 2013, 109, 47–55 Search PubMed.
- R. Xue, J. Zhang, Y. Li and Y. Li, Organic solar cell materials toward commercialization, Small, 2018, 14(41), 1801793 Search PubMed.
- R. Sun, Q. Wu, J. Guo, T. Wang, Y. Wu, B. Qiu, Z. Luo, W. Yang, Z. Hu and J. Guo, A layer-by-layer architecture for printable organic solar cells overcoming the scaling lag of module efficiency, Joule, 2020, 4(2), 407–419 Search PubMed.
- S. Badgujar, G. Y. Lee, T. Park, C. E. Song, S. Park, S. Oh, W. S. Shin, S. J. Moon, J. C. Lee and S. K. Lee, High-performance small molecule via tailoring intermolecular interactions and its application in large-area organic photovoltaic modules, Adv. Energy Mater., 2016, 6(12), 1600228 Search PubMed.
- Y. Lin, Y. Li and X. Zhan, Small molecule semiconductors for high-efficiency organic photovoltaics, Chem. Soc. Rev., 2012, 41(11), 4245–4272 CAS.
- C. Xu, Z. Zhao, K. Yang, L. Niu, X. Ma, Z. Zhou, X. Zhang and F. Zhang, Recent progress in all-small-molecule organic photovoltaics, J. Mater. Chem. A, 2022, 10(12), 6291–6329 RSC.
- C. Yang, Q. An, M. Jiang, X. Ma, A. Mahmood, H. Zhang, X. Zhao, H. F. Zhi, M. H. Jee and H. Y. Woo, Optimized crystal framework by asymmetric core isomerization in selenium-substituted acceptor for efficient binary organic solar cells, Angew. Chem., 2023, 135(49), e202313016 Search PubMed.
- E. Bundgaard and F. C. Krebs, Low band gap polymers for organic photovoltaics, Sol. Energy Mater. Sol. Cells, 2007, 91(11), 954–985 CrossRef CAS.
- G. J. Hedley, A. J. Ward, A. Alekseev, C. T. Howells, E. R. Martins, L. A. Serrano, G. Cooke, A. Ruseckas and I. D. Samuel, Determining the optimum morphology in high-performance polymer-fullerene organic photovoltaic cells, Nat. Commun., 2013, 4(1), 2867 Search PubMed.
- X. Zhan and D. Zhu, Conjugated polymers for high-efficiency organic photovoltaics, Polym. Chem., 2010, 1(4), 409–419 RSC.
- H.-R. Bai, H. Zhang, H. Meng, Y. Li, X. Xu, M.-Q. Liu, Y. Chen, Z.-F. Yao, H.-F. Zhi and A. Mahmood, Electron-deficient fused dithieno-benzothiadiazole-bridged polymer acceptors for high-efficiency all-polymer solar cells with low energy loss, Mater. Sci. Eng., R, 2025, 163, 100916 Search PubMed.
- Y. Lin and X. Zhan, Oligomer molecules for efficient organic photovoltaics, Acc. Chem. Res., 2016, 49(2), 175–183 Search PubMed.
- J. L. Segura, N. Martín and D. M. Guldi, Materials for organic solar cells: the C 60/π-conjugated oligomer approach, Chem. Soc. Rev., 2005, 34(1), 31–47 Search PubMed.
- T. Duan, Q. Chen, D. Hu, J. Lv, D. Yu, G. Li and S. Lu, Oligothiophene-based photovoltaic materials for organic solar cells: rise, plateau, and revival, Trends Chem., 2022, 4(9), 773–791 CAS.
- H. R. Bai, Q. An, M. Jiang, H. S. Ryu, J. Yang, X. J. Zhou, H. F. Zhi, C. Yang, X. Li and H. Y. Woo, Isogenous asymmetric–symmetric acceptors enable efficient ternary organic solar cells with thin and 300 nm thick active layers simultaneously, Adv. Funct. Mater., 2022, 32(26), 2200807 Search PubMed.
- X. Kong, T. He, H. Qiu, L. Zhan and S. Yin, Progress in organic photovoltaics based on green solvents: from solubility enhancement to morphology optimization, Chem. Commun., 2023, 59(81), 12051–12064 CAS.
- X. Yang, Y. Shao, S. Wang, M. Chen, B. Xiao, R. Sun and J. Min, Processability Considerations for Next-Generation Organic Photovoltaic Materials, Adv. Mater., 2023, 2307863 Search PubMed.
- F. Zhang, D. Wu, Y. Xu and X. Feng, Thiophene-based conjugated oligomers for organic solar cells, J. Mater. Chem., 2011, 21(44), 17590–17600 CAS.
- W. Tang, J. Hai, Y. Dai, Z. Huang, B. Lu, F. Yuan, J. Tang and F. Zhang, Recent development of conjugated oligomers for high-efficiency bulk-heterojunction solar cells, Sol. Energy Mater. Sol. Cells, 2010, 94(12), 1963–1979 CrossRef CAS.
- Q. Zhao, Y. Shan, H. Zhou, G. Zhang and W. Liu, Machine learning-assisted performance prediction and molecular design of all-small-molecule organic solar cells based on the Y6 acceptor, Sol. Energy, 2023, 265, 112115 CrossRef CAS.
- W. Sun, Y. Zheng, K. Yang, Q. Zhang, A. A. Shah, Z. Wu, Y. Sun, L. Feng, D. Chen and Z. Xiao, Machine learning–assisted molecular design and efficiency prediction for high-performance organic photovoltaic materials, Sci. Adv., 2019, 5(11), eaay4275 Search PubMed.
- S. Nagasawa, E. Al-Naamani and A. Saeki, Computer-aided screening of conjugated polymers for organic solar cell: classification by random forest, J. Phys. Chem. Lett., 2018, 9(10), 2639–2646 CAS.
- K. Kranthiraja and A. Saeki, Experiment-oriented machine learning of polymer: non-fullerene organic solar cells, Adv. Funct. Mater., 2021, 31(23), 2011168 CAS.
- Q. Yang, A. Vriza, C. A. Castro Rubio, H. Chan, Y. Wu and J. Xu, Artificial Intelligence for Conjugated Polymers, Chem. Mater., 2024, 36(6), 2602–2622 CAS.
- J. Munshi, W. Chen, T. Chien and G. Balasubramanian, Transfer learned designer polymers for organic solar cells, J. Chem. Inf. Model., 2021, 61(1), 134–142 CAS.
- N. Meftahi, M. Klymenko, A. J. Christofferson, U. Bach, D. A. Winkler and S. P. Russo, Machine learning property prediction for organic photovoltaic devices, npj Comput. Mater., 2020, 6(1), 166 CrossRef CAS.
- B. Cao, L. A. Adutwum, A. O. Oliynyk, E. J. Luber, B. C. Olsen, A. Mar and J. M. Buriak, How to optimize materials and devices via design of experiments and machine learning: Demonstration using organic photovoltaics, ACS Nano, 2018, 12(8), 7434–7444 CrossRef CAS PubMed.
- P. Malhotra, K. Khandelwal, S. Biswas, F.-C. Chen and G. D. Sharma, Opportunities and challenges for machine learning to select combination of donor and acceptor materials for efficient organic solar cells, J. Mater. Chem. C, 2022, 10(47), 17781–17811 RSC.
- Y. Miyake and A. Saeki, Machine learning-assisted development of organic solar cell materials: issues, analyses, and outlooks, J. Phys. Chem. Lett., 2021, 12(51), 12391–12401 CAS.
- V. Bhat, C. P. Callaway and C. Risko, Computational approaches for organic semiconductors: from chemical and physical understanding to predicting new materials, Chem. Rev., 2023, 123(12), 7498–7547 Search PubMed.
- P. Shetty, A. Adeboye, S. Gupta, C. Zhang and R. Ramprasad, Accelerating materials discovery for polymer solar cells: Data-driven insights enabled by natural language processing, Chem. Mater., 2024, 36(16), 7676–7689 Search PubMed.
- A. Mahmood, A. Irfan and J.-L. Wang, Machine learning for organic photovoltaic polymers: a minireview, Chin. J. Polym. Sci., 2022, 40(8), 870–876 CAS.
- A. Mahmood and J.-L. Wang, A time and resource efficient machine learning assisted design of non-fullerene small molecule acceptors for P3HT-based organic solar cells and green solvent selection, J. Mater. Chem. A, 2021, 9(28), 15684–15695 Search PubMed.
- Z. Liu, L. Lin, Q. Jia, Z. Cheng, Y. Jiang, Y. Guo and J. Ma, Transferable multilevel attention neural network for accurate prediction of quantum chemistry properties via multitask learning, J. Chem. Inf. Model., 2021, 61(3), 1066–1082 Search PubMed.
- J. Lee and R. Asahi, Transfer learning for materials informatics using crystal graph convolutional neural network, Comput. Mater. Sci., 2021, 190, 110314 CAS.
- E. King-Smith, Transfer learning for a foundational chemistry model, Chem. Sci., 2024, 15(14), 5143–5151 Search PubMed.
- H. Wang, J. Feng, Z. Dong, L. Jin, M. Li, J. Yuan and Y. Li, Efficient screening framework for organic solar cells with deep learning and ensemble learning, npj Comput. Mater., 2023, 9(1), 200 Search PubMed.
- C.-K. Lee, C. Lu, Y. Yu, Q. Sun, C.-Y. Hsieh, S. Zhang, Q. Liu and L. Shi, Transfer learning with graph neural networks for optoelectronic properties of conjugated oligomers, J. Chem. Phys., 2021, 154(2), 024906 CrossRef CAS PubMed.
- M. Nakata and T. Shimazaki, PubChemQC project: a large-scale first-principles electronic structure database for data-driven chemistry, J. Chem. Inf. Model., 2017, 57(6), 1300–1308 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields, J. Phys. Chem., 1994, 98(45), 11623–11627 CrossRef CAS.
- P. Hariharan and J. A. Pople, Accuracy of AH n equilibrium geometries by single determinant molecular orbital theory, Mol. Phys., 1974, 27(1), 209–214 Search PubMed.
- O. D. Abarbanel and G. R. Hutchison, Using genetic algorithms to discover novel ground-state triplet conjugated polymers, Phys. Chem. Chem. Phys., 2023, 25(16), 11278–11285 CAS.
- G. Landrum, RDKit: Open-source cheminformatics, https://www.rdkit.org/, (accessed 2023-07-20) Search PubMed.
- K. T. Schütt, H. E. Sauceda, P.-J. Kindermans, A. Tkatchenko and K.-R. Müller, Schnet–a deep learning architecture for molecules and materials, J. Chem. Phys., 2018, 148(24), 241722 Search PubMed.
- P. Cheng and Y. Yang, Narrowing the band gap: the key to high-performance organic photovoltaics, Acc. Chem. Res., 2020, 53(6), 1218–1228 CAS.
- T. Otsubo, Y. Aso and K. Takimiya, Functional oligothiophenes as advanced molecular electronic materials, J. Mater. Chem., 2002, 12(9), 2565–2575 CAS.
- G. Turkoglu, M. E. Cinar and T. Ozturk, Thiophene-based organic semiconductors, Sulfur Chem., 2019, 79–123 Search PubMed.
- L. Zhang, N. S. Colella, B. P. Cherniawski, S. C. Mannsfeld and A. L. Briseno, Oligothiophene semiconductors: synthesis, characterization, and applications for organic devices, ACS Appl. Mater. Interfaces, 2014, 6(8), 5327–5343 CAS.
- R. Fitzner, E. Mena-Osteritz, K. Walzer, M. Pfeiffer and P. Bäuerle, A–D–A-Type Oligothiophenes for Small Molecule Organic Solar Cells: Extending the π-System by Introduction of Ring-Locked Double Bonds, Adv. Funct. Mater., 2015, 25(12), 1845–1856 Search PubMed.
- Y. Zou, Y. Wu, H. Yang, Y. Dong, C. Cui and Y. Li, The effect of alkylthio side chains in oligothiophene-based donor materials for organic solar cells, Mol. Syst. Des. Eng., 2018, 3(1), 131–141 RSC.
- Z. Luo, R. Ma, J. Yu, H. Liu, T. Liu, F. Ni, J. Hu, Y. Zou, A. Zeng and C.-J. Su, Heteroheptacene-based acceptors with thieno [3, 2-b] pyrrole yield high-performance polymer solar cells, Natl. Sci. Rev., 2022, 9(7), nwac076 Search PubMed.
- C. Bulumulla, R. Gunawardhana, P. L. Gamage, J. T. Miller, R. N. Kularatne, M. C. Biewer and M. C. Stefan, Pyrrole-containing semiconducting materials: synthesis and applications in organic photovoltaics and organic field-effect transistors, ACS Appl. Mater. Interfaces, 2020, 12(29), 32209–32232 Search PubMed.
- P. Gao, D. Cho, X. Yang, V. Enkelmann, M. Baumgarten and K. Müllen, Heteroheptacenes with fused thiophene and pyrrole rings, Chem. – Eur. J., 2010, 16(17), 5119–5128 CrossRef CAS PubMed.
- D. S. Matteson and H. Snyder, A Practical Synthesis of Thieno [3, 2-b] pyrrole, J. Org. Chem., 1957, 22(11), 1500–1504 Search PubMed.
- S.-L. Chang, K.-E. Hung, F.-Y. Cao, K.-H. Huang, C.-S. Hsu, C.-Y. Liao, C.-H. Lee and Y.-J. Cheng, Isomerically Pure Benzothiophene-Incorporated Acceptor: Achieving Improved V OC and J SC of Nonfullerene Organic Solar Cells via End Group Manipulation, ACS Appl. Mater. Interfaces, 2019, 11(36), 33179–33187 Search PubMed.
- A. L. Capodilupo, E. Fabiano, L. De Marco, G. Ciccarella, G. Gigli, C. Martinelli and A. Cardone, [1] Benzothieno [3, 2-b] benzothiophene-based organic dyes for dye-sensitized solar cells, J. Org. Chem., 2016, 81(8), 3235–3245 Search PubMed.
- I. Shafiq, M. Khalid, G. Maria, N. Raza, A. A. Braga, S. Bullo and M. Khairy, Use of benzothiophene ring to improve the photovoltaic efficacy of cyanopyridinone-based organic chromophores: a DFT study, RSC Adv., 2024, 14(18), 12841–12852 CAS.
- C. B. Nielsen, R. S. Ashraf, N. D. Treat, B. C. Schroeder, J. E. Donaghey, A. J. White, N. Stingelin and I. McCulloch, 2, 1, 3-Benzothiadiazole-5, 6-Dicarboxylic Imide–A Versatile Building Block for Additive-and Annealing-Free Processing of Organic Solar Cells with Efficiencies Exceeding 8%, Adv. Mater., 2015, 27(5), 948–953 Search PubMed.
- Z. Wu, B. Fan, F. Xue, C. Adachi and J. Ouyang, Organic molecules based on dithienyl-2, 1, 3-benzothiadiazole as new donor materials for solution-processed organic photovoltaic cells, Sol. Energy Mater. Sol. Cells, 2010, 94(12), 2230–2237 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4me00188e |
| This journal is © The Royal Society of Chemistry 2025 |