
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mitigating triplet loss in 2D WSe2/non-fullerene heterostructures using halogenated acceptors†
Sreelakshmi Chandrabose *ab,
Ana M. Valenciacd,
Meysam Raoufia,
Nisreen Alshehrieg,
Tracey M. Clarkef,
Frédéric Laquaie,
Caterina Cocchicd and
Dieter Neher*a
*ab,
Ana M. Valenciacd,
Meysam Raoufia,
Nisreen Alshehrieg,
Tracey M. Clarkef,
Frédéric Laquaie,
Caterina Cocchicd and
Dieter Neher*a
aInstitute of Physics and Astronomy, University of Potsdam, Karl-Liebknecht-Straße 24/25, 14476, Germany. E-mail: neher@uni-potsdam.de
bLaboratoire Interactions, Dynamiques et Lasers (LIDYL), CEA-Paris Saclay, 91191 Gif-Sur-Yvette Cedex, France. E-mail: sreelakshmi.chandrabose@cea.fr
cCarl von Ossietzky Universität Oldenburg, Institute of Physics and Center for Nanoscale Dynamics, 26129 Oldenburg, Germany
dInstitut für Physik & IRIS Adlershof, Humboldt-Universität zu Berlin, 12489 Berlin, Germany
eKAUST Solar Center, Physical Science and Engineering Division (PSE), Materials Science and Engineering Program (MSE), King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia
fDepartment of Chemistry, University College London, London WC1H 0AJ, UK
gPhysics and Astronomy Department, College of Sciences, King Saud University, Riyadh 12372, Kingdom of Saudi Arabia
First published on 6th January 2025
Abstract
Two-dimensional transition metal dichalcogenides (2D TMDCs) can be combined with organic semiconductors to form hybrid van der Waals heterostructures. Specially, non-fullerene acceptors (NFAs) stand out due to their excellent absorption and exciton diffusion properties. Here, we couple monolayer tungsten diselenide (ML-WSe2) with two well performing NFAs, ITIC, and IT-4F (fluorinated ITIC) to achieve hybrid architectures. Using steady state and time resolved spectroscopic techniques, we reveal sub-picosecond free charge generation in the heterostructure of ML-WSe2 with ITIC, where however, bimolecular recombination of spin uncorrelated charge carriers with possible contributions from geminate charge recombination cause rapid formation of low-lying triplet (T1) states in ITIC. Importantly, this unwanted process is effectively suppressed when the fluorinated derivative of ITIC, IT-4F, is deposited on ML-WSe2. We observe a similar scenario when replacing the ML-TMDC with copper thiocyanate (CuSCN) as the hole acceptor meaning that triplet state formation is not driven by the spin–orbit coupling of ML-WSe2. From ab initio calculations based on density functional theory, we interpret the high triplet formation in the ML-WSe2/ITIC hybrid bilayer due to changes in the nature and energies of the interfacial charge transfer (CT) levels. Our results highlight the delicate balance between excitons and charges in such inorganic/NFA heterostructures.
New conceptsSo far, there have been numerous investigations on the charge transfer process in heterostructures of 2D transition metal dichalcogenides (TMDCs) and organic semiconductors, where in most of the cases, organic materials functioned as hole acceptors. Herein, we add non-fullerene based organic electron acceptors to the list for the first time, which have high as well as complementary absorption with 2D TMDCs, and thus the hybrids can span the whole UV-Vis-NIR region of the solar spectrum. This manuscript presents a systematic study of the charge transfer mechanism at the interfaces of 2D WSe2 and two non-fullerene acceptors ITIC and IT-4F using ultrafast transient absorption spectroscopy (TAS). The importance of this study lies especially in combining non-fullerene acceptors with 2D TMDCs, as they have received significant interest recently in the field of organic photovoltaics due to their attractive properties. The mechanism behind triplet generation and mitigation in WSe2/non-fullerene heterostructures is carefully addressed with appropriate experimental evidence and theoretical support. Furthermore, this study opens a way to the realization of photovoltaic devices using 2D TMDCs as the non-fullerene acceptors solve the problem of low absorption and thereby low exciton harvesting exhibited by TMDCs. |
Introduction
Owing to their unique electronic and optical properties, two-dimensional (2D) transition metal dichalcogenides (TMDCs) have become promising candidates in the field of optoelectronics to achieve thin layer devices such as photovoltaic cells,1,2 photodetectors,3 field effect transistors4 and light emitting diodes.5 In contrast to traditional inorganic semiconductors, 2D TMDCs are excitonic materials possessing relatively large exciton binding energies of several hundreds of meV, arising from the high quantum confinement effect and reduced dielectric screening.6 This brings the necessity of combining them with a second material with suitable energetics to form type-II van der Waals (vdW) heterostructures especially for photovoltaic applications.Organic semiconductors are of particular interest for interfaces with 2D TMDCs as they are able to interact with them via vdW forces due to the lack of surface dangling bonds.7 Additionally, the good tunability of organic semiconductors’ frontier orbital energies combined with high optical absorption in the UV-Vis region make them ideal partners for 2D semiconductors. For instance, Shastry et al. reported the first experimental realization of the photovoltaic effect in the type-II heterojunction of molybdenum disulphide (MoS2) and an organic π-donor polymer PTB7.2 The MoS2/PTB7 cell exhibited a record current density per thickness, much higher than the competing photovoltaic technologies, and thus established TMDC's potential in the field of ultrathin solar cells.2 Later, Zhong et al. explored ultrafast transient absorption spectroscopy to study the charge transfer mechanism at the MoS2/PTB7 junction where they could probe the generation of photocarriers in two distinct time scales: <250 fs, due to the electron transfer process from PTB7 to electronically hot states of MoS2 and 1–5 ps due to both hole transfer from photoexcited MoS2 to PTB7 and exciton diffusion-limited electron transfer from PTB7 to MoS2.8 Since then, there have been numerous investigations on the charge transfer process in heterostructures of 2D TMDCs and various organic semiconductors such as polymers (P3HT, PCDTBT,9 PTB78), small molecules (pentacene,10 tetracene,11 CuPc, H2Pc,12 ZnPc,13 PDI,14 C6015), organic dyes (PTCDA,16,17 PTCDI18) and carbon nanotubes (SWCNT).19 In fact, in most of the studied 2D TMDC/organic combinations, organic materials functioned as hole accepting or electron donating components, while only in a limited number of reported heterostructures, they could act as electron acceptors. For instance, Rijal et al. observed ultrafast photoinduced electron transfer from either monolayer WSe2 or MoS2 to organic acceptors such as C60 and PTCDI using time resolved microwave conductivity (TRMC) where they addressed the effect of energy level offsets on free charge formation.18 However, from a photovoltaic device perspective, these acceptors are not ideal for combining with TMDCs as they possess very low absorption especially in the near infrared (NIR) region. Herein, we add non-fullerene based organic electron acceptors to the list for the first time, which have high as well as complementary absorption with 2D TMDCs, and thus the hybrids can span the whole UV-Vis-NIR region of the solar spectrum.
Unlike in 2D TMDC/TMDC heterostructures, exciton diffusion length has a prime role in efficient exciton harvesting at the sharp interface formed by 2D TMDC/organic hybrids, as the photo-generated exciton in the thick organic layer must travel a large distance to find the interface where it can be dissociated. Recently, non-fullerene acceptors (NFAs), a novel class of organic electron acceptors, have received significant interest in the field of organic photovoltaics due their attractive properties including tunable bandgap, strong absorption coefficients and importantly Förster resonance energy transfer (FRET) mediating long exciton diffusion lengths exceeding 30 nm.20–22 Recent work demonstrated high power conversion efficiencies (PCEs) in so-called pseudo-bilayers based on an NFA and an electron-donating polymer.22–24 This gives the motivation to combine NFAs with 2D TMDCs to achieve novel hybrid architectures that benefit from the advantageous properties of both the materials.
In this work, we selected two best performing NFAs, ITIC and IT-4F (fluorinated ITIC), exhibiting long exciton diffusion lengths of >30 nm to form type-II heterojunctions with a well-known 2D TMDC, ML-WSe2, acting as an acceptor for photo-generated holes in the NFA layer (Fig. 1a). The chosen NFAs are characterized by large absorption coefficients of >105 M−1 cm−1 and broad absorption ranging from the visible to near infrared regions, making them ideal systems to couple with WSe2 to efficiently harvest the solar photons (Fig. S1 and Table S1, ESI†). The energetic landscapes of all the materials are shown in Fig. 1b where all the energy values are taken from the literature.25–27 The figure reveals that each donor and acceptor combination exhibits favorable thermodynamic driving forces to provide a photovoltaic effect. Moreover, it has been established that due to the strong electron withdrawing nature of halide atoms, fluorination in ITIC simultaneously increases the ionization potential (IP) and electron affinity (EA) without causing severe steric hindrance in the resulting molecules.28
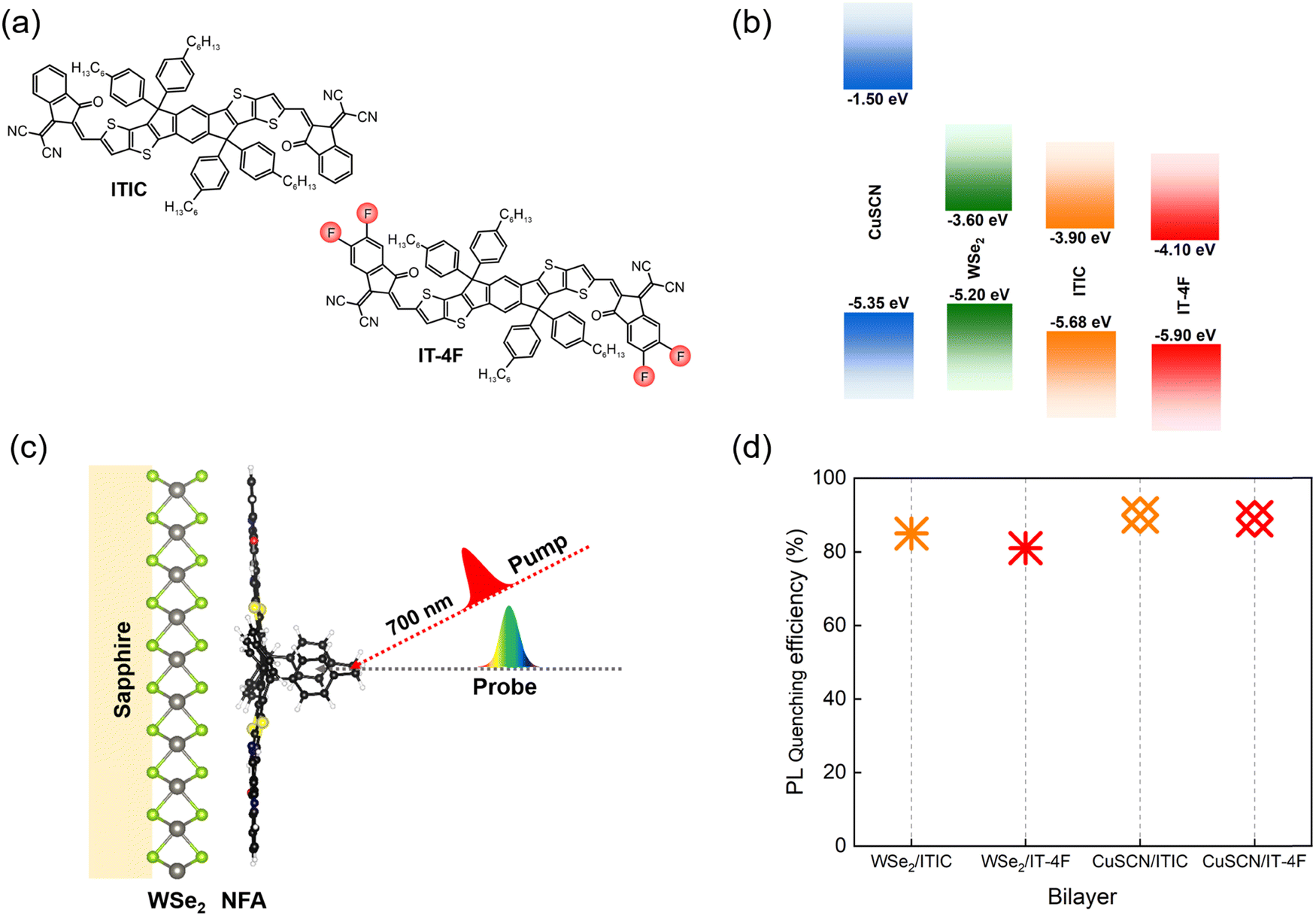 | ||
| Fig. 1 Active layer components and photoluminescence quenching. (a) Molecular structures of the NFAs, ITIC and IT-4F; (b) energy band diagram of donor materials (ML-WSe2 and CuSCN) and NFAs where all the energy values are taken from the literature;25–27 (c) schematic structure of the active layer; (d) photoluminescence quenching efficiencies in ML-WSe2/NFA and CuSCN/NFA bilayers (the corresponding PL spectra are given in Fig. S2, ESI†). | ||
We explore steady state photoluminescence and ultrafast broadband transient absorption spectroscopy (TAS) to probe the charge transfer mechanism at ML-WSe2/NFA interfaces (the active layer structure is given in Fig. 1c). For the ML-WSe2/ITIC interface, it is found that the optically excited excitons undergo fast dissociation and generate polarons in sub-picosecond time scales, which is in line with the strong photoluminescence quenching observed in the system. From pump fluence dependent TAS measurements we demonstrate that the polaron recombination follows bimolecular kinetics, which then populates the low-lying triplet (T1) states of the acceptor, and thus opens an additional non-radiative loss channel in the heterostructure. To prove that the heavy atoms in the donor WSe2 have no role in the spin flipping process, we conducted parallel TAS measurements on a bilayer comprising CuSCN, a large bandgap inorganic semiconductor with nearly the same ionization energy as WSe2, where we could observe a similar phenomenon. The triplet loss channel is strongly suppressed in the bilayers with the fluorinated derivative of ITIC, IT-4F, on WSe2 and CuSCN. Supported by ab initio results from density functional theory (DFT) calculations, we ascribe the suppression of triplet formation to changes in the nature and energies of the interfacial charge transfer states. Overall, by elucidating the excitation dynamics in 2D TMDC/NFA hybrids, we emphasize the delicate balance between excitons and charges in such inorganic/organic heterostructures.
Methodology
Material preparation
The organic non-fullerene acceptors, ITIC and IT-4F, were purchased from 1-Material Inc. and Sigma Aldrich, respectively. The monolayer WSe2 grown on c-cut sapphire substrate via chemical vapor deposition (CVD) was purchased from the 2D Semiconductors Inc. For the films of NFAs, the NFA molecules were dissolved in chlorobenzene (concentration of 5 mg mL−1). For neat organic samples, this solution was spin-coated statically onto micro-glass substrates in a nitrogen atmosphere and at room temperature at a rotation speed of 4500 rpm for 40 seconds. The films of NFAs on monolayer WSe2 were prepared under the same conditions. Here, all the bilayer films were fabricated using the same WSe2 monolayer where each time it is washed with chloroform several times to remove the organic layer on top. We measured steady state absorption of TMDC to confirm that no remnant of NFA was present on WSe2 after each washing with chloroform. For reference measurements, CuSCN (100 nm)/NFA bilayer films were prepared using the same procedure for NFAs while the CuSCN layer was prepared via spin casting the filtered solution of CuSCN in diethyl sulphide (26 mg mL−1) at 2500 rpm for 30 s. All the films were annealed at 110 °C (105 °C for CuSCN) for 10 minutes. The NFA layer thicknesses were measured using atomic force microscopy (AFM) and were ∼15 nm and ∼11 nm for ITIC and IT-4F, respectively. Prior to the measurements, the samples were stored in a nitrogen glovebox under dark conditions to prevent contact with air and light.Steady state optical measurements
Time resolved spectroscopic measurements
Multivariate curve resolution by alternating least squares (MCR-ALS) is used for spectral decomposition. This iterative tool works by optimizing a set of initial guesses of spectral and kinetic profiles under the action of specific constraints until a convergence criterion is achieved. The method is based on the following bilinear decomposition,
| ΔA(λ,t) = C{opt}(t)·S{opt}(λ)T + Error |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in chlorobenzene. Then, the solutions were spin-coated on pre-cleaned quartz substrates in a nitrogen environment at 1000 rpm for 30 s.
:1 in chlorobenzene. Then, the solutions were spin-coated on pre-cleaned quartz substrates in a nitrogen environment at 1000 rpm for 30 s.Quasi-steady-state PIA experiments were performed with a pump probe setup consisting of a 100 W quartz tungsten halogen (QTH) light source (Oriel Instruments-Newport) used as probe and the output of narrow linewidth continuous-wave laser (Cobolt 08-DPL-515 nm) used as the pump. The transmitted probe light was detected after dispersion by a monochromator by a silicon-based photodetector (Thorlabs DET10A/M) in the wavelength range from 200 to 1100 nm, and InbS detector. The pump light was modulated at approximately 368 Hz by a lock-in, and the change in transmission ΔT induced by the pump was extracted by using the lock-in technique (Stanford Research Systems, Model SR830 DSP). After the PIA measurement, the total transmission was measured to calculate ΔT/T. PIA measurement was performed in a nitrogen-cooled optical cryostat (Oxford Instruments Optistat CF) at 80 K in a nitrogen atmosphere. The total probe area was approximately 0.25 cm2.
For the 1 ns to 100 μs delay (long delay) TA measurement, a fraction of the output signal of titanium:sapphire amplifier (Coherent LEGEND DUO, 4.5 mJ, 3 kHz, 100 fs) was focused into a c-cut 3 mm thick sapphire window, thereby generating a white-light super-continuum from 500 to 1600 nm. Whilst the excitation light (pump pulse) was provided by an actively Q-switched Nd:YVO4 laser (InnoLas picolo AOT) frequency-doubled to provide pulses at 532 nm. The laser was triggered by an electronic delay generator (Stanford Research Systems DG535), itself triggered by the TTL sync from the legend DUO, allowing control of the delay between pump and probe with a jitter of roughly 100 ps. Pump and probe beams were focused on the sample with the aid of appropriate optics, which was kept under a dynamic vacuum of <10−5 mbar. The transmitted fraction of the white light was guided to a custom-made prism spectrograph (Entwicklungsburo Stresing) where it was dispersed by a prism onto a 512-pixel NMOS linear image sensor (Hamamatsu S8381-512). The probe pulse repetition rate was 3 kHz, while the excitation pulses were mechanically chopped to 1.5 kHz (100 fs to 8 ns delays) or directly generated at 1.5 kHz frequency (1 ns to 100 μs delays), while the detector array was read out at 3 kHz. Adjacent diode readings corresponding to the transmission of the sample after excitation and in the absence of an excitation pulse were used to calculate ΔT/T. Measurements were averaged over several thousand shots to obtain a good signal-to-noise ratio.
Ab initio calculations
The calculations presented in this work are based on DFT,30,31 implemented in the all-electron, numeric atom-centered code FHI-aims.32,33 We employ tight integration grids and tier2 basis sets,34 and relax the atomic positions until the Hellmann–Feymann forces are smaller than 10−3 eV Å−1. We use the Monkhorst–Pack scheme35 (Γ-point included) for sampling the Brillouin zone with a 4 × 4 × 1 k-point grid. In the structural optimization runs, the Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation36 is employed for the exchange–correlation potential while the hybrid functional HSE0637 including spin–orbit coupling (SOC) is used to calculate electronic properties with the optimized geometries to obtain an accurate description of charge transfer and energy level alignment. All calculations include the Tkatchenko–Scheffer scheme with many body dispersions to account for vdW interactions.38 To avoid the spurious electrostatic couplings between periodic images, a vacuum region of 42 Å is included in the supercell and a dipole correction is employed.39 The partial charge analysis in the ground state is performed with the Hirshfeld scheme embedded in FHI-aims. The software VESTA is used to produce the charge-density plots.Results
Steady state absorption and photoluminescence
The steady state absorption spectra of the neat donor materials (2D WSe2 and CuSCN) along with that of the bilayers of ITIC and IT-4F with the monolayer WSe2 are displayed in Fig. S1 (ESI†). For the donor WSe2, the absorption peaks appear at 422 nm, 505 nm, 595 nm and 745 nm which represent the C, A′, B and A excitonic transitions, respectively.40,41 The low energy excitonic peaks A and B are well studied and originate from the transitions between the spin split valence band and degenerate conduction band at the K point of the Brillouin zone. The strong A′ and C band at higher photon energies arise from the transitions between the deep parallel bands where the absorption is amplified due to the so-called ‘band nesting effect’.42 In contrast to WSe2, CuSCN exhibits negligible absorption within the wavelength range above 400 nm, while the background in the spectrum arises from interference effects.43 The bilayer spectra include the absorption features from NFAs with two extra absorption bands peaking at 425 and 595 nm which correspond to the C and A′ excitons in WSe2, respectively. Here the absorption maxima of all the NFAs are well matched with the previously reported values.44,45 We observed similar absorption profiles of neat NFAs and bilayer films, confirming that the spin casting of solution processed NFAs was not affected by the different surfaces of the glass and the ML-WSe2 covered substrate.Fig. 1d includes the results of photoluminescence quenching measurements in ML-WSe2/NFA bilayers. As the figure shows, the PL of both the NFAs underwent significant reduction on interfacing with the TMDC, which reflects the strong rate of exciton dissociation and thereby hole transfer towards the donor WSe2. Here, very thin layers of NFAs (∼15 nm) were used in the bilayer films to make sure that the PL quenching and the charge transfer could occur prior to NFA exciton relaxation back to the ground state, given that the inorganic/organic bilayers create sharp interfaces, in contrast to the partially intermixed organic/organic bilayer interfaces.46 The PL quenching efficiencies were reproducible at various spots of the bilayer films, suggesting homogeneous coverage of the substrate with the ML-WSe2 within the special resolution of our experiments.
Triplet formation in ITIC based heterostructures
A series of transient absorption spectra for neat NFA films at various pump–probe time delays are shown in Fig. S3a and b (ESI†). All the measurements are carried out at an excitation wavelength of 700 nm and fluence of ∼2 μJ cm−2. The spectra give negative differential absorption features around the corresponding ground state absorption wavelengths, which can be largely attributed to the ground state bleaching (GSB). The spectra also feature positive, red shifted sub-gap excitonic photoinduced absorption (PIA) features. The similar decay rates of GSB and PIA features confirm the presence of single species, singlet excitons (insets of Fig. S3c and d, ESI†). Additionally, the exciton lifetimes in both NFA films were found to be shorter compared to that of the corresponding monomolecular decay times (Table S1 (ESI†), all the parameters are collected from the literature). This reveals that singlet–singlet exciton annihilation (SSA), a common loss channel in NFAs, fosters exciton decays in our TAS measurements.47 However, the GSB and the exciton PIA remain the only species over the entire delay time, meaning that SSA does not produce any other excitation.Fig. 2b gives the transient absorption spectra of a ML-WSe2/ITIC bilayer with excitation at 700 nm and at a pump fluence of 2.2 μJ cm−2. At early time scales, the spectral features resemble that of the singlet excitons generated in the pristine ITIC film (Fig. 2a). Note that the ML-WSe2 exhibits a weak absorption at the excitation wavelength, which is the reason why we do not detect a notable signal from the excited WSe2. Given the very good spectral overlap of the WSe2 emission and the ITIC absorption (see Fig. S1 for the steady state absorption spectra, ESI†), we speculate rapid depopulation of any exciton population on WSe2 due to resonant energy transfer.48–50 After 0.6 ps, a new negative, red shifted feature is recorded at 745 nm, overlapping with the retaining GSB of ITIC. This secondary feature is attributed to the GSB of the A band of WSe2, which we interpret as a signature of hole transfer to the 2D monolayer, and which persists beyond the 7 ns time window of the experiment. The pump fluence dependent measurements on the bilayer confirm that the transferred species undergo bimolecular recombination, representing the formation of a charge separated state (Fig. S4a, ESI†). On the other hand, the electron PIA expected at ∼980–1000 nm is only weak, and a new PIA band gradually builds up at ∼1220 nm.51 To identify this spectral feature, we did triplet sensitization measurements where the TAS measurement is carried out on a heavy metal doped ITIC film (ITIC: PtOEP). The triplet induced spectrum of ITIC (overlaid on Fig. 2b) reveals that the photoinduced feature at ∼1220 nm is due to triplet excitons on ITIC. The triplet population (obtained after spectral decomposition using MCR-ALS, see details in the methodology section) then increase up to ∼100 ps after which it undergoes fast recombination with a time constant of ∼4 ns (amplitude weighted lifetime) (Fig. 2c).
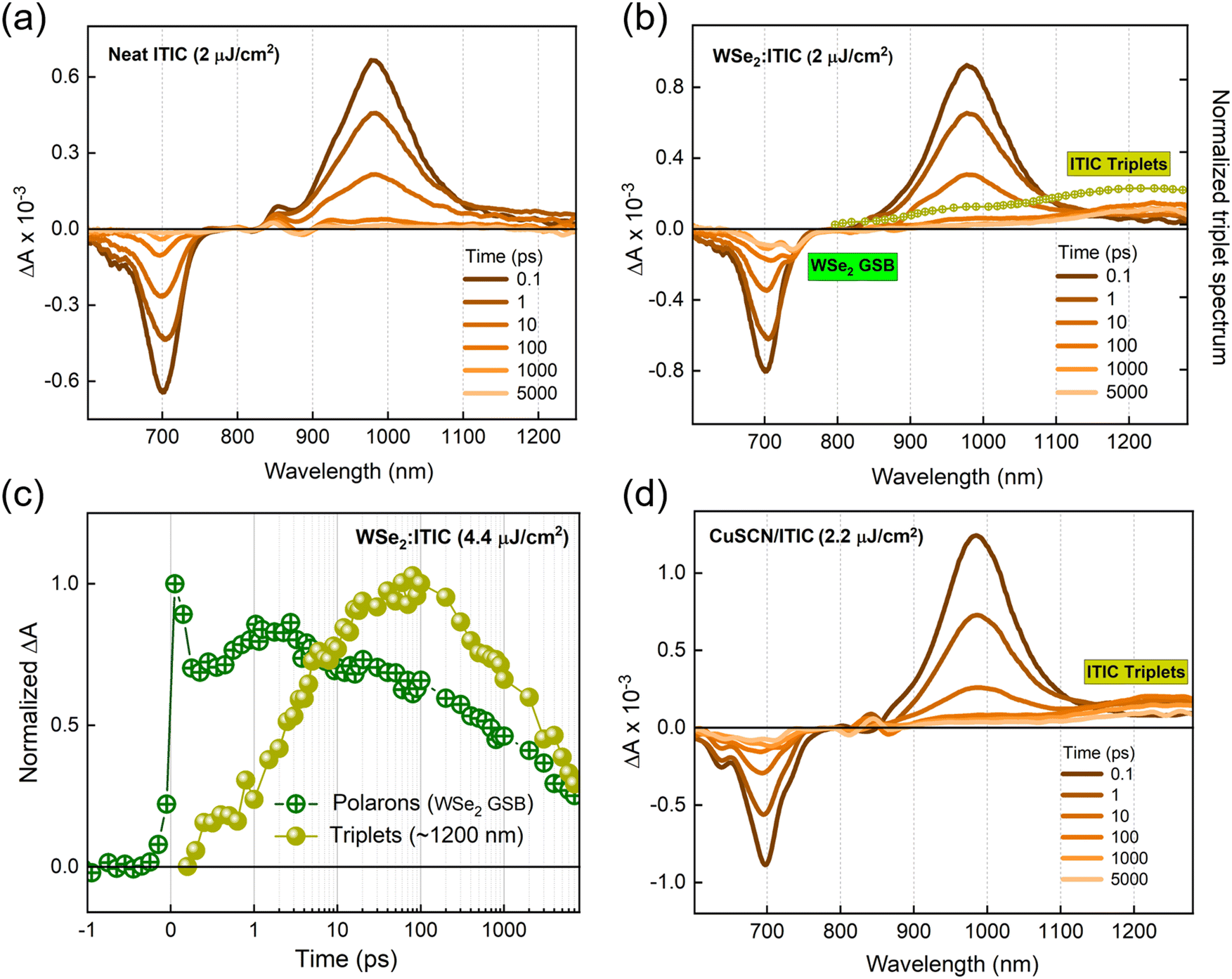 | ||
| Fig. 2 Excitation dynamics in ITIC-based hybrid systems. Transient absorption spectra of (a) a neat ITIC film and (b) the WSe2/ITIC bilayer (both excited at 700 nm at pump fluence of 2 μJ cm−2); the normalized triplet induced spectrum is overlaid, (c) kinetics of WSe2 hole GSB along with the triplet exciton dynamics (acquired from MCR-ALS), and (d) transient absorption spectra of the CuSCN/ITIC film excited at 700 nm and at a pump fluence of 2.2 μJ cm−2. | ||
The population of triplet levels (T1) in a material can happen via three channels: singlet fission, intersystem crossing, and recombination of photo-generated charge carriers or CT states. Here the possibility of singlet fission in the NFA phase is ruled out as the singlet excitons in the neat ITIC film deactivate mainly through singlet–singlet exciton annihilation (SSA) and monomolecular recombination; as evident from the exciton decays plotted for different pump fluence (Fig. S3c and d, ESI†). The fast-initial exciton decay which depends on the excitation fluence is mostly due to SSA, while the slower intensity independent decay at longer time scales arises from monomolecular recombination of singlet excitons. On the other hand, the TAS spectra miss the characteristic absorption of triplet excitons, meaning that the decay of singlets did not result in the detectable generation of any new excited species. For the same reason, intersystem crossing in the ITIC layer can be excluded too.
Most organic materials are characterized by weak spin–orbit coupling and thereby long spin relaxation times (100 ps–1 ms).52 In our ML-WSe2/ITIC bilayer, a significant triplet population is created within a few picoseconds after photoexcitation (Fig. 2c). There are recent reports showing that the strong spin orbit coupling associated with the heavy metal atoms in TMDCs can expedite a spin flip to picosecond time scale. For instance, using time-resolved photoemission spectroscopy (TR-TPPE), Kafle et al. studied the ultrafast CT dynamics at the MoS2/ZnPc interface where they could detect sub-picosecond triplet exciton formation in ZnPc domains followed by the electron transfer to the acceptor MoS2; the triplets were generated due to the back transfer of electrons in MoS2 whose spins were flipped to the donor ZnPc.13 To check if the heavy atom effect in the WSe2 has a role in populating the triplet states in ITIC, we replaced the TMDC by CuSCN, a well-known inorganic hole transporting material (HTL)43 with similar ionization energy as ML-WSe2. Here CuSCN is chosen as the donor material as it does not include any heavy atoms which can induce a spin flipping. Efficient exciton dissociation in the bilayer is confirmed by the quenching of ITIC exciton PL by ∼90% in the bilayer (Fig. 1d).
Fig. 2d presents the TAS spectra of the CuSCN/ITIC bilayer excited at 700 nm and at a pump fluence of 2.2 μJ cm−2, where the bilayer is fabricated by spin coating a 15 nm layer of ITIC on a 100 nm thick CuSCN layer. The TAS spectra at early time scales are dominated by the ITIC singlet excitons due to the negligible optical absorption of CuSCN at the pump wavelength (Fig. S1, ESI†). Interestingly, within a few picoseconds, the triplet band of ITIC at ∼1220 nm is populated, retaining the GSB at ∼700 nm, which persists beyond 7 ns. Here the exact timescale of the hole transfer process is imperceptible as the hole GSB of CuSCN should appear in the deep UV region according to its steady state absorption spectrum (Fig. S1, ESI†). The above observations suggest the possibility of a common mechanism behind the triplet generation process in both WSe2/ITIC and CuSCN/ITIC bilayers and thereby obviates the role of the heavy atom effect from the scenario.
Finally, we consider the possibility of triplet exciton generation from photogenerated free carriers and CT states. The mechanism is depicted in the Jablonski diagram shown in Fig. 3b. The process involves the bimolecular recombination of the spin-uncorrelated electrons and holes, filling the low-lying triplet states in a material via intermediate charge transfer triplet states (CT3). Indeed, this is a common non-radiative loss mechanism in non-fullerene acceptor based organic solar cells, which occurs mainly due to the large energy difference between the singlet and triplet excitons in these materials, placing the triplet state even below the CT3.53–55 Furthermore, geminate recombination of bound charge pairs or CT states prior to charge separation can also serve as the source of triplet excitons.56,57 To prove the contribution of the aforementioned processes in triplet generation in the considered bilayers, we performed TAS measurements at different fluences. As pointed out earlier, the decay kinetics of the generated free carriers in the ML-WSe2/ITIC bilayer is determined by bimolecular recombination (Fig. S4a, ESI†). Fig. 3a shows the TAS spectra at ∼100 ps time delay (where the triplet population is maximum). The T1 state absorption spectra at ∼1220 nm become more pronounced with increasing fluence, going along with the strong reduction of the hole GSB of WSe2 at ∼745 nm. In addition, the TAS spectrum measured at the lowest fluence (1.1 μJ cm−2) includes a very clear PIA signature from electrons at ∼1000 nm along with a relatively low contribution from triplets, meaning that triplet excitons grow mainly with the bimolecular recombination of charges. Additionally, we present the fluence dependent kinetics of triplet excitons obtained after spectral decomposition of the 800–1270 nm region using the MCR-ALS algorithm, in Fig. S4b (ESI†) (the corresponding spectra are displayed in Fig. S4c, ESI†). The fluence dependent generation of the triplets emphasizes the fact that they originate from the bimolecular recombination of charge carriers, especially at high fluences. However, we also note that at low fluences, geminate recombination could also be a source of triplets as seen from the close similarity of triplet rise kinetics at 1.1 and 2.0 μJ cm−2 fluences. Even at these low fluences, the triplet population grow immediately at very early times, which then maximizes at the same time scale irrespective of fluence – all hallmarks of geminate recombination. The localized triplet state then undergoes recombination in ∼4 ns time scale which is much faster than the monomolecular triplet lifetime of 120–160 μs reported for ITIC in polystyrene.58 This implies that the triplet state in the bilayer deactivates via higher order recombination pathways, most probably triplet-charge annihilation, resulting in an additional non-radiative loss.55 Importantly, our study shows that the combination of WSe2 with ITIC is unfavorable for its use in photodiodes and photovoltaic cells.
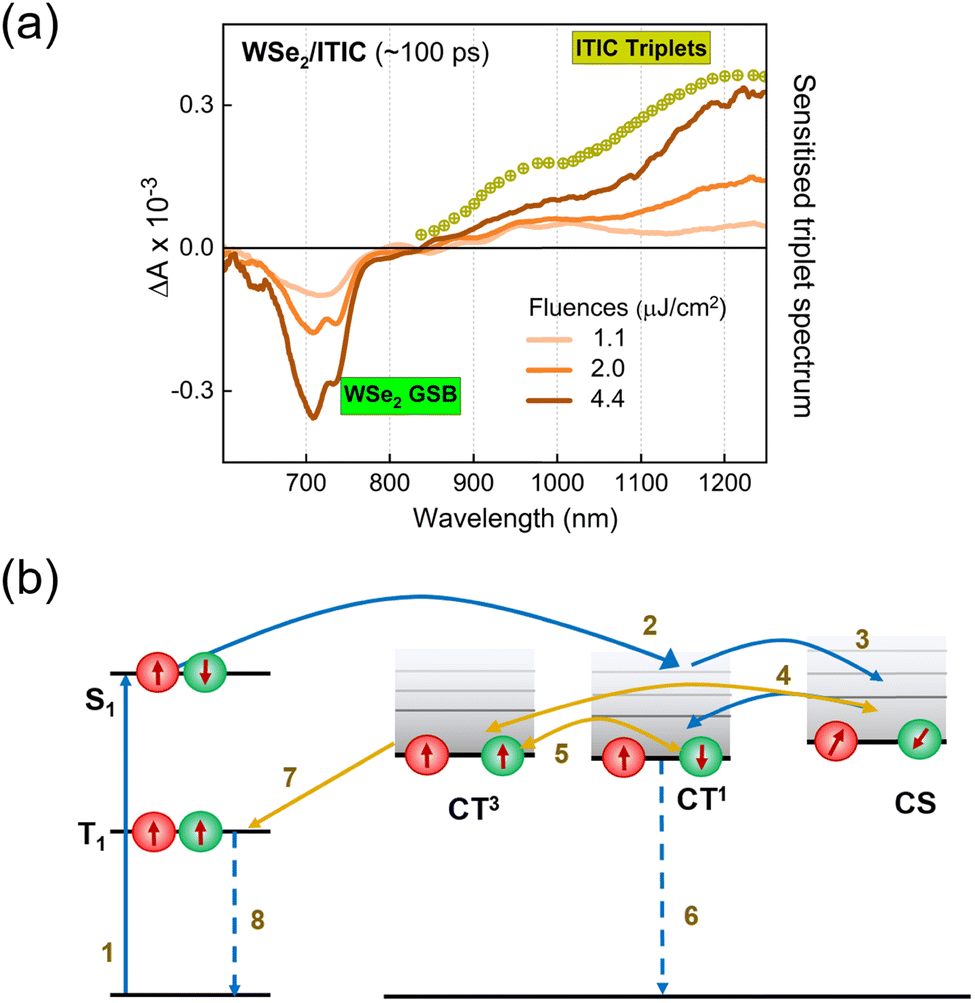 | ||
| Fig. 3 Mechanism of triplet exciton generation in the ML-WSe2/ITIC bilayer. (a) Fluence dependent transient absorption spectra of the ML-WSe2/ITIC film probed at ∼100 ps after photoexcitation and (b) Jablonski diagram showing the process of generation of triplet excitons in the bilayer. (1) Photoexcitation creates singlet (S1) excitons, (2) formation of CT1 states at the heterojunction that separates into charges (3), bimolecular recombination of charge carriers generates CT1 and CT3 states (4), direct formation of the CT3 state from the CT1 state (5), recombination of the CT1 state to the ground state (6), energetically favorable relaxation of the CT3 state to the T1 state (7), and T1 state return to the ground state via triplet-charge annihilation (8). In our case, the triplets are formed in ITIC, the acceptor. | ||
To quantify the triplet exciton formation in ITIC, we used the recently published value for the molar extinction coefficient of the excited triplet, εT,58 following the equation,
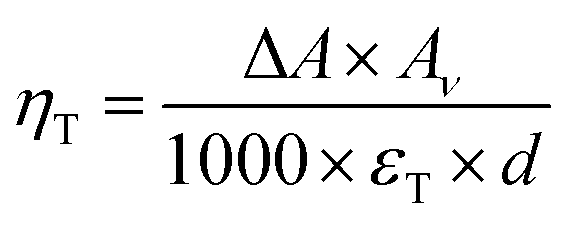
Fluorination suppresses triplet generation
As mentioned before, triplet formation through free charge recombination is a rather common process in organic materials. Recently, Souza et al. established that the fluorination and chlorination of NFAs can serve as facile, but highly efficient ways to suppress the triplet generation channel in organic bulk heterojunction devices.59 They attribute this to the reduction of the singlet–triplet energy gap, ΔEST, upon fluorine and chlorine substitution, which reduces the binding energy of the triplet exciton and thereby favors their re-dissociation into free charges. Alternatively, Wang et al. attributed the suppression of triplet formation in organic photovoltaic blends upon fluorination to a reduction of the energy of the CT3 excited states at the heterojunction below the triplet exciton in the neat NFA, favoring their re-splitting into free charges.60 Based on these findings, we selected a halogenated derivative of ITIC, IT-4F with four fluorine atoms introduced into the terminal acceptor moieties of ITIC (molecular structure is shown in Fig. 1a).45 It has been reported that the fluorinated (X) derivative exhibits improved crystallinity due to the non-covalent interactions of F⋯H and S⋯F groups which facilitate exciton and charge transport.61Photoluminescence quenching measurements reveal that both the ML-WSe2/IT-4F and CuSCN/IT-4F bilayers exhibit very similar PL quenching efficiencies of ∼90% (Fig. 1d). The results of the TAS measurement on a ML-WSe2/IT-4F bilayer after excitation at 700 nm are shown in Fig. 4a. The singlet exciton bleach of IT-4F at early times exhibits a redshift in comparison with that of ITIC and is in accordance with its ground state absorption spectrum. The bleach signal lives longer throughout the delay stage, representing the formation of secondary excitations. Importantly, the PIA region at later times is dominated by a new long-lived spectral band at ∼1000 nm, overlapping with the excitonic signals at ∼970 nm, which we attribute to polarons formed after exciton dissociation at the donor–acceptor junction.62 On the other hand, the spectral region near ∼1200 nm where the triplet absorption in IT-4F should occur, undergoes continues decay meaning that no triplets are generated in the bilayer from charge recombination (the triplet spectrum of IT-4F is overlaid, and detailed triplet spectra are presented in Fig. S5, ESI†). Here the WSe2 GSB formation due to hole transfer is not clearly resolved as it is buried inside the red-shifted IT-4F GSB band.
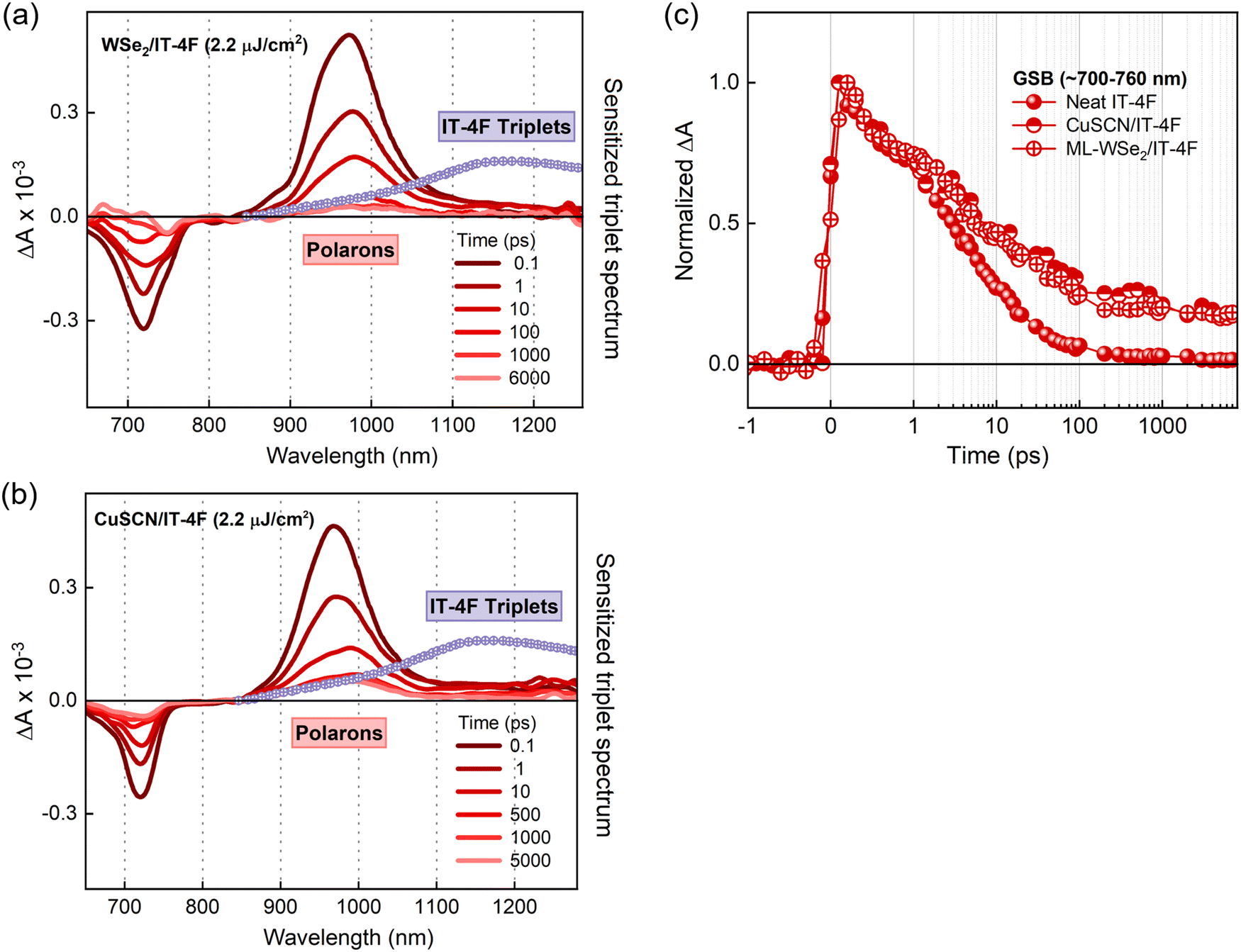 | ||
| Fig. 4 Fluorination suppresses triplet generation. Series of transient absorption spectra of (a) ML-WSe2/IT-4F and (b) CuSCN/IT-4F bilayers (both excited at 700 nm and at a pump fluence of 2.2 μJ cm−2) with the sensitized triplet spectrum of IT-4F overlaid, and (c) the corresponding transient kinetics probed at the ground state bleach (GSB) signals. | ||
We observed a similar scenario when IT-4F is interfaced with the donor CuSCN. The corresponding TAS spectra shown in Fig. 4b feature a strong polaron-based PIA at ∼1000 nm without any triplets generated. The above results suggest that the fluorination in ITIC strongly suppressed the triplet generation channel in the bilayer. Furthermore, the charge recombination in the IT-4F bilayers occurs at a slower rate than that of the ITIC bilayer which is ascribed to the better ordered intermolecular arrangements in IT-4F in comparison with the non-fluorinated counterpart.63
We also carried out charge generation studies in bilayers comprising CuSCN as the donor and a chlorinated derivative of ITIC, IT-4Cl (ITIC with four chlorine atoms), as the acceptor.64 IT-4Cl possesses similar energies of the highest occupied molecular orbital (HOMO) and lowest occupied molecular orbital (LUMO) as IT-4F (Fig. S6a, ESI†). In fact, the PL of IT-4Cl is efficiently quenched in the bilayer. TAS measurements reveal efficient polaron formation in the CuSCN/IT-4Cl bilayer, similar to the CuSCN/IT-4F bilayer (a summary of TAS measurement results in the CuSCN/IT-4F bilayer is presented in Fig. S6b–d, ESI†). On the other hand, the TAS spectra miss the characteristic feature of triplet excitons (see Fig. S5 for the triplet spectrum of IT-4Cl, ESI†).
Discussion
We probed efficient photoinduced charge transfer across the ML-WSe2/NFA interface using ultrafast transient absorption spectroscopy. A significant population of triplet excitons are observed in the ML-WSe2/ITIC bilayer, which are formed through the bimolecular recombination of spin uncorrelated free charge carriers. This pathway was identified through its characteristic fluence dependence. Our findings are distinctly different from the results of earlier studies in a hybrid sample formed by a MoS2 single crystal and a thin ZnPc layer.13 There, triplet formation in the organic part is proposed to be due to the spin flip of an interfacial CT state, mediated by the strong spin–orbit coupling in the TMDC crystals. In support to our interpretation, similar results were obtained when the WSe2 donor was replaced by CuSCN, which has very small spin–orbit coupling. This unwanted non-radiative loss channel is suppressed when halogenated derivatives of ITIC are used as acceptors. Here, we address the possible mechanism underlying the halogenation effect on the triplet loss channel in the bilayers.We, therefore, propose that the different properties seen for the non-halogenated and halogenated NFA originate in the different energetic landscapes. To this end, ab initio calculations were carried out on the electronic structure of the heterojunctions formed by the non-fullerene acceptors ITIC and IT-4F adsorbed on ML-WSe2. To this end, we built a 5 × 10 supercell of the TMDC accommodating one molecule each. With the application of periodic boundary conditions, we effectively obtain a molecular monolayer on top of WSe2. Notice that after full optimization of the hybrid system, the lateral carbon rings in the molecule lie on the same side of the backbone assuming therefore a different geometry in comparison with the gas-phase configuration (Fig. S7, ESI†).
We investigate the electronic structure of the WSe2/ITIC and WSe2/IT-4F interfaces with the molecules adsorbed flat on top of the monolayer (Fig. 1c). We checked that this is the energetically most stable structure (see the ESI,† Fig. S9) with the adsorbed molecules forming a monolayer on top of WSe2 (see Fig. S10 and Table S2, ESI†). First, we inspect the energy level alignment of the hybrid systems in comparison with the isolated constituents (see Fig. 5a), and find that fluorination leads to an almost rigid downshift of the HOMO and the LUMO of IT-4F compared to ITIC such that the gap of the former molecule is reduced by 180 meV with respect to the latter, in agreement with previous calculations.65–67 The weak influence of this functionalization on the charge distribution of the conjugated backbone can be further appreciated in the electrostatic potential maps calculated for ITIC and IT-4F (Fig. S8, ESI†).
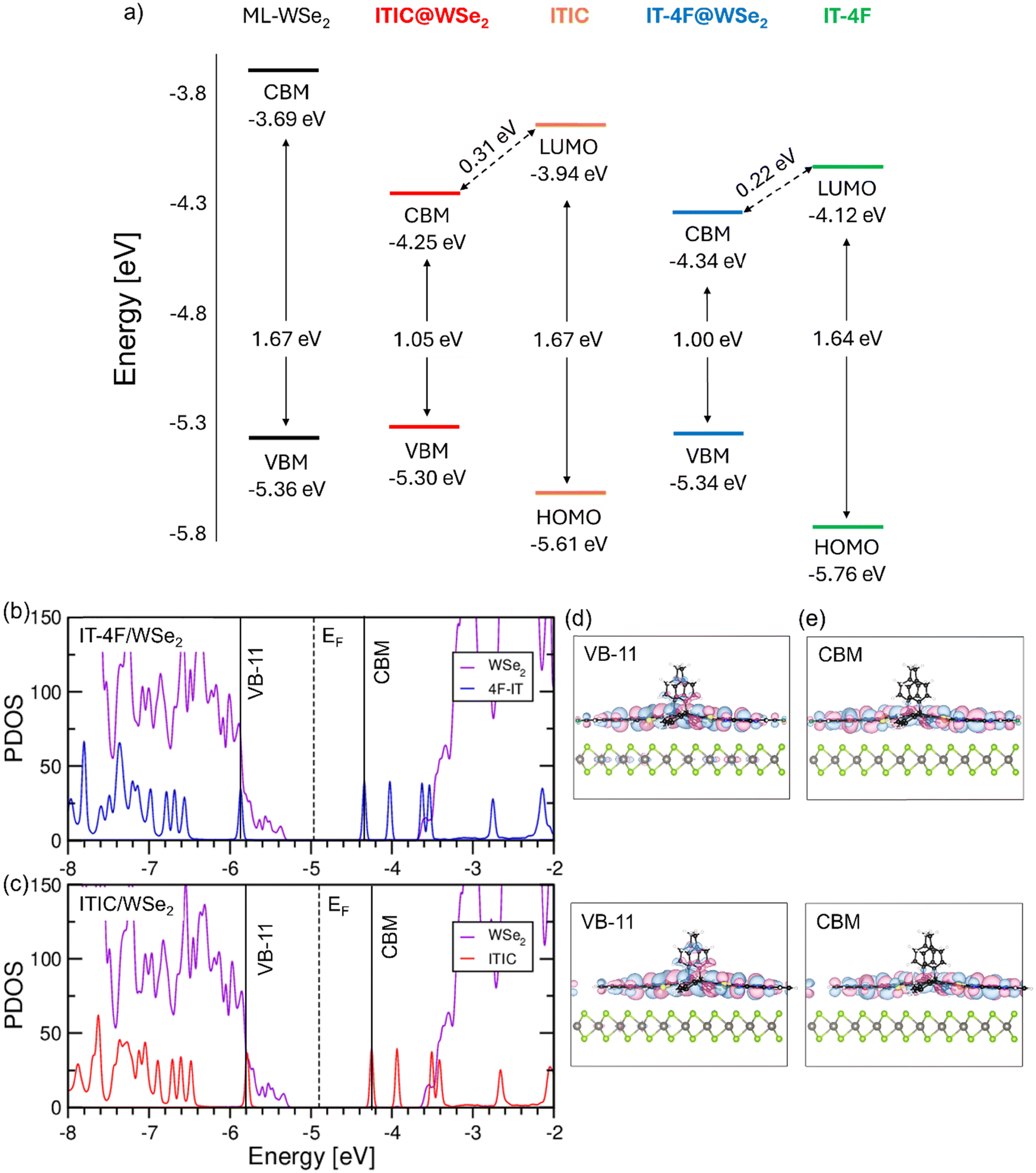 | ||
| Fig. 5 (a) Energy level alignment of the frontier states of WSe2, ITIC, IT-4F and their heterostructures, ITIC/WSe2 and IT-4F/WSe2, calculated from DFT (HSE06+SOC); projected density of states (PDOS) with contributions of each subsystem of (b) IT-4F/WSe2 and (c) ITIC/WSe2 visualized with a Gaussian broadening of 20 meV; real-space representation of (d) VB-11 and (e) CBM marked in the PDOS (iso-surfaces equal to 10% of the maximum value) calculated at the Γ-point. | ||
Considering now the energy level alignment of WSe2 with respect to ITIC and IT-4F, we find a type-II line up in both cases, with the HOMO (LUMO) of the molecules being energetically lower (higher) than the valence band maximum (conduction band minimum) of the monolayer, see Fig. 5(a). This picture is reflected in the projected density of states representing the electronic structure of the heterostructures. As shown in Fig. 5(b) and (c), the valence band maximum (VBM) corresponds to the occupied band edge of the monolayer while the lowest unoccupied state coincides with the LUMO of the molecules. The HOMO of the molecules, showing very similar contributions in the visualized energy range, is found ∼0.5 eV below the VBM and is the 12th occupied state (VB-11) of the hybrid system, see Fig. 5(b) and (c). While being energetically comprised in the valence band manifold of WSe2, it appears as a localized state with almost no signatures of hybridization with the wave function of the underlying monolayer, see Fig. 5(d). Clear signatures of hybridization, on the other hand, appear at lower energies in the valence region, as testified by a broadening of the molecular levels. In the unoccupied region of the PDOS, the LUMO of the molecules pinning the CBM gives rise to distinct peaks in Fig. 5(b) and (c) and their character remains unaltered compared to the gas phase, see Fig. 5(e) and Fig. S13 (ESI†). It is worth noting that the electron withdrawing character of both ITIC and IT-4F is so pronounced, that both the LUMO and the LUMO+1 are below the conduction band maximum of WSe2 and the higher unoccupied orbitals almost coincide in energy with the conduction band onset of the monolayer, see Fig. 5(b) and (c).
The electronic structure of the hybrid systems confirms the strong electronic coupling between the monolayer and the molecules. Due to the intrinsic properties of the molecular acceptors, 0.48 electrons are predicted to be transferred in the ground state from the monolayer to both ITIC and IT-4F in the respective interfaces. This is an indication of the effectiveness of these molecules acting as electron acceptors: for comparison, recent DFT calculations of F4TCNQ adsorbed on a MoS2 monolayer revealed negligible charge transfer in the ground state of the hybrid system.68,69 It is worth noting that in contrast to all-organic interfaces exhibiting similar values of charge transfer as those reported here,70,71 the characteristics of the frontier orbitals of the NFA are not affected upon adsorption and their share remains substantially unaltered compared to the gas phase, see Fig. S14 (ESI†). Here, charge transfer manifests itself through a downshift of the LUMO energies in the heterostructures: 0.31 eV in WSe2/ITIC and 0.22 eV in WSe2/IT-4F (see Table S3, ESI†).
On top of this effect, the dielectric polarization of the substrate contributes to the reduction of the HOMO–LUMO gap of the adsorbed molecule. Considering the different configuration assumed by the moieties on top of WSe2 in comparison with their shape in the gas-phase, we additionally checked the role of corresponding structural changes to the band-gap renormalization. To do so, we computed the electronic structure of the gas-phase molecules in the geometry they assume when adsorbed on WSe2 and found an increase (lowering) of the HOMO (LUMO) level on the order of 5 meV, leading to a total effect on the fundamental gap of less than 10 meV (see Fig. S13, ESI†). From this analysis, we can conclude that charge-transfer in the ground state and dielectric screening from the substrate are mainly responsible for the electronic structure of the two hybrid interfaces. With this knowledge, we can go back to Fig. 5a and consider the band edge of the heterostructures with respect to the frontier states of the isolated components. Given the type-II character of the bilayers, it is remarkable that the VBM of WSe2 experiences an upshift of 0.06 eV upon adsorption of ITIC, while it is only of 0.02 eV with IT-4F. The lowest unoccupied state undergoes an energy reduction of 0.32 eV (0.2 eV) with respect to the LUMO energy of ITIC (IT-4F) modelled in the gas phase. In combination with two effects, the fundamental bandgap is significantly reduced in the hybrid structure compared to energy levels of the neat components. Here, WSe2/IT-4F has an overall lower fundamental gap (1.00 eV vs. 1.05 eV in WSe2/ITIC) but also a lower LUMO energy compared to neat IT-4F (0.24 eV vs. 0.32 eV). These results suggest not only an energy reduction of the hybrid CT state but also a smaller binding energy in WSe2/IT-4F, enabling a more rapid charge separation competing with triplet formation. Unfortunately, the energy of the triplet state in ITIC or IT-4F has not been measured yet. Based on our calculations for the NFA in the gas phase performed in the framework of time-dependent density functional theory (see Table S4, ESI†), fluorination reduces the singlet–triplet energy difference by 0.02 eV in agreement with previous theoretical results.57 Interestingly, Han et al., reported that fluorination affects the energy of the local singlet exciton energy to a much larger extent that that of the triplet state energy, and that the singlet–triplet gap gets significantly reduced in aggregated IT-4F.59 Given the strongly reduced triplet formation yield in WSe2/IT-4F as proven by our TAS experiments, we, therefore, speculate that the NFA triplet exciton lies only a little below (or even above) the hybrid CT in this system. This would allow CT re-dissociation and decay to efficiently compete with NFA triplet exciton formation via BET (back electron transfer).
Finally, we note that IT-4F (and probably also IT-4Cl) has a larger quadrupole moment than ITIC.72 The role of molecular quadrupole moments has been recently discussed with regard to energy offsets and free charge formation in organic photovoltaic blends.72,73 It has been shown that the presence of such quadrupole moments alters the energies of interfacial states and this effect may also contribute to the observed differences in our hybrid bilayer systems. This will be the topic of future work.
Conclusion
Herein we investigated the mechanism of charge transfer at 2D WSe2/NFA bilayer interfaces using ultrafast transient absorption spectroscopy. For the ML-WSe2/ITIC bilayer, transient absorption experiments reveal fast generation of triplet excitons on the NFA. A similar scenario was observed when the NFA is combined with an inorganic HTL layer CuSCN, ruling out spin-flipping induced by the heavy atoms in ML-WSe2 as the main reason for the triplet formation. Instead, our data suggest that bimolecular recombination of the spin-uncorrelated photogenerated charge carriers with possible contributions from geminate charge recombination populate the low-lying triplet level of ITIC. This is suppressed when replacing ITIC by its fluorinated version, IT-4F, where instead we observe the formation of long-lived charges. DFT calculations showed that fluorination lowers the fundamental gap at the interface but also lowers the energy offset between the LUMO of the hybrid state and that of the neat NFA. In combination with two effects, fluorination renders triplet formation less likely but also causes a smaller barrier for free charge formation. Overall, by studying the excitation dynamics in 2D TMDC/NFA systems, we highlighted the delicate balance between excitons and charges in such inorganic/organic heterostructures.Data availability
Datasets for ultrafast transient absorption and steady state spectroscopy. Chandrabose, S. (2024). Mitigation of triplet loss in 2D WSe2/non-fullerene heterostructures using halogenated acceptors [Data set]. Zenodo, https://doi.org/10.5281/zenodo.12646283. MCR-ALS GUI program package for ultrafast transient absorption data analysis, https://mcrals.wordpress.com/download/mcr-als-2-0-toolbox/ developed by Roma Tauler. Zenodo: https://doi.org/10.5281/zenodo.6334791. Datasets for ab initio calculations. Valencia, Ana M. (2024). Electronic properties of ITIC-WSe2 and F4\_IT-WSe2 [Data set], Zenodo, https://doi.org/10.5281/zenodo.11171242.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was funded by the German Science Foundation through the Collaborative Research Center HIOS (Project number 182087777 – SFB 951). A. M. V. and C. C. acknowledge additional support from the German Federal Ministry of Education and Research (Professorinnenprogramm III), and from the State of Lower Saxony (Professorinnen für Niedersachsen). The computational resources were provided by the North-German Supercomputing Alliance (HLRN) through the project nic00076. This publication is based upon work supported by King Abdullah University of Science and Technology (KAUST).References
- M. M. Furchi, A. Pospischil, F. Libisch, J. Burgdörfer and T. Mueller, Nano Lett., 2014, 14, 4785 CrossRef CAS PubMed.
- T. A. Shastry, I. Balla, H. Bergeron, S. H. Amsterdam, T. J. Marks and M. C. Hersam, ACS Nano, 2016, 10, 10573 CrossRef CAS PubMed.
- J. F. Gonzalez Marin, D. Unuchek, K. Watanabe, T. Taniguchi and A. Kis, npj 2D Mater. Appl., 2019, 3, 14 CrossRef.
- X. Chen, C. Liu and S. Mao, Nano-Micro Lett., 2020, 12, 95 CrossRef CAS PubMed.
- C. Wang, F. Yang and Y. Gao, Nanoscale Adv., 2020, 2, 4323 RSC.
- S. Park, N. Mutz, T. Schultz, S. Blumstengel, A. Han, A. Aljarb, L. J. Li, E. J. W. List-Kratochvil, P. Amsalem and N. Koch, 2D Mater., 2018, 5, 5865 CrossRef.
- F. Liu, W. L. Chow, X. He, P. Hu, S. Zheng, X. Wang, J. Zhou, Q. Fu, W. Fu, P. Yu, Q. Zeng, H. J. Fan, B. K. Tay, C. Kloc and Z. Liu, Adv. Funct. Mater., 2015, 25, 5865 CrossRef CAS.
- C. Zhong, V. K. Sangwan, C. Wang, H. Bergeron, M. C. Hersam and E. A. Weiss, J. Phys. Chem. Lett., 2018, 9, 2484 CrossRef CAS PubMed.
- C. E. Petoukhoff, S. Kosar, M. Goto, I. Bozkurt, M. Manish and K. M. Dani, Mol. Syst. Des. Eng., 2019, 4, 929 RSC.
- P. Pentacene, M. van der Waals, S. B. Homan, V. K. Sangwan, I. Balla, H. Bergeron, E. A. Weiss, M. C. Hersam, G. M. Carroll, R. Limpens, N. R. Neale, J. S. Bhosale, J. E. Moore, X. Wang, P. Bermel, M. S. Lundstrom and Lumenistic LLC, Nano Lett., 2018, 17, 164 Search PubMed.
- T. Zhu, L. Yuan, Y. Zhao, M. Zhou, Y. Wan, J. Mei and L. Huang, Sci. Adv., 2018, 4, 1 Search PubMed.
- S. Padgaonkar, S. H. Amsterdam, H. Bergeron, K. Su, T. J. Marks, M. C. Hersam and E. A. Weiss, J. Phys. Chem. C, 2019, 123, 13337 CrossRef CAS.
- T. R. Kafle, B. Kattel, S. D. Lane, T. Wang, H. Zhao and W. L. Chan, ACS Nano, 2017, 11, 10184 CrossRef CAS PubMed.
- X. Yu, A. Rahmanudin, X. A. Jeanbourquin, D. Tsokkou, N. Guijarro, N. Banerji and K. Sivula, ACS Energy Lett., 2017, 2, 524 CrossRef CAS.
- K. Rijal, F. Rudayni, T. R. Kafle and W. L. Chan, J. Phys. Chem. Lett., 2020, 11, 7495 CrossRef CAS PubMed.
- S. Park, N. Mutz, S. A. Kovalenko, T. Schultz, D. Shin, A. Aljarb, L. J. Li, V. Tung, P. Amsalem, E. J. W. List-Kratochvil, J. Stähler, X. Xu, S. Blumstengel and N. Koch, Adv. Sci., 2021, 8, 2100215 CrossRef CAS PubMed.
- X. Liu, J. Gu, K. Ding, D. Fan, X. Hu, Y. W. Tseng, Y. H. Lee, V. Menon and S. R. Forrest, Nano Lett., 2017, 17, 3176 CrossRef CAS PubMed.
- K. Rijal, F. Rudayni, T. R. Kafle and W. L. Chan, J. Phys. Chem. Lett., 2020, 11, 7495 CrossRef CAS PubMed.
- D. B. Sulas-Kern, H. Zhang, Z. Li and J. L. Blackburn, Mater. Horiz., 2019, 6, 2103 RSC.
- A. Armin, W. Li, O. J. Sandberg, Z. Xiao, L. Ding, J. Nelson, D. Neher, K. Vandewal, S. Shoaee, T. Wang, H. Ade, T. Heumüller, C. Brabec and P. Meredith, Adv. Energy Mater., 2021, 11, 2003570 CrossRef CAS.
- Y. Firdaus, V. M. Le Corre, S. Karuthedath, W. Liu, A. Markina, W. Huang, S. Chattopadhyay, M. M. Nahid, M. I. Nugraha, Y. Lin, A. Seitkhan, A. Basu, W. Zhang, I. McCulloch, H. Ade, J. Labram, F. Laquai, D. Andrienko, L. J. A. Koster and T. D. Anthopoulos, Nat. Commun., 2020, 11, 5220 CrossRef CAS PubMed.
- D. B. Riley, O. J. Sandberg, N. Zarrabi, Y. R. Kim, P. Meredith and A. Armin, Adv. Mater., 2023, 35, 2211174 CrossRef CAS PubMed.
- K. Jiang, J. Zhang, Z. Peng, F. Lin, S. Wu, Z. Li, Y. Chen, H. Yan, H. Ade, Z. Zhu and A. K. Y. Jen, Nat. Commun., 2021, 12, 468 CrossRef CAS PubMed.
- S. Liu, Y. Zhou, Z. Liang, B. Zhao, W. Wang, Z. Xue, K. Ding, Z. Cong, H. Wu, G. Lu and C. Gao, ACS Appl. Energy Mater., 2023, 6, 5047 CrossRef CAS.
- J. Bertrandie, J. Han, C. S. P. De Castro, E. Yengel, J. Gorenflot, T. Anthopoulos, F. Laquai, A. Sharma and D. Baran, Adv. Mater., 2022, 34, 2202575 CrossRef CAS PubMed.
- N. Chaudhary, R. Chaudhary, J. P. Kesari, A. Patra and S. Chand, J. Mater. Chem. C, 2015, 3, 11886 RSC.
- J. Kang, S. Tongay, J. Zhou, J. Li and J. Wu, Appl. Phys. Lett., 2013, 102, 012111 CrossRef.
- M. Zhang, X. Guo, S. Zhang and J. Hou, Adv. Mater., 2014, 26, 1118 CrossRef CAS PubMed.
- J. Jaumot, R. Gargallo, A. De Juan and R. Tauler, Chemom. Intell. Lab. Syst., 2005, 76, 101 CrossRef CAS.
- I. E. Gaa, P. Hohenbergt Ecole, X. Superzeure, F. I’aris and W. Konnt, Phys. Rev., 1965, 136, B864 Search PubMed.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Comput. Phys. Commun., 2009, 180, 2175 CrossRef CAS.
- S. V. Levchenko, X. Ren, J. Wieferink, R. Johanni, P. Rinke, V. Blum and M. Scheffler, Comput. Phys. Commun., 2015, 192, 60 CrossRef CAS.
- V. Havu, V. Blum, P. Havu and M. Scheffler, J. Comput. Phys., 2009, 228, 8367 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207 CrossRef CAS.
- J. Hermann and A. Tkatchenko, Phys. Rev. Lett., 2020, 124, 146401 CrossRef CAS PubMed.
- A. Tkatchenko, R. A. Distasio, R. Car and M. Scheffler, Phys. Rev. Lett., 2012, 108, 1 CrossRef PubMed.
- W. Zhao, Z. Ghorannevis, L. Chu, M. Toh, C. Kloc, P. H. Tan and G. Eda, ACS Nano, 2013, 7, 791 CrossRef CAS PubMed.
- H. L. Liu, C. C. Shen, S. H. Su, C. L. Hsu, M. Y. Li and L. J. Li, Appl. Phys. Lett., 2014, 105, 201905 CrossRef.
- A. Carvalho, R. M. Ribeiro and A. H. Castro Neto, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 115205 CrossRef.
- N. Chaudhary, R. Chaudhary, J. P. Kesari, A. Patra and S. Chand, J. Mater. Chem. C, 2015, 3, 11886 RSC.
- Y. Lin, J. Wang, Z. Zhang, H. Bai, Y. Li and D. Zhu, Adv. Mater., 2015, 1170 CrossRef CAS PubMed.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148 CrossRef CAS PubMed.
- S. Y. Park, S. Chandrabose, M. B. Price, H. S. Ryu, T. H. Lee, Y. S. Shin, Z. Wu, W. Lee, K. Chen, S. Dai, J. Zhu, P. Xue, X. Zhan, H. Y. Woo, J. Y. Kim and J. M. Hodgkiss, Nano Energy, 2021, 84, 105924 CrossRef CAS.
- S. Chandrabose, K. Chen, A. J. Barker, J. J. Sutton, S. K. K. Prasad, J. Zhu, J. Zhou, K. C. Gordon, Z. Xie, X. Zhan and J. M. Hodgkiss, J. Am. Chem. Soc., 2019, 141, 6922–6929 CrossRef CAS PubMed.
- N. Zorn Morales, N. Severin, J. P. Rabe, S. Kirstein, E. List-Kratochvil and S. Blumstengel, Adv. Opt. Mater., 2023, 11, 2301057 CrossRef CAS.
- M. Pranav, A. Shukla, D. Moser, J. Rumeney, W. Liu, R. Wang, B. Sun, S. Smeets, N. Tokmoldin, F. Jaiser, T. Hultzsch, S. Shoaee, W. Maes, L. Lüer, C. Brabec, K. Vandewal, D. Andrienko, S. Ludwigs and D. Neher, Energy Environ. Sci., 2024, 17, 6676–6697 RSC.
- S. Karuthedath, J. Gorenflot, Y. Firdaus, N. Chaturvedi, C. S. P. De Castro, G. T. Harrison, J. I. Khan, A. Markina, A. H. Balawi, T. A. Dela Peña, W. Liu, R. Z. Liang, A. Sharma, S. H. K. Paleti, W. Zhang, Y. Lin, E. Alarousu, D. H. Anjum, P. M. Beaujuge, S. De Wolf, I. McCulloch, T. D. Anthopoulos, D. Baran, D. Andrienko and F. Laquai, Nat. Mater., 2021, 20, 378 CrossRef CAS PubMed.
- T. Umeyama, K. Igarashi, D. Sasada, Y. Tamai, K. Ishida, T. Koganezawa, S. Ohtani, K. Tanaka, H. Ohkita and H. Imahori, Chem. Sci., 2020, 11, 3250 RSC.
- C. Boehme and J. M. Lupton, Nat. Nanotechnol., 2013, 8, 612 CrossRef CAS PubMed.
- R. Wang, J. Xu, L. Fu, C. Zhang, Q. Li, J. Yao, X. Li, C. Sun, Z. G. Zhang, X. Wang, Y. Li, J. Ma and M. Xiao, J. Am. Chem. Soc., 2021, 143, 4359 CrossRef CAS PubMed.
- L. Xue, Y. Yang, J. Xu, C. Zhang, H. Bin, Z. G. Zhang, B. Qiu, X. Li, C. Sun, L. Gao, J. Yao, X. Chen, Y. Yang, M. Xiao and Y. Li, Adv. Mater., 2017, 29, 1703344 CrossRef PubMed.
- A. Rao, P. C. Y. Chow, S. Gélinas, C. W. Schlenker, C. Z. Li, H. L. Yip, A. K. Y. Jen, D. S. Ginger and R. H. Friend, Nature, 2013, 500, 435 CrossRef CAS PubMed.
- T. R. Kafle, B. Kattel, S. D. Lane, T. Wang, H. Zhao and W. L. Chan, ACS Nano, 2017, 11, 10184 CrossRef CAS PubMed.
- S. D. Dimitrov, S. Wheeler, D. Niedzialek, B. C. Schroeder, H. Utzat, J. M. Frost, J. Yao, A. Gillett, P. S. Tuladhar, I. McCulloch, J. Nelson and J. R. Durrant, Nat. Commun., 2015, 6, 6501 CrossRef CAS PubMed.
- J. Guo, B. Moss and T. M. Clarke, J. Mater. Chem. A, 2022, 10, 20874–20885 RSC.
- J. P. A. Souza, L. Benatto, G. Candiotto, L. S. Roman and M. Koehler, J. Phys. Chem. A, 2022, 126, 1393 CrossRef CAS PubMed.
- R. Wang, J. Xu, L. Fu, C. Zhang, Q. Li, J. Yao, X. Li, C. Sun, Z. G. Zhang, X. Wang, Y. Li, J. Ma and M. Xiao, J. Am. Chem. Soc., 2021, 143, 4359 CrossRef CAS PubMed.
- K. Reichenbächer, H. I. Süss and J. Hulliger, Chem. Soc. Rev., 2005, 34, 22 RSC.
- Y. Dong, H. Cha, H. L. Bristow, J. Lee, A. Kumar, P. S. Tuladhar, I. McCulloch, A. A. Bakulin and J. R. Durrant, J. Am. Chem. Soc., 2021, 143, 7599 CrossRef CAS PubMed.
- K. Reichenbächer, H. I. Süss and J. Hulliger, Chem. Soc. Rev., 2005, 34, 22 RSC.
- H. Zhang, H. Yao, J. Hou, J. Zhu, J. Zhang, W. Li, R. Yu, B. Gao, S. Zhang and J. Hou, Adv. Mater., 2018, 30, 1 CAS.
- C. Cocchi, D. Prezzi, A. Ruini, E. Benassi, M. J. Caldas, S. Corni and E. Molinari, J. Phys. Chem. Lett., 2012, 3, 924 CrossRef CAS PubMed.
- J. P. A. Souza, L. Benatto, G. Candiotto, L. S. Roman and M. Koehler, J. Phys. Chem. A, 2022, 126, 1393 CrossRef CAS PubMed.
- L. Benatto and M. Koehler, J. Phys. Chem. C, 2019, 123, 6395 CrossRef CAS.
- S. Park, H. Wang, T. Schultz, D. Shin, R. Ovsyannikov, M. Zacharias, D. Maksimov, M. Meissner, Y. Hasegawa, T. Yamaguchi, S. Kera, A. Aljarb, M. Hakami, L. J. Li, V. Tung, P. Amsalem, M. Rossi and N. Koch, Adv. Mater., 2021, 33, 2008677 CrossRef CAS PubMed.
- G. Melani, J. P. Guerrero-Felipe, A. M. Valencia, J. Krumland, C. Cocchi and M. Iannuzzi, Phys. Chem. Chem. Phys., 2022, 24, 16671 RSC.
- H. Méndez, G. Heimel, S. Winkler, J. Frisch, A. Opitz, K. Sauer, B. Wegner, M. Oehzelt, C. Röthel, S. Duhm, D. Többens, N. Koch and I. Salzmann, Nat. Commun., 2015, 6, 8560 CrossRef PubMed.
- A. M. Valencia and C. Cocchi, J. Phys. Chem. C, 2019, 123, 9617 CrossRef CAS.
- S. Karuthedath, J. Gorenflot, Y. Firdaus, N. Chaturvedi, C. S. P. De Castro, G. T. Harrison, J. I. Khan, A. Markina, A. H. Balawi, T. A. Dela Peña, W. Liu, R. Z. Liang, A. Sharma, S. H. K. Paleti, W. Zhang, Y. Lin, E. Alarousu, D. H. Anjum, P. M. Beaujuge, S. De Wolf, I. McCulloch, T. D. Anthopoulos, D. Baran, D. Andrienko and F. Laquai, Nat. Mater., 2021, 20, 378 CrossRef CAS PubMed.
- R. C. I. MacKenzie, Handbook of Optoelectronic Device Modeling and Simulation: Fundamentals, Materials, Nanostructures, LEDs, and Amplifiers, 2017, vol. 1, p. 191 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mh00894d |
| This journal is © The Royal Society of Chemistry 2025 |