
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid 3D printing of unlayered, tough epoxy–alcohol resins with late gel points via dual-color curing technology†
Florian
Mayer
 a,
Dominik
Laa
b,
Thomas
Koch
b,
Jürgen
Stampfl
b,
Robert
Liska
a and
Katharina
Ehrmann
*a
a,
Dominik
Laa
b,
Thomas
Koch
b,
Jürgen
Stampfl
b,
Robert
Liska
a and
Katharina
Ehrmann
*a
aInstitute of Applied Synthetic Chemistry, Technische Universität Wien, Vienna, Austria. E-mail: katharina.ehrmann@tuwien.ac.at
bInstitute of Materials Science and Technology, Technische Universität Wien, Vienna, Austria
First published on 26th November 2024
Abstract
Additive manufacturing technologies and, in particular, vat photopolymerization promise complex structures that can be made in a fast and easy fashion for highly individualized products. While the technology has upheld this promise many times already, some polymers are still out of reach or at least problematic to print reliably. High-performance epoxide-based resins, which are regulated by chain transfer via multifunctional alcohols, are a typical example of resins with late gel points, which require long irradiation times and high light intensities to print. Therefore, we have developed a dual-colour printing approach where rapid radical curing of a soft, wide-meshed polymer network facilitates fast and easy 3D structuring of the subsequently slow curing step-growth formulation at an orthogonal initiation-wavelength regime. Thereby the methacrylate system acts as a scaffold for an uncured epoxide alcohol system during the printing process, which is then cured with UV light post-printing. This way tough alcohol-regulated epoxy-systems become accessible to vat photopolymerization achieving outstanding high-resolution 3D printed parts without significant layering effects. The demonstrated wide-meshed matrix-assisted printing approach has the potential to make a multitude of slowly curing resins accessible to vat photopolymerization techniques, at low irradiation intensities and high curing speeds.
New conceptsWe pioneer multi-wavelength scaffold-aided 3D printing as a means of printing previously inaccessible material classes with late gel points. We use a fast curing, loose methacrylate network for 3D printing at longer wavelengths, acting as a scaffold for semi-orthogonal cationically initiated monomers, which remain unaffected by the printing process. Subsequent post-curing at shorter UV-wavelengths triggers the photoacid generator and induces cationic polymerization of a polymer network with late gel point. The geometric stability during the printing process has been optimized to print complex shapes at excellent resolution despite the use of low irradiation intensities and fast printing speeds. We have demonstrated this new concept for alcohol-regulated epoxy networks, in which alcohols act as chain transfer agents, significantly delaying the gel point. This enhances the network homogeneity, resulting in tougher epoxide thermosets without lowering the onset of the glass transition temperature. The toughening effect is a direct result of the delayed gel point and can therefore only be exploited to its full extent if scaffold-aided 3D printing is applied, demonstrating the method's advantage over the standard direct printing approach and even opening the door to facile printing of currently inaccessible polymer types from slow polymerization reactions in the future. |
Introduction
The invention of additive manufacturing has provided academia and industry with a new set of tools to access complex and customized three-dimensional (3D) structures more easily and cheaper than with conventional manufacturing methods. While several materials such as metals and ceramics are printable, polymers have become a main focus.1–4 Since the first patent for vat photopolymerization in 1984 was filed to utilize light-curing for 3D patterning, a great variety of printing techniques have emerged,5 however, the utilized resins remain largely confined to (meth)acrylic resins because the reactivity of the resin is the limiting factor for fast printing speeds. Furthermore, resulting printed parts are often rather brittle because their fast curing requires rather high crosslinking density and early gelation, which causes shrinkage stresses that further deteriorate their performance. The mechanical properties of printed parts are currently not only limited by the material properties but also by the layer-by-layer printing technique, which typically causes earlier material failure compared to bulk-cured parts without this layering effect.In a first step towards better material properties for vat photopolymerized objects, epoxides or oxetanes were copolymerized with (meth)acrylates.6–9 While the reactivity of pure epoxide resins was originally too low to be printed, partial substitution of (meth)acrylates with ring-opening monomers already reduced shrinkage stress and therefore resulted in better mechanical performance.10–12 With the advent of hot lithography, a stereolithographic printing method, which utilizes a precisely heated vat and building platform of up to 140 °C, the possibility arose to broaden the material chemistry horizon. Higher reactivities and lower viscosities of the resin at elevated temperatures13–15 have allowed precise printing of a variety of materials with enhanced thermomechanical properties compared to traditional free radical photopolymer networks: highly viscous (meth)acrylate formulations containing monomers of higher molecular weights to lower the crosslinking density16,17 and more slowly curing chain-regulated (meth)acrylic resins18,19via radical photoinitiators such as ethyl(2,4,6-trimethylbenzoyl)-phenyl phosphinate (TPO-L), as well as pure cationic photopolymerization of epoxides,6,10,15,20–24 oxetanes,24 cyclic esters,25 oxazolines26 and cyclic carbonates25,27via photoacid generators (PAGs) such as the onium-salt UV6976 have been printed at elevated temperatures. The ring-opening mechanism for such monomers has lowered the volume decrease during the sol–gel process and hence shrinkage stresses in the respective materials, which are known to decrease mechanical performance.
Another approach to achieve enhanced material properties is the printing of interpenetrating networks (IPNs), which consist of at least two independent polymer networks that are irreversibly entangled with each other. To synthesize an IPN, two orthogonal curing mechanisms are needed that do not interfere with each other during curing and therefore do not form copolymers but independent networks. This bears the advantage that separating the networks would only be possible by breaking bonds.4,6 The predominant combination of IPN curing mechanisms is the combination of radical and cationic polymerization of e.g., epoxide and (meth-)acrylate6,20,28–31 or oxetane and acrylate9,32–34 systems, respectively. While first IPNs were initially fabricated by soaking a cured polymer with a monomer mixture and curing it in a second step, modern IPNs utilize monomers, for which curing of the two networks can be initiated simultaneously9,32,33 or even sequentially.4,6,35 Simultaneously cured IPNs have not only made epoxide and other slower curing monomers accessible for 3D printing10,27,36,37 but also decreased volumetric shrinkage compared to pure methacrylate resins.10 Additionally, less oxygen inhibition and even faster polymerisation rates were observed.4,6,23 For sequential IPN curing it is important that at least one of the monomers can be polymerized orthogonally, i.e. under conditions where polymerization of the second monomer does not occur. This can be controlled particularly well via photoinitiators in photopolymerization and has been exploited in 3D printing applications to manufacture hard IPN and soft methacrylic sections within one print by switching the illumination mode from visible to UV light during printing.28,29,38–42 The two-stage curing approach has also been utilized to enhance printing speeds by utilizing a fast photopolymerization reaction for the printing step and continue curing of the second network via thermal or light-triggered post-curing.43–45
Although such IPNs can exhibit high Tg and tensile strength, the low toughness of the epoxide system can be a limitation for the toughness of the final polymer.46 To transcend this limitation, multifunctional alcohols have been employed as chain extenders, effectively introducing chain transfer reactions to the polymerization process.15,47 This increases the network homogeneity and improves mechanical properties, especially toughness.47,48 Although such systems have already been printed stereolithographically,47 increased mechanical performance correlates with increasing printing times due to later onsets of gelation. Furthermore, the regulating effect of the alcohol cannot fully evolve in such a printing process due to fast vitrification if curing is significantly sped up via high intensity irradiation and heating.15 On the other end of the spectrum, significantly enhanced mechanical performance can be achieved through slow printing speeds.11
Herein, we enable facile and fast 3D printing of a slow curing epoxide alcohol system, with good mechanical properties by combining it with a soft polyester network in a dual cure IPN. For this purpose, we decouple the printing process from the slow chain-regulated epoxide curing process via sequential IPN formation (Fig. 1A). A fast-curing wide-meshed methacrylic scaffold network, that already contains the epoxide and alcohol monomers for the hard network, is printed with visible light at much lower irradiation intensities and exposure times compared to the epoxide alcohol network. Critically, the epoxide alcohol network is not cured during the printing process but in a post-printing step, due to the orthogonal curing mechanism and the semi-orthogonal initiation system. It is only after the post-processing step that the final IPN is yielded, giving it sufficient time to form the network in a step-growth type polymerization (Fig. 1B and C). Compared to previous systems, the wide-mesh approach requires elevated temperatures to lower the viscosity sufficiently for printing, and therefore a thermally stable initiator for the second curing stage. This enables the formation of a mechanically robust network throughout the printed layers of the first network. Therefore, the geometric object is still printed in a conventional layer-by-layer approach while the architecture of the loadbearing hard network resembles that of layerless printing techniques (volumetric 3D printing). The low crosslinking density and high mobility of uncured components for the second curing stage allow full exploitation of the chain transfer regulatory effect in such IPNs.
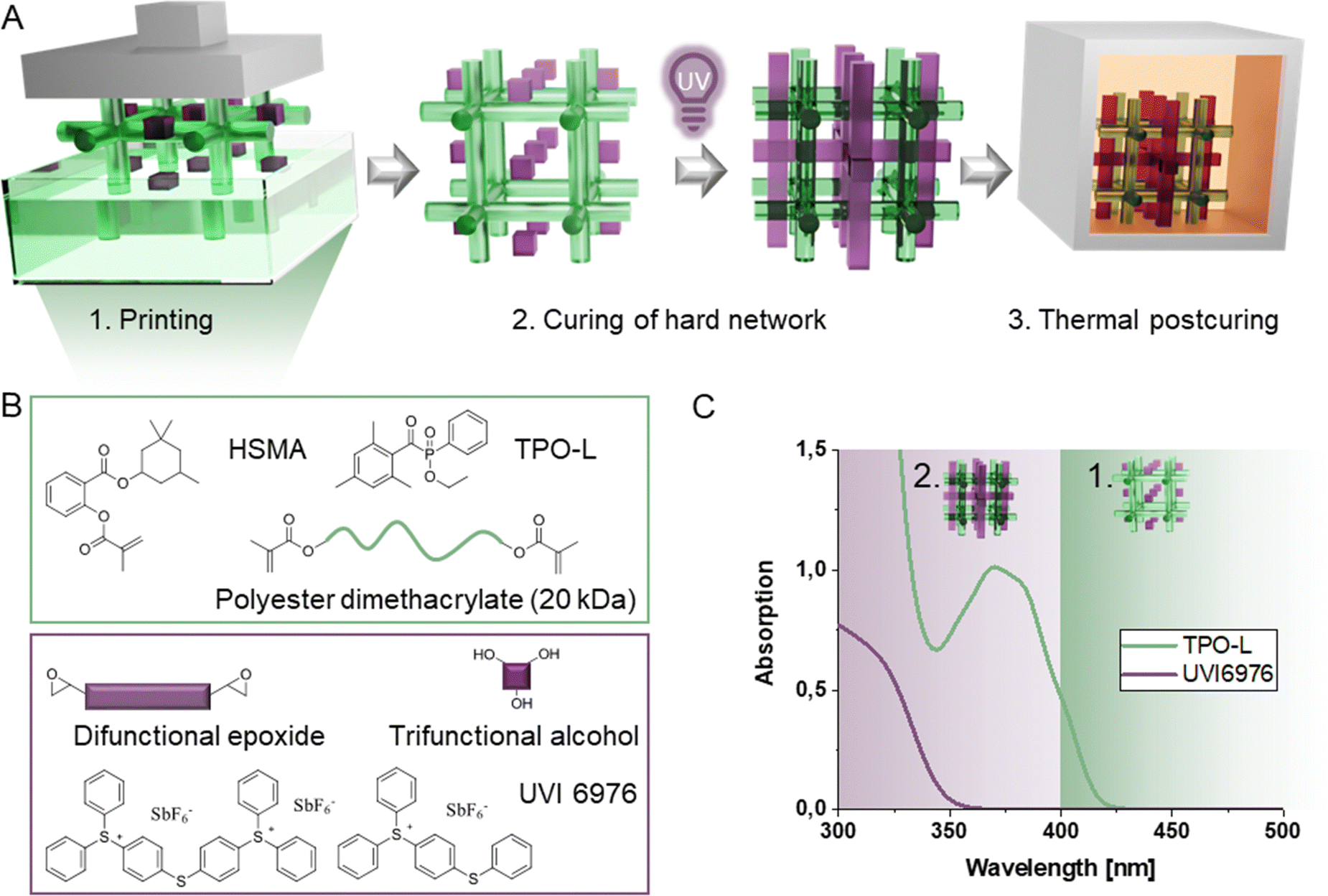 | ||
| Fig. 1 (A) Fast 3D printing via sequential interpenetrating network formation where a wide-meshed soft methacrylic system is printed in the presence of the hard network building blocks and the layerless hard network is cured post-printing. (B) Formulation components of the interpenetrating network (IPN). (C) Absorption spectra of utilized radical (TPO-L) and cationic (UVI6976) photoinitiators. | ||
Results and discussion
To obtain a high load-bearing photopolymer network, epoxides were chosen as monomers for network formation. However, classical epoxide networks are known for their pronounced brittleness due to their high crosslinking density as well as early gel point of the highly reactive formulation. We aimed to alleviate this effect through the addition of multifunctional alcohols, which effectively act as chain transfer agents, leading to slightly wider, more homogeneous networks (Fig. 2A). This effect is accompanied by a delay of the gel point, which renders additive manufacturing of such monomer formulations more difficult and slower, limiting the accessibility of high-strength epoxy networks for fast and high-resolution 3D printing. Moreover, the accessibility of step-growth type polymer classes with late gel points in general remains a challenge for light-based 3D printing to this day. Herein, we have first developed the optimal alcohol–epoxide network for high load bearing applications without imposing restrictions concerning reactivity. This hard network was then complimented by fast-curing macromolecular methacrylates with a visible-light responsive radical photoinitiator, which enable fast printing of a soft, wide-meshed network containing the unreacted epoxy monomers and alcohol chain-transfer agents (single curing, denoted as radical single curing, ‘rs’ for samples only cured with visible light hereafter). The chain-regulated epoxide network was cured into the final alcohol–epoxy material with UV light post-printing, resulting in an interpenetrating network (IPN) of equal and arguably slightly enhanced performance compared to the undiluted network (double curing, denoted as ‘d’ for samples cured with UV light hereafter).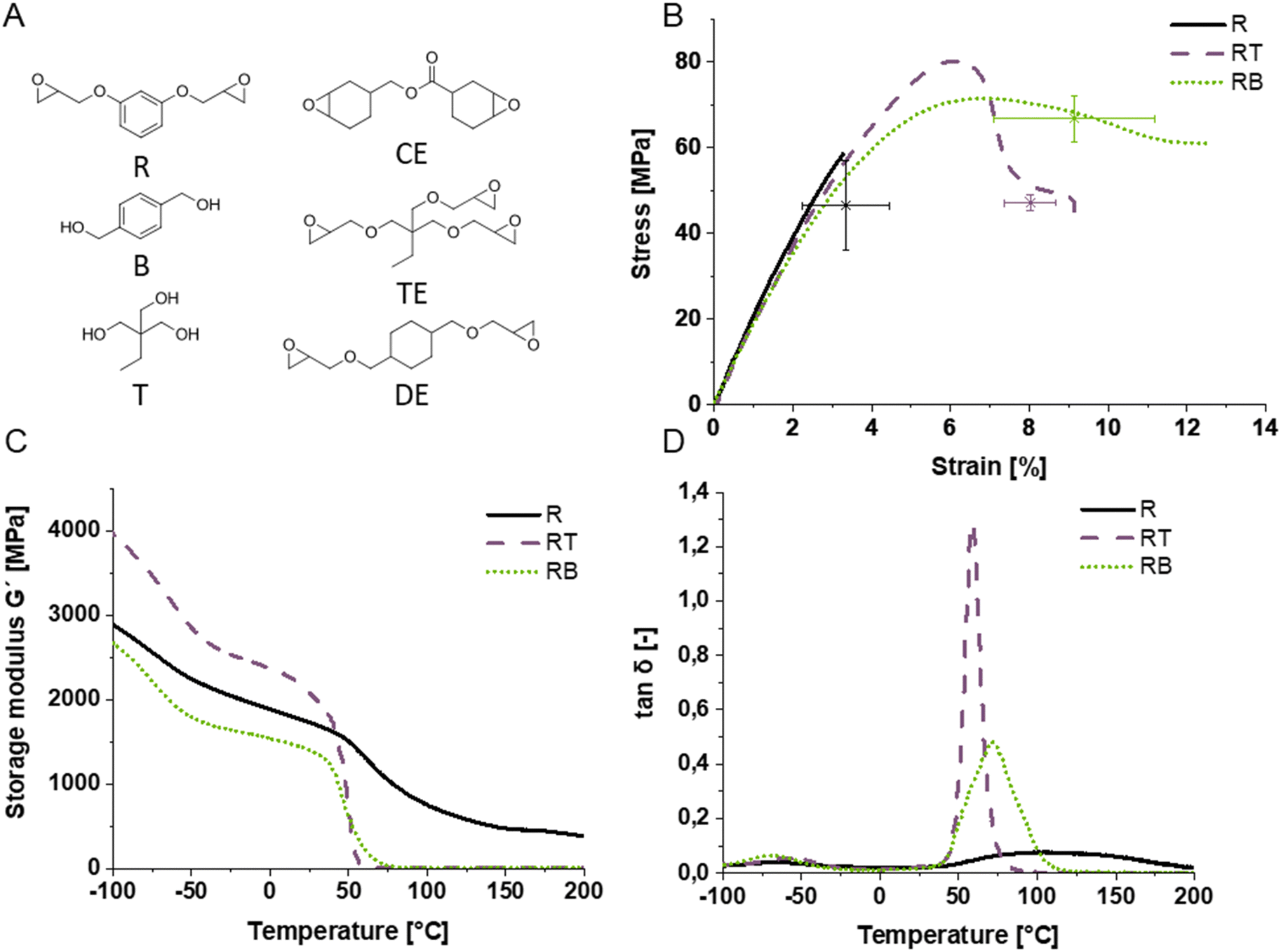 | ||
Fig. 2 (A) Monomer structures of utilized epoxides and alcohols (resorcinol diglycidylether R, 3,4-epoxycyclohexylmethyl 3,4-epoxycyclohexanecarboxylate CE, benzene dimethanol B, trimethylolpropane triglycidylether TE, trimethylolpropane T, cyclohexane dimethanol diglycidylether DE) and (thermo)mechanical testing of R alone and in combination with T (RT) and B (RB) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 functional group ratio of epoxide to alcohol moiety: (B) tensile tests, (C) storage modulus (G′), and (D) tanδ. :1 functional group ratio of epoxide to alcohol moiety: (B) tensile tests, (C) storage modulus (G′), and (D) tanδ. | ||
Development of a high-strength epoxy-based network
To investigate epoxide alcohol systems, four different epoxides were first tested for their mechanical performance in combination with trimethylolpropane (T; Fig. 2A and Fig. S3, ESI†). T was chosen because it is one of the simplest trifunctional alcohols, melts at 60 °C and is miscible with tested epoxides at this temperature. The tested epoxides were di- or trifunctional, with aliphatic, cycloaliphatic or aromatic core structures. For the aliphatic epoxides trimethylolpropane triglycidylether (TE) was chosen. Aromatic epoxides were represented by resorcinol diglycidylether (R). The group of cycloaliphatic epoxides was investigated through cyclohexanedimethanol diglycidylether (DE), which bears a cyclohexane core, and 3,4-epoxycyclohexylmethyl-3,4-epoxycyclohexanecarboxylate (CE), which has a cyclohexane ring directly connected to the epoxide. Epoxide alcohol mixtures are named with the starting letter for the epoxide, followed by the starting letter for the alcohol. To investigate the (thermo-) mechanical properties, dynamic mechanical thermal analysis (DMTA) and tensile testing were employed. Generally, the addition of the alcohols to the epoxide in an equimolar amount leads to an increase in mechanical performance. The most rigid epoxide R exhibited the best performance, especially upon alcohol addition, which gives slightly wider meshed networks (Fig. S3, ESI†), and was therefore used in all subsequent experiments.To compare the performance of di- and trifunctional alcohols, benzene dimethanol (B), was employed as a chain transfer agent in combination with R in addition to T and their effect on the thermomechanical performance was investigated (Fig. 2). B was chosen as an alternative alcohol to T because the aromatic core was expected to increase the mechanical performance and because of its miscibility with R at 70 °C.
Tensile tests revealed improved strains at break indicating an increase in homogeneity of both alcohol-regulated networks (Fig. 2B). Most importantly, all alcohol-containing specimens exhibited yielding and an increase in strain at break without significant decreases in tensile strength compared to the pure epoxide system, thus toughening the resulting photopolymer. Decreased confidence intervals of the strain at break compared to the pure epoxide additionally indicate increased homogeneity. The strain at break was chosen over the yield point to compare mean performance values in the diagram because it is the most distinct measure for the polymers. While there was no drastic dependence of material performance on the structure or functionality of alcohols, the polymer network RT containing the trifunctional alcohol as chain transfer agent exhibits the most pronounced yield point and also the highest strength at the yield point (78 MPa). The polymer with the highest elongation at break is the combination of the epoxide R with the diol B with 9.5% on average. Since the tensile tests only indicate thermomechanical performance at room temperature, determining storage (G′) and loss moduli (G′′) within a broad temperature range and the glass transition temperature (Tg) is important to understand the limitations for application temperatures above room temperature. Furthermore, the toughening effect of different alcohols as transfer agents is not very pronounced in the tensile testing results and therefore network regulation efficiency should be further investigated.
For this purpose, dynamic mechanic thermal analysis (DMTA) allows the analysis of the glass transition range, which becomes narrower for more homogeneous networks as anticipated for the alcohol-containing epoxide networks. Indeed, a steeper slope of the storage modulus curve (Fig. 2C) and a narrower loss factor curve (Fig. 2D) are obtained for the epoxide alcohol systems compared to the reference network without alcohol chain transfer reactions. Looking at the pure epoxide R, a broad flat peak is observed with its maximum at 110 °C. In comparison, the polymers RB and RT have a smaller full width at half-maximum (FWHM), i.e. are more defined. Their maxima lie at 75 °C and 55 °C, respectively. Additionally, the onset of the peak in the tanδ curve at 45 °C is the same in all polymers, indicating that the regulation did not lead to softening of the polymer due to wider mesh size, a critical aspect when photopolymers are toughened by chain transfer mechanisms, indicating applicability beyond room temperature.
The higher maximum peak height of RT in the loss factor curve is typical for non-softening polymer networks and its narrower peak shape indicates better network homogeneity compared to RB. Therefore, RT was subjected to IPN studies in the following.
Preparation and curing of epoxide–alcohol mixtures
The optimized hard network was combined with a wide-meshed methacrylate network to obtain faster printing speeds and unlayered network architecture. For this purpose, a macromolecular methacrylate-terminated polyester was synthesized from Dynapol L208, a commercial polyester with a molecular weight of 20 kDa (Scheme S1, ESI†). The modified polyester was subsequently combined with the reactive diluent homosalate methacrylate (3,3,5-trimethylcyclohexyl salicylate methacrylate, HSMA,75 wt%) to obtain a structural scaffold at high printing speed in the presence of epoxides and alcohols, which can be cured subsequently (Fig. 1A, B and Fig. 3A). The total amount of radical network in wt% in the final IPN is added to the end of the IPN name as a number. The methacrylic network contains 25 wt% of the modified polyester and 75 wt% of the reactive diluent HSMA in all cases. The epoxide alcohol network contains R and an equimolar ratio of T in terms of reactive groups as determined previously. | ||
| Fig. 3 (A) Monomers used in this study. (B) Exemplary photo-DSC curve measured with broad band UV light (320–500 nm) at 70 °C with characteristic values as determined for the epoxide alcohol systems and IPNs. (C) Time until maximum heat is measured (tmax). R: resorcinol diglycidyl ether, T: trimethylol propane. RT: polymer of R and T in a 1:1 functional group ratio, IPN-X: polymer consisting of X wt% methacrylate network (X = 75/65/50 wt% for homosalate methacrylate and 25/35/50 wt% of the modified polyester) and 100 − X wt% RT. Characteristic values in IPN-formulations correspond to methacrylate-reactivity only in appropriate dilution with the epoxide system, which was not cured during photo-DSC experiments due to the absence of the photoacid generator (PAG). | ||
To allow for sequential IPN formation, the acylphosphine oxide TPO-L was chosen as the radical initiator in the visible light range and UVI6976 was chosen as the solely UV-reactive initiator, a sulfonium antimonate acting as cationic photoinitiator through light-triggered release of a super acid (Fig. 1C). This allows printing of the radical network at 385 nm, where UVI6976 is not active and thus does not initiate the cationic epoxide alcohol reaction. To cure the second network based on the cationically responsive monomers, post-curing is conducted with lower wavelength UV light (320 nm). Critically, the form-stability of the wide-meshed network must be sufficient for high resolution printing in the presence of a significant amount of uncured epoxide monomers. Subsequently, a reactivity study was conducted to demonstrate the effect of sequential curing on printing speed and the thermomechanical properties of resulting IPNs have been determined as an indicator for the mechanical performance of final printed specimens.
Reactivity is measured as the heat development during the exothermic photopolymerization reaction. The time until maximum heat evolution (tmax) was chosen as indicator for the reaction speed (Fig. 3B). Other characteristic time points during this polymerization process like the time point when the heat evolution starts (tonset) or has reached 95% completion (t95) were also investigated. The results demonstrate that the methacrylate-containing IPN formulations (rs) expectedly react much faster than the pure epoxy–alcohol system (cs).
All rs experiments for IPN formulations performed similarly irrespective of applied methacrylate concentrations, which indicates that the dilution in the epoxide alcohol system does not affect the reaction speed of the macromonomer severely (Fig. 3C). tmax for rs curing of methacrylate in the IPN formulation is reached after only 40% of cs curing time of the epoxide alcohol system. Higher amounts of methacrylate seem to lower the double bond conversion for rs curing calculated from the theoretical heat of polymerization. This can be attributed to the higher mobility of the methacrylate groups in the IPNs with less methacrylate content and thus lower macromonomer content (Fig. S4C, ESI†).
The maximum rate of polymerisation derived from the peak height is higher for the radical network of the IPNs (rs) than it is for the epoxy alcohol system (cs). The difference between the IPNs is negligible (Fig. S4D, ESI†).
Since the 3D printer utilizes an LED for digital light projection of each layer onto the vat bottom, photo-DSC measurements of the epoxy/alcohol-diluted radical system (rs) in the presence of both initiators were also performed using an LED centered around 385 nm. The experiments indicate equal reactivity under these irradiation conditions (Fig. S5, ESI†). The same experiment for the epoxide–alcohol system alone (cs), however, did not lead to significant heat evolution, proofing its inactivity during printing.
 | ||
| Fig. 4 (A) Demonstration of form stability upon curing of only the soft network (radical network, green) in the presence of the uncured hard network (epoxy/alcohol network, blue) for formulations with varying concentrations of the soft network. (B) tanδ curve of the dynamic mechanical thermal analysis (storage and loss modulus curves can additionally be found in the ESI,† Fig. S6). (C) Representative tensile curves, and (D) tensile toughness calculated from the area under the tensile curve of R, RT, IPN 25 wt%, IPN 35 wt% and IPN 50 wt% from DMTA. R: resorcinol diglycidyl ether, T: trimethylol propane, RT: polymer of R and T in a 1:1 functional group ratio, IPN X wt%: polymer of X wt% methacrylate network (consisting of 25 wt% of homosalate methacrylate, and 75 wt% of the modified polyester) and 100 − X wt% RT. | ||
The glass transition is a particularly suitable network characteristic to determine the presence of interpenetrating networks and was therefore elicited by DMTA (Fig. 4B). As expected, the tanδ curve of the IPNs (d) are broadened and even exhibit peak shoulders, indicating the formation of two separate networks with deviating glass transition temperatures (Tgs). The bimodal peaks in IPN 35 wt% (d) and IPN 50 wt% (d) indicate the two Tgs for the two networks. The Tg of the cationic network RT (cs) is at around 55 °C, which renders the second peak to be that of the methacrylate network at around 70 °C. For IPN 35 wt% (d) the higher peak of the bimodal distribution, which is at about 55 °C, is in alignment with the RT (cs) peak, which indicates that for IPN 35 wt% (d) the polymer properties lean towards those of the epoxide alcohol network. For the IPN 50 wt% (d) the higher peak in the tanδ curve is shifted to higher values signifying that the epoxide/alcohol influence starts to dominate the mechanical properties in IPN 50 wt% (d). For IPN 25 wt% (d), there is no bimodal peak, but a slight shift towards a higher Tg indicating that the concentration of methacrylate network is not yet high enough to show a pronounced shoulder in the tanδ curve. These observations are in alignment with the tensile test curves and thereof derived tensile toughness values.
The methacrylate peak may have a higher Tg than the cationic network but the properties in the tensile tests are mainly dominated by the crosslinking density of these two networks.
The impact of alcohol-induced chain transfer reactions on the mechanical properties is clearly demonstrated by the comparison of the tensile testing results of the pure epoxide matrix with the alcohol-containing epoxide matrix (Fig. 4B and C): Upon addition of alcohols the originally brittle material R turns into a yielding material RT with quadrupled tensile toughness from approximately 1 MJ m−3 to 4 MJ m−3. Compared to both of these materials, IPN 35 wt% (d) outperforms even RT with equal tensile strength and increased elongation at break, effectively increasing tensile toughness. IPN 50 wt% (d) also demonstrates increased elongation at break but its tensile strength decreased significantly, however, arguably still exhibits high values. This behaviour is attributed to significant contribution of the flexible polyester-based methacrylic network. While it must be mentioned that the yield strength is lowered in contrast to the purely cationic epoxy–alcohol network RT, this is underproportional compared to the increase in elongation at break for the material, still resulting in an overall toughness increase compared to R. Interestingly, the toughening effect of the hard network RT does not occur for IPN 25 wt% (d), likely due to the significant impact of the hard network on the material properties, paired with an inhomogeneous microstructure of the IPN at this hard/soft network ratio.
In light of the application in the dual curing approach for 3D printing, the form stability of an object, which has only been cured with visible light, i.e. contains the cured soft methacrylate network and the uncured epoxide alcohol monomers (rs), was evaluated and deemed sufficient for 3D printing from 35 wt% soft network content on (Fig. 4A).
The printer used in these experiments is a DLP printer with a DLP-light engine that irradiates with a wavelength of 385 nm with a maximum intensity of 75 mW cm−2. Pixel sizes of 50 μm are projected on an area of 96 mm × 54 mm. As this printer was custom-made, it can work with volumes of as little as 1.5 mL in a custom vat, up to standard amounts of formulation in a larger vat. The printed parts are built on a building platform of 42 × 38 mm. The size of the printer is convenient for small printing jobs and the testing of new systems because only small amounts of formulation are needed.
To find the perfect printing parameters, different irradiation times and intensities as well as different cleaning methods for the green body were tested. It was shown that it is beneficial for the resolution to use longer irradiation times and less intensity, compared to higher intensity with corresponding shorter irradiation time: an irradiation time of 6 s with 10 mW cm−2 yielded a better result than irradiating for 3 s with 20 mW cm−2. The best results were achieved using an irradiation time of 8 s and an intensity of 7.5 mW cm−2, which was the lowest possible intensity achievable by the printer hardware. These experiments already indicate that the printing process is highly energy efficient despite the dilution of the methacrylic components. In comparison to this, the pure regulated hard network RT could not be printed under these conditions and required the maximum printer light source intensity of 75 mW cm−2 and a minimum irradiation time of 32.5 s. To print pieces of comparable quality, it took an irradiation time of 35 s. Therefore, printing of IPN 50 wt% (rs) took only 10% of the intensity and a quarter of the time to print RT alone. These results demonstrate impressively how the developed strategy could make a range of previously unprintable materials accessible to the technology.
Next, test chips were designed and printed to investigate the maximally achievable resolution for IPN 50 wt% (rs) compared to RT (Fig. 5A and C). The voxels of the printer are 50 μm small, leading the lines and dots to increase in steps of 50 μm for every voxel added in the STL file. Comparison of the printed part (d) in light microscopic images with intended feature sizes in STL files gave chip deviations between 2 and 10% as determined from digital microscopy images of various features.
 | ||
| Fig. 5 Light- (LM) or scanning electron microscopic (SEM) images of structures printed from IPN 50 wt%: resolution test chip to compare printing resolution between (A) designed dimensions and LM images of (B) RT and (C) IPN 50 wt%, including enlarged images of the marked areas: (a) printed lines (width 100 μm), (b) grid (2 × 2 voxel size 100 μm), (c) best resolution (Res.) measurement of IPN grid with target size 150 μm, and (d) worst resolution measurement of IPN lines with target size 200 μm. Hollow sphere: (D) full size, LM, (E) detail (bar), LM, (F) full size, SEM, (G) detail, SEM; hollow pyramid: (H) full size, LM, (I) full size, SEM; full pyramid: (J) detail (tip), SEM, (K) side view, SEM; benchy: (L) side view, LM, (M) detail (bow), LM, (N) top view, SEM, (O) side view, SEM. | ||
Furthermore, efforts were made to achieve a similar resolution for the epoxide alcohol network. Since printing this network already required the highest intensity the printer could deliver, the only remaining irradiation variable was irradiation time. Therefore, a screening from 30 s to 40 s irradiation time was performed. The yielded parts ranged from under- to over polymerized. Features obtained with 32.5 s and 35 s irradiation time were closest to the achieved resolution of the IPN. From these experiments it was found that the chip printed with IPN 50 wt% (d) displays better resolution than the RT part. This was the closest possible resolution to the IPN, based on the overpolymerization or underpolymerization of all other samples.
To demonstrate the ability to print complex objects with the reported mild and fast printing conditions, a range of 3D objects were printed and imaged by light and scanning electron microscopy (Fig. 5D–O and Fig. S8, ESI†). These images also demonstrate how the layers of the printed objects are interconnected because the hard network is only cured post-printing.
Conclusions
We have demonstrated light-based 3D printing of a tough epoxy polymer network via a dual-cure IPN approach. Toughening of the epoxy network was achieved by chain-regulation with alcohols and thus delaying the gel point, which makes 3D printing more challenging. By printing a wide-meshed soft methacrylate network with high reactivity (rs, 385 nm) in the presence of the epoxide and alcohol monomers of the hard cationic network, the curing thresholds were lowered, and faster printing speeds could be achieved than if the cationic network was printed directly (cs). Subsequent post-curing of the alcohol and epoxy monomers in the green body (d, 320 nm) afforded an inter-layer penetrating, load-bearing network. It was shown that utilization of the soft, fast curing network does not lower the onset temperature of the glass transition (Tg = 45 °C) and even has a slightly positive effect on the tensile toughness of the IPN (t(cs) = 4.1 MJ m−3vs. t(d) = 7.2 MJ m−3). This approach allows faster and more energy-efficient 3D printing of high load bearing, tough constructs with excellent inter-layer connectivity. The irradiation time during the printing process could be reduced from 32.5 s to 8 s, while the light intensity could be lowered from 75 mW cm−2 to 7.5 mW cm−2. We argue that the established dual-cure printing technique has the potential to make other photoacid generator-initiated polymers accessible to fast and mild 3D printing, which would otherwise be of insufficient reactivity for printing. With the herein introduced printing concept, delayed gel points of resins are no longer problematic to obtain form-stable 3D constructs in short time frames and with low energy input.While we believe that the presented printing concept opens the door to new material chemistry in 3D printed specimens, we would simultaneously like to emphasize that several challenges associated with creating such IPNs will need to be addressed by the community in the future. The most important limitation for the presented concept is the solubility/mixability of the components at processing temperature. To yield a homogeneous sample, a homogeneous formulation must be achieved, otherwise domains will form that can influence the mechanical properties. This directly plays into the second limitation of IPNs: Phase separation can be beneficial for the mechanical properties and cause a symbiotic effect, or it can cause premature material failure. Finally, the orthogonality of the network formation reactions must be considered. If the initiators are not chosen as presented in this paper but rather allow for some reactivity of the cationic photopolymerization already during 3D printing, one polymerization type can interfere with the other, which we expect to impose a negative effect on the mechanical properties due to reduced step-growth behaviour.
The presented IPN printing concept could be used to enable printing for different kinds of slow curing polymer networks. Especially polyaddition reactions, that tend to have a later gel point than chain growth reactions, might benefit from these kinds of materials. Another possible application field might be to integrate thermoplastic materials into these kinds of networks to create semi-IPNs and therefore enable easy DLP printing for thermoplastic materials.
While we expect easy transferability of the concept to different cationically cured networks utilizing the same supporting radical network, a certain degree of adaptation is naturally expected. For example, the IPNs in this work yielded the best mechanical properties for 35 wt% of the printable methacrylic network, while 50 wt% of the methacrylic network enabled us to print complex structures. When using this kind of methacrylic network as a scaffold for other polymer networks, these ratios might change.
Author contributions
Florian Mayer: data curation, formal analysis, investigation, methodology, project administration, validation, visualization, writing – original draft, writing – review & editing. Dominik Laa: data preparation, formal analysis, investigation, methodology, validation, writing – review & editing. Thomas Koch: data curation, formal analysis, writing – review & editing. Jürgen Stampfl: conceptualization, formal analysis, funding acquisition, methodology, resources, writing – review & editing. Robert Liska: conceptualization, formal analysis, funding acquisition, methodology, project administration, resources, supervision, writing – review & editing. Katharina Ehrmann: conceptualization, data curation, formal analysis, investigation, methodology, project administration, supervision, validation, visualization, writing – original draft, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.† Raw data for this article beyond the scope of the ESI,† including raw data of all presented graphs are available at TU Wien Research Data at https://doi.org/10.48436/tyab4-bzn33.Conflicts of interest
There are no conflicts to declare.Notes and references
- T. T. Wohlers, W. Associates, I. Campbell, T. Caffrey, O. Diegel and J. Kowen, Wohlers Report 2018: 3D Printing and Additive Manufacturing State of the Industry: Annual Worldwide Progress Report, Wohlers Associates, 2018 Search PubMed.
- J. Stampfl and R. Liska, Macromol. Chem. Phys., 2005, 206, 1253–1256 CrossRef CAS.
- G. Mitteramskogler, R. Gmeiner, R. Felzmann, S. Gruber, C. Hofstetter, J. Stampfl, J. Ebert, W. Wachter and J. Laubersheimer, Addit. Manuf., 2014, 1–4, 110–118 Search PubMed.
- B. Suthar, H. X. Xiao, D. Klempner and K. C. Frisch, Polym. Adv. Technol., 1996, 7, 221–233 CrossRef CAS.
- C. W. Hull, US Pat., US4575330A, 1984.
- C. Decker, T. Nguyen Thi Viet, D. Decker and E. Weber-Koehl, Polymer, 2001, 42, 5531–5541 CrossRef CAS.
- O. Konuray, J. M. Salla, J. M. Morancho, X. Fernandez-Francos, M. Garcia-Alvarez and X. Ramis, Thermochim. Acta, 2020, 692, 178754 CrossRef CAS.
- O. Konuray, F. Di Donato, M. Sangermano, J. Bonada, A. Tercjak, X. Fernandez-Francos, A. Serra and X. Ramis, eXPRESS Polym. Lett., 2020, 14, 881–894 CrossRef CAS.
- J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 594–601 CrossRef CAS.
- M. Sangermano, W. Carbonaro, G. Malucelli and A. Priola, Macromol. Mater. Eng., 2008, 293, 515–520 CrossRef CAS.
- A. Fantoni, J. Ecker, M. Ahmadi, T. Koch, J. Stampfl, R. Liska and S. Baudis, ACS Sustainable Chem. Eng., 2023, 11, 12004–12013 CrossRef CAS.
- S. Lantean, I. Roppolo, M. Sangermano, C. F. Pirri and A. Chiappone, Inventions, 2018, 3, 29 CrossRef.
- M. Pfaffinger, Laser Tech. J., 2018, 15, 45–47 CrossRef CAS.
- Y. Chen, P. Dorfinger, C. Gorsche, G. Harakaly, S. Kaza, M. Kury, C. Li, R. Liska and J. Stampfl, US Pat., US2019338067A1, 2018.
- C. Dall'Argine, A. Hochwallner, N. Klikovits, R. Liska, J. Stampf and M. Sangermano, Macromol. Mater. Eng., 2020, 305, 2000325 CrossRef.
- B. Steyrer, B. Busetti, G. Harakály, R. Liska and J. Stampfl, Addit. Manuf., 2018, 21, 209–214 CAS.
- C. Gorsche, K. Seidler, R. Harikrishna, M. Kury, T. Koch, N. Moszner and R. Liska, Polymer, 2018, 158, 149–157 CrossRef CAS.
- K. Seidler, M. Griesser, M. Kury, R. Harikrishna, P. Dorfinger, T. Koch, A. Svirkova, M. Marchetti-Deschmann, J. Stampfl, N. Moszner, C. Gorsche and R. Liska, Angew. Chem., Int. Ed., 2018, 57, 9165–9169 CrossRef CAS.
- M. Kury, K. Ehrmann, C. Gorsche, P. Dorfinger, T. Koch, J. Stampfl and R. Liska, Polym. Int., 2022, 71, 897–905 CrossRef CAS.
- L. Lecamp, C. Pavillon, P. Lebaudy and C. Bunel, Eur. Polym. J., 2005, 41, 169–176 CrossRef CAS.
- J. Joy, K. Winkler, K. Joseph, S. Anas and S. Thomas, New J. Chem., 2019, 43, 9216–9225 RSC.
- M.-S. Lin and S.-T. Lee, Polymer, 1995, 36, 4567–4572 CrossRef CAS.
- J. R. Nowers, J. A. Costanzo and B. Narasimhan, J. Appl. Polym. Sci., 2007, 104, 891–901 CrossRef CAS.
- L. Pezzana, R. Wolff, G. Melilli, N. Guigo, N. Sbirrazzuoli, J. Stampfl, R. Liska and M. Sangermano, Polymer, 2022, 254, 125097 CrossRef CAS.
- D. Kojic, K. Ehrmann, R. Wolff, Y. Mete, T. Koch, J. Stampfl, S. Baudis and R. Liska, Polym. Chem., 2023, 14, 4809–4818 RSC.
- N. Klikovits, L. Sinawehl, P. Knaack, T. Koch, J. Stampfl, C. Gorsche and R. Liska, ACS Macro Lett., 2020, 9, 546–551 CrossRef CAS PubMed.
- S. Schandl, T. Koch, J. Stampfl, K. Ehrmann and R. Liska, React. Funct. Polym., 2023, 182, 105460 CrossRef CAS.
- J. J. Schwartz and A. J. Boydston, Nat. Commun., 2019, 10, 791 CrossRef CAS PubMed.
- N. D. Dolinski, Z. A. Page, E. B. Callaway, F. Eisenreich, R. V. Garcia, R. Chavez, D. P. Bothman, S. Hecht, F. W. Zok and C. J. Hawker, Adv. Mater., 2018, 30, 1800364 CrossRef PubMed.
- M. Sangermano, W. D. Cook, S. Papagna and S. Grassini, Eur. Polym. J., 2012, 48, 1796–1804 CrossRef CAS.
- C. Rocco, F. Karasu, C. Croutxé-Barghorn, X. Allonas, M. Lecompère, G. Riess, Y. Zhang, A. C. C. Esteves, L. G. J. van der Ven, R. A. T. M. van Benthem and G. de With, Mater. Today Commun., 2016, 6, 17–27 CrossRef CAS.
- M. Sangermano, G. Malucelli, R. Bongiovanni and A. Priola, Eur. Polym. J., 2004, 40, 353–358 CrossRef CAS.
- E. Hasa, J. P. Scholte, J. L. P. Jessop, J. W. Stansbury and C. A. Guymon, Macromolecules, 2019, 52, 2975–2986 CrossRef CAS.
- E. Hasa, J. W. Stansbury and C. A. Guymon, Polymer, 2020, 202, 122699 CAS.
- M. J. Allen, H.-M. Lien, N. Prine, C. Burns, A. K. Rylski, X. Gu, L. M. Cox, F. Mangolini, B. D. Freeman and Z. A. Page, Adv. Mater., 2023, 35, 2210208 CAS.
- A. P. Dhand, M. D. Davidson, J. H. Galarraga, T. H. Qazi, R. C. Locke, R. L. Mauck and J. A. Burdick, Adv. Mater., 2022, 34, 2202261 CrossRef CAS PubMed.
- K. Chen, J. A. Campbell and D. A. Lewis, ACS Appl. Polym. Mater., 2022, 4, 9076–9084 CAS.
- I. Cazin, M. O. Gleirscher, M. Fleisch, M. Berer, M. Sangermano and S. Schlögl, Addit. Manuf., 2022, 57, 102977 CAS.
- Y. Ma, V. Kottisch, E. A. McLoughlin, Z. W. Rouse, M. J. Supej, S. P. Baker and B. P. Fors, J. Am. Chem. Soc., 2021, 143, 21200–21205 CrossRef CAS PubMed.
- B. Wang, E. Engay, P. R. Stubbe, S. Z. Moghaddam, E. Thormann, K. Almdal, A. Islam and Y. Yang, Nat. Commun., 2022, 13, 367 CrossRef CAS.
- E. Rossegger, J. Strasser, R. Höller, M. Fleisch, M. Berer and S. Schlögl, Macromol. Rapid Commun., 2023, 44, 2200586 CrossRef CAS PubMed.
- N. D. Dolinski, E. B. Callaway, C. S. Sample, L. F. Gockowski, R. Chavez, Z. A. Page, F. Eisenreich, S. Hecht, M. T. Valentine, F. W. Zok and C. J. Hawker, ACS Appl. Mater. Interface, 2021, 13, 22065–22072 CrossRef CAS.
- Y. Li, F. Zhang, Y. Liu and J. Leng, Chem. Eng. J., 2024, 491, 152196 CrossRef CAS.
- I. Cazin, K. Plevová, W. Alabiso, E. Vidović and S. Schlögl, Adv. Eng. Mater., 2024, 26, 2301699 CrossRef CAS.
- X. Kuang, Z. Zhao, K. Chen, D. Fang, G. Kang and H. J. Qi, Macromol. Rapid Commun., 2018, 39, 1700809 CrossRef PubMed.
- M. Ahmadi, K. Ehrmann, T. Koch, R. Liska and J. Stampfl, Chem. Rev., 2024, 124, 3978–4020 CrossRef CAS PubMed.
- A. Fantoni, T. Koch, S. Baudis and R. Liska, ACS Appl. Polym. Mater., 2023, 5, 731–742 CrossRef CAS.
- M. T. Kiker, A. Uddin, L. M. Stevens, K.-Y. Chung, P. Lu and Z. A. Page, Polym. Chem., 2023, 14, 3843–3850 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mh01261e |
| This journal is © The Royal Society of Chemistry 2025 |