DOI:
10.1039/D4MH01356E
(Communication)
Mater. Horiz., 2025,
12, 973-986
Hydrogen sulfide-generating semiconducting polymer nanoparticles for amplified radiodynamic–ferroptosis therapy of orthotopic glioblastoma†
Received
30th September 2024
, Accepted 7th November 2024
First published on 8th November 2024
Abstract
A variety of therapeutic strategies are available to treat glioblastoma (GBM), but the tumor remains one of the deadliest due to its aggressive invasiveness, restrictive blood–brain barrier (BBB), and exceptional resistance to drugs. In this study, we present a hydrogen sulfide (H2S)-generating semiconducting polymer nanoparticle (PFeD@Ang) for amplified radiodynamic–ferroptosis therapy of orthotopic glioblastoma. Our results show that in an acidic tumor microenvironment (TME), H2S donors produce large amounts of H2S, which inhibits mitochondrial respiration and alleviates cellular hypoxia, thus enhancing the radiodynamic effect during X-ray irradiation; meanwhile, Fe3+ is reduced to Fe2+ by tannic acid in an acidic TME, which promotes an iron-dependent cell death process in tumors. H2S facilitates the ferroptosis process by increasing the local H2O2 concentration via inhibiting catalase activity. This kind of amplified radiodynamic–ferroptosis therapeutic strategy could remarkably inhibit glioma progression in an orthotopic GBM mouse model. Our study demonstrates the potential of PFeD@Ang for GBM treatment via targeted delivery and combinational therapeutic actions of RDT and ferroptosis therapy.
New concepts
In this work, a semiconducting polymer nanoparticle (PFeD@Ang) that can produce hydrogen sulfide (H2S) is reported for orthotopic glioblastoma therapy via amplifying the radiodynamic–ferroptosis effect. A H2S donor and Fe3+–tannic acid (TA) chelates were co-loaded into semiconductor polymer nanoparticles with surface embellishing of angiopep-2 (Ang) to form PFeD@Ang. Due to surface conjugation of Ang, the nanoparticle could overcome the blood–brain barrier (BBB) and target orthotopic glioblastoma to realize effective tumor accumulation. In the tumor microenvironment, the H2S donor generated a large amount of H2S for inhibiting mitochondrial respiration to alleviate tumor hypoxia, and suppressing catalase activity to increase local H2O2 concentration. As such, PFeD@Ang mediated an amplified radiodynamic effect upon X-ray irradiation after hypoxia alleviation, as well as enabled an enhanced ferroptosis effect due to the increased H2O2 level. This amplified antitumor efficacy was found to greatly inhibit the growth of orthotopic glioblastoma in mouse models. This study offers a valuable scheme for overcoming various limitations of traditional therapeutics to enable amplified and combinational actions for deep-seated tumor therapy. Overall, this proposed therapeutic strategy holds great promise for treating brain tumors and other central nervous system diseases.
|
Introduction
Glioblastoma (GBM), which is a common intracranial tumor, has a poor prognosis.1–5 Current treatments mainly include surgical resection and chemotherapy.6,7 However, due to the aggressive nature of the tumors and the blurred borders, complete resection of glioma tumors is often difficult to achieve, leading to high tumor recurrence.8 Chemotherapy is another major treatment of malignant GBM. However, a large proportion of GBM patients are highly resistant to the most commonly used chemotherapeutic agent temozolomide.9,10 The blood–brain barrier (BBB) significantly limits chemotherapy drug penetration into brain tissue, reducing the therapeutic effects of many anti-glioblastoma drugs.11–13 Furthermore, the systemic toxicity of chemotherapeutic agents causes detrimental side effects on the patient's immune system and other normal tissues. Thus, despite surgery and chemotherapy being the primary treatments for managing GBM, they possess notable limitations and have not succeeded in substantially improving the overall prognosis of GBM patients. These challenges have promoted researchers to explore novel therapeutic strategies.
Radiotherapy is also a treatment modality of GBM.14–16 Traditional radiation therapy relies mainly on the direct interaction of ionizing radiation with DNA to cause fatal damage to cancer cells.17 However, the non-specific nature of this method often causes severe collateral damage to the surrounding healthy tissues, leading to a large number of undesirable side effects.18,19 Advances in nanotechnology and materials science, including X-ray-based radiodynamic therapy (RDT), have promoted the development of new therapeutic methods.20–22 During RDT, photosensitizers, typically conjugated to nanoparticles, are activated by X-ray irradiation to produce reactive oxygen species (ROS), which selectively induce cytotoxic effects within the tumor microenvironment (TME).23,24 X-ray-based RDT has shown promising applications in tumor treatment in recent years. It has been shown that gold nanoparticles (AuNPs) conjugated with photosensitizers can significantly enhance ROS production under X-ray irradiation, leading to improved tumor control.25–27 Similarly, the efficacy of other nanomaterials such as titanium dioxide and hafnium oxide in RDT has been investigated, showing favorable results in terms of biocompatibility and therapeutic efficacy.28–30 However, RDT alone still suffers from insufficient efficacy, which can be ascribed to the following factors: (1) RDT is highly dependent on oxygen, and the tumor site is a highly hypoxic environment and (2) cancer cells can resist RDT through mechanisms such as increased antioxidant enzyme expression, enhanced DNA repair, and altered apoptotic pathways.31,32 Therefore, more and more studies have attempted to combine RDT with other therapies to improve anti-tumor efficacy.
In recent years, ferroptosis-based therapeutic strategies have shown great potential in treating glioblastoma and have attracted significant attention.33–37 For example, the ferroptosis response can specifically target cancer cells by inducing iron-dependent lipid peroxidation, leading to cell death.38,39 This mechanism is particularly effective for GBM due to the high metabolic activity of tumors and associated oxidative stress.40 However, despite these advantages, the efficacy of ferroptosis therapies in GBM is often affected by certain factors inherent in the TME. One such factor is the limited effectiveness of using hydrogen peroxide (H2O2) in the TME to induce iron oxidation.41 Endogenous levels of H2O2 in tumors are typically limited, which leads to insufficient induction of ferroptosis.42–44 Moreover, the presence of glutathione (GSH) in the tumor microenvironment acts as a potent antioxidant, scavenging free radicals and further compromising the efficacy of ferroptosis-based therapies. This dual limitation results in suboptimal antitumor responses. To address these challenges, we have developed strategies aimed at both increasing H2O2 production within the tumor microenvironment and reducing GSH levels, thereby enhancing the induction of ferroptosis and improving overall antitumor efficacy.45 In addition, the presence of antioxidant defense mechanisms in tumor cells, such as the overexpression of glutathione peroxidase 4 (GPX4), would further reduce the effects of H2O2-mediated iron porphyrin poisoning.46,47 Hydrogen sulfide (H2S) is a gaseous molecule with diverse regulatory functions in the TME. It has been demonstrated to inhibit mitochondrial respiration, which in turn mitigates tumor hypoxia.48–50 In addition, H2S inhibits catalase activity, leading to an increase in the local concentration of H2O2. Elevated levels of H2O2 increase the efficiency of the Fenton reaction, which promotes iron oxidation more efficiently.51,52 Chen et al. demonstrated in their study that H2S donors significantly enhanced iron-based nanoparticle-induced lipid peroxidation and cell death by increasing the levels of H2O2.53 This combined strategy exploits the unique properties of H2S and overcomes the limitations of conventional ferroptosis therapies, such as substandard H2O2 levels and poor antioxidant defenses. By enhancing the Fenton reaction and disrupting key cellular processes, this dual approach may provide a more effective and targeted means of inducing cancer cell death.
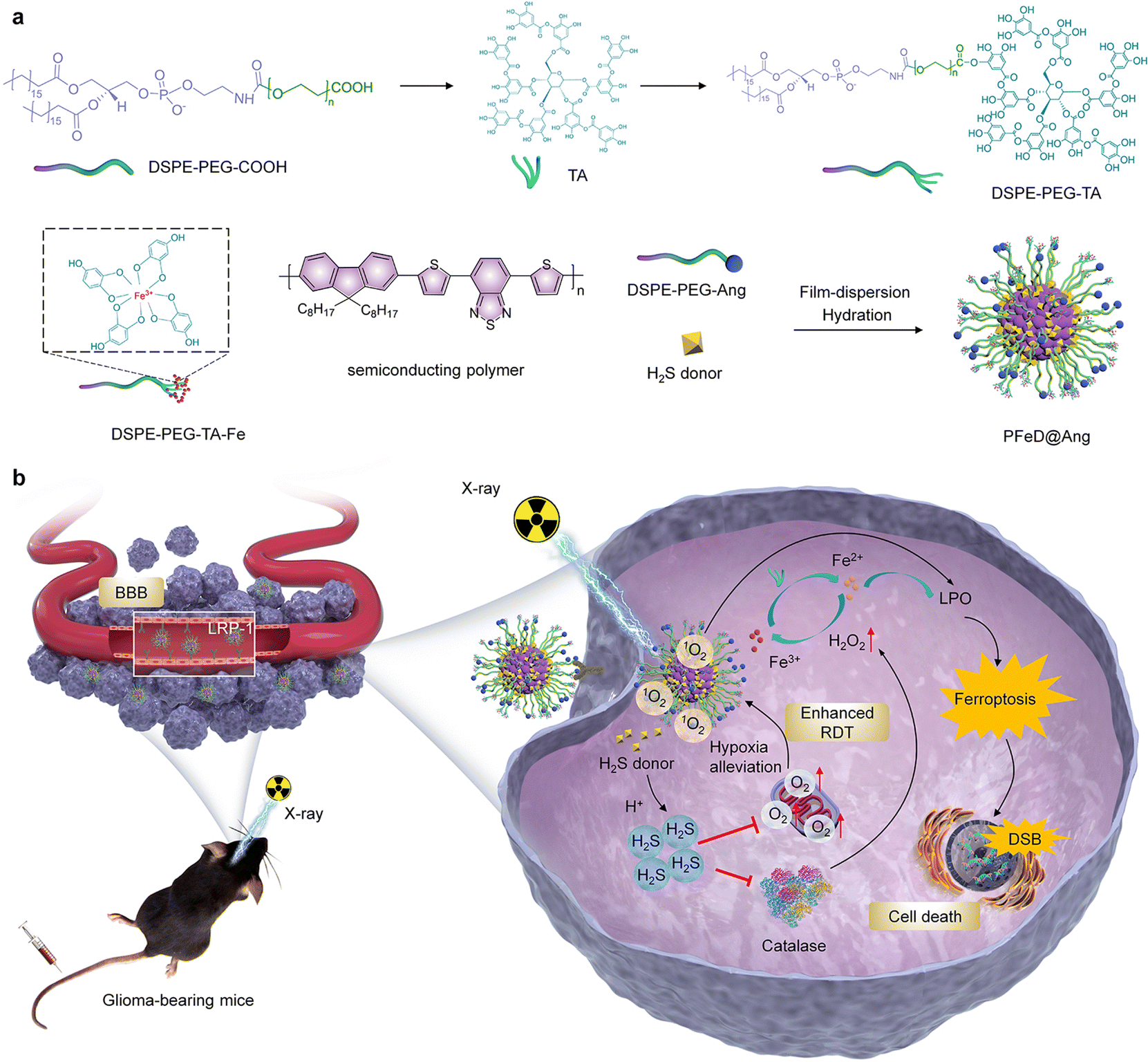
Herein, a hydrogen sulfide-generating semiconducting polymer nanoparticle (PFeD@Ang) was developed for amplified radiodynamic–ferroptosis therapy of orthotopic glioblastoma. PFeD@Ang was designed by loading a H2S donor and Fe3+–tannic acid (TA) chelates into semiconductor polymer nanoparticles and modifying the surface with angiopep-2 (Fig. 1a). The inherent physicochemical properties of nanoparticles can significantly influence their cellular uptake. In general, smaller particle size and negative surface charge tend to facilitate cellular internalization. However, the presence of the BBB limits the efficiency of drug delivery to the brain.12 To address this, the addition of targeting molecules can further enhance nanoparticle uptake, thereby improving their targeting efficiency. PFeD@Ang nanoparticles constructed by low-density lipoprotein receptor-related protein (LRP-1) can effectively traverse the blood–brain barrier and accurately reach the tumor site through the mediation of LRP-1 targeting receptor angiopep-2.54–56 Although numerous angiopep-2-modified nanoparticles have been developed, most previous formulations primarily loaded a single drug, limiting their capability for multimodal therapy.57,58 In this study, we simultaneously loaded both drug and sonosensitizer, enabling multiple therapeutic modalities and suggesting improved efficacy. In the TME, the H2S donor generates a large amount of H2S gas, which inhibits mitochondrial respiration and alleviates cellular hypoxia, thus enhancing the radiodynamic effect upon X-ray irradiation; meanwhile, in the acidic TME, Fe3+ is reduced to Fe2+ by TA, which promotes the intratumor iron-dependent cell death process. Moreover, semiconductor polymer nanoparticles generate a large amount of ROS under X-ray irradiation. The produced singlet oxygen (1O2) rapidly depletes glutathione (GSH), thereby leading to ferroptosis. This ferroptosis process can be induced by both RDT and the iron-based Fenton reaction.59 Through inhibiting the activity of catalase, H2S causes an increase in the local concentration of H2O2 and thus promotes the ferroptosis process more effectively. The prepared PFeD@Ang exhibits potent anti-tumor efficacy in an orthotopic glioma mouse model (Fig. 1b). Our nanosystem thus offers a responsive platform to address multiple challenges in the management of GBM.
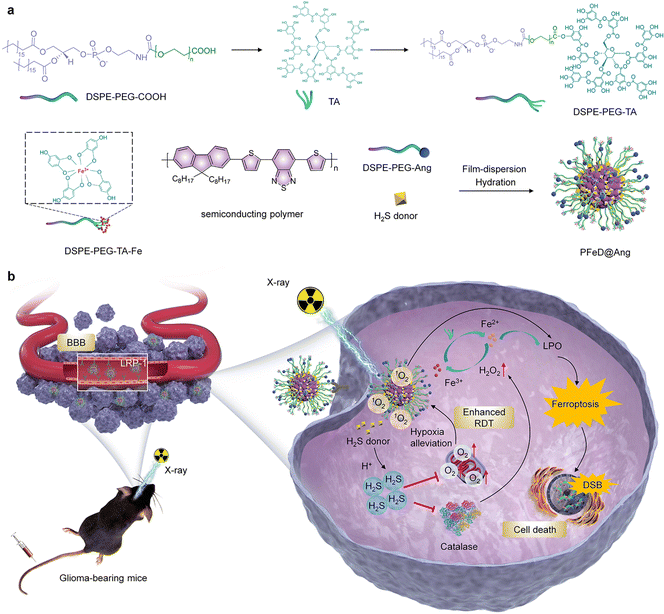 |
| | Fig. 1 Development of H2S-generating semiconducting polymer nanoparticles for orthotopic glioma therapy. (a) Synthesis route of PFeD@Ang. (b) A schematic diagram of PFeD@Ang for orthotopic glioma therapy via radiodynamic–ferroptosis therapy. | |
Results and discussion
Nanoparticle preparation and characterization
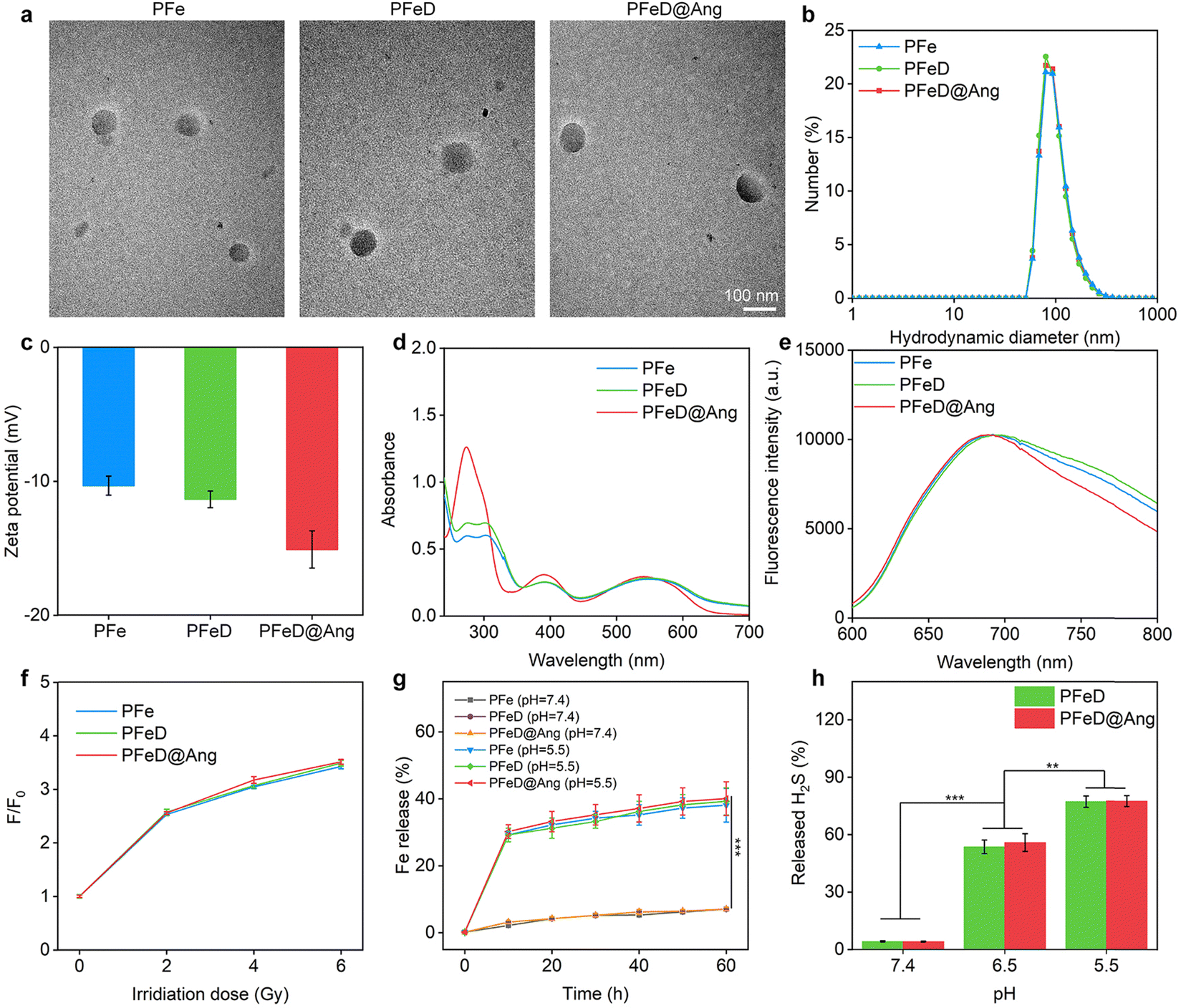
To enhance BBB crossing, brain-targeting peptide angiopep-2 was conjugated with DSPE–PEG through alkylation between the peptide's sulfhydryl group and DSPE–PEG–Mal's maleimide group. The characteristic absorption peak of the maleimide group in DSPE–PEG–Mal was observed at 7.0 ppm in the 1H NMR spectrum (Fig. S1a, ESI†). When the maleimide reacted with angiopep-2 to obtain Ang-modified DSPE–PEG–Mal (DSPE–PEG–Ang), the absorption peak at 7.0 ppm disappeared, which proved the successful synthesis of DSEP–PEG–Ang (Fig. S1b, ESI†).60 The successful synthesis of DSPE–PEG–Ang was also verified using MALDI-TOF-MS spectroscopy, confirming that the synthesized DSPE–PEG–Ang had a molecular weight of 5699.398 (Fig. S2, ESI†). To synthesize DSPE–PEG–TA–Fe, DSPE–PEG–TA was obtained by esterification reaction between DSPE–PEG-COOH and TA (Fig. S3, ESI†). Subsequently, DSPE–PEG–TA–Fe was synthesized through the chelation of TA with Fe3+, with the Fe content measured at 5.1 ± 0.2 wt% using ICP-OES. PFeD@Ang was constructed by nanoprecipitation of DSPE–PEG–Ang, DSPE–PEG–TA–Fe, PFODBT, and H2S donor. For comparison, nanoparticles without angiopep-2 ligand modification (named PFeD) and without angiopep-2 ligand modification as well as H2S donor-loaded nanoparticles were also synthesized (named PFe). TEM analysis revealed consistent, symmetrical spherical morphologies for PFe, PFeD, and PFeD@Ang nanoparticles (Fig. 2a). The hydrodynamic diameters of PFe, PFeD, and PFeD@Ang measured by DLS were 91.4, 92.2, and 94.6 nm, respectively (Fig. 2b), which were in agreement with the data from TEM analysis (∼85 nm). Upon storage in an aqueous solution for 2 weeks, no significant change in particle size was observed, indicating excellent stability of the synthesised PFe, PFeD and PFeD@Ang (Fig. S4, ESI†). The zeta potentials of PFe, PFeD and PFeD@Ang were −10.3, −11.3 and −15.1 mV, respectively (Fig. 2c). The reduced zeta potential of PFeD@Ang compared to PFe and PFeD may be due to its surface conjugation with angiopep-2. Following incubation of erythrocytes with PFe, PFeD and PFeD@Ang, it was demonstrated that these nanoparticles exhibited negligible hemolytic effects, thereby confirming their suitability for intravenous injection (Fig. S5, ESI†). We also detected the absorbance and fluorescence properties of PFe, PFeD, and PFeD@Ang. Characteristic peaks of PFODBT at 380 and 545 nm were observed in PFe, PFeD, and PFeD@Ang absorption spectra, confirming successful PFODBT loading (Fig. 2d). Fluorescence spectra showed similar 700 nm peaks for PFe, PFeD, and PFeD@Ang, indicating the minimal impact of drug encapsulation and surface modification on their optical properties (Fig. 2e). The radio dynamic effects of PFe, PFeD and PFeD@Ang were assessed using the 1O2 probe. In solution systems containing PFe, PFeD and PFeD@Ang, the fluorescence intensity of singlet oxygen sensor green (SOSG) exhibited a gradual enhancement with increasing doses of X-ray irradiation (Fig. S6, ESI†). It was found that the 1O2 generation efficiencies for PFe, PFeD and PFeD@Ang were similar upon X-ray irradiation. The fluorescence intensity at an X-ray dose of 10 Gy increased by about 3.5-fold higher compared to the unirradiated groups. These results showed that PFe, PFeD and PFeD@Ang can consistently produce 1O2 under X-ray irradiation (Fig. 2f). Moreover, it was found that the 1O2 generation efficiencies for PFODBT and PFeD@Ang were similar upon X-ray irradiation, suggesting that the radiosensitization effect of the nanoparticle was solely ascribed to PFODBT (Fig. S7, ESI†). TA could chemically reduce released Fe3+ to Fe2+ in an acidic environment, driving redox cycling of Fe and efficiently generating lipid peroxides necessary for ferroptosis.52 Fe release from PFeD@Ang was evaluated at different pH levels including 7.4, 6.5, and 5.5. As shown in Fig. 2g, at pH 7.4, the Fe release was less than 10%. Conversely, at pH 5.5, up to 40% of iron was released. These results suggest that the prepared nanoparticles can readily release Fe in an acidic TME.61 The ability of the liberated Fe to produce a hydroxy radical (˙OH) through the Fenton reaction was confirmed by employing the ˙OH indicator 3,3′,5,5′-tetramethylbenzidine (TMB). After incubation with H2O2 alone, the absorbance values of the ˙OH indicator in PFe, PFeD, and PFeD@Ang solutions showed no significant changes. PFe, PFeD, and PFeD@Ang solutions did not show significant absorbance decreases after incubation with H2O2 alone. In contrast, PFe, PFeD, and PFeD@Ang showed a significant increase in absorbance after incubation with TMB and H2O2 in a solution at pH 5.5. The absorbance of the ˙OH indicator increased 9.5-fold in the nanoparticles + TMB + H2O2 group compared to the nanoparticles + H2O2 and nanoparticles alone groups (Fig. S8, ESI†), suggesting that the prepared nanoparticles could produce ˙OH. The ability of PFeD and PFeD@Ang to release H2S under different pH conditions was then verified using a H2S detection kit. At pH 5.5, PFeD and PFeD@Ang could release approximately 78% H2S, demonstrating that PFeD and PFeD@Ang could successfully release H2S in the acidic TME (Fig. 2h). We investigated the release profiles of PFeD@Ang and PFeD at pH 5.5 over time. As shown in Fig. S9 (ESI†), the release of H2S reached 77% within 30 minutes.
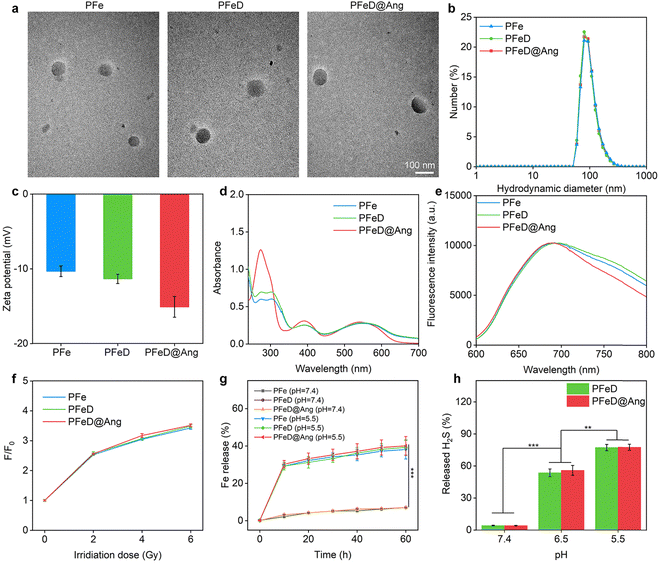 |
| | Fig. 2 PFe, PFeD and PFeD@Ang characterization. (a) TEM images of PFe, PFeD and PFeD@Ang. (b) Hydrodynamic diameters of PFe, PFeD and PFeD@Ang. (c) Zeta potentials of PFe, PFeD and PFeD@Ang (n = 3). (d) UV-vis absorbance spectra of PFe, PFeD and PFeD@Ang in aqueous solutions. (e) Fluorescence spectra of PFe, PFeD and PFeD@Ang. (f) 1O2 generation evaluation of PFe, PFeD and PFeD@Ang solutions after X-ray irradiation (n = 3). (g) Cumulative release of Fe from PFe, PFeD and PFeD@Ang at pH 7.4 or 5.5 (n = 3). (h) H2S donor release percentages from PFeD and PFeD@Ang at pH 7.4, 6.5 or 5.5 (n = 3). | |
Investigations of nanoparticles in cells
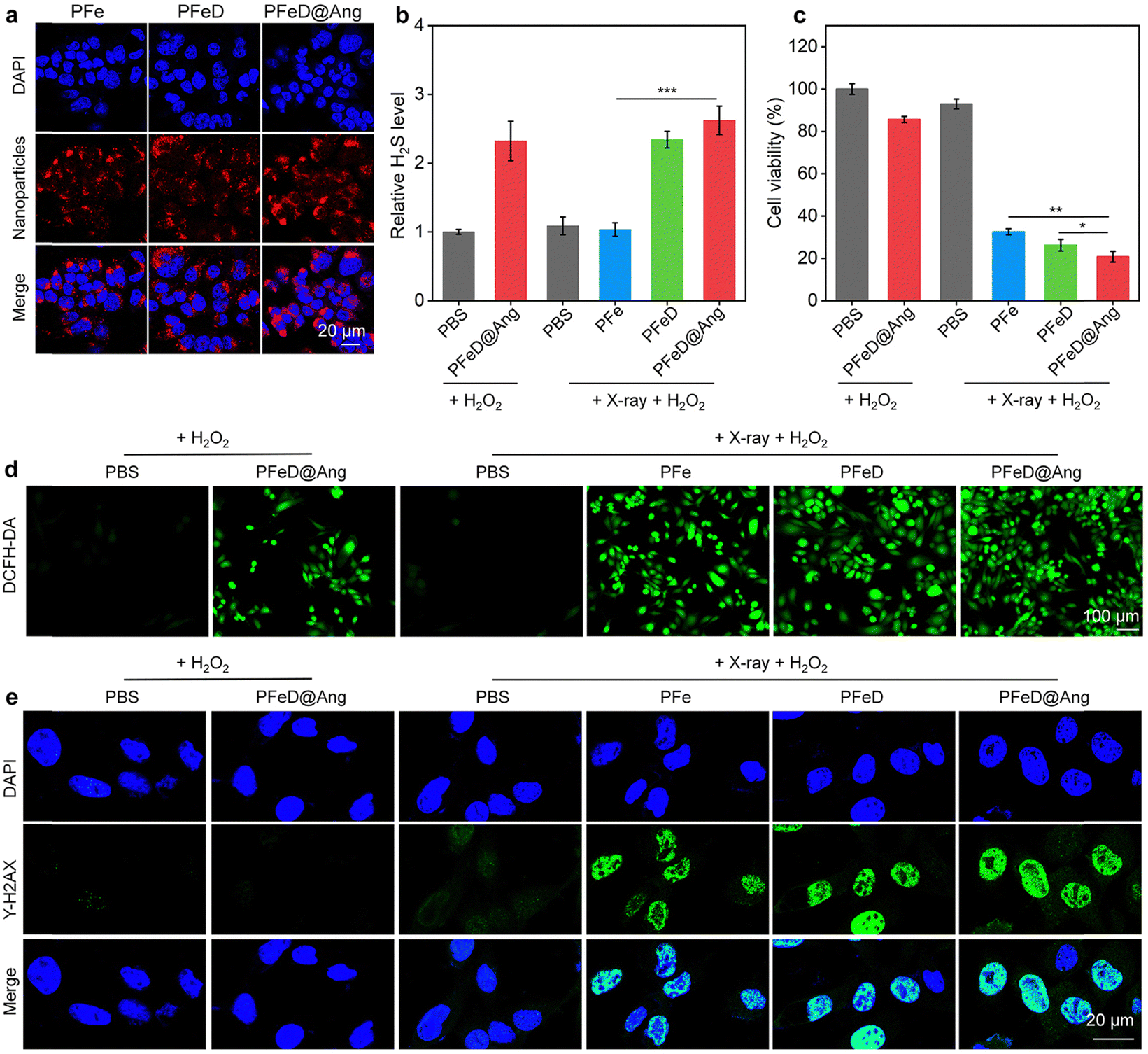
The cytoplasm of GL261-luc cancer cells displayed a distinct red fluorescence signal when they were co-incubated with PFe, PFeD and PFeD@Ang for 24 hours (Fig. 3a), confirming the effective phagocytosis of PFe, PFeD and PFeD@Ang by GL261-luc cancer cells. Based on quantitative intracellular fluorescence analysis, PFeD@Ang treatment increased the fluorescence intensity in GL261-luc cancer cells by about 1.2-fold more compared with PFe or PFeD treatment (Fig. S10, ESI†). This finding suggested that angiopep-2 effectively targets the LRP-1 receptor expressed on GL261-luc cancer cells, facilitating enhanced internalization of PFeD@Ang in these cells compared to PFe and PFeD. A H2S detection kit was used to validate the production of H2S by GL261-luc cancer cells. As shown in Fig. 3b, the PFeD and PFeD@Ang groups exhibited elevated levels of H2S in GL261-luc cancer cells compared to the PBS group, confirming the intracellular H2S generation by the nanoparticles. The cytotoxicity and therapeutic effects of PFe, PFeD and PFeD@Ang were investigated by the CCK-8 assay following co-incubation with GL261-luc cancer cells for 24 hours. It was discovered that even at a concentration of 100 μg mL−1, the cellular activity of GL261-luc cancer cells treated with PFe, PFeD, and PFeD@Ang was more than 80% (Fig. S11, ESI†), which indicated that the nanoparticles had negligible cytotoxicity. The therapeutic efficacy of the nanoparticles was assessed by irradiating the treated cells with X-ray. The results indicated that PFe, PFeD, and PFeD@Ang significantly reduced cell survival under X-ray irradiation compared to the PBS control group. Specifically, the cell survival rates were reduced to 20.7%, 26.3%, and 32.6% for the PFeD@Ang + X-ray + H2O2 group, PFeD + X-ray + H2O2 group, and PFe + X-ray + H2O2 group, respectively (Fig. 3c). Moreover, we conducted a comparative study to evaluate the efficacy of H2S-releasing nanoparticles against angiopep-2 conjugated nanoparticles loaded with doxorubicin (DOX) and temozolomide (TMZ). The cell survival rates in the PFeD@Ang + X-ray + H2O2 group exhibited a 2.7-fold decrease compared to the DOX@Ang group and a 2.3-fold decrease compared to the TMZ@Ang group, demonstrating that the H2S-releasing nanoparticle system provides superior therapeutic efficacy over other drug delivery carriers (Fig. S12, ESI†). These results underscore the enhanced cytotoxicity of the PFeD@Ang formulation in the presence of X-ray irradiation and H2O2, highlighting its potential as an effective therapeutic strategy against gliomas. Fluorescence imaging of intracellular ROS production using 2′,7′-dichlorofluorescein diacetate (H2DCFDA) was used to verify ROS production. While no fluorescent signal was detected in the PBS + H2O2 group, green fluorescent signals were observed in the PFeD@Ang+ H2O2, PFe + X-ray + H2O2, PFeD + X-ray + H2O2 and PFeD@Ang + X-ray + H2O2 groups (Fig. 3d). Compared to the PFeD@Ang + H2O2 group, the ROS signal intensity in the PFe + X-ray + H2O2, PFeD + X-ray + H2O2, and PFeD@Ang + X-ray + H2O2 groups increased by 2.1-fold, 2.8-fold, and 3.1-fold, respectively. These results demonstrated that X-ray irradiation significantly enhances the radiosensitizing effects of the nanoparticles. The ROS signal intensity in the PFeD@Ang + X-ray + H2O2 group increased by 1.4-fold and 1.2-fold compared to the PFe + X-ray + H2O2 group and the PFeD + X-ray + H2O2 group, respectively (Fig. S13, ESI†). By identifying phosphorylated γ-H2AX, DNA double-strand breaks (DSBs) were detected to validate the radiosensitization effects. Following PFe, PFeD and PFeD@Ang incubations and X-ray (4 Gy) exposure, the γ-H2AX signals were detected within GL261-luc cancer cells (Fig. 3e). Cells treated with PBS + H2O2 or PBS + X-ray + H2O2 only have slight γ-H2AX fluorescence. In comparison to the PBS + H2O2 group, the γ-H2AX signal intensity was about 18.0-fold higher in the PFe + X-ray + H2O2 and PFeD + X-ray + H2O2 groups, and about 20.0-fold higher in the PFeD@Ang + X-ray + H2O2 group (Fig. S14, ESI†). Consequently, a substantial radiosensitization effect was demonstrated by PFe, PFeD and PFeD@Ang to generate DNA DSBs.
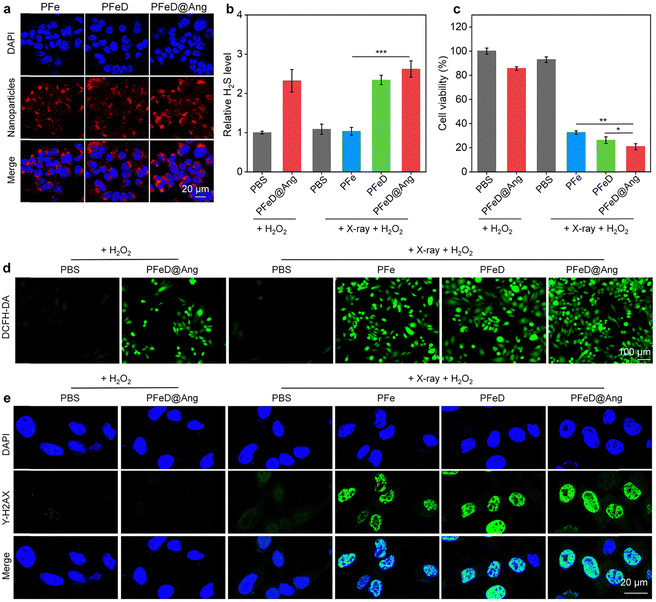 |
| | Fig. 3 Investigations of nanoparticles in cells. (a) Fluorescence images of GL261-luc cancer cells treated with PFe, PFeD and PFeD@Ang for 12 hours. (b) The amounts of H2S released in GL261-luc cancer cells after various treatments (n = 3). (c) Viability of GL261-luc cancer cells after treatment (n = 3). (d) ROS fluorescence images of GL261-luc cancer cells. (e) γ-H2AX assay of GL261-luc cells treated with PBS, PFe, PFeD, and PFeD@Ang under X-ray irradiation and H2O2. | |
Studies on anti-tumor mechanisms
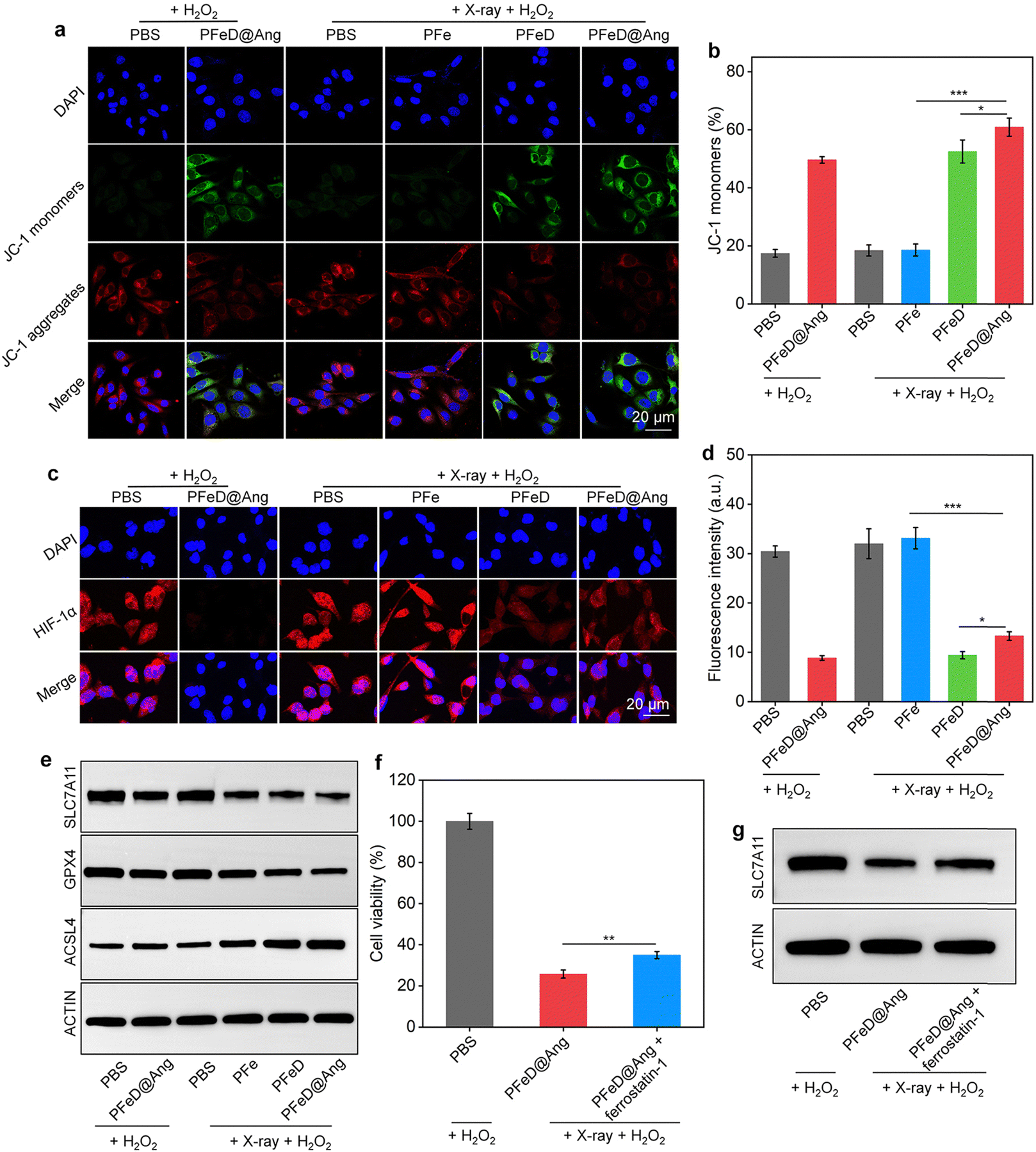
The mitochondrial damage generated by the H2S donor was assessed by JC-1 staining. Notably, distinct green fluorescent signals (JC-1 monomer) were observed in the PFeD@Ang + H2O2, PFeD + X-ray + H2O2, and PFeD@Ang + X-ray + H2O2 groups. This observation indicated a significant reduction in mitochondrial membrane potential and severe mitochondrial damage (Fig. 4a). Compared to the PFe + X-ray + H2O2 group, the JC-1 monomer levels in the PFeD + X-ray + H2O2 group were approximately 3-fold higher, confirming that the H2S donor effectively releases H2S within the tumor, inhibits mitochondrial respiration, and induces changes in mitochondrial membrane potential (Fig. 4b). In addition, the content of the JC-1 monomer in the PFeD@Ang + H2O2 group was 2.8-fold higher than that in the PBS + H2O2 group (Fig. 4b). This suggested that the H2S donor could generate H2S in the TME without X-ray irradiation, subsequently impairing mitochondrial function and demonstrating the acid-responsive nature of the prepared nanoparticles. Furthermore, it has been suggested that H2S can alleviate hypoxia by inhibiting cellular respiration.62 PFeD@Ang-induced mitochondrial dysfunction was assessed by oxygen content and hypoxic conditions. The oxygen indicator (Ru(dpp)3Cl2) fluorescence, quenched by oxygen, showed weaker signals in the PFeD and PFeD@Ang groups than in the controls, indicating increased oxygen levels from inhibited respiration. This oxygen boost could enhance oxygen-dependent RDT. After X-ray irradiation, fluorescence further increased due to RDT-mediated oxygen consumption. The fluorescence intensity in the PFeD and PFeD@Ang-treated cells was over 10.3-fold lower than in the PFe-treated cells (Fig. S15, ESI†). Therefore, the hypoxic microenvironment in cancer cells was evaluated via immunofluorescence staining of HIF-1α. The PFeD and PFeD@Ang treatments significantly decreased the fluorescence signals of HIF-1α staining in comparison to the PBS + H2O2 control group. The intensity of HIF-1α fluorescence signals was higher in the PBS + H2O2, PFe + X-ray + H2O2, and PBS + X-ray + H2O2 groups than in the PFeD- and PFeD@Ang-treated groups. Notably, the PFeD@Ang + H2O2 group exhibited almost no fluorescence. This phenomenon could be attributed to the ability of PFeD@Ang to release H2S in the TME to relieve hypoxia (Fig. 4c). Compared to the PFeD + X-ray + H2O2 and PFeD@Ang + X-ray + H2O2 groups, the HIF-1α signal intensity was about 3.0-fold higher in the PBS + H2O2 group. The hypoxia staining signal was more pronounced in the nanoparticle-treated groups with X-ray irradiation than in the nanoparticle-treated groups without X-ray irradiation, attributed to oxygen depletion by the RDT effect (Fig. 4d). Ferroptosis was confirmed by measuring glutathione levels (GSH), acyl coenzyme A synthase long chain member 4 (ACSL4), solute carrier family 7 member 11 (SLC7A11) and glutathione peroxidase 4 (GPX4) expression levels. The intracellular GSH levels were approximately 0.3 μmol in the PFeD@Ang + X-ray + H2O2 groups, 0.4 μmol in the PFeD + X-ray + H2O2, 0.6 μmol in the PFe + X-ray + H2O2 group, 0.5 μmol in the PFeD@Ang + H2O2 group, and approximately 0.7 μmol in the other groups (Fig. S16a, ESI†). Malondialdehyde (MDA) levels in the various treatment groups were assessed using the MDA kit. The levels of MDA in the PFeD + X-ray + H2O2 group and PFeD@Ang + X-ray + H2O2 group were about 2.4- and 2.7-fold higher, respectively, compared to the PBS + H2O2 group (Fig. S16b, ESI†). Moreover, the PFeD@Ang + H2O2, PFe + X-ray + H2O2, PFeD + X-ray + H2O2, and PFeD@Ang + X-ray + H2O2 groups exhibited significantly reduced expression levels of GPX4 and SLC7A11 but the ACSL4 expression level was notably higher in the PFeD@Ang + H2O2, PFe + X-ray + H2O2, PFeD + X-ray + H2O2, and PFeD@Ang + X-ray + H2O2 groups compared to the PBS + H2O2 group (Fig. 4e and Fig. S17a, ESI†). The expression of SLC7A11 in the PFeD@Ang + X-ray + H2O2 group decreased by approximately 2.3- and 1.4-fold compared to the PFe + X-ray + H2O2 and PFeD + X-ray + H2O2 groups, respectively. The expression of GPX4 in the PFeD@Ang + X-ray + H2O2 group decreased by about 2.4- and 1.4-fold compared to the PFe + X-ray + H2O2 and PFeD + X-ray + H2O2 groups. In contrast, the expression of ACSL4 in the PFeD@Ang + X-ray + H2O2 group increased by about 1.4- and 1.2-fold compared to the PFe + X-ray + H2O2 and PFeD + X-ray + H2O2 groups (Fig. S17b, ESI†). We also evaluated the effect of the ferroptosis inhibitor ferrostatin-1. Compared to the PFeD@Ang + X-ray + H2O2 group, the PFeD@Ang + X-ray + H2O2 + ferrostatin-1 significantly increased cell viability (Fig. 4f). Moreover, the PFeD@Ang + X-ray + H2O2 + ferrostatin-1 group exhibited significantly higher expression levels of SLC7A11 compared to the PFeD@Ang + X-ray + H2O2 group (Fig. 4g and Fig. S18a, ESI†). Specifically, the expression of SLC7A11 in the PFeD@Ang + X-ray + H2O2 + ferrostatin-1 group increased by about 1.3-fold compared to the PFe PFeD@Ang + X-ray + H2O2 group, confirming that the ferroptosis inhibitor effectively rescued the cells from death (Fig. S18b, ESI†).
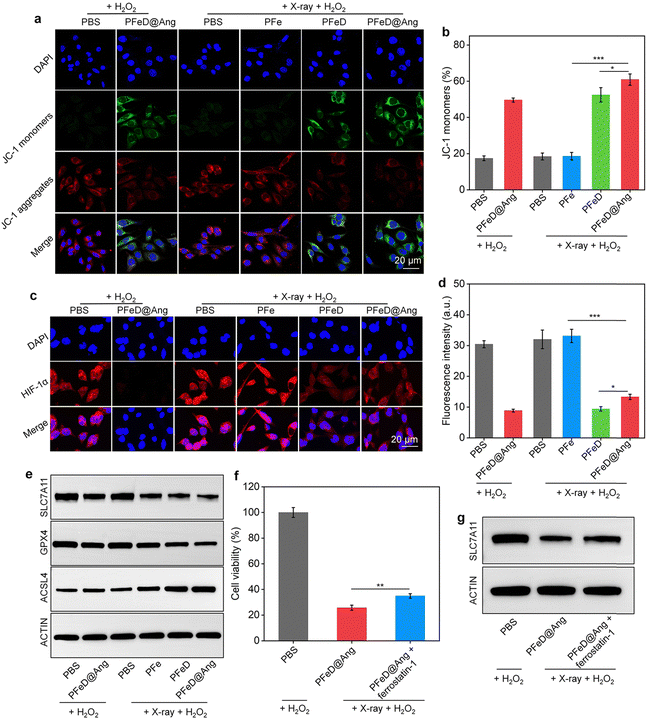 |
| | Fig. 4 Studies on anti-tumor mechanisms. (a) Confocal images showing JC-1 staining in the treated GL261-luc cells. (b) Quantified JC-1 fluorescence intensity in the treated cells (n = 5). (c) Confocal images of HIF-1α staining in the treated cells. (d) Quantified HIF-1α fluorescence intensity post-treatment (n = 5). (e) Representative WB images of SLC7A11, GPX4, and ACSL4 in GL261-luc cancer cells after various treatments. (f) Cell viability after PFeD@Ang or PFeD@Ang + ferrostatin-1 with X-ray or H2O2 treatment (n = 3). (g) Representative WB images of SLC7A11 in GL261-luc cancer cells after various treatments. | |
Evaluation of the efficacy of BBB crossing
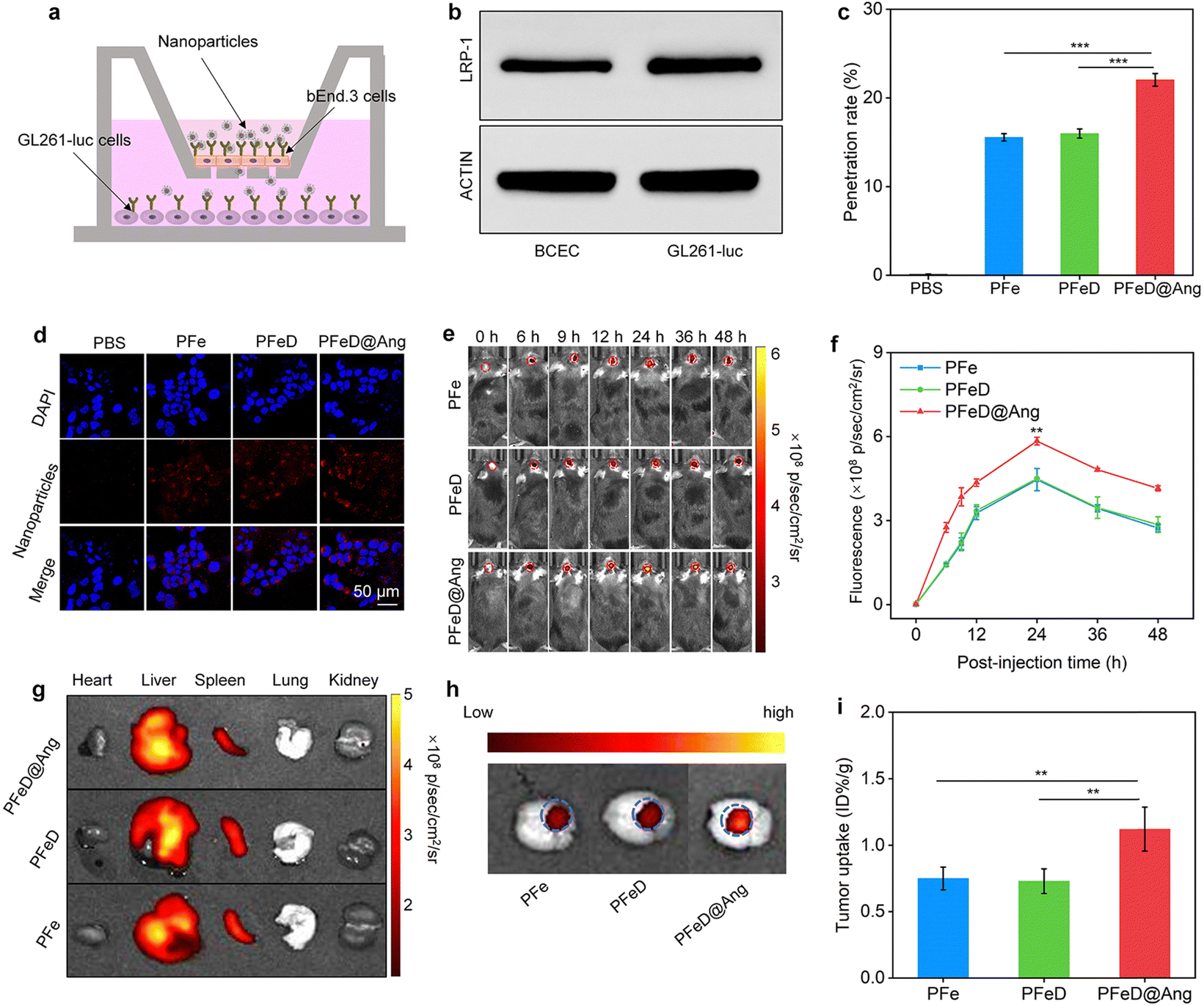
In vitro BBB penetration abilities of various nanoparticles were assessed by replicating a monolayer transwell system using bEnd.3 cells (Fig. 5a). To verify the integrity of our constructed BBB model, we assessed the expression of LRP-1 on the surface of BCEC and GL261-luc cells. Our results showed that LRP-1 was expressed on the surface of both BCEC and GL261-luc cells (Fig. 5b and Fig. S19, ESI†). The penetration rate of PFe and PFeD nanoparticles was approximately 15%, whereas PFeD@Ang achieves nearly 22%. This enhanced penetration of PFeD@Ang was attributed to angiopep-2, which targets the LPR-1 receptor on the surface of bEnd.3 cells (Fig. 5c). In the bottom chambers, GL261-Luc cancer cells exposed to the penetrated PFe, PFeD, and PFeD@Ang nanoparticles exhibited red fluorescence signals, whereas the PBS group showed no fluorescence signals (Fig. 5d).
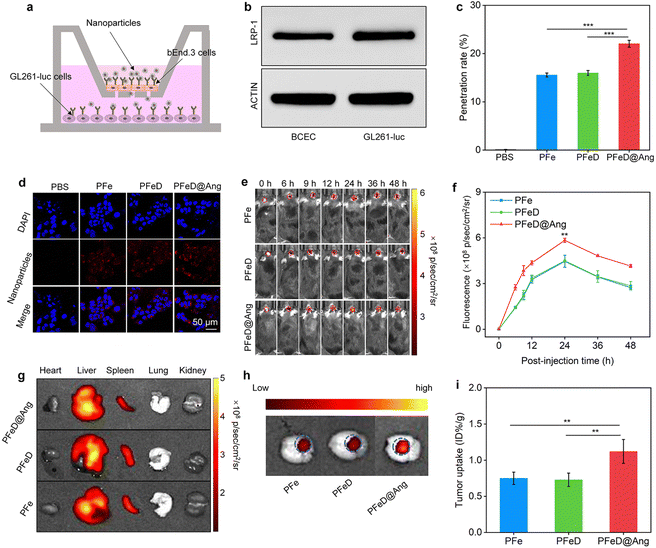 |
| | Fig. 5 Evaluation of the efficacy of BBB crossing. (a) Transwell model schematic illustration. (b) Representative WB images of LRP-1 in BCEC and GL261-luc cancer cells. (c) PFe, PFeD, and PFeD@Ang penetrating efficacy analysis in a transwell culture system (n = 5). (d) Confocal fluorescence imaging of GL261-luc cancer cells in transwell chambers. (e) Fluorescence images of orthotopic gliomas after the injection of PFe, PFeD, and PFeD@Ang (tumors are indicated by red dotted circles). (f) Fluorescence signals were analyzed at glioma tumor sites in mice after the injection of PFe, PFeD, and PFeD@Ang (n = 5). (g) Fluorescence images of organs. (h) Fluorescence images of a tumor. (i) Determination of nanoparticle content in tumors (n = 3). | |
Various nanoparticles were evaluated for their capacity to penetrate the BBB and target the brain in an orthotopic glioma mouse model. Fluorescence signals at the orthotopic glioma tumor sites progressively increased following intravenous injections of PFe, PFeD, and PFeD@Ang, peaking at 24 hours post-injection for all three groups (Fig. 5e). PFeD@Ang exhibited significantly higher accumulation in tumor areas compared to PFe and PFeD. The fluorescence intensities in the brain tumor areas of the PFeD@Ang group were approximately 1.5-fold higher than that in the PFe and PFeD groups (Fig. 5f). 24 hours post-injection, major organs were harvested for biodistribution analysis. Fluorescence signals were detected in the tumor-bearing brains, livers, and spleens of the PFe, PFeD, and PFeD@Ang injected groups (Fig. 5g and Fig. S20, ESI†). As shown in Fig. 5h, PFeD@Ang exhibited better accumulation at the tumor site (indicated by the blue circle) compared to PFeD and PFe (Fig. 5h), with the content of PFeD@Ang in the glioma tumor being approximately 1.5-fold higher than that of PFeD and PFe (Fig. 5i). These results indicate that PFeD@Ang can effectively penetrate the BBB and accumulate at the tumor site, facilitating effective treatment of orthotopic gliomas.
In vivo assessment of anti-glioma effects
Orthotopic GL261-luc GBM-bearing C57BL/6 mouse models were applied here for in vivo antitumor effect evaluation, which has been widely recognized as a representative glioma model.63 Using GL261-luc glioma models, therapeutic nanoparticles and X-ray effects on tumors were evaluated by quantifying tumor growth through bioluminescence signals (BL) (Fig. 6a).64 Orthotopic GL261-luc glioblastoma-bearing C57BL/6 mice were randomly divided into six groups: PBS, PFeD@Ang, PBS + X-ray, PFe + X-ray, PFeD + X-ray, and PFeD@Ang + X-ray. Since the effects of the H2S donor have already been validated at the cellular level by comparing the PFe and PFeD groups, we included the PFeD@Ang + X-ray group as a control to further assess the ability of angiopep-2 to cross the BBB. Compared to the PBS group, BL signals in the PFe + X-ray, PFeD + X-ray, and PFeD@Ang + X-ray groups decreased steadily. PFeD@Ang + X-ray showed the lowest BL signal, indicating the highest anticancer efficacy due to enhanced nanoparticle accumulation in glioma tumors (Fig. 6b and Fig. S21, ESI†). After 14 days of therapy, BL signal intensities in the PBS group were 1.7-, 2.3-, and 7.7-fold higher than in the PFe + X-ray, PFeD + X-ray, and PFeD@Ang + X-ray groups (Fig. 6c). The PFeD@Ang + X-ray group showed the highest anti-glioma efficacy. Mice in the PBS and PBS + X-ray groups lost weight as tumors developed, indicating low treatment efficacy. In contrast, mice in the PFe + X-ray, PFeD + X-ray, and PFeD@Ang + X-ray groups gained weight (Fig. 6d). Examination of animal survival rates revealed that the mice in the PFeD@Ang + X-ray group maintained a 100% survival rate, while all the mice in the PBS, PFeD@Ang, PBS + X-ray, and PFe + X-ray groups perished after 30 days (Fig. 6e). Hematoxylin–eosin (H&E) staining was utilized to evaluate apoptosis/necrosis in tumor locations. Due to their anti-tumor activity, brain tumors in the PFeD + X-ray and PFeD@Ang + X-ray groups appeared smaller than those in the control group (Fig. 6f). Apoptosis/necrosis was observed at tumor sites in the PFe + X-ray, PFeD + X-ray, and PFeD@Ang + X-ray groups, with the PFeD@Ang + X-ray group showing the highest apoptosis/necrosis and the fewest tumor cells. These findings underscored the superior anti-glioma efficacy of the PFeD@Ang + X-ray group.
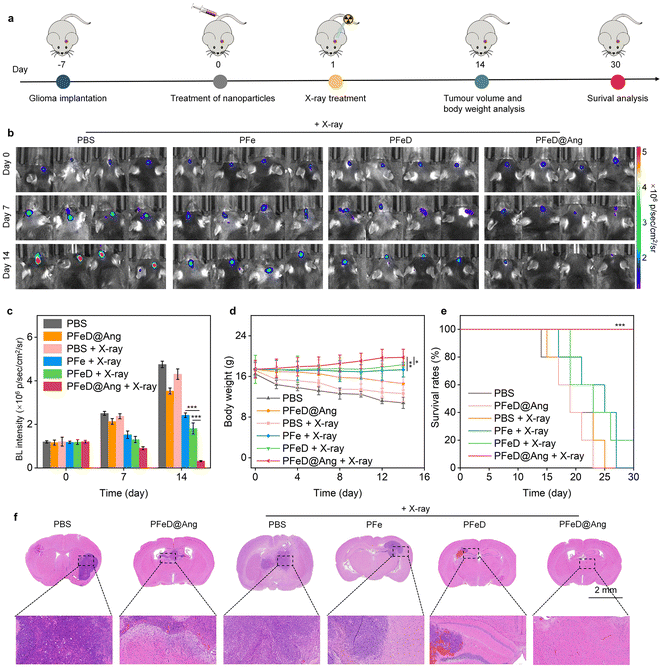 |
| | Fig. 6
In vivo assessment of anti-glioma effects. (a) Treatment plan using orthotopic GL261-luc glioma mouse models. (b) Bioluminescence (BL) pictures of mice with orthotopic GL261-luc glioma on day 0, 7 and 14 following different treatment (n = 4). (c) Luminescence levels in orthotopic GL261-luc glioma mice (n = 4). (d) Body weight changes in glioma-bearing mice (n = 4). (e) Survival curves of treated glioma mice (n = 10). (f) H&E-stained brain slices from orthotopic GL261-luc glioma mice. | |
Antitumor mechanisms in vivo
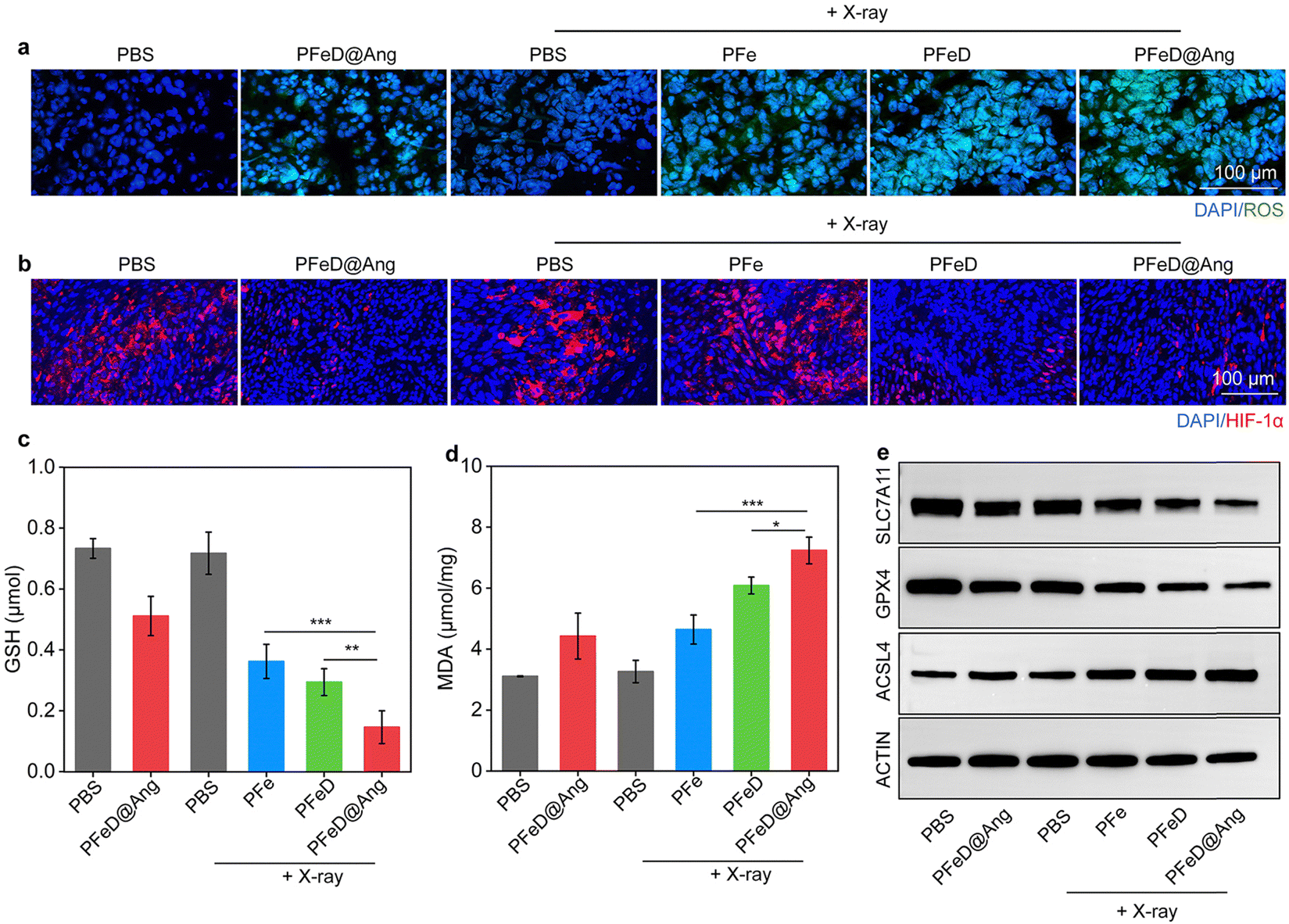
We then studied anti-tumor mechanisms in vivo. As illustrated in Fig. 7a, tumor ROS generation was assessed by using the DHE staining method. The PBS and PBS + X-ray groups generated minimal ROS signals, while the PFeD@Ang and PFe + X-ray groups exhibited weak ROS fluorescence signals. A significant increase in ROS signals was observed in the PFeD + X-ray and the PFeD@Ang + X-ray groups. According to Fig. S22 (ESI†), the fluorescence signal intensity in the PFeD@Ang + X-ray group was 1.3-fold higher compared to the PFeD + X-ray group and 1.7-fold higher compared to the PFe + X-ray group. Immunofluorescence HIF-1α staining was employed to assess tumor hypoxia following treatment (Fig. 7b). There was a significant reduction in the red fluorescence signals of HIF-1α in the PFeD and PFeD@Ang treatment groups, indicating that the nanoparticles effectively alleviated tumor hypoxia through inhibition of cellular respiration. The PFeD@Ang treatment group exhibited the weakest HIF-1α fluorescence signal. The HIF-1α fluorescence signal increased in the PFeD + X-ray and PFeD@Ang + X-ray groups as a result of further oxygen depletion caused by the RDT effect. When HIF-1α fluorescence signal intensity was compared to PBS, the fluorescence signal intensity decreased by 3.1-fold and 2.5-fold in the PFeD + X-ray and PFeD@Ang + X-ray groups, respectively (Fig. S23, ESI†). Intratumoral GSH levels were significantly lower in the PFeD@Ang + X-ray group (0.2 μmol) compared to the PFeD + X-ray (0.3 μmol) and PFe + X-ray (0.4 μmol) groups (Fig. 7c). Furthermore, the PFeD@Ang + X-ray group (7.2 μmol mg−1) showed noticeably higher MDA levels in tumor tissues compared to the PFe + X-ray (4.6 μmol mg−1) and PFeD + X-ray (6.1 μmol mg−1) groups (Fig. 7d). Intratumoral expression levels of GPX4 and SLC7A11 were significantly decreased, but ACSL4 levels were markedly increased in the PFe + X-ray, PFeD + X-ray, and PFeD@Ang + X-ray groups. The lowest GPX4 and SLC7A11 expression and the highest ACSL4 expression were observed in the PFeD@Ang + X-ray group (Fig. 7e and Fig. S24a, ESI†). The expression of SLC7A11 in the PFeD@Ang + X-ray group decreased by about 2.1- and 1.5-fold compared to the PFe + X-ray and PFeD + X-ray groups respectively. The expression of GPX4 in the PFeD@Ang + X-ray group decreased by about 2.7- and 1.9-fold compared to the PFe + X-ray and PFeD + X-ray groups. In contrast, the expression of ACSL4 in the PFeD@Ang + X-ray group increased by about 1.5- and 1.1-fold compared to the PFe + X-ray and PFeD + X-ray groups (Fig. S24b, ESI†). This is attributed to the enhanced targeting and enrichment of PFeD@Ang at the tumor site compared to PFe and PFeD. These results suggested that PFe, PFeD, and PFeD@Ang could successfully induce ferroptosis within the TME.
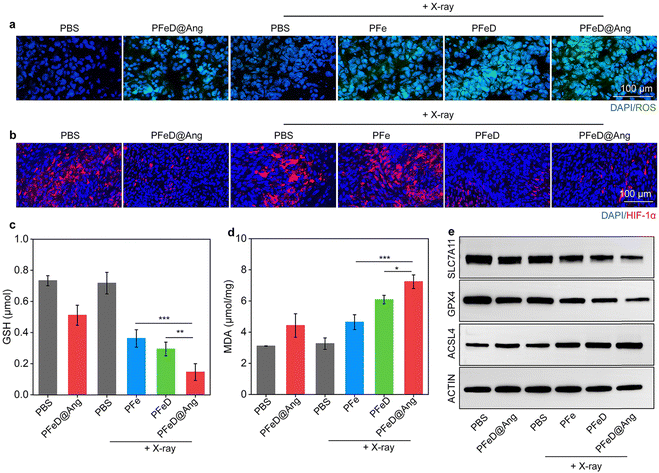 |
| | Fig. 7 Antitumor mechanisms in vivo. (a) ROS signal images in orthotopic glioma tumor tissues. (b) HIF-1α signal images in orthotopic glioma tumor tissues. (c) GSH levels in orthotopic glioma tumors of each group (n = 5). (d) MDA levels in orthotopic glioma tumors (n = 5). (e) Representative WB images of SLC7A11, GPX4, and ACSL4 in tumors after various treatments. | |
H&E staining of PFeD@Ang + X-ray group tissues after 14 days showed no significant differences from the PBS control group (Fig. S25, ESI†). Blood biochemistry and routine indices were similar between the treated and control mice (Fig. S26 and S27, ESI†), confirming the biosafety of PFeD@Ang treatment in vivo.
Conclusion
In this study, we developed a H2S-generating semiconducting polymer nanoparticle (PFeD@Ang) for amplified RDT-ferroptosis therapy of orthotopic glioblastoma. The PFeD@Ang nanoparticles were engineered by incorporating a H2S donor and Fe3+-TA chelates into semiconducting polymer nanoparticles, with surface modification of angiopep-2. These nanoparticles were designed to traverse the BBB and specifically target tumor sites via LRP-1-mediated endocytosis. Upon reaching the TME, the H2S donors within the PFeD@Ang nanoparticles release substantial quantities of H2S, inhibiting mitochondrial respiration and alleviating cellular hypoxia. This action enhances the RDT effect under X-ray irradiation. Concurrently, within the acidic TME, Fe3+ was reduced to Fe2+ by TA, thereby promoting ferroptosis in tumor cells. Additionally, H2S inhibits catalase activity, resulting in increased local H2O2 concentration, which further augments the iron oxidation process. The prepared PFeD@Ang exhibited potent anti-tumor efficacy in an orthotopic glioma mouse model. Overall, the proposed therapeutic strategy has great promise for treating brain tumors and other central nervous system diseases.
Experimental section
Materials
H2S donor was supplied by MedChemExpress (USA). Angiopep-2 (TFFYGGSRGKRNNFKTEEYC) was obtained from China peptide Co., Ltd. (Suzhou, China). Tannic acid (TA) and 4-dimethylaminopyridine (DMAP) were purchased from Aladdin (Shanghai, China). Sigma-Aldrich Corporation (USA) provided poly[2,7-(9,9′-dioctylfluorenyl)-alt-4,7-bis(thiophen-2-yl)benzo-2,1,3-thiadiazole] (PFODBT). Molecular Probes (USA) supplied the cell counting kit-8 (CCK-8) and the single-line-state oxygen sensor green (SOSG). Anti-γ H2A (phosphorylated S139) antibody was provided by Beyotime (Beijing, China). The H2S content assay kit was purchased from Solarbio (Beijing, China). All western blot (WB) antibodies were purchased from Abcam or Biolegend (USA). Ponsure Biotechnology Co., Ltd. (Shanghai, China) and J&K Scientific Co., Ltd. (Beijing, China) provided the remaining chemical reagents.
Evaluation of 1O2 generation
After mixing SOSG with a solution of PFe, PFeD, or PFeD@Ang (3 mL, PFODBT concentration = 15 μg mL−1), the mixture was subjected to 4 Gy of X-ray irradiation. A fluorescence spectrophotometer was used to detect the fluorescence intensities of SOSG to evaluate the generation of 1O2.
Kinetic study of Fe release
PFeD@Ang, PFeD, and PFe were dispersed in 1.0 mL PBS (pH 5.5 and 7.4) to study pH-responsive Fe release, quantified by ICP-OES.
H2S release evaluation
To explore the pH response of the H2S donor, PFeD and PFeD@Ang were dispersed in 1 mL PBS. H2S release was measured using an H2S content assay kit (Solarbio, Beijing) at pH values of 5.5, 6.5, and 7.4.
Evaluation of hydroxyl radical (˙OH) generation
The 3,3′,5,5′-tetramethylbenzidine (TMB) was added to solutions containing H2O2 (100 μM) and PFe, PFeD, or PFeD@Ang (Fe concentration = 20 μg mL−1). After 4 Gy X-ray irradiation, TMB solution absorbance at 650 nm was measured to assess ˙OH generation.
Hemolysis assay
Freshly obtained mouse erythrocytes were co-incubated in PBS containing PFe, PFeD, or PFeD@Ang for 2 hours at room temperature. Hemolysis rates were determined by measuring the absorbance of the samples after centrifugation and incubation. PBS or water were used as the erythrocytes’ negative and positive controls, respectively.
In vitro cell uptake evaluation
GL261-luc cells were treated with PFe, PFeD, and PFeD@Ang (PFODBT concentration = 25 μg mL−1) for 12 hours. The cell nucleus was stained with DAPI following PBS washing. A confocal laser scanning microscope (CLSM, Carl Zeiss LSM 700, Jena, Germany) was used to measure the uptake of PFe, PFeD, and PFeD@Ang by cells. The intensity of the cells’ fluorescence was semi-quantified using Image J.
Assessment of the therapeutic effect of ferroptosis inhibitors
The therapeutic efficacy of the ferroptosis inhibitor ferrostatin-1 was evaluated using the CCK-8 assay and Western blot (WB) analysis. Briefly, GL261-luc cells were treated with PBS, PFeD@Ang, and PFeD@Ang + ferrostatin-1 (PFODBT concentration = 25 μg mL−1), followed by 4 Gy X-ray irradiation and 100 μM H2O2. Subsequently, CCK-8 assays were performed, and protein expression levels were assessed by WB analysis.
Assessment of intracellular ROS levels
GL261-luc cells were incubated with PBS, PFe, PFeD, and PFeD@Ang (25 μg mL−1 PFODBT) in the presence of H2DCFDA (10 μM). Subsequently, the cells underwent 4 Gy X-ray irradiation. Intracellular ROS levels were evaluated by observing cell fluorescence using fluorescence microscopy (Leica DMi8, Germany).
Detection of mitochondrial membrane potential
For a whole 12-hour period, GL261-luc cells were treated with PFe, PFeD, and PFeD@Ang (PFODBT concentration = 25 μg mL−1). Cells were stained for 20 minutes at 37 °C with JC-1 (C2006, Beyotime, China) and DAPI (C1002, Beyotime, China), followed by two washes with JC-1 dye buffer. Fluorescence was then observed using a laser scanning confocal microscope, and fluorescence intensity was quantified using Image J.
The O2 content detection in vitro
GL261-luc cells were treated with PBS, PFe, PFeD, and PFeD@Ang (25 μg mL−1 PFODBT), followed by 4 Gy X-ray irradiation. Post-treatment, cells were stained with an oxygen indicator and Hoechst, and O2 levels were measured via flow cytometry.
Assessment of intracellular hypoxia level
GL261-luc cells were cultured with PBS, PFe, PFeD, and PFeD@Ang (25 μg mL−1 PFODBT) for 12 hours, then exposed to 4 Gy X-ray irradiation. Cells were collected at 8 hours of treatment and immunofluorescence staining was performed with HIF-1α antibody (Abcam, ab179483).
Detection of in vitro LRP-1 expression
GL261-luc cells and BCEC were collected for protein extraction and then rinsed with PBS. WB analysis was employed to determine the LRP-1 expression levels in cells. Antibodies for LRP-1 and ACTIN were purchased from Abcam.
Modeling orthotopic gliomas in mice
All animal experiments followed the guidelines approved by Donghua University's Animal Care and Use Ethics Committee (approval number: DHUEC-STCSM-2021-24). Mice were housed in SPF-level rooms with food and water, at 26 °C, 50% humidity, and a 12-hour light/dark cycle. Orthotopic glioma mouse models were constructed based on our previous work.65
In vivo evaluation of tumor accumulation and biodistribution
C57BL/6 mice bearing orthotopic GL261-luc gliomas were injected intravenously with PFe, PFeD and PFeD@Ang (PFODBT concentration = 300 μg mL−1). In vivo real-time fluorescence imaging using an IVIS spectrum imaging system was used to assess the accumulation of the nanoparticles in the tumors at 0, 6, 9, 12, 24, 36 and 48 hours after injection. To assess biodistribution, orthotopic GL261-luc gliomas mice injected with nanoparticles were euthanized and their organs collected for fluorescence imaging 24 hours later.
In vivo evaluation of drug concentration
Tumor tissue from mice with orthotopic gliomas was homogenized in 1 mL of PBS using a tissue homogenizer. The homogenate was then centrifuged at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 minutes, and the supernatant was collected and filtered through a 0.22 μm filter. The fluorescence intensity of PFODBT was measured using a fluorescence spectrophotometer (RF-6000, SHIMADZU, Japan), and the in vivo nanoparticle uptake was normalized to the percentage of the injected dose per gram of tumor tissue (%ID/g), following established field protocols.
000 rpm for 20 minutes, and the supernatant was collected and filtered through a 0.22 μm filter. The fluorescence intensity of PFODBT was measured using a fluorescence spectrophotometer (RF-6000, SHIMADZU, Japan), and the in vivo nanoparticle uptake was normalized to the percentage of the injected dose per gram of tumor tissue (%ID/g), following established field protocols.
In vivo evaluation of anti-glioma efficacy
GL261-luc glioma-bearing C57BL/6 mice were divided into six groups randomly: PBS, PFeD@Ang, PBS + X-ray, PFe + X-ray, PFeD + X-ray, and PFeD@Ang + X-ray. Mice were intravenously administered with PBS, PFe, PFeD, or PFeD@Ang (300 μg mL−1 and 200 μL per mouse, respectively) via the tail vein. 24 hours later, mice with orthotopic gliomas tumors were treated with 6 Gy of X-ray irradiation targeting tumor sites. Through BL imaging on the IVIS imaging system, the growth of tumors containing orthotopic gliomas was monitored. We investigated the apoptosis/necrosis of tumor cells in mouse brains stained with H&E after therapy. The survival rates of each group were evaluated over a 30-day period.
In vivo ROS level evaluation
C57BL/6 mice with orthotopic GL261-luc gliomas were injected with PBS, PFe, PFeD, and PFeD@Ang (PFODBT concentration = 300 μg mL−1, 200 μL per mouse), and the tumor sites of the gliomas were irradiated with X-rays. To measure ROS levels, GL261-luc glioma tumors from mice were cryosectioned and stained with appropriate antibodies to analyze ROS levels.
In vivo ferroptosis effect evaluation
Following intravenous injection via the tail vein of PBS, PFe, PFeD, and PFeD@Ang (PFODBT concentration = 300 μg mL−1, 0.2 mL), mice were exposed to 6 Gy X-ray irradiation 24 hours later. Mice were euthanized and orthotopic glioma tumors were removed. The amounts of GSH and MDA in the tumors were assessed using MDA and GSH assay kits. WB analysis was employed to determine ACSL4, GPX4 and SLC7A11 expression levels in the tumors.
Evaluation of hypoxia in tumors
Mice inoculated with GL261-luc tumors were injected with PBS or nanoparticles, and then irradiated with X-rays (6 Gy). Tumors were collected from mice after 12 hours of treatment, and HIF-1α expression levels were detected using immunofluorescence staining.
Biosafety assessment
The organs of orthotopic glioma tumor-bearing mice were excised for histological morphology evaluation using H&E staining. Blood samples were taken from treated mice for routine parameter and blood biochemistry assessments.
Statistical analysis
To evaluate statistical differences between groups, a two-tailed unpaired Student's t-test was used. Significance levels were denoted as * for p < 0.05, ** for p < 0.01, and *** for p < 0.001. Statistical analysis was performed using GraphPad Prism 8. In the figure captions, sample sizes (n) are provided for all experiments.
Author contributions
W. T., J. L., Q. Z. and J. L. conceived the idea, designed the experiments and supervised the research. A. Z., S. S., J. H., Z. S. and Y. L. conducted the experiments, analysed the data and drew the scheme. A. Z., S. S. and J. H. drafted the manuscript. W. T., J. L., Q. Z. and J. L. revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
The authors declare no competing financial interests.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52203155 and 22308120), and the Science and Technology Commission of Shanghai Municipality (22490760700, 22ZR1401100 and 20DZ2254900).
References
- K. Aldape, K. M. Brindle, L. Chesler, R. Chopra, A. Gajjar, M. R. Gilbert, N. Gottardo, D. H. Gutmann, D. Hargrave and E. C. Holland, Nat. Rev. Clin. Oncol., 2019, 16, 509–520 CrossRef CAS PubMed.
- J. Yang, L. Xu, Y. Ding, C. Liu, B. Wang, Y. Yu, C. Hui, S. Ramakrishna, J. Zhang and Y. Long, Adv. Fiber Mater., 2023, 5, 209–222 CrossRef CAS.
- S. Pinel, N. Thomas, C. Boura and M. Barberi-Heyob, Adv. Drug Delivery Rev., 2019, 138, 344–357 CrossRef CAS.
- C. Qiao, J. Yang, Q. Shen, R. Liu, Y. Li, Y. Shi, J. Chen, Y. Shen, Z. Xiao, J. Weng and X. Zhang, Adv. Mater., 2018, 30, 1705054 CrossRef PubMed.
- Y. Zou, X. Sun, Y. Wang, C. Yan, Y. Liu, J. Li, D. Zhang, M. Zheng, R. S. Chung and B. Shi, Adv. Mater., 2020, 32, 2000416 CrossRef CAS.
- P. Y. Wen and D. A. Reardon, Nat. Rev. Neurol., 2016, 12, 69–70 CrossRef CAS PubMed.
- M. Weller, T. Cloughesy, J. R. Perry and W. Wick, Neuro-Oncology, 2012, 15, 4–27 CrossRef.
- C. N. de Leeuw and M. A. Vogelbaum, Neuro-Oncology, 2018, 21, 179–188 CrossRef.
- M. Lim, Y. Xia, C. Bettegowda and M. Weller, Nat. Rev. Clin. Oncol., 2018, 15, 422–442 CrossRef CAS.
- M. Touat, A. Idbaih, M. Sanson and K. L. Ligon, Ann. Oncol., 2017, 28, 1457–1472 CrossRef CAS.
- D. Furtado, M. Björnmalm, S. Ayton, A. I. Bush, K. Kempe and F. Caruso, Adv. Mater., 2018, 30, 1801362 CrossRef PubMed.
- A. Galstyan, J. L. Markman, E. S. Shatalova, A. Chiechi, A. J. Korman, R. Patil, D. Klymyshyn, W. G. Tourtellotte, L. L. Israel and O. Braubach, Nat. Commun., 2019, 10, 3850 CrossRef CAS.
- K. Hynynen, N. Engl. J. Med., 2024, 390, 82–85 CrossRef CAS.
- J. Kuang, Z.-Y. Rao, D.-W. Zheng, D. Kuang, Q.-X. Huang, T. Pan, H. Li, X. Zeng and X.-Z. Zhang, ACS Nano, 2023, 17, 13333–13347 CrossRef CAS.
- L. Su, K. Zhu, X. Ge, Y. Wu, J. Zhang, G. Wang, D. Liu, L. Chen, Q. Li, J. Chen and J. Song, Nano Lett., 2024, 24, 3727–3736 CrossRef CAS PubMed.
- M. Zuckermann, C. He, J. Andrews, A. Bagchi, R. Sloan-Henry, B. Bianski, J. Xie, Y. Wang, N. Twarog, A. Onar-Thomas, K. J. Ernst, L. Yang, Y. Li, X. Zhu, J. K. Ocasio, K. M. Budd, J. Dalton, X. Li, D. Chepyala, J. Zhang, K. Xu, L. Hover, J. T. Roach, K. C.-H. Chan, N. Hofmann, P. J. McKinnon, S. M. Pfister, A. A. Shelat, Z. Rankovic, B. B. Freeman, J. Chiang, D. T. W. Jones, C. L. Tinkle and S. J. Baker, Mol. Cancer, 2024, 23, 123 CrossRef CAS.
- M. A. Knoll, R. Jagsi and K. Rosenzweig, JAMA Oncol., 2019, 5, 442 CrossRef PubMed.
- G. Ratnayake, A. L. Bain, N. Fletcher, C. B. Howard, K. K. Khanna and K. J. Thurecht, Cancer Lett., 2018, 439, 14–23 CrossRef CAS.
- E. T. Vitti and J. L. Parsons, Cancers, 2019, 11, 946 CrossRef CAS.
- S. Goel, D. Ni and W. Cai, ACS Nano, 2017, 11, 5233–5237 CrossRef CAS.
- G. Song, L. Cheng, Y. Chao, K. Yang and Z. Liu, Adv. Mater., 2017, 29, 1700996 CrossRef PubMed.
- H. Wei, J. Gu, F. Ren, L. Zhang, G. Xu, B. Wang, S. Song, J. Zhao, S. Dou and Y. Li, Small, 2021, 17, 2100446 CrossRef CAS.
- C. Zhang, X. Wang, X. Dong, L. Mei, X. Wu, Z. Gu and Y. Zhao, Biomaterials, 2021, 276, 121023 CrossRef CAS PubMed.
- Y. Li, M. Jiang, Z. Deng, S. Zeng and J. Hao, Adv. Sci., 2021, 8, 2004391 CrossRef CAS.
- Y. Yang, Y. Lin, D. Di, X. Zhang, D. Wang, Q. Zhao and S. Wang, J. Colloid Interface Sci., 2017, 508, 323–331 CrossRef CAS PubMed.
- T. Liu, S. Shi, C. Liang, S. Shen, L. Cheng, C. Wang, X. Song, S. Goel, T. E. Barnhart, W. Cai and Z. Liu, ACS Nano, 2015, 9, 950–960 CrossRef CAS PubMed.
- X.-D. Zhang, D. Wu, X. Shen, J. Chen, Y.-M. Sun, P.-X. Liu and X.-J. Liang, Biomaterials, 2012, 33, 6408–6419 CrossRef CAS.
- L. Cheng, D. Jiang, A. Kamkaew, H. F. Valdovinos, H.-J. Im, L. Feng, C. G. England, S. Goel, T. E. Barnhart, Z. Liu and W. Cai, Adv. Funct. Mater., 2017, 27, 1702928 CrossRef.
- Y. Liu, K. Ai, J. Liu, M. Deng, Y. He and L. Lu, Adv. Mater., 2013, 25, 1353–1359 CrossRef CAS PubMed.
- H. Yu, Y. Yang, T. Jiang, X. Zhang, Y. Zhao, G. Pang, Y. Feng, S. Zhang, F. Wang, Y. Wang, Y. Wang and L. W. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 27536–27547 CrossRef CAS PubMed.
- J. Bailleul, Y. Ruan, L. Abdulrahman, A. J. Scott, T. Yazal, D. Sung, K. Park, H. Hoang, J. Nathaniel, F.-I. Chu, D. Palomera, A. Sehgal, J. E. Tsang, D. A. Nathanson, S. Xu, J. O. Park, J. ten Hoeve, K. Bhat, N. Qi, H. I. Kornblum, D. Schaue, W. H. McBride, C. A. Lyssiotis, D. R. Wahl and E. Vlashi, Neuro-Oncology, 2023, 25, 1989–2000 CrossRef CAS PubMed.
- Y. Liu, J. Zhang, J. Du, K. Song, J. Liu, X. Wang, B. Li, R. Ouyang, Y. Miao, Y. Sun and Y. Li, Acta Biomater., 2021, 129, 280–292 CrossRef CAS.
- Z. Cao, X. Liu, W. Zhang, K. Zhang, L. Pan, M. Zhu, H. Qin, C. Zou, W. Wang, C. Zhang, Y. He, W. Lin, Y. Zhang, D. Han, M. Li and J. Gu, ACS Nano, 2023, 17, 23746–23760 CrossRef CAS.
- B. Li, X. Chen, W. Qiu, R. Zhao, J. Duan, S. Zhang, Z. Pan, S. Zhao, Q. Guo, Y. Qi, W. Wang, L. Deng, S. Ni, Y. Sang, H. Xue, H. Liu and G. Li, Adv. Sci., 2022, 9, 2105451 CrossRef CAS PubMed.
- Y. Zhang, X. Fu, J. Jia, T. Wikerholmen, K. Xi, Y. Kong, J. Wang, H. Chen, Y. Ma, Z. Li, C. Wang, Q. Qi, F. Thorsen, J. Wang, J. Cui, X. Li and S. Ni, ACS Appl. Mater. Interfaces, 2020, 12, 43408–43421 CrossRef CAS.
- Y. Zhang, K. Xi, X. Fu, H. Sun, H. Wang, D. Yu, Z. Li, Y. Ma, X. Liu, B. Huang, J. Wang, G. Li, J. Cui, X. Li and S. Ni, Biomaterials, 2021, 278, 121163 CrossRef CAS PubMed.
- Y. Zhu, X. Niu, C. Ding, Y. Lin, W. Fang, L. Yan, J. Cheng, J. Zou, Y. Tian, W. Huang, W. Huang, Y. Pan, T. Wu, X. Chen and D. Kang, Adv. Sci., 2024, 11, 2402516 CrossRef CAS.
- M. Zhou, M. Yuan, Y. Jin, Q. Zhou, Y. Yu, J. Li, X. Dai, X. Zheng, H. Yu and W. Wang, Adv. Funct. Mater., 2023, 33, 2303899 CrossRef CAS.
- G. Lei, L. Zhuang and B. Gan, Cancer Cell, 2024, 42, 513–534 CrossRef CAS PubMed.
- S. J. Dixon, K. M. Lemberg, M. R. Lamprecht, R. Skouta, E. M. Zaitsev, C. E. Gleason, D. N. Patel, A. J. Bauer, A. M. Cantley and W. S. Yang, Cell, 2012, 149, 1060–1072 CrossRef CAS.
- Y. Yang, P. Wang, R. Shi, Z. Zhao, A. Xie, Y. Shen and M. Zhu, Chem. Eng. J., 2022, 441, 136042 CrossRef CAS.
- H. Liu, M. Mu, Y. Hou, Y. Gong, C. Wang, G. Ma, K. Guo, L. Ma and X. Sun, Adv. Funct. Mater., 2024, 34, 2401370 CrossRef CAS.
- Y.-W. Mao, K.-F. Chu, P. Song, A.-J. Wang, T. Zhao and J.-J. Feng, Biomater. Adv., 2024, 162, 213919 CrossRef CAS.
- T. Yang, M. Zhou, M. Gao, W. Qin, Q. Wang, H. Peng, W. Yao, L. Qiao and X. He, Small, 2023, 19, 2205692 CrossRef CAS.
- A. Bansal and M. C. Simon, J. Cell Biol., 2018, 217, 2291–2298 CrossRef CAS.
- M. A. Badgley, D. M. Kremer, H. C. Maurer, K. E. DelGiorno, H.-J. Lee, V. Purohit, I. R. Sagalovskiy, A. Ma, J. Kapilian, C. E. M. Firl, A. R. Decker, S. A. Sastra, C. F. Palermo, L. R. Andrade, P. Sajjakulnukit, L. Zhang, Z. P. Tolstyka, T. Hirschhorn, C. Lamb, T. Liu, W. Gu, E. S. Seeley, E. Stone, G. Georgiou, U. Manor, A. Iuga, G. M. Wahl, B. R. Stockwell, C. A. Lyssiotis and K. P. Olive, Science, 2020, 368, 85–89 CrossRef CAS.
- K. Bersuker, J. M. Hendricks, Z. Li, L. Magtanong, B. Ford, P. H. Tang, M. A. Roberts, B. Tong, T. J. Maimone, R. Zoncu, M. C. Bassik, D. K. Nomura, S. J. Dixon and J. A. Olzmann, Nature, 2019, 575, 688–692 CrossRef CAS.
- W. Sun, C. Zhu, J. Song, S.-C. Ji, B.-P. Jiang, H. Liang and X.-C. Shen, Adv. Healthcare Mater., 2023, 12, 2300385 CrossRef CAS.
- M. Wan, Z. Liu, T. Li, H. Chen, Q. Wang, T. Chen, Y. Tao and C. Mao, Angew. Chem., Int. Ed., 2021, 60, 16139–16148 CrossRef CAS.
- H. Zheng, B. Ma, Y. Shi, Q. Dai, D. Li, E. Ren, J. Zhu, J. Liu, H. Chen, Z. Yin, C. Chu, X. Wang and G. Liu, Chem. Eng. J., 2021, 406, 126888 CrossRef CAS.
- O. Kabil and R. Banerjee, J. Biol. Chem., 2010, 285, 21903–21907 CrossRef CAS PubMed.
- R. Yin, C. Fan, J. Sun and C. Shang, Chem. Eng. J., 2018, 336, 587–594 CrossRef CAS.
- H.-f Yan, T. Zou, Q.-z Tuo, S. Xu, H. Li, A. A. Belaidi and P. Lei, Signal Transduction Targeted Ther., 2021, 6, 49 CrossRef CAS.
- S. Lara, A. Perez-Potti, L. M. Herda, L. Adumeau, K. A. Dawson and Y. Yan, ACS Nano, 2018, 12, 4930–4937 CrossRef CAS PubMed.
- H. Zhang, W. Jiang, T. Song, M. Song, S. Liu, J. Zhou, H. Cheng and Y. Ding, Adv. Sci., 2024, 11, 2309314 CrossRef CAS.
- Z. Zhu, Y. Zhai, Y. Hao, Q. Wang, F. Han, W. Zheng, J. Hong, L. Cui, W. Jin, S. Ma, L. Yang and G. Cheng, J. Extracell. Vesicles., 2022, 11, e12255 CrossRef CAS PubMed.
- A. Kadari, D. Pooja, R. H. Gora, S. Gudem, V. R. M. Kolapalli, H. Kulhari and R. Sistla, Eur. J. Pharm. Biopharm., 2018, 132, 168–179 CrossRef CAS.
- S. Habib and M. Singh, Polymers, 2022, 14, 712 CrossRef CAS.
- B. Zhang, H. Liu, Y. Wang, Y. Zhang and J. Cheng, J. Mater. Chem. B, 2023, 11, 9685–9696 RSC.
- K. Shao, R. Huang, J. Li, L. Han, L. Ye, J. Lou and C. Jiang, J. Controlled Release, 2010, 147, 118–126 CrossRef CAS.
- Z. Wang, Y. Guo, Y. Fan, J. Chen, H. Wang, M. Shen and X. Shi, Adv. Mater., 2022, 34, 2107009 CrossRef CAS PubMed.
- M. Li, Y. Liu, Y. Zhang, N. Yu and J. Li, Adv. Sci., 2023, 10, 2305150 CrossRef CAS.
- T. Szatmári, K. Lumniczky, S. Désaknai, S. Trajcevski, E. J. Hídvégi, H. Hamada and G. Sáfrány, Cancer Sci., 2006, 97, 546–553 CrossRef.
- A. Zhu, W. Tu, M. Ding, Y. Zhang, J. Liu, X. Chen, L. Wang, Y. Liu and J. Li, Chem. Eng. J., 2024, 497, 154652 CrossRef CAS.
- M. Ding, A. Zhu, Y. Zhang, J. Liu, L. Lin, X. Wang and J. Li, Nano Today, 2024, 57, 102398 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *f and
Jingchao
Li
*f and
Jingchao
Li