
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual organic structure-directing agents in zeolite synthesis: cooperation or competition?
Amirhossein
Javdani
 ,
Juna
Bae
,
Gleb
Ivanushkin
and
Michiel
Dusselier
*
,
Juna
Bae
,
Gleb
Ivanushkin
and
Michiel
Dusselier
*
Center for Sustainable Catalysis and Engineering (CSCE), KU Leuven, 3001 Leuven, Belgium. E-mail: michiel.dusselier@kuleuven.be
First published on 3rd April 2025
Abstract
Organic structure-directing agents (OSDAs) play a vital role in the structural arrangement and compositional diversity of zeolites. The synthesis procedure and properties of zeolites can be improved through the “dual-OSDA” approach, which involves cooperation and/or competition of different OSDAs in the synthesis media. Two OSDAs achieving results that neither of the OSDAs can deliver on its own is referred to as the “cooperative OSDA” approach. In this manner, one can enhance zeolite properties by stabilizing different parts of the framework and altering the synthesis outcome, such as acidity and/or Al distribution. However, cooperation can easily be disrupted, and one of the challenges in dual-OSDA zeolite synthesis is determining the conditions under which OSDA molecules can function in harmony to affect zeolite properties and control phase selection. On the other hand, competition does not always result in negative outcomes (e.g., intergrowth materials). In this review, we discussed the importance of cooperative dual-OSDA synthesis in certain cases, explained the outcomes of this approach, and described the different behaviors key to cooperative systems.
 Amirhossein Javdani | Amirhossein Javdani is a PhD researcher in the Center for Sustainable Catalysis and Engineering, under the supervision of Prof. Michiel Dusselier at KU Leuven (Belgium). He earned his BSc (2015) in Chemical Engineering from Tehran Polytechnique (Iran) and afterward completed his master studies in Chemical Engineering with a sub-discipline of process design at Noshirvani University of Technology (Iran, 2019). He joined the Dusselier group in 2021 as a doctoral student for the European Project “Z-EURECA”, which focuses on unconventional routes for zeolite synthesis through reactor development. |
 Juna Bae | Juna Bae received her PhD in Chemical Engineering from POSTECH (South Korea) in 2020 under the guidance of Prof. Suk Bong Hong, studying the synthesis of zeolites with novel framework structures and/or compositions using simple organic structure-directing agents. In 2020–2021, she stayed with the same group and focused her research on the effect of fluoride concentration in the initial synthesis mixture on zeolite crystallization. Since 2021, she has been a postdoctoral researcher in the Dusselier group, working on the synthesis of small- and medium-pore zeolites with controlled acid sites. |
 Gleb Ivanushkin | Gleb Ivanushkin is a postdoctoral researcher at the Center for Sustainable Catalysis and Engineering at KU Leuven (Belgium). He obtained his MSc from Moscow State University (Russia) in 2018 and completed his PhD in 2023 under the supervision of Prof. Michiel Dusselier at KU Leuven, focusing on non-conventional zeolite synthesis. During his PhD, he contributed to pioneering research on the Electro-Assisted zeolite Synthesis approach. Currently, his work focuses on advanced characterization of microporous materials, employing operando and synchrotron-based techniques to explore their structural and functional properties for environmental catalysis. |
 Michiel Dusselier | Prof. Michiel Dusselier obtained his PhD (Bioscience Engineering Catalytic Technology) at KU Leuven, Belgium and did postdoctoral work at Caltech, USA. He is now an associate professor at KU Leuven and cofounded the Center for Sustainable Catalysis and Engineering. His focus is on zeolite synthesis (interzeolite conversion and nonconventional routes) and reactor design for such syntheses (subject of an ERC grant), degradable plastics, heterogeneous catalysis, and CO2 conversion. Recently, he received the Young Researcher Award of the International Zeolite Association (2022) and the N3C Award of the Dutch Catalysis Society (2023). |
Wider impact“Cooperative-competitive” behavior of organic structure-directing agents in zeolite synthesis was addressed and studied thoroughly in dual-OSDA systems. This work will be of interest to researchers in the field of zeolite science and, more broadly to specialists in (micro)porous material synthesis and will provide insights into certain ambiguous aspects of in/organic interactions in the field of material design. The concept of “cooperation and competition” has not been previously addressed, and this study will provide answers to some critical questions. We investigated “real cooperation” cases to understand the empirical question: why do two OSDAs sometimes not work together? In the end, we presented challenges related to the field, some guidelines on how to assess cooperation, and an outlook for future research in this field. |
Introduction
Zeolites are crystalline microporous materials, composed of TO4 (T = Si, Al) tetrahedral units, with a wide range of industrial applications in catalysis, adsorption, and separation.1,2 Organic structure-directing agent (OSDA) molecules play a vital role in zeolite science. They function as an internal skeleton (although flexible, often charged, and thus not only space-filling in nature), on top of which, by the interaction of inorganic precursors, a different topology can be synthesized via a hydrothermal method.3,4 It is well known that the size, shape, flexibility, hydrophilicity, and charge of the employed OSDAs not only direct the framework topology of the produced zeolite but also alter zeolite crystallization kinetics.5–8 Besides acting as templates (space filling with a tight fit), OSDAs also function as more loose space fillers and charge balancers within zeolite structures, making them multi-functional and unique in zeolite science.9,10 The use of organics in zeolite synthesis can sometimes be considered an environmental burden (in combustion calcination with treatment of the gases or in their production), and so OSDA-free synthesis has also been extensively considered.11–13 However, such approaches – which often need a large dose of seed crystals – are often limited in their phase selectivity or in terms of the final product's composition.14 OSDAs are still trending significantly due to their efficiency and better control over the zeolite synthesis outcome in terms of zeolite structure, acid site density, and crystal morphology.Besides the conventional method using a single OSDA, combinations of OSDAs have also been employed in zeolite synthesis. The “dual-OSDA” systems are very compelling to study due to the cooperative or competitive behavior of OSDAs. Cooperation of OSDAs could lead to a certain topology (which otherwise is not achievable15), reduce the synthesis cost,16 fasten the synthesis,17 or alter the Al distribution/siting,7 while the competition of OSDAs may cause phase selectivity issues or intergrowth of different structures.18 The charge density mismatch (CDM) approach is a practical example of the cooperation of OSDAs, where they act as charge balancers and make use of a temporary mismatch in the synthesis media, which leads to a particular structure that could not be obtained with a single-OSDA system.19 On the other hand, novel intergrowth zeolites, which have shown superior catalytic activity, could be obtained by controlling the competition of OSDAs in synthesis media.20
There have been several review articles on the organic and inorganic SDAs and their effect on the characterization and catalytic performance of zeolites.16,21–24 In this concise review, we focus on dual-OSDA systems to address the “cooperative-competitive” role of OSDAs in zeolite synthesis. Here, we also investigate “real cooperation” cases attempting to answer an empirical question: why do two OSDAs sometimes not work together harmoniously? In the end, we discuss challenges related to controlling the cooperation of OSDAs and offer an outlook for future research in this field.
Dual-OSDA approach: cooperation
What is the definition of the “dual-OSDA” approach?
When it comes to zeolite synthesis, it's crucial to distinguish between “dual-OSDA” and other (more broadly definable) “dual-organic” approaches. In the former, both organic compounds play a role in directing the synthesis towards a particular phase or structure with a specific feature. Conversely, the latter approach can involve using a mesopore director or morphology modifier (e.g., surfactants) to achieve a hierarchical or mesoporous structure.25–29 This kind of dual-organic synthesis isn't discussed here, as one of the organic compounds doesn't contribute to the direction of synthesis towards a specific phase or alter the aluminum location within a phase.Various zeolite structures (such as CHA, FER, LTA, MOR, and UFI) have been synthesized using the dual-OSDA approach. The main idea behind this approach is the collaboration of organics to generate different types of secondary building units (SBUs) in a zeolite structure.16 However, this is not the only explanation. After reviewing the literature (Table 1), it can be concluded that there are four main motivations for using dual OSDAs. These motivations (i to iv) are systematically coming back in Table 1 while select examples are explaining the principle here:
| Ref. | OSDA #1 | OSDA #2 | Topology | Remarks |
|---|---|---|---|---|
| 30 |
|
|
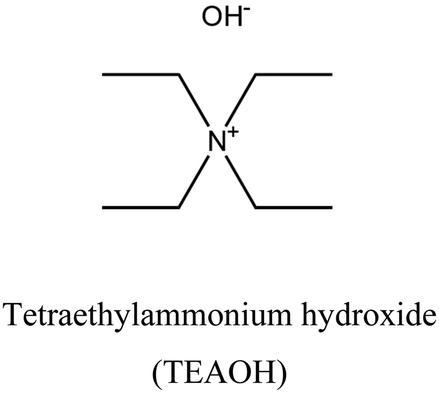
UFI BPH | i: UFI and BPH topologies with Si/Al ratio lower than 10 have been discovered only through the cooperation of OSDAs. |
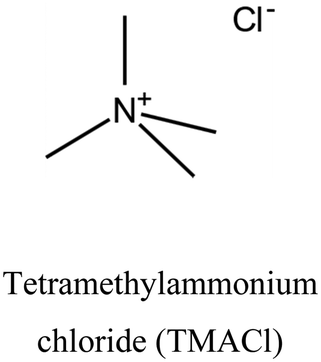
| 31 | LTA | i: LTA topology with Si/Al ratio of 3.3 could be obtained through the cooperation of OSDAs. It is interesting to note that TEA+ and TMA+ are responsible for building different composite building units (CBUs) of LTA topology. | ||
| 32 and 33 | UFI | i: UFI structure with less impurities could be obtained through the cooperation of TEA+/TMA+, while a formation pathway was proposed for this system via a mechanistic study. The molar composition of OSDAs proved to be effective in reducing impurity phases like SOD in this system. | ||
| 19 and 34 | UFI BPH | i: The impact of temperature on the cooperative role of OSDAs during the nucleation and crystal growth stages of UFI and BPH frameworks has been investigated, suggesting temperature-driven control on the cooperation and phase selectivity. | ||
| 35 | BEA | i: BEA topology could be synthesized via a dual-OSDA route. The study concluded that an optimum temperature should be employed to avoid “TEA+ decomposition” in the TEA+/TMA+ system. Nanocrystalline beta zeolite crystallized at 155 °C, whereas heating at higher temperatures (ranging from 160 to 200 °C) gave no crystalline products due to the instability of OSDAs. | ||
| 36 and 37 | UFI | i: Changing synthetic variables (e.g., starting Si/Al, temperature, and alkalinity) can shift the system toward different modes of cooperation (as discussed in the text below): “Co-construction”, where both OSDAs are incorporated in the structure, and/or “charge balancing”, where one OSDA is not incorporated but provides a necessary situation for other OSDA toward crystallization. | ||
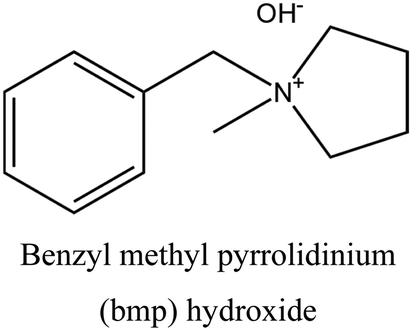
| 38 |
|
|
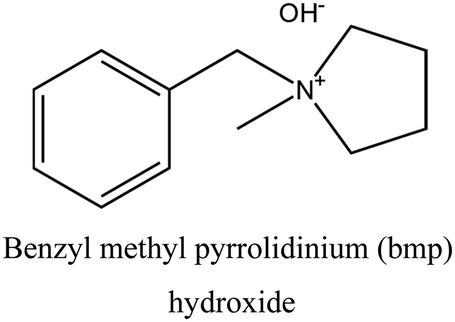
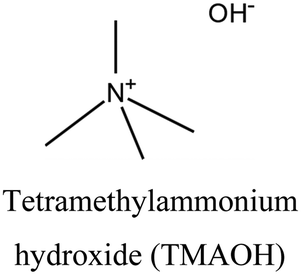
FER | i: FER topology could be obtained through the cooperative role of TMA+ and a bulky compound (bmp), as shown in Fig. 2. Both OSDAs are necessary during the nucleation of FER topology, and the final structure will not form without either of them. |
| 39 |

|
FER | i: Layered ferrierite-like material could be obtained through the cooperation of bulky molecules like bmp and quinuclidine. When bmp was employed alone as an OSDA, a mixture of phases was obtained, and the crystallization rate was rather slow. The quinuclidine molecule is accommodated inside the ferrierite cages, while the bulkier bmp is located in the 10-membered-ring zeolite channels. | |
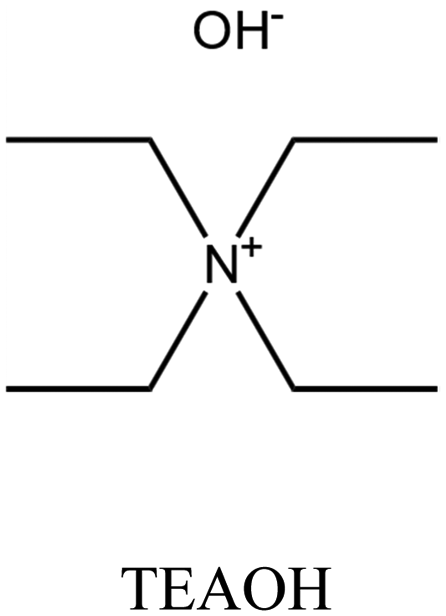
| 40 and 41 |
|

|
MOR | ii: Seed-induced high silica MOR zeolite (Si/Al: 37) was synthesized in the absence of fluoride media by using a dual-templating method. Both TEAOH and HMI participated in the crystallization of MOR. Synthesizing with a single OSDA (TEAOH or HMI) would result in MOR with lower Si/Al (<13), an impurity, or another topology. |

|
||||
|
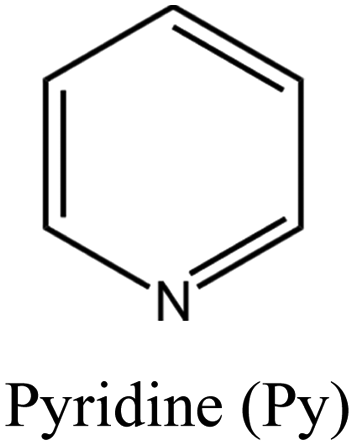
ii: High silica MOR zeolite (Si/Al: 30) could be obtained through the cooperation of TEA+ and different compounds (i.e., hexamethyleneimine, cyclohexylamine, pyridine, and piperidine). Bulky TEA+ cation and neutral heterocycle molecules served as space filler and cooperatively squeezed the occupation of sodium cation. This cooperative behavior is strongly influenced by alkalinity level and can be negatively impacted if it exceeds the optimal range. | |||

|
||||
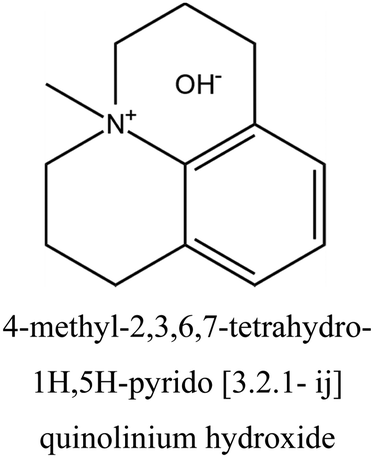
| 42 |

|
|
LTA | ii: Pure silica LTA (named ITQ-29) could be obtained using a supramolecular assembly of a quinolinium derivative OSDA. This supramolecule self-assembles and facilitates the formation of large cavities in the structure. Moreover, it could cooperate with TMA+, resulting in an acid catalyst with LTA topology. TMA+ was incorporated into the structure and compensated the framework charges generated by introducing Al. |
| 43–47 |

|
FER | ii: It was found that the combination of TMA+ and the mentioned OSDAs allows control of the aluminum siting and, subsequently, the density of acid sites in the ferrierite zeolite. Tailoring the acidity of FER zeolite is possible by choosing a suitable cooperative system. For instance, using Pyr as a co-OSDA results in the selective population of acid sites in the 8-membered ring channels, whereas using HMI generates FER zeolites with an increased concentration of acid sites in the 10-membered ring channels. | |
|
||||

|
||||
| 48 |

|

|
FAU | ii: It was found that the cooperation of OSDAs is responsible for lowering the aluminum content of FAU (increasing the Si/Al ratio from 3 to 6) and tuning its acid site distribution. The addition of 15-Crown-5 (CE) as a cooperative OSDA greatly promotes the fraction of trans choline cation (Ch+) in the sod cage at a fixed Ch+ amount in each unit cell. Meanwhile, CE and gauche Ch+ fully occupy the supercage. As a result, less Na+ is found in the sod (and others), which leads to less Al content and, thus, higher Si/Al ratios. This high silica FAU (>6) could not be synthesized using a single OSDA. |
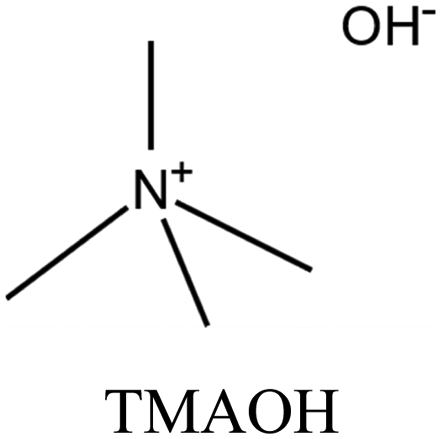
| 49 |
|

|
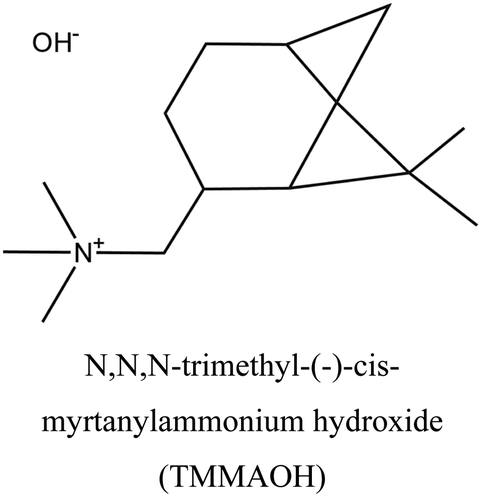
CON | iii: The synthesis of CON zeolite, a promising catalyst for methanol to olefins reactions, can be achieved using highly expensive TMMAOH as an OSDA. Substituting it with commercially available TEAOH would similarly result in CON framework but with a lower yield and longer synthesis time. Hence, the synthesis economy could improve, but there must be a minimum amount of TMMAOH present in the synthesis. Two main routes were investigated in this study to simultaneously reduce both the synthesis time and OSDA costs: replacing the majority of TMMAOH with NaOH or TEAOH. NaOH-TMMAOH system led to a layered phase, while the intermediate addition of TEAOH resulted in CON zeolite with low TMMAOH usage (up to 80% reduced compared to the parent). |
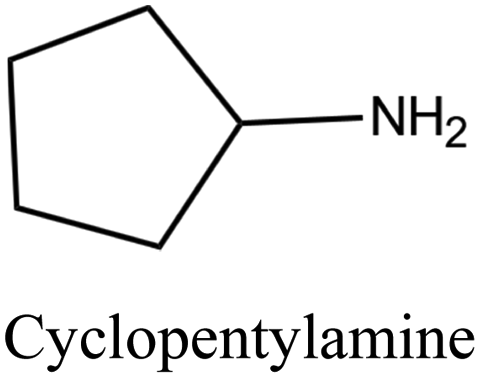
| 50 |

|
|
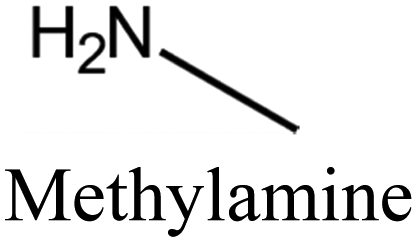
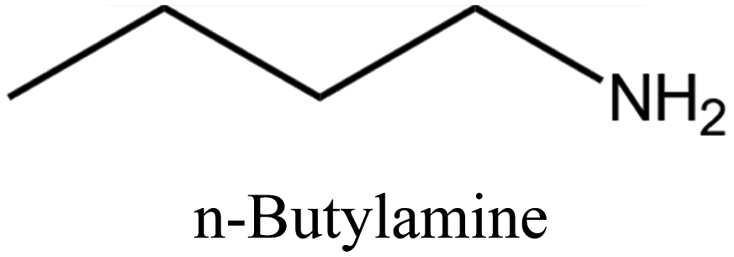
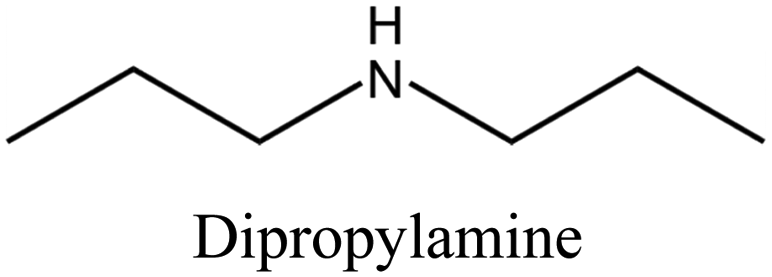
MTT | iii: Al-rich MTT structure (named SSZ-32) could be synthesized through the “dual-OSDA” route by introducing different amines into the synthesis media. This could reduce the use of costly OSDAs and provide flexibility by allowing various types of suitable amines. It should be noted these amines will work as long as a small amount of the diisopropylimidazolium cation is present. This benefits the zeolite manufacturer since the imidazole OSDA could be completely used and incorporated into the growing zeolite structure and there would be none to treat in the reaction waste stream, while the excess amines could be recycled and reused. |
|
||||

|
||||
|
||||

|
||||
|
||||
| 51 and 52 |

|

|
CHA | iii: The dual-OSDA approach could reduce the consumption of expensive OSDA (TMAdaOH) to a great extent by introducing an economically sourced quaternary amine as a cooperative OSDA. Besides cost reduction, it was found TEA+/TMA+ acts as a “bridging function” or “nucleation promoter” in the structure formation. The results indicate that only the first OSDA (TMAda+) was mainly incorporated into the framework. |
|
||||
| 53 |
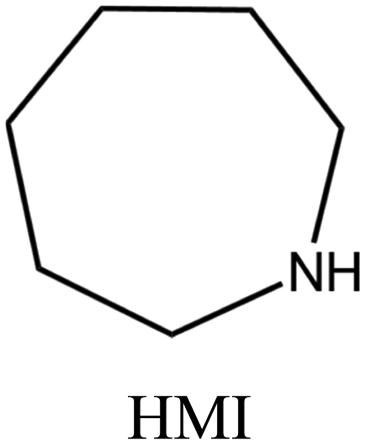
|
MWW | iv: It was found that when HMI is used together with TMAda+, the crystallization time of pure silica MWW type zeolite (named ITQ-1) is shortened (3 days compared to 14–17 days where TMAda+ was used as a single OSDA). Moreover, the reproducibility of the synthesis, as well as the quality of the materials obtained, is greatly improved through the dual-OSDA approach. | |
| 54 |
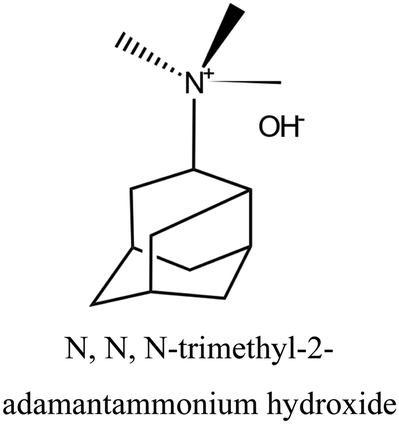
|
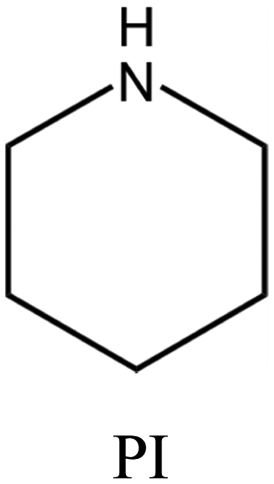
|
MWW | iv: The use of smaller amounts of a complex OSDA like N,N,N-trimethyl-2-adamantammonium hydroxide coupled with piperidine or isobutylamine in the dual-OSDA system produces MWW topology faster than the complex OSDA alone (4–5 days compared to 10 days). |

|
||||
| 55 |
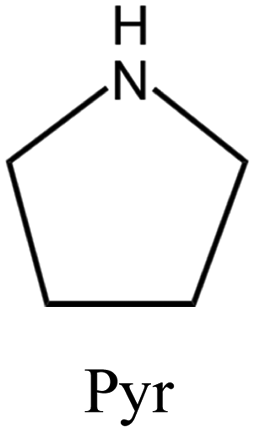
|
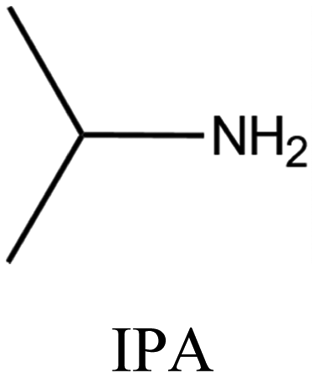
|
MTT | iv: MTT topology was synthesized by employing the dual OSDAs pyrrolidine and isopropylamine. The presence of these OSDAs widened the range of chemical composition (e.g., Si/Al ratio). Meanwhile, adding a small amount of IPA could significantly speed up the crystallization to 3 d, while the crystallization time took up 6 d when Pyr was acting as the single SDA. In this system, each organic amine agent serves as a supplement for the structure-directing effect of another one. Nevertheless, neither Pyr nor IPA can be totally replaced by the other. |
i. Novel structure: achieving specific zeolite topologies or particular structural arrangements may not be possible using only one OSDA. To address this challenge, two different OSDAs could be used together to facilitate structure formation. There are different behaviors when these OSDAs cooperate. BEA, BPH, LTA, and UFI topologies could be obtained through the cooperation of tetraethylammonium (TEA+) and tetramethylammonium (TMA+) via the CDM approach.19,30–33,35–37 The CDM concept was introduced to foster cooperation between OSDAs by researchers at UOP.36Fig. 1 depicts the primary steps involved in the CDM approach. Initially, a low Si/Al mixture of silica and alumina source is prepared along with a low-charge density OSDA (e.g. TEA+), known as the CDM mixture ①. This OSDA cannot efficiently balance the high charge on the potential aluminosilicate framework (calculated from the precursor ratios). In other words, the formation of crystalline material is hindered by an electrostatic barrier (CDM barrier) ②. Subsequently, a mixture of a higher-charge density OSDA (e.g. TMA+) called the crystallization mixture, is added ③. It's worth noting that the CDM barrier is so robust that no solid phase (not just zeolite crystals) will form without adding a crystallization OSDA.33 Finally, the barrier can be eliminated by providing charge balance and sufficient heat to achieve zeolite crystallization ④. Interestingly, temperature could direct the synthesis toward different structures in the CDM procedure. LTA and UFI topologies could be obtained from the same mixture gel at 100 and 150 °C, respectively.19,31,33
 | ||
| Fig. 1 Cartoon scheme of charge density mismatch (CDM) procedure: ① the CDM mixture consists of a low-charge density OSDA (OSDA1), ② OSDA1 cannot overcome the CDM barrier, ③ the crystallization mixture consists of a higher-charge density OSDA (OSDA2) is added to the system, and ④ the barrier can be eliminated. | ||
The FER topology is a great example of how different OSDAs can work together in the structure formation process. In this case, TMA+ is responsible for forming the FER cavities, which are then assembled around a bulkier OSDA like benzyl methyl pyrrolidinium (bmp). As shown in Fig. 2, bmp is too large to fit inside the FER cavities (7 Å along the a-direction and approximately 3.5 Å in the b-direction). Instead, it accommodates the 10-membered ring channels of the structure, making it an interesting medium-pore material for catalytic reactions. It should be noted that both OSDAs are required to be present in the synthesis medium to allow FER formation. In the absence of the TMA, a mixture of phases is obtained, while the absence of bmp yields a crystalline product different from ferrierite.38
 | ||
| Fig. 2 Location of TMA and bmp OSDAs inside the framework in the cooperative strategy of FER synthesis, as determined by molecular mechanics calculations and corroborated by NMR, which was reported by the authors in ref. 38. | ||
ii. Altering aluminum distribution: the “dual-OSDA” approach is a promising method to exert effective control over aluminum distribution, Si/Al ratio, and acidity of zeolites. The cooperative role of OSDAs in Al distribution in FER and FAU topologies has been thoroughly discussed elsewhere.7 It has been revealed that the combination of OSDAs could change Al siting, manipulate the density of acid sites located at the 10-membered ring channels of FER topology, and subsequently improve catalytic performance.43–45
Achieving MOR zeolites with high Si/Al ratios (more than 20) through conventional routes is challenging due to the lack of thermal stability during post-treatments, as well as the hazardous use of fluoride source.40,41,56 Nevertheless, high silica MOR (Si/Al: 37) could be achieved through a fluoride-free route via the cooperation of TEA+ and heterocycle compounds like hexamethyleneimine (HMI). TEA+ plays the dominant role in this system, and no MOR can be obtained in its absence (e.g. when replaced with TMA+ and TPA+).40,41 Both TEA+ and HMI molecules were present within the pores of the as-made MOR zeolite, and both molecules likely contributed to the process of crystallization. Additionally, the morphology of the MOR zeolites was altered through a dual-OSDA approach, resulting in smaller crystal sizes compared to synthesis using fluoride media.41,57
iii. Cost-effective synthesis: interesting structures have been developed in zeolite science by introducing homemade and complicated OSDAs.6,58,59 However, the higher cost of these OSDAs has put a restriction on the industrial usage of the new zeolites. Thus, developing methods to overcome this obstacle is highly desirable and critical. This can be done by replacing part of the costly OSDA with less expensive compounds.60 The dual-OSDA approach could be a cost-effective solution for those zeolites synthesized using an expensive OSDA. In this approach, small amounts of an expensive OSDA can initiate nucleation, while adding more portions of inexpensive OSDA enables the full crystallization process and reduces zeolite synthesis costs. For instance, the high cost of TMAdaOH can limit the use of CHA zeolites for certain commercial processes; however, it is possible to significantly reduce the consumption of this costly OSDA without compromising textural properties or catalytic performance.51,61 While a small quantity of TMAdaOH (TMAdaOH/SiO2: 0.05) is needed to achieve CHA topology in a cooperative route, most OSDA consumption is replaced with inexpensive tetraethylammonium hydroxide (TEAOH). As another example, LTA topology could be obtained through a dual-OSDA approach (CDM) using commercial TEAOH and TMAOH organics rather than applying complex homemade-OSDA62–64 in an HF-aided system. Using cost-effective and commercially available OSDAs would enhance the synthesis economy, simplify the system, and eliminate the hazards of HF. The limits of the CDM LTA are however a lower Si/Al.
iv. Fast synthesis: the dual-OSDA approach could speed up the crystallization of MWW and MTT topologies (as mentioned in Table 1).53–55 ITQ-1 zeolite with MWW structure comprises two different 10-membered ring channels and a large 12-membered ring supercage. However, the size of TMAda+ prevents it from fitting into the sinusoidal 10-membered ring channels. The conventional synthesis process is lengthy and challenging to reproduce because crystallization would be facilitated only through “accidentally present” organics of suitable size to fill the sinusoidal 10-membered ring. These organic fragments come from partial decomposition of the TMAda+ during heating or residual contaminants on PTFE liners.53 It is possible to use HMI along with TMAdaOH in a cooperative approach to stabilize the void spaces. HMI stabilizes the 10-membered ring channel, while TMAdaOH stabilizes the 12-membered ring supercage. This approach not only significantly shortens the crystallization time (from 14–17 to just 3 days) but also greatly improves the reproducibility of the synthesis process. In a similar approach, the MWW topology could be achieved after 10 days in the presence of another adamantyl component (N,N,N-trimethyl-2-adamantammonium hydroxide) but cooperation with a second OSDA, such as piperidine or isobutylamine, reduces synthesis time to 5 days.54
How do OSDAs cooperate in the “dual-OSDA” approach?
In “real cooperation”, both distinct organic compounds actively participate in zeolite formation, exhibiting different behaviors in structure formation:I. Co-construction: in this type of cooperation, each OSDA seems responsible for constructing a specific structural component. The combination of OSDAs results in a synergistic effect where each OSDA stabilizes different features of the framework to achieve the most favorable interaction energy. For instance, in the dual-OSDA synthesis of FAU zeotype (SAPO-37), TPA+ forms the supercage while the smaller TMA+ fits into sod cages.16,65 Another well-elaborated example of co-construction could be the synthesis of high silica FAU (Si/Al: 6) using choline ion (Ch+) and 15-crown-5 (CE) OSDAs.48 This [charged OSDA]/[neutral OSDA] system clearly demonstrates a co-construction behavior, where the trans Ch+ conformer was captured in the sod cage, while the gauche Ch+ conformer and mainly the CE complex formed the supercage. That means Ch+ plays a key role in zeolite formation, occluding its different isomers into the structure. However, the presence of CE is essential since no trans Ch+ was found without CE, and the fraction of trans Ch+ is closely related to the Si/Al ratio of FAU. This kind of cooperation where structural isomer forms of one OSDA are seemingly influenced by the other OSDA is quite unique.
II. Charge balancing: the cooperation happens due to balancing the mismatch between charges of the OSDAs and aluminosilicate solution in the synthesis media. In this case, both OSDAs are responsible for compensating the negative charge of the aluminosilicate solution. A classic example is CDM, where solid formation is motivated by the coulombic stabilization enabled by the crystallization OSDA. The crystallization OSDA dominates during the nucleation stage, while the CDM OSDA plays a more significant role in crystal growth.31 Both OSDAs can be incorporated into the framework, otherwise, CDM OSDA can force the crystallization OSDA to incorporate into the structure in a “forced cooperative templating” (i.e., only one of the OSDAs is incorporated).33,36 For example, LTA topology could be obtained through a TMA+-TEA+ dual OSDA approach. Charge balancing process leads initially to the formation of LTA cages, primarily involving TMA+. This process then transitions into a construction behavior, in which both OSDAs are integrated into the structure. It's slightly different from co-construction since both OSDAs are simultaneously responsible for forming structural units, such as sod cages.
III. Promoting nucleation: it has been observed that both OSDAs can initiate nucleation together through a synergistic effect, and only one of them gets incorporated into the structure. In the case of synthesizing CHA using a dual-OSDA approach, TEA+ does not get occluded in the framework and only helps to promote nucleation, while TMAda+ gets incorporated into the framework structure.51 Sometimes the nucleation process begins with the first OSDA, while the second OSDA helps facilitate particle–particle aggregation. In the TMAda+–TMA+ system, the nucleation starts with the construction of the cha cage, which involves the incorporation of the TMAda+, and the primary particles form by subsequent cha cages connection. Simultaneously, TMA+ is said to play a role in bridging neighboring particles through electrostatic interactions, which promotes crystal formation.52
Competition of OSDAs: phase selectivity
How does cooperation turn into competition?
One crucial problem in a dual-OSDA system is rather a fundamental question: how to achieve cooperation between OSDAs, while preventing them from directing the synthesis toward multiple (or even amorphous) phases. The cooperation depends on various parameters, but the synthesis temperature, OSDAs relative ratio, alkalinity, and inorganic cations are the main parameters underlined in the literature.34,35,40 It is essential to carefully consider the synthesis temperature and molar ratios of OSDAs to control the cooperation and phase selectivity in the CDM synthesis (examples in Table 1). Specifically, different structures can be obtained by varying the synthesis temperature in the fixed OSDAs ratio (TEA/TMA:8/1),34 while the cooperation takes place at 100 and 150 °C, leading to the formation of LTA and UFI structures, respectively. However, increasing the temperature to 175 °C results in amorphous material due to the decomposition of TEA+.35 The relative amount of OSDAs is also very important in initiating synergy between them, since cooperation could be turned into competition by manipulating this ratio. In the FAU synthesis using the CE–Ch+ system, control experiments were performed while varying the Ch+/CE ratio or synthesis temperature and keeping other variables constant. Fig. 3A shows some of these results where cooperation could be spoiled in the presence of inadequate conditions. It was observed that sod units with trans Ch+ conformer occlusion preferentially aggregated into the SOD phase rather than cooperated with CE to form pure FAU zeolite.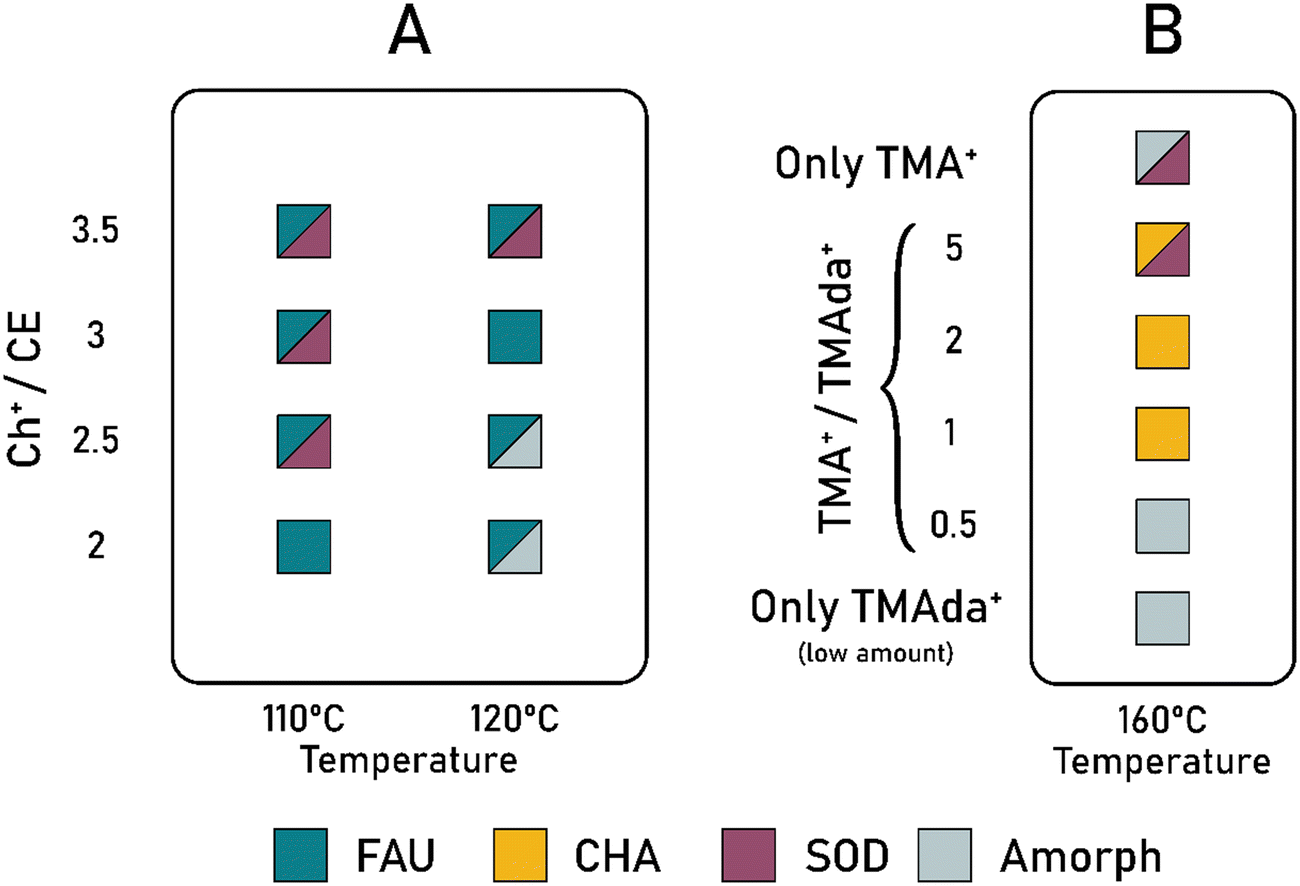 | ||
| Fig. 3 Effect of OSDAs relative ratio on the cooperative behavior of (A) Ch+–CE system toward the synthesis of pure FAU (Si/Al ratio is 7 and total OSDA/Si ratio varies between 0.4–0.7 in synthesis mixture) and (B) TMA+–TMAda+ system toward the synthesis of CHA (Si/Al ratio is 10 and total OSDA/Si ratio varies between 0.06–0.25 in the synthesis mixture). Based on data in ref. 48 for FAU and ref. 52 for CHA. | ||
As another example, CHA zeolite could be obtained through a wide range of TMAdaOH/TMAOH in a cooperative manner, while too low TMAOH concentration leads to an amorphous product, and excessive TMAOH concentration results in a denser SOD phase (mixed with CHA).52 In a cost-effective approach, the amount of costly TMAdaOH was kept low, and the amount of inexpensive counterpart varied, as shown in Fig. 3B. A very low amount of TMAdaOH, the well-established OSDA for synthesizing CHA, could not direct the synthesis towards pure CHA, thus a synergy between OSDAs is needed for this purpose. On the other hand, sole use of TMAOH would yield a mixture of amorphous and SOD phases.
In addition to the relative ratio of OSDAs, alkalinity and inorganic cations could enhance or disrupt the cooperation. A dual-OSDA approach has been implemented to obtain high silica MOR (Si/Al > 30), as mentioned earlier in Table 1: cooperative behavior was easily spoiled by the high alkalinity ratio (OH/Si > 0.5). In the presence of inorganic cations, organic species are more difficult to incorporate in zeolite formation and result in a framework with a relatively low Si/Al ratio.40 In addition to the Si/Al ratio, inorganic cations significantly influenced the formation of the pure MOR phase, where a slight increase in the Na+/Si ratio resulted in the formation of quartz impurity in strongly basic media.41 It was found that a certain amount of alkalinity is needed for the synthesis of high silica pure MOR zeolites.
Timed addition, or intermediate addition, can be an unconventional way to control cooperation/competition among OSDAs. N,N,N-trimethyl-(−)-cis-myrtanylammonium hydroxide (TMMAOH) is the primary OSDA used for the formation of CON zeolite.49 However, TMMAOH is relatively expensive, which led to the addition of TEAOH as an alternative OSDA to develop a more cost-effective approach. Interestingly, the timing of TEAOH addition is crucial in achieving cooperation towards the pure CON phase. Introducing TEAOH in the early stages of synthesis can create competition between OSDAs, and the strong structure-directing effect of TEAOH can direct the synthesis toward other phases, like MFI, rather than achieving CON topology. On the other hand, adding TEAOH in later stages results in cooperation and promotes the formation of CON zeolites. 1H NMR, 13C MAS NMR, and CHN analyses indicate the presence of TEA+ and suggest that even a small amount of TEA+ occluded in the framework enables the crystallization of CON zeolite.
Is competition always undesirable?
As discussed earlier, the competition of OSDAs may result in phase selectivity issues and lead to obtaining either amorphous or crystalline materials with mixed topology. However, this competition can also result in some intergrowth zeolites, which have shown remarkable catalytic activity.18,20,66–68 An interesting example is the synthesis of the AEI/CHA intergrowth using TMAda+ and N,N-diethyl-2,6-dimethylpiperidinium (DEDMP), which are two well-known OSDAs for synthesizing pure CHA and AEI topologies, respectively, along with its performance in the methanol to olefin reaction (MTO) and the selective catalytic reduction of NOx (SCR).18,69,70 The silicoaluminophosphate form of this intergrowth is a competitive catalyst compared to SAPO-34 (pure CHA) for converting methanol or dimethyl ether (DME) into olefins due to weaker acid sites and a longer catalyst lifetime.68,71,72 However, it is necessary and simultaneously challenging to fine-tune their synthesis conditions to avoid the crystallization of independent zeolite phases.18 For more detailed information, we would like to refer the reader to the recent review focused on the synthesis and catalysis aspects of these materials.73Among the OSDAs frequently used in zeolite synthesis, tetraethylammonium (TEA+, tt.tt and tg.tg forms) and (2-hydroxyethyl)trimethylammonium (choline, gauche and trans forms) are known to exist as mixtures of isomers.74,75 The structure-directing effect through cooperation (or competition) of these isomers has been clearly demonstrated by leading to the synthesis of several frameworks. However, the competitive formation of two or more framework structures can also be influenced by geometrically different isomers of the organic compounds used as OSDAs. Fig. 4 shows representative isomeric OSDAs used in zeolite synthesis. Remarkable changes in phase selectivity can be seen in a pair of isomers of N,N-diethyldecahydroquinolinium76 (Fig. 4a). For example, the use of cis isomer of this compound led the formation of four different framework structures: SSZ-26 (CON), SSZ-31 (intergrowth SSZ-31), SSZ-35 (STF), and SSZ-48 (SFE). However, the trans isomer yielded cage-based zeolites: SSZ-13 (CHA) and SSZ-36 (intergrowth RTH-ITE). As expected, when using bulkier bicyclic OSDAs, such as N,N-diethyl-2-methyldecahydroquinolinium and 3-ethyl-1,3,8,8-tetramethyl-3-azoniabicyclo-[3.2.1]octane cations, the two isomers of each OSDA exhibited completely different phase selectivities (Fig. 4b and c).77–79 The competition between the formation of channel-based CIT-9 (GME) and caged-based SSZ-39 (AEI) zeolites was also observed in the cis and trans isomers of monocyclic N,N-dimethyl-3,5-dimethylpiperidinium cation with relatively minor difference in OSDA shape compared to bicyclic compounds (Fig. 4d).80
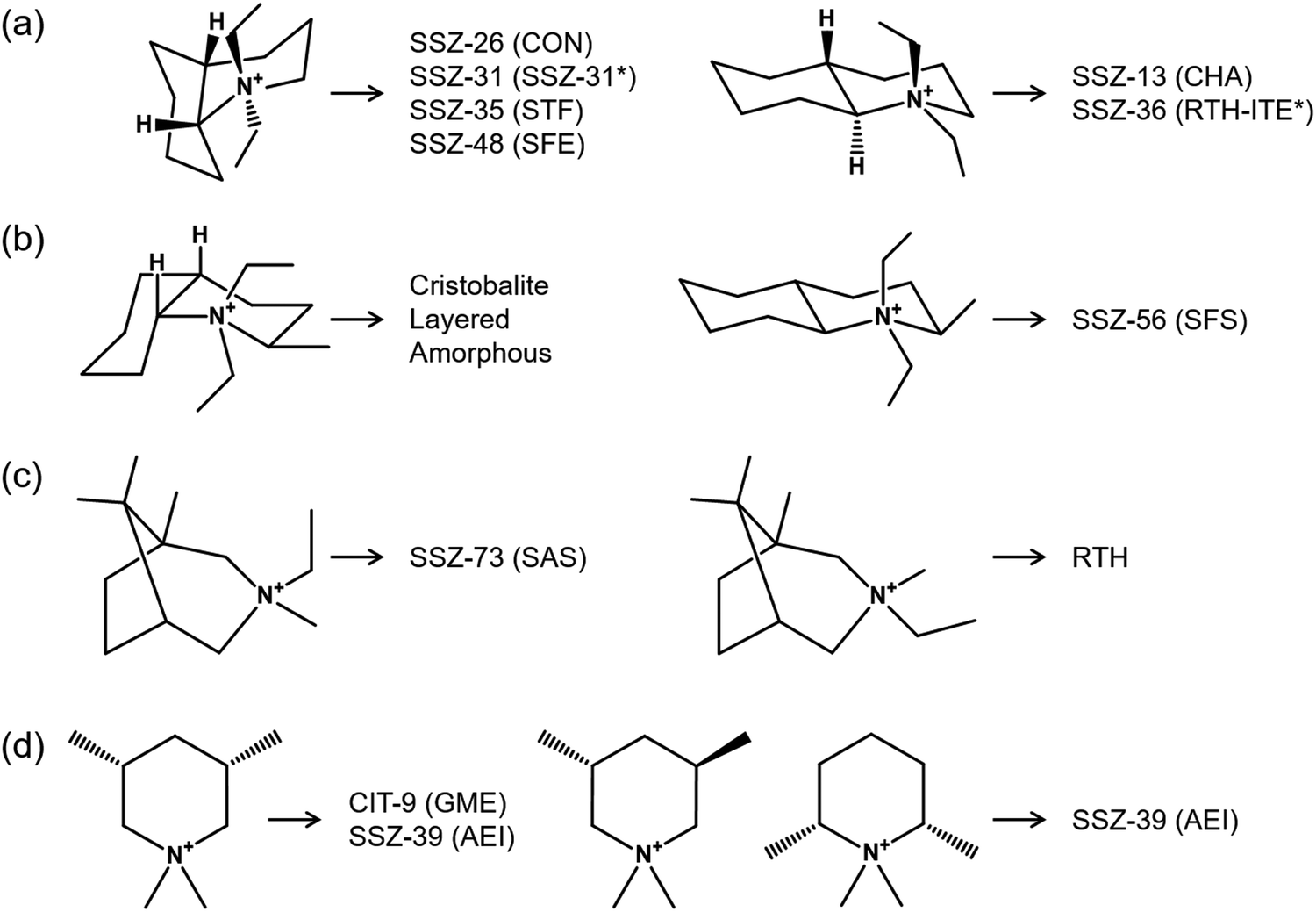 | ||
| Fig. 4 Representative effect of geometric isomers on phase selectivity. The cis (left) and trans (right) isomers of (a) N,N-diethyldecahydroquinolinium, (b) N,N-diethyl-2-methyldecahydroquinolinium, and (c) 3-ethyl-1,3,8,8-tetramethyl-3-azoniabicyclo-[3.2.1]octane cations. (d) The effect of diastereo- and structural isomers of dimethylpiperidinium-based OSDAs: N,N-dimethyl-cis-3,5- (left), N,N-dimethyl-trans-3,5- (middle), and N,N-dimethyl-cis-2,6-dimethylpiperidinium (right). Asterisk (*) indicates the disordered framework structure (i.e., intergrowth family). | ||
It should be noted here that, unlike the above cases, organic isomers can compete with each other for the structure-direction of the identical zeolite building unit. This can lead to differences in the crystallization kinetics as well as the material properties. Dusselier and co-workers examined the effect of diastereo- and structural isomers of dimethylpiperidinium-based OSDAs, which include N,N-dimethyl-cis-3,5- (cis-3,5), N,N-dimethyl-trans-3,5- (trans-3,5), and N,N-dimethyl-cis-2,6-dimethylpiperidinium (cis-2,6), on the crystallization of SSZ-39 (Fig. 4d).81 The authors reported optimum synthesis conditions where pure SSZ-39 can be synthesized with the cis-3,5, trans-3,5, or cis-2,6 isomers and mixtures thereof. All as-synthesized SSZ-39 solids were calculated to have 4 organic molecules per unit cell, i.e., 1 OSDA per cage, regardless of the cis-3,5/trans-3,5/cis-2,6 isomer ratio in the initial gel. When isomers were in competition in the synthesis media, there were remarkable isomer-dependent trends in the relative crystallization rate (trans-3,5 > cis-3,5 > cis-2,6), as well as in the organic to be occluded preferentially (trans-3,5 > cis-3,5 and cis-2,6 > cis-3,5). The same phenomenon, i.e., faster crystallization kinetics induced by trans isomer, can be also confirmed in the similar study by the Shantz's group.82 In addition, they found that the presence of trans isomer in Si-rich (Si/Algel: 45 vs. 15, 30) gels affects the Si/Al ratio and local structure (Al zoning) of SSZ-39, resulting in the different deNOx activity after Cu-exchange.82,83
Similarly, the competitive structure direction of dual OSDAs for the same framework structure can occur in organics with similar geometric shape but different charge distributions. For example, phosphonium-based SDAs have higher positive charge concentration on P than N-based counterparts. Alonso and Blasco investigated the structure-directing properties of TEA, tetraethylphosphonium (TEP), and mixtures of them with different ratios in the formation of pure-silica ZSM-5 (MFI).84 All ZSM-5 products contained ca. 4 organic molecules per unit cell. When both organics were present simultaneously in the initial gel (i.e., [TEP/(TEP + TEA)]gel = 0.12 and 0.25), TEA and TEP competed with each other to preferentially occupy the 4 channel intersections (i.e., [TEP/(TEP + TEA)]solid = 0.26 and 0.42). The favourable incorporation of TEP over TEA resulted in the increase in the crystallization rate as rising the [TEP/(TEP + TEA)]gel ratio. Additionally, the presence of P-containing OSDA led to control of the crystal size and local structural disorder (SiO− or SiOH defects). However, it is difficult to control the removal of P species because most of the P remains inside the zeolite as extra-framework oxidized phosphorous species upon calcination.
On the other hand, several studies have demonstrated that the location of Al atoms in ZSM-5 (i.e., straight and/or sinusoidal channels or channel intersections) can be controlled using various branched/straight-chain alcohols as non-charged, pore-filling agents together with Na+ ions.85–87 This suggests that the synthetic concept using competition between organics with similar geometric shapes can be extended to competition between positively charged organics (e.g., tetrapropylammonium for ZSM-5) and neutral alcohols with similar shapes for the synthesis of fine-tuned aluminosilicate zeolites. To apply this competitive strategy widely, a further understanding of their multiple chemical interactions in complex media is still required. The timed additional synthesis method via a (semi)continuous reactor system may provide clues to control the competitive directing effect of dual OSDAs.88,89
Conclusion
Improved zeolite synthesis can be achieved through a dual-OSDA approach, which involves cooperation and/or competition of different OSDAs in the synthesis media. When OSDA cooperation seems to occur, this is noticed from co-inclusion in the resulting zeolite, (e.g. by stabilizing different parts of the framework) and/or altering the synthesis outcome, such as acidity, Al distribution, and/or the synthesis kinetics. This approach can sometimes be regarded as more cost-effective (less consumption of expensive organics) and time-efficient (faster kinetics) than conventional syntheses using a single OSDA. However, cooperation can easily be disrupted, and one of the challenges in dual-OSDA zeolite synthesis is determining the conditions under which OSDA species can function in harmony to affect zeolite properties and control phase selection. In this featured article, we discussed the importance of cooperative dual-OSDA synthesis in certain cases, explained the outcomes of this approach, and described the different behaviors of cooperative systems. As illustrated in Fig. 5, it reveals that the cooperation can be feasible by (I) controlling the molar ratio of OSDAs, (II) having a suitable synthesis temperature, and (III) providing a sufficient alkalinity in the system. The timing of the addition of OSDAs could also be considered as an unconventional strategy to achieve cooperation toward our desired phase. When cooperation is overwhelmed by the competition of OSDAs, amorphous or mixed crystalline phases can be obtained. Still, an intergrowth of zeolites can also be formed in a targeted way, gaining a lot of momentum recently.18,73,90 It is clear that the proper selection and combination of the OSDAs with the conditions mentioned above will promote cooperation toward zeolites with well-tailored properties. However, this selection procedure remains a tedious task that involves trial and error methods. Thus, computational and data-driven approaches are needed to develop an efficient roadmap. | ||
| Fig. 5 Schematic representation of cooperative and competitive synthetic strategies with their outcomes. OSDA molar ratio, synthesis temperature, and alkalinity are the most effective parameters to control cooperation in the dual-OSDA approach. | ||
Perspective and outlook
• Intergrowths are zeolite-type materials that (in some cases) possess remarkable catalytic activity. Instead of combining two OSDAs and creating a competition that requires monitoring and control, researchers have developed “bi-selective OSDAs”.8,90,91 These compounds can form different topologies in a facile and efficient one-pot synthesis. These multifunctional OSDAs were designed through computational modeling, data mining, and data-driven screening. Although these materials are not yet industrialized, achieving cost-effective and bi-selective OSDAs that reduce the usage of expensive OSDAs will be a big challenge.• Industrial zeolite catalysts require a large amount of expensive OSDAs during their manufacturing process. Not only their synthesis is costly, but also the calcination step (burning the organic) often requires considerable and careful environmental consideration (e.g. dealing with off-gases). Although there have been efforts to recycle OSDAs in zeolite synthesis, this approach has not yet been commercialized. Therefore, it is necessary to significantly reduce the usage of these expensive OSDAs in the industry, which is counter to promoting the addition of two such organics. Yet, a cooperative dual-OSDA approach could be a good solution when a significant portion of more expensive or burdensome OSDAs is substituted by a cheaper and more benign one. Unconventional methods such as intermediate addition (or timed addition) of OSDAs, e.g., via the use of fed-batch reactors, could have significant potential to reduce the needed amount of a specific OSDA (and even perhaps in single OSDA systems) and lower the synthesis cost.
• It has been shown that cooperation between OSDAs could speed up the crystallization and reduce the synthesis time for specific structures. This suggests the possibility of using some OSDAs as “accelerators” in zeolite production, potentially impacting both production time and cost, especially when scaling up the synthesis.
• Many studies lack proper control experiments and a clear mechanistic understanding. Researchers could gain insights into the individual roles of OSDAs in cooperative strategies by conducting more control experiments. For one, the ratios of OSDAs should be varied in both directions (above and below 1) while for single OSDA experiments, multiple controls are needed. For example the synthesis medium either needs to be carefully adjusted to have a control at the same OH/Si level, or, at the same inorganic content. Additionally, identifying the main drivers of cooperation (e.g., according to one or more of the modes i–iv) as well as addressing the nature of cooperative behavior (e.g. co-construction or promoting nucleation) would help ongoing research in this area. Ultimately, most solutions for dual-OSDAs arise from amine chemistry, but there is potential to explore beyond that limitation.
• Intermediate additions of OSDAs or changing OSDA ratios midway have mitigated undesired interactions and enabled “cost-effective” synthesis by replacing costly OSDAs with commercially available ones.49,52 Developing reactor designs to facilitate these midway changes without interrupting the operational parameters (e.g., temperature and pressure) could be an intriguing option for studying the cooperation behavior and controlling the “cooperation-competition” in zeolite science.
• Machine learning and data-driven approaches are revolutionizing science in many fields today, including the world of zeolites. Traditional trial and error methods for finding suitable OSDA candidates for the cooperative formation of zeolites may not be always the way to go in the near future. Recent studies have explored the potential of using data-driven methods to identify optimal OSDAs for cooperation.8,9,18,92,93 Their approaches involve computational modeling based on the binding energy of OSDAs. These new techniques94 allow for the design of more efficient and commercially viable syntheses to produce finely tuned zeolite and zeotype materials. Some hurdles here might be the modeling of interaction of charges of OSDA and framework and its impact on zeolite synthesis kinetics.
Data availability
No primary research results, software, or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. J., G. I., and M. D. thank the European Research Council (ERC) for funding ERC Starting Grant 948449 to M. D. named Z-EURECA: ZEolite synthesis in Unusual Reactors for Enhanced CAtalysts. J. B. acknowledges the financial support of the Research Foundation-Flanders (FWO Vlaanderen) for personal postdoc funding (12E6923N).References
- C. Martínez and A. Corma, Coord. Chem. Rev., 2011, 255, 1558–1580 Search PubMed.
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef PubMed.
- C. S. Cundy and P. A. Cox, Microporous Mesoporous Mater., 2005, 82, 1–78 Search PubMed.
- C. S. Cundy and P. A. Cox, Chem. Rev., 2003, 103, 663–701 Search PubMed.
- J. Li, A. Corma and J. Yu, Chem. Soc. Rev., 2015, 44, 7112–7127 Search PubMed.
- M. Moliner, F. Rey and A. Corma, Angew. Chem., Int. Ed., 2013, 52, 13880–13889 Search PubMed.
- J. Bae and M. Dusselier, Chem. Commun., 2023, 59, 852–867 Search PubMed.
- D. Schwalbe-Koda, S. Kwon, C. Paris, E. Bello-Jurado, Z. Jensen, E. Olivetti, T. Willhammar, A. Corma, Y. Román-Leshkov, M. Moliner and R. Gómez-Bombarelli, Science, 2021, 374, 308–315 Search PubMed.
- Z. Jensen, S. Kwon, D. Schwalbe-Koda, C. Paris, R. Gómez-Bombarelli, Y. Román-Leshkov, A. Corma, M. Moliner and E. A. Olivetti, ACS Cent. Sci., 2021, 7, 858–867 CrossRef PubMed.
- J. Jiang, J. Yu and A. Corma, Angew. Chem., Int. Ed., 2010, 49, 3120–3145 CrossRef PubMed.
- K. Iyoki, K. Itabashi and T. Okubo, Microporous Mesoporous Mater., 2014, 189, 22–30 CrossRef.
- A. Javdani, J. Ahmadpour and F. Yaripour, Microporous Mesoporous Mater., 2019, 284, 443–458 Search PubMed.
- M. H. Nada and S. C. Larsen, Microporous Mesoporous Mater., 2017, 239, 444–452 CAS.
- D. Schwalbe-Koda, O. A. Santiago-Reyes, A. Corma, Y. Román-Leshkov, M. Moliner and R. Gómez-Bombarelli, Chem. Mater., 2022, 34, 5366–5376 CAS.
- R. W. Broach, E. P. Boldingh, D. Y. Jan, G. J. Lewis, J. G. Moscoso and J. C. Bricker, J. Catal., 2013, 308, 142–153 CAS.
- Z. Asgar Pour and K. O. Sebakhy, Chem, 2022, 4, 431–446 CAS.
- M. A. Camblor, A. Corma, M. J. Díaz-Cabañas and C. Baerlocher, J. Phys. Chem. B, 1998, 102, 44–51 CrossRef CAS.
- E. Bello-Jurado, D. Schwalbe-Koda, M. Nero, C. Paris, T. Uusimäki, Y. Román-Leshkov, A. Corma, T. Willhammar, R. Gómez-Bombarelli and M. Moliner, Angew. Chem., Int. Ed., 2022, 61, e202201837 CrossRef CAS.
- M. B. Park, D. Jo, H. C. Jeon, C. P. Nicholas, G. J. Lewis and S. B. Hong, Chem. Mater., 2014, 26, 6684–6694 CrossRef CAS.
- Y. Naraki, K. Ariga, K. Nakamura, K. Okushita and T. Sano, Microporous Mesoporous Mater., 2017, 254, 160–169 CAS.
- R. F. Lobo, S. I. Zones and M. E. Davis, J. Inclusion Phenom. Mol. Recognit. Chem., 1995, 21, 47–78 CAS.
- M. Gálvez-Llompart, A. Cantín, F. Rey and G. Sastre, Z. Kristallogr. - Cryst. Mater., 2019, 234, 451–460 CrossRef.
- K. Egeblad, C. H. Christensen, M. Kustova and C. H. Christensen, Chem. Mater., 2008, 20, 946–960 CAS.
- R. G. Chitac, M. D. Shannon, P. A. Cox, J. Mattock, P. A. Wright and A. Turrina, AI-Guided Design and Property Prediction for Zeolites and Nanoporous Materials, 2023, 113–143 Search PubMed.
- K. Liu, M. Çağlayan, A. Dikhtiarenko, X. Zhang, O. Sayidov, E. Abou-Hamad, J. Gascon and A. Dutta Chowdhury, Catal. Today, 2023, 408, 22–35 CAS.
- P. Rani, R. Srivastava and B. Satpati, Cryst. Growth Des., 2016, 16, 3323–3333 CAS.
- L. Emdadi and D. Liu, J. Mater. Chem. A, 2014, 2, 13388–13397 CAS.
- H. Dai, J. Claret, E. L. Kunkes, V. Vattipalli, N. Linares, C. Huang, M. Fiji, J. García-Martinez, A. Moini and J. D. Rimer, Angew. Chem., Int. Ed., 2022, 61, e202117742 CAS.
- M. Kumar, H. Luo, Y. Román-Leshkov and J. D. Rimer, J. Am. Chem. Soc., 2015, 137, 13007–13017 CAS.
- C. S. Blackwell, R. W. Broach, M. G. Gatter, J. S. Holmgren, D. Y. Jan, G. J. Lewis, B. J. Mezza, T. M. Mezza, M. A. Miller, J. G. Moscoso, R. L. Patton, L. M. Rohde, M. W. Schoonover, W. Sinkler, B. A. Wilson and S. T. Wilson, Angew. Chem., Int. Ed., 2003, 42, 1737–1740 CAS.
- M. B. Park, Y. Lee, A. Zheng, F. S. Xiao, C. P. Nicholas, G. J. Lewis and S. B. Hong, J. Am. Chem. Soc., 2013, 135, 2248–2255 CAS.
- R. Yao, W. Zhang, Y. Peng, H. Song, C. Zhu, L. Shu and W. Yang, Microporous Mesoporous Mater., 2022, 334, 111776 CrossRef CAS.
- M. B. Park, N. H. Ahn, R. W. Broach, C. P. Nicholas, G. J. Lewis and S. B. Hong, Chem. Mater., 2015, 27, 1574–1582 CrossRef CAS.
- S. H. Kim, M. B. Park, H. K. Min and S. B. Hong, Microporous Mesoporous Mater., 2009, 123, 160–168 CrossRef.
- M. B. Park, S. H. Ahn, C. P. Nicholas, G. J. Lewis and S. B. Hong, Microporous Mesoporous Mater., 2017, 240, 159–168 Search PubMed.
- G. J. Lewis, M. A. Miller, J. G. Moscoso, B. A. Wilson, L. M. Knight and S. T. Wilson, Stud. Surf. Sci. Catal., 2004, 154, 364–372 CrossRef.
- D. Y. Jan, G. J. Lewis, T. M. Mezza, J. G. Moscoso, R. L. Patton, M. P. Koljack and P. V. Tota, Stud. Surf. Sci. Catal., 2004, 154, 1332–1340 Search PubMed.
- A. B. Pinar, L. Gomez-Hortiguela and J. Perez-Pariente, Chem. Mater., 2007, 19, 5617–5626 CrossRef.
- R. García, L. Gómez-Hortigüela, I. Díaz, E. Sastre and J. Pérez-Pariente, Chem. Mater., 2008, 20, 1099–1107 Search PubMed.
- L. Wang, H. Pan, J. Qian, K. Yan, X. Yang, L. Liu, G. Song and H. Zhu, Microporous Mesoporous Mater., 2023, 354, 112569 Search PubMed.
- A. Lv, H. Xu, H. Wu, Y. Liu and P. Wu, Microporous Mesoporous Mater., 2011, 145, 80–86 Search PubMed.
- A. Corma, F. Rey, J. Rius, M. J. Sabater and S. Valencia, Nature, 2004, 431, 287–290 Search PubMed.
- A. B. Pinar, C. Márquez-Álvarez, M. Grande-Casas and J. Pérez-Pariente, J. Catal., 2009, 263, 258–265 Search PubMed.
- A. B. Pinar, L. Gómez-Hortigüela, L. B. McCusker and J. Pérez-Pariente, Chem. Mater., 2013, 25, 3654–3661 CrossRef.
- Y. Román-Leshkov, M. Moliner and M. E. Davis, J. Phys. Chem. C, 2011, 115, 1096–1102 CrossRef.
- A. B. Pinar, R. Verel, J. Pérez-Pariente and J. A. Van Bokhoven, Microporous Mesoporous Mater., 2014, 193, 111–114 CrossRef.
- C. Márquez-Alvarez, A. B. Pinar, R. García, M. Grande-Casas and J. Pérez-Pariente, Top. Catal., 2009, 52, 1281–1291 Search PubMed.
- Q. Ke, I. Khalil, B. Smeyers, Z. Li, R. de Oliveira-Silva, B. Sels, D. Sakellariou and M. Dusselier, Angew. Chem., Int. Ed., 2021, 60, 24189–24197 CAS.
- T. Shibuya, K. Iyoki, H. Onozuka, M. Takemoto, S. Tsutsuminai, T. Takewaki, T. Wakihara and T. Okubo, Cryst. Growth Des., 2023, 23, 3509–3517 CAS.
- S. I. Zones and S. J. Hwang, Chem. Mater., 2002, 14, 313–320 CAS.
- Z. Zhang, Y. Li, Z. Chen, F. Ji, X. Liang, H. Xuan, L. Han and P. Han, Fuel, 2024, 362, 130885 Search PubMed.
- Y. Guo, T. Sun, X. Liu, Q. Ke, X. Wei, Y. Gu and S. Wang, Chem. Eng. J., 2019, 358, 331–339 CAS.
- M. A. Camblor, A. Corma, M. J. Díaz-Cabañas and C. Baerlocher, J. Phys. Chem. B, 1998, 102, 44–51 CAS.
- S. I. Zones, S. Hwang and M. E. Davis, Chem. – Eur. J., 2001, 7, 1990–2001 Search PubMed.
- Y. Chen, C. Li, X. Chen, Y. Liu and C. Liang, Microporous Mesoporous Mater., 2018, 268, 216–224 Search PubMed.
- H. K. Beyer, I. M. Belenykaja, I. W. Mishin and G. Borbely, Stud. Surf. Sci. Catal., 1984, 18, 133–140 CrossRef CAS.
- B. Lu, Y. Oumi, K. Itabashi and T. Sano, Microporous Mesoporous Mater., 2005, 81, 365–374 Search PubMed.
- M. E. Davis and S. I. Zones, Synthesis of Porous Materials, Marcel-Dekker Inc, Basel, 1997, vol. 1 Search PubMed.
- M. Moliner, F. Rey and A. Corma, Angew. Chem., Int. Ed., 2013, 52, 13880–13889 Search PubMed.
- S. I. Zones, US8007763B2, 2011.
- S. I. Zones, US8298511B2, 2012.
- D. Jo, T. Ryu, G. T. Park, P. S. Kim, C. H. Kim, I. S. Nam and S. B. Hong, ACS Catal., 2016, 6, 2443–2447 CrossRef CAS.
- J. E. Schmidt, S. I. Zones, D. Xie and M. E. Davis, Microporous Mesoporous Mater., 2014, 200, 132–139 CrossRef CAS.
- B. W. Boal, J. E. Schmidt, M. A. Deimund, M. W. Deem, L. M. Henling, S. K. Brand, S. I. Zones and M. E. Davis, Chem. Mater., 2015, 27, 7774–7779 CAS.
- L. S. de Saldarriaga, C. Saldarriaga and M. E. Davis, J. Am. Chem. Soc., 1987, 109, 2686–2691 Search PubMed.
- S. P. Sree, E. Verheyen, M. De Prins, T. Van Der Donck, L. Van Tendeloo, F. Schuetze and J. A. Martens, ACS Mater. Lett., 2021, 3, 658–662 CAS.
- B. Wang, Z. Tian, P. Li, L. Wang, Y. Xu, W. Qu, H. Ma, Z. Xu and L. Lin, Mater. Res. Bull., 2009, 44, 2258–2261 CAS.
- D. Zhao, Y. Zhang, Z. Li, Y. Wang and J. Yu, Chem. Eng. J., 2017, 323, 295–303 Search PubMed.
- ExxonMobil Chemical Patents Inc Symyx Solutions Inc., US Pat., 7094389B2, 2004.
- P. N. R. Vennestrøm, J. R. Thøgersen, P. L. T. Gabrielsson, L. Van Tendeloo, F. W. Schütze and M. Moliner, Microporous Mesoporous Mater., 2023, 358, 112336 Search PubMed.
- D. Zhao, Y. Zhang, Z. Li, Y. Wang and J. Yu, RSC Adv., 2017, 7, 939–946 Search PubMed.
- W. Jiao, J. Su, H. Zhou, S. Liu, C. Liu, L. Zhang, Y. Wang and W. Yang, Microporous Mesoporous Mater., 2020, 306, 110444 Search PubMed.
- Y. Wang, C. Tong, Q. Liu, R. Han and C. Liu, Chem. Rev., 2023, 123, 11664–11721 CAS.
- J. Bae and S. B. Hong, Chem. Sci., 2018, 9, 7787–7796 Search PubMed.
- J. E. Schmidt, D. Fu, M. W. Deem and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 16044–16048 Search PubMed.
- G. S. Lee, Y. Nakagawa, S. J. Hwang, M. E. Davis, P. Wagner, L. Beck and S. I. Zones, J. Am. Chem. Soc., 2002, 124, 7024–7034 Search PubMed.
- C. Kim, S. J. Hwang, A. W. Burton and S. I. Zones, Microporous Mesoporous Mater., 2008, 116, 227–232 Search PubMed.
- D. S. Wragg, R. Morris, A. W. Burton, S. I. Zones, K. Ong and G. Lee, Chem. Mater., 2007, 19, 3924–3932 Search PubMed.
- S. Elomari, A. Burton, R. C. Medrud and R. Grosse-Kunstleve, Microporous Mesoporous Mater., 2009, 118, 325–333 CAS.
- M. Dusselier, J. H. Kang, D. Xie and M. E. Davis, Angew. Chem., Int. Ed., 2017, 56, 13475–13478 Search PubMed.
- M. Dusselier, J. E. Schmidt, R. Moulton, B. Haymore, M. Hellums and M. E. Davis, Chem. Mater., 2015, 27, 2695–2702 CAS.
- R. Ransom, J. Coote, R. Moulton, F. Gao and D. F. Shantz, Ind. Eng. Chem. Res., 2017, 56, 4350–4356 CrossRef CAS.
- R. Ransom, R. Moulton and D. F. Shantz, J. Catal., 2020, 382, 339–346 Search PubMed.
- J. Martinez-Ortigosa, J. Simancas, J. A. Vidal-Moya, P. Gaveau, F. Rey, B. Alonso and T. Blasco, J. Phys. Chem. C, 2019, 123, 22324–22334 CrossRef.
- S. Park, T. Biligetu, Y. Wang, T. Nishitoba, J. N. Kondo and T. Yokoi, Catal. Today, 2018, 303, 64–70 Search PubMed.
- T. Biligetu, Y. Wang, T. Nishitoba, R. Otomo, S. Park, H. Mochizuki, J. N. Kondo, T. Tatsumi and T. Yokoi, J. Catal., 2017, 353, 1–10 Search PubMed.
- T. Yokoi, H. Mochizuki, T. Biligetu, Y. Wang and T. Tatsumi, Chem. Lett., 2017, 46, 798–800 CrossRef.
- A. Deneyer, Q. Ke, J. Devos and M. Dusselier, Chem. Mater., 2020, 32, 4884–4919 CrossRef.
- A. Javdani, G. Ivanushkin, A. Deneyer and M. Dusselier, React. Chem. Eng., 2025, 10, 379–391 Search PubMed.
- S. Kwon, E. Bello-Jurado, E. Ikonnikova, H. Lee, D. Schwalbe-Koda, A. Corma, T. Willhammar, E. A. Olivetti, R. Gomez-Bombarelli, M. Moliner and Y. Román-Leshkov, ACS Appl. Mater. Interfaces, 2024, 16, 14661–14668 CrossRef PubMed.
- D. Schwalbe-Koda, A. Corma, Y. Román-Leshkov, M. Moliner and R. Gómez-Bombarelli, J. Phys. Chem. Lett., 2021, 12, 10689–10694 Search PubMed.
- D. Schwalbe-Koda, O. A. Santiago-Reyes, A. Corma, Y. Román-Leshkov, M. Moliner and R. Gómez-Bombarelli, Chem. Mater., 2022, 34, 5366–5376 Search PubMed.
- W. Chaikittisilp and T. Okubo, Science, 2021, 374, 257–258 Search PubMed.
- OSDB, https://zeodb.mit.edu/index, (accessed 21 November 2024).
| This journal is © The Royal Society of Chemistry 2025 |