
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advancing colorectal cancer research through lipidomics
Pedro Santiago ab,
Tânia Meloab,
Maria Barceló-Nicolauc,
Gwendolyn Barceló-Coblijnc,
Pedro Dominguesa and
Rosário Domingues*ab
ab,
Tânia Meloab,
Maria Barceló-Nicolauc,
Gwendolyn Barceló-Coblijnc,
Pedro Dominguesa and
Rosário Domingues*ab
aMass Spectrometry Center, LAQV-REQUIMTE, Department of Chemistry, University of Aveiro, Santiago University Campus, 3810-193 Aveiro, Portugal
bCESAM—Centre for Environmental and Marine Studies, Department of Chemistry, University of Aveiro, Santiago University Campus, 3810-193 Aveiro, Portugal. E-mail: mrd@ua.pt
cLipids in Human Pathology, Institut d'Investigació Sanitària Illes Balears (IdISBa, Health Research Institute of the Balearic Islands), Palma, Balearic Islands, Spain
First published on 4th June 2025
Abstract
Colorectal cancer (CRC) is currently a global health burden, with staggering worldwide prevalence. CRC is ranked as the third most common and second deadliest cancer worldwide. With rising life expectancy population growth, CRC incidence and mortality are projected to increase, particularly among individuals under 50. This underscores the need to improve early detection of CRC. Although colonoscopy remains the preferred diagnostic technique, due to its high sensitivity and specificity for CRC its invasive nature and cost result in low adherence rates. Consequently, the scientific community is actively exploring alternative diagnostic methods, primarily through biomarkers, molecules exhibiting dysregulated levels associated with specific diseases. Lipidomics has become crucial in cancer research, as lipids play key roles in metabolic pathways driving cancer development. Recent investigations have revealed decreased levels of lipid classes such as lysophosphatidylcholine (LPC) in CRC patients compared to healthy controls, alongside an increase in specific sphingolipid species across multiple studies. In the context of CRC progression, triglycerides (TGs) stand out as the lipids that display the most pronounced differentiation among different disease stages. These lipid dysregulations present promising avenues for identifying potential therapeutic targets and innovative diagnostic methods, however, a comprehensive understanding of these processes requires further exploration.
1. Introduction
Over the last decade, the global incidence of noncommunicable diseases (NCDs), such as cardiovascular diseases, cancer, diabetes, and chronic respiratory diseases, has increased significantly, resulting in major societal and economic repercussions. Currently, NCDs account for 74% of all deaths worldwide, corresponding to a total of 41 million fatalities, making them the most prevalent cause of mortality in the globe. Within NCDs, cancer stands out, ranking as the second most common cause of disease-related mortality in the world, only behind cardiovascular diseases.1 In 2020 alone, an estimated 19.3 million new cancer cases were reported, contributing to over 10 million deaths, reflecting the increasing global burden of this disease.2Cancer exhibits a pronounced heterogeneity, comprising a diverse array of cancer types that can manifest in different regions of the human body. This heterogeneity also extends to variations in the geographical distribution of the disease, etiological factors, and pathological features.3 Among the most prevalent cancer types, lung cancer, breast cancer, and colorectal cancer (CRC), present a particularly high incidence (Fig. 1). CRC holds the third position in global cancer incidence, and the second position in terms of cancer-associated mortality, recording 1.9 million new cases and nearly 935![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths in 2020.2
000 deaths in 2020.2
 | ||
| Fig. 1 Worldwide incidence of several types of cancer in 2020.3 | ||
As life expectancy continues to increase and the global population exhibits sustained growth, it is anticipated that the incidence of various diseases, including cancer, will significantly rise. Projections suggest that the total number of new cancer cases will reach approximately 29.4 million by 2040,4 with CRC expected to account for around 3.2 million of those new diagnoses.5 Moreover, there has been a noteworthy and concerning increase in CRC incidence among individuals below 50 years of age, a phenomenon whose underlying causes are yet to be comprehensively understood.6 This statistical trend underscores that, although effective screening methods are available, there is still the need to enhance the early detection of this disease. Currently, colonoscopy is by far the most common and well-established diagnostic method for CRC. However it is an invasive procedure with limited accessibility, particularly for restrictive populations. Therefore, alternative methods have been developed, e.g. computed tomography colonography, an imaging method that creates a virtual colonoscopy by using appropriate gut wall distention and three-dimensional reconstruction of the colon. This technique presents high sensitivity in detecting colon tumors that have spread beyond the intestinal wall and is, therefore, particularly valuable in the identification of metastases.7 Despite their efficacy, the invasiveness, procedural complexity, and post-therapy consequences resulting from surgery and chemotherapy, lead to a poor patient adherence. In turn, this can contribute to misdiagnosis and an increased risk of CRC-related mortality.
Thus, there is an urgent need to develop new and less invasive diagnostic techniques for CRC. In this context, the omics technologies are emerging as powerful tools for identifying reliable biomarkers associated with CRC pathogenesis, as they allow high-throughput analysis of biological data, providing insights into the structure, function, and interactions of biomolecules. Among these technologies, lipidomics has gained increasing attention due to the crucial roles lipids play in multiple biological processes, such as cell survival, proliferation, interaction, and apoptosis. Given these functions, dysregulated lipid metabolism has been increasingly associated with cancer pathogenesis.8
In this work, we provide a comprehensive overview of the current state of the art research on the application of lipidomics in CRC. For that, we will start with a brief overview of the main mechanisms of CRC, its etiology and known risk factors. Subsequently, we examine the literature that reported the use of lipidomic tools to unravel the CRC pathology and search for biomarkers of the disease, while also elucidating about the adaptation of lipids in CRC evolution, which may contribute to providing possible therapeutic targets and suggesting new screening methods that enable earlier and more accurate diagnosis.
2. Overview of CRC: etiology, pathology and therapy
CRC is a type of cancer that can arise in every part of the colon, including the ascending, transverse, descending, sigmoid sections and the rectum. This malignancy usually originates from abnormal tissue growths known as polyps, which can develop on the inner lining of the colon or rectum and may develop into cancer over time.9 As common symptoms, CRC can present changes in bowel habits, rectal bleeding, abdominal pain, unintentional weight loss, and fatigue. However, in its early stages, CRC is often asymptomatic, which underlines the importance of developing less complex and invasive screening methods, to enable an early detection of the disease, even prior the onset of symptoms.102.1. Characteristics and metabolic/pathogenic pathways of CRC
CRC carcinogenesis is a complex and multi-factorial process, characterized by significant alterations at both genetic and molecular levels. To comprehensively elucidate these complex pathogenic pathways, it is firstly necessary to understand the structural organization of the human intestinal epithelium. The normal colonic mucosa consists of a sheet-like layer of epithelial cells that fold into cylindrical protrusions known as crypts, which are supported by the lamina propria, a layer of interstitial matrix and loose connective tissue that is situated directly beneath the epithelium (Fig. 2). These crypts represent the fundamental functional units of the intestinal system.11 The colonic crypt can be divided into three distinct zones (Fig. 2): the stem-cell (SC) niche at the base of the crypt, the transit-amplifying compartment in the middle, and the differentiated cell zone, at the top the crypt. The lower crypt layer is composed mainly by SCs and surrounding mesenchymal cells. SCs exhibit two distinctive functional characteristics: the ability of self-renewal through division, thus maintaining an undifferentiated state, and totipotency, the capacity to generate any differentiated cell type present in the tissue of origin. When the SCs divide in an asymmetrical way, they renew themselves and generate transit-amplifying cells, an intermediate cell type that is not yet fully differentiated and that populate the intermediate zone of the crypt. As these cells move up the crypt, they multiply and differentiate into one of the three types of colon epithelial lineages: the colonocytes, the mucus-secreting goblet cells or the enteroendocrine cells.12 The specific cellular compositions of each zone contributes to the continuous renewal of the colonic epithelium, as reviewed by Ricci-Vitiani et al.12 An illustration of the composition and layout of the human colonic crypt can be found in Fig. 2.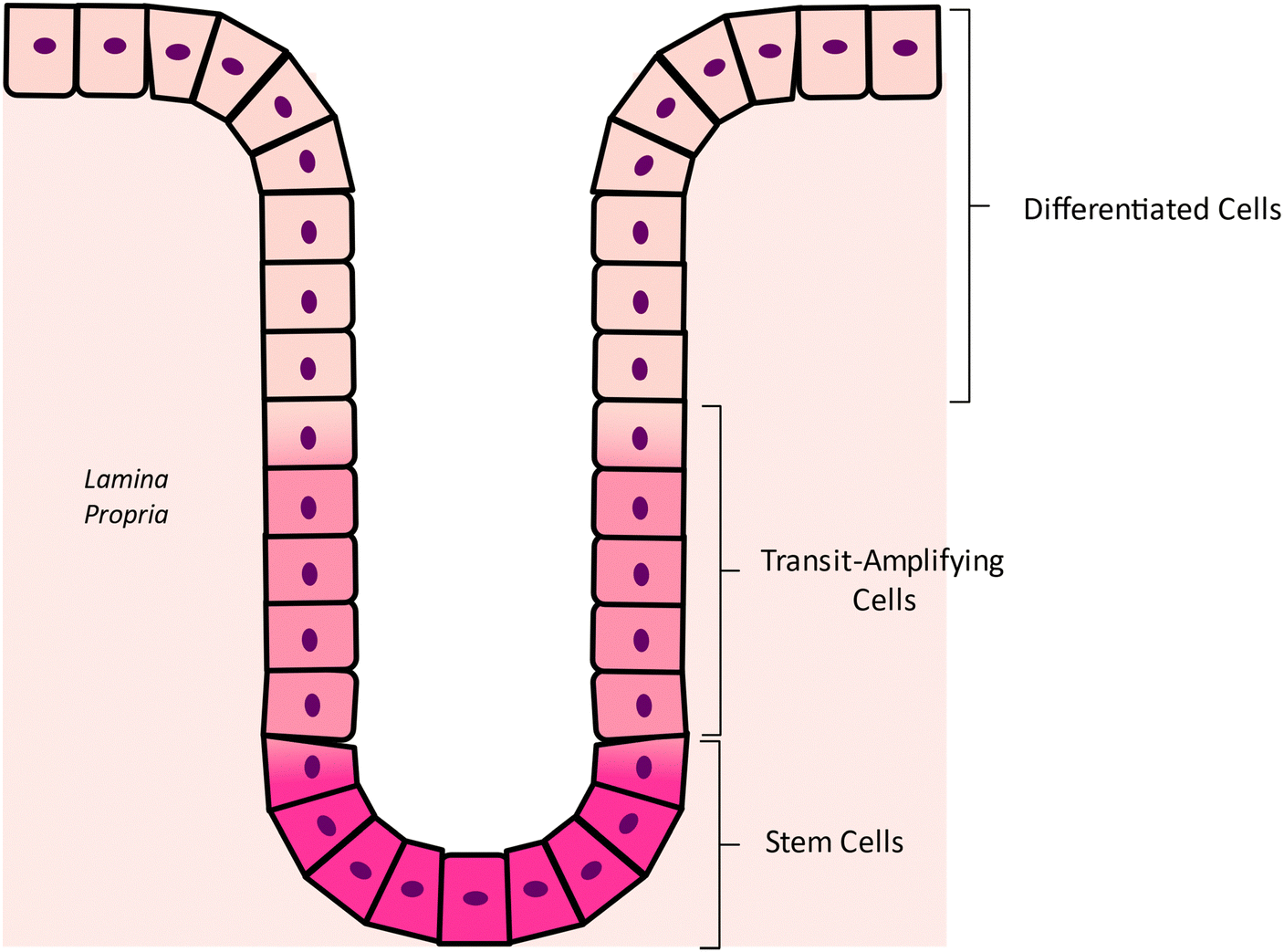 | ||
| Fig. 2 Illustration of the colonic crypt composition and layout. | ||
While SCs play a pivotal role in the homeostasis of the intestinal self-renewal process, they can also be one of the main causes for the onset and development of CRC. The normal processes of maintenance, proliferation and differentiation of stem-cells are tightly regulated by signaling molecules, presumably produced by the surrounding mesenchymal cells, and belonging to the Wnt/β-catenin signaling pathway.12 Any disruption or aberrant activation of this pathway can lead to dysregulated stem cell behavior.
The Wnt/β-catenin pathway is often associated with a large multitude of diseases, especially cancer, due to its regulatory role in critical cellular processes, such as cell migration, differentiation, survival, and proliferation, as reviewed by Zhao et al.13 In the absence of Wnt ligands, the β-catenin protein, usually found in the cytoplasm, undergoes phosphorylation, marking it for degradation. This process is facilitated by a complex comprising glycogen synthase kinase 3β (GSK3β), casein kinase I (CK I), Axin, and the tumor-suppressor protein adenomatous polyposis coli (APC). In this complex, GSK3β stimulates the phosphorylation of β-catenin, while APC mediates its interaction with the ubiquitin-mediated proteolytic pathway in the cytoplasm. Axin, in turn, serves as the scaffold for the development and maintenance of this entire protein complex.13 However, upon binding of Wnt ligands to their respective receptors, the previously described complex is deactivated, resulting in an accumulation of cytoplasmatic β-catenin. The stabilized β-catenin translocates to the nucleus, where it activates transcription factors capable of modulating genes such as MYC, a proto-oncogene that stimulates the cell to enter the S-phase of the cell cycle, where DNA synthesis occur14 (Fig. 3). As a result, the dysregulation of MYC and other transcription factors expression can disrupt normal cell cycle regulation, leading to increased cell division and uncontrolled proliferation.
 | ||
| Fig. 3 Schematic representation of activated and inhibited Wnt/β-catenin pathway. | ||
After the acquisition of mutations by a basally situated crypt stem cell, it originates a mutant clone that sets off phenomenon known as niche succession. During this process, the mutant clone colonizes the base of the crypt, replacing the unmutated stem cells in this region.15
As the mutant stem cells proliferate, they will, eventually, fully occupy the entirety of the niche, marking an event termed monoclonal conversion,16 which gives rise to the first neoplastic precursor lesion, the monocryptal adenoma. The progression of this first lesion involves its expansion through crypt fission, the process by which colonic crypts in humans divide. This generates adenomas, which can acquire further mutations in key regulatory genes, promoting their transformation into malignant lesions known as adenocarcinomas. Upon this conversion, adenocarcinomas invade the submucosa layer and acquire metastatic capabilities, hence, in the context of the pathophysiology of CRC, marking a critical point in the cancer development process.
Due to the crucial participation of SCs, this type of cancer can be defined as a stem cell disease, as they are the only cells to have the intrinsic self-renewal ability and the capacity to attain the quantity of consecutive mutations required for the transformation of a cell from a normal state to a cancerous one. In CRC, the most common sequence of mutations, and thus the responsible for the vast majority of cases, is the adenoma–carcinoma sequence, firstly described by Vogelstein et al. in 1988,17 who state that during the development of the solid tumor, mutations are acquired in a specific sequence, increasing the genetic instability of the neoplasm. Usually, the APC gene is the first to suffer a “hit”, or mutation, followed by the activation of RAS oncogene and function loss of the tumor suppressor gene TP53. Of course, CRC can have different genetic backgrounds, and its pathogenesis may not always follow the same order of events and other genetic pathways have been established, namely the serrated neoplasia pathway, and the microsatellite instability pathway, both affecting different genetic and epigenetic factors and, also, with much smaller incidences, as reviewed by other studies.18,19
2.2. Etiology and associated risk factors
Colorectal cancer is a complex disease with multifaceted origins that are intrinsically intertwined with the patient's environmental exposures and lifestyle practices. The primary habits and associated risks contributing to the development of colorectal cancer are illustrated in Fig. 4. | ||
| Fig. 4 Compilation of the various risk factors for the development of colorectal cancer. | ||
A important category of factors that heavily influences the probability of developing CRC is the one that includes the hereditary factors, as it is estimated that 10 to 20% of individuals diagnosed with CRC exhibit a positive familial history regarding this particular malignancy.20 The degree of risk is contingent upon the number and genealogical proximity of the affected relatives, resulting in the heritability of CRC ranging from 12 to 35%, underscoring a substantial genetic contribution.21,22 Some studies have correlated single nucleotide polymorphisms with cancer susceptibility, however it is estimated that they only account for a small fraction of CRC heritability and that most of the causative factors are still unknown.23–26
Age and gender also constitute additional risk factors for CRC development. The intricate relationship between age and CRC emerges from the alterations in DNA repair and cellular regulatory mechanisms that naturally occur with advancing age. Consequently, colorectal cancer exhibits a pronounced association with older individuals, with a heightened incidence observed in those aged over 50 years.27 However, this paradigm seems to be shifting due to the escalating incidence of young-onset CRC diagnostics witnessed over the past two decades.28 Regarding gender as a risk factor, males present a 1.5 times greater susceptibility to developing colorectal cancer and exhibit an almost 25% higher likelihood to die from the disease when compared to their female counterparts.29 This effect has been associated with the distinct dietary choices and risk behaviors associated to men. Due to this discrepancy of incidence between males and females, the creation of gender-individualized screening guidelines for colonoscopy has been suggested, in an attempt to enhance early detection in both males and females.30
Tobacco use has been consistently associated with an increased risk of CRC.31 Although the smoke is not in direct contact with the colorectal mucosa, procarcinogens and carcinogens, such as polycyclic aromatic hydrocarbons and nitrosamines, present in cigarette smoke are absorbed into the bloodstream by the lungs. Once systemically distributed, these compounds can form DNA adducts and induce mutations in cells of several organs and tissues.32 In the context of CRC, these compounds damage the colorectal mucosa and are linked to the development of colorectal adenomatous polyps, which are known to be CRC precursor lesions.33 In the same paradigm of substance abuse, alcohol has also been identified as a risk factor for the development of CRC, mainly due to the metabolites arising from ethanol breakdown, notably acetaldehyde. The enzymatic activity of alcohol dehydrogenases mediates this breakdown, yielding metabolites with pronounced carcinogenic properties, particularly implicated in the induction of irreversible DNA damage.34,35
In addition to all the factors already mentioned, the dietary choices and nutritional status of individuals or populations play a pivotal role in determining the risk of developing CRC. Therefore, the adoption of a healthful dietary regimen assumes significance not only in mitigating the risk of CRC development but also in serving as a preventive measure against its occurrence. A dietary pattern characterized by a high intake of red meat, sugars, and processed foods is notably associated with an elevated predisposition of CRC development, primarily attributed to the induction of oxidative stress and inflammation by these dietary components. The consumption of red and processed meat is specifically linked to the formation of heme iron compounds, known to generate reactive oxygen species that instigate oxidative stress. In turn, this environment leads to DNA damage and the initiation of carcinogenesis through the triggering of mutagenic events. Also, during meat cooking and processing, the generation of carcinogenic and mutagenic compounds, such as heterocyclic amines, polycyclic aromatic hydrocarbons, and N-nitroso compounds, may occur.36 Moreover, high consumption of sugars may lead to insulin resistance and provoke type 2 diabetes, which, due to the widely recognized effect of insulin in stimulating colonic development, can further prompt cancer carcinogenesis.37,38 Furthermore, a dietary regimen that does not include an adequate intake of foods rich in fiber, antioxidants, calcium, vitamin D, and sulforaphane may also be regarded as another risk factor for CRC development. These essential nutrients play a crucial role in the prevention of oxidative stress and inflammation, both of which are pivotal contributors to the carcinogenic process. When the calcium intake is adequate, the free calcium present in the gut can bind to the bile acids produced during fat digestion and thus neutralize them and their mutagenic properties, preventing cellular damage and hyperproliferation. Extracellular calcium and calcium-sensing receptors are also linked to pathways involved in the control of cellular proliferation, differentiation, and apoptosis. In the regulation of colon epithelial cells, both compounds work together in the suppression of the β-catenin binding to transcription factors.39 In turn, mechanisms associated with vitamin D appear to be complementary to the ones influenced by calcium. For example, vitamin D is involved in bile acid catabolism, as, together with lithocholic acid (LCA), activate vitamin D receptors (VDR), which performs a detoxification of LCA in the intestine.40 Additionally, ligand bound VDR can also induce cell arrest in G1 phase, which is an important way to proportion protection against CRC, as it promotes cell apoptosis.41 Furthermore, vitamin D signaling interacts with several growth-factor pathways, which results in growth suppression and has important effects in immunity and inflammation control, which may be of extreme importance in colorectal carcinogenesis and its prevention.42 Similarly, sulforaphane (SFN), a naturally occurring compound found in cruciferous vegetables, also exhibits cancer-inhibitory properties. In the context of CRC, SFN exerts its inhibitory effects by impeding one of the central pathways associated with the disease, the Wnt/β-catenin signaling pathway. Specifically, SFN inhibits the formation of active β-catenin-based transcription complexes within the nucleus, which results in the cessation of stimulated cell proliferation, while it is also observed an increase in cell apoptosis.43 In Table 1, we can find a summary of the above-mentioned dietary factors that, in one way or another, influence CRC carcinogenesis, as well as the corresponding mechanisms.
| Dietary factor | Associated mechanism | Implications on CRC |
|---|---|---|
| Red meat consumption | Heme iron compounds generate reactive oxygen species (ROS), leading to oxidative stress. | Trigger for mutagenic events that can start carcinogenesis. |
| Meat cooking and processing | Generation of heterocyclic amines (HCAs), polycyclic aromatic hydrocarbons (PAHs), and N-nitroso compounds (NOCs). | Promotion of mutagenic events. |
| Sugar | Insulin resistance and type-2 diabetes. | Insulin growth factor 1 (IGF-1) stimulates cell proliferation. |
| Calcium | Bile acids binding; suppression of β-catenin binding to transcription factors. | Prevention of cellular damage and control of proliferation, differentiation, and apoptosis. |
| Vitamin D | Bile acids catabolism; ligand bound VDR; vitamin D signaling. | Intestine detoxification; cell arrest in G1 phase; growth suppression, immunity, and inflammation control. |
| Sulforaphane | Inhibition of active β-catenin formation. | Cessation of stimulated cell proliferation and increase in cell apoptosis. |
In recent years, the lack of physical activity and an overall sedentary lifestyle, has become significant contributors to the onset of various diseases. This lifestyle, coupled with poor dietary choices, contributes to the emergence of an overweight population that, ultimately, may progress into obesity. Obesity, characterized by intrinsic features such as chronic inflammation, is identified as a triggering factor for colon cell carcinogenesis. This exacerbated inflammatory response is strongly associated with the endocrine functions of the adipose tissue, as it secretes free fatty acids (FFAs) and pro-inflammatory molecules, such as adipokines and chemokines, which leads to a large dysregulation of inflammatory pathways, thus creating a well-suited environment for tumor cells proliferation.44
2.3. Intervention for the prevention and treatment of CRC
As CRC is mainly associated with modifiable risk factors, the incidence and mortality of this disease can be lowered by raising awareness for certain lifestyle changes, as having a well-balanced diet, minimal consumption of alcohol and tobacco, and regular physical activity. To ensure that prevention strategies are reaching the ones that need the most, certain risk calculations models that consider the genetic background and the environmental factors of each individual have been in development, establishing the risk to develop CRC, as well as the most appropriate to start screening for the disease.45Concerning treatment modalities, CRC can be managed through various procedures or a combination of them. Surgery remains the primary approach for CRC treatment, employing specific methods for the removal of tumors and associated lymph nodes, as comprehensively reviewed by Dorudi et al.46 Additionally, radiotherapy has demonstrated promising outcomes when employed as a preoperative intervention, effectively mitigating the overall risk of local tumor recurrence.47 In cases where the disease is at its initial stages, endoscopy emerges as a viable option for the resection of malignant polyps.48
3. Lipidomics in colorectal cancer research
Despite significant advancements in understanding colorectal cancer (CRC) pathophysiology, current diagnostic and screening methods remain challenging due to their invasiveness and complexity. These limitations contribute to reduced patient adherence, increased levels of misdiagnosis, and, ultimately, to a rise in CRC's incidence and mortality. In response, researchers have tried to overcome these obstacles through the identification of possible biomarkers, however, finding specific and trustworthy ones is a main issue in cancer research.Lipids are a diverse group of biomolecules that, according to the LIPID MAPS, are organized into eight main categories: fatty acyls, glycerolipids, glycerophospholipids, sphingolipids, sterol lipids, prenol lipids, saccharolipids, and polyketides.49 These groups comprise several classes and sub classes contributing to the complexity of the lipidome. Lipids have essential roles in cellular physiology, functioning as structural components of membranes, energy reservoirs, and key mediators in signaling pathways. Under healthy conditions, the lipidome remains relatively stable; however, it undergoes significant alterations in various pathological states. This context-dependent modulation of the lipidome not only reflects disease-specific biochemical changes but also presents a valuable opportunity to elucidate pathophysiological mechanisms and identify potential biomarkers. Consequently, lipidomics has emerged as a powerful and promising platform for the investigation of disease processes and biomarker discovery. Lipidomics aims for the comprehensive analysis of the lipid profile within a certain biological system, the lipidome, through the use of mass spectrometry (MS) analytical solutions, usually coupled with liquid chromatography, are the most common analytical solutions and allowing the identification and quantification of the lipid species in biological samples.50 Differences in methodologies and analytical measures across different laboratories lead to significant challenges in data interpretation, result comparison and clinical translations between different studies.
Given that the dysregulation of normal lipid metabolism is associated with the onset and development of several diseases and is already considered a hallmark of cancer,51 clinical lipidomics has emerged has a crucial subfield of lipidomics. Its primary goal is to translate lipidomic research into clinical applications, particularly to improve disease prediction, diagnostic and management.52
In the case of CRC, altered lipid metabolism and its association with the onset, progression and metastasis of this disease has been increasingly studied.53,54 To fulfill the demands of sustained growth and proliferation, cancer cells suffer a metabolic reprogramming, often associated with lipid metabolism. In fact, upregulation of de novo fatty acid synthesis, driven by increased activity of enzymes such as fatty acid synthase (FASN), is associated with the incorporation of saturated fatty acids into the membrane, enhancing the protection of cancer cells against free radicals and therapeutics.55 FASN can also be associated with lipid β-oxidation and promotion of cellular respiration, giving another survival advantage to CRC cells.56 Furthermore, anti-inflammatory lipid mediators such as lipoxin A4 (LXA4), that are reported to have an important inflammatory pro-resolving effect in intestinal inflammation, are usually found at lower level in CRC.57
Henceforth, CRC has increasingly become the focus of investigations employing lipidomic approaches to comprehensively explore various aspects of the disease. To better understand the key characteristics of the existing body of literature associated to CRC and lipidomics, we performed a comprehensive overview of such literature. To this end, the following query string of keywords was inserted into the Scopus database: “TITLE-ABS-KEY (lipidomic*) AND (colorectal AND cancer OR crc OR colon AND cancer)”. This retrieved a total of 201 documents. Several filters were then applied to narrow these documents to original research articles, thereby excluding reviews (47), editorials (5), book chapters (3), notes (2), letters (2), and errata (1). Additionally, articles not written in English were excluded, resulting in the removal of one article in Chinese and another in Russian. This process resulted in a universe of 139 articles. Further inspection was performed to exclude any other articles that may have evaded the applied filtering or did not meet the requirements for this study, e.g. studies that did not employ mass-spectrometry-based approaches, studies that were not CRC-specific and studies that did not present lipidomic-related results. This refinement process resulted in a reduced total of 68 articles. Subsequently, these articles were categorized based on specific characteristics, including the matrix used in the studies, the primary objectives of the study concerning CRC, and whether a global lipidome characterization was undertaken. The graphical representations of these literature characteristics were compiled into Fig. 5.
 | ||
| Fig. 5 (A) Graph illustrating the distribution of literature, regarding colorectal cancer (CRC) and lipidomics, based on the type of matrix utilized in the respective studies. The distinct colors represent the frequency of studies employing different matrices such as animal models, plasma (which comprises both plasma and blood serum), tissue, cell lineages and fecal samples. It was also included the “multiple matrices” category for studies that used two or more of the mentioned matrices. (B) Graph illustrating the published studies classified according with the objective related to CRC. Light blue color denotes the studies that focused on comparing CRC samples with healthy controls (HC) while the darkest blue represents the studies that focused on studying the dysregulations associated with CRC progression. The green color corresponds to studies that, instead, focused on the response of a specific treatment, the dysregulation of a specific pathway or the action of a certain risk factor. (C) Graph representing a distinction between studies that focused their lipidomic research in a specific lipid class or specie and the ones that performed a global overview of the lipidome. | ||
As illustrated in Fig. 5(A), there exists considerable variability in the matrices employed in different studies. The most prevalent matrices include animal models, human CRC tissue, cell lineages and plasma, with latter comprising studies that used either plasma or blood serum. Regarding the study objectives concerning CRC, Fig. 5(B) shows that a predominant portion of studies focused on either conducting direct comparisons between samples derived from CRC-related specimens and those from healthy controls or, alternatively, aimed at investigating the metabolic and lipidic dysregulations that may manifest during CRC progression. Nevertheless, a significant number of papers did not fall into either of these categories, as they explored diverse aspects such as the response to a specific treatment, the dysregulation of a particular pathway, or the influence of a specific risk factor, among other variables. Lastly, in Fig. 5(C), it is represented the number of papers that have performed either untargeted lipidomic analyses, characterizing the entire lipidome, or those using a target approach, concentrating on specific lipid classes or species. By interpreting the graphic representation, it is hereby established that the majority of the lipidomic and CRC associated papers (59%) performed a global overview of the lipidome, while the remaining studies performed targeted analysis, focusing on specific classes or species of lipids.
As previously stated, this work aims to review the main works that, using lipidomic approaches, described a lipid dysregulation associated with CRC. However, it is also our objective to enhance studies that establish such dysregulation using human matrices. For this reason, when analyzing articles to then be organized in Tables 2 and 3, articles in which human matrices were used, such as plasma and tissue, were prioritized over the ones that used cell lines or animal models. Therefore, from each article whose methods fit our parameters, information such as the analyzed sample, which lipid extraction method was used, what was the lipidomic analytical approach used and which lipid species were dysregulated, among other details, were retrieved and organized in Tables 2 and 3. In the manuscript, we have adopted the nomenclature of LIPID MAPS,58 and the level of identification was aligned with what was described in the original papers.
| Plasma/serum | |||||
|---|---|---|---|---|---|
| Methodology | Study details | Results | Article ref. | ||
| Lipid extraction method | Analytical method | ↑ Increased | ↓ Decreased | ||
| ESI – electrospray ionization; MS – mass spectrometry; LC – liquid chromatography; Q-ToF – quadrupole time-of-flight; UHPLC – ultra high performance liquid chromatography; CRC – colorectal cancer; AD – adenomatous polyps; LPC – lysophosphatidylcholine; SPC – sphingosylphosphorylcholine; FFA – free fatty acids; TG – triglycerides; GluCer – glucosylceramide; LacCer – lactosylceramide; PG – phosphatidylglycerol; PE O- – phosphatidylethanolamine plasmalogen; SM – sphingomyelin; FAHFA – fatty acid ester of hydroxy fatty acid; CE – cholesteryl ester; LPI – lyso-phosphatidylinositol; Cer – ceramide; PUFAs – polyunsaturated fatty acids; PC – phosphatidylcholine. | |||||
| HC vs. CRC | |||||
| Chloroform/H2O (1:1, v/v) |
Triple QqQESI-MS (targeted) | 133 CRC patients | — | Total LPC, saturated LPC, unsaturated LPC, LPC(16:0), LPC(18:0), LPC(18:1), LPC(18:2), total 18-LPC, and LPC(20:0) |
Zhao et al.59 |
| Folch method chloroform/methanol (2:1, v/v) |
2D LC-ESI-QToF (untargeted) | 25 CRC patients | FFA(22:2), FFA(22:4), FFA(20:1), FFA (20:2); TG(56:6), TG(52:2), TG(52:1); GluCer(42:3;O2), GluCer(42:2;O2), GluCer(36:4;O2), GluCer(34:1;O2), GluCer (33:2;O2), LacCer(42:4;O2), LacCer(40:1;O2), LacCer(40:2;O2), LacCer(40:4;O2), LacCer(38:1;O2), LacCer(35:1;O2); PG(34:0); SM(42:2;O2) |
LPC(20:3), LPC(18:3), LPC(18:2), LPC(14:0); PE O-(36:3), PE O-(38:3); SM(38:8;O2) |
Shen et al.60 |
| CRC progression | |||||
| Chloroform/H2O (1:1, v/v) |
LC-ESI-QTrap (untargeted) | 120 HC, 120 AP patients and 120 CRC patients | — | Total LPC, saturated LPC, unsaturated LPC, LPC(16:0), LPC(18:0), LPC(18:1), LPC(18:2), and LPC(22:6); SM(16:0;O2) and SM(18:0;O2) in AP; SPC in CRC |
Li et al.61 |
| Modified bligh and dyer method | UHPLC-ESI-Q-orbitrap (untargeted) | 50 HC, 46 CAA patients and 50 CRC patients | PC 36:1e and PC 38:6; SM 36:1;O2, SM 36:2;O2 and SM 40:3;O2 |
PC 32:3, PC O-35:6/PC P-35:5, PC 37:7, PC 39:6 and PC 44:5 |
Chen et al.62 |
| Bligh and dyer method | UHPLC – ESI-TripleTOF (untargeted) | 20 stage I/II CRC patients and 20 stage III/IV CRC patients | CE(20:4); TG 40:0 (TG 12:0_12:0_16:0), TG 42:0 (TG 12:0_14:0_16:0), TG 44:0 (TG 14:0_14:0_16:0), TG 46:0 (TG 14:0_16:0_16:0), TG 48:0 (TG 16:0_16:0_16:0), and TG 54:0 (TG 16:0_18:0_20:0) |
FAHFA 27:1 (FAHFA 9:0_18:1) |
Liu et al.63 |
| Tissue | |||||
|---|---|---|---|---|---|
| Methodology | Study details | Results | Article ref. | ||
| Lipid extraction method | Analytical method | ↑ Increased | ↓ Decreased | ||
| FIA – flow injection analysis; ESI – electrospray ionization; MS – mass spectrometry; LC – liquid chromatography; Q-ToF – quadrupole time-of-flight; UHPLC – ultra high performance liquid chromatography; CRC – colorectal cancer; AD – adenomatous polyps; LPS – lysophosphatidylserine; TG – triglycerides; LPG – lysohosphatidylglycerol; LPI – lysophosphatidylinositol; LPA – lysophosphatidic acid; Cer – ceramide; PUFAs – polyunsaturated fatty acids. | |||||
| HC vs. CRC | |||||
| Bligh and dyer method | FIA-ESI-TripleQ and FIA-FTMS (untargeted) | Discovery (n = 106) and validation cohorts (n = 28 and n = 26) | TG(56:4), (56:5), and (56:6) containing PUFA (FA 18:2, 20:2, 20:3, 20:4, 20:5, 22:4, 22:5, 22:6) |
— | Ecker et al.93 |
| Methanol with zirconium beads | LC-ESI-TripleQ (targeted) | 11 CRC patients | Total LPI and LPS; LPI 18:0, LPI 20:4; LPG 18:1, LPG 18:2, LPG 22:6; LPS 18:0, LPS 18:1, LPS 20:3, LPS 20:4, LPS 22:6 |
Total LPA, LPA 18:1, LPA 18:2 |
Kitamura et al.94 |
| HC vs. CRC and CRC progression | |||||
| MTBE/MeOH (5:1.5; v/v) |
NanoESI-Q-Orbitrap (untargeted) | 20 patients with sporadic CRC | LPI(18:1) and LPI(18:0); Cer(32:1;O2), Cer(34:2;O2), Cer(34:1;O2), Cer(40:2;O2), Cer(41:2;O2), Cer(42:3;O2), Cer(42:2;O2), Cer(43:2;O2) |
T3/T4 compared to Tis/T1/T2: total PE plasmalogens | Wang et al.86 |
3.1. Profiling of the lipidome in CRC human plasma/serum
The analysis of biofluids, such as plasma, is crucial to achieve a comprehensive understanding of the dynamic physiological and pathological mechanisms of diseases. Although plasma lipid composition is influenced by dietary factors, it also reflects systemic and organ-specific changes, and may provide insights into cancer's impact and potentially uncover biomarkers for diagnosing and understanding CRC in turn, could open new paths in the diagnosis and therapy of this disease.With the objective of characterizing the lipidome alterations associated with CRC, a study found in the literature performed untargeted analysis of the lipid profile of plasma from patients with CRC and healthy volunteers.60 This study showed that some lipid species presented significant content differences, which marked them as possible biomarkers for CRC. For example, some FFAs (FFA(22:2), FFA(22:4), FFA(20:1), FFA (20:2)) and TGs (TG(56:6), TG(52:2) and TG(52:1)) were found to be upregulated in CRC, which seems to be related with a dysfunction in the fatty acid β-oxidation metabolism, an important pathway where ATP is produced through the β-oxidation of FFAs that, in turn, were originated by the breakdown of TGs. The increase of TGs and FFAs levels may suggest that CRC cells are adjusting the lipid metabolism to guarantee the energy source. In an opposite trend, several LPC species (LPC(20:3), LPC(18:3), LPC(18:2) and LPC(14:0)) were found to be with lower levels in CRC patients. Herein, the decrease of LPC species can be correlated with the increase of expression of acyl-CoA:lyso-phospholipid acyltransferases that catalyze the acylation reaction of LPCs to form phosphatidylcholine (PC). Although the relationship of these enzymes with CRC is still not fully understood, their overexpression in this disease has already been reported in the literature.64 However, the lowering of LPC levels can also be related with other enzymes involved in lysophospholipid metabolism, such as for lysophospholipase D (lyso-PLD) and autotaxin (ATX). These enzymes convert LPC to lysophosphatidic acid (LPA), which has been linked with the regulation of processes such as cell migration, adhesion, proliferation and angiogenesis, all crucial mechanisms to cancer pathogenesis.65 Sphingolipids were also found to be dysregulated. Previous studies have correlated sphingomyelin (SM) with roles in cellular growth, apoptosis, cell adhesion, differentiation, and migration processes.66,67 Also, ceramide (Cer) which is characterized by having proapoptotic and anti-proliferative properties, was found to be downregulated in human colorectal adenomas, while the levels of sphingosine-1-phosphate (S1P), a pro-proliferative molecule, was found to be increased in CRC.68 Ceramide is also a precursor for the synthesis of more complex sphingolipids, such as glucosylceramide (GluCer) and lactosylceramide (LacCer). In fact, several forms of both of these lipid species were found to be significantly increased in CRC, namely GluCer(42:3;O2), GluCer(42:2;O2), GluCer(36:4;O2), GluCer(34:1;O2), GluCer (33:2;O2), LacCer(42:4;O2), LacCer(40:1;O2), LacCer(40:2;O2), LacCer(40:4;O2), LacCer(38:1;O2), and LacCer(35:1;O2). This dysregulation was justified by the possible amplified activity of the glucosylceramide synthase and of the LacCer synthetic pathway, which can be relevant to CRC due to their roles in sphingolipid metabolism, which has implications for cancer progression, metastasis, and treatment resistance. These results marked both metabolic pathways as possible targets for further studies that aim to discover more about their relationship with CRC.
A targeted analysis of the lipidome focused on evaluating the viability of lysophosphatidylcholines (LPCs) as cancer biomarkers compared the lipid profile of the plasma of 133 CRC patients and 125 healthy controls.59 It was noticed that in cancer samples there were significantly reduced levels of LPCs, in specific, total LPC content, saturated LPC, unsaturated LPC and LPC molecular species, namely LPC(16:0), LPC(18:0), LPC(18:1), LPC(18:2), total 18-LPC, and LPC(20:0). These results are in agreement with data from untargeted analysis published in other studies that have described the decrease of LPC(18:2).60 These altered levels of LPCs are likely due to the altered expression of the already discussed enzymes acyl-CoA:lyso-phospholipid acyltransferases, lyso-PLD and ATX.64,65 However, it is also important to acknowledge that decreased levels of LPCs are not exclusive to CRC. This trend of downregulated levels of LPCs has been reported in other metabolic disorders, including obesity and type 2 diabetes mellitus,69 as well as hyperlipidemia and cardiovascular diseases.70 As such, these alterations may reflect dysregulations associated with the overall metabolic health, rather than being specific to CRC pathophysiology. In fact, both studies reviewed above did not discriminate variables such as body mass index (BMI), type 2 DM or age and, therefore, influence of these factors on the observed lipid profiles cannot be ruled out.
In addition to directly comparing the lipidomic profiles of healthy individuals and patients diagnosed with CRC, it is also crucial to consider cancer as a progressive and evolving disease. Therefore, important insights can be gained when studying disease progression, particularly in its earliest stages, before cancer is fully established. As such, it is important to evaluate whether lipidomic variations are already detectable in precancerous lesions, such as colorectal advanced adenoma (CAA) and, more broadly, adenomatous polyps (AP). Usually, the cohorts in these studies consist of three different groups: a control group of healthy subjects, a group comprising individuals with pre-cancerous lesions, and a group of patients diagnosed with cancer. The results are often analyzed through comparisons among all three groups simultaneously or, alternatively, by comparing two of the three groups, depending on the specific findings.
As previously stated in this review, the adenoma–carcinoma sequence is one of the main mechanisms through which CRC develops, with colorectal adenomas being the type of lesions most associated to further develop into cancer.17 In this context, an untargeted lipidomic analysis on serum samples from a cohort comprising 50 healthy controls, 46 CAA patients, and 50 CRC patients have identified lipid species with strong discriminatory power between CAA and CRC, which could hold significant utility in the early diagnosis of CRC, while also contributing to the exploration of lipid metabolic pathways associated with disease progression.62 This study emphasized a strong correlation between the dysregulation of PC and SM metabolism and the malignant development of CAA into CRC, as 67% of the identified differential lipids belonged to these two lipid classes. In fact, the 10 lipid species with the highest discriminatory power between the CAA and CRC groups were either PCs or SMs. Specifically, the levels of PC O-36:1/PC P-36:0, PC 38:6, SM 36:1;O2, SM 36:2;O2, and SM 40:3;O2 were noticeably upregulated, while the levels of PC 32:3, PC O-35:6/PC P-35:5, PC 37:7, PC 39:6, and PC 44:5 were significantly downregulated in CRC samples compared to CAA group. Of these, PC O-35:6/PC P-35:5, PC 44:5, and SM 40:3;O2 were considered the most promising biomarkers to differentiate CAA from CRC. Notably, an earlier study had already identified PC O-35:6/PC P-35:5 and PC 44:5 as lipid species with good diagnostic accuracy for CAA, a finding that this study extends by also showing their strong discriminatory capacity between CAA and CRC.71 PCs are essential phospholipids involved in the regulation of several key cellular processes, including cell growth, survival, proliferation and membrane structure maintenance. As such, any dysregulation within PC metabolism can disrupt these processes and contribute to the development of pathological conditions, such as cancer. While PCs are often reported to be upregulated in cancer, reflecting the increased membrane biosynthesis required for the rapid proliferation of tumor cells,72 this study found many PC species to be downregulated in CRC. This may be correlated with their known anti-inflammatory potential, as exogenous PCs have been shown to inhibit cytokine tumor necrosis factor (TNF-α)-induced pro-inflammatory signaling and gene expression in intestinal epithelial cells by interfering with nuclear factor-κB (NF-κB) activity.73,74 Given that TNF-α is a regulator of epithelial function during inflammatory processes, reduced PC levels could facilitate pro-inflammatory microenvironment in CRC. Notably, levels of TNF-α were found to be increased in human inflammatory bowel disease, a well-established risk factor for CRC,75 which suggests that PCs may have a protective effect through the modulation of inflammatory pathways.73 Additionally, the level of PCs may also be downregulated due to their role as precursors to other major phospholipid classes, such as LPCs and SMs. Regarding LPCs, multiple studies have found this class of lipids to be downregulated in CRC.59–61 As previously described, this decrease may be associated with the overexpression of enzymes such as LPCAT, which catalyzes the conversion of LPCs to PCs, highlighting the complex and not yet fully understood modulation of the lipid metabolism of both classes in cancer.64 Furthermore, enzymes like lyso-PLD and ATX, responsible for the production of LPA through LPCs, can also come into play.65 On the other hand, all SMs of the ten initial potential biomarkers identified were found to be upregulated in CRC patients. As reviewed elsewhere, disturbances in sphingolipid metabolism may contribute significantly to the development and progression of CRC, mainly due to their influence in important cellular processes, such as cell growth, proliferation and programmed cell death.76 In this case, the elevated levels of SM can be correlated to reduced activity of the alkaline sphingomyelinase (SMase), an enzyme that catalyzes the conversion of SM to ceramide (Cer). Evidence suggests that this family of enzymes exhibits decreased activity in CRC, leading to an accumulation of SM and a simultaneous reduction in Cer levels.77 This is in accordance with existing literature on the anti-tumorigenic properties of ceramides, which have been reported to be capable of inducing the activation of the apoptotic cascade and inhibiting tumor growth.78,79
An additional untargeted study carried out a plasma lipidome profiling across three groups: 120 healthy individuals, 120 patients with AP, and 120 patients diagnosed with CRC.61 This study aimed to evaluate the molecular and biochemical changes that may occur within the time gap that AP, a precursor lesion strongly associated with CRC, takes to fully evolve into cancer. Among its findings, this study described a decrease in the total levels of LPC, total saturated LPC, total unsaturated LPC and LPC molecular species, such as LPC(16:0), LPC(18:0), LPC(18:1), and LPC(18:2), similarly to what was previously described.59,60 This information may be relevant since LPCs can be a biomarker not only for the presence of CRC but also for the development of this disease, opening the possibility of early diagnosis when symptoms are very mild or even before symptoms start. However, levels of other lipids, such as sphingomyelin (SM) and sphingosylphosphorylcholine (SPC), did not show such gradual alterations. In fact, a decrease in plasma levels of SM was only found in individuals with AP, while SPC displayed a down-regulation of their levels exclusively in CRC patients. Both SM and SPC can be converted into S1P, a biomolecule that has been shown to regulate key processes for cancer progression, such as angiogenesis, cell growth, cell survival and cell invasion.80 Moreover, SM can undergo transformation into Cer, a pro-apoptotic molecule. Therefore, a decrease in SM may withhold the synthesis of Cer and thus contribute to the evasion of cell apoptosis and promoting malignant cell survival. Although the sensitivity and specificity of these biomarkers have not yet reached the required threshold, the findings in this study, conducted within a Chinese population, demonstrated high consistency with those reported in an American population by Zhao et al.,59 which suggests that these potential biomarkers may not be influenced by racial factors.
In a different study focused specifically on CRC progression, plasma samples were collected from patients categorized according to the TNM clinical staging system, which attributes a stage (I to IV) according to the extent of the tumor (T), the extent of spread to the lymph nodes (N) and presence of metastasis (M).63 The comparison between two groups of patients with stage I/II and stage III/IV of CRC, reported eight different lipid species that help distinguish between different stages of cancer. The cholesteryl ester (CE) CE(20:4) and TGs such as TG 40:0 (TG 12:0_12:0_16:0), TG 42:0 (TG 12:0_14:0_16:0), TG 44:0 (TG 14:0_14:0_16:0), TG 46:0 (TG 14:0_16:0_16:0), TG 48:0 (TG 16:0_16:0_16:0), and TG 54:0 (TG 16:0_18:0_20:0) were found to be increased, while the fatty acid ester of hydroxy fatty acid species (FAHFA) 27:1 (FAHFA 9:0_18:1) presented decreased levels. For the first time, TGs, particularly those containing saturated fatty acids, were reported to be the lipids with the most pronounced differentiation between distinct stages of CRC, with increased levels in plasma of advanced-stage CRC patients. TGs are mostly present in lipid droplets, a cytoplasmatic organelle that is involved in lipid storage and are mainly expressed in fat-storing cells.81 An increase in lipid droplets count constitutes a pathologic phenomenon that has been described in both cancer and inflammatory cells, as reviewed elsewhere.81 Also, the production of lipid droplets, facilitated by lysophosphatidylcholine acyltransferase 2, is associated with chemoresistance in CRC.82 Additionally, research propose that the knockdown of adipose triglyceride lipase (ATGL), the enzyme that sets a rate limit in TG hydrolysis, could be an important factor in the prevention of cell proliferation and invasion.83 The dysregulation of TG observed in conjunction with other studies on CRC corroborates the involvement of TG in the pathogenesis of this cancer type. In patients with advanced-stage CRC there was also an CE(20:4) increase, which is linked with the upregulation of the acylcoenzyme A:cholesterol acyltransferase gene family in CRC, whose suppression is correlated with the inhibition of cell proliferation, migration and invasion.84 On the other hand, FAHFA are lipid species frequently associated with both anti-inflammatory and antidiabetic properties. Consistent with reports of decreased levels observed in breast cancer patients,85 the findings of lower levels of FAHFA 27:1 (FAHFA 9:0_18:1) in this study, specifically among CRC stage III/IV patients, suggests that individuals with advanced-stage colorectal cancer exhibit lower plasma FAHFA levels, which may have increased the body's inflammatory response.
We should keep in mind that the dissimilar lipid classes reported in the original studies (as shown in Table 2) may also be influenced by differences in sample preparation and analytical methodologies. Therefore, findings must be validated using standardized methods.
3.2. Profiling of the lipidome in CRC tissue
As previously stated, the analysis of plasma is of crucial importance to monitor the dynamic lipid alterations associated with CRC. On the other hand, the lipidomic analysis of CRC tissue provides the unveiling of localized molecular signatures, such as altered lipid profiles and dysregulated lipid pathways within the tumor microenvironment (TME).The variation of the lipid profile of tumor tissue from different locations, namely from the rectum and the ascending, transverse, descending and sigmoid colon, was evaluated in comparison with normal tissue from the same individuals.86 Also, the lipidome differences seen throughout CRC progression were asserted by comparing early staged tumors with more advanced ones. The levels of the lipid species LPI(18:1) and LPI(18:0) were found to be elevated in CRC tissue, an event that is most likely associated with a possible up-regulation of the G-protein coupled receptor 55 (GPR55). This LPI receptor is known for being involved in the Ca2+ mobilization, and in the activation of signaling proteins that promote migration, therefore playing an essential role in cancer metastasis.87,88 Also, the total amount of Cer was also increased in tumors, mostly due to the increase of Cer(32:1;O2), Cer(34:2;O2), Cer(34:1;O2), Cer(40:2;O2), Cer(41:2;O2), Cer(42:3;O2), Cer(42:2;O2), Cer(43:2;O2). An increase in the lipid contents of Cer is probably due to an increase of the expression of synthases (CerS) in CRC, which was already proved in previous works, however the role of this lipid class in colorectal cancer is still elusive due to the different effects that some species inside this class present.55 Considering the differences between different stages of CRC progression, a decrease in ether phospholipids, namely phosphatidylethanolamine plasmalogens (PE P-) was noticeable in advanced tumors with increased invasion. Plasmalogens are important endogenous antioxidant molecules, and their downregulation indicates an imbalance probably due to oxidative stress conditions. Although oxidative stress has been detected in almost every type of cancer, tumor cells usually express antioxidants in an attempt to detoxify the ambient from ROS and thus develop freely, which suggests that there is a delicate homeostasis between ROS and cancer cell function.89 With this into account, the lower levels of PE P- found suggest that oxidative stress may be enhanced in more advanced stages of the disease and that PE P- may have a regulatory role in tumor growth and further progression, if capable of reducing ROS. Additionally, LPC levels were, once again, found to be decreased, which is in accordance with other studies.59,61
Across several studies, some specific lysophospholipid (LPL) species and/or classes have been reported to be dysregulated in CRC. Additionally, although LPLs are usually present in smaller amounts than other lipid classes, they still play critical functional roles within cellular processes. Thus, to evaluate specific changes in lysophospholipid levels associated with CRC, a study performed a targeted lipidomic approach in both normal and tumor tissue of 11 patients diagnosed with sigmoid colon cancer. Consistently with what was earlier detailed, LPI 18:0 and LPI 20:4 were found to be increased in CRC. In a similar trend, lysophosphatidylglycerol (LPGs) species were also found to be increased in CRC tissue, and, as well as LPIs, can influence the Ca2+ influx into the GPR55, suggesting that both LPIs and LPGs may play important roles in cell migration and, consequently, cancer metastasis.90,91 Lysophosphatidylserine (LPS) species were also found to be increased in CRC tissue. The upregulation of certain LPS species may be primarily due to an already established relationship between LPSs and a migration mechanism of colorectal cancer cells, since GPR34, a LPS receptor, can activate the PI3K/Akt pathway, a major signaling pathway involved in the chemotactic migration of cells.92 In contrast, this study found LPA species to be downregulated in CRC tissue, an interesting trend given that LPA is often associated with the promotion of carcinogenesis. This downregulation is most likely due to the upregulation of lysophospholipid receptors, such as the LPA2 receptor, which is known to be overexpressed in CRC and to be involved in malignant CRC phenotypes.95,96 The increased presence of LPA receptors may lead to significant binding of LPA molecules, consequently leading to a decrease in their free form.
The comparison of healthy mucosa and tumor tissue across three independent cohorts (one discovery cohort and two validation cohorts),93 using untargeted lipidomics analysis, showed that the TG profile stood out as the more robust and reproductible across all tested cohorts. The results found elevated proportions of TG(56:4), TG(56:5), and TG(56:6) containing PUFA (FA 18:2, 20:2, 20:3, 20:4, 20:5, 22:4, 22:5, 22:6) in tumor samples. TGs have already been described as promising biomarkers for cancer progression in previous studies that performed lipid profiling in plasma samples.63 However, the TGs reported in the present study appear to contain unsaturated fatty acids, in contrast to the ones that were found to be increased in plasma. With further studies, this information may be of great importance as it suggests that TGs with saturated fatty acids may be good markers for the progression of CRC while TGs with PUFAs may help in the distinction of healthy and diseased mucosa, which confers prognostic significance to them.
On a different note, it has always been established that the tumor microenvironment (TME) plays a critical role in the development and progression ability of solid tumors. Because of this, techniques that enable the profiling of TME have been gaining more and more notoriety in the world of cancer research. Mass spectrometry imaging (MSI) coupled with techniques such as matrix-assisted laser-desorption ionization (MALDI) has been emerging in this area, as it allows biomolecules to be simultaneously localized and quantified in several histological regions of interest. Studies using this technique have already reported differences in some lipid species in human colonoscopic sections, namely PIs, arachidonic acid-containing lipids, and ethanolamine plasmalogens, which varied along the colon crypt axis, and concomitant to the differentiation process.97,98
3.3. Profiling of the lipidome in alternative matrices: insights from fecal samples
Although this review primarily focuses on lipidomic findings from plasma and tissues samples, it is important to acknowledge that other biological samples have also been studied in the context of CRC. Due to its non-invasive nature and vast biochemical information, human fecal matter stands out. As feces interact directly with the colorectal mucosa and result from the interplay between dietary components and gut microbiota, they offer a unique perspective on the colorectal cancer pathophysiology. Therefore, this sample matrix should not be overlooked in the spectrum of lipidomic research, as they can hold promising results in biomarker discovery.Despite being less established than plasma and tissue analysis, lipidomic approaches have also started to be employed in fecal samples of patients with CRC.99–102 One of the studies discriminates CE(18:2) and CE(20:4) as the most significantly altered lipids in CRC feces.99 The increase of these lipid species is consistent with in silico analysis of tumoral tissue that reported elevated levels of the gene that encodes for phosphatidylcholine-sterol acyltransferase, an enzyme responsible for the CE synthesis.103
In a different study, differences were found at the sphingolipid level, namely in the increase of sphingosine and sphinganine levels in adenomas.102 Although these molecules are usually correlated with antiproliferative and proapoptotic molecules, they can also be phosphorylated into S1P, which is a molecule inductive of proliferation and antiapoptotic.80 In that same study, both n − 3 and n − 6 PUFAs were also upregulated in adenomas, which is interesting since these molecules work in a competitive way and have opposite effects, with n − 6 PUFAs being pro-inflammatory and promoters of carcinogenesis, and n − 3 PUFAs being the exact opposite.104,105 A n − 3:n − 6 PUFA ratio would be interesting to further understand the pathophysiological relevance of this data, however, such ratio is not reported by the article.
Even though fecal samples are proving to be of value in the research of biomarkers for CRC, a study that performed an integrative metabolomic approach of both plasma and fecal samples in adenoma-colorectal cancer progression also examined the diagnostic potential of both matrices.100 Although significantly altered metabolites were found in plasma and fecal samples, this study established that plasma metabolites have better performance, i.e. are more accurate, sensitive, and specific, than fecal metabolites in CRC diagnosis.
3.4. Plasticity of lipid profiles in CRC plasma and tissue: a comparative overview
Both plasma and tissue lipidomic studies reveal interesting patterns of lipid metabolism-associated disruptions, with some specific lipid species and classes standing out as potential biomarkers, either for the disease itself or for stages of progression. Although the lipidome of both matrixes are mostly different, the comparison of both CRC matrices shows some complementary results and even some similarities at class level.Plasma analyses have highlighted the upregulation of lipid species of TGs and FFAs groups, which suggests a modulation of the energy metabolism to cope with the energy demands of CRC cells.60,63 With a different trend, but equally significant, LPCs presented decreased levels in CRC plasma, likely due to the overexpression of enzymes, such as LPCAT, that catalyze the conversion of LPCs into PCs, or lyso-PLD or ATX, that produce LPA through LPCs.59–61 Additionally, PCs showed that even among lipid species from the same class, different trends may occur. PCs can be upregulated in response to increased cellular proliferation demands, or alternatively, downregulated, possibly due to their anti-inflammatory properties or their role as precursors for other major lipid classes.62
In tissue samples, the lipid profile highlights the local metabolic fluctuations that may occur within the tumor. Although the reasons remain unclear, Cer presented a significant trend of increased levels in tumor tissue, which can be correlated with increased expression of CerS.86 Similarly, LPIs were also upregulated, likely linked to signaling pathways involved in cell migration and metastasis.94 Furthermore, certain TGs, specifically those with PUFAs, were found to be increased in tumor tissue compared to healthy tissue, contrasting with the upregulated TGs in plasma, that only contained saturated fatty acids.93 In turn, PE plasmalogens were reported to be downregulated in advanced-stage tumors, which suggests an environment of enhanced oxidative stress environment, where these lipid species may play a regulatory role.86
Comparing plasma and tissue lipid profiles provides a broader view of the underlying metabolic changes that occur during CRC onset and development. Plasma lipidomics focuses on circulating biomarkers that could assist in early detection, while tissue studies illuminate localized changes that may enhance understanding of TME interactions and cancer progression. Together, these findings could guide future biomarker research and CRC treatment strategies.
4. Conclusion
Altered levels of certain lipids and dysregulation of metabolic pathways, are intrinsically connected with the onset and development of CRC. Lipidomics has affirmed itself as a pivotal area in the research for alternative CRC diagnostic methods. With these approaches, it is possible to provide insights into the role of the lipidome in biological metabolic regulation by comparing alterations in lipid metabolism between normal and pathological conditions, finding important lipid structures, activities, and interactions with other lipids, proteins, and other metabolites. However, several lipids, with different regulation mechanisms, are involved in the trigger and development pathways of cancer, which makes the already complex study of the lipidome even more challenging. While significant progress has been made, there are still knowledge gaps that persist. Many lipid species remain unexplored and therefore, novel biomarkers are yet to be discovered. Because of this, there is still a need for a more comprehensive and detailed lipidome profiling, as well as for a deeper understanding of how such changes in lipid composition influence the cancer initiation, progress, and metastasis. Also, this translation of the lipidomics discoveries into clinical applications requires rigorous validation, standardization, and integration with other research areas as only then would it be possible to obtain a holistic view of colorectal cancer. Therefore, future research should focus on elucidating the timeline of lipidomic alterations, on the understanding of the lipid functional roles in CRC pathogenesis and also conduct studies that are able to enhance the robustness and reproducibility of these findings.Data availability
This study is a review of existing literature and does not include original data. As such, no new data were generated or analyzed in this work. All information is derived from previously published studies, which are appropriately cited within the manuscript.Conflicts of interest
During the preparation of this work, the authors used CHATGPT v4o to improve the quality and clarity of the English language. After using this tool, the authors reviewed and edited the content as needed and takes full responsibility for the content of the publication.Acknowledgements
The authors thank the University of Aveiro, Fundação para a Ciência e Tecnologia (FCT), and Ministério da Ciência Tecnologia e Ensino Superior (MCTES) for the financial support to the research units CESAM [UIDB/50017/2020 + UIDP/50017/2020 + LA/P/0094/2020] and LAQV-REQUIMTE (UIDP/50006/2020 + UIDB/50006/2020) through national funds and, where applicable, co-funded by ERDF, within Portugal 2020 Partnership Agreement and Compete 2020. This publication is based upon work from COST Action Pan-European Network in Lipidomics and EpiLipidomics (EpiLipidNET), CA19105, supported by COST (European Cooperation in Science and Technology). Tânia Melo acknowledges FCT/MCTES for individual funding in the scope of the Individual Call to Scientific Employment Stimulus (2020.03278.CEECIND).References
- Non communicable diseases, https://www.who.int/news-room/fact-sheets/detail/noncommunicable-diseases, (accessed 30 October 2023).
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, CA-Cancer J. Clin., 2021, 71, 209–249 CrossRef.
- Cancer Today, https://gco.iarc.who.int/today/, (accessed 6 March 2025).
- C. P. Wild, Nat. Rev. Cancer, 2019, 19, 123–124 CrossRef CAS.
- Y. Xi and P. Xu, Transl. Oncol., 2021, 14, 101174 CrossRef PubMed.
- M. Araghi, I. Soerjomataram, A. Bardot, J. Ferlay, C. J. Cabasag, D. S. Morrison, P. De, H. Tervonen, P. M. Walsh, O. Bucher, G. Engholm, C. Jackson, C. McClure, R. R. Woods, N. Saint-Jacques, E. Morgan, D. Ransom, V. Thursfield, B. Møller, S. Leonfellner, M. G. Guren, F. Bray and M. Arnold, Lancet Gastroenterol. Hepatol., 2019, 4, 511–518 CrossRef.
- E. Nerad, M. J. Lahaye, M. Maas, P. Nelemans, F. C. H. Bakers, G. L. Beets and R. G. H. Beets-Tan, Am. J. Roentgenol., 2016, 207, 984–995 CrossRef.
- M. G. Vander Heiden, L. C. Cantley and C. B. Thompson, Science, 2009, 324, 1029–1033 CrossRef CAS PubMed.
- What Is Colorectal Cancer? https://www.cancer.org/cancer/types/colon-rectal-cancer/about/what-is-colorectal-cancer.html, (accessed 3 January 2024).
- Colorectal cancer, https://www.who.int/news-room/fact-sheets/detail/colorectal-cancer, (accessed 3 January 2024).
- E. Marshman, C. Booth and C. S. Potten, BioEssays, 2002, 24, 91–98 CrossRef PubMed.
- L. Ricci-Vitiani, E. Fabrizi, E. Palio and R. De Maria, J. Mol. Med., 2009, 87, 1097–1104 CrossRef PubMed.
- H. Zhao, T. Ming, S. Tang, S. Ren, H. Yang, M. Liu, Q. Tao and H. Xu, Mol. Cancer, 2022, 21, 144 CrossRef CAS PubMed.
- T.-C. He, A. B. Sparks, C. Rago, H. Hermeking, L. Zawel, L. T. da Costa, P. J. Morin, B. Vogelstein and K. W. Kinzler, Science, 1998, 281, 1509–1512 CrossRef CAS.
- Y. Yatabe, S. Tavaré and D. Shibata, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10839–10844 CrossRef CAS.
- S. A. C. McDonald, S. L. Preston, L. C. Greaves, S. J. Leedham, M. A. Lovell, J. A. Z. Jankowski, D. M. Turnbull and N. A. Wright, Cell Cycle, 2006 DOI:10.4161/cc.5.8.2641.
- B. Vogelstein, E. R. Fearon, S. R. Hamilton, S. E. Kern, A. C. Preisinger, M. Leppert, A. M. M. Smits and J. L. Bos, N. Engl. J. Med., 1988, 319, 525–532 CrossRef CAS.
- E. Dekker, P. J. Tanis, J. L. A. Vleugels, P. M. Kasi and M. B. Wallace, The Lancet, 2019, 394, 1467–1480 CrossRef.
- M. J. Arends, Appl. Immunohistochem. Mol. Morphol., 2013, 21, 97 CrossRef CAS PubMed.
- N. B. Henrikson, E. M. Webber, K. A. Goddard, A. Scrol, M. Piper, M. S. Williams, D. T. Zallen, N. Calonge, T. G. Ganiats, A. C. J. W. Janssens, A. Zauber, I. Lansdorp-Vogelaar, M. van Ballegooijen and E. P. Whitlock, Genet. Med., 2015, 17, 702–712 CrossRef.
- R. E. Schoen, A. Razzak, K. J. Yu, S. I. Berndt, K. Firl, T. L. Riley and P. F. Pinsky, Gastroenterology, 2015, 149(6), 1438–1445.e1 CrossRef.
- K. Czene, P. Lichtenstein and K. Hemminki, Int. J. Cancer, 2002, 99, 260–266 CrossRef CAS PubMed.
- S. Jiao, U. Peters, S. Berndt, H. Brenner, K. Butterbach, B. J. Caan, C. S. Carlson, A. T. Chan, J. Chang-Claude, S. Chanock, K. R. Curtis, D. Duggan, J. Gong, T. A. Harrison, R. B. Hayes, B. E. Henderson, M. Hoffmeister, L. N. Kolonel, L. L. Marchand, J. D. Potter, A. Rudolph, R. E. Schoen, D. Seminara, M. L. Slattery, E. White and L. Hsu, Hum. Mol. Genet., 2014, 23, 3898–3905 CrossRef CAS.
- U. Peters, S. Jiao, F. R. Schumacher, C. M. Hutter, A. K. Aragaki, J. A. Baron, S. I. Berndt, S. Bézieau, H. Brenner, K. Butterbach, B. J. Caan, P. T. Campbell, C. S. Carlson, G. Casey, A. T. Chan, J. Chang–Claude, S. J. Chanock, L. S. Chen, G. A. Coetzee, S. G. Coetzee, D. V. Conti, K. R. Curtis, D. Duggan, T. Edwards, C. S. Fuchs, S. Gallinger, E. L. Giovannucci, S. M. Gogarten, S. B. Gruber, R. W. Haile, T. A. Harrison, R. B. Hayes, B. E. Henderson, M. Hoffmeister, J. L. Hopper, T. J. Hudson, D. J. Hunter, R. D. Jackson, S. H. Jee, M. A. Jenkins, W. Jia, L. N. Kolonel, C. Kooperberg, S. Küry, A. Z. LaCroix, C. C. Laurie, C. A. Laurie, L. Le Marchand, M. Lemire, D. Levine, N. M. Lindor, Y. Liu, J. Ma, K. W. Makar, K. Matsuo, P. A. Newcomb, J. D. Potter, R. L. Prentice, C. Qu, T. Rohan, S. A. Rosse, R. E. Schoen, D. Seminara, M. Shrubsole, X. Shu, M. L. Slattery, D. Taverna, S. N. Thibodeau, C. M. Ulrich, E. White, Y. Xiang, B. W. Zanke, Y. Zeng, B. Zhang, W. Zheng and L. Hsu, Gastroenterology, 2013, 144, 799–807.e24 CrossRef CAS.
- R. S. Houlston, E. Webb, P. Broderick, A. M. Pittman, M. C. Di Bernardo, S. Lubbe, I. Chandler, J. Vijayakrishnan, K. Sullivan, S. Penegar, L. Carvajal-Carmona, K. Howarth, E. Jaeger, S. L. Spain, A. Walther, E. Barclay, L. Martin, M. Gorman, E. Domingo, A. S. Teixeira, D. Kerr, J.-B. Cazier, I. Niittymäki, S. Tuupanen, A. Karhu, L. A. Aaltonen, I. P. M. Tomlinson, S. M. Farrington, A. Tenesa, J. G. D. Prendergast, R. A. Barnetson, R. Cetnarskyj, M. E. Porteous, P. D. P. Pharoah, T. Koessler, J. Hampe, S. Buch, C. Schafmayer, J. Tepel, S. Schreiber, H. Völzke, J. Chang-Claude, M. Hoffmeister, H. Brenner, B. W. Zanke, A. Montpetit, T. J. Hudson, S. Gallinger, H. Campbell, M. G. Dunlop, COGENT Study, Group 1, Colorectal Cancer Association Study Consortium, Group 2, CoRGI Consortium, Group 3 and International Colorectal Cancer Genetic Association Consortium, Nat. Genet., 2008, 40, 1426–1435 CrossRef CAS PubMed.
- U. Peters, C. M. Hutter, L. Hsu, F. R. Schumacher, D. V. Conti, C. S. Carlson, C. K. Edlund, R. W. Haile, S. Gallinger, B. W. Zanke, M. Lemire, J. Rangrej, R. Vijayaraghavan, A. T. Chan, A. Hazra, D. J. Hunter, J. Ma, C. S. Fuchs, E. L. Giovannucci, P. Kraft, Y. Liu, L. Chen, S. Jiao, K. W. Makar, D. Taverna, S. B. Gruber, G. Rennert, V. Moreno, C. M. Ulrich, M. O. Woods, R. C. Green, P. S. Parfrey, R. L. Prentice, C. Kooperberg, R. D. Jackson, A. Z. LaCroix, B. J. Caan, R. B. Hayes, S. I. Berndt, S. J. Chanock, R. E. Schoen, J. Chang-Claude, M. Hoffmeister, H. Brenner, B. Frank, S. Bézieau, S. Küry, M. L. Slattery, J. L. Hopper, M. A. Jenkins, L. Le Marchand, N. M. Lindor, P. A. Newcomb, D. Seminara, T. J. Hudson, D. J. Duggan, J. D. Potter and G. Casey, Hum. Genet., 2012, 131, 217–234 CrossRef PubMed.
- A. Howren, E. C. Sayre, J. M. Loree, S. Gill, C. J. Brown, M. J. Raval, A. Farooq and M. A. De Vera, JNCI, J. Natl. Cancer Inst., 2021, 113, 863–868 CrossRef PubMed.
- K. S. El Din, J. M. Loree, E. C. Sayre, S. Gill, C. J. Brown, H. Dau and M. A. De Vera, BMC Cancer, 2020, 20, 288 CrossRef.
- A. Lewandowska, G. Rudzki, T. Lewandowski, A. Stryjkowska-Góra and S. Rudzki, Cancer Control, 2022, 29, 10732748211056692 CrossRef PubMed.
- I. Lansdorp-Vogelaar, M. van Ballegooijen, A. G. Zauber, R. Boer, J. Wilschut, S. J. Winawer and J. D. F. Habbema, Gastrointest. Endosc., 2009, 70, 96–108 CrossRef PubMed , 108.e1–24.
- J. M. Ordóñez-Mena, V. Walter, B. Schöttker, M. Jenab, M. G. O’Doherty, F. Kee, B. Bueno-de-Mesquita, P. H. M. Peeters, B. H. Stricker, R. Ruiter, A. Hofman, S. Söderberg, P. Jousilahti, K. Kuulasmaa, N. D. Freedman, T. Wilsgaard, A. Wolk, L. M. Nilsson, A. Tjønneland, J. R. Quirós, F. J. B. van Duijnhoven, P. D. Siersema, P. Boffetta, A. Trichopoulou, H. Brenner and Consortium on Health and Ageing: Network of Cohorts in Europe and the United States (CHANCES), Ann. Oncol., 2018, 29, 472–483 CrossRef.
- Q. Li, S. Geng, H. Luo, W. Wang, Y.-Q. Mo, Q. Luo, L. Wang, G.-B. Song, J.-P. Sheng and B. Xu, Signal Transduction Targeted Ther., 2024, 9, 1–48 CrossRef PubMed.
- E. Botteri, S. Iodice, S. Raimondi, P. Maisonneuve and A. B. Lowenfels, Gastroenterology, 2008, 134, 388–395.e3 CrossRef PubMed.
- S. Cai, Y. Li, Y. Ding, K. Chen and M. Jin, Eur. J. Cancer Prev., 2014, 23, 532–539 CrossRef CAS.
- R. Baan, K. Straif, Y. Grosse, B. Secretan, F. E. Ghissassi, V. Bouvard, A. Altieri and V. Cogliano, Lancet Oncol., 2007, 8, 292–293 CrossRef.
- Red and Processed Meat and Colorectal Cancer Incidence: Meta-Analysis of Prospective Studies | PLOS ONE, https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0020456, (accessed 6 November 2023).
- M. Pollak, Nat. Rev. Cancer, 2012, 12, 159–169 CrossRef CAS PubMed.
- D. S. Michaud, C. S. Fuchs, S. Liu, W. C. Willett, G. A. Colditz and E. Giovannucci, Cancer Epidemiol., Biomarkers Prev., 2005, 14, 138–147 CrossRef CAS.
- E. Kállay, E. Bajna, F. Wrba, S. Kriwanek, M. Peterlik and H. S. Cross, Cancer Detect. Prev., 2000, 24, 127–136 Search PubMed.
- M. Makishima, T. T. Lu, W. Xie, G. K. Whitfield, H. Domoto, R. M. Evans, M. R. Haussler and D. J. Mangelsdorf, Science, 2002, 296, 1313–1316 CrossRef CAS.
- B. A. Scaglione-Sewell, M. Bissonnette, S. Skarosi, C. Abraham and T. A. Brasitus, Endocrinology, 2000, 141, 3931–3939 CrossRef CAS PubMed.
- R. M. Bostick, M. Goodman and E. Sidelnikov, in Genetics of Colorectal Cancer, ed. J. D. Potter and N. M. Lindor, Springer, New York, NY, 2009, pp. 277–298 Search PubMed.
- D. B. Bernkopf, G. Daum, M. Brückner and J. Behrens, Oncotarget, 2018, 9, 33982–33994 CrossRef.
- E. Yehuda-Shnaidman and B. Schwartz, Obes. Rev., 2012, 13, 1083–1095 CrossRef CAS.
- J. Jeon, M. Du, R. E. Schoen, M. Hoffmeister, P. A. Newcomb, S. I. Berndt, B. Caan, P. T. Campbell, A. T. Chan, J. Chang-Claude, G. G. Giles, J. Gong, T. A. Harrison, J. R. Huyghe, E. J. Jacobs, L. Li, Y. Lin, L. Le Marchand, J. D. Potter, C. Qu, S. A. Bien, N. Zubair, R. J. Macinnis, D. D. Buchanan, J. L. Hopper, Y. Cao, R. Nishihara, G. Rennert, M. L. Slattery, D. C. Thomas, M. O. Woods, R. L. Prentice, S. B. Gruber, Y. Zheng, H. Brenner, R. B. Hayes, E. White, U. Peters and L. Hsu, Gastroenterology, 2018, 154, 2152–2164.e19 CrossRef.
- S. Dorudi, R. J. Steele and C. S. McArdle, Br. Med. Bull., 2002, 64, 101–118 CrossRef PubMed.
- B. Ma, P. Gao, H. Wang, Q. Xu, Y. Song, X. Huang, J. Sun, J. Zhao, J. Luo, Y. Sun and Z. Wang, Int. J. Cancer, 2017, 141, 1052–1065 CrossRef CAS.
- M. Ferlitsch, A. Moss, C. Hassan, P. Bhandari, J.-M. Dumonceau, G. Paspatis, R. Jover, C. Langner, M. Bronzwaer, K. Nalankilli, P. Fockens, R. Hazzan, I. M. Gralnek, M. Gschwantler, E. Waldmann, P. Jeschek, D. Penz, D. Heresbach, L. Moons, A. Lemmers, K. Paraskeva, J. Pohl, T. Ponchon, J. Regula, A. Repici, M. D. Rutter, N. G. Burgess and M. J. Bourke, Endoscopy, 2017, 49, 270–297 CrossRef PubMed.
- LIPID MAPS, https://www.lipidmaps.org/databases/lmsd/browse, (accessed 21 April 2025).
- B. Burla, M. Arita, M. Arita, A. K. Bendt, A. Cazenave-Gassiot, E. A. Dennis, K. Ekroos, X. Han, K. Ikeda, G. Liebisch, M. K. Lin, T. P. Loh, P. J. Meikle, M. Orešič, O. Quehenberger, A. Shevchenko, F. Torta, M. J. O. Wakelam, C. E. Wheelock and M. R. Wenk, J. Lipid Res., 2018, 59, 2001–2017 CrossRef CAS PubMed.
- M. T. Snaebjornsson, S. Janaki-Raman and A. Schulze, Cell Metab., 2020, 31, 62–76 CrossRef CAS PubMed.
- T. G. Meikle, K. Huynh, C. Giles and P. J. Meikle, J. Lipid Res., 2021, 62, 100127 CrossRef CAS.
- G. Yan, L. Li, B. Zhu and Y. Li, Oncotarget, 2016, 7, 33429–33439 CrossRef.
- A. Pakiet, J. Kobiela, P. Stepnowski, T. Sledzinski and A. Mika, Lipids Health Dis., 2019, 18, 29 CrossRef.
- E. Rysman, K. Brusselmans, K. Scheys, L. Timmermans, R. Derua, S. Munck, P. P. Van Veldhoven, D. Waltregny, V. W. Daniëls, J. Machiels, F. Vanderhoydonc, K. Smans, E. Waelkens, G. Verhoeven and J. V. Swinnen, Cancer Res., 2010, 70, 8117–8126 CrossRef CAS PubMed.
- Y. Y. Zaytseva, J. W. Harris, M. I. Mitov, J. T. Kim, D. A. Butterfield, E. Y. Lee, H. L. Weiss, T. Gao and B. M. Evers, Oncotarget, 2015, 6, 18891–18904 CrossRef PubMed.
- H. Liu, J. Zeng, W. Huang, Q. Xu, D. Ye, R. Sun and D. Zhang, J. Cancer, 2019, 10, 4719–4730 CrossRef CAS PubMed.
- LIPID MAPS, https://www.lipidmaps.org/, (accessed 23 April 2025).
- Z. Zhao, Y. Xiao, P. Elson, H. Tan, S. J. Plummer, M. Berk, P. P. Aung, I. C. Lavery, J. P. Achkar, L. Li, G. Casey and Y. Xu, J. Clin. Oncol., 2007, 25, 2696–2701 CrossRef CAS.
- S. Shen, L. Yang, L. Li, Y. Bai, C. Cai and H. Liu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1068–1069, 41–48 CrossRef CAS.
- S. Li, B. Guo, J. Song, X. Deng, Y. Cong, P. Li, K. Zhao, L. Liu, G. Xiao, F. Xu, Y. Ye, Z. Zhao, M. Yu, Y. Xu, J. Sang and J. Zhang, Metabolomics, 2013, 9, 202–212 CrossRef CAS.
- H. Chen, H. Zhou, Y. Liang, Z. Huang, S. Yang, X. Wang, Z. She, Z. Wei and Q. Zhang, J. Pharm. Biomed. Anal., 2023, 234, 115582 CrossRef CAS.
- T. Liu, F. Peng, J. Yu, Z. Tan, T. Rao, Y. Chen, Y. Wang, Z. Liu, H. Zhou and J. Peng, Anal. Bioanal. Chem., 2019, 411, 5079–5088 CrossRef CAS.
- F. Mansilla, K.-A. da Costa, S. Wang, M. Kruhøffer, T. M. Lewin, T. F. Ørntoft, R. A. Coleman and K. Birkenkamp-Demtröder, J. Mol. Med., 2009, 87, 85–97 CrossRef CAS PubMed.
- S. Sengupta, Z. Wang, R. Tipps and Y. Xu, Semin. Cell Dev. Biol., 2004, 15, 503–512 CrossRef CAS PubMed.
- R. P. Rao, C. Yuan, J. C. Allegood, S. S. Rawat, M. B. Edwards, X. Wang, A. H. Merrill, U. Acharya and J. K. Acharya, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11364–11369 Search PubMed.
- R. C. Dickson, E. E. Nagiec, M. Skrzypek, P. Tillman, G. B. Wells and R. L. Lester, J. Biol. Chem., 1997, 272, 30196–30200 Search PubMed.
- K. Kurek, B. Łukaszuk, A. Świdnicka-Siergiejko, P. Rogalski, E. Wróblewski, A. Chabowski, A. Dąbrowski and M. Żendzian-Piotrowska, Lipids, 2015, 50, 349–358 Search PubMed.
- M. N. Barber, S. Risis, C. Yang, P. J. Meikle, M. Staples, M. A. Febbraio and C. R. Bruce, PLoS One, 2012, 7, e41456 Search PubMed.
- S. Rai and S. Bhatnagar, OMICS, 2017, 21, 132–142 Search PubMed.
- H. Chen, J. Zhang, H. Zhou, Y. Zhu, Y. Liang, P. Zhu and Q. Zhang, Front. Oncol., 2022, 28(12), 934145 Search PubMed.
- R. de F. Saito, L. N. de S. Andrade, S. O. Bustos and R. Chammas, Front. Immunol., 2022, 13, 768606 CrossRef.
- I. Treede, A. Braun, R. Sparla, M. Kühnel, T. Giese, J. R. Turner, E. Anes, H. Kulaksiz, J. Füllekrug, W. Stremmel, G. Griffiths and R. Ehehalt, J. Biol. Chem., 2007, 282, 27155–27164 CrossRef CAS PubMed.
- I. Treede, A. Braun, P. Jeliaskova, T. Giese, J. Füllekrug, G. Griffiths, W. Stremmel and R. Ehehalt, BMC Gastroenterol., 2009, 9, 53 CrossRef PubMed.
- S. C. Shah, J. R. Ten Hove, D. Castaneda, C. Palmela, E. Mooiweer, J.-F. Colombel, N. Harpaz, T. A. Ullman, A. A. van Bodegraven, J. M. Jansen, N. Mahmmod, A. E. van der Meulen-de Jong, C. Y. Ponsioen, C. J. van der Woude, B. Oldenburg, S. H. Itzkowitz and J. Torres, Clin. Gastroenterol. Hepatol., 2018, 16, 1106–1113.e3 CrossRef.
- M. Machala, J. Procházková, J. Hofmanová, L. Králiková, J. Slavík, Z. Tylichová, P. Ovesná, A. Kozubík and J. Vondráček, Int. J. Mol. Sci., 2019, 20, 6051 CrossRef CAS.
- E. Hertervig, Å. Nilsson, M. Nilbert and R.-D. Duan, Int. J. Colorectal. Dis., 2003, 18, 309–313 CrossRef PubMed.
- M. Selzner, A. Bielawska, M. A. Morse, H. A. Rüdiger, D. Sindram, Y. A. Hannun and P.-A. Clavien, Cancer Res., 2001, 61, 1233–1240 CAS.
- E. H. Ahn and J. J. Schroeder, Exp. Biol. Med., 2002, 227, 345–353 CrossRef CAS.
- S. Milstien and S. Spiegel, Cancer Cell, 2006, 9, 148–150 CrossRef CAS PubMed.
- P. T. Bozza and J. P. B. Viola, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2010, 82, 243–250 CrossRef CAS PubMed.
- A. K. Cotte, V. Aires, M. Fredon, E. Limagne, V. Derangère, M. Thibaudin, E. Humblin, A. Scagliarini, J.-P. P. de Barros, P. Hillon, F. Ghiringhelli and D. Delmas, Nat. Commun., 2018, 9, 322 CrossRef.
- G. Chen, G. Zhou, S. Aras, Z. He, S. Lucas, I. Podgorski, W. Skar, J. G. Granneman and J. Wang, Sci. Rep., 2017, 7, 13021 CrossRef.
- K. Ye, Y. Wu, Y. Sun, J. Lin and J. Xu, Life Sci., 2016, 155, 133–139 CrossRef CAS.
- Q.-F. Zhu, J.-W. Yan, Y. Gao, J.-W. Zhang, B.-F. Yuan and Y.-Q. Feng, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1061–1062, 34–40 CrossRef CAS.
- Y. Wang, S. Hinz, O. Uckermann, P. Hönscheid, W. von Schönfels, G. Burmeister, A. Hendricks, J. M. Ackerman, G. B. Baretton, J. Hampe, M. Brosch, C. Schafmayer, A. Shevchenko and S. Zeissig, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2020, 1865, 158579 CrossRef CAS PubMed.
- J. Kargl, L. Andersen, C. Hasenöhrl, D. Feuersinger, A. Stančić, A. Fauland, C. Magnes, A. El-Heliebi, S. Lax, S. Uranitsch, J. Haybaeck, A. Heinemann and R. Schicho, Br. J. Pharmacol., 2016, 173, 142–154 Search PubMed.
- C. M. Henstridge, N. A. B. Balenga, L. A. Ford, R. A. Ross, M. Waldhoer and A. J. Irving, FASEB J., 2009, 23, 183–193 Search PubMed.
- G.-Y. Liou and P. Storz, Free Radical Res., 2010, 44, 479–496 CrossRef CAS.
- S. Oka, K. Nakajima, A. Yamashita, S. Kishimoto and T. Sugiura, Biochem. Biophys. Res. Commun., 2007, 362, 928–934 Search PubMed.
- K. Makide, A. Uwamizu, Y. Shinjo, J. Ishiguro, M. Okutani, A. Inoue and J. Aoki, J. Lipid Res., 2014, 55, 1986–1995 CrossRef CAS PubMed.
- Y. Iida, N. H. Tsuno, J. Kishikawa, K. Kaneko, K. Murono, K. Kawai, T. Ikeda, S. Ishihara, H. Yamaguchi, E. Sunami, J. Kitayama, Y. Yatomi and T. Watanabe, Anticancer Res., 2014, 34, 5465–5472 Search PubMed.
- J. Ecker, E. Benedetti, A. S. D. Kindt, M. Höring, M. Perl, A. C. Machmüller, A. Sichler, J. Plagge, Y. Wang, S. Zeissig, A. Shevchenko, R. Burkhardt, J. Krumsiek, G. Liebisch and K.-P. Janssen, Gastroenterology, 2021, 161, 910–923.e19 CrossRef CAS.
- C. Kitamura, H. Sonoda, H. Nozawa, K. Kano, S. Emoto, K. Murono, M. Kaneko, M. Hiyoshi, K. Sasaki, T. Nishikawa, Y. Shuno, T. Tanaka, K. Hata, K. Kawai, J. Aoki and S. Ishihara, Tumour Biol., 2019, 41, 1010428319848616 CrossRef PubMed.
- D. Shida, J. Kitayama, H. Yamaguchi, Y. Okaji, N. H. Tsuno, T. Watanabe, Y. Takuwa and H. Nagawa, Cancer Res., 2003, 63, 1706–1711 CAS.
- S. Lin, D. Wang, S. Iyer, A. M. Ghaleb, H. Shim, V. W. Yang, J. Chun and C. C. Yun, Gastroenterology, 2009, 136, 1711–1720 CrossRef CAS PubMed.
- J. Bestard-Escalas, J. Garate, A. Maimó-Barceló, R. Fernández, D. H. Lopez, S. Lage, R. Reigada, S. Khorrami, D. Ginard, J. Reyes, I. Amengual, J. A. Fernández and G. Barceló-Coblijn, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2016, 1861, 1942–1950 CrossRef CAS.
- D. H. Lopez, J. Bestard-Escalas, J. Garate, A. Maimó-Barceló, R. Fernández, R. Reigada, S. Khorrami, D. Ginard, T. Okazaki, J. A. Fernández and G. Barceló-Coblijn, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2018, 1863, 928–938 CrossRef CAS.
- J. Cubiella, M. Clos-Garcia, C. Alonso, I. Martinez-Arranz, M. Perez-Cormenzana, Z. Barrenetxea, J. Berganza, I. Rodríguez-Llopis, M. D’Amato, L. Bujanda, M. Diaz-Ondina and J. M. Falcón-Pérez, Cancers, 2018, 10, 300 CrossRef.
- Y. Sun, X. Zhang, D. Hang, H. C.-H. Lau, J. Du, C. Liu, M. Xie, Y. Pan, L. Wang, C. Liang, X. Zhou, D. Chen, J. Rong, Z. Zhao, A. H.-K. Cheung, Y. Wu, H. Gou, C. C. Wong, L. Du, J. Deng, Z. Hu, H. Shen, Y. Miao and J. Yu, Cancer Cell, 2024, 42, 1386–1400.e8 CrossRef CAS.
- B. A. Baxter, K. D. Parker, M. J. Nosler, S. Rao, R. Craig, C. Seiler and E. P. Ryan, World J. Gastroenterol., 2020, 26, 335–352 CrossRef CAS PubMed.
- M. Kim, E. Vogtmann, D. A. Ahlquist, M. E. Devens, J. B. Kisiel, W. R. Taylor, B. A. White, V. L. Hale, J. Sung, N. Chia, R. Sinha and J. Chen, mBio, 2020, 11, e03186-19 CrossRef.
- Myofibroblast-Derived SFRP1 as Potential Inhibitor of Colorectal Carcinoma Field Effect|PLOS One, https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0106143, (accessed 22 April 2025).
- M. Azrad, C. E. Turgeon and W. Demark-Wahnefried, Front. Oncol., 2013, 4(3), 224 Search PubMed.
- J. K. Innes and P. C. Calder, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2018, 132, 41–48 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |