
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic transfer hydrogenation of furfural using mechanically activated MgO as catalyst†
Antonio Manuel Pérez-Merchánab,
Benjamín Torres-Oleaab,
Marcella Scala cd,
Nikolaos Dimitratoscd,
Irene Malpartidae,
Cristina García-Sanchoab,
Josefa M. Mérida-Roblesab,
Pedro Maireles-Torresab,
Ramón Moreno-Tostab and
Juan Antonio Cecilia*ab
cd,
Nikolaos Dimitratoscd,
Irene Malpartidae,
Cristina García-Sanchoab,
Josefa M. Mérida-Roblesab,
Pedro Maireles-Torresab,
Ramón Moreno-Tostab and
Juan Antonio Cecilia*ab
aDepartamento de Química Inorgánica, Cristalografía y Mineralogía, Universidad de Málaga, Facultad de Ciencias, Campus de Teatinos, 29071 Málaga, Spain. E-mail: jacecilia@uma.es
bInstituto de Investigación en Biorrefinerías "I3B", Universidad de Málaga, Facultad de Ciencias, Campus de Teatinos s/n 29071, Malaga, Spain
cDipartimento di Chimica Industriale “Toso Montanari”, Università di Bologna, Viale Risorgimento 4, 40136 Bologna, Italy
dCenter for Chemical Catalysis – C3, Alma Mater Studiorum, Università di Bologna, Viale Risorgimento 4, 40136 Bologna, Italy
eDeasyl S.A., Plan-les-Ouates, Geneva, 1228, Switzerland
First published on 28th January 2025
Abstract
Several MgO materials have been prepared throughout wet flow semi-continuous mechanochemical treatment of the Mg(OH)2 precursors and their subsequent calcination. This mechanochemical treatment of the precursors has shown a clear influence on the textural properties and the amount and strength of basic sites of the MgO catalysts after its calcination, obtaining higher values than that observed in a MgO sample synthesized without mechanochemical treatment. An in-depth investigation was conducted on the effects of the mechanochemical treatment on the catalytic performance in the catalytic transfer hydrogenation of furfural, and a positive effect on the activity of the catalysts was found after short mechanochemical treatment times. The highest conversion values at shorter reaction times were obtained after a mechanochemical treatment of 15 min, reaching a furfuryl alcohol yield of 79% after 2 h of reaction at 90 °C, using 2-propanol as both hydrogen donor and solvent. This data notably improves that obtained for the untreated material, which only reaches a conversion of 48% under the same experimental conditions. The stability of the material in the reaction media as well as their reusability were also investigated, and the interaction nature of 2-propanol with the MgO surface has been elucidated by attenuated total reflection spectroscopy and 2-propanol adsorption studies.
1. Introduction
Lignocellulosic biomass has emerged as an alternative to traditional fossil fuels because of its renewable character, useful derived chemicals and potentially usable stored energy by thermochemical or enzymatic processes.1–3 The most abundant fractions of lignocellulose (cellulose, hemicellulose and lignin) can be isolated and purified4 and then each of them can be used as a starting material to obtain a wide range of high value-added products, which were previously synthesized by other routes from fossil fuels. Hemicellulose, a branched polysaccharide, is mainly composed of C5 sugars (xylose and arabinose). Hemicellulose can be depolymerized into their respective monomers through acid treatment under controlled conditions.5 Both xylose and arabinose are dehydrated to obtain furfural (FUR).6,7FUR is considered, together with bioethanol, one of the main products obtained from the sugar platform. The high interest in FUR is attributed to its high reactivity and versatility, being considered as a building block molecule due to the broad spectrum of products that are obtained by different chemical treatments.7 Among them, the production of furfuryl alcohol (FOL) from FUR through hydrogenation is the most important. Traditionally, this reaction has been carried out using copper chromite as the catalyst of choice.8,9 However, the toxicity of the Cr species has led to the search and development of Cr-free catalysts, mainly Cu- and Ni-based catalysts.7–16
In the last few years, catalytic transfer hydrogenation (CTH) has emerged as a cheaper and safer alternative.17–22 This reaction can take place in the absence of metal phases (M0), using catalysts with Lewis acid or basic a active sites and sacrificing alcohol, mainly a secondary alcohol, which donates a hydrogen to the FUR molecule to form FOL through a six-membered intermediate.18 Most studies have been performed with Zr-based catalysts, where the presence of Lewis acid sites promotes the reduction of FUR to FOL.23–27 In this sense, several authors have incorporated Zr species in silica or zeolite framework forming Brønsted acid sites, which are involved in consecutive reactions to obtain alkyl furfuryl ethers, alkyl levulinates or γ-valerolactone, although the yield and non-detected products are more limited.24,26,28–31 Other authors have also pointed out the role of Lewis acid sites in Al-32,33 and Fe-based catalysts34,35 as active phase in the reduction of FUR to FOL.
Basic catalysts have also been studied in the CTH of FUR. These catalysts have a higher catalytic activity than acid catalysts, obtaining high conversion values even at lower temperatures. Among them, most studies focus on MgO-based catalysts as the active phase;36–38 however, the main disadvantage of these basic materials is related to their regeneration, as several authors have observed.37,39
The present study focuses on the role of MgO crystallinity on the amount of basic sites and thus on the catalytic behavior of this catalyst in the hydrogenation of FUR to FOL via CTH. For this purpose, Mg(OH)2, used as a precursor, has been treated in a semi-continuous mechanochemical reactor at different reaction times and subsequently calcined to obtain MgO. Traditionally, mechanochemistry has been used in the metallurgy field to form alloys.40 More recently, the mechanochemical studies are focused on other fields such as inorganic, organic, or organometallic chemistry as well as material sciences,41 or the synthesis of materials with catalytic properties.42 Generally, ball-milling promotes the formation of crystalline defects in solid acids through impacts, friction and shear processes.42 These defects are directly related to the active phases involved in the reaction. On the other hand, this strategy also allows the synthesis of materials in shorter treatment times and milder conditions. Furthermore, in this work, the analysis of the basic sites involved in the hydrogenation of FUR and its stability is also carried out.
2. Experimental section
A semi-continuous flow mechanochemical reactor, DYNO®-MILL RESEARCH LAB (Willy A. Bachofen AG, Switzerland), was used for the treatment of Mg(OH)2 (Merck), using deionized water as carrier (10 wt% Mg(OH)2). The procedure was as follows: 8 g of Mg(OH)2 was added to 80 mL of deionized water, and this suspension was fed to the reactor (operating in loop mode by recirculating the reaction suspension), with a rotation speed of 8.5 m s−1, using 1 mm beads (ZrO2 doped with Y2O3) at 25 °C (measured in the outlet of the machine) at different times, for up to 240 min. For each study, the reactor of 80 mL was filled in a 60% of the total volume with the ZrO2 microbeads. The suspended material was then separated by centrifugation at 8000 rpm for 15 min before drying at 70 °C for 24 h. Finally, the solid was calcinated at a temperature of 450 °C with a heating rate of 5 °C min−1, maintaining this temperature for 2 h to obtain MgO as active phase.The different materials were characterized by X-ray diffraction, transmission electron microscopy, particle size analyzer, attenuated total reflection, N2 adsorption–desorption isotherms at −196 °C, CO2 temperature-programmed desorption, X-ray photoelectron spectroscopy, FTIR spectroscopy analysis coupled to 2-propanol adsorption and 2-propanol adsorption–desorption isotherms at 25 °C. A description of the equipment used in the characterization of the samples is detailed in the ESI.†
The catalytic study was carried out in a glass reactor, with thread bushing (Ace, 15 mL, pressure rated to 10 bar). In a typical experiment, 96 μg of FUR was dissolved in 2-propanol, maintaining a 2-propanol/FUR molar ratio of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and 100 mg of catalyst was added. Prior to the reaction, the samples were purged with a He flow for 30 seconds. Reaction time was investigated up to 3 h under continuous stirring (300 rpm), at temperatures between 90 and 135 °C using a thermally-controlled aluminum block. After the reaction, the reactor was immediately cooled in a water bath. Samples were microfiltered and analyzed by gas chromatography (Shimadzu GC-14A), using a flame ionization detector and a CP-Wax 52 CB capillary column. The FUR conversion and yields of the products were determined as follows (eqn (1) and (2)):
:1, and 100 mg of catalyst was added. Prior to the reaction, the samples were purged with a He flow for 30 seconds. Reaction time was investigated up to 3 h under continuous stirring (300 rpm), at temperatures between 90 and 135 °C using a thermally-controlled aluminum block. After the reaction, the reactor was immediately cooled in a water bath. Samples were microfiltered and analyzed by gas chromatography (Shimadzu GC-14A), using a flame ionization detector and a CP-Wax 52 CB capillary column. The FUR conversion and yields of the products were determined as follows (eqn (1) and (2)):
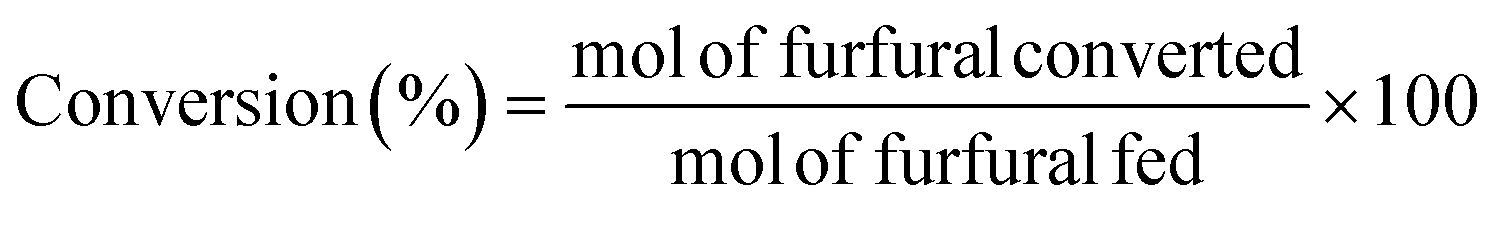 | (1) |
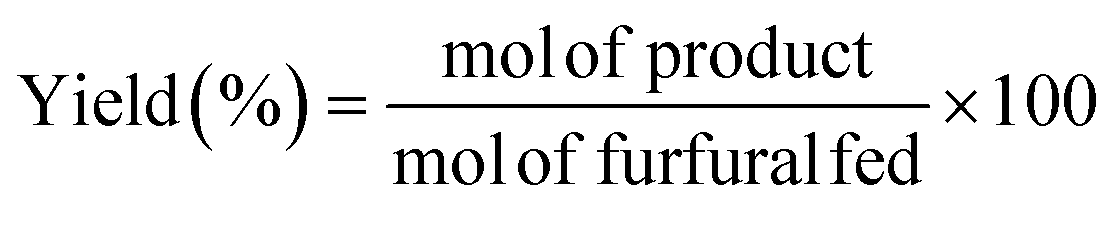 | (2) |
3. Characterization of the samples
The analysis of precursors obtained by mechano-treatment at different times was performed by X-ray diffraction (XRD) (Fig. 1A). All diffractograms display the same profile, showing diffraction peaks attributed to the presence of hexagonal Mg(OH)2 brucite structure as a unique crystalline phase (PDF: 00-044-1482). The diffractogram shows a slight broadening of the characteristic diffraction peaks of Mg(OH)2, suggesting a decrease in crystallinity. After calcination at 450 °C (Fig. 1B), the typical diffraction peaks ascribed to the (111), (200) and (220) planes of the periclase structure (MgO) (PDF: 01-087-0653) are observed, which agree with the literature.43 In addition, small peaks located about 2θ(°) of 30 and 50 are detected, mainly after longer treatments in the reactor. These peaks are assigned to the presence of tetragonal ZrO2 (PDF: 01-079-1769), caused by the slight degradation of the Y-doped ZrO2 microbeads used in the mechanochemical treatment. The determination of crystal size of MgO nanoparticles was carried out by the Williamson–Hall method by analyzing the main peak (2θ of 42.7°), associated with the (002) planes (ESI, Fig. S1†). These data reveal that all MgO catalysts show a rather small average crystal size, below 10 nm. In this sense, the mechanochemical treatment causes a progressive decrease in the average crystal size from 9.2 nm, after 1 min of mechanochemical treatment, to 5.8 nm after 240 min.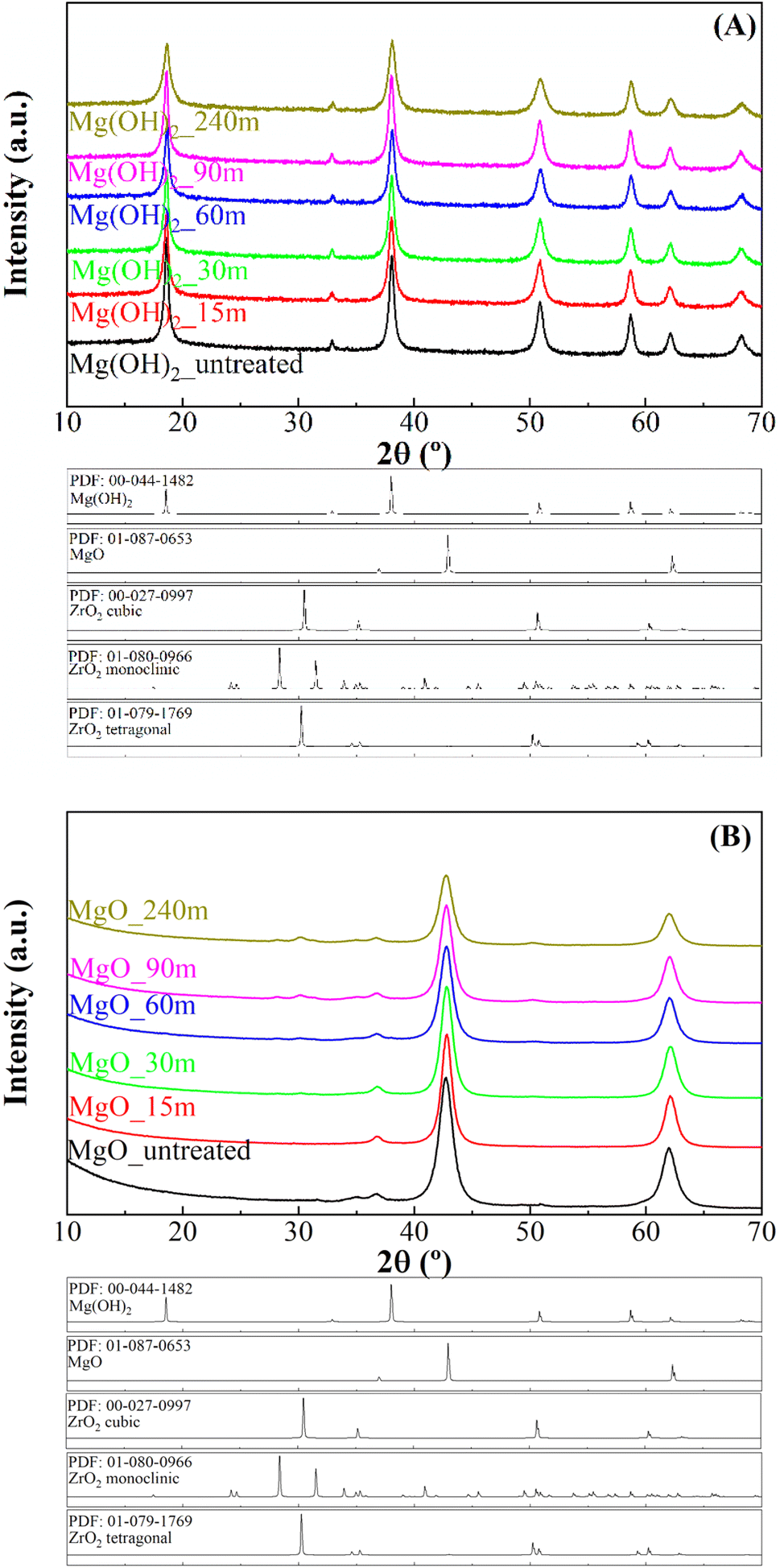 | ||
| Fig. 1 X-ray diffraction patterns of Mg(OH)2 precursors (A) and MgO catalysts (B). | ||
The morphology of the MgO catalysts was analyzed by TEM (Fig. 2). The MgO_untreated sample is composed of large particles. Mechanochemical treatment breaks down the particles into smaller fragments. Despite the treatment, not all the particles suffer a breakdown, as fragments and large particles coexist after mechanochemical treatment. High resolution images (ESI, Fig. S2†) hardly shows this trend, since the mechanochemical treatment also seems to promote the agglomeration of the MgO crystals. This should provide a greater number of structural defects at the grain boundary, which must be related to the active sites and the catalytic behavior. In the same way, the particle size distribution was also evaluated (ESI, Fig. S3†). The obtained results show how the mechanochemical treatment causes a progressive decrease in the particle size, which seems to agree with the decrease in crystallinity observed by XRD (Fig. 1B). On the other hand, the analysis of the MgO catalysts subjected to the mechanochemical treatment by EDX also confirm that the use of a prolonged treatment also provokes a progressive fragmentation of the Y-doped ZrO2 microbeads, in such a way that the higher proportion of Zr is observed for the MgO_240m catalyst (ESI, Fig. S4†).
 | ||
| Fig. 2 TEM micrographs of MgO_untreated (A), MgO_15m (B), MgO_60m (C) and MgO_240m (D) catalysts. | ||
The determination of the textural properties was conducted by N2 adsorption–desorption isotherms at −196 °C (ESI, Fig. S5†). The isotherms show that the N2 adsorbed at low relative pressure is very low, which indicates that the obtained materials display low microporosity (Table 1). The specific surface area increases according to the mechanochemical treatment, ranging from 126 m2 g−1 for the MgO_untreated sample to 202 m2 g−1 for the MgO_240m sample.
| Sample | SBET (m2 g−1) | t-plot (m2 g−1) | Total Vp (cm3 g−1) | Vmicrop (cm3 g−1) |
|---|---|---|---|---|
| MgO_untreated | 126 | 6 | 0.2673 | 0.0021 |
| MgO_15m | 168 | 8 | 0.3175 | 0.0009 |
| MgO_60m | 195 | 5 | 0.4116 | 0.0005 |
| MgO_240m | 203 | 3 | 0.4290 | 0.0001 |
This increase is accompanied by an increment of the pore volume, from 0.267 to 0.429 cm3 g−1. At higher relative pressure, the data reveal that those samples subjected to long periods of mechanochemical treatment have a greater N2 adsorption capacity, giving rise to isotherms classified as Type II, which are associated with macroporous materials.44 In this sense, the study of the pore size distribution, estimated by the DFT method,45 shows that the MgO_untreated sample displays pores with a maximum size of 6.8 nm (Fig. S1†). However, the mechanochemical treatment provokes a shift of pore size distribution, appearing with a maximum value close to 5 nm, which is related to the increase of the SBET values (Table 1). On the other hand, it is also striking the apparition of another maximum in the pore size distribution of about 20 nm for those samples subjected to longer mechanochemical treatment. This data could be related to a decrease in the size of the MgO nanoparticles, which generates space between adjacent particles, leading to macropores. The increase in the surface area of the material as well as the pore volume could be directly related to the inverse of the time that the materials were mechanically treated (Fig. S1†).
The estimation of the basic sites, involved in the catalytic transfer hydrogenation of FUR, has been performed by temperature-programmed desorption of CO2 (CO2-TPD) (Fig. 3 and Table 2). In all MgO catalysts, two separate desorption bands denote the presence of two types of basic sites that interact with CO2. The main contribution takes place from 70 to 260 °C, with a maximum at 160–170 °C. Moreover, a smaller contribution of sites with greater basicity is also observed at higher temperatures. XRD analysis post CO2-TPD shows that the MgO materials are regenerated after desorption (ESI, Fig. S6†). As could be expected, those MgO catalysts with smaller crystal sizes and higher surface area display a higher amount of available basic sites. In addition, the number of basic sites increases with the treatment time, existing a direct relationship between treatment time and basicity. This relationship must be associated to an increase at the grain boundaries by the mechanochemical treatment.
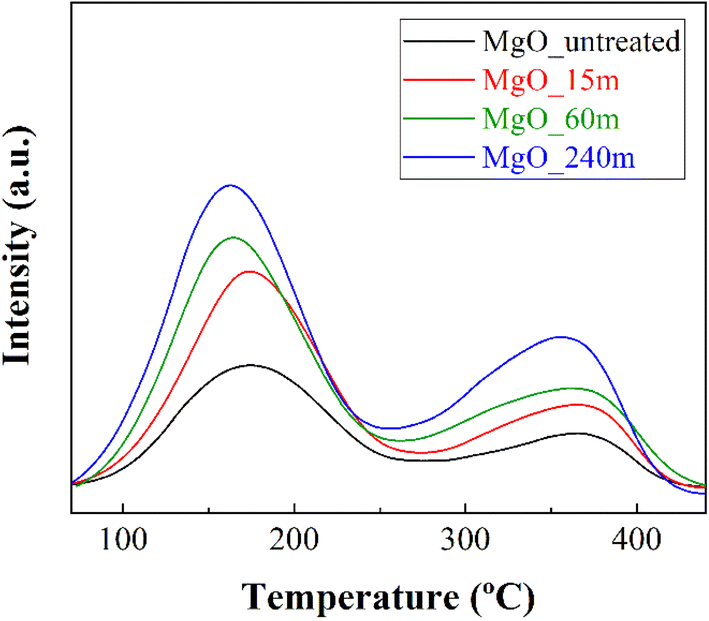 | ||
| Fig. 3 CO2-TPD of MgO_untreated, MgO_15m, MgO_60m and MgO_240m catalysts. | ||
| Sample | Weak basic sites (μmol g−1) | Strong basic sites (μmol g−1) | Total basic sites (μmol g−1) |
|---|---|---|---|
| MgO_untreated | 189 | 89 | 278 |
| MgO_15m | 302 | 125 | 427 |
| MgO_60m | 357 | 161 | 518 |
| MgO_240m | 417 | 310 | 727 |
The chemical characterization of the catalyst surface was carried out by XPS (Fig. 5 and Table 3). The C 1s core level spectra display two contributions: a main band at 284.8 eV, associated with adventitious carbon, and another one at 289.5 eV, which can be assigned to carbonate species, due to the partial carbonation of MgO in contact with atmospheric CO2 (Fig. 5A). Nevertheless, the intensity of this latter contribution is very low and similar for all samples, since the Mg(OH)2 precursors were calcined prior to their analysis.46 On the other hand, the O 1s core level spectra are also composed of two bands: the most intense contribution located at 529.6 eV is typical of oxide species and the less intense appearing at higher binding energy values, about 532 eV, is attributed to the presence of carbonate or hydroxide species, caused by MgO reactivity when exposed to atmospheric CO2 and H2O (Fig. 5B).46 Finally, Mg 2p core level spectra display a single contribution located at 49.4 eV, typical of Mg2+ species (Fig. 5C).46
| Sample | Atomic concentrations (%) | Molar ratio | ||
|---|---|---|---|---|
| C 1s | O 1s | Mg 2p | O/Mg | |
| MgO_untreated | 13.25 | 50.23 | 36.51 | 1.38 |
| MgO_15m | 13.32 | 48.59 | 38.68 | 1.28 |
| MgO_60m | 13.84 | 49.06 | 37.09 | 1.33 |
| MgO_240m | 15.85 | 48.17 | 35.97 | 1.34 |
Regarding the surface atomic concentration, all MgO catalysts exhibit similar O/Mg ratios, the atomic concentration of Mg being 35.97–38.68%, while the concentration of O is 48.17–50.23%. Thus, the calculated O/Mg atomic ratios were between 1.28 and 1.38, which is above the expected theoretical value, probably due to the presence of carbonate and hydroxyl groups on the surface of the MgO, as mentioned before.
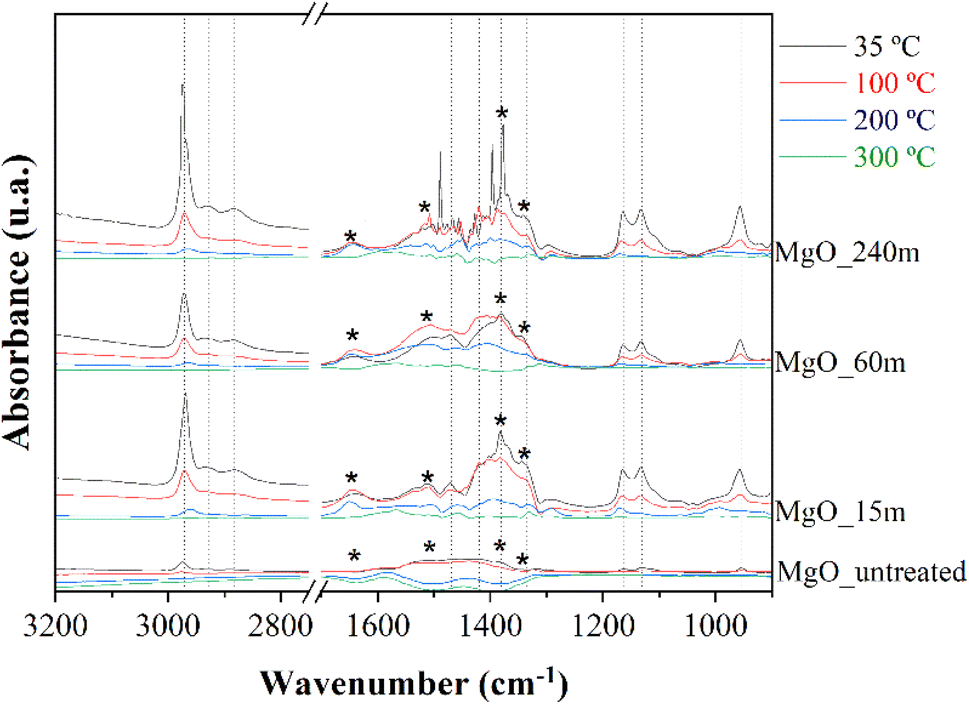
Fig. 6 shows the FTIR-ATR spectra of the MgO catalysts synthesized as a function of mechanochemical treatment time and subsequent calcination. All samples display a broad band between 3900 and 3200 cm−1, which is assigned to the stretching vibration mode of OH groups. In the same way, the vibration band located about 1640 cm−1 corresponds to the bending vibration mode of adsorbed H2O molecules,47,48 suggesting that MgO samples exposed to longer mechanochemical treatment are more prone to H2O adsorption. In addition, another band is observed at 1385 cm−1,49 which can be assigned to carbonate species, confirming that MgO is sensitive to be carbonation due to its basicity. Finally, the band at 840 cm−1 is attributed to the Mg–O stretching vibration mode.50
4. Catalytic tests
Once the MgO catalysts were obtained, these were studied in the catalytic transfer hydrogenation of FUR to FOL (Scheme 1).18 The catalytic results show that FUR conversion in all MgO catalysts increases with reaction time, although the behavior of the catalysts differs between them (Fig. 7A). Thus, the catalyst synthesized without the mechanochemical treatment (MgO_untreated) reaches a FUR conversion of 68% after 3 h of reaction at 90 °C. However, the use of the mechanochemical treatment, which causes a decrease in particle size, improves the FUR conversion notably. In this way, MgO_15m achieves a conversion of 79% after only 2 h at 90 °C, while MgO_untreated only displays a conversion of 48% under these experimental conditions. However, it is remarkable that even though the particle size decreases and the amount of basic sites increases clearly, the catalytic conversion diminishes noticeably for those catalysts treated for longer times. Thus, the catalyst subjected to mechanochemical treatment for 240 min only achieves a FUR conversion of 32% after 2 h at 90 °C. | ||
| Scheme 1 Reaction scheme proposed from the products obtained. | ||
The comparison of the obtained data with other reported in the literature for basic catalysts evidences that a higher FUR conversion is attained at lower temperatures, compared to those with Lewis acid sites, such as Al- and Zr-based catalysts, for which the reaction temperature had to increase by at least 120 °C.51 These data suggest that hydrogen transfer is favored on basic sites. In this sense, it has been previously reported the use of a similar reaction temperature with other basic catalysts, such as CaAlOx obtained from hydrocalumites, for the reduction of benzaldehyde and cyclohexanone.52 However, Xiao has performed the reduction of cinnamaldehyde with MgAlOx obtained from hydrotalcites at 80 °C.53
Despite the different catalytic behavior of the MgO catalysts (Fig. 7B), their selectivity patterns are similar (Scheme 1). Thus, the main product is FOL, obtaining yields very close to 78% for those catalysts previously subjected to mechanochemical treatment between 1 and 30 min. Among them, the most active catalyst is that treated for 15 min, which achieves a yield of 72% after 2 h at 90 °C. The use of longer time of mechanochemical treatment worsens the FOL yield. In this sense, Zr- or Hf-based catalysts also promoted consecutive reactions,54 in such a way that other products, such as alkyl furfuryl ether, alkyl levulinates or valerolactone can be formed from FOL. Nevertheless, all these products possess commercial interest, although the existence of consecutive reactions would limit the yield of the target product and undesired reactions can take place facilitating the formation of carbonaceous deposits which can block the active sites. With the use of basic catalysts, the CTH process largely avoids the detrimental effects of secondary processes in FOL production.37 Similar results have been obtained with catalysts that have exclusively Lewis acid sites, even though they require higher reaction temperature.32 However, the coexistence of Lewis and Brønsted acid sites could promote consecutive reactions.55 On the other hand, small proportions of 4-(2-furyl)-3-buten-2-one (FAc) are also detected in all cases. Based on these results, FOL is obtained by the hydride transfer from the sacrificing alcohol (2-propanol) to FUR,18 while FAc is formed via condensation of FUR with the acetone generated in the CTH process from 2-propanol under basic conditions.56,57 However, the maximum FAc yield barely exceeds 7% in the best of cases, after 3 h at 90 °C.
In the following study, the influence of the reaction temperature was evaluated for MgO_15m (Fig. 8), which was selected due to its high FUR conversion in ashort reaction time. As it was observed in previous studies, the FUR conversion increases with the reaction temperature, although higher values were obtained at lower temperatures and shorter times than with other catalysts with Lewis acid sites.18 Thus, the results reported in Fig. 8 point out that total FUR conversion can be achieved after only 30 min at 135 °C. The analysis of the obtained products reveals that an increase in the reaction temperature did not promote the formation of by products, being FOL the main product throughout the temperature range with a maximum yield of 80% at 120 °C. In the same way, the formation of FAc was also detected, although the maximum yield was only 13% for the reactions carried out at higher temperatures. In this sense, the high activity of the catalysts at higher temperatures favors the formation of high quantities of acetone in short reaction times, which reacts with FUR to form FAc under basic conditions.56
A key parameter in the use of heterogeneous catalysts is their reusability (Fig. 9). The study of the MgO_15m catalyst after the first catalytic cycle shows that the FUR conversion decreases notably, from 82 to 25%, after 3 h at 90 °C. These data agree with other previously reported in the literature for basic catalysts, where a strong deactivation was observed, in such a way that the catalysts could not be regenerated, even at high temperatures.37 Regarding the obtained products, both FOL and FAc decrease proportionally, so it is expected the blockage of the active sites by FUR, reaction products, or the solvent used in the reaction. In this sense, Gyngazova et al. have reported that alkaline earth metal oxides interact strongly with methanol.39 In addition, the thermal treatment of used catalysts during the regeneration process leads to the formation of CO2, which can react with metal oxides to form the corresponding metal carbonates, which are highly stable.39
Considering the previous study (Fig. 9), in an additional test, the MgO_15m catalyst was treated with 2-propanol at 90 °C for 3 h, and then the reactor was cooled down before FUR was added to carry out the reaction under the same experimental conditions (Fig. 10). Interestingly, the prior treatment of the catalyst with 2-propanol caused a drastic loss of catalytic activity, diminishing the FUR conversion from 82 to 21%. This strong deactivation by the reaction of alcohols with a solid basic catalyst is in agreement with previous studies reported by other authors.37,39
The nature of the deactivation of the catalyst surface was studied by controlled adsorption and desorption of 2-propanol monitored by FTIR spectroscopy analysis (Fig. 10). After the adsorption of 2-propanol, three narrow signals, at 2971, 2926 and 2884 cm−1, were identified, which are assigned to the stretching vibration modes of the C–H bond in –CH3 (asymmetric and symmetric) and –CH groups of 2-propanol, respectively. This would confirm the presence of adsorbed 2-propanol on the catalyst surface. However, these bands were only barely perceptible in MgO_untreated, indicating a smaller quantity of 2-propanol was adsorbed on the surface of the mechanically untreated MgO, due to a smaller number of available basic centers. On the other hand, all spectra show absorption bands located at 1469, 1383, 1330, 1165 and 1130 cm−1, which are assigned to symmetric and asymmetric δ (CH3), δ (CH), ν (C–O), ν (C–C) and r (CH3) modes of 2-propoxide species, due to the strong, dissociative adsorption of 2-propanol on the surface of MgO.58 On the opposite, the absence of vibration bands at 1260, 1251 and 1241 cm−1, assigned to δ (C–H), δ (O–H), discards a non-dissociative adsorption of 2-propanol. Thus, these data would confirm that the adsorption of 2-propanol on the MgO surface occurs through a dissociative mechanism in which 2-propanol molecules are adsorbed in the form of 2-propoxide onto the basic sites, while the proton has been captured by the surface of the catalyst. The band found at 950 cm−1 belongs to r (CH2) of 2-propanol.59,60
Previous works have demonstrated that the surface of high crystalline MgO displays a (100) thermodynamically stable plane. In this case, the 5-coordinated ions have been unable to dissociate water from alcohol due to weak interactions. However, the presence of defects, such as monoatomic or diatomic steps, convex positions, corners, or vacancies, generates low coordination sites that are highly reactive. Low coordination sites can interact strongly leading to the proton abstraction in alcohols, or water, onto the surface of the MgO, forming alkoxides and hydroxyl groups.61,62 This is in agreement with the FTIR spectroscopy analysis, where the untreated surface by mechano-treatment shows weaker interaction with the alcohol molecules due to the existence of a smaller amount of defects, which would imply a lower amount of chemisorption sites and higher crystallinity, as was suggested by CO2-TPD (Fig. 4) and XRD (Fig. 1). On the other hand, the exertion of mechanical force on the Mg(OH)2 samples have exposed low coordination sites, as indicated by the increase in the surface area (Table 1) and the amount of basic sites (Table 2) of the resulting MgO, as well as the decrease in size of the MgO crystals, promoting the adsorption of 2-propoxide anions.
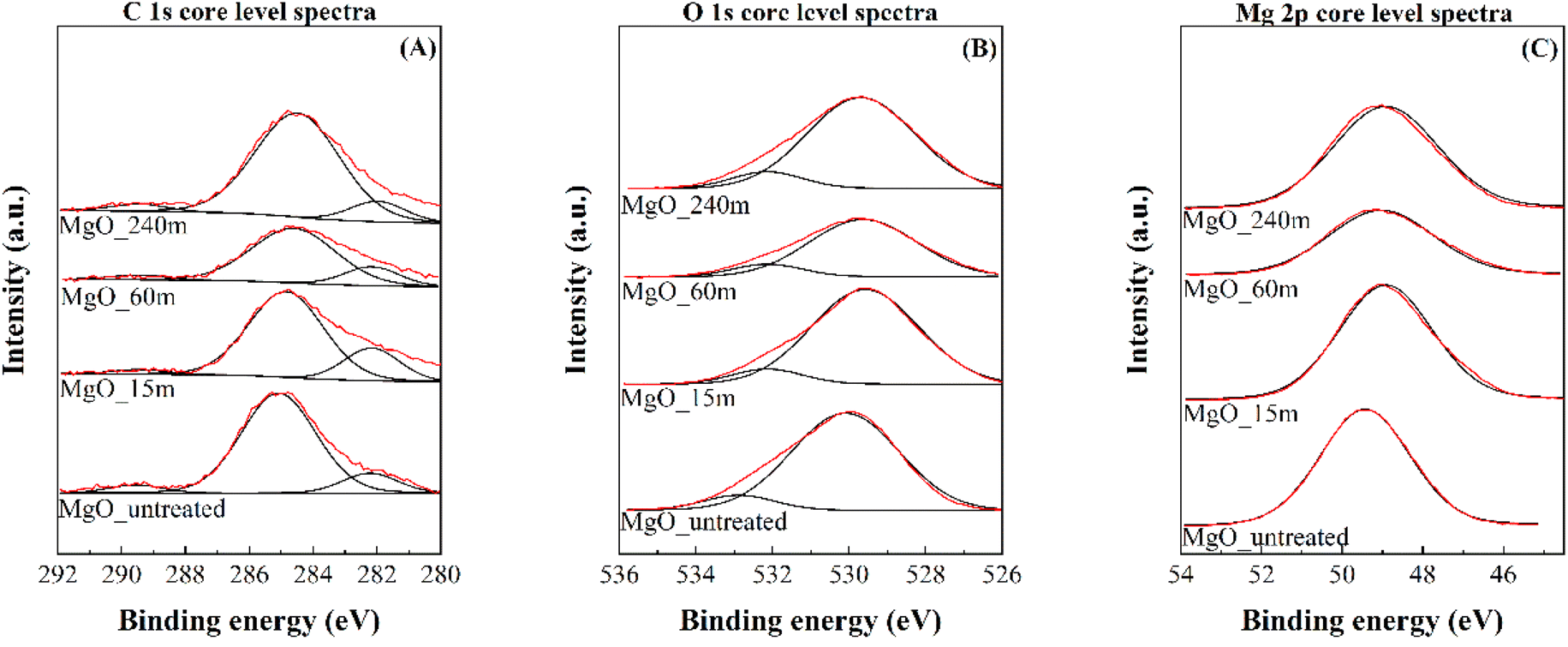 | ||
| Fig. 4 C 1s (A), O 1s (B) and Mg 2s (C) core level spectra obtained by XPS of MgO_untreated, MgO_15m, MgO_60m and MgO_240m catalysts. | ||
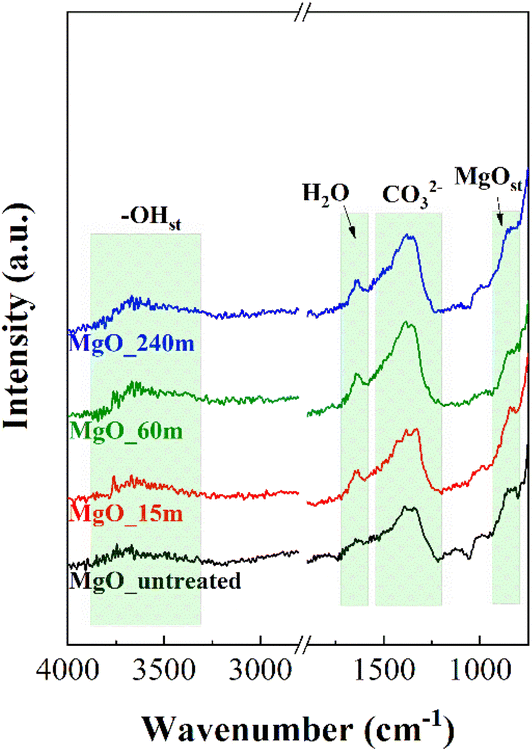 | ||
| Fig. 5 ATR-spectra of MgO_untreated, MgO_15m, MgO_60m and MgO_240m catalysts. | ||
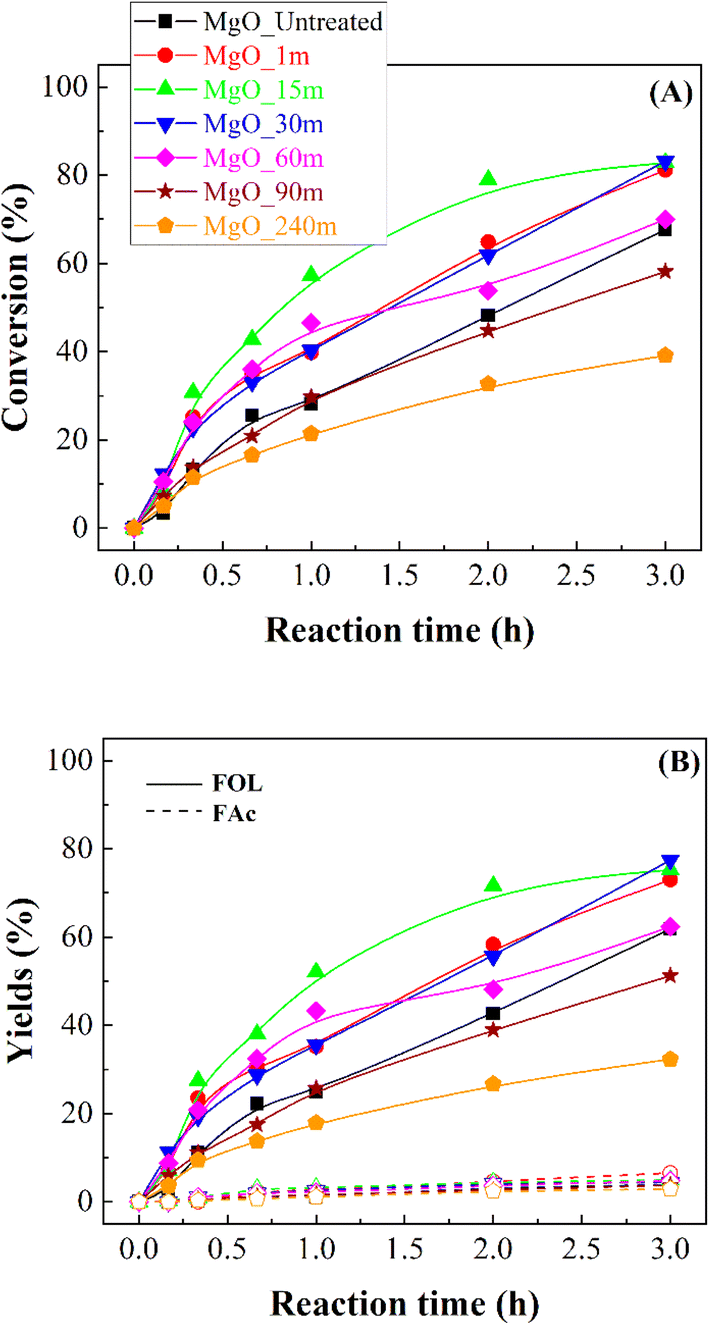 | ||
| Fig. 6 FUR conversion (A) and yields (B) in the CTH of FUR using MgO catalysts (experimental conditions: temperature: 90 °C; 2-propanol/FUR molar ratio: 50; 0.1 g catalyst; FUR/catalyst weight ratio: 1). FOL: furfuryl alcohol, FAc: 4-(2-furyl)-3-buten-2-one. | ||
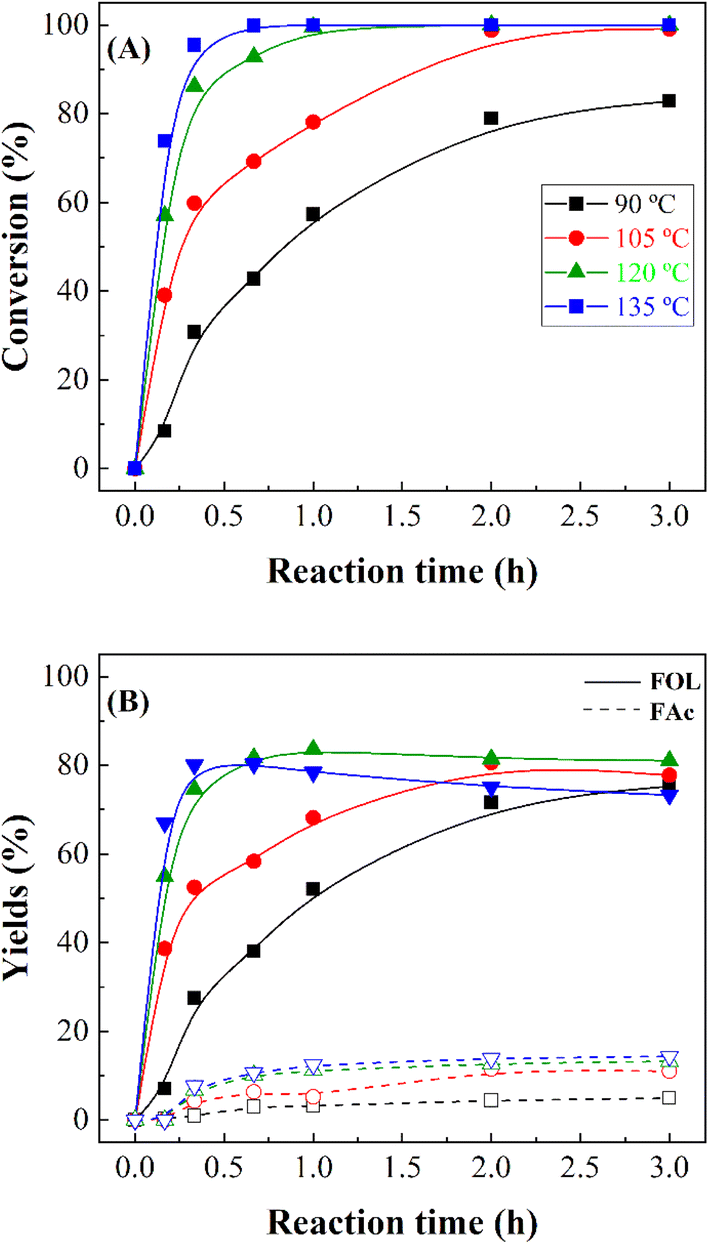 | ||
| Fig. 7 FUR conversion (A) and yields (B) in the CTH of FUR using MgO_15m catalyst (experimental conditions: temperature: 90–135 °C; 2-propanol/FUR molar ratio: 50; 0.1 g catalyst; FUR/catalyst weight ratio: 1). | ||
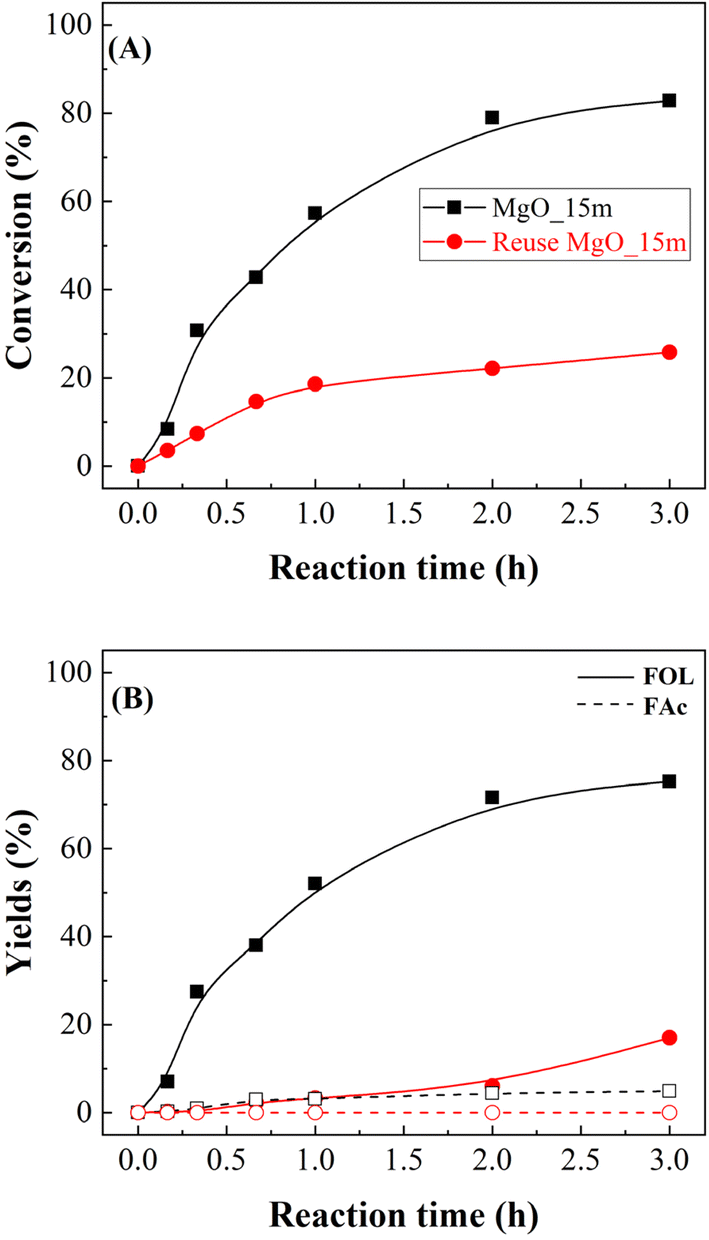 | ||
| Fig. 8 FUR conversion (A) and yields (B) in the CTH of FUR using MgO_15m catalyst after 2 cycles of reaction (experimental conditions: temperature: 90 °C; 2-propanol/FUR molar ratio: 50; 0.1 g catalyst; FUR/catalyst weight ratio: 1). | ||
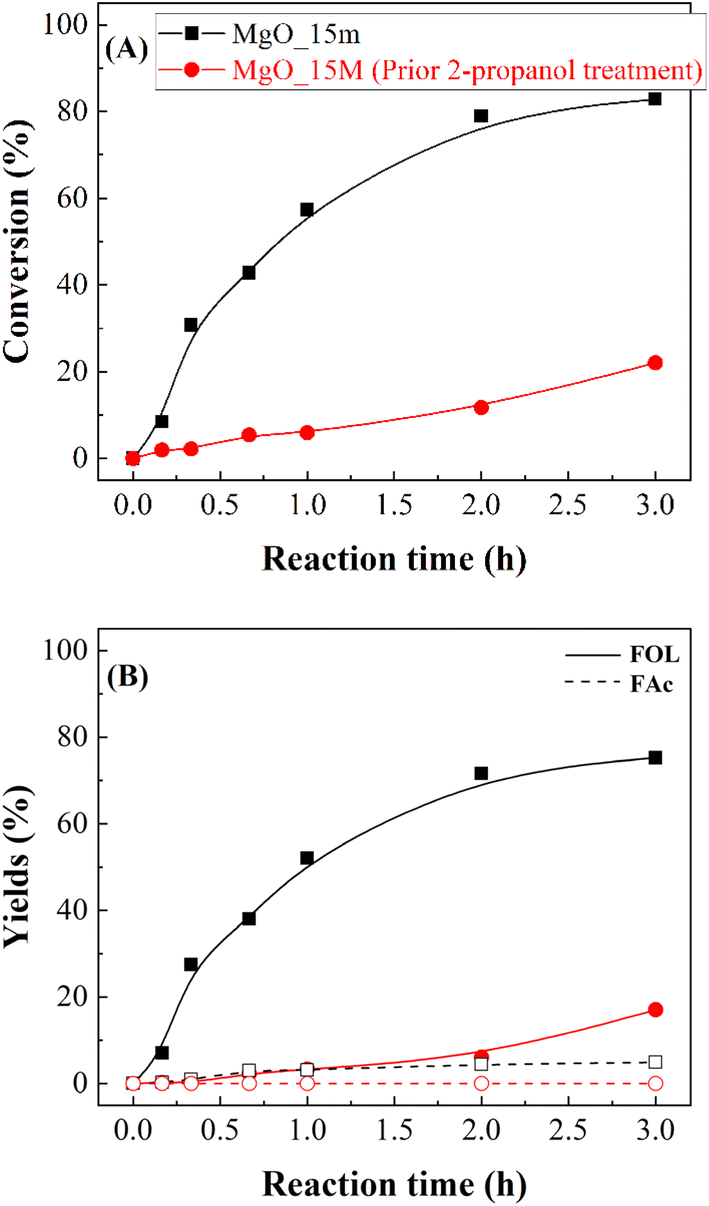 | ||
| Fig. 9 FUR conversion (A) and yields (B) in the CTH of FUR using fresh MgO_15m catalyst and after pretreatment in 2-propanol at 90 °C for 3 h (experimental conditions: temperature: 90 °C; 2-propanol/FUR molar ratio: 50; 0.1 g catalyst; FUR/catalyst weight ratio: 1). | ||
 | ||
| Fig. 10 FTIR spectroscopy analysis coupled with 2-propanol adsorption for MgO catalysts. | ||
Finally, some carbonate species can be identified in the spectra of Fig. 10. Thus, the signals at 1650 and 1340 cm−1 correspond to bidentate carbonate ions over plane surfaces, while the absorption bands at 1510 and 1340 cm−1 are assigned to monodentate carbonate species. In both cases, the band at lower wavenumber corresponds to the symmetric stretching, while the band at higher values is associated with the asymmetric stretching of the O–C–O bonds.58,63
It is noteworthy to remark on the stability of such 2-propoxide species on the surface of the catalyst, since they are detectable even after evacuation at 300 °C.
The study of fresh catalysts shows that mechanical treatment for a longer time to obtain MgO nanoparticles favors the adsorption of a higher amount of 2-propanol as monolayer, as suggested by the linearity at a relative pressure between 0.05 and 0.45 (Fig. 11 and Table 2). The increase must be related to the increase in the number of basic sites due to the longer mechanochemical treatment of the materials, which is related to a decrease in the MgO crystal size, or an improvement of its textural properties (Table 1). The 2-propanol adsorption–desorption profiles in a second cycle decays notably in comparison to the first adsorption. This decrease is ascribed to the strong adsorption of propoxide species on the surface of the MgO, as was inferred from FTIR spectroscopy analysis coupled with 2-propanol adsorption (Fig. 10), causing partial blockage of the available basic sites.
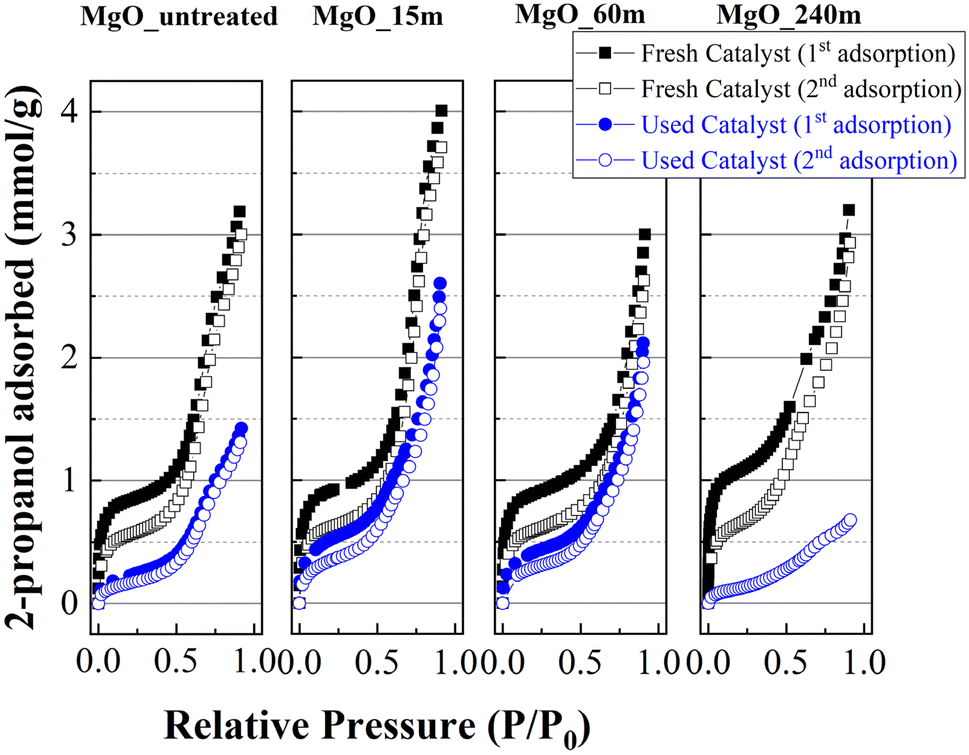 | ||
| Fig. 11 2-Propanol adsorption isotherms at 25 °C for MgO catalysts, before and after the reaction at 90 °C for 3 h. | ||
In the same way, the catalysts were collected after carrying out a reaction at 90 °C for 1 h to study their ability to adsorb 2-propanol. The analysis of the adsorption at a relative pressure between 0.05 and 0.45, where takes place the adsorption on the monolayer, reveals a lower adsorption of 2-propanol in comparison to that observed for fresh catalysts, confirming that the basic sites have been blocked. In this sense, it is important to note that the catalyst with the worst catalytic performance(MgO_240m) also displays the lowest 2-propanol adsorption after the reaction, confirming that those catalysts with a high proportion of available and stronger basic sites are prone to be deactivated with 2-propanol more easily. In the same way, the presence of higher meso- and macroporosity for MgO_240m catalyst (ESI, Fig. S5†) can also promote a faster interaction between the active phase and the alcohol and FUR promoting a faster deactivation. In the case of MgO catalysts subjected to a shorter mechanochemical treatment time, the surface values are lower. However, the CO2-TPD data indicate that the strength of the basic centers is weaker. This implies the presence of more labile centers where propoxide species can be desorbed more easily, so that the basic centers should become available faster, obtaining better conversion values and in shorter times than those catalysts with more available basic sites and better textural properties.
Finally, the comparison of the obtained results with those reported in the literature for other basic catalyst (Table 4) reveals that the use of mechanochemical treatment for the synthesis of the precursors allows to achieve higher conversion values and yields than other basic catalysts, even in shorter times. From the data obtained in the present study, it can be inferred that the mechanochemical treatment generates a greater amount of grain boundary defects, which lead to more active catalytic sites in the catalytic transfer of hydrogen for the reduction of FUR to FOL.
| Catalyst | Alcohol | Temp (°C) | P (MPa) | H donor/FUR mole ratio | T (h) | XFUR (%) | YFOL (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| La2O3 | iPOH | 180 | 0.8 | 35.6 | 17 | 98 | 94 | 64 |
| La(OH)3 | iPOH | 180 | 0.8 | 35.6 | 17 | 60 | 53 | 65 |
| MgO | iPOH | 170 | — | 23.8 | 5 | 100 | 74 | 66 |
| MgO | — | 180 | — | — | — | 99 | 98 | 38 |
| MgO | iPOH | 100 | 0.1 | 10.8 | 20 | 15 | 8 | 36 |
| MgO | iPOH | 150 | 0.8 | 62.5 | 1 | 44 | 40 | 67 |
| MgO–Al2O3 | iPOH | 110 | 0.1 | 50 | 4 | 100 | 90 | 37 |
| MgO–Fe2O3 | iPOH | 170 | 0.1 | 50 | 6 | 100 | 90 | 68 |
| MgO | iPOH | 90 | 0.1 | 50 | 2 | 79 | 72 | This work |
5. Conclusions
Several MgO catalysts have been prepared from previously mechanochemically treated Mg(OH)2. Wet mecanochemical treatment has been demonstrated to completely alter the behavior of the final catalysts due to modifications upon the surface. Linear correlations were found between mechanochemical treatment time and surface area. Also, the treatment distorts the surface of the catalysts, generating defects, strong basic sites, as found by FTIR spectroscopy and CO2-TPD. Treating the precursors for 1 to 60 min was found to have positive effects on the catalytic activity in comparison to the untreated MgO, while longer treatment times were detrimental for both FUR conversion and FOL yield of the CTH process. The catalyst prepared after a mechanochemical treatment of 15 min, MgO_15m, was the best performing catalyst, with a conversion of 79% of FUR (from 48% in the MgO_untreated) and a FOL yield of 72% (from 39% in the MgO_untreated). Optimum reaction temperature was found to be 120 °C, and the reusability and poisoning of the catalyst with 2-propoxide species was evaluated by using ATR and 2-propanol adsorption studies.Data availability
The data presented in this study are available on request from the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Spanish Ministry of Science and Innovation (PID2021-122736OB-C42) and FEDER (European Union) funds. B. Torres-Olea thanks Ministerio de Universidades for his predoctoral contract (FPU20/02334).References
- G. Kumar, J. Dharmaraja, S. Arvindnarayan, S. Shoban, P. Bakonyi, G. D. Saratale, N. Nemestóthy, K. Bélafi-Bakó, J. J. Yoon and S. H. Kim, Fuel, 2019, 251, 352–357 CrossRef CAS.
- I. K. M. Yu, H. Chen, F. Abeln, H. Auta, J. Fan, V. L. Dudarin, J. H. Clark, S. Parsons, C. J. Chuck, S. Zhang, G. Luo and D. C. W. Tsang, Crit. Rev. Environ. Sci. Technol., 2021, 51, 1479–1532 CrossRef CAS.
- A. S. Jatoi, S. A. Abbasi, Z. Hashmi, A. K. Shah, M. S. Alam, Z. A. Bhatti, G. Maitlo, S. Hussain, G. A. Khandro, M. A. Usto and A. Iqbal, Biomass Convers. Biorefin., 2021, 13, 6457–6469 CrossRef.
- A. K. Kumar and S. Sharma, Bioresour. bioprocess., 2017, 4, 7 CrossRef PubMed.
- F. Delbecq, Y. Wang, A. Muralidhara, K. El Ouardi, G. Marlair and C. Len, Front. Chem., 2018, 6, 146 CrossRef PubMed.
- E. Cousin, K. Namhaed, Y. Pérès, P. Cognet, M. Delmas, H. Hermansyah, M. Gozan, P. A. Alaba and M. K. Aroua, Sci. Total Environ., 2022, 847, 157599 CrossRef CAS PubMed.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. López-Granados, Energy Environ. Sci., 2016, 9, 1144–1189 RSC.
- K. Yan, G. Wu, T. Lafleur and C. Jarvis, Renewable Sustainable Energy Rev., 2014, 38, 663–676 CrossRef CAS.
- Y. Wang, D. Zhao, D. Rodríguez-Padrón and C. Len, Catalysts, 2019, 9, 796 CrossRef.
- C. P. Jiménez-Gómez, J. A. Cecilia, D. Durán-Martín, R. Moreno-Tost, J. Santamaría-González, J. Mérida-Robles, R. Mariscal and P. Maireles-Torres, J. Catal., 2016, 336, 107–115 CrossRef.
- C. P. Jiménez-Gómez, J. A. Cecilia, R. Moreno-Tost and P. Maireles-Torres, ChemSusChem, 2016, 10, 1448–1459 CrossRef PubMed.
- C. P. Jiménez-Gómez, J. A. Cecilia, A. C. Alba-Rubio, A. Cassidy, R. Moreno-Tost, C. García-Sancho and P. Maireles-Torres, Fuel, 2022, 319, 123827 CrossRef.
- Y. Nakagawa, H. Nakagawa, H. Watanabe and K. Tomishige, ChemCatChem, 2012, 4, 1791–1797 CrossRef CAS.
- C. P. Jiménez-Gómez, J. A. Cecilia, R. Moreno-Tost and P. Maireles-Torres, ChemCatChem, 2017, 9, 2881–2889 CrossRef.
- C. P. Jiménez-Gómez, C. Defilippi, J. A. Cecilia, R. Moreno-Tost, P. Maireles-Torres and C. Giordano, Mol. Catal., 2020, 487, 110889 CrossRef.
- X. Meng, Y. Yang, L. Chen, M. Xu, X. Zhang and M. Wei, ACS Catal., 2019, 9, 4226–4235 CrossRef CAS.
- Z. An and J. Li, Green Chem., 2022, 24, 1780–1808 RSC.
- M. J. Gilkey and B. Xu, ACS Catal., 2016, 6, 1420–1436 CrossRef CAS.
- M. Wu, L. Bai, F. Deng, J. He, K. Song and H. Li, Coord. Chem. Rev., 2025, 523, 216259 CrossRef CAS.
- K. Wang, Z. Li, Z. Guo, J. Huang, T. Liu, M. Zhou, J. Hu and H. Li, Green Chem., 2024, 26, 2454–2475 RSC.
- Z. Guo, P. Zhou, L. Jiang, S. Liu, Y. Yang, Z. Li, P. Wu and Z. Zhang, Adv. Mater., 2024, 4, 2311149 CrossRef PubMed.
- H. Li, J. He, A. Riisager, S. Saravanamurugan, B. Song and S. Yang, ACS Catal., 2016, 6, 7722–7727 CrossRef CAS.
- R. López-Asensio, C. P. Jiménez-Gómez, C. García-Sancho, R. Moreno-Tost, J. A. Cecilia and P. Maireles-Torres, Int. J. Mol. Sci., 2019, 20, 828 CrossRef PubMed.
- R. Maderuelo-Solera, S. Richter, C. P. Jiménez-Gómez, C. García-Sancho, F. J. García-Mateos, J. M. Rosas, R. Moreno-Tost, J. A. Cecilia and P. Maireles-Torres, Ind. Eng. Chem. Res., 2021, 6, 18791–18805 CrossRef PubMed.
- J. Iglesias, J. A. Melero, G. Morales, J. Moreno, Y. Segura, M. Paniagua, A. Cambra and B. Hernández, Catalysts, 2015, 5, 1911–1927 CrossRef CAS.
- Z. Liu, Z. Zhang, R. Fu, J. Xu, J. Lu, Z. Wen and B. Xue, ACS Appl. Nano Mater., 2023, 6, 13196–13207 CrossRef.
- S. Kumaravel, J. K. Alagarasan, A. K. Yadav, W. Ali, M. Lee, M. E. Khan, S. K. Ali, A. H. Bashiri, W. Zakri and K. Balu, J. Phys. Chem. Solids, 2014, 186, 111831 CrossRef.
- R. Barakov, N. Shcherban, O. Petrov, D. N. Rainer, M. Kubu, J. Cejka, M. Shamzhy and M. Opanasenko, Catal. Today, 2024, 426, 114406 CrossRef CAS.
- J. A. Melero, G. Morales, J. Iglesias, M. Paniagua and C. López-Aguado, Ind. Eng. Chem. Res., 2018, 57, 11592–11599 CrossRef CAS.
- W. Li, M. Li, H. Liu, W. Jia, X. Yu, S. Wang, X. Zeng, Y. Sun, J. Wei, X. Tang and L. Lin, Mol. Catal., 2021, 506, 111538 CrossRef CAS.
- H. Zhang, W. Yang, I. I. Roslan, S. Jaenicke and G. K. Chuah, J. Catal., 2019, 375, 56–67 CrossRef CAS.
- R. López-Asensio, J. A. Cecilia, C. P. Jiménez-Gómez, C. García-Sancho, R. Moreno-Tost and P. Maireles-Torres, Appl. Catal., A, 2018, 556, 1–9 CrossRef.
- C. García-Sancho, C. P. Jiménez-Gómez, N. Viar-Antuñano, J. A. Cecilia, R. Moreno-Tost, J. M. Mérida-Robles, J. Requies and P. Maireles-Torres, Appl. Catal., A, 2021, 609, 117905 CrossRef.
- F. Wang and Z. Zhang, ACS Sustainable Chem. Eng., 2017, 5, 942–947 CrossRef CAS.
- F. Li, S. Jiang, J. Huang, Y. Wang, S. Lu and C. Li, New J. Chem., 2020, 44, 478–486 RSC.
- J. Hidalgo-Carrillo, A. Parejas, M. J. Cuesta-Rioboo, A. Marinas and F. J. Urbano, Catalysts, 2018, 8, 539 CrossRef.
- R. López-Asensio, J. A. Cecilia-Buenestado, C. Herrera-Delgado, M. A. Larrubia-Vargas, C. García-Sancho, P. J. Maireles-Torres and R. Moreno-Tost, Catalysts, 2023, 13, 45 CrossRef.
- K. S. Koppadi, R. R. Chada, S. S. Enumula, R. K. Marella, S. R. R. Kamaraju and D. R. Burri, Catal. Lett., 2017, 147, 1278–1284 CrossRef CAS.
- M. S. Gyngazova, L. Grazia, A. Lolli, G. Innocenti, T. Tabanelli, M. Mella, S. Albonetti and F. Cavani, J. Catal., 2019, 372, 61–73 CrossRef CAS.
- C. Suryanarayana, Mechanical alloying and milling, Prog. Mater. Sci., 2001, 46, 1–184 CrossRef CAS.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Frisic, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Mechanochemistry: opportunities for new and cleaner synthesis, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- A. P. Amrute, J. De Bellis, M. Felderhoff and F. Schüth, Mechanochemical synthesis of catalytic materials, Chem.–Eur. J., 2021, 27, 6819–6847 CrossRef CAS PubMed.
- M. Kitagawa, S. Misu, J. Ichikawa and H. Matsuhashi, Preparation of active MgO by short-time thermal decomposition of Mg(OH)2, Res. Chem. Intermed., 2015, 41, 9463–9473 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodríguez-Reinoso, J. Rouquerol and K. S. W. King, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- J. Landers, G. Y. Gor and A. V. Neimark, Colloids Surf., A, 2013, 43, 3–32 CrossRef.
- J. F. Moulder, W. F. Stickle, P. E. Sobol, et al., in Handbook of X-Ray Photoelectron Spectroscopy, ed. J. Chastain, Perkin-Elmer Corporation, Physical Electronics Division, Eden Prairie, 1992 Search PubMed.
- N. Sutradhar, A. Sinhamahapatra, S. K. Pahari, P. Pal, H. C. Bajaj, I. Mukhopadhyay and A. B. Panda, J. Phys. Chem., 2011, 115, 12308–12316 CAS.
- W. A. Khaleel, S. A. Sadeq, I. A. M. Alani and M. H. M. Ahmed, Opt. Laser Technol., 2019, 115, 331–336 CrossRef CAS.
- C. M. Janet, B. Viswanathan, R. P. Viswanathand and T. K. Varadarajan, J. Phys. Chem. C, 2007, 111, 10267–10272 CrossRef CAS.
- A. T. Vu, S. Jiang, K. Ho, J. B. Lee and C. H. Lee, Chem. Eng. J., 2015, 269, 82–93 CrossRef CAS.
- J. F. Miñambres and J. Cejka, Catal. Rev., 2024, 2111–2152 CrossRef.
- M. Mora, M. I. López, C. Jiménez-Sanchidrián and J. R. Ruíz, Catal. Lett., 2010, 136, 192–198 CrossRef CAS.
- Z. Xiao, Mol. Catal., 2017, 436, 1–9 CrossRef CAS.
- W. Sun, H. Li, X. Wang and A. Liu, Front. Chem., 2022, 10, 863674 CrossRef CAS PubMed.
- L. Bui, H. Luo, W. R. Gunther and Y. Román-Leshkov, Angew. Chem., Int. Ed., 2013, 52, 8022–8025 CrossRef CAS PubMed.
- R. Huang, J. Chang, H. Choi, J. M. Vohs and R. J. Gorte, Catal. Lett., 2022, 152, 3833–3842 CrossRef CAS.
- L. Hora, V. Kelbichova, O. Kikhtyanin, O. Bortnovskiy and D. Kubicka, Catal. Today, 2014, 223, 138–147 CrossRef CAS.
- D. Carriazo, C. Martín and V. Rives, Catal. Today, 2007, 126, 153–161 CrossRef CAS.
- S. A. Fuente, C. A. Ferretti, N. F. Domancich, V. K. Díez, C. R. Apesteguía, J. I. Di Cosimo, R. M. Ferullo and N. J. Castellani, Appl. Surf. Sci., 2015, 327, 268–276 CrossRef CAS.
- P. R. Rossi, G. Busca, V. Lorenzelli, O. Saur and J. C. Lavalley, Langmuir, 1987, 3, 52–58 CrossRef CAS.
- M. M. Branda, P. G. Belelli, R. M. Ferullo and N. J. Castellani, Catal. Today, 2003, 85, 153–165 CrossRef CAS.
- H. Petitjean, K. Tarasov, F. Delbecq, P. Sautet, J. M. Krafft, P. Bazin, M. C. Paganini, E. Giomello, M. Che, H. Lauron-Pernot and G. Costentin, J. Phys. Chem. C, 2010, 114, 3008–3016 CrossRef CAS.
- C. Chizallet, G. Costentin, M. Che, F. Delbecq and P. Sautet, J. Phys. Chem. B, 2006, 110, 15878–15886 CrossRef CAS PubMed.
- T. A. Natsir, T. Hara, N. Ichikuni and S. Shimazu, Chem. Lett., 2017, 46, 1580–1583 CrossRef CAS.
- T. A. Natsir, T. Hara, N. Ichikuni and S. Shimazu, Bull. Chem. Soc. Jpn., 2018, 91, 1561–1569 CrossRef CAS.
- N. S. Biradar, A. M. Hengne, S. S. Sakate, R. K. Swami and C. V. Rode, Catal. Lett., 2016, 146, 1611–1619 CrossRef CAS.
- Z. Liu, Z. Zhang, Z. Wen and B. Xue, Catal. Lett., 2022, 152, 3537–3547 CrossRef CAS.
- R. Maderuelo-Solera, R. López-Asensio, J. A. Cecilia, C. P. Jiménez-Gómez, C. García-Sancho, R. Moreno-Tost and P. Maireles-Torres, Appl. Clay Sci., 2019, 183, 105351 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mr00128a |
| This journal is © The Royal Society of Chemistry 2025 |