
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect arylation of gem-difluorostyrenes using in situ mechanochemically generated calcium-based heavy Grignard reagents†
Xihong Wanga,
Yamato Fukuzawab,
Pan Gao a,
Julong Jianga,
Satoshi Maedaac,
Koji Kubota*ab and
Hajime Ito*ab
a,
Julong Jianga,
Satoshi Maedaac,
Koji Kubota*ab and
Hajime Ito*ab
aInstitute for Chemical Reaction Design and Discovery (WPI-ICReDD), Hokkaido University, Sapporo, Hokkaido, Japan. E-mail: hajito@eng.hokudai.ac.jp; kbt@eng.hokudai.ac.jp
bDivision of Applied Chemistry, Faculty of Engineering, Hokkaido University, Sapporo, Hokkaido, Japan
cDepartment of Chemistry, Faculty of Science, Hokkaido University, Sapporo, Hokkaido, Japan
First published on 25th December 2024
Abstract
In this study, we disclosed that calcium-based heavy Grignard reagents, prepared in situ through a mechanochemical method, reacted with gem-difluorostyrenes in the absence of transition-metal catalysts to afford thermodynamically less favorable (E)-monofluorostilbenes with good to high stereoselectivity. To the best of our knowledge, this is the first example of nucleophilic substitution of a C(sp2)–F bond by an arylcalcium compound.
Organocalcium compounds have gained significant attention owing to their unique structures and reactivities.1–5 However, there has been limited exploration of synthetic applications involving calcium-based carbon nucleophiles.6 This is largely due to the absence of straightforward and convenient methods for generating these highly reactive nucleophiles from commercially available starting materials, which may hinder a comprehensive investigation of their reactivity profiles.1–5 As a result, basic reactions involving organocalcium nucleophiles are currently underdeveloped.
Building on this background, we recently developed a straightforward mechanochemical protocol that employs ball milling to generate calcium-based heavy Grignard reagents (R–CaX)6–13 from unactivated calcium metal.14–16 Conventionally, such direct synthesis requires the preparation of activated calcium metal, such as Rieke calcium.10–12 In contrast, our mechanochemical approach employs commercial calcium metal and does not require inert gas protection, simplifying the operating process for the generation of organocalcium nucleophiles.14,15 This simple protocol is expected to enable synthetic chemists to more easily explore novel reactivities of organocalcium nucleophiles. Indeed, we found that mechanochemically generated aryl calcium species react smoothly with unactivated alkyl halides,14,15 including alkyl fluorides, in the absence of transition metal catalysts (Scheme 1A).16,17
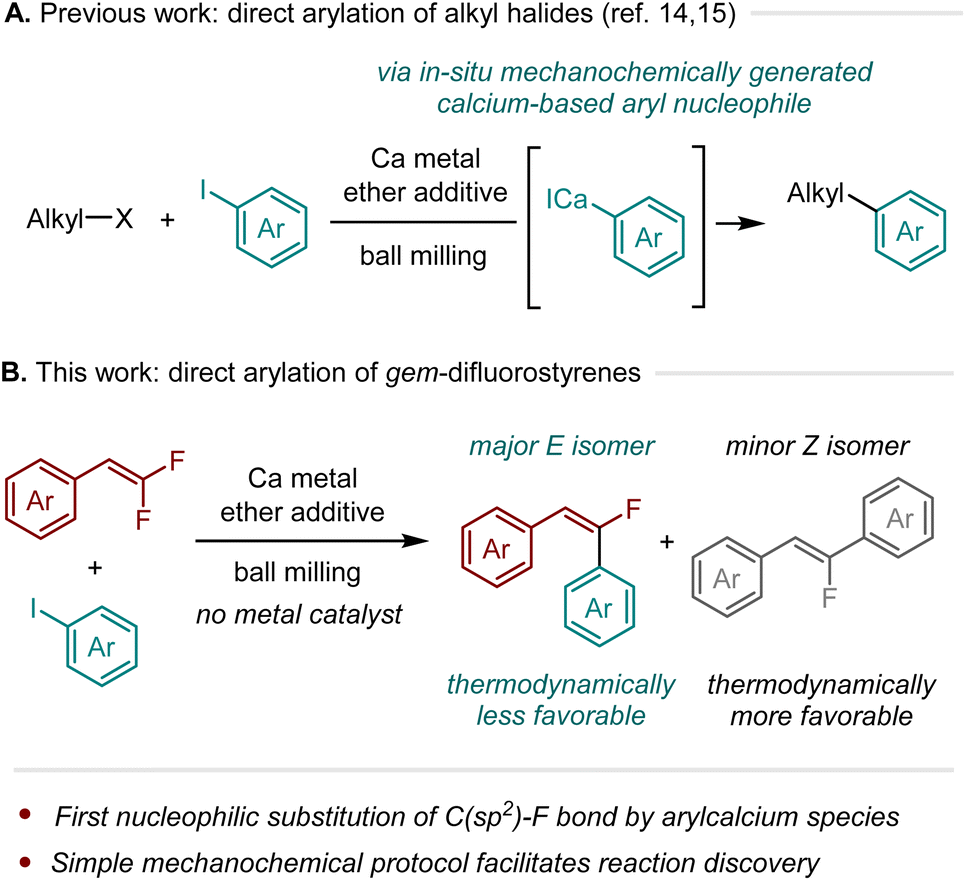 | ||
| Scheme 1 Discovery of new reactions involving organocalcium compounds by a mechanochemical method. | ||
Based on these achievements, particularly the C(sp3)–F bond arylation by organocalcium compounds,15 we investigated the feasibility of a direct organocalcium-mediated C(sp2)–F bond arylation reaction.18 Herein, we report that calcium-based heavy Grignard reagents, which were mechanochemically generated in situ, reacted with gem-difluorostyrenes in the absence of transition-metal catalysts to afford thermodynamically less favorable (E)-monofluorostilbenes with good to high stereoselectivity (Scheme 1B).19–28 To the best of our knowledge, this is the first example of nucleophilic substitution of a C(sp2)–F bond by an arylcalcium compound.29,30 Furthermore, this represents a rare instance where thermodynamically less favorable stereoisomers are formed from the defluorofunctionalization of gem-difluoroalkenes.19–28 The utility of this protocol was demonstrated by the stereoselective synthesis of a mono-fluorinated combretastatin A-4 analog.31 This study illustrates the effectiveness of a straightforward mechanochemical method for exploring a previously overlooked reactivity of calcium-based heavy Grignard reagents.
Initially, we optimized the conditions of the defluoroarylation of gem-difluorostyrene (1a) using an in situ mechanochemically generated aryl calcium nucleophile (Table 1). All reactions were conducted in a Retsch MM400 mixer mill (stainless-steel milling jar; 30 Hz; stainless-steel balls). Commercially available gem-difluorostyrene (1a), unactivated calcium metal (1.0 equiv.), and iodobenzene (2a, 3.0 eq.) were sequentially weighed in air and added to a 1.5 mL stainless-steel jar along with two 7 mm stainless-steel balls. Upon the addition of tetrahydropyran (THP, 4.0 equiv.), which is an essential additive for the generation of calcium-based aryl nucleophiles,14,15 the jar was sealed and then placed in a Retsch MM400 mixer mill. After ball milling at 30 Hz for 1 h, (E)-monofluorostilbene [(E)-3aa] was obtained in 60% yield with high stereoselectivity towards the thermodynamically less favorable E isomer (E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z = 81:19) (entry 1).19 It was found that the use of tetrahydrofuran (THF) instead of THP afforded (E)-3aa in 68% yield with a slightly higher E selectivity (E:Z = 83:17). Notably, the pure E isomer of 3aa (E:Z = >99:1) was isolated by silica gel column chromatography (entry 2). When the milling frequency was reduced to 20 Hz, the yield of 3aa decreased (entry 3, 58%; E:Z = 85:15). The use of 1.0 equivalent of calcium metal afforded a lower yield (54%) with a slightly increased E:Z selectivity (entry 4, E:Z = 86:14). The use of 2.0 equivalents of calcium metal significantly decreased both the yield and E:Z ratio (entry 5, 42% yield, E:Z = 74:26). We also examined the effect of the amount of THF additive (entries 6 and 7). When using 8.0 equivalents of THF, the E:Z ratio was similar to that of the reaction using 4.0 equivalents (entry 6, E:Z = 85:15), whereas 2.0 equivalents of THF resulted in a poor E:Z ratio (entry 7, E:Z ratio = 77:23).
:Z = 81:19) (entry 1).19 It was found that the use of tetrahydrofuran (THF) instead of THP afforded (E)-3aa in 68% yield with a slightly higher E selectivity (E:Z = 83:17). Notably, the pure E isomer of 3aa (E:Z = >99:1) was isolated by silica gel column chromatography (entry 2). When the milling frequency was reduced to 20 Hz, the yield of 3aa decreased (entry 3, 58%; E:Z = 85:15). The use of 1.0 equivalent of calcium metal afforded a lower yield (54%) with a slightly increased E:Z selectivity (entry 4, E:Z = 86:14). The use of 2.0 equivalents of calcium metal significantly decreased both the yield and E:Z ratio (entry 5, 42% yield, E:Z = 74:26). We also examined the effect of the amount of THF additive (entries 6 and 7). When using 8.0 equivalents of THF, the E:Z ratio was similar to that of the reaction using 4.0 equivalents (entry 6, E:Z = 85:15), whereas 2.0 equivalents of THF resulted in a poor E:Z ratio (entry 7, E:Z ratio = 77:23).
|
|||||
|---|---|---|---|---|---|
| Entry | Equiv. Of Ca | Hz | Additive | Yieldb of 3aa (%) | E:Zb |
| a Conditions: 1a (0.5 mmol), 2a (1.5 mmol), Ca, and additive in a stainless-steel ball-milling jar (1.5 mL) with two stainless-steel balls (diameter: 7 mm).b Yields were determined by 1H NMR spectroscopy using dibromomethane as an internal standard.c Isolated yield.d After repeated column chromatographic separations, the E:Z ratio was >99:1. |
|||||
| 1 | 1.5 | 30 | THP (4.0 equiv.) | 60 | 81:19 |
| 2 | 1.5 | 30 | THF (4.0 equiv.) | 68 (45c,d) | 83:17 |
| 3 | 1.5 | 20 | THF (4.0 equiv.) | 58 | 85:15 |
| 4 | 1.0 | 30 | THF (4.0 equiv.) | 54 | 86:14 |
| 5 | 2.0 | 30 | THF (4.0 equiv.) | 42 | 74:26 |
| 6 | 1.5 | 30 | THF (8.0 equiv.) | 54 | 85:15 |
| 7 | 1.5 | 30 | THF (2.0 equiv.) | 56 | 77:23 |

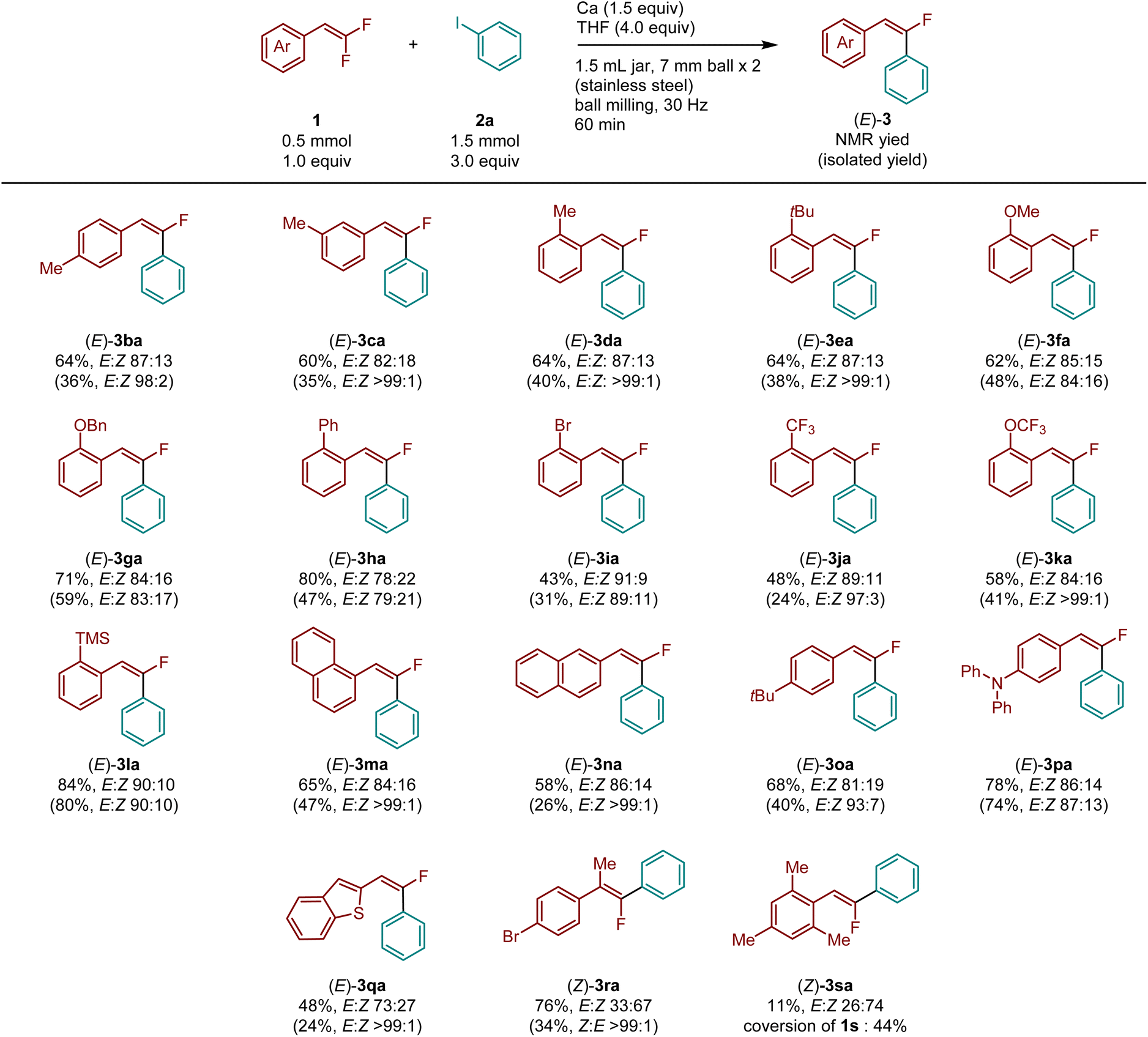
With the optimized conditions in hand, we explored the substrate scope for the defluoroarylation of various gem-difluorostyrenes (Table 2). gem-Difluorostyrenes containing a methyl group in the para, meta, and ortho positions on the phenyl ring (1b–1d) reacted smoothly to afford the corresponding products (3ba–3da) in moderate to good yields (60–64%) with high E selectivity. Almost pure E isomers were obtained via purification by either silica gel column chromatography or gel-permeation column chromatography (for details, see ESI†). Substrates bearing ortho-substituents such as tert-butyl- (1e), methoxy- (1f), benzyloxy- (1g), phenyl- (1h), bromo- (1i), trifluoromethyl- (1j), trifluoromethoxy- (1k), or trimethylsilyl- (1l) were fully tolerated under defluoroarylation reaction conditions, resulting in the corresponding monofluorostilbene derivatives in moderate to good yields (43–84%) with high E selectivity (3ea–3la). Naphthalene-containing substrates (1m and 1n) underwent defluoroarylation to form the desired products (3ma and 3na) in good yields (65 and 58%, respectively) with high E selectivity. The reactions of substrates bearing tert-butyl (1o) or diphenylamino (1p) groups at the para position afforded the desired products (3oa and 3pa) in good yields (68 and 78%, respectively) with high E selectivity. A benzo[b]thiophene-containing substrate (1q) was also converted to the desired product 3qa with good E selectivity (E:Z = 73:27). Interestingly, the reaction of 1-bromo-4-(1,1-difluoroprop-1-en-2-yl)benzene (1r) afforded (Z)-3ra as the major isomer (E:Z = 33:67) in high yield (76%). Unfortunately, 2-(2,2-difluorovinyl)-1,3,5-trimethylbenzene (1s), which contains two methyl groups at ortho position of the phenyl ring, afforded only an 11% yield of 3sa with good Z selectivity (E:Z = 26:74). It should be noted that the relatively low isolated yields compared to their NMR yields were primarily due to the difficulty of separating the E and Z arylated products. To isolate stereochemically pure E products, we performed multiple rounds of purification using silica gel column chromatography and gel-permeation chromatography. However, this process led to relatively low isolated yields of the desired E products.
| a Reaction conditions: gem-difluoroalkene 1 (0.5 mmol, 1.0 equiv.), iodobenzene 2a (1.5 mmol, 3.0 equiv.), Ca (0.75 mmol, 1.5 equiv.), THF (4.0 mmol) in a stainless-steel ball-milling jar (1.5 mL) with two stainless-steel balls (diameter: 7 mm), ball milling (30 Hz) for 1 h. 1H NMR yields are shown. Isolated yields are shown in parentheses. The E/Z ratio was determined by 19F NMR analysis. |
|---|
 |
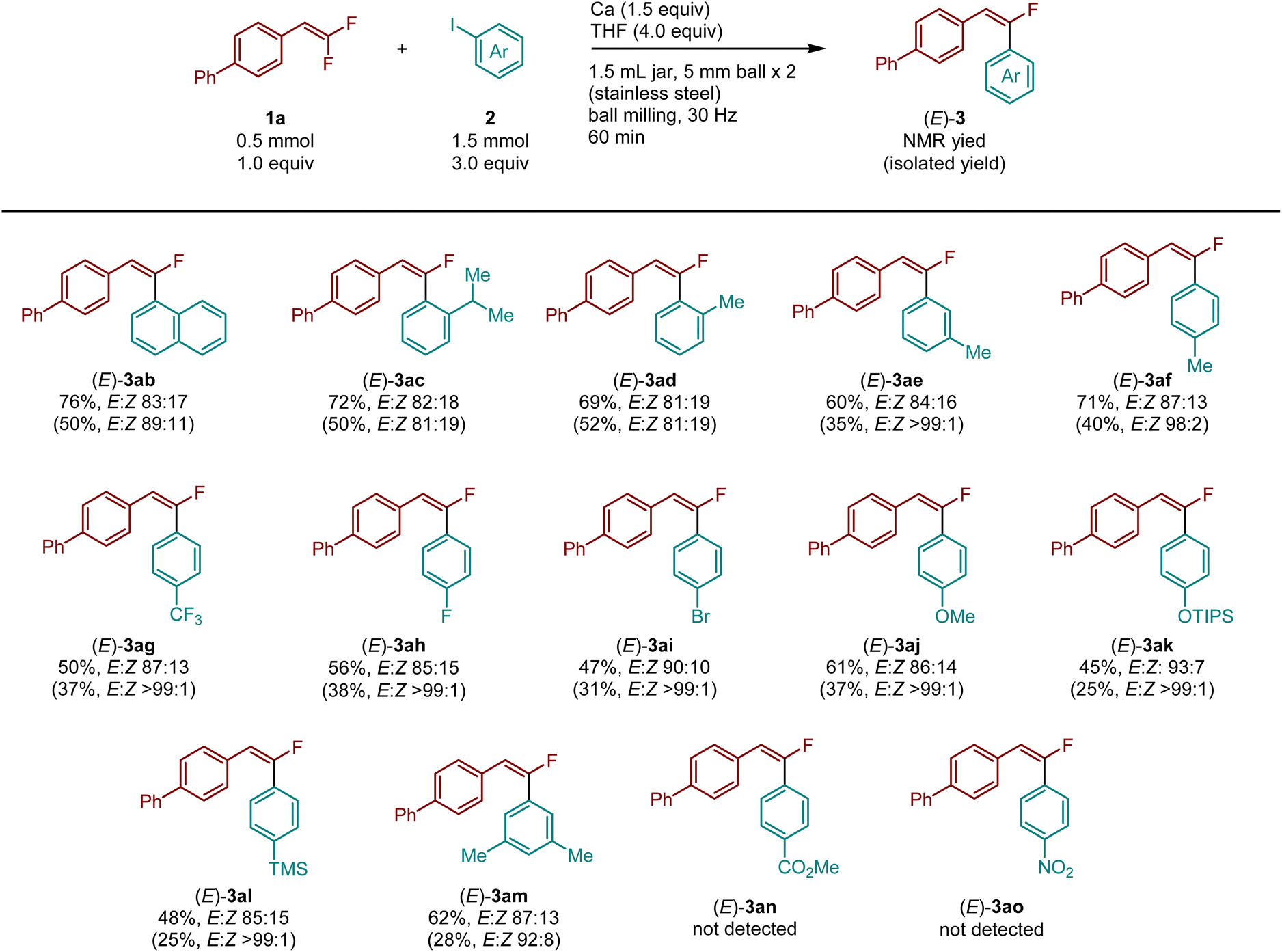
Subsequently, the reactions were investigated using various aryl iodides (Table 3). We found that 1-iodonaphthalene (2b), o-isopropyl- (2c)-, and o-methyl- (2d)-substituted iodobenzene derivatives furnished the desired products (3ab–3ad) in high yields (69–76%) with high E stereoselectivity. However, separation of the E/Z isomers by silica gel chromatography proved challenging in these cases. Methyl groups at different positions did not significantly affect the reactivity and selectivity (3ae: 60%, E:Z = 84:16, 3af: 71%, E:Z = 87:13). Aryl iodides bearing either electron-withdrawing or electron-donating substituents at the para position and two methyl groups at the meta position were also suitable for this reaction (3ag–3am). Unfortunately, ester and nitro groups were incompatible under these conditions.
| a Reaction conditions: gem-difluoroalkene 1 (0.5 mmol, 1.0 equiv.), iodobenzene 2a (1.5 mmol, 3.0 equiv.), Ca (0.75 mmol, 1.5 equiv.), THF (4.0 mmol) in a stainless-steel ball-milling jar (1.5 mL) with two stainless-steel balls (diameter: 7 mm), ball milling (30 Hz) for 1 h. 1H NMR yields are shown. Isolated yields are shown in parentheses. The E/Z ratio was determined by 19F NMR analysis. |
|---|
 |
To demonstrate the potential applicability of this method to the synthesis of bioactive compounds, we synthesized a mono-fluorinated combretastatin A-4 analogue [(E)-3tp] with anticancer activity (Scheme 2).31 The reaction between 5-(2,2-difluorovinyl)-1,2,3-trimethoxybenzene (1t) and 2-fluoro-4-iodo-1-methoxybenzene (2p) in the presence of calcium metal furnished the desired product 3tq with excellent E selectivity. Pure (E)-3tq was obtained by flash silica gel chromatography.
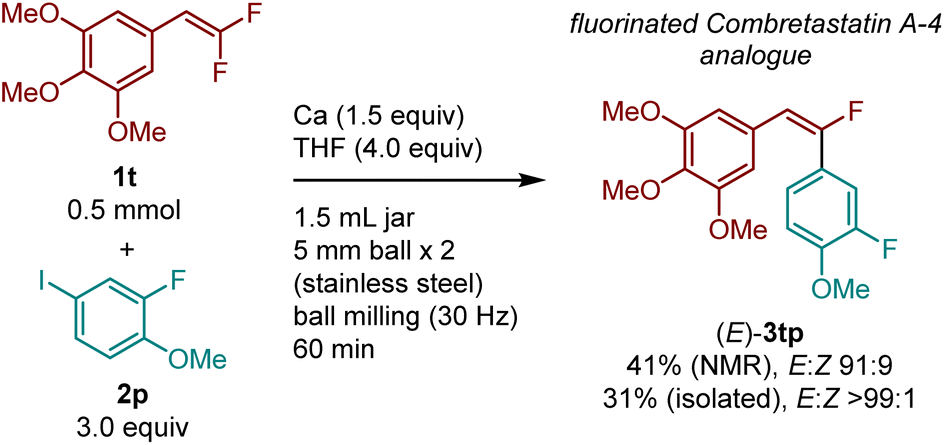 | ||
| Scheme 2 Synthesis of fluorinated Combretastatin A-4 analog (E)-3tp via E-selective defluoroarylation using a calcium-based heavy Grignard reagent. The details of the conditions are shown in the ESI.† | ||
Next, the reaction using Rieke calcium under solution-based conditions was investigated to obtain a mechanistic insight (Scheme 3). The Rieke method using lithium biphenylide was employed to generate the corresponding aryl calcium species from 2a, and the desired product (E)-3aa was obtained with high stereoselectivity (21%, E:Z = 89:11). This result suggests that the C(sp2)–F bond arylation under mechanochemical conditions most likely occurs through the selective formation of aryl calcium nucleophiles via direct insertion of calcium metal into a C(sp2)–I bond, followed by defluoroarylation of a gem-difluorostyrene. At this stage, the detailed mechanism of the nucleophilic substitution of the C(sp2)–F bond remains unclear.
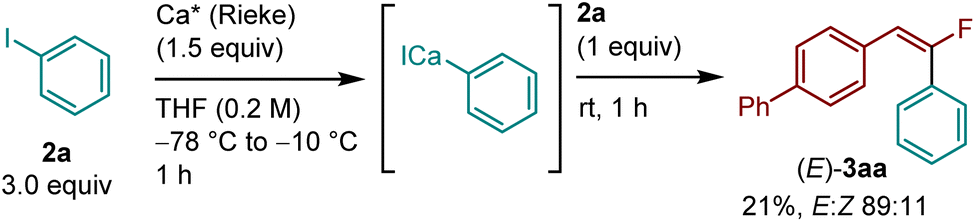 | ||
| Scheme 3 Reaction using Rieke calcium in solution. The details of the conditions are shown in the ESI.† | ||
Next, we tested the defluoroarylation reaction using conventional Grignard reagents (Scheme 4). Cao et al. previously reported the defluoroalkylation of gem-difluorostyrenes using bulky alkyl Grignard reagents in the absence of transition-metal catalysts to form thermodynamically more stable Z stereoisomers. In contrast, the use of sterically less hindered primary alkyl Grignard reagents afforded E/Z mixtures (Scheme 4A).26 Interestingly, it was found that the use of a THF solution of Ph–MgCl in the defluoarylation of 1a at room temperature led to thermodynamically less stable (E)-3aa in 73% yield with high stereoselectivity (E:Z = 84:16) (Scheme 4B). The reaction with Ph–MgBr also exhibited the E selectivity, however, the product yield was low (E:Z = 81:19, 18%). When these reactions were carried out at 80 °C, the yields were improved while the E selectivity was slightly decreased (Cl: E:Z = 79:21, 79%, Br: E:Z = 79:21, 70%). Interestingly, excellent Z selectivity was obtained when Ph–MgI was used at 80 °C (E:Z = 1:>99, 21%). Overall, these results show that the E stereoselectivity is not unique to calcium-based heavy Grignard reagents. In the case of traditional Grignard reagents, the stereoselectivity of this transformation is highly sensitive to the choice of organic groups (alkyl or aryl) and halides on magnesium.
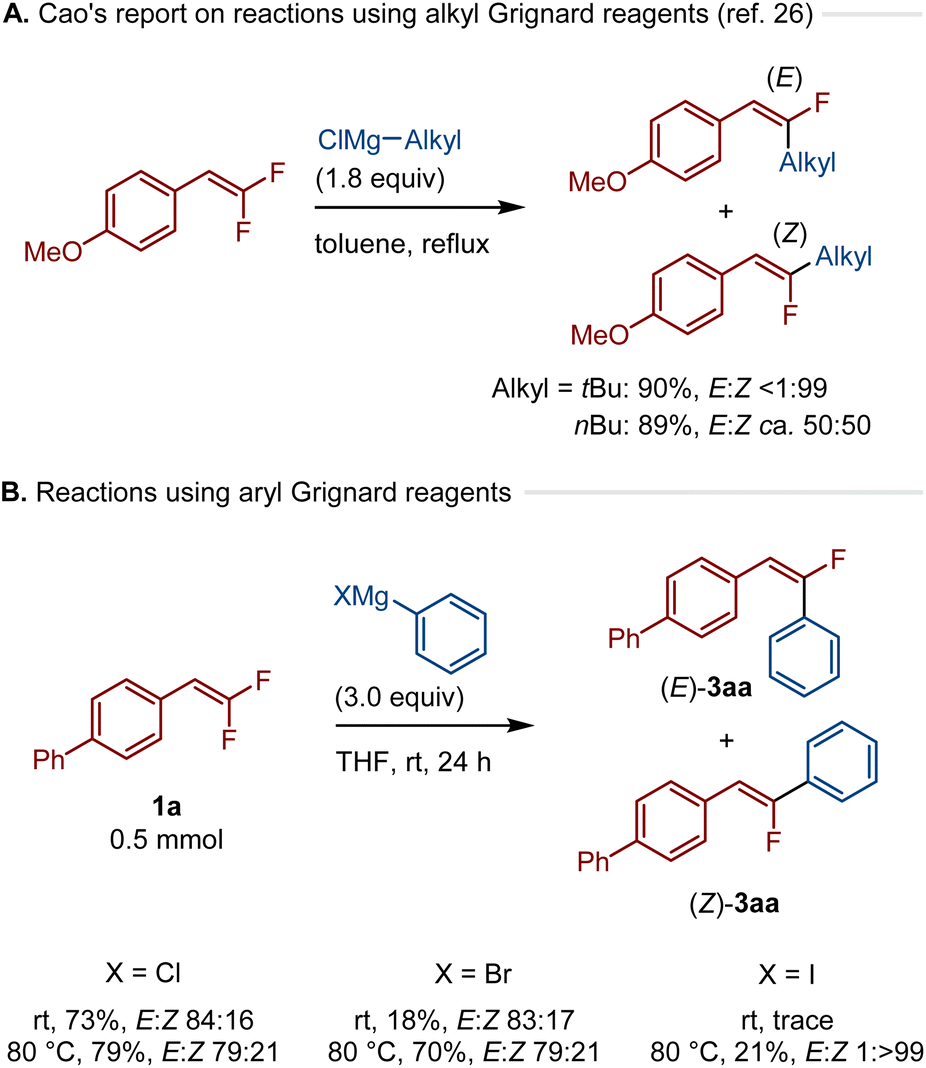 | ||
| Scheme 4 Reactions using Grignard reagents in solution. The details of the conditions are shown in the ESI.† | ||
In conclusion, we found a new reaction between calcium-based heavy Grignard reagents, which were generated in situ via a simple mechanochemical method, and gem-difluorostyrenes in the absence of transition-metal catalysts, resulting in moderate to high yields of the corresponding C(sp2)–F bond arylation products with good to high stereoselectivity. Notably, this is the first reported instance of an arylcalcium species engaging in nucleophilic substitution of a C(sp2)–F bond. These findings confirm that our operationally simple mechanochemical protocol using commercial calcium metal provides a valuable platform for discovering new reactions involving calcium-based carbon nucleophiles. Further studies to elucidate the mechanism of the C(sp2)–F bond substitution reaction as well as the observed stereoselectivity are ongoing in our laboratory.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Japan Society for the Promotion of Science (JSPS) via the KAKENHI grants 22H00318, 22K18333, 24H00453, 24H01832, and 24H01050; by the JST via the CREST grant JPMJCR19R1 and FOREST grant JPMJFR201I; and by the Institute for Chemical Reaction Design and Discovery (ICReDD) established by the World Premier International Research Initiative (WPI), MEXT, Japan. We thank Ms. Hina Shoji for her help with cross-checking the experiments.Notes and references
- For selected reviews on organocalcium chemistry, see (a) T. P. Hanusa, Chem. Rev., 1993, 93, 1023 CrossRef CAS; (b) M. Westerhausen, Angew. Chem., Int. Ed., 2001, 40, 2975 CrossRef CAS; (c) J. S. Alexander, Eur. J. Inorg. Chem., 2002, 2002, 2761 CrossRef.
- Selected examples and reviews of the reactions using organocalcium compounds as catalysts: (a) S. Harder, Chem. Rev., 2010, 110, 3852 CrossRef CAS PubMed; (b) S. Kobayashi and Y. Yamashita, Acc. Chem. Res., 2011, 44, 58 CrossRef CAS PubMed; (c) M. S. Hill, D. J. Liptrota and C. Weetman, Chem. Soc. Rev., 2016, 45, 972 RSC; (d) M. Magre, S. Marcin and M. Rueping, Chem. Rev., 2022, 122, 8261 CrossRef CAS PubMed; (e) L. Zhao, P. Deng, X. Gong, X. Kang and J. Cheng, ACS Catal., 2022, 12, 7877 Search PubMed.
- Selected examples of reactions using organocalcium compounds as carbon nucleophiles: (a) A. S. Wilson, M. S. Hill, M. F. Mahon, C. Dinoi and L. Maron, Science, 2017, 358, 1168 CrossRef CAS PubMed; (b) B. M. Wolf, C. Stuhl, C. Maichle-Mössmer and R. Anwander, J. Am. Chem. Soc., 2018, 140, 2373 CrossRef CAS PubMed.
- Selected examples of organocalcium-mediated polymerization are as follows: (a) S. Harder, F. Feil and A. Weeber, Organometallics, 2001, 20, 1044 CrossRef CAS; (b) S. Harder and F. Feil, Organometallics, 2002, 21, 2268 CrossRef CAS; (c) M. Westerhausen, S. Schneiderbauer, A. N. Kneifel, Y. Söltl, P. Mayer, H. Nöth, Z. Zhong, P. J. Dijkstra and J. Feijen, Eur. J. Inorg. Chem., 2003, 2003, 3432 CrossRef; (d) Y. Li, H. Deng, W. Brittain and M. S. Chisholm, Polym. Bull., 1999, 42, 635 CrossRef CAS; (e) M.-W. Hsiao and C.-C. Lin, Dalton Trans., 2013, 42, 2041 RSC.
- Selected reviews of calcium-based heavy Grignard reagents: (a) M. Westerhausen, M. Gärtner, R. Fischer, J. Langer, L. Yu and M. Reiher, Chem.–Eur. J., 2007, 13, 6292 CrossRef CAS PubMed; (b) M. Westerhausen, M. Gärtner, R. Fischer and J. Langer, Angew. Chem., Int. Ed., 2007, 46, 1950 CrossRef CAS PubMed; (c) M. Westerhausen, Coord. Chem. Rev., 2008, 252, 1516 CrossRef CAS; (d) M. Westerhausen, A. Koch, H. Görls and S. Krieck, Chem.–Eur. J., 2017, 23, 1456 CrossRef CAS PubMed; (e) A. Koch, Q. Dufrois, M. Wirgenings, H. Görls, S. Krieck, M. Etienne, G. Pohnert and M. Westerhausen, Chem.–Eur. J., 2018, 24, 16840 CrossRef CAS PubMed; (f) S. Harder and J. Langer, Nat. Rev. Chem, 2023, 7, 843 CrossRef PubMed; (g) P. Schüler, S. Sengupta, S. Krieck and M. Westerhausen, Chem.–Eur. J., 2023, 29, e202300833 CrossRef PubMed.
- (a) Organometallics in Synthesis: A Manual, ed. M. Schlosser, Wiley, Chester, 2nd edn, 2013 Search PubMed; (b) T. Banno, Y. Hayakawa and M. Umeno, J. Organomet. Chem., 2002, 653, 288 CrossRef CAS; (c) Handbook of Grignard Reagents, ed. G. S. Silverman and P. E. Rakita, Marcel Dekker, 1996 Search PubMed; (d) The Chemistry of Organozinc Compounds, ed. Z. Rappoport and I. Marek, John Wiley & Sons, Chichester, U.K., 2006 Search PubMed; (e) U. Wietelmann and J. Klett, Z. Anorg. Allg. Chem., 2018, 644, 194 CrossRef CAS PubMed.
- (a) M. Gärtner, H. Görls and M. Westerhausen, Organometallics, 2007, 26, 1077 CrossRef; (b) M. Gärtner, H. Görls and M. Westerhausen, J. Organomet. Chem., 2008, 693, 221 CrossRef; (c) M. Westerhausen, Angew. Chem., Int. Ed., 2009, 48, 5741 CrossRef PubMed; (d) J. Langer, M. Köhler, H. Görls and M. Westerhausen, Chem.–Eur. J., 2014, 20, 3154 CrossRef CAS PubMed.
- (a) R. Fischer, M. Gärtner, H. Görls, L. Yu, M. Reiher and M. Westerhausen, Angew. Chem., Int. Ed., 2007, 46, 1618 CrossRef CAS PubMed; (b) P. Schüler, S. Sengupta, A. Koch, H. Görls, S. Krieck and M. Westerhausen, Chem.–Eur. J., 2022, 28, e202201897 CrossRef PubMed; (c) P. Schüler, S. Sengupta, S. Krieck and M. Westerhausen, Chem.–Eur. J., 2023, 29, e20230083 CrossRef PubMed.
- A. Koch, M. Wirgenings, S. Krieck, H. Görls, G. Pohnert and M. Westerhausen, Organometallics, 2017, 36, 3981 CrossRef CAS.
- (a) T.-C. Wu, H. Xiong and R. D. Rieke, J. Org. Chem., 1990, 55, 5045 CrossRef CAS; (b) K. Mochida and H. Ogawa, J. Organomet. Chem., 1983, 243, 131 CrossRef CAS; (c) R. D. Rieke, T.-C. Wu and L. I. Rieke, Org. Synth., 1995, 72, 147 CrossRef CAS; (d) D. Bryce-Smith and A. C. Skinner, J. Chem. Soc., 1963, 577 RSC; (e) M. L. Hays and T. P. Hanusa, Tetrahedron Lett., 1995, 36, 2435 CrossRef CAS.
- J. Langer, M. Köhler, H. Görls and M. Westerhausen, J. Organomet. Chem., 2014, 751, 563 CrossRef CAS.
- (a) R. Fischer, M. Gärtner, H. Görls and M. Westerhausen, Organometallics, 2006, 25, 3496 CrossRef CAS; (b) M. Gärtner, H. Görls and M. Westerhausen, Synthesis, 2007, 725 Search PubMed; (c) M. Westerhausen, M. Gärtner, R. Fischer and J. Langer, Angew. Chem., Int. Ed., 2007, 46, 1950 CrossRef CAS; (d) N. Kawabata, A. Matsumura and S. Yamashita, Tetrahedron, 1973, 29, 1069 CrossRef CAS.
- (a) M. Köhler, J. Langer, H. Görls and M. Westerhausen, Organometallics, 2014, 33, 6381 Search PubMed; (b) M. Gärtner, H. Görls and M. Westerhausen, J. Organomet. Chem., 2008, 693, 221 CrossRef.
- P. Gao, J. Jiang, S. Maeda, K. Kubota and H. Ito, Angew. Chem., Int. Ed., 2022, 61, e202207118 CrossRef CAS PubMed.
- P. Gao, J. Jiang, Y. Fukuzawa, S. Maeda, K. Kubota and H. Ito, RSC Mechanochem., 2024, 1, 486 RSC.
- For selected reviews on reaction development using mechanochemistry, see: (a) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413 RSC; (b) G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7668 RSC; (c) J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13 CrossRef CAS PubMed; (d) J. G. Hernández and C. Bolm, J. Org. Chem., 2017, 82, 4007 CrossRef PubMed; (e) J. G. Hernández, Chem.–Eur. J., 2017, 23, 17157 CrossRef PubMed; (f) T. K. Achar, A. Bose and P. Mal, Beilstein J. Org. Chem., 2017, 13, 1907 CrossRef CAS PubMed; (g) J.-L. Do and T. Friščić, Synlett, 2017, 28, 2066 CrossRef CAS; (h) D. Tan and T. Friščić, Eur. J. Org Chem., 2018, 18, 18 CrossRef; (i) J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080 RSC; (j) J. Andersen and J. Mack, Green Chem., 2018, 20, 1435 RSC; (k) N. R. Rightmire and T. P. Hanusa, Dalton Trans., 2016, 45, 2352 RSC; (l) M. Leonardi, M. Villacampa and J. C. Menéndez, Chem. Sci., 2018, 9, 2042 RSC; (m) D. Tan, L. Loots and T. Friščić, Chem. Commun., 2016, 52, 7760 RSC; (n) D. Tan and F. García, Chem. Soc. Rev., 2019, 48, 2274 RSC; (o) C. Bolmm and J. G. Hernández, Angew. Chem., Int. Ed., 2019, 58, 3285 CrossRef PubMed; (p) T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018 CrossRef PubMed; (q) I. N. Egorov, S. Santra, D. S. Kopchuk, I. S. Kovalev, G. V. Zyryanov, A. Majee, B. C. Ranu, V. L. Rusinov and O. N. Chupakhin, Green Chem., 2020, 22, 302 RSC; (r) A. Pocheddu, E. Colacino, L. De Luca and F. Delogu, ACS Catal., 2020, 10, 8344 CrossRef; (s) K. Kubota and H. Ito, Trends Chem., 2020, 2, 1066 CrossRef CAS; (t) P. Ying, J. Yu and W. Su, Adv. Synth. Catal., 2021, 363, 1246 CrossRef CAS; (u) K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145 Search PubMed; (v) V. Martinez, T. Stolar, B. Karadeniz, I. Brekalo and K. Užarević, Nat. Rev. Chem, 2023, 7, 51 CrossRef CAS PubMed; (w) N. Fantozzi, J.-N. Volle, A. Porcheddu, D. Virieux, F. García and E. Colacino, Chem. Soc. Rev., 2023, 52, 6680 RSC; (x) K. Kubota, Bull. Chem. Soc. Jpn., 2023, 96, 913 CrossRef CAS.
- Selected examples of transition-metal-free nucleophilic substitution of alkyl fluorides using organometallic reagents: (a) K. Matsubara, T. Ishibashi and Y. Koga, Org. Lett., 2009, 11, 1765 CrossRef CAS PubMed; (b) J. Terao, S. A. Begum, Y. Shinohara, M. Tomita, Y. Naitoh and N. Kambe, Chem. Commun., 2007, 855 RSC; (c) L. Kane, B. C. Figula, K. Balaraman, J. A. Bertke and C. Wolf, Nat. Commun., 2024, 15, 1866 CrossRef PubMed.
- For selected reviews on C–F bond activation in organic synthesis, see: (a) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119 CrossRef CAS PubMed; (b) J.-D. Hamel and J.-F. Paquin, Chem. Commun., 2018, 54, 10224 Search PubMed; (c) T. Stahl, H. F. T. Klare and M. Oestreich, ACS Catal., 2013, 3, 1578 CrossRef CAS; (d) G. Coates, F. Rekhroukh and M. R. Crimmin, Synlett, 2019, 30, 2233 Search PubMed; (e) T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931 CrossRef CAS PubMed; (f) Q. Shen, Y.-G. Huang, C. Liu, J.-C. Xiao, Q.-Y. Chen and Y. Guo, J. Fluorine Chem., 2015, 179, 14 CrossRef CAS.
- DFT calculations [B3LYP-D3/6-311G(d,p)//B3LYP-D3/6-311G(d,p)] revealed that (Z)-3a is thermodynamically more stable than (E)-3a by 15.0 kJ mol−1.
- Reviews on the synthesis and applications of monofluoroalkenes: (a) S. Oishi, H. Kamitani, Y. Kodera, K. Watanabe, K. Kobayashi, T. Narumi, K. Tomita, H. Ohno, T. Naito, E. Kodama, M. Matsuoka and N. Fuji, Org. Biomol. Chem., 2009, 7, 2872 RSC; (b) H. Yanai and T. Taguchi, Eur. J. Org Chem., 2011, 2011, 5939 CrossRef CAS; (c) G. Landelle, M. Bergeron, M.-O. Turcotte-Savard and J.-F. Paquin, Chem. Soc. Rev., 2011, 40, 2867 RSC; (d) M. Drouin, J.-D. Hamel and J.-F. Paquin, Synthesis, 2018, 50, 881 CrossRef CAS; (e) A. Niida and N. Fujii, Org. Lett., 2006, 8, 613 CrossRef CAS PubMed.
- Transition-metal-catalyzed direct defluoroarylation of gem-difluorostyrenes to form (Z)-monofluorostilbenes: (a) R. T. Richard and F. D. Toste, Angew. Chem., Int. Ed., 2016, 55, 11629 CrossRef PubMed; (b) Y. Xiong, T. Huang, X. Ji, J. Wu and S. Cao, Org. Biomol. Chem., 2015, 13, 7389 RSC; (c) P. Tian, C. Feng and T.-P. Loh, Nat. Commun., 2015, 6, 7472 CrossRef PubMed.
- Stepwise synthesis of (Z)-monofluorostilbenes via borylation/palladium-catalyzed cross-coupling methodology: (a) H. Sakaguchi, Y. Uetake, M. Ohashi, T. Niwa, S. Ogoshi and T. Hosoya, J. Am. Chem. Soc., 2017, 139, 12855 CrossRef CAS PubMed; (b) J. Zhang, W. Dai, Q. Liu and S. Cao, Org. Lett., 2017, 19, 3283 CrossRef CAS PubMed.
- Stepwise synthesis of (Z)-monofluorostilbenes via defluoroarylation/palladium-catalyzed cross-coupling: S. Porey, Y. Bairagi, S. Guin, X. Zhang and D. Maiti, ACS Catal., 2023, 13, 14000 CrossRef CAS.
- Silyl-group-directed stereoselective synthesis of tri- and tetrasubstituted fluoroalkenes: G. Landelle, P. A. Champagne, X. Barbeau and J.-F. Paquin, Org. Lett., 2009, 11, 681 CrossRef CAS PubMed.
- Wittig reaction with α-fluorobenzylphosphonium salt to give (E)-monofluorostilbenes: D. Mandal, R. Gupta and R. D. Young, J. Am. Chem. Soc., 2018, 140, 10682 CrossRef CAS PubMed.
- Direct defluoroalkylation of gem-difluorostyrenes using Grignard reagents to form Z-monofluoroalkenes: W. Dai, H. Shi, X. Zhao and S. Cao, Org. Lett., 2016, 18, 4284 CrossRef CAS PubMed.
- Palladium-catalyzed chelation-assisted arylation of ester-substituted gem-difluoroalkenes: (a) Y. Wang, X. Qi, Q. Ma, P. Liu and G. C. Tsui, ACS Catal., 2021, 11, 4799 CrossRef CAS; (b) Y. Wang and G. C. Tsui, Org. Lett., 2024, 26, 5822 CrossRef CAS PubMed.
- Selected examples of stereoselective functionalization of ester-substituted gem-difluoroalkenes: (a) M. Li, Y. Wang and G. C. Tsui, Org. Lett., 2021, 23, 8072 CrossRef CAS PubMed; (b) Q. Ma, Y. Wang and G. C. Tsui, Angew. Chem., Int. Ed., 2020, 59, 11293 CrossRef CAS PubMed; (c) Z. Luo, Y. Zong and G. C. Tsui, Org. Lett., 2023, 25, 4406 CrossRef CAS PubMed; (d) Y. Wang, Y. Tang, Y. Zong and G. C. Tsui, Org. Lett., 2022, 24, 4087 CrossRef CAS PubMed.
- An example of intramolecular aromatic C(sp2)–F bond alkenylation using alknenyl calcium species: H. Li, X.-Y. Wang, B. Wei, L. Xu, W.-X. Zhang, J. Pei and Z. Xi, Nat. Commun., 2014, 5, 4508 CrossRef CAS PubMed.
- An example of direct arylation of an aromatic C(sp2)–Br bond using aryl calcium nucleophiles: K. G. Pearce, C. Dinoi, M. S. Hill, M. F. Mahon, L. Maron, R. S. Schwamm and A. S. S. Wilson, Angew. Chem., Int. Ed., 2022, 61, e202200305 CrossRef CAS PubMed.
- D. Alloatti, G. Giannini, W. Cabri, I. Lustrati, M. Marzi, A. Ciacci, G. Gallo, M. O. Tinti, M. Marcellini, T. Riccioni, M. B. Guglielmi, P. Carminati and C. Pisano, J. Med. Chem., 2008, 51, 2708 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mr00135d |
| This journal is © The Royal Society of Chemistry 2025 |