
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemoselectivity switch by mechanochemistry in the base-catalysed dione-acylation†
Sally Nijema,
Alexander Kaushanskya,
Svetlana Pucovskib,
Elisa Ivrya,
Evelina Colacino c,
Ivan Halaszb and
Charles E. Diesendruck*a
c,
Ivan Halaszb and
Charles E. Diesendruck*a
aSchulich Faculty of Chemistry, Resnick Sustainability Center for Catalysis, Technion – Israel Institute of Technology, Haifa, 32000, Israel. E-mail: charles@technion.ac.il
bRuđer Bošković Institute, Bijenička c. 54, 10000 Zagreb, Croatia
cICGM, Univ Montpellier, CNRS, ENSCM, 34293 Montpellier, France
First published on 11th February 2025
Abstract
The mechanochemistry of small molecules is an exponentially growing area of investigation relevant to developing sustainable synthesis to reduce waste and energy consumption, with great potential in large-scale chemical manufacturing. Occasionally, mechanochemical processes exhibit different reactivities, resulting in varying product selectivity compared to solution processes. In this study, we investigate the catalytic mechanism of a solvent-free one-pot acylation reaction of dimedone and 3-phenylpropanoic acid using a solvent-free ball-mill approach. The mechanochemical procedure afforded complete chemoselectivity towards a single acylation product after short milling, contrary to solution studies that previously reported product mixtures. Selectivity towards a single acylation product is controlled by the choice of the catalytic base. Under these mechanical process conditions, 4-dimethylaminopyridine (DMAP) is the only base that promotes the formation of the more desirable C-acylation product, whereas other bases exclusively afford the O-acylation product. Based on experimental findings, supported by theoretical modeling, we provide a mechanistic understanding of the base-dependent chemoselectivity, which leads to an enolate esterification that, in the case of DMAP, is converted to the thermodynamic product via Fries rearrangement. Finally, we explore the reaction scope with additional dicarbonyl compounds and carboxylic acids.
Introduction
Carbonyls are among the most useful functional groups for creating new C–C and C–O bonds. Due to their high polarization, different sites with partial charges are available to undergo reactions with carbon nucleophiles and electrophiles. An increasing number of reactive sites naturally raises the question of selectivity, which is further heightened in the case of diones. For example, the acylation of 1,3-diones using activated esters has been extensively investigated in solution,1,2 typically providing mixtures of dicarbonyl compounds obtained through C- and O-acylation pathways to form a β-diketone or an enol ester. Occasional selectivity towards one product or the other has been observed, but the reaction is quite sensitive to the nature of the catalytic base and the reaction conditions (e.g., temperature and solvent). The underlying reasons for the irregular selectivity observed and how to achieve better selectivity remain unclear. Importantly, these acylation pathways are vital preliminary steps in the synthesis of several natural products and drug compounds.3–5 Moreover, this process is not atom-economical, and the use of heat and solvent further increases its negative environmental impact.6Recently, the use of solvent-free mechanochemical approaches has garnered considerable attention from both academic and industrial research,7–9 as it can potentially reduce the environmental cost of chemical processes,6,10,11 while enabling new selective chemical transformations.12–14 In mechanochemistry, a solid mixture of reactants is ground together under ambient conditions using diverse milling devices, each providing different types of mechanical stress that can be adjusted/modulated by changing technical and process parameters. If adequately designed, the output of the reaction (product) requires no further purification. Given the success of mechanochemistry in making chemical processes more efficient, green, and selective,6,13 and the importance of the acylation of dicarbonyl compounds in the preparation of drug-like molecules,15–17 we report the solid-state acylation of dimedone with 3-phenylpropanoic acid as a model in the presence of various catalytic bases, using a time-efficient solvent-free ball-milling procedure. By using solvent-free conditions, we obtained distinct selectivities depending on the base used in the milling process (selectivity switch),14 allowing us to study the mechanistic pathways leading to the two dominant products in the solid state and support such studies with in silico modeling. The formation of the C-acylated product proceeds through a Fries rearrangement18,19 via a base-mediated acyl transfer from the O-acylated product. The scope of the target reaction under mechanochemical conditions is also explored.
Results and discussion
The acylation reactions of dimedone with 3-phenylpropanoic acid were carried out in the presence of different bases in catalytic amounts (10 mol%) and N,N′-dicyclohexylcarbodiimide (DCC) as the coupling agent (Fig. 1). The reaction was investigated using a home-made ball-mill vortex mixer.20 The solid mixture and 25 stainless-steel 3/16-inch diameter balls were placed in a 50 mL stainless steel cylindrical jar, kept under air, and milled at 1470 rpm (24.5 Hz) for 1 h.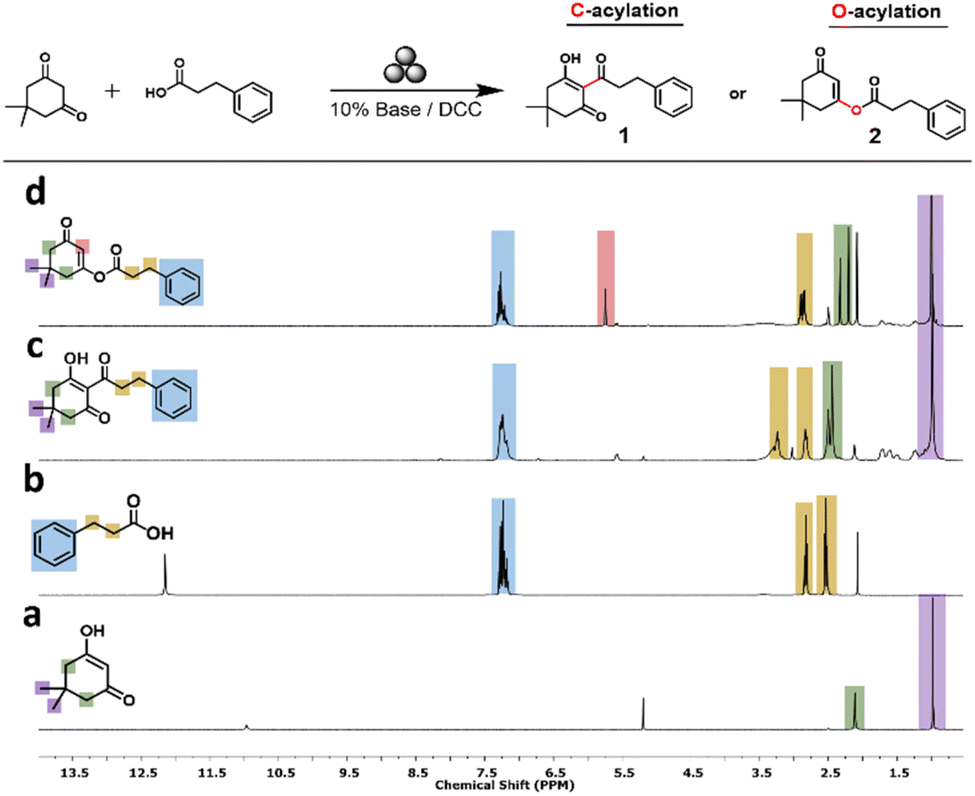 | ||
| Fig. 1 1H-NMR (DMSO-d6, 400 MHz) spectra of the reactants (a) dimedone and (b) 3-phenylpropanoic acid compared to the main products (c) C-acylation product (1) and (d) O-acylation product (2). | ||
The base-catalysed acylation was evaluated using a variety of organic and inorganic bases while maintaining other parameters (e.g., vibration frequency and atmosphere) constant. Product formation was monitored via 1H and 13C NMR in DMSO-d6. Several distinguishable peaks enabled us to differentiate between the two dominant products: the aryl ketone product 1 resulting from the C-acylation and the O-acylated product enol ester 2 (Fig. 1). As seen in Fig. 1, both 1 and 2 present similar aromatic peaks at 7.2 ppm (blue) and the two methyl group signals at 0.9 ppm (violet). These peaks are also present in the reactant phenyl propionic acid and dimedone, respectively. However, the methylene groups present in dimedone (green) and the aliphatic chain of the phenyl propionate (yellow) are shifted after the reaction, making them distinct for each product in both chemical shift and splitting. Moreover, a characteristic singlet peak resulting from a vinylic proton in the O-acylation product 2 appears at 5.7 ppm (red). The 13C NMR further supports these significant differences in chemical shifts (see the ESI†).
Products 1 and 2 were obtained with high conversions after less than 1 h of milling and with complete selectivity depending on the added base (Table 1). Full conversion (reactant peaks not visible in NMR) was obtained with 1 equivalent of 2,2′-bipyridine (2,2′-bipy) and ethylamine (EtNH2) (entries 9, 10). Importantly, good conversion was achieved even in the absence of a base (entry 12). These conversions are higher than previously reported solution experiments, which yielded 62–88% after 14–16 h,1,2 highlighting the efficiency of this mechanochemical, solvent-free procedure. Interestingly, contrary to the reactions in solution, clear chemoselectivity is observed under mechanochemical conditions (Table 1).1,2
| Entry | Base | pKab | Conversionc (%) | O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C :C |
|---|---|---|---|---|
| a Acylation conditions: base (0.1 equiv.) and DCC (1 equiv.) were added and milled together with the reactants using a ball-mill vortex for 1 h at r.t under air.b pKa values of the corresponding conjugated acids in aqueous solution.21–24c Conversion was determined via 1H NMR integrals for the O-acylated product (singlet at 5.75 ppm) or the C-acylated product (triplet at 3.2 ppm) relative to the residual dimedone (singlet at 5.19 ppm).d 1 equiv. of the base was used. | ||||
| 1 | K2CO3 | 6.3 | 92.1 | 1:0 |
| 2 | NaOH | 13.9 | 84.7 | 1:0 |
| 3 | DBU | 11.5 | 89.8 | 1:0 |
| 4 | Et3N | 10.7 | 86.9 | 1:0 |
| 5 | tBuOK | 20.0 | 70.6 | 1:0 |
| 6 | Aniline | 4.6 | 78.7 | 1:0 |
| 7 | Pyridine | 5.3 | 91.7 | 1:0 |
| 8 | DMA | 5.1 | 90.9 | 1:0 |
| 9 | 2,2′-bipy | 4.3 | 99.0d | 1:0 |
| 10 | EtNH2 | 10.7 | 99.0d | 1:0 |
| 11 | DMAP | 9.6 | 88.5 | 0:1 |
| 12 | None | — | 81.9 | 1:0 |
The C-acylation aryl ketone product 1 is obtained with complete selectivity only when utilizing N,N-dimethylamino pyridine (DMAP) as a base (Table 1, entry 1), while the O-acylation enol ester product 2 is formed with complete selectivity when using any other base (or no base), regardless of its pKa.
For comparison, in previous solution studies,1 DMAP was also selective towards the C-selective product, although other bases, such as Et3N, N-methyl-2-pyrrolidone (NMP), and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), were also able to produce such a compound but resulted in mixtures of C- and O-acylated products. However, the one-pot DMAP-catalyzed acylation mechanistic studies have not reached a consensus regarding the role of the catalytic base. Xu et al. demonstrated the nucleophilic attack of DMAP at the carbonyl group, resulting in an acetylpyridinium-acetate ion pair, which, after the acyl transfer step required an auxiliary base (NH3) to regenerate DMAP from the subsequent DMAP–acid complex.25 Kohout et al. showed that in the case of cyclic 1,3-diones, the base plays a crucial role only in forming a dimedone enolate, which then attacks the activated acid in a direct C-acylation pathway.1 However, neither of the suggested mechanisms explains why such chemoselectivity is not observed with other bases, or why some bases produce mixtures of products.
Given that our results provided full chemoselectivity, we recognize the opportunity to use this experimental protocol to further investigate the role of the base in defining the reaction chemoselectivity by combining experimental results and in silico modelling. Clearly, the electronic structure of DMAP is suitable for the C-acylation selectivity, as other bases containing similar amine functionalities and the same steric conditions (pyridine and N,N-dimethylaniline, DMA), as well as bases with a variety of electronic properties, produced product 2 exclusively. Scheme 1 presents plausible base-catalyzed pathways for the C- and O-acylation processes of dimedone, starting from deprotonated dimedone and a DCC-activated ester of the phenyl propionic acid. The path marked in red (path 1) is based on the nucleophilic attack of a carbanion species on the activated ester, leading to the direct formation of the C-acylation product 1, as proposed by Kohout et al.1 Alternatively, the second path marked in blue starts with a nucleophilic attack by the enolate anion to form the O-acylation product 2, which may then undergo DMAP-promoted acyl transfer (Fries rearrangement)18,19 to yield the C-acylated product 1.
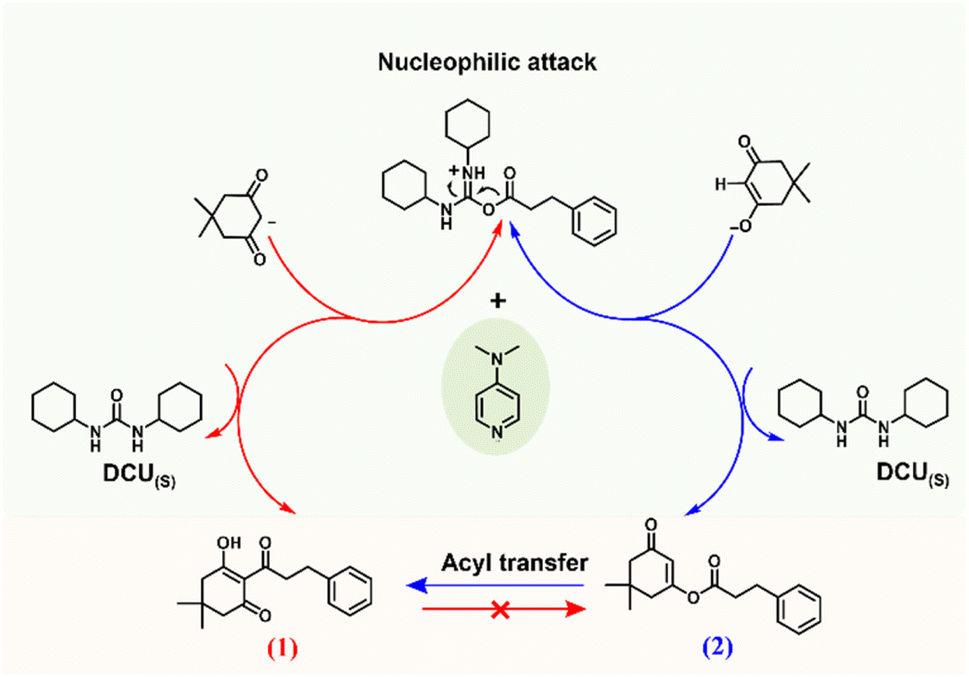 | ||
| Scheme 1 Two possible DMAP-catalysed acylation pathways: nucleophilic attack of a carbanion on the activated ester leads to direct formation of the C-acylation product 1 (red), while nucleophilic attack through the enolate forms the O-acylation product 2, which can be followed by acyl group transfer leading to the indirect formation of product 1 (blue). | ||
DMAP presents a unique combination of basicity and nucleophilicity centered on endocyclic nitrogen, imparted by the lone pair donation from the exo-nitrogen into the pyridine ring,26–28 and has found numerous applications in group-transfer reactions, especially acylation reactions.25,29–32 To investigate the role of DMAP within these possible mechanochemical pathways, the acylation was examined as a two-step process: deprotonation of dimedone and the subsequent nucleophilic attack. We conducted two solid-state reactions in which these steps were performed separately (Table 2). In the first experiment, dimedone and 3-phenylpropionic acid were milled with DCC in the absence of a base, followed by milling with DMAP (entry 1). In the second, the reactants were milled with DMAP, without DCC, followed by the addition of DCC and further milling (entry 2).
| Entry | Step | Additivea | Conversionc (%) | O:C |
|---|---|---|---|---|
| a Additives amount: DCC (1.0 equiv.), and base (0.1 equiv.).b 0.05 equiv. of each base.c Conversion was determined via 1H NMR integrals of the O-acylated product (singlet at 5.75 ppm) and C-acylated product (triplet at 3.2 ppm) relative to residual dimedone (singlet at 5.19 ppm). | ||||
| 1 | 1.1 | DCC | 81.9 | 1:0 |
| 1.2 | DMAP | 86.7 | 0:1 | |
| 2 | 2.1 | DMAP | 0.0 | — |
| 2.2 | DCC | 69.6 | 0:1 | |
| 3 | 3.1 | DMAP + NaOHb + DCC | 72.1 | 0.44:0.56 |
| 4 | 4.1 | NaOH + DCC | 81.3 | 1:0 |
| 4.2 | DMAP | 79.9 | 0.12:0.88 | |
| 5 | 5.1 | DMAP + DCC | 59.5 | 0:1 |
| 5.2 | NaOH | 62.0 | 0:1 | |
As described above, the first step does not require a base (entry 1, step 1.1) when the reaction is carried out in a solvent-free grinding protocol, producing the enol ester 2. Upon the addition of DMAP and further milling (entry 1, step 1.2), all of 2 was converted into 1, supporting DMAP's role as an acyl-transfer agent in a Fries rearrangement reaction rather than as a base catalyst for the C-acylation reaction. Conversely, in the absence of DCC (entry 2, step 2.1), DMAP simply neutralizes the propanoic acid, and no acylation occurs. However, the addition of DCC to the pre-milled mixture leads to the direct formation of product 1 (entry 2, step 2.2). DCC is clearly necessary for the activation of the acid as an electrophile; without it, only an acid–base reaction occurs.
Further experiments were conducted to probe the chemoselectivity, using NaOH as an O-selective base (the strongest base and nucleophile in aqueous conditions) in competition with DMAP. As shown in Table 2, when NaOH and DMAP are used together at equal loadings in the presence of DCC (entry 3), a mixture of 1 and 2 was obtained after 1 h, with a slight preference for product 1. Next, in the presence of DCC, we performed an experiment using NaOH as a base but added DMAP after the initial hour of milling with NaOH and continued milling for an additional hour (entry 4). As expected, after the first step, the O-acylated product 2 was obtained with full selectivity. However, after milling for another hour with DMAP, most of compound 2 was converted into the C-acylated product 1, supporting DMAP's capability to induce the Fries rearrangement.
The reaction was repeated inverting the order of the bases (entry 5, step 5.1). After milling with DMAP for 1 h, the C-acylation product 1 was obtained with complete selectivity. The addition of NaOH and milling for an additional hour (entry 5, step 5.2) slightly increased the conversion but did not change the reaction selectivity, confirming the irreversibility of the acyl transfer reaction.
Similarly, the 2 to 1 conversion could not be achieved with NaOH, with or without surplus DCC. While these experiments support that 2 can be converted into 1 in the presence of DMAP, they do not rule out the direct formation of 1 when DMAP is used (red path).1 Therefore, to gain further insights into the reaction mechanism, computational studies were conducted to estimate the energetic aspects of the different pathways.
The transition state Gibbs free energy (ΔG‡) and enthalpy change (ΔH‡) for the two possible direct acylation pathways towards products 1a and 2a (using acetic acid as a model reactant to reduce the number of possible conformers) were calculated at the ωB97M-V/def2-TZVP(cpcm[hexane])//BP86-D3bj/def2 SVP(cpcm[hexane]) level of theory. All key intermediates and transition states were thoroughly investigated by CREST33 to find the lowest energy conformers. Many possible pathways were examined (see the ESI†), but the lowest energy pathways towards each product are shown in Scheme 2. Starting from 3a (representing the individually optimized reactants), in which dimedone is in its enol form (lower in energy according to calculations), the O-acylated product 2a can be obtained through a lower activation barrier (19.0 kcal mol−1) than the C-acylated product 1a (24.4 kcal mol−1) in the absence of any base, although it is less thermodynamically favourable (−14.2 kcal mol−1 vs. −23.4 kcal mol−1). Indeed, C-acylated product 1a is the thermodynamic product, while O-acylated product 2a is kinetically preferred. The energy gap between the two suggested transition states is likely due to the higher ring strain of the cyclooctene-like transition state obtained for the C-acylation reaction.
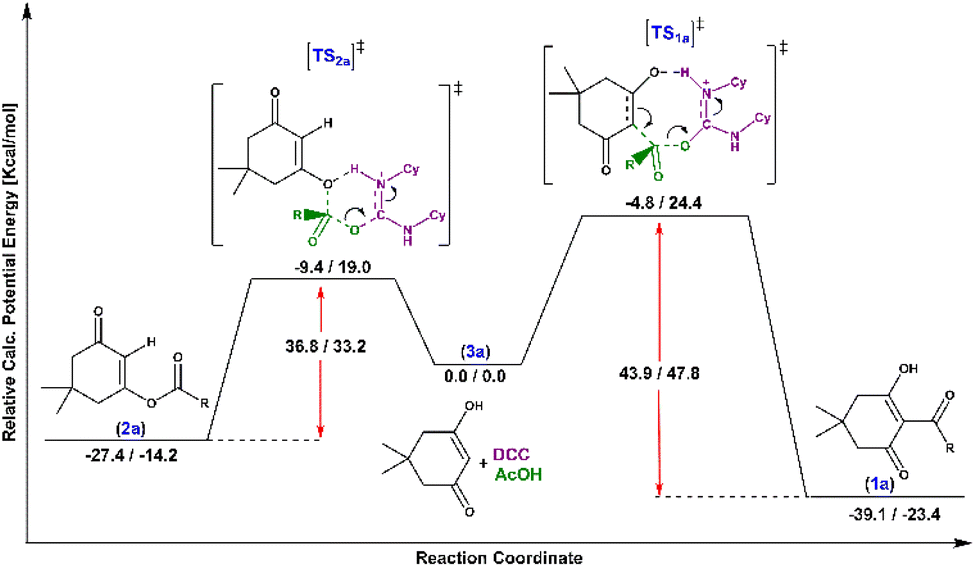 | ||
| Scheme 2 Enthalpy/free energy profiles for the acylation of dimedone with acetic acid towards direct C-acylation reaction to product 1a and O-acylation reaction to product 2a. | ||
Additional calculations were performed to study the Fries rearrangement from the kinetic product 2a into the thermodynamic product 1a using DMAP and pyridine (Scheme 3). First, the O-acylation product 2a undergoes a nucleophilic attack by the base on the ester carbonyl. The activation barrier for DMAP is 24.6 kcal mol−1, which makes this reaction plausible even at room temperature, whereas the same step using pyridine has a higher transition state of 29.8 kcal mol−1. These results indicate that the reaction catalysed by pyridine is three orders of magnitude slower at room temperature, although it might occur at higher temperatures, explaining the loss of selectivity in reactions conducted while heating in solution. According to the transition state geometric models (see the ESI†), the different behaviour of the two bases may be explained by the electron donation from the exocyclic nitrogen atom, which makes DMAP more nucleophilic,34 and also allows it to bend towards the dimedone ring through the deplanarization of the nitrogen from the aromatic plane.
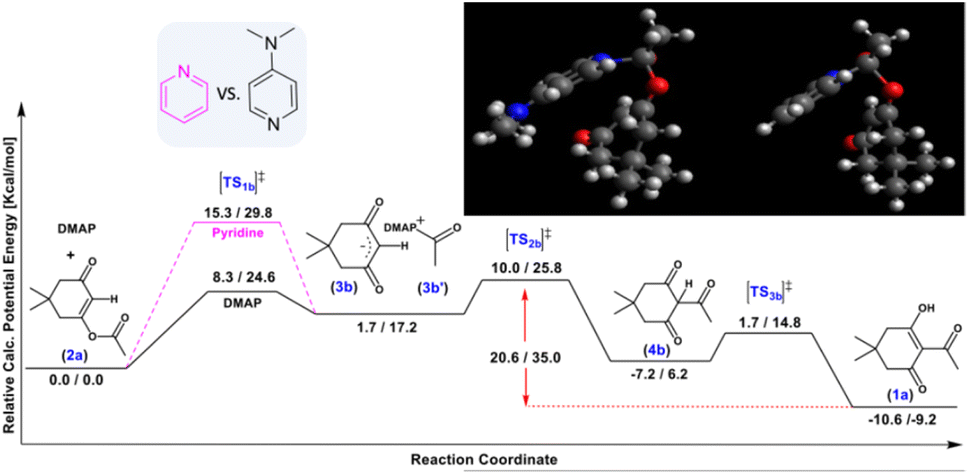 | ||
| Scheme 3 Enthalpy/free energy profiles for the DMAP/pyridine-catalysed conversion of O-acylated product 2 into C-acylated product 1 through three different transition states [TS1-3b]‡. Top right inset: [TS1b]‡ for DMAP (left) and pyridine (right). | ||
This spatial phenomenon is not observed in the case of pyridine (see the ESI†), as a change in nitrogen hybridization is required. The subsequent carbanion attack occurs through transition state [TS2b]‡, which is 25.8 kcal mol−1 relative to the starting reactant 2a, but remains reachable at room temperature. Finally, exothermic reactivation of DMAP and tautomerization lead to the production of 1a. The computational studies show that the overall reaction is exergonic for the direct production of either product (ΔG(1a) = −23.4 kcal mol−1 for the thermodynamic process and ΔG(2a) = −14.2 kcal mol−1 for the kinetic reaction). Moreover, the conversion of the kinetic product 2a to the stable product 1a is also exergonic (ΔG(1a) = −9.2 kcal mol−1) through multiple stable intermediates, whereas the reverse reaction is virtually impossible at r.t (35.0 kcal mol−1), as observed experimentally (Table 2, entry 5).
To extend the scope and relevance of this mechanochemical process, additional mechanically induced acylation reactions were tested under the same reaction conditions using a broad set of carbonyl compounds and carboxylic acids. Each pair of reactants was tested using DMAP or NaOH as catalysts. The ratio between the final C- and O-acylated products was determined using 1H-NMR spectroscopy and GC-MS (see ESI†). Interestingly, full selectivity towards the O-acylated products was again achieved when using NaOH as the catalyst (Table 3). However, when DMAP was used, the Fries rearrangement was less efficient, with only a few cases leading to significant amounts of C-acylated products (entries 4, 7, and 11). No significant optimization was conducted towards these specific products. Notably, no rearrangement was observed with benzoic acid (entry 13).
| Entry | Acid | Base | Conversiona (%) | O:C |
|---|---|---|---|---|
| a Conversions were determined via 1H NMR.b 0.5 equiv. of adipic acid was used.c The mixture was milled for an additional hour. | ||||
| 1 | 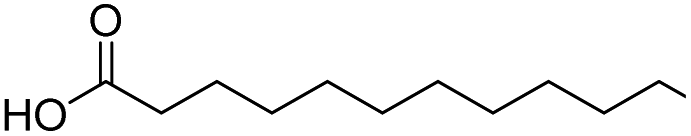 |
DMAP | 96.3 | 0.58:0.42 |
| 2 | NaOH | 94.3 | 1:0 | |
| 3 | 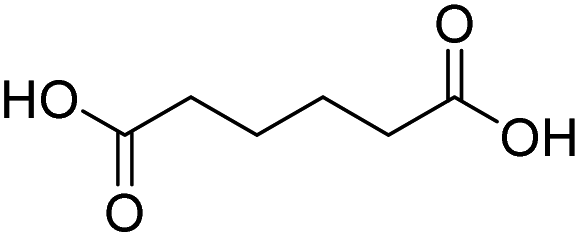 |
DMAP | 100.0 | 1:0 |
| 4 | DMAPb | 89.9 | 0.25:0.75 | |
| 5 | NaOH | 85.8 | 1:0 | |
| 6 | 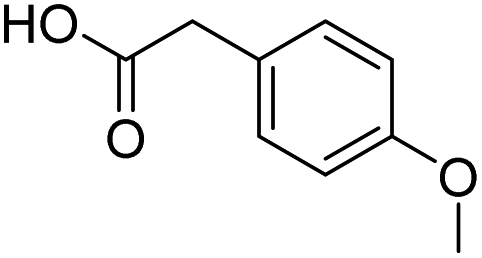 |
DMAP | 89.1 | 0.33:0.67 |
| 7 | DMAPc | 76.4 | 0.07:0.93 | |
| 8 | NaOH | 85.1 | 1:0 | |
| 9 | 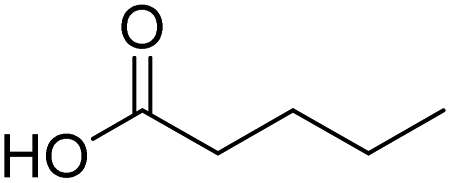 |
DMAP | 75.1 | 0.42:0.58 |
| 10 | NaOH | 89.5 | 1:0 | |
| 11 | 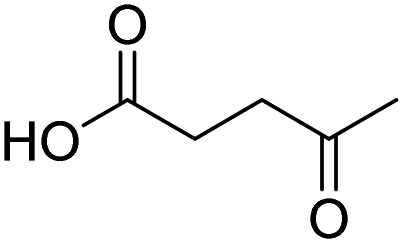 |
DMAP | 42.4 | 0.1:0.9 |
| 12 | NaOH | 56.2 | 1:0 | |
| 13 | 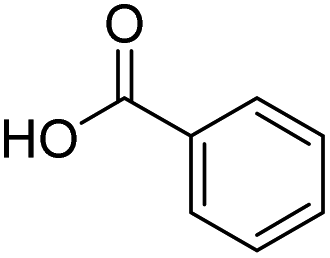 |
DMAP | 70.9 | 1:0 |
| 14 | NaOH | 69.4 | 1:0 | |
Different carbonyl reagents were also exploited in the reaction with phenyl propionic acid (Table 4). Apart from acetophenone (entries 9 and 10), which has a single carbonyl and did not react at all, all dicarbonyl compounds tested progressed selectively toward one of the acylation products using NaOH or DMAP as a base. 1,3-Cyclohexanedione, which is similar in structure to dimedone, led to the expected selectivity for both NaOH and DMAP (entries 1 and 2). In the case of ethyl acetoacetate (entries 7 and 8), the only unsymmetric dicarbonyl used, a mixture of constitutional isomers formed due to the O-acylation reaction through different sites of the dicarbonyl molecule when using NaOH, while DMAP promoted only the reaction and Fries rearrangement to the C-acylated product (see Fig. S48 and S49 in the ESI†). Conversely, both acetylacetone (entries 3 and 4) and 2,2,6,6-tetramethylheptane-3,5-dione (entries 5 and 6) promoted the selective formation of the corresponding O-acylated product, regardless of the base employed (i.e., in this case, DMAP was unable to catalyze the Fries rearrangement).
| Entry | Ketone | Base | Conversiona (%) | O:C |
|---|---|---|---|---|
| a Conversions were calculated via 1H-NMR.b A mixture of constitutional isomers was formed when using each base. | ||||
| 1 | 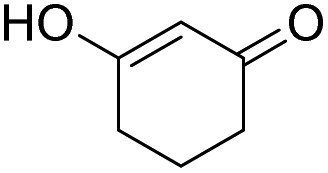 |
DMAP | 43.2 | 0:1 |
| 2 | NaOH | 5.6 | 1:0 | |
| 3 | 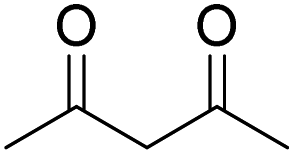 |
DMAP | 14.6 | 1:0 |
| 4 | NaOH | 10.4 | 1:0 | |
| 5 | 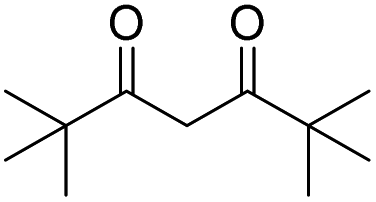 |
DMAP | 85.4 | 1:0 |
| 6 | NaOH | 78.9 | 1:0 | |
| 7 | 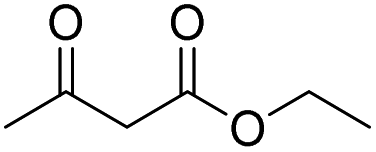 |
DMAP | 60.9 | 0:1 |
| 8 | NaOH | 22.5 | 0:1b | |
| 9 | 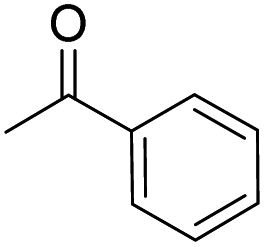 |
DMAP | 0.0 | — |
| 10 | NaOH | 0.0 | — | |
Given the importance of this reaction for the preparation of pharmaceutical products, we decided to scale up the mechanochemical acylation process (30 g dimedone loading in the presence of DMAP) using different types of mills typically found in sustainable chemical synthesis in the industry:2,35,36 attritor, planetary, and hammer mill. The three techniques differ in the type of mechanical force delivered, milling media, and grinding frequencies (see details in Table 5). The experiment with the attritor mill was stopped after 10 min as the material became too viscous and did not allow the continuous movement of the small balls (1 mm diameter), resulting in a mere 23% conversion. The planetary mill, on the other hand, provided 65% conversion in only 30 min. Finally, the hammer mill was less effective, yielding only 35% conversion in the same processing time. Importantly, all test reactions resulted in the C-acylation exclusively, regardless of the milling technique and apparatus. Therefore, these larger mills, which are more common on a larger scale than vibrational mills, while less efficient than the lab-scale mill, clearly maintain chemical selectivity, and given enough time, can achieve full conversion.
| Mill type | Jar (composite, volume, frequency) | Balls (weight, composite, diameter) | O:C |
Conversionb (%) |
|---|---|---|---|---|
| a Conditions: dimedone (30 g) was loaded in each reaction and milled for up to 30 min with all other components at stoichiometric conditions.b Conversion was calculated via 1H-NMR. | ||||
| Attritor | Teflon, 0.5 L, 1400 rpm | 600 g zirconia 1 mm | 0:1 |
22.8 |
| Planetary mill | Alumina, 0.3 L, 400 rpm | 50 g alumina 1 cm | 0:1 |
65.3 |
| Hammer mill | Alumina, 12 L, 140 rpm | None | 0:1 |
34.6 |
Conclusions
We investigated the chemoselectivity of the one-pot mechanochemical acylation of dimedone using ball-milling devices. The solid-state reactions, catalyzed by a base, demonstrated a complete chemoselectivity switch after short milling processes, outperforming solution-based processes, which require longer times and heating. When DMAP was used as a catalyst, there was complete selectivity towards the thermodynamic C-acylated product, while the kinetic enol ester selectively produced the O-acylated product in the presence of all other tested base catalysts. Experimental and theoretical mechanistic studies suggest that the unique chemoselectivity with DMAP occurs through a two-step mechanism: first producing the kinetic O-acylated intermediate, similar to all other bases, followed by a Fries rearrangement leading to the thermodynamic C-acylated product. It is essential to consider, however, that the entire study, particularly the computational modelling, was conducted presuming a classical view of ions and their solvating molecules.37 This interaction and the dynamics of ions might be entirely different in a solid-state reaction, where ions coordinate reactants/products. This key point remains an open question in the field of mechanochemical synthesis. Moreover, the water content in these reactions might affect reactivity as well. Once the selectivity of the reaction was explained, the scope of the reaction and its selectivity were further explored. Variations of the carboxylic acid yielded Fries rearrangement reactions with lower chemoselectivity; however, this can be improved through reaction optimization. Changes to the dicarbonyl nucleophile proved to be chemoselective in the presence of NaOH or DMAP, but again, Fries rearrangement was not always accessible under room-temperature milling. As the reaction time, selectivity, efficiency, and possibly environmental impact improve over the same reaction in solution, this study highlights the advantages of mechanochemistry in providing a sustainable solvent-free protocol, enabling experimental exploration of reaction mechanisms, and supplying evidence for pathways that lead to chemoselectivity.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Sally Nijem: writing – original draft, investigation, validation, visualization. Alexander Kaushansky: formal analysis, data curation, validation. Svetlana Pucovski: investigation, validation. Elisa Ivry: writing – review & editing. Evelina Colacino: conceptualization, review & editing. Ivan Halasz: conceptualization, supervision, review & editing. Charles Diesendruck: conceptualization, project administration, supervision, resources, visualization, writing – review & editing. All the authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Israel Science Foundation (Grant No. 174/23) for financial support and the Neubauer Family Foundation for the “Neubauer Doctoral Fellowship”. The Croatian Science Foundation (grant No. IP-2020-02-1419) is also acknowledged for its financial support. This article is based on work from COST Action CA18112 Mechanochemistry for Sustainable Industry,7,38,39 supported by COST (European Cooperation in Science and Technology). COST is a funding agency for research and innovation networks. Our actions help connect research initiatives across Europe and enable scientists to grow their ideas by sharing them with their peers, thereby enhancing their research, careers, and innovation (https://www.cost.eu/).Notes and references
- M. Kohout, B. Bielec, P. Steindl, G. Trettenhahn and W. Lindner, Tetrahedron, 2015, 71, 2698–2707 CrossRef CAS.
- S. Goncalves, M. Nicolas, A. Wagner and R. Baati, Tetrahedron Lett., 2010, 51, 2348–2350 CrossRef CAS.
- Y. A. Chabbert and M. R. Scavizzi, Antimicrob. Agents Chemother., 1976, 9, 36–41 CrossRef CAS PubMed.
- I. Chopra, Antimicrob. Agents Chemother., 1994, 38, 637–640 CrossRef CAS PubMed.
- B. Safak, I. H. Ciftci, M. Ozdemir, N. Kiyildi, Z. Cetinkaya, O. C. Aktepe, M. Altindis and G. Asik, Phytother. Res., 2009, 23, 955–957 CrossRef CAS PubMed.
- N. Fantozzi, J.-N. Volle, A. Porcheddu, D. Virieux, F. García and E. Colacino, Chem. Soc. Rev., 2023, 52, 6680–6714 RSC.
- F. Gomollón-Bel, ACS Cent. Sci., 2022, 8, 1474–1476 CrossRef PubMed.
- X.-Z. Lim, Chem. Eng. News, 2020, 98(38), 11 CAS.
- F. Gomollon-Bel, Chem. Eng. News, 2022, 100, 21–22 Search PubMed.
- E. Colacino, V. Isoni, D. Crawford and F. Garcia, Trends Chem., 2021, 3, 335–339 CrossRef CAS.
- O. Galant, G. Cerfeda, A. S. McCalmont, S. L. James, A. Porcheddu, F. Delogu, D. E. Crawford, E. Colacino and S. Spatari, ACS Sustain. Chem. Eng., 2022, 10, 1430–1439 CrossRef CAS.
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef PubMed.
- F. Cuccu, L. De Luca, F. Delogu, E. Colacino, N. Solin, R. Mocci and A. Porcheddu, ChemSusChem, 2022, 15, e202200362 CrossRef CAS PubMed.
- J. G. Hernández and C. Bolm, J. Org. Chem., 2017, 82, 4007–4019 CrossRef PubMed.
- D. Mauzerall and F. H. Westheimer, J. Am. Chem. Soc., 1955, 77, 2261–2264 CrossRef CAS.
- T. Kobayashi, K. Yamanoue, H. Abe and H. Ito, Eur. J. Org. Chem., 2017, 2017, 6693–6699 CrossRef CAS.
- I. Antonini, P. Polucci, A. Magnano, D. Cacciamani, M. T. Konieczny, J. Paradziej-Łukowicz and S. Martelli, Bioorg. Med. Chem., 2003, 11, 399–405 CrossRef CAS PubMed.
- K. Fries and G. Finck, Ber. Dtsch. Chem. Ges., 1908, 41, 4271–4284 CrossRef CAS.
- D. Virieux, F. Delogu, A. Porcheddu, F. García and E. Colacino, J. Org. Chem., 2021, 86, 13885–13894 CrossRef CAS PubMed.
- J. Stojaković, B. S. Farris and L. R. MacGillivray, Chem. Commun., 2012, 48, 7958–7960 RSC.
- I. Kaljurand, A. Kütt, L. Sooväli, T. Rodima, V. Mäemets, I. Leito and I. A. Koppel, J. Org. Chem., 2005, 70, 1019–1028 CrossRef CAS PubMed.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- A. M. Hyde, R. Calabria, R. Arvary, X. Wang and A. Klapars, Org. Process Res. Dev., 2019, 23, 1860–1871 CrossRef CAS.
- D. D. Perrin, Aust. J. Chem., 1964, 17, 484–488 CrossRef CAS.
- S. Xu, I. Held, B. Kempf, H. Mayr, W. Steglich and H. Zipse, Chem.–Eur. J., 2005, 11, 4751–4757 CrossRef CAS PubMed.
- F. Brotzel, B. Kempf, T. Singer, H. Zipse and H. Mayr, Chem.–Eur. J., 2007, 13, 336–345 CrossRef CAS PubMed.
- N. De Rycke, F. Couty and O. R. P. David, Chem.–Eur. J., 2011, 17, 12852–12871 CrossRef CAS PubMed.
- A. C. Spivey and S. Arseniyadis, Angew. Chem., Int. Ed., 2004, 43, 5436–5441 CrossRef CAS PubMed.
- Y. Okuno, S. Isomura, T. Kamakura, F. Sano, K. Tamahori, T. Goto, T. Hayashida, Y. Kitagawa, A. Fukuhara and K. Takeda, ChemSusChem, 2015, 8, 1711–1715 CrossRef CAS PubMed.
- H. Tabuchi and A. Ichihara, J. Chem. Soc., Perkin Trans. 1, 1994, 125–133 RSC.
- A. Berkessel and H. Gröger, in Asymmetric Organocatalysis, 2005, pp. 347–392 Search PubMed.
- A. C. Spivey, T. Fekner and H. Adams, Tetrahedron Lett., 1998, 39, 8919–8922 CrossRef CAS.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- V. Lutz, J. Glatthaar, C. Würtele, M. Serafin, H. Hausmann and P. R. Schreiner, Chem.–Eur. J., 2009, 15, 8548–8557 Search PubMed.
- S. R. Chhabra, A. N. Khan and B. W. Bycroft, Tetrahedron Lett., 1998, 39, 3585–3588 CrossRef CAS.
- B. Kellam, B. W. Bycroft, W. C. Chan and S. R. Chhabra, Tetrahedron, 1998, 54, 6817–6832 CrossRef CAS.
- D. R. Dekel, M. Amar, S. Willdorf, M. Kosa, S. Dhara and C. E. Diesendruck, Chem. Mater., 2017, 29, 4425–4431 CrossRef CAS.
- M. Baláž, L. Vella-Zarb, J. G. Hernández, I. Halasz, D. E. Crawford, M. Krupička, V. André, A. Niidu, F. García, L. Maini and E. Colacino, Chim. Oggi, 2019, 37, 32–34 Search PubMed.
- J. G. Hernández, I. Halasz, D. E. Crawford, M. Krupicka, M. Baláž, V. André, L. Vella-Zarb, A. Niidu, F. García, L. Maini and E. Colacino, Eur. J. Org Chem., 2020, 2020, 8–9 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, supporting chromatograms and spectra, computational details and data. See DOI: https://doi.org/10.1039/d4mr00141a |
| This journal is © The Royal Society of Chemistry 2025 |