
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational screening of milling parameters for Ru–Na/Al2O3 dual-function materials for integrated CO2 capture and methanation†
Andrea Braga ,
Maila Danielis*,
Sara Colussi and
Alessandro Trovarelli
,
Maila Danielis*,
Sara Colussi and
Alessandro Trovarelli
Dipartimento Politecnico e INSTM, Università degli Studi di Udine, Via del Cotonificio 108, 33100 Udine, Italy. E-mail: maila.danielis@uniud.it
First published on 5th May 2025
Abstract
The interest in the use of mechanochemistry as a green alternative to conventional solution-based synthesis methods has been steadily growing in recent years. Recently, Dual-Function Materials (DFMs) have been explored for the preparation of multicomponent systems which combine a sorbent and a catalytic phase co-supported on a support oxide for the capture of CO2 from flue gases and its subsequent conversion into added-value products when exposed to H2 (or CH4) in a chemical-looping-type reaction. However, the complexity of setting the right milling parameters, which are interconnected and strongly dependent on the precursor materials, is exacerbated in the multi-component system. In this work, we address this issue by employing a Design of Experiments (DoE) statistical approach for the screening of the most relevant milling parameters for the synthesis of Ru–Na/Al2O3 DFMs for integrated CO2 capture and methanation (ICCU-MET). The milling intensity and the organic precursors proved to be the key factors positively affecting the DFMs' capture capacity and CH4 conversion, respectively.
Introduction
Among the several strategies available in the field of CO2 emission abatement,1 the recent implementation of the so-called integrated CO2 capture and utilisation (ICCU)2,3 technologies is being studied as an alternative to classical processes such as amine sorption4 and CaO carbonation,5 which are very efficient in CO2 capture but require an additional process for its valorisation. The ICCU process, instead, is based on cyclic CO2 capture alternated with a CO2 conversion step on the same reactive bed, performed by switching the reaction atmosphere between the CO2-containing flue gas and the reactive gas.6The key components of this technology are the materials used to capture and valorise CO2, called Dual-Function Materials (DFMs).7 These materials belong to the family of heterogeneous catalysts and are solid nanomaterials composed of two functionalities dispersed on a support oxide: the sorbent function, usually linked to elements with basic characteristics such as alkaline metals, and the catalytic function, normally associated with transition metals.2,8 The two functions need to work in synergy to be able to first capture CO2 and then efficiently convert it into useful products such as CH4, CO and H2 (in syngas).9 Indeed, one of the most interesting uses of CO2 is to recycle it as a feedstock for e-fuels.10 In principle, renewable energy can be used to produce green hydrogen by electrolysis, which is then combined with CO2 for the synthesis of CH4 to store energy and be used as a sustainable fuel.
The main advantage of the ICCU process for DFMs is that, after the capture step directly from the emission source, the CO2 valorisation to useful products (such as CH4, CO + H2, and CH3OH) is performed in the same reactor, thus removing the necessity to regenerate the sorbent material and transport the CO2 to another facility. Hence, energy and transport costs can be reduced.
In this regard, the most commonly studied DFM formulations combine Al2O3 as the support oxide, since it has a relatively high surface area and good mechanical and thermal stability, Na, Ca, K, and Ba oxides as sorbent phases due to their basic properties, and Ni and Ru as the elements for the catalytic phase.2,3,8,11 The most suitable DFMs for the methanation of CO2 are composed of Ru or Ni, and Ca and Na as sorbent phases or a mixture of them to optimise the CO2 capture and release at different temperatures.3,12–14 The most commonly used methods for the preparation of DFMs are based on wet chemistry, usually by impregnating metal solutions on support oxides, followed by co-precipitation and sol–gel methods.2 These standard methods are suitable for producing materials with strong interactions among the different components, although they often require several impregnation and drying steps, long thermal treatments, and calcination steps at high temperatures to decompose and remove the element precursors. Dry mechanochemical methods are a more sustainable alternative for the preparation of heterogeneous catalysts for several reasons:15,16 they are solventless methods, they can be scaled up to industrial production,17 and in principle, they can reduce or remove the need for thermal treatments yielding the final working materials with quick syntheses at room temperature and reduced energy consumption.18–23 Successful attempts have been reported in several fields,16,24 but limited research has been conducted on the application of dry milling to the synthesis of dual-function materials.
Our previous work25 explored the feasibility of mechanochemically preparing DFMs based on RuNi/(Na2O or CaO)/CeO2–Al2O3 for CO2 capture and conversion to CO by the reverse water-gas shift reaction. In this work, we expand the use of dry ball milling with a rational investigation of the effect of the milling parameters on the CO2 capture and methanation performance in the integrated carbon capture and methanation process (ICCU-MET). The DFMs prepared in this study are composed of Ru + Na2O/Al2O3, which is a reference composition2,3,12,26,27 mostly suitable for medium-temperature (250–400 °C) CO2 capture and methanation.28,29 The simple composition allows us to focus our attention on the milling parameters and their effects on the materials' performance. The capture properties of Na-based DFMs are influenced by the nature of the Na precursor,29 and, similarly, different precursors used in dry ball milling can affect the final catalytic properties.30,31 For these reasons, the choice of Ru and Na precursors and three different milling regimes were thoroughly studied to account for the different nature of the precursors and the effect of milling on the obtained DFMs. A full factorial experimental design was used to efficiently combine these parameters and to analyse the results, highlighting the most relevant parameters affecting the performance of DFMs.
Experimental
Synthesis of dual-function materials (DFMs) by dry ball milling
The DFMs were prepared by mixing both Na and Ru precursors with 5%La2O3-γ-Al2O3 (MI386, Grace) in a single-step dry ball milling process using a Fritsch Pulverisette 23 Mini-mill. The MI386 support was previously calcined at 550 °C for 2 h in air. With 890 mg of MI386, appropriate amounts of Ru acetylacetonate (RuAc, Strem Chemicals), metallic Ru black powder (RuM, Strem Chemicals), Na2CO3 (NaC, Carlo Erba) and NaNO3 (NaN, VWR Chemicals) were mixed to obtain respectively 1 wt% metallic Ru and 10 wt% Na2O equivalent. The powders were loaded into a 15 mL ZrO2 jar with a single ZrO2 ball (m = 10 g, Ø = 15 mm), and the ball-to-powder mass ratio varied between 9 and 10 depending on the different precursors used (Table 1).| # | Sample | Time (min) | Frequency (Hz) | Ru precursor | Na precursor | BPR |
|---|---|---|---|---|---|---|
| 1 | RuMNaC/MI386-15 | 15 | 15 | Ru metal | Na2CO3 | 10 |
| 2 | RuMNaC/MI386-35 | 35 | 35 | Ru metal | Na2CO3 | 10 |
| 3 | RuMNaN/MI386-15 | 15 | 15 | Ru metal | NaNO3 | 9 |
| 4 | RuMNaN/MI386-35 | 35 | 35 | Ru metal | NaNO3 | 9 |
| 5 | RuAcNaC/MI386-15 | 15 | 15 | RuAc | Na2CO3 | 9 |
| 6 | RuAcNaC/MI386-35 | 35 | 35 | RuAc | Na2CO3 | 9 |
| 7 | RuAcNaN/MI386-15 | 15 | 15 | RuAc | NaNO3 | 9 |
| 8 | RuAcNaN/MI386-35 | 35 | 35 | RuAc | NaNO3 | 9 |
| 9 | RuMNaC/MI386-27 | 27 | 27 | Ru metal | Na2CO3 | 10 |
| 10 | RuMNaN/MI386-27 | 27 | 27 | Ru metal | NaNO3 | 9 |
| 11 | RuAcNaC/MI386-27 | 27 | 27 | RuAc | Na2CO3 | 9 |
| 12 | RuAcNaN/MI386-27 | 27 | 27 | RuAc | NaNO3 | 9 |
The milling intensity was set at low, medium, and high intensity regimes by coupling the milling time and milling frequency: the low-intensity samples were milled for 15 min at 15 Hz, the medium-intensity ones were milled for 27 min at 27 Hz, and the high-intensity samples were milled at 35 Hz for 35 min. The milling was paused after 15 min to clean the jar and ball surfaces and manually mix the powders. The loose powder was first removed from the jar, and then the milling medium was scratched with a brush to avoid the formation of large amounts of unmixed powders on the walls of the jar. The samples were ready and used directly after the milling procedure.
A full factorial experimental design,32,33 generated using Minitab 21.4, was used to create the sample series to study the combination of different parameters and their combined effects: the milling intensity, the Ru precursor and the Na precursor. Given the categorical nature of both Ru and Na precursors, a total of 12 samples were generated. The sample series contained all the combinations of the three parameters to study the effect of each one on different DFM characteristics, such as the CO2 adsorption capacity and the CH4 production rates. The samples were prepared and tested in a random order to minimise and distribute the environmental effects.31
The prepared DFMs, as reported in Table 1, were named x-Ru(M,Ac)Na(C,N)/MI386-(15,27,35), where x represents the identification number, (M,Ac) represents the Ru precursor, (C,N) represents the Na precursor, and the last number represents both the milling time and frequency. Several Na/MI386 samples were also prepared by milling different loadings of the two Na precursors with MI386 at the three different milling regimes as preliminary samples to find the experimental design ranges.
Characterisation
Powder X-ray diffraction (XRD) measurements were carried out with a Philips X'Pert diffractometer with a Cu cathode (Cu Kα λ = 1.5406 Å, 40 kV and 40 mA) collecting the diffractograms with a step of 0.02° and 40 s per step; higher resolution patterns were collected on the spent DFMs from 30 to 50° with a step of 0.01° and 320 s per step. The patterns were fitted with pseudo-Voigt functions in OriginLab 2021 to isolate the metallic Ru peak FWHM at 44° and estimate the crystallite size with Scherrer's equation:34where λ = 1.5406 Å is the X-ray wavelength and
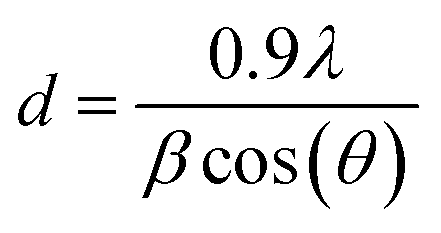
is the peak broadening corrected for the instrumental broadening, measured on a Si standard, at a given 2θ.
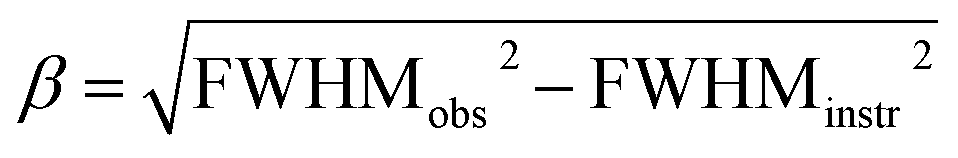
Thermogravimetric analysis (TGA) was conducted using a TGA Q5500 (TA Instruments) on as-prepared samples in 25 mL min−1 of synthetic air (i.e., dry air in a commercial cylinder, with no moisture or CO2) to observe the precursors' decomposition patterns by loading about 10 mg of powders on platinum pans and heating from room temperature to 900 °C (10 °C min−1). Additionally, TGA was used to study the activation of the DFMs in 5% H2/N2 (25 mL min−1) by loading about 10 mg of DFM on platinum pans and heating from room temperature to 400 °C and holding for 30 min (10 °C min−1).
Specific surface area and pore size distribution values were obtained using a Nova 800 analyser (Anton Paar) to record physisorption isotherms at −196 °C in N2. The specific surface area and pore size were estimated using the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods, respectively. Prior to the analysis, all samples were outgassed in a vacuum at 150 °C for 90 min. Selected samples were reduced ex situ for 1 h at 400 °C in 10% H2/N2 (50 mL min−1, 10 °C min−1) to measure the surface area and pore distribution of the activated DFMs.
Temperature Programmed Reduction (H2-TPR) measurements were performed from the activation step inside the reactor used for the ICCU-MET tests. Briefly, 250 mg of DFM were placed inside a fixed bed quartz reactor (12 mm internal diameter) and activated in 50 mL min−1 of 10% H2/N2 from room temperature to 400 °C and holding for 1 h (10 °C min−1). The evolution of H2, CO2, CO and CH4 was monitored with an ABB AO2020 online gas analyser recording data every 5 s and integrated with OriginLab.
Integrated CO2 capture and methanation testing
The DFMs were tested for the cyclic capture and methanation of CO2 (ICCU-MET) in a fixed-bed quartz reactor with a 12 mm internal diameter. The reaction apparatus and the testing procedure are reported in Scheme 1. The DFM powders were supported on a quartz wool bed and a thermocouple was placed above the catalytic bed. 250 mg of DFM was activated in situ with 50 mL min−1 of 10% H2/N2 from room temperature to 400 °C for 1 h (10 °C min−1) and cooled to 350 °C in N2. After the temperature was stable, ICCU-MET cyclic testing was conducted as follows: (i) 10 min of CO2 capture with 50 mL min−1 of 5% CO2/N2; (ii) a 10 min purge with 100 mL min−1 of N2; (iii) 10 min of methanation with 50 mL min−1 of 10% H2/N2; (iv) a final 10 min purge with 100 mL min−1 of N2. In total, five cycles were performed for each test. The evolution of H2, CO2, CO and CH4 was followed online with an ABB AO2020 analyser recording data points every 5 s. The molar flows were integrated with OriginLab to obtain the moles of CO, CO2 and CH4:where i = (CO, CO2, CH4), mDFM is the DFM mass loaded in the reactor, and Fi is the molar flow.

 | ||
| Scheme 1 (a) Reaction apparatus scheme composed of independently controlled mass flow controllers connected to a 4-way valve leading to the reactor or a bypass; (b) ICCU-MET reaction procedure. The CO2 capture and methanation steps represent one cycle; 5 cycles are performed per experiment. | ||
To estimate the amount of CO2 captured, blank measurements were taken at 350 °C in a reactor loaded with 250 mg of coarse quartz grains previously calcined at 1200 °C. Three injections of 10 min of 50 mL min−1 of 5% CO2/N2 were performed, similar to the CO2 capture steps to obtain the reference area. The blank measurement was repeated after the testing campaign to check the reproducibility and stability of the reaction system. The CO2 capture capacity at each step was estimated by subtracting the area of the integrated CO2 signal from the reference CO2 area (see Fig. S1†). In addition, considering that all the DFMs produced some amounts of CO during the capture steps and that during the purge some weakly physisorbed CO2 was released, the captured CO2 was corrected by subtracting these two contributions as well:

The CO2 conversion to CH4 during the methanation step was calculated considering the release of CO2 during the methanation step:

The methanation selectivity was calculated considering the production of CH4 and CO and the release of CO2:

Results and discussion
Evaluation of alkali-supported oxide binary systems
A preliminary investigation by BET, XRD, and TG analyses was carried out on metal-free Na(C,N)/Al2O3 systems to ensure significant variability among the different milling parameters and precursor choices for the factorial design of experiments. The results so obtained, reported in Fig. 1, also provided reference samples for the fully formulated DFMs investigated in the following.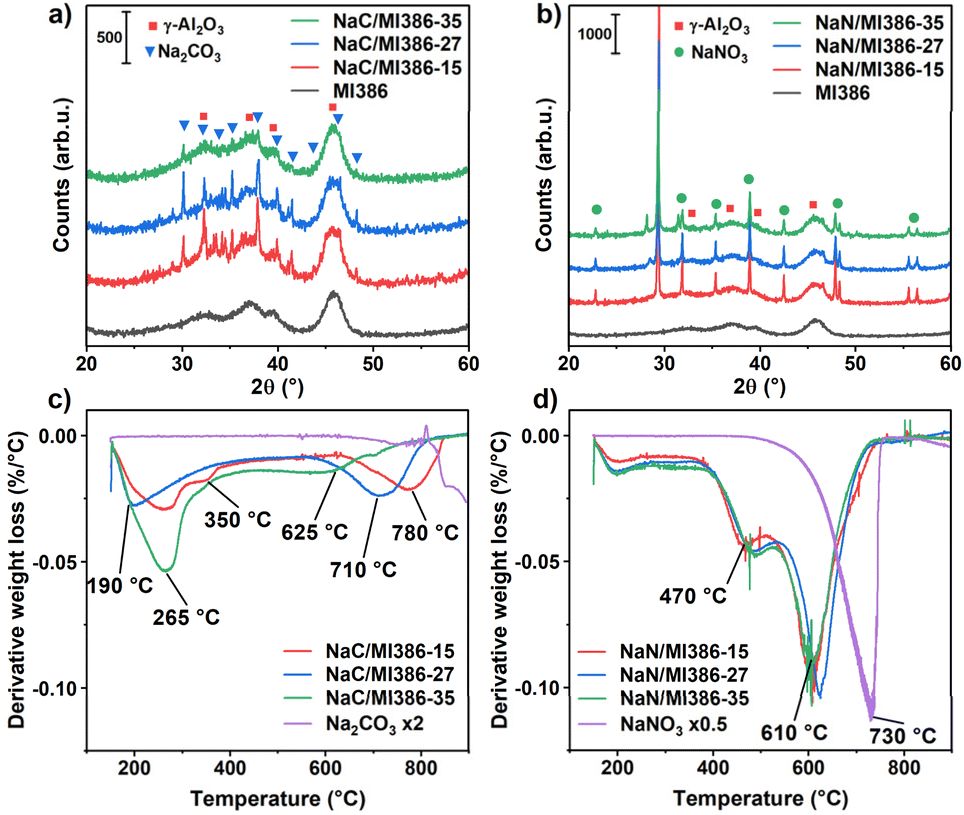 | ||
| Fig. 1 XRD diffractograms of the (a) Na2CO3 and (b) NaNO3 precursors milled with MI386 at different intensities. Derivative mass loss signals during the TGA in air of (c) NaC/MI386 and (d) NaN/MI386. Derivative signals for the bare salts are reported either doubled (Na2CO3, ×2) or halved (NaNO3, ×0.5) in intensity for clarity. | ||
The crystalline structure of the as-prepared Na/MI386 samples was studied by XRD to observe the modifications induced on the Na salts by milling at different regimes. The XRD patterns of NaC/MI386 and NaN/MI386 are reported in Fig. 1a and b. The identified phases are Al2O3 (PDF: 00-010-0425), Na2CO3 (PDF: 00-037-0451 and 00-019-1130), and NaNO3 (PDF: 01-085-0850). The intensity of the Na2CO3 pattern decreased with increasing milling intensity, indicating a progressive reduction in the crystallinity degree. Remarkably, a significant loss in crystallinity was observed in the sample milled at 35 Hz for 35 min, while less striking differences were observed between the sample milled at 15 Hz for 15 min and the one milled at 27 Hz for 27 min. Regarding the NaN/MI386 samples, the NaNO3 pattern was less affected by the increasing milling intensity and large crystallites were still present in the sample NaN/MI386-35. The NaNO3 crystallinity decreased gradually by about 33% from the lowest to the highest milling intensity. The peak shape and FWHM did not change, suggesting minor modifications to the NaNO3 structure. In all samples, the Al2O3 crystalline structure did not change upon increasing the milling intensity.
In parallel, the BET specific surface areas, the BJH pore diameters and volumes are reported in Table S1,† while the N2 isotherms and the pore size distributions are shown in Fig. S2.† In general, adding both precursors to MI386 by milling decreased the support surface area and pore volume; at higher milling intensities, the BET area decreased further, and the pore structure collapsed. The use of NaNO3 led to a lower BET surface area and pore volume compared to Na2CO3, likely because of the higher amount (23 vs. 16 wt%, respectively) to result in 10 wt% Na2O. However, similar trends were observed for the pores' diameter and volume, with significant losses, especially at the highest milling intensity.
To investigate whether the changes in crystallinity would affect the decomposition behaviour, the weight of the Na precursors was followed in TGA measurements by calcining the samples from room temperature to 900 °C (10 °C min−1) in air. The derivative weight loss profiles of the NaC/MI386 and NaN/MI386 samples are reported in Fig. 1c and d and the respective weight loss signals are in Fig. S3,† together with the pure Na2CO3 and NaNO3 samples reported as references. It clearly appears that the weight loss of the Na(C,N)/MI386 samples was influenced by the presence of Al2O3, which shifted the salt reduction at lower temperatures. Indeed, pure Na2CO3 decomposition started after 850 °C, as it is a very stable compound,28,35 while pure NaNO3 decomposed in a single step peaking at 730 °C. When supported on Al2O3, the characteristic decomposition occurred at much lower temperatures, most noticeably for NaC/MI386. On the latter, the milling intensity also plays a relevant role, while NaN/MI386 samples were mainly unaffected. In fact, the shift towards lower temperatures of the Na2CO3 decomposition steps observed with increasing milling intensity can be ascribed to a larger fraction of Na2CO3 species in close contact with Al2O3, promoted by milling, which is reported to favour the decomposition of the carbonate.35,36
Overall, the TGA results were in good accordance with the XRD patterns: for Na2CO3, higher milling intensity resulted in more amorphous Na2CO3 and enhanced decomposition at low temperatures (Fig. 1a and c);35,36 for NaNO3, the change in milling parameters barely affected its crystalline structure as well as the decomposition rate (Fig. 1b and d).
Phase composition and thermal stability of the DFMs
First, the morphological and redox properties of the 12 DFM samples of the statistical design were investigated. A focus of the XRD patterns of the as-prepared DFMs is reported in Fig. 2, while full-range patterns are reported in Fig. S4.† In Fig. S4e,† the XRD diffractograms of the four different precursors are also reported in the range 35–50°. In agreement with what observed for NaC/MI386 samples (Fig. 1a), on Ru(M,Ac)NaC/MI386 samples, the intensity of the Na2CO3 pattern decreased with increasing milling intensity (Fig. 2a and S4a†) and the peaks were broader; this could be related to the gradual amorphisation of the Na2CO3 crystallites with increasing milling intensity. No Ru-related patterns could be identified for both RuM and RuAc DFMs; in particular, the metallic Ru peak at 44.0° was not observed, indicating a high dispersion for the RuM precursor. The NaNO3 pattern was less affected by the increasing milling intensity: the area of the main peak, centred at 29.3° 2θ (Fig. 1b), decreased by 34% on the samples milled at 35 Hz for 35 min compared to the ones milled at 15 Hz for 15 min, and no differences were found by milling with RuM or RuAc, confirming the low influence of the milling intensity on the NaNO3 crystallinity. Similarly to the Na2CO3-based DFMs, no patterns related to Ru species were observed suggesting a high dispersion of both Ru precursors (Fig. 2). On all samples, the γ-Al2O3 crystalline phase did not show any modification compared with the bare support and at the different milling intensities. Overall, the as-prepared DFMs were similar to the binary Na(C,N)/MI386 samples reported in Fig. 1 from a structural point of view, suggesting that the presence of ruthenium has a negligible effect on the structural properties of the materials. | ||
| Fig. 2 Details of the XRD patterns of the as-prepared (a) Na2CO3-based DFMs and (b) NaNO3-based DFMs. | ||
However, the presence of Ru influenced the decomposition behaviour of the Na precursors in air, as shown in Fig. S5 and S6,† compared with the Ru-free supports shown in Fig. 1c and d. In Na2CO3-based DFMs, the samples milled at medium and high intensity displayed similar weight loss signals occurring earlier than the respective DFMs milled at 15 Hz and 15 min. The presence of Ru promoted the low-temperature decomposition in both sample subgroups, reducing the decomposition temperature by 20–70 °C compared with the metal-free supports (compare Fig. 1c with S5c and d†). DFMs based on RuAc showed a sharper and more intense decomposition at 190–240 °C associated with the RuAc decomposition, which accelerated the Na2CO3 decomposition. At high temperatures, low-intensity milled samples displayed higher weight loss than metal-free NaC/MI386-15, with peak temperatures reduced by 30–50 °C due to Ru. The samples milled at higher intensities showed minor differences compared to the metal-free supports at high temperatures.
Regarding the NaNO3 samples, as observed in XRD (Fig. 1 and S4†) and the TGA analysis of the metal-free supports (Fig. S1d†), even the presence of Ru has a minimal impact on the decomposition properties observed across milling intensities. RuM-based DFMs showed the same weight loss profiles; compared with the metal-free supports, the peak at 480 °C was only slightly anticipated at 320 °C, while the peak at about 600 °C remained unchanged. The RuAc-based DFMs showed a sharp peak at 280 °C with a shoulder at 190 °C corresponding to the acetylacetonate and part of the NO3 decomposition. The high-temperature decomposition was unaffected by both the milling intensity and the Ru precursor for the samples milled at medium-high regimes, while only for the sample 7-RuAcNaN/MI386-15 it shifted to 560 °C.
However, in the current application, no prior calcination treatment in air was needed on the DFM samples, and their activation was carried out directly in the reactor in H2 up to 400 °C to decompose the Ru and Na precursors. Hence, additional TGA measurements were used to study their reactivity under H2 atmosphere. The DFMs' weight profiles in H2 and the derivative signals are reported in Fig. 3 and S7,† respectively. The four samples milled at low intensity showed a higher residual weight after the activation compared to the samples milled at medium-high intensity, indicating a lower degree of Na salt decomposition. For comparison, in Fig. S8,† the weight loss at 400 °C under H2 is reported alongside the weight loss values measured in air at 400 °C and 900 °C, where Na precursor decomposition was completed for all the samples (from Fig. S5 and S6†). The samples milled at low intensity showed a smaller weight loss in H2 compared with the weight loss at 900 °C in air, while the samples milled at medium and high intensity showed comparable weight losses suggesting the complete decomposition at 400 °C in H2. For comparison, all the samples showed 1.5–3 times smaller weight losses at 400 °C in air, highlighting the role of Ru in activating H2 to enhance the low-temperature Na precursors' decomposition. These results show that medium-high milling intensities and H2 activated by Ru allowed the complete decomposition of the Na precursors at 400 °C.
 | ||
| Fig. 3 TGA weight profiles in 5% H2/N2 up to 400 °C of DFMs based on (a) RuM + Na2CO3, (b) RuM + NaNO3, (c) RuAc + Na2CO3 and (d) RuAc + NaNO3. | ||
Redox behaviour of the DFMs
The derivative signals in Fig. S7† can be compared to the H2-TPR signals obtained during the in situ activation in the reactor reported in Fig. 4. The H2-TPR step carried out in the reactor allowed us to follow the gas evolution of the RuNa/MI386 samples, which also provides insights into the passive Direct Air Capture (DAC) capacity of the DFMs.25 | ||
| Fig. 4 H2-TPR signals measured during the activation procedure inside the reactor with 50 mL min−1 of 10% H2/N2. | ||
On Na2CO3-based DFMs, as the temperature increased the CO2 adsorbed from the atmosphere was released and gradually converted to CH4 starting from 250 °C. The samples based on RuAc showed a more intense and sharper CH4 release peak around 350 °C suggesting the formation of more active sites, while the samples based on RuM showed a longer tail at 400 °C, indicating the presence of less active Ru species. Furthermore, the CH4 production slowed with increasing milling intensity. The maximum CH4 production rate was achieved at 340–350 °C, which was consequently taken as the testing temperature. Similarly, the NaNO3 samples showed comparable profiles in both the TGA and the reactor (see Fig. S7b, d and 4), corresponding to the decomposition of NaNO3. The samples based on RuM showed a broader temperature range for NaNO3 decomposition, starting at 200–250 °C and ending with a sharp event after 5 min at 400 °C in both the TGA and in the reactor. The two samples milled at medium-high intensity showed a very similar profile and during the H2-TPR the O2 signal was recorded, which probably originated from the NOx species detected from the O2 sensor. Regarding the RuAcNaN/MI386 samples, the decomposition of NaNO3 started at about 280 °C and a quick release of both CO2 and O2 was observed at about 320 °C with an exothermic reaction, followed by a sharp consumption of H2 at 350–360 °C. Also in this sample subgroup, the two DFMs milled at medium-high intensity showed the same decomposition pattern, suggesting that similar structures and reactivity were obtained already by milling the Ru and Na precursors for 27 min at 27 Hz.
In Table 2, the quantitative analysis of the evolution of CH4, CO2 and CO released during the in situ DFM activation shown in Fig. 4 is reported. The evolution of carbon species can be related both to the presence of the precursors, as in the case of the Na2CO3-based and RuAc-based DFMs, and to the atmospheric CO2 captured at room temperature.25 Overall, all the samples showed higher total C species than the amount loaded with the precursors, suggesting that they are all characterised by some passive CO2 capture capacity. The evolution of the carbon species by passive Direct Air Capture (DAC) and the CH4/CO2 ratio are also summarised in Fig. S9.† The samples based on NaC and RuAc release the highest amount of carbon, both due to the precursors and DAC, followed by the RuMNaC/MI386 and RuAcNaN/MI386 samples. The least performing were the ones with no carbon contribution from the precursors (RuMNaN/MI386), which also exhibited the lowest CH4 production. Conversely, the RuAc-based DFMs displayed the highest methane release, suggesting higher reactivity.
| # | Sample | CO2 | CH4 | CO | Total C | C from precursors | C from passive DAC |
|---|---|---|---|---|---|---|---|
| 1 | RuMNaC/MI386-15 | 293 | 598 | 0 | 891 | 151 | 740 |
| 9 | RuMNaC/MI386-27 | 709 | 776 | 21 | 1506 | 151 | 1355 |
| 2 | RuMNaC/MI386-35 | 534 | 931 | 4 | 1468 | 151 | 1317 |
| 3 | RuMNaN/MI386-15 | 182 | 49 | 0 | 232 | 0 | 232 |
| 10 | RuMNaN/MI386-27 | 289 | 50 | 0 | 339 | 0 | 339 |
| 4 | RuMNaN/MI386-35 | 287 | 78 | 0 | 365 | 0 | 365 |
| 5 | RuAcNaC/MI386-15 | 724 | 813 | 24 | 1561 | 328 | 1233 |
| 11 | RuAcNaC/MI386-27 | 916 | 1224 | 57 | 2196 | 328 | 1868 |
| 6 | RuAcNaC/MI386-35 | 872 | 1287 | 59 | 2218 | 328 | 1890 |
| 7 | RuAcNaN/MI386-15 | 640 | 239 | 0 | 879 | 165 | 714 |
| 12 | RuAcNaN/MI386-27 | 1029 | 372 | 10 | 1412 | 165 | 1247 |
| 8 | RuAcNaN/MI386-35 | 958 | 436 | 12 | 1406 | 165 | 1241 |
All the samples showed the same trend of both the total C and the CO2 release as a function of the milling intensity: the lowest values were related to the samples milled for 15 min at 15 Hz, while the highest amounts of CO2 released and CH4 production were achieved respectively at medium and high intensities.
ICCU-MET performance and DoE analysis
Finally, the ICCU-MET performance of the prepared DFMs was evaluated by performing five cycles of CO2 capture and methanation at 350 °C (5% CO2/N2 and 10% H2/N2, 50 mL min−1), in order to investigate the effects of the Ru and Na precursors and the milling parameters. The CO2 capture capacity and the CH4 production for all the cycles are reported in Fig. 5, while the transient profiles of CO, CO2 and CH4 during the 5th methanation step are shown in Fig. 6. The summary of the DFMs' performance quantitative analyses is reported in Table S2.† The reactions involved in the ICCU-MET process can be described by the following chemical equations:37| CO2 capture: Na2O + CO2 ⇌ Na2CO3 |
| NaOH + CO2 ⇌ NaHCO3 |
| CH4 formation: Na2CO3 ⇌ Na2O + CO2 |
| CO2 + 4H2 ⇌ CH4 + 2H2O |
| Na2O + H2O ⇌ 2NaOH |
 | ||
| Fig. 5 (a) CO2 capture capacity and (b) CH4 production for each cycle of the DFMs tested at 350 °C under 5% CO2/N2 and 10% H2/N2 (50 mL min−1, 250 mg, and 10 min each cycle). | ||
 | ||
| Fig. 6 Transient gas profiles of CO, CO2 and CH4 during the 5th methanation step (350 °C, 10% H2/N2, 50 mL min−1, and 250 mg). | ||
In parallel, the RuAc samples produced more CH4 during the methanation step. A table comparing the activity of the best performing dry milled samples compared to wet-based DFMs from the literature is reported in the ESI (Table S3).† The samples milled at medium and low intensity showed a more stable performance within five cycles, while the highest milling regime resulted in a constant decrease in the CH4 production.
At the beginning of each methanation step, on all samples some CO2 and CO were released together with CH4, as can be observed in Fig. 6. The CO2 release is likely due to the presence of weakly bound CO2 or the decomposition of carbonate sites far away from the Ru catalytic sites. Another CO2 release mechanism can be triggered by the local increase in temperature due to the reduction of oxidised surface species.38
The Ru precursor played a major role in the DFMs' performance considering both the CH4 production and the release of unwanted products like CO2 and CO. The RuM DFMs showed higher amounts of CO2 and CO released at the beginning of the methanation step, and the transient evolution reached the maximum CH4 production only after both CO and CO2 release decreased to zero. In addition, the higher the milling intensity, the slower the CH4 production rate. On the other side, the samples based on RuAc released very small amounts of CO and CO2 and the CH4 production was rapid (see Table S2†), characterised by a quick production of CH4 at the beginning of the methanation step. Similar to the RuM DFMs, by increasing the milling intensity the transients flattened, and the profile showed a longer tail, suggesting the presence of slower active sites formed at higher milling regimes in line with the CH4 profiles during the H2-TPR in Fig. 4.
The cumulative CH4 production during the 5th methanation cycle is shown in Fig. S10† and is helpful to highlight both the total CH4 production and the kinetics of the methane release. It is possible to appreciate the separation of the two groups based on the Ru precursor, where RuAc samples not only produced more CH4 but also showed the steepest increase, as highlighted by the difference in the slope at the beginning of the methanation step. Indeed, it is possible to estimate the initial methanation release rate by integrating the amount of CH4 produced in the first 60 s of the 5th methanation step, considering that the cumulative release of CH4 is almost linear.
In addition to the most evident results, statistical analysis of the experimental design results was carried out to reveal additional correlations between milling parameters, DFM precursors, and performance. Several performance indicators (CO2 capture capacity, total CH4 production, CO2 conversion, CH4 selectivity, CH4 release rate, CO release during the capture step, and CO2 release during the methanation step) were analysed to evaluate the influence of each parameter. Details of the catalytic DFMs' performance are reported in full in Table S2,† while the statistical analysis of the experimental design results is reported as ANOVA tables in Tables S4–10.† The most influential milling parameter for each performance indicator is the one with the lowest p-value.
From the statistical analysis, it clearly appears that the CO2 capture capacity was mostly influenced by the milling intensity: the p-value associated with the milling intensity is 0.05, while the Na and Ru precursor choice showed p-values of 0.27 and 0.17 respectively (see Table S4†), suggesting a limited effect of the precursors on the capture capacity.
To better understand the effect of each parameter on the capture capacity, for each sample the captured CO2 amount was grouped and plotted against the different milling parameter values, as reported in Fig. 7a. These plots are built by plotting the median value of a given response versus a given factor at different levels. In other words, the CO2 capture capacity of all the samples is reported grouped for each milling parameter depending on the parameter's values. Therefore, the effect of the milling intensity on the CO2 capture capacity appears very clear: the higher the milling intensity, the higher the capture capacity. Regarding the Na precursor, it seems that on average the Na2CO3-based DFMs performed slightly better than the NaNO3 ones, although the wide distribution of values around the median limits the significance. A similar conclusion can be drawn for the effect of the Ru precursor. In Fig. 7a, the response values at different milling intensities are also grouped by the Ru precursor; in fact, the positive effect of increasing the milling intensity was mostly associated with the combination of higher intensity and the RuAc precursor. This is also evident from the response values in the Ru precursor plot, where the values are grouped by milling intensity.
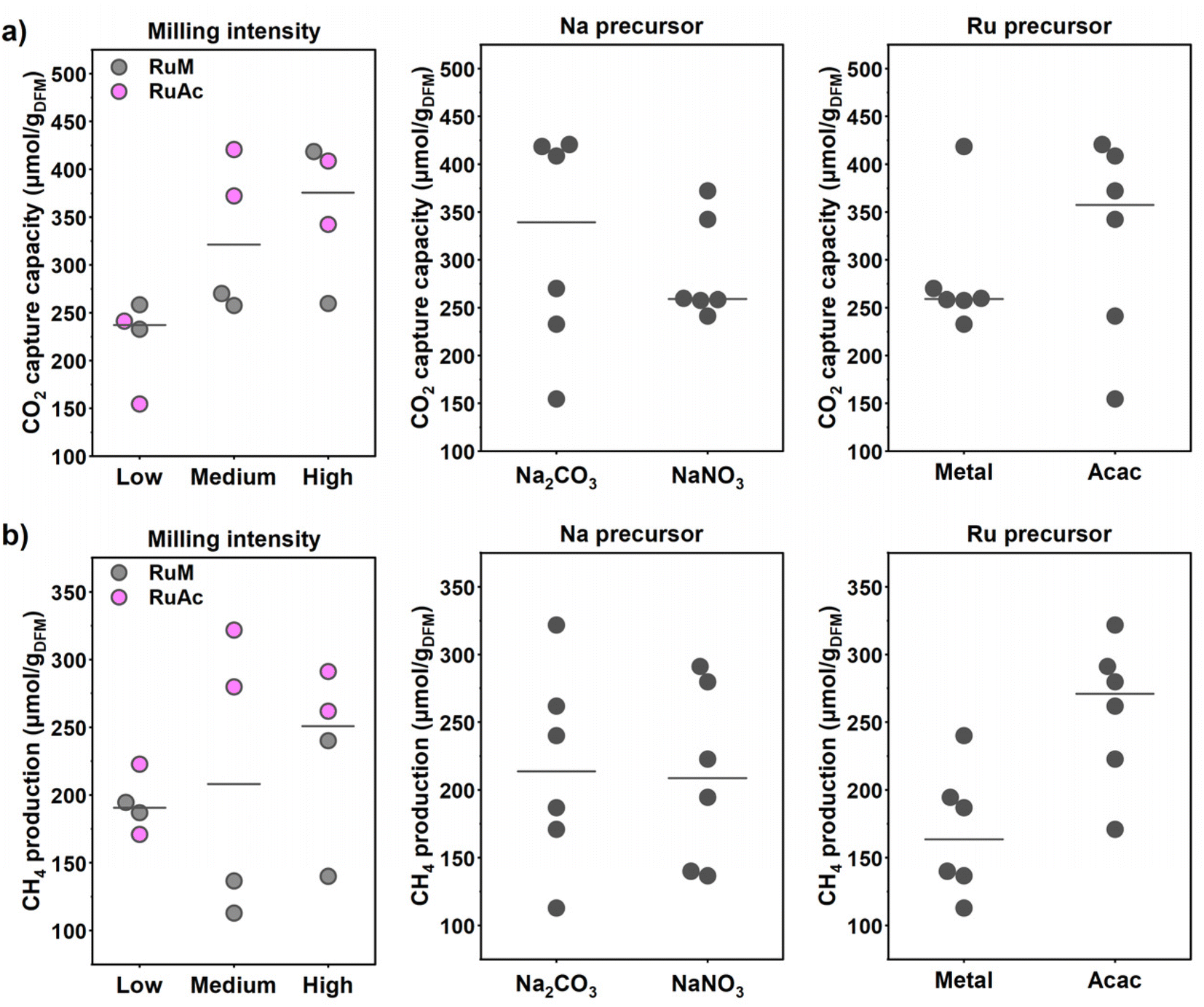 | ||
| Fig. 7 Main effect plots of the (a) CO2 capture capacity and (b) average CH4 production at the different milling parameters. Each dot represents the response of one sample prepared with the corresponding milling parameter value. | ||
On the other hand, the average CH4 production was solely affected by the choice of Ru precursor; in particular, high CH4 production was obtained for the samples based on RuAc. In Table S5† the ANOVA table is reported: the Ru precursor p-value is the smallest at 0.08, and the other p-values are much higher, indicating no significant effect on the methane production. The main effect plots in Fig. 7b show the positive effect related to the use of RuAc, while the type of Na precursor clearly had no effect on the production of CH4. Interestingly, there was an interaction between the milling intensity and the Ru precursor, with RuM samples showing a negative interaction upon increasing the milling intensity and the RuAc samples instead displaying a positive interaction.
Other relevant metrics to evaluate the DFMs' performance are the CO2 conversion, the CH4 selectivity and release rate, the CO produced during the capture step and the CO2 released during the methanation (see Table S2†).
The median CO2 conversion for the RuM samples was 61% and for the RuAc DFMs it was 86%, showing a higher ability of the latter to convert the CO2 captured in the previous steps (Fig. S11†). The most influential parameter was the Ru precursor with a p-value of 0.07, followed by the milling intensity with a p-value of 0.11, associated with a higher CO2 conversion at lower milling intensities (Table S6†). For both RuM and RuAc samples, there was no difference between the subgroups of samples based on Na2CO3 and NaNO3 on average, indicating that the conversion was only affected by the Ru active sites. Thus, the spillover of the adsorbed CO2 on different Na sorbent sites formed from different Na precursors is not a limiting factor. Higher CO2 conversions were achieved at the lowest milling regimes, although this was mostly due to the lower CO2 capture capacity, i.e., a lower amount of reacting CO2, as shown in Fig. 7a. The sample 5-RuAcNaC/MI386-15 stood out as an outlier with a CO2 conversion of 115%: this was probably due to the conversion of residual Na2CO3 precursor species, which were not decomposed during the DFM activation.
Similar results were obtained for the CH4 selectivity, considering both the production of CO as a byproduct and the release of CO2 from weak basic sites or from sorbent sites that might be too far from the Ru active sites to be converted. The only parameter with a low p-value of 0.11 was the Ru precursor (Table S7†). The RuAc DFMs showed higher selectivity with an average of 95.6% of CH4, 1.0% of CO and 3.4% of CO2 released at the beginning of the methanation step. No differences were observed between the Na precursors, and a slightly higher selectivity towards methane was associated with lower milling intensity. The CH4 selectivity is closely related to the CO2 released during the methanation step. A higher amount of CO2 was released for the samples based on RuM, and considering the CO2 captured, the RuM samples lost about 5–10% of the CO2 adsorbed, while the RuAc DFMs lost just 3% of it. This was probably linked to a better dispersion of the Ru active sites which covered more efficiently the surface of the DFMs intercepting the release of CO2 from the sorbent sites.
Regarding the CH4 release rate, high values are important because faster DFMs can allow for shorter methanation steps and more efficient capture-conversion cycles. The related main effect plots are shown in Fig. S12.† The RuAc samples were twice as fast compared to the RuM DFMs, with a median of 38 and 18 μmol CH4 gDFM−1 min−1, respectively. Furthermore, faster DFMs were obtained by milling at lower intensities, although this effect is only related to RuAc DFMs. This can be clearly seen in Fig. 6 with the shape of the CH4 transient that flattens in the RuM samples and by increasing the milling regimes. The best performing samples were the ones prepared with RuAc and NaC (5-RuAcNaC/MI386-15, 7-RuAcNaC/MI386-15 and 11-RuAcNaC/MI386-27); the first one, despite producing the lowest amount of CH4 for the RuAc samples (170.8 μmol gDFM−1), released 50% of the total CH4 in the first minute.
The last metric to consider is the unwanted production of CO during the CO2 capture step. This phenomenon is often linked to the presence of reducing species (either H2 or oxygen vacancies, Ov) on the DFM's surface coming from the prior reduction step. A low amount of CO is desirable, as this molecule is a pollutant itself. Statistically, low significance was obtained for this metric for each milling parameter (Table S9†), although the smallest p-value of 0.20 was associated with both the Ru precursor and the two-way interaction between Na and Ru precursors. Indeed, by looking at Fig. S13,† the use of RuAc resulted in lower amounts of CO during the capture step compared to the use of RuM. As the ANOVA analysis suggested, there is a combination between Ru and Na precursors, in particular coupling RuAc and Na2CO3, resulting in 25% less CO release compared to samples based on RuAc and NaNO3, while no differences are observed in the RuM samples.
Overall, the best performing DFM was the sample 11-RuAcNaC/MI386-27. This sample showed both the highest CO2 capture capacity and CH4 production (respectively 420.6 and 321.8 μmol gDFM−1), with a rather fast and sustained CH4 production rate. The combination of RuAc and Na2CO3 also resulted in lower amounts of CO produced during the capture phase and the least CO2 lost at the beginning of the methanation step.
To compare the overall performance of the DFMs studied here, radar plots summarising all main activity indicators are represented in Fig. 8, where a larger area corresponds to better global performance. The F and G axes, corresponding to the CO and CO2 releases, are inverted, as lower values are preferred. The green dashed line represents the sample 11-RuAcNaC/MI386-27 as a comparison with the other DFM subgroups.
 | ||
| Fig. 8 Radar plots showing the performance indicators used to evaluate the DFMs. A = CO2 capture capacity (μmol gDFM−1), B = CH4 production (μmol gDFM−1), C = CO2 conversion (%), D = CH4 selectivity (%), E = CH4 release rate (μmol gDFM−1 min−1), F = CO released during the capture (μmol gDFM−1), and G = CO2 released during the methanation step. The green dashed line represents the sample 11-RuAcNaC/MI386-27 as a comparison with the other DFM subgroups. | ||
Post-test characterisation and activity correlation
The textural and morphological properties of the post-reaction samples were analysed in order to understand the most important factors leading to the differences in reactivity observed during ICCU-MET tests and thus their correlation with the milling parameters.First, BET and XRD analyses were carried out to investigate changes in the DFM structure. The evolution of the surface area and the pore structure at different stages of the study is reported in Table 3 for the samples based on RuAc, which are also considered representative of the ones prepared with RuM. As previously observed (Table S1†), with increasing milling intensity a lower surface area was obtained for the samples based on both Na2CO3 and NaNO3 due to the structural modifications induced by stronger mechanical forces. After reduction, the surface area of 5-RuAcNaC/MI386-15 remained unchanged, while it decreased for the two Na2CO3-based DFMs milled at higher intensities (samples 11-RuAcNaC/MI386-27 and 6-RuAcNaC/MI386-35). This can be explained by the lower degree of Na2CO3 decomposition observed at the low milling intensity, which resulted in smaller modifications of the sample. Regarding the NaNO3 samples, for all the DFMs the area increased after the reduction treatment, likely due to the increase in the size and volume of the pores resulting from the evolution of NOx species. After ICCU-cycling tests, all the spent DFMs showed a lower surface area compared to the activated (reduced) DFMs, probably due to the dynamic surface reorganisation occurring under switching oxidising/reducing atmospheres.
| Sample | As-prepareda | Reduced | Spent |
|---|---|---|---|
| a reported BET values of the NaC/MI386 and NaN/MI386 samples, to avoid RuAc decomposition during the outgassing pretreatment. | |||
| 5-RuAcNaC/MI386-15 | 138 | 139 | 137 |
| 11-RuAcNaC/MI386-27 | 120 | 102 | 80 |
| 6-RuAcNaC/MI386-35 | 73 | 70 | 61 |
| 7-RuAcNaN/MI386-15 | 120 | 138 | 112 |
| 12-RuAcNaN/MI386-27 | 77 | 89 | 72 |
| 8-RuAcNaN/MI386-35 | 34 | 78 | 65 |
It is interesting to notice that the samples with higher CO2 capture capacity, namely those milled at higher intensities, exhibited lower surface areas, suggesting that the chemical interaction between Na and the Al2O3 surface plays a key role in the CO2 capture rather than the surface area itself. Furthermore, the decrease in surface area can partially explain the slower reactivity of the Ru species observed with increasing milling intensity, as the Ru species are probably less dispersed.
Thus, XRD analysis was carried out to investigate the structure of the spent DFMs, including the sorbent, support, and ruthenium sites. The full diffractograms are reported in Fig. S14,† while the detailed 35–50° 2θ range is reported in Fig. 9. Compared with the XRD of the as-prepared DFMs in Fig. 2 and S4,† in general for the Na2CO3-based DFMs the intensity of the Na2CO3 pattern decreased after the ICCU-MET cyclic testing, especially for the samples milled at medium-high intensity, while the two samples milled at the lowest intensity still showed a similar intensity compared to the as-prepared ones. This is in agreement with the TGA results shown in Fig. 4 where the samples milled at the lowest intensity experienced a lower weight loss at 400 °C in H2, possibly due to an insufficient mechanical activation and decomposition of the Na2CO3 precursor. Furthermore, the Na2CO3 species observed in the spent NaC-DFMs correspond to natrite, a monoclinic dehydrated form of Na2CO3 (PDF: 00-037-0451). The samples based on NaNO3 instead showed the complete decomposition of the nitrate precursor and the appearance of a weak peak at 20.5°, which could be attributed to the formation of NaAlOx species, although other peaks could not be clearly detected at these concentrations. For some NaN-DFMs, it was also possible to see weak peaks corresponding to residual Na2CO3 formed during the CO2 capture.
 | ||
| Fig. 9 XRD diffractograms of the spent DFMs: (a) Na2CO3-based DFMs and (b) NaNO3-based DFMs. | ||
Considering the Ru species, on the as-prepared samples no metallic Ru or RuO2 reflections could be identified, inferring the presence of highly dispersed Ru species (Fig. 2). After the ICCU-MET testing, all six RuAc samples did not show any Ru-related phases suggesting that they maintained a highly dispersed Ru phase. The samples based on RuM, instead, showed a clear peak centred at 44.0°, which corresponds to the main peak of metallic Ru (PDF: 01-088-1734). The Ru crystallite size, estimated with the Scherrer equation after the deconvolution of the complex peak structure (see Fig. S15†), ranged from 11 to 17 nm. Within this subgroup, no correlation was found between the Ru crystallite and the performance of the DFMs (CO production and CH4 selectivity). However, the absence of the Ru metal peak in the as-prepared DFMs and the rather large crystallite size identified after the ICCU-MET testing suggest that the RuM precursor is not stable on the surface of the DFMs, regardless of the milling intensity, leading to the sintering of the Ru active sites. This could explain the better performance of the RuAc DFMs, which in turn showed high Ru dispersion even after the ICCU-MET tests. The high Ru dispersion coupled with the enhanced CO2 sorption capacity of Na2CO3-based samples is likely the key for the peak in activity exhibited by RuAcNaC/MI386 samples, especially when milled under intermediate energy conditions (sample 11).
Conclusions
In this work, the mechanochemical preparation of RuNa/Al2O3 dual-function materials for integrated CO2 capture and conversion to CH4 was investigated based on a full factorial design, with the aim of rationally analysing the effect of different milling parameters, such as the milling intensity and the choice of Ru and Na precursors, on the DFM performance.Seven performance indicators were chosen as representatives of the complex DFM behaviour, i.e., the CO2 capture capacity, the CH4 production, the CH4 release rate and selectivity, and the CO and CO2 release during the CO2 capture and the methanation step. The statistical analysis of the obtained results highlighted that all the milling parameters affected the DFMs' properties to a certain extent, but the strongest effects were observed specifically for the milling intensity on CO2 capture capacity and Na and Ru precursors on methanation.
In fact, higher milling intensities were associated with improved CO2 capture capacity by creating stronger chemical interactions between Na and Al2O3, while the combination of Na2CO3 and Ru acetylacetonate resulted in faster DFMs with higher CH4 production and selectivity and a decreased CO release during the CO2 capture step. The use of RuAc also resulted in smaller and more stable Ru nanoparticles, likely contributing to the improved CO2 conversion performance compared to the Ru metal precursor. It clearly appears that an interplay between all the components of the DFMs is crucial, with a balance between the easier Na2CO3 formation and decomposition, achieved at medium-high milling regimes, and the formation of rapid and active Ru sites obtained under milder milling conditions. The DoE approach allowed the isolation of the main effects of the chosen milling parameters on optimal DFM performance, paving the way for the rational preparation of DFMs by mechanochemistry. Nonetheless, further characterisation will be needed to understand in detail the structure and reactivity of these DFMs, as the complex structure is not easily characterised and the structure–activity relationships are not straightforward, as evinced by the fact that higher CO2 capture is associated with a lower surface area.
Data availability
Data for this article, including original data for all main and supplementary figures, are available at Zenodo at https://doi.org/10.5281/zenodo.14415918.Author contributions
A. B.: conceptualization; investigation; methodology; data curation; formal analysis; validation; visualization; writing – original draft. M. D.: conceptualization; investigation; methodology; validation, visualization; supervision; writing – review & editing. S. C.: funding acquisition; resources; supervision; writing – review & editing. A. T.: funding acquisition; resources; supervision; project administration; writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was carried out within the Interconnected Nord-Est Innovation Ecosystem (iNEST) and received funding from the European Union Next-GenerationEU (PIANO NAZIONALE DI RIPRESA E RESILIENZA (PNRR) – MISSIONE 4 COMPONENTE 2, INVESTIMENTO 1.5 – D.D. 1058 23/06/2022, ECS00000043, CUP: G23C22001130006). M.D. is grateful for funding under the REACT EU Italian PON 2014–2020 Program – Action IV.4 – Innovation (DM 1062, 10/08/2021, CUP G51B21006040007).Notes and references
- W. Gao, S. Liang, R. Wang, Q. Jiang, Y. Zhang, Q. Zheng, B. Xie, C. Y. Toe, X. Zhu, J. Wang, L. Huang, Y. Gao, Z. Wang, C. Jo, Q. Wang, L. Wang, Y. Liu, B. Louis, J. Scott, A. C. Roger, R. Amal, H. He and S. E. Park, Chem. Soc. Rev., 2020, 49, 8584–8686 RSC.
- I. S. Omodolor, H. O. Otor, J. A. Andonegui, B. J. Allen and A. C. Alba-Rubio, Ind. Eng. Chem. Res., 2020, 59, 17612–17631 CrossRef CAS.
- X. G. Zhang, A. Buthiyappan, J. Jewaratnam, H. S. C. Metselaar and A. A. A. Raman, J. Environ. Chem. Eng., 2024, 12, 111799 CrossRef CAS.
- D. J. Heldebrant, J. Kothandaraman, N. Mac Dowell and L. Brickett, Chem. Sci., 2022, 13, 6445–6456 RSC.
- Y. Hu, W. Liu, H. Chen, Z. Zhou, W. Wang, J. Sun, X. Yang, X. Li and M. Xu, Fuel, 2016, 181, 199–206 CrossRef CAS.
- S. Sun, H. Sun, P. T. Williams and C. Wu, Sustainable Energy Fuels, 2021, 5, 4546–4559 RSC.
- P. Melo Bravo and D. P. Debecker, Waste Disposal Sustainable Energy, 2019, 1, 53–65 CrossRef.
- A. Porta, R. Matarrese, C. G. Visconti, L. Castoldi and L. Lietti, Ind. Eng. Chem. Res., 2021, 60, 6706–6718 CrossRef CAS.
- Y. Zhang, S. Zhao, L. Li, J. Feng, K. Li, Z. Huang and H. Lin, Catal. Sci. Technol., 2024, 14, 790–819 RSC.
- L. P. Merkouri, T. Ramirez Reina and M. S. Duyar, Nanoscale, 2022, 14, 12620–12637 RSC.
- A. Bermejo-López, B. Pereda-Ayo, J. A. González-Marcos and J. R. González-Velasco, Sustainable Energy Fuels, 2021, 5, 1194–1210 RSC.
- A. Bermejo-López, B. Pereda-Ayo, J. A. Onrubia-Calvo, J. A. González-Marcos and J. R. González-Velasco, J. CO2 Util., 2022, 58, 1–11 CrossRef.
- S. Cimino, E. M. Cepollaro and L. Lisi, Appl. Catal., B, 2022, 317, 121705 CrossRef CAS.
- M. A. Arellano-Treviño, N. Kanani, C. W. Jeong-Potter and R. J. Farrauto, Chem. Eng. J., 2019, 375, 121953 CrossRef.
- K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145–2162 CrossRef CAS PubMed.
- P. Baláž, M. Achimovicová, M. Baláž, P. Billik, C. Z. Zara, J. M. Criado, F. Delogu, E. Dutková, E. Gaffet, F. J. Gotor, R. Kumar, I. Mitov, T. Rojac, M. Senna, A. Streletskii and W. C. Krystyna, Chem. Soc. Rev., 2013, 42, 7571–7637 RSC.
- E. Colacino, V. Isoni, D. Crawford and F. García, Trends Chem., 2021, 3, 335–339 CrossRef CAS.
- A. P. Amrute, J. De Bellis, M. Felderhoff and F. Schüth, Chem.–Eur. J., 2021, 27, 6819–6847 CrossRef CAS PubMed.
- M. Danielis, S. Colussi, C. de Leitenburg, L. Soler, J. Llorca, A. Trovarelli, C. de Leitenburg, L. Soler, J. Llorca and A. Trovarelli, Angew. Chem., Int. Ed., 2018, 57, 10212–10216 CrossRef CAS PubMed.
- A. Braga, M. Armengol-Profitós, L. Pascua-Solé, X. Vendrell, L. Soler, I. Serrano, I. J. Villar-Garcia, V. Pérez-Dieste, N. J. Divins and J. Llorca, ACS Appl. Nano Mater., 2023, 6, 7173–7185 CrossRef CAS PubMed.
- N. J. Divins, A. Braga, X. Vendrell, I. Serrano, X. Garcia, L. Soler, I. Lucentini, M. Danielis, A. Mussio, S. Colussi, I. J. Villar-Garcia, C. Escudero, A. Trovarelli and J. Llorca, Nat. Commun., 2022, 13, 1–11 Search PubMed.
- J. De Bellis, H. Petersen, J. Ternieden, N. Pfänder, C. Weidenthaler and F. Schüth, Angew. Chem., Int. Ed., 2022, 61, e202208016 CrossRef CAS PubMed.
- X. Liu, Y. Li, L. Zeng, X. Li, N. Chen, S. Bai, H. He, Q. Wang and C. Zhang, Adv. Mater., 2022, 34, 1–30 Search PubMed.
- L. Yang, Z. Pan and Z. Tian, ChemCatChem, 2024, 16, e202301519 CrossRef CAS.
- M. Danielis, L. P. Merkouri, A. Braga, A. Trovarelli, M. S. Duyar and S. Colussi, J. CO2 Util., 2024, 86, 102895 CrossRef CAS.
- S. Wang, E. T. Schrunk, H. Mahajan and R. J. Farrauto, Catalysts, 2017, 7, 1–13 Search PubMed.
- H. Liu, L. Cen, X. Xie, L. Liu, Z. Sun and Z. Sun, J. Energy Chem., 2025, 100, 779–791 CrossRef CAS.
- M. T. Dunstan, F. Donat, A. H. Bork, C. P. Grey and C. R. Müller, Chem. Rev., 2021, 121, 12681–12745 CrossRef CAS PubMed.
- A. I. Tsiotsias, A. G. Georgiadis, N. D. Charisiou, A. G. S. Hussien, A. A. Dabbawala, K. Polychronopoulou and M. A. Goula, ACS Omega, 2024, 9, 11305–11320 CrossRef CAS PubMed.
- M. Danielis, S. Colussi, C. de Leitenburg and A. Trovarelli, Catal. Commun., 2020, 135, 105899 CrossRef CAS.
- M. Danielis, A. Braga, N. J. Divins, J. Llorca, A. Trovarelli and S. Colussi, Crystals, 2023, 13, 1–16 CrossRef.
- R. Leardi, Anal. Chim. Acta, 2009, 652, 161–172 CrossRef CAS PubMed.
- M. Gorbounov, J. Taylor, B. Petrovic and S. Masoudi Soltani, S. Afr. J. Chem. Eng., 2022, 41, 111–128 Search PubMed.
- A. L. Patterson, Phys. Rev., 1939, 56, 978–982 CrossRef CAS.
- T. S. Nguyen, L. Lefferts, K. B. Saisankargupta and K. Seshan, ChemCatChem, 2015, 7, 1833–1840 CrossRef CAS.
- C. J. Keturakis, F. Ni, M. Spicer, M. G. Beaver, H. S. Caram and I. E. Wachs, ChemSusChem, 2014, 7, 3459–3466 CrossRef CAS PubMed.
- A. Bermejo-López, B. Pereda-Ayo, J. A. González-Marcos and J. R. González-Velasco, Appl. Catal., B, 2019, 256, 117845 CrossRef.
- S. Sun, Y. Zhang, C. Li, Y. Wang, C. Zhang, X. Zhao, H. Sun and C. Wu, Sep. Purif. Technol., 2023, 308, 122956 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mr00147h |
| This journal is © The Royal Society of Chemistry 2025 |