
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudying the synthesis, antimicrobial activity, and phenol red removal of gelatin-stabilized copper nanoparticles
Trung Dien
Nguyen
 *,
Sang Thanh
Ngo
,
Yen Hai
Hoang
,
Nhung Thi Tuyet
Thai
,
Huong Thi Thu
Nguyen
and
Gia Thi Ngoc
Trinh
*,
Sang Thanh
Ngo
,
Yen Hai
Hoang
,
Nhung Thi Tuyet
Thai
,
Huong Thi Thu
Nguyen
and
Gia Thi Ngoc
Trinh
Can Tho University, 3/2 Street, Ninh Kieu, Can Tho, 94000, Vietnam. E-mail: ndtrung@ctu.edu.vn
First published on 19th November 2024
Abstract
This study presents a synthesis method for environmentally friendly copper nanoparticles using ascorbic acid and gelatin as key components. The influence of precursor concentration, reductant amount, and stabilizer on the process was systematically investigated to obtain optimal results for the synthesis. The optimal parameters for forming copper nanoparticles, including 20 g per L gelatin, 19.3 mM (AcO)2Cu, and 41.5 mM ascorbic acid, were determined using a central composite design of the response surface methodology. Successful generation of pure copper nanoparticles with both spherical and cylindrical shapes, whose sizes were 43.1 and 105.2 nm, respectively, was confirmed by X-ray diffraction analysis and transmission electron microscopy. The synthesized nanomaterial was stable for a two-week storage time after which they gradually oxidized into Cu2+ ions. During antimicrobial activity testing, the synthesized nanoparticles displayed distinctive ability to inhibit the growth of Gram-positive bacteria (Lactobacillus fermentum, Bacillus subtilis, and Staphylococcus aureus), Gram-negative bacteria (Escherichia coli), and cancer cells (A549, Hep-G2, KB, and MCF7). Copper nanoparticles synthesized by chemical reduction demonstrated notable inhibitory activity against various pathogenic fungi that affect plants, including Fusarium solani, Rhizoctonia solani, and Colletotrichum gloeosporioides. Additionally, the catalytic activity of the produced nanomaterial with a bandgap energy of 2.14 eV and a specific surface area of 40.6 m2 g−1 was explored in the degradation of phenol, a common dye used in laboratories and industries. An optimized phenol red removal of 94.4% was achieved after a 540 second reaction time using response surface methodology, specifically a central composite design with an optimal dosage of copper nanoparticles at 31.5 ppm, a NaBH4 concentration of 53.1 mM, and a pH of 7.5.
1. Introduction
Nanotechnology has played a significant role in research and development, particularly metallic nanoparticles. Noble metals like gold and silver have been extensively studied for their potential applications in areas such as antimicrobial activity,1–3 catalysis,4–6 and medicine.7,8 Besides them, copper nanoparticles (CuNPs) have emerged as a cheap promising alternative with comparable or even superior properties. CuNPs have demonstrated excellent antifungal and antibacterial effects against common pathogens,9,10 as well as efficient catalytic ability in organic chemical processes.11,12 Due to their affordability, non-toxicity, and a large surface area, CuNP materials have been extensively applied in the fields of biomedicine and catalysis.13–15 As a result, the synthesis of CuNPs has recently attracted considerable research attention. Diverse CuNP synthesis methods have been explored, including physical, chemical, and biological approaches.16 Biological methods demand extensive time and effort, while physical methods can be cost-prohibitive. Therefore, the chemical method has emerged as a more effective and cost-efficient option for CuNP production.17 However, surmounting challenges like the oxidation susceptibility of pure CuNPs and high-energy nanoparticle aggregation is vital for synthesizing highly pure, stable CuNPs, unlocking their multifaceted potential. To address these issues, ascorbic acid and gelatin were chosen as non-toxic agents in CuNP synthesis by chemical reduction. Ascorbic acid, found in various fruits and vegetables, possesses excellent antioxidant properties and acts as a reducing agent.18,19 Gelatin, derived from collagen obtained from different sources, offers ecological advantages and strong binding capabilities.20 The successful and enduring synthesis and stabilization of CuNPs have been achieved through effective utilization of ascorbic acid and gelatin together, capitalizing on their beneficial properties and synergistic impacts.21 Traditional chemical reduction utilized toxic chemical agents, complex procedures, and high energy consumption.22–24 Conversely, incorporating green components in synthesizing CuNPs offered several benefits regarding efficiency and environmental impact. Green-synthesis methods typically use non-toxic, renewable resources and eco-friendly conditions, resulting in fewer harmful by-products and lower energy consumption. These methods also enhanced efficiency by simplifying the synthesis process and reducing the need for extensive purification steps.CuNPs are recognized for extensive applications across various fields, especially in the realms of biological and catalytic applications.25 Their antimicrobial properties have been extensively studied, demonstrating the potential for combating microbial infections.26,27 Additionally, ongoing studies suggest promising biological properties of CuNPs that may extend to potential applications in treating numerous cancer cell lines.28,29 These research studies underscore the broad applications and favorable biological attributes. This prompts the notion of assessing our CuNPs against microbial infections and their potential in treating cancer, particularly with more prevalent strains. In addition, CuNPs have been studied for their catalytic activity in the degradation of organic dyes which are commonly used in many industries.30–32 Specifically, phenol red (PR) is a triphenylmethane dye utilized as a pH indicator, particularly in cell biology laboratories, and can be found in industrial effluents from textiles, paper, printing, and leather. Unexpectedly, this dye poses risks to the skin, eye, and respiratory system, potentially becoming carcinogenic upon prolonged or concentrated exposure.33 Given their stability, removing dyes from wastewater is crucial for water pollution control, and eco-friendly nanocatalysts offer a promising solution. Numerous studies have delved into PR degradation, utilizing the catalytic activity of metallic nanoparticles. These investigations often focus on the absence and presence of nanoparticles as homogeneous catalysts, examining PR degradation under varying pH conditions.34–37 However, the impact of factors such as nanoparticle dosages acting as heterogeneous catalysts and their interaction with different reductant quantities remains insufficiently addressed in the previous research.
In light of these statements, our study provided a straightforward and eco-friendly CuNP synthesis approach, stabilizing the nanoparticles with gelatin and reducing the precursor with ascorbic acid. A systematic investigation was conducted into how reductant, stabilizer, and precursor concentrations affected the effectiveness of CuNP synthesis using a response surface design. The critical features of the produced CuNPs such as phase components, morphology, optical properties, and catalytic characteristics were carefully examined. The biological activities of CuNPs were tested on Gram-positive bacteria (Lactobacillus fermentum, Bacillus subtilis, and Staphylococcus aureus), Gram-negative bacteria (Escherichia coli, Pseudomonas aeruginosa, and Salmonella enterica), cancer cells (A549, Hep-G2, KB, and MCF7), and plant pathogenic fungi (Fusarium solani, Rhizoctonia solani, and Colletotrichum gloeosporioides). The catalytic potential of CuNPs has also been focused on the degradation of PR with comprehensively optimizing the catalyst dosage, NaBH4 concentration, and pH of PR solution for PR conversion.
2. Experimental
2.1. Materials
Sigma-Aldrich provided the chemicals that were utilized for the manufacture and analysis of the characteristics of CuNPs. Copper(II) acetate monohydrate (Cu(OAc)2·H2O), L-ascorbic acid (C6H8O6), and gelatin were employed as the precursor, reductant, and stabilizer to produce CuNPs, respectively. For investigating the photocatalytic activity, phenol red (C19H14O5S), sodium borohydride (NaBH4), and a buffer solution pH 10 were used to measure effective conversion. The point of zero charge of the as-prepared nanomaterial was determined by adjusting the pH of hydrochloric acid (HCl), sodium chloride (NaCl), and sodium hydroxide (NaOH). Potato dextrose agar and agar ((C12H18O9)n) were employed for treating fungi. Dimethyl sulfoxide (DMSO, C2H6OS), L-glutamine (C5H10N2O3), sodium bicarbonate (NaHCO3), glucose (C6H12O6), HEPES (C8H18N2O4S), sodium pyruvate (C3H3NaO3), and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT, C18H16BrN5S) were used for evaluating the anticancer activity of the synthesized CuNPs-G against human breast cancer cell lines. Ampicillin, cefotaxime, and ellipticine were used as control samples for testing the antimicrobial activity.2.2. Synthesis of CuNPs-G
The formation of CuNPs involved major components, including stabilizers, precursors, and reductants, playing a crucial role in determining the size, shape, stability, and overall quality of the produced nanoparticles. The precursor provides the copper ions necessary for nanoparticle formation and influences nucleation and growth processes. The reductant donates electrons to reduce copper ions to metallic copper and control the reduction reaction rate, impacting the distribution size of nanoparticles. The stabilizer prevents aggregation and controls the nucleation and growth rate, ensuring the production of uniformly sized and shaped nanoparticles. This study investigated the effects of the precursor Cu(AcO)2, the reducing agent ascorbic acid, and the stabilizing agent gelatin on the CuNP formation. The synthesis procedure is described in detail in the following.Gelatin was dissolved in water with continuous magnetic stirring and heating until complete dissolution on a magnetic stirrer at a speed of 1000 rpm at 60 °C for 60 min. Subsequently, (AcO)2Cu·H2O was added to the resulting solution to obtain a homogeneous blue-green solution. Ascorbic acid was then introduced to reduce Cu2+ ions to nanoparticles, and the reaction occurred for 30 min. Three continuous factors were examined by varying gelatin concentrations (X1 = 5, 12.5, and 20 g L−1), (AcO)2Cu concentrations (X2 = 10, 20, and 30 mM), and ascorbic acid concentrations (X3 = 30, 45, and 60 mM), with the response variables being maximum absorbance (Y1) and minimized surface plasmon resonance (SPR) (Y2).
A response surface design (RSD) was used to estimate the interactions of separate variables with the response variables. The central composite full (CCF) design was used to optimize experimental conditions by considering factors and interactions. A randomized experimentation of 20 runs was considered with six center points: X1 = 12.5 g L−1, X2 = 20 mM, and X3 = 45 mM. The values of factors for producing CuNPs are listed in Table 1. Model terms including linear, squares, and interactions were used to describe the influence of synthesis parameters (X1, X2, and X3) on the response variables (Y1 and Y2) corresponding to formula (1).
| Yi = α0 + ∑αiXi + ∑αiiXi2 + ∑αijXiXj | (1) |
| Run order | X 1, g L−1 | X 2, mM | X 3, mM | Y 1 | Y 2, nm |
|---|---|---|---|---|---|
| 1 | 20 | 30 | 60 | 0.785 | 575.6 |
| 2 | 12.5 | 20 | 45 | 1.292 | 574.6 |
| 3 | 5 | 10 | 30 | 0.245 | 573.6 |
| 4 | 12.5 | 20 | 45 | 1.272 | 574.6 |
| 5 | 5 | 20 | 45 | 1.055 | 575.6 |
| 6 | 12.5 | 20 | 45 | 1.279 | 574.6 |
| 7 | 5 | 10 | 60 | 0.138 | 568.8 |
| 8 | 12.5 | 20 | 45 | 1.283 | 574.6 |
| 9 | 20 | 10 | 60 | 0.213 | 568.4 |
| 10 | 20 | 10 | 30 | 0.492 | 570.8 |
| 11 | 5 | 30 | 30 | 0.816 | 581.8 |
| 12 | 12.5 | 20 | 45 | 1.301 | 574.4 |
| 13 | 12.5 | 20 | 30 | 1.325 | 577.2 |
| 14 | 12.5 | 10 | 45 | 0.344 | 571.6 |
| 15 | 20 | 20 | 45 | 1.281 | 571.4 |
| 16 | 12.5 | 20 | 45 | 1.276 | 574.6 |
| 17 | 12.5 | 30 | 45 | 0.982 | 579.0 |
| 18 | 20 | 30 | 30 | 1.469 | 576.8 |
| 19 | 12.5 | 20 | 60 | 0.857 | 573.8 |
| 20 | 5 | 30 | 60 | 0.643 | 579.8 |
The appropriateness of the generated models was estimated from the coefficient of determination (indicated by R-square, R2) and probability value p. The values were considered to be statistically meaningful for the models with p < 0.05 and R2 approximately approaching 1. Optimized responses were utilized to determine the optimal values (X1, X2, and X3) for CuNP synthesis, reaching the highest value of Y1 and the lowest value of Y2. Experiment design, model generation, and optimization were performed by manipulating the OriginPro 2024 software. CuNPs manufactured under optimal operating conditions were denoted as CuNPs-G. Additionally, CuNPs synthesized using the CuNPs-G process but without the presence of gelatin were designated as CuNPs-wG for comparison of antibacterial and cell inhibition activities with the CuNPs-G sample.
2.3. Characteristics of CuNPs-G
To monitor the formation and stability of copper nanoparticles, UV-vis spectrophotometry was performed, specifically using a V730 model from Jasco with a double-beam spectrophotometer. The instrument used a 1 cm quartz cell to hold the samples during analysis. The wavelength range used for measurement was 500–700 nm. The scanning speed was set to 1000 nm min−1 with a data increment of 0.2 nm. For morphological analysis, transmission electron microscopy (TEM) was employed using a JEM-1400 instrument from JEOL, which allowed for detailed morphological analysis and particle size distribution data of the synthesized CuNPs-G. The test sample containing CuNPs-G was deposited onto a 300-mesh carbon-coated copper grid. This grid served as the substrate for the nanoparticles during TEM imaging. The TEM images were captured using a JEM-2100 instrument from JEOL at an acceleration voltage of 200 kV. To calculate the size distribution of the generated CuNPs-G, both ImageJ and OriginPro 2024 were employed. Using a Thermo Nicolet 6700 Fourier transform infrared (FTIR) spectrometer in the wavenumber range of 4000–400 cm−1 enabled detailed characterization of the functional groups present on the nanomaterial surface. The zeta potential (ZP) of the formed CuNPs-G was measured using an SZ-100 nanoparticle analyzer at 25 °C. The mean ZP value of the as-synthesized CuNPs-G was measured using a Malvern Zetasizer Nano ZS. The isoelectric point charge (PZC) of CuNPs-G was controlled by acid–base titration. 100 mL solutions with pH of approximately 2, 4, 6, 8, 10, and 12 were prepared from 0.1 M solutions of NaOH, NaCl, and HCl. The prepared solutions (denoted as pHi) were introduced into a beaker containing 0.10 g of CuNPs-G and magnetically stirred for 48 h. The suspensions were filtered to obtain transparent solutions and, then the pH values (designated as pHf) were recorded. The PZC value of CuNPs-G was determined by the dependence of pHi on (pHf − pHi). The pH values of the solutions were measured using a Horiba pH meter F-71. The crystalline phase of the synthesized nanomaterial was analyzed using a Bruker D2 Phaser X-ray diffractometer with Cu Kα radiation operated at 40 mA and 40 kV. The XRD measurements covered a range of 2θ angles from 10 to 80°. The estimation of the average crystallite size of the as-synthesized CuNPs-G based on the broadening of diffraction peaks in the XRD pattern was accomplished using Debye Scherrer's formula (2). | (2) |
To evaluate the synthesis efficiency, the concentration of Cu2+ ions in the samples was determined using inductively coupled plasma optical emission spectroscopy (ICP-OES). The estimation included the initial solution consisting solely of the precursor, 19.3 mM (AcO)2Cu, and the resulting CuNPs-G. Prior to analysis, CuNPs-G synthesized using optimal parameters underwent centrifugation at 6800 rpm for a 5 h period to remove solid components. The synthesis efficiency, expressed as H, was calculated by applying formula (3).
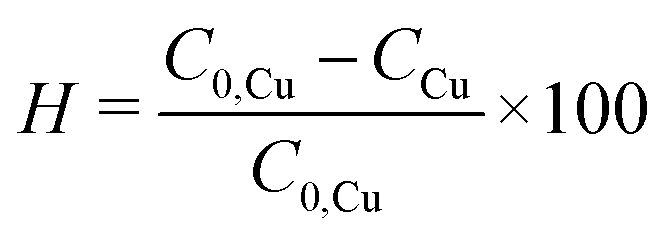 | (3) |
Nitrogen adsorption–desorption isotherms were recorded using a Nova 2200e instrument. The specific surface area of CuNPs-G was calculated according to the Brunauer–Emmett–Teller (BET) nitrogen adsorption isotherms. The measurement of the absorption wavelength was conducted through utilization of diffuse reflectance spectroscopy (DRS) on a Cary 5000 UV-vis-NIR spectrophotometer. Subsequently, these data were calculated to determine the band gap energy (Eg) of CuNPs-G, employing Tauc's relation, expressed by formula (4).
 | (4) |
To investigate the stability of the synthesized CuNPs-G sample, UV-vis measurements were conducted at different storage times, with weekly intervals. The CuNPs-G sample was stored at room temperature. After one week, UV-vis measurements were used to record variations in absorption values and SPR of CuNPs. The stability assessment experiment continued until the absorption value of CuNPs in the CuNPs-G sample decreased by approximately 30% compared to the initial absorption.
2.4. Antibacterial activity of CuNPs-G
To assess the antibacterial activity of the synthesized CuNPs-G, the well diffusion method was employed to investigate against various bacteria, including Lactobacillus fermentum (L. fermentum) N4, Bacillus subtilis (B. subtilis) ATCC 6633, and Staphylococcus aureus (S. aureus) ATCC 12600 for Gram-positive bacteria, as well as Escherichia coli (E. coli) ATCC 25922, Salmonella enterica (S. enterica) ATCC 35664, and Pseudomonas aeruginosa (P. aeruginosa) ATCC 27853 for Gram-negative bacteria. Following MacFarland's standard guidelines, the pathogenic cultures were subcultured and incubated at 37 °C for 24 h to achieve a concentration of 5 × 105 CFU mL−1. Gram-positive bacteria were exposed to ampicillin as the control, while Gram-negative bacteria were exposed to cefotaxime. The chemical-mediated CuNPs were prepared at four concentrations of 2, 4, 8, and 16 ppm. The concentration at which 50% inhibition was reached, indicated by the IC50 value, was observed for the synthesized CuNPs on 96-well microplates, and the absorbance was measured at a wavelength of 630 nm.2.5. Antiproliferative activity of CuNPs-G on cancer cell lines
The cytotoxicity assay of CuNPs-G was carried out on four cancer cell lines, namely lung cancer A549, hepatic cancer HepG2, epidermal carcinoma KB, and breast cancer MCF7, using the MTT method. The MTT method involved culturing the cancer cell lines in a 96-well plate with a cell density of 5000 cells per well, using DMEM (Dulbecco's modified Eagle's medium) supplemented with the following components: 2 mM L-glutamine, 1.5 g per L sodium bicarbonate, 4.5 g per L glucose, 10 mM HEPES, 1.0 mM sodium pyruvate, and 10% FBS (fetal bovine serum). The culture was incubated at 37 °C with 5% CO2. After 24 h of incubation, the 96-well plate was switched to FBS-free DMEM. CuNPs-G diluted with DMSO solvent at varying concentrations: 2, 4, 8, and 16 ppm, were added to the wells, ensuring that the final concentration of DMSO in the culture medium did not exceed 0.05% to avoid solvent toxicity. After 48 h, 20 μL of MTT solution with a concentration of 2 mg mL−1 was added to each well, followed by a further 4 h incubation period. The surviving cells' cellular enzymes converted MTT to formazan crystals that were insoluble in the culture medium. Subsequently, DMSO was used to dissolve the formazan crystals, and the absorbance was measured at 550 nm. The percentage of surviving cells represented the toxicity of CuNPs-G, with higher toxicity leading to a lower number of surviving cells. The percentage toxicity was determined by the absorbance in the test wells compared to the control wells. In this experiment, ellipticine was used as the control sample.2.6. Antifungal activity of CuNPs-G
The poison plate technique was employed to evaluate the antifungal characteristics of the chemically assisted CuNPs-G. Five concentrations of CuNPs were prepared to assess the antifungal efficacy. For Fusarium solani (F. solani), CuNPs were used at numerous concentrations: 250, 300, 350, 400, and 450 ppm. The values of CuNPs were fixed at 200, 250, 300, 350, and 140 ppm for Colletotrichum gloeosporioides (C. gloeosporioides). Additionally, CuNP concentrations reached 200, 400, 600, 800, and 1000 ppm on samples containing Rhizoctonia solani (R. solani). A solution containing 20 g per L potato dextrose agar and 20 g per L agar in water was subjected to microwave irradiation and used as the culture medium for control samples. The culture medium for the test samples was prepared similarly to the controls but included synthesized CuNPs at various concentrations. Each CuNP concentration was added separately to sterilized Petri plates. Active mycelial growth, measuring 5.0 mm in diameter, was placed at the center of each plate in a sterile laminar flow cabinet. The plates were incubated at 25 °C for a week for F. solani, two weeks for R. solani, and three weeks for C. gloeosporioides. All experiments were conducted five times. The suppression of mycelial growth (S) was calculated using formula (5).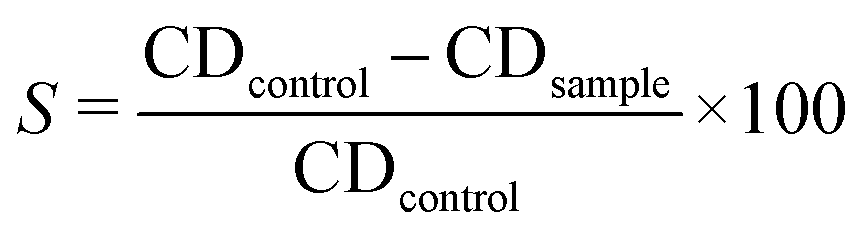 | (5) |
2.7. Photocatalytic activity of CuNPs-G
For preparing the catalyst for PR removal, CuNPs-G was centrifuged at 6800 rpm for 4 h to obtain precipitates. The resulting precipitates were effectively washed with water and ethanol three times, respectively, removing any impurities or contaminants present on the surface and then vacuum dried at 60 °C overnight. The red-brown product prepared by drying in a vacuum oven was used as a catalyst in the reaction for PR removal. The experiments were conducted at 25 °C using a 100 mL solution of 10 ppm PR with a pH of 6–10. The suspension with the presence of CuNPs was stirred for 40 min to establish adsorption/desorption equilibrium. After continuous agitation, varying amounts of NaBH4 reductant were introduced. The reaction process was monitored for 540 s. The reaction solution was then filtered to eliminate CuNPs and analyzed the resulting solution on a UV-visible spectrophotometer at a wavelength of 560 nm. PR removal efficiency (RE) was measured from the PR concentration and calculated using formula (6).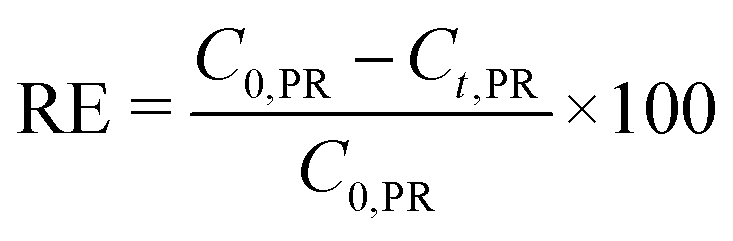 | (6) |
To evaluate the efficiency of PR removal, three operating parameters were varied: pH of PR (X4 = 6, 8, and 10), CuNPs-G dosage (X5 = 10, 25, and 40 ppm), and NaBH4 concentration (X6 = 30, 50, and 70 mM). The response variable measured was PR conversion (Y3). RSD and CCF were designed to calculate the interactions of the separate variables with the response variable. A randomized experiment of 20 runs was considered with six center points: X4 = 8, X5 = 25 ppm, and X6 = 50 mM. The optimal values for investigating the catalytic activity of CuNPs-G are listed in Table 3. Model terms including main and quadratic were employed to illustrate the influence of X4, X5, and X6 on Y3, according to formula (1).
2.8. Data analysis
The statistical analysis for bacteria, cancer cells, and fungi was performed using Descriptive Statistics in Excel with the Data Analysis ToolPak. The resulting data were expressed as the mean value ± standard deviation of triplicate measurements for bacteria and cancer cells and from five individual experiments for fungi. Significance levels were tested at p < 0.05. Similar to the investigation of antibacterial, antifungal, and cancer cell inhibition, the values obtained from the central composite design using the response surface design for CuNPs-G synthesis as well as PR degradation were significant when p was less than 0.05 and R2 approached approximately 1.3. Results and discussion
3.1. Impact of operating factors on CuNPs-G formation
The mechanism for forming CuNPs with the reducing agent N2H4 in polyethylene terephthalate was proposed. The obtained findings indicated that ultrasonic waves are more effective than traditional heating methods in facilitating the CuNP formation which relied on generating reactive radicals.38 In this study, the proposed formation of CuNPs used ascorbic acid as a reducing agent and gelatin as a stabilizing agent. Initially, a complex was formed between Cu2+ and gelatin.21,39 This was followed by the reduction of Cu2+ to Cu0 within the gelatin–Cu2+ complex. The reduction process occurred in two stages.40 The first stage involved the formation of intermediate ascorbate radicals and electrons that reduced gelatin–Cu2+ to gelatin–Cu+. Subsequently, another electron led to the formation of dehydroascorbic acid and further reduced gelatin–Cu+ to gelatin–Cu0. The chemical reduction mechanism involving ascorbic acid and gelatin is depicted in Fig. 1. | ||
| Fig. 1 The mechanism of CuNP formation from ascorbic acid and gelatin. | ||
The CuNP formation was determined by observing the UV-vis spectrum in the wavelength range from 530 to 590 nm.41 Metal nanoparticles with small size exhibited a low wavelength-shifted SPR, while large-sized metal nanoparticles showed a higher wavelength-shifted SPR. In addition, the absorption intensity is proportional to the concentration of metal nanoparticles formed in the solution.42Table 1 presents the dependent variable values, including the absorbance Y1 and surface plasmon resonance peak Y2, obtained from experiments investigating independent variables, including gelatin, Cu(AcO)2, and ascorbic acid. The values of Y1 in the experimental samples were recorded in the range from 0.138 to 1.469, and values of Y2 ranged from 568.4 to 581.8 nm. Fig. 2 shows the response surface plot of the influence of the precursor, stabilizer, and reducing agent on the absorbance and position of the surface plasmon resonance peak.
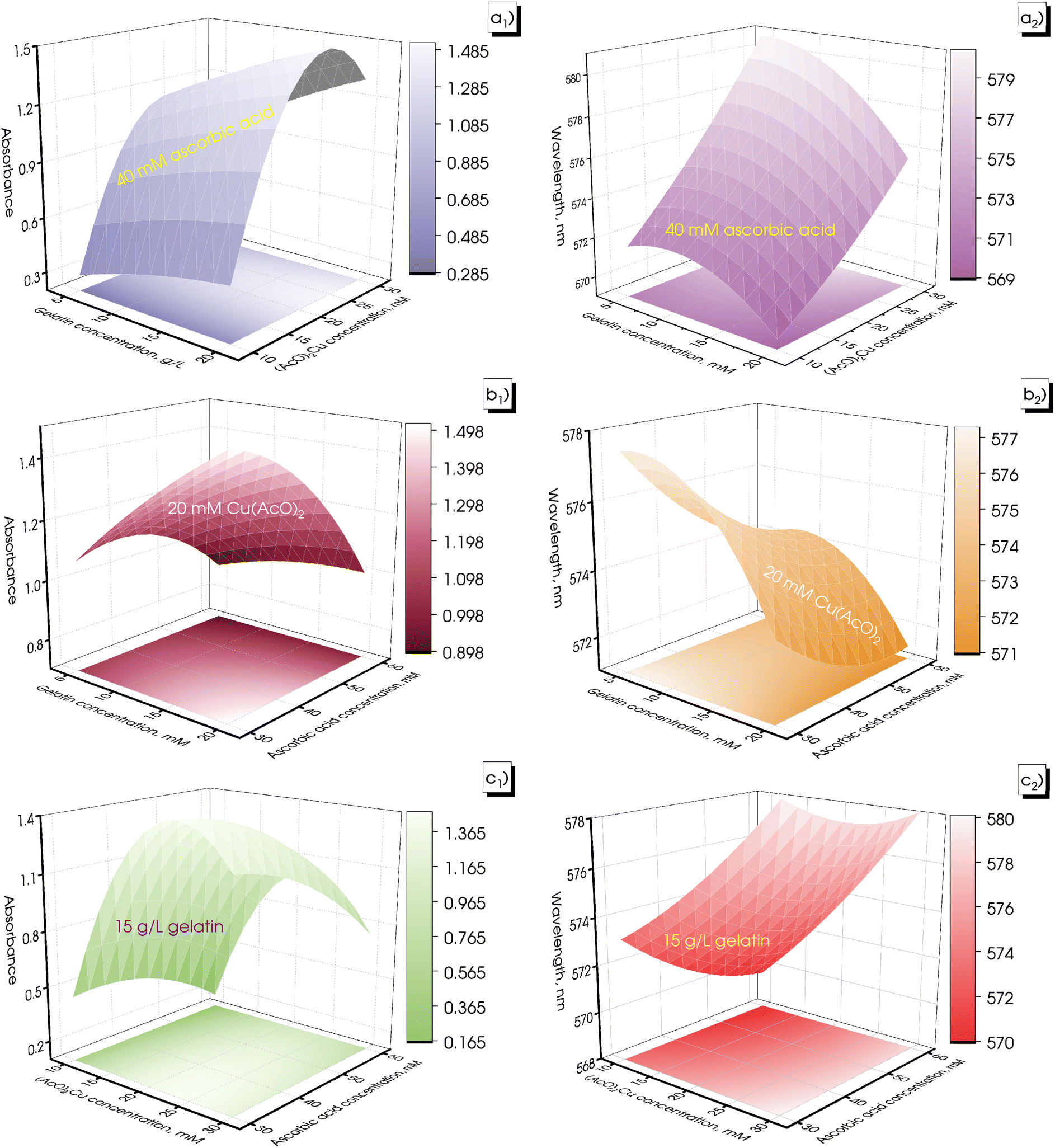 | ||
| Fig. 2 Surface plot with projection for the absorbance (a1–c1) and SPR (a2–c2) in the green-synthesized CuNPs at numerous extract volumes and (AcO)2Cu concentrations. | ||
With the increase of the gelatin concentration from 5 to 20 g L−1, absorbance of the as-synthesized samples increased with a decrease in the value of SPR, as illustrated in Fig. 2a1, a2, b1, and b2. This demonstrates that high gelatin concentration is beneficial for material formation with numerous small-sized CuNPs. The gelatin stabilizer on the surface of CuNPs played an important role in limiting agglomeration to develop large particles as well as protecting the resulting CuNPs from oxidation in the presence of air. At a low gelatin concentration, the stabilizing role was ineffective, and then the particles were easy to agglomerate due to the lack of a stabilizing agent. However, using a high gelatin concentration increased the osmotic pressure of the colloidal solution, causing CuNPs to agglomerate more easily.43
Fig. 2a1, a2, c1, and c2 illustrate the influence of precursor concentration on Y1 and Y2 values. With the increase of the Cu(AcO)2 precursor concentration from 10 to 30 mM, the obtained Y1 and Y2 values also increased. Typically, the precursor concentration greatly affected the size of the formed nanoparticles as well as increasing the number of the produced nanoparticles.16 It is more difficult to produce well-dispersed nanoparticles with high precursor concentrations due to obstacles of precursor solubilization, which reduced the amount of the formed product. On the other hand, particle agglomeration was observed at high concentrations of precursor, causing a shift to higher wavelengths.44
In the present study, ascorbic acid concentrations were investigated at values ranging from 30 to 60 mM. There was a gradual decrease in SPR of the synthesized samples corresponding to increasing ascorbic acid concentration, as illustrated in Fig. 2b1, b2, c1, and c2. However, the absorbance values of the investigated samples only increased when the ascorbic acid concentration increased from 30 to 45 mM, then gradually decreased with the further increase of ascorbic acid concentration to 60 mM. In the CuNP synthesis reaction, ascorbic acid acted as a reducing agent as well as an effective protective agent to inhibit the CuNPs from being oxidized.45 As the reducing agent concentration increased, more precursor ions were reduced at the same time, leading to the formation of more nuclei and growth into smaller particles.46 However, when the ascorbic acid concentration was high, the pH of the solution dropped, decreasing the reduction potential of ascorbic acid. This reason slowed down the reduction of copper ions to copper atoms, leading to a decrease in the amount of the formed CuNPs.47
The results of the regression analysis of model (1) are presented in Table 2. In particular, p values <0.05 show that the interactions of factors X1, X2, and X3 (interactions of Xi, XiXi, and XiXj) were significant. In addition, the R2 values of 0.985 for Y1 and 0.993 for Y2 confirm the suitability of the proposed model. The optimal values for the synthesis were determined at the points with the highest absorbance values Y1 and the shortest absorption wavelengths Y2. Calculation results show that the optimal conditions for synthesizing CuNPs included 20 g per L gelatin, 19.3 mM Cu(AcO)2, and 41.5 mM ascorbic acid. A CuNP sample synthesized with optimal parameters was denoted as CuNPs-G.
| α 0 | α 1 | α 2 | α 3 | α 11 | α 22 | α 33 | α 12 | α 13 | α 23 | R 2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Y 1 | 1.248 | 0.134 | 0.326 | −0.171 | −0.026 | −0.531 | −0.103 | 0.059 | −0.085 | −0.059 | 0.985 |
| Y 2 | 574.7 | −1.7 | 4.0 | −1.4 | −1.3 | 0.5 | 0.7 | −0.8 | 0.4 | 0.5 | 0.993 |
| p of Y1 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | 0.039 | 0.044 | 0.008 | 0.044 | ||
| p of Y2 | <0.001 | <0.001 | <0.001 | 0.004 | 0.008 | 0.017 | <0.001 | 0.016 | 0.005 |
The effectiveness of the ideal synthesis was ascertained using ICP-OES analysis. The measured Cu2+ concentrations were 248.3 ppm for the initial solution containing only 19.3 mM precursor Cu(OAc)2 and 60.7 ppm for the solution produced after 6 h of centrifugation at 6800 rpm to eliminate CuNPs. The synthesis efficiency was determined from formula (7). Consequently, the calculated concentration of CuNPs was 187.6 ppm, and a synthesis efficiency of 75.6% was attained under optimal parameters.
 | (7) |
With the parameters obtained, the synthesized CuNPs-G sample was stored for different durations at room temperature to investigate its stability over time. The results of the investigation are illustrated in Fig. 3. Fig. 3a displays the UV-vis spectra of these samples at the time of synthesis and at evenly spaced time intervals of 1 w. During the initial 2 week period, the absorption peak increased and reached a maximum value of 1.654. This increase can be attributed to residual reductants and stabilizers remaining in the solution samples. Throughout the remaining survey period, the recorded spectra exhibited a continuous decrease in peak intensities and an insignificant increase in SPR, indicating a clear reduction. The final peak value of 1.129 was observed on the 7th week, and the values of SPR fluctuated from 579.2 to 581.6 nm during storage time (Fig. 3b). This phenomenon can be attributed to the reversion of Cu0 to Cu2+ in solution.48 These findings confirmed the excellent durability of CuNPs-G for up to two weeks, with CuNPs starting to convert into Cu2+ after being extended beyond two weeks.
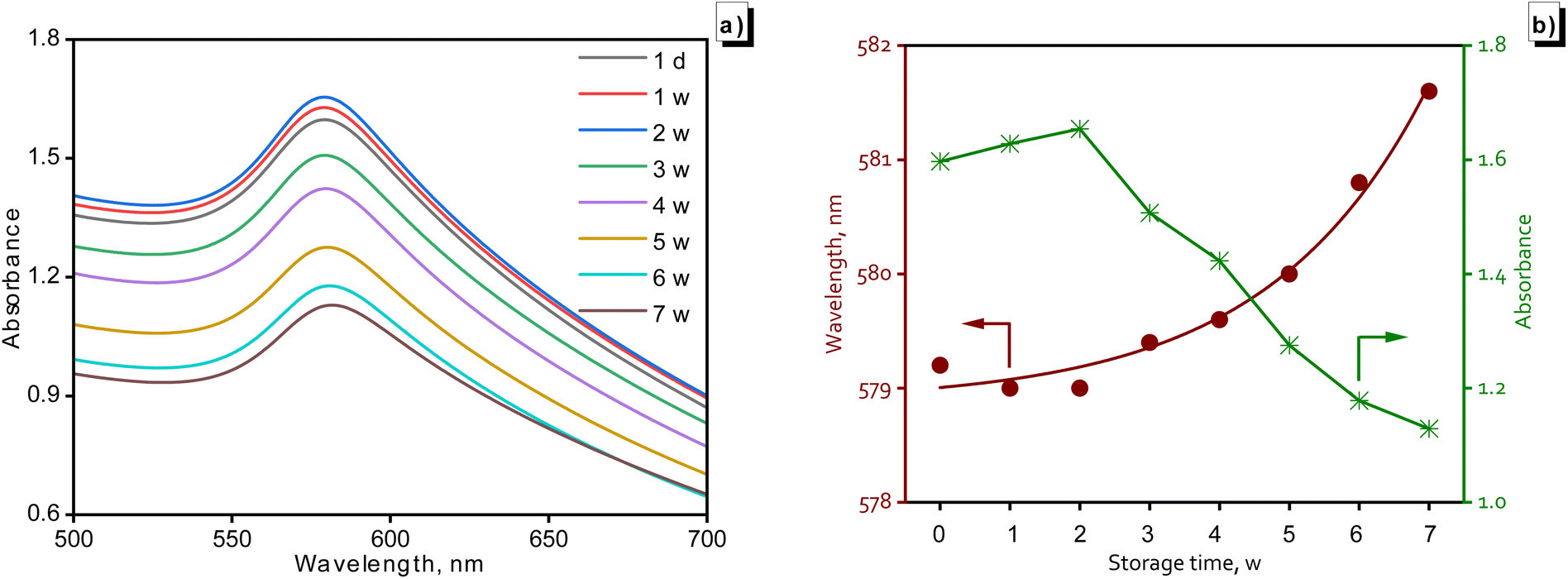 | ||
| Fig. 3 Stability of CuNPs-G over time through UV-vis spectroscopy: the variation in absorbance over storage time (a) and the change in SPR-absorbance with extended investigation time (b). | ||
3.2. Characterization of CuNPs-G
Fig. 4 presents the phase composition and presence of functional groups on the surface of the obtained material. The XRD pattern of generated CuNPs-G is shown in Fig. 4a. Three prominent peaks in the pattern with 2θ values of 43.34, 50.46, and 74.23° served as indicators of the copper face-centered cubic (fcc) lattice. These peaks correspond to the Miller indices (111), (200), and (220), respectively, with reference to the Joint Committee on Powder Diffraction Standards (JCPDS) No. 04-0836. As the (111) plane had a peak intensity that was significantly higher than the others, it was concluded that the predominant orientation for the crystal structure of CuNPs-G was the (111) plane. Additionally, there were no significant peaks of impurities in the XRD pattern, making it a quite clean sample. The average size of the crystallites in the produced CuNPs-G, as reported in Table 3, was 17.6 nm. Additionally, the characteristic peaks of copper were retained in the CuNPs-Greact, indicating that the phase composition of the synthesized CuNPs-G material remained unchanged after PR removal.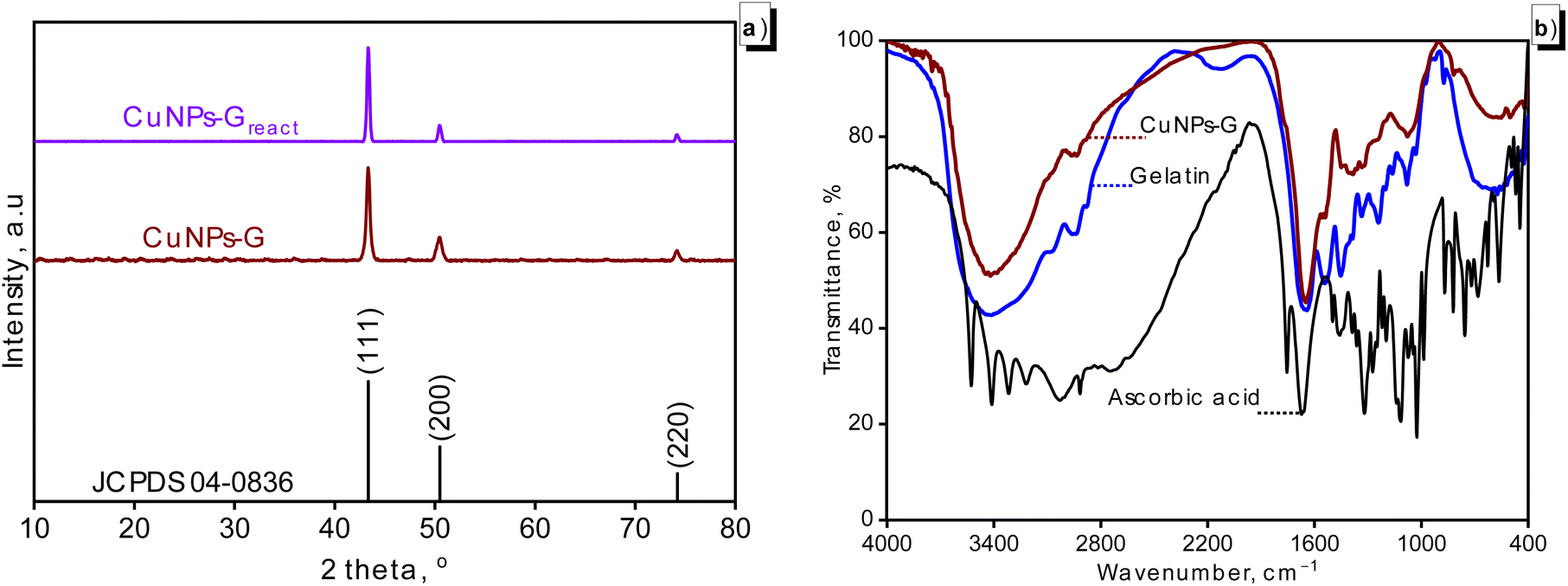 | ||
| Fig. 4 XRD pattern analysis of the generated CuNPs-G/CuNPs-G after the PR degradation reaction (CuNPs-Greact) (a) and the FTIR spectrum of the prepared samples (b). | ||
| Position 2θ, ° | FWHM | Size of nanoparticles, nm | Average size, nm |
|---|---|---|---|
| 43.34 | 0.459 | 18.6 | 17.6 |
| 50.46 | 0.637 | 13.8 | |
| 74.23 | 0.492 | 20.3 |
Fig. 4b compares the FTIR spectra of the produced CuNPs-G, pure gelatin and ascorbic acid. Spectra of CuNPs-G and gelatin showed a wide peak in the 3200–3600 cm−1 region, indicating the presence of O–H stretching vibration that originates in gelatin. The peak measured at 2946 cm−1 served as the sign for the C–H stretching vibrations of the alkane groups in the chain of the gelatin polymer.49 The characteristic peaks for the bond in amide groups in gelatin were also observed in the 1200–1600 cm−1 region, where amide-I was seen at 1647 cm−1 and amide-II was present at 1543 cm−1.50 The presence of C–N and N–H bond stretching in gelatin was attributed to peaks at 1451 and 1243 cm−1, respectively.51 The weak interactions between CuNPs and gelatin, like the van der Waals force, were primarily responsible for the modest shifts in peak magnitude of the characteristic vibrations of gelatin relative to those observed for the produced CuNPs.52 The FTIR spectra of gelatin and CuNPs-G were almost similar, supporting that gelatin effectively served as a stabilizer on the particle surfaces.
The FTIR spectrum of ascorbic acid showed characteristic valence vibrations of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond in the lactone ring and the C–O bond of the enol hydroxyl groups at 1670 and 1321 cm−1.53 However, these signals did not appear in the FTIR spectrum of CuNPs-G due to the oxidation of enol hydroxyl groups to form carbonyl groups. Additionally, the peaks recorded at 3527, 3412, 3317, and 3217 cm−1 characterized the O–H bonds in the hydroxyl groups, along with a signal at 1755 cm−1, which is attributed to the CO bond of the carbonyl groups on the lactone ring of ascorbic acid.54 Owing to the existence of gelatin on the surface of CuNPs-G and the oxidized enol hydroxyl groups, the hydroxyl peaks of ascorbic acid were not observed overlapping in the range 3600–3200 cm−1.55 The signal of carbonyl groups on the lactone ring of ascorbic acid was also not recorded for the carbonyl groups bound to the surface of CuNPs.54
C double bond in the lactone ring and the C–O bond of the enol hydroxyl groups at 1670 and 1321 cm−1.53 However, these signals did not appear in the FTIR spectrum of CuNPs-G due to the oxidation of enol hydroxyl groups to form carbonyl groups. Additionally, the peaks recorded at 3527, 3412, 3317, and 3217 cm−1 characterized the O–H bonds in the hydroxyl groups, along with a signal at 1755 cm−1, which is attributed to the CO bond of the carbonyl groups on the lactone ring of ascorbic acid.54 Owing to the existence of gelatin on the surface of CuNPs-G and the oxidized enol hydroxyl groups, the hydroxyl peaks of ascorbic acid were not observed overlapping in the range 3600–3200 cm−1.55 The signal of carbonyl groups on the lactone ring of ascorbic acid was also not recorded for the carbonyl groups bound to the surface of CuNPs.54
TEM images of CuNPs-G were collected to examine the shape and size distribution of the particles to learn more about the morphology of the created CuNPs-G (Fig. 5). Fig. 5a demonstrates that the particles were both spherical and cylindrical, with comparable particle sizes for each kind of shape. Spherical particles' diameters varied from 15 to 85 nm, while cylindrical particles' sizes tend to be distributed across the range between 75 and 125 nm (Fig. 5b and c). With an error of 8.0 nm, the typical diameter of spherical particles was measured to be 43.1 nm. However, the average size of cylindrical particles was higher, at 105.2 nm, with an analogous variation of 7.5 nm.
 | ||
| Fig. 5 TEM image of the generated CuNPs-G (a) and size distribution of spherical particles (b) and cylindrical particles (c). | ||
The generated CuNPs-G in this study were recorded on the surface with both positive and negative charge signals, as shown in Fig. 6. The zeta potential of metallic nanoparticles was supposed to be greatly influenced by the presence of a stabilizer, and particles with positive or negative values higher than ±30 mV for zeta potential are regarded to cause stable dispersion.56 The zeta potential of the produced CuNPs-G sample was −36.7 mV for the negative portion and +72.2 mV for the positive portion. The negative charge matched the recorded range reported in the research by Musa et al. in 2016, where copper nanoparticles were also synthesized from copper(II) acetate and ascorbic acid and stabilized by gelatin. The research showed that a negative zeta potential was developed in all of the samples in the aqueous solution at different doses of gelatin as a result of the carboxylic and amino groups adhering to the surface of the CuNPs.21 However, the positive charge was also noted on the generated CuNPs-G. Different from Musa et al.,21 ascorbic acid was utilized on its own in the synthesis of CuNPs-G without neutralization by NaOH, lowering the pH of the solution. On the surface of the gelatin, the acidity produced fewer negatively charged pieces –COO− than positively charged species [–NH–C(NH2)–NH2]+ and –NH3+, suggesting that the surface of gelatin molecules has a net positive charge indicated by the zeta potential value.57 The positive charge shown on the surface of CuNPs-G is therefore a result of the used type-A gelatin, which has an isoelectric point larger than 7. The validity and precise determination of the relationship between these arguments require more research. Despite that, the assertion is partially supported by the significantly high recorded intensity of the positive zeta potential observed on CuNPs-G.
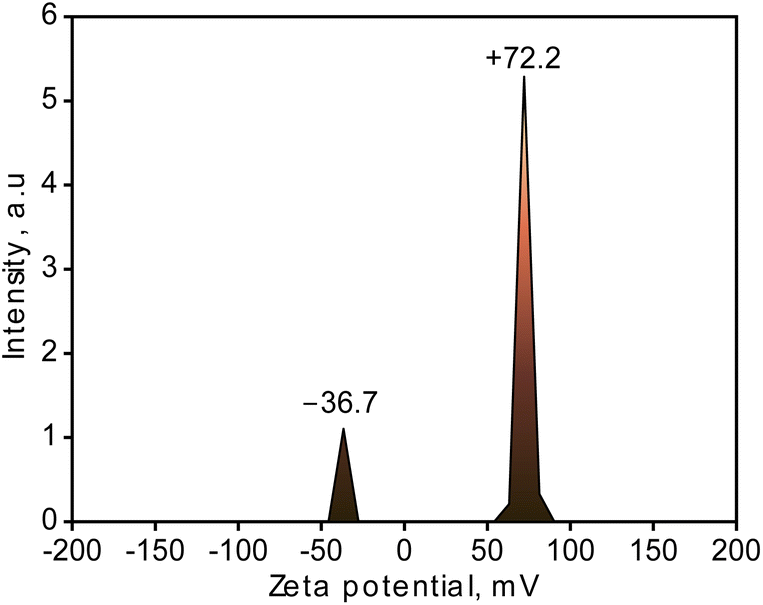 | ||
| Fig. 6 The measured zeta potential of the generated CuNPs-G. | ||
Fig. 7 illustrates the optical properties and surface properties of the synthesized CuNPs-G. UV-vis diffuse reflectance spectra were recorded to gain a more precise indication of the purity of the synthesized CuNPs-G. As depicted in Fig. 7a, the material exhibited a distinct absorption edge at approximately 580 nm, consistent with the UV-vis absorption analysis. Using Tauc's relation, the analysis yielded an estimated band gap energy (Eg) of 2.14 eV, which aligned with the observed absorption wavelength (λ). This value of energy indicates the presence of pure copper and signifies the material's exceptional level of purity. This finding is consistent with the majority of results reported in a comprehensive review conducted by Al-Hakkani.41 Additionally, the synthesized CuNPs-G absorbed visible light.
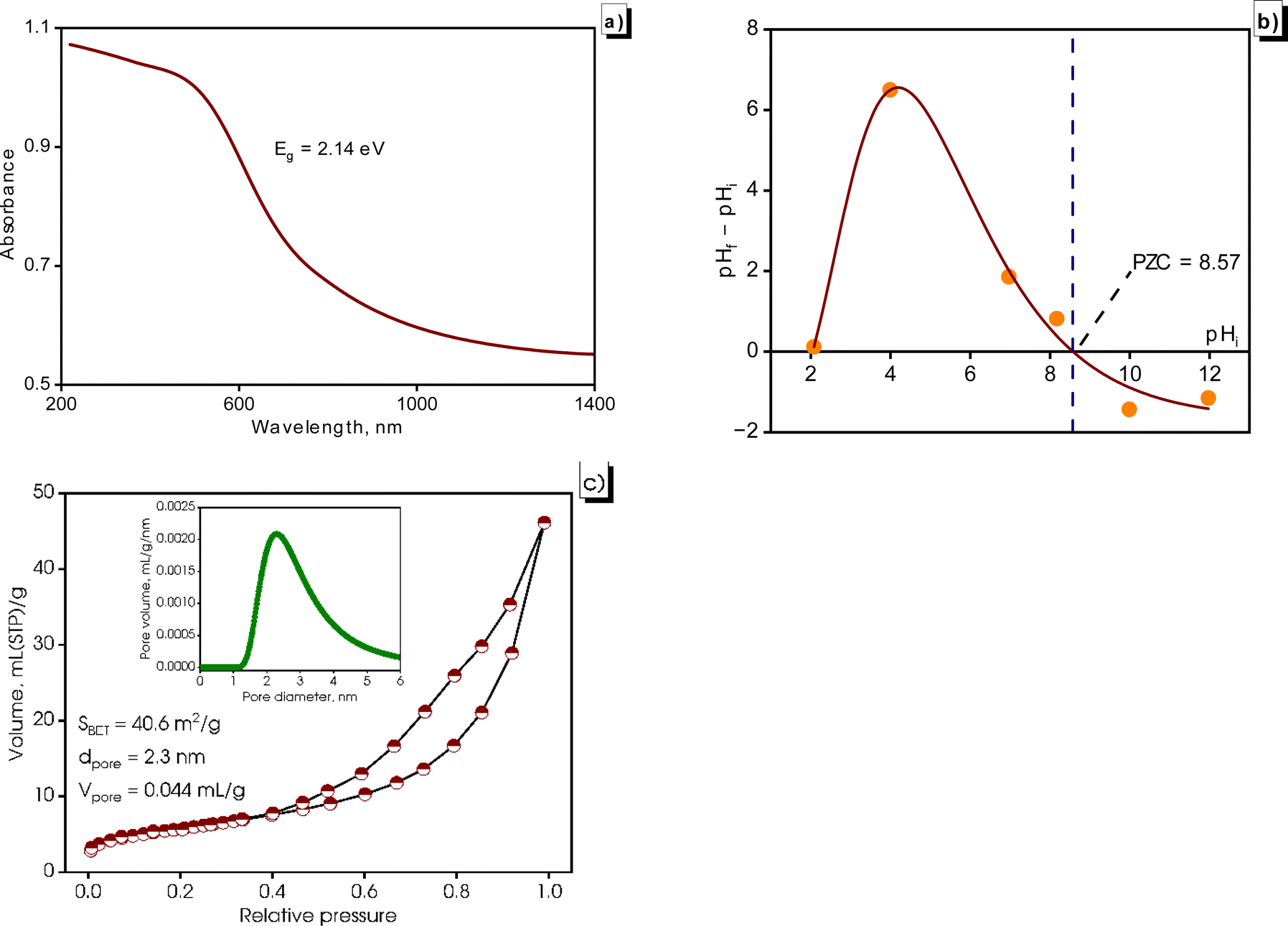 | ||
| Fig. 7 DRS spectra (a), PZC (b), and adsorption–desorption isotherms (c) of the generated CuNPs-G. | ||
The graph ΔpH = f(pHi) with pHi values adjusted from 2 to 12 is depicted in Fig. 7b. The intersection of the graph with the horizontal axis was determined at pHi = 8.57, which was the PZC value of CuNPs-G. Before PZC, the surface carried a positive charge and the surface charge changed to negative after PZC. The high PZC value shows that the resulting CuNPs-G material had good ability to capture anions at pH < 8.57.58
In Fig. 7c, the adsorption isotherm obtained from the BET analysis of CuNPs-G shows a characteristic Type II behavior as per the IUPAC classification. It also displays a hysteresis loop of type H3, commonly associated with mesoporous solids. These solids often consist of uniform aggregates of plate-like particles, creating slit-like pores.59 The observed adsorption isotherm shape suggests the presence of mesoporous properties and indicates the potential existence of microporosity in CuNPs-G. By applying the BET equation to the acquired adsorption isotherm, the specific surface area was calculated to be 40.6 m2 g−1 for the synthesized CuNPs-G. This significant value indicates a larger exposure of the material to the external environment, enhancing its adsorption capacity, interaction, and reactivity. As a result, CuNPs-G demonstrates a strong potential for interacting with other substances during chemical processes, adsorption, or reactions.
3.3. Antimicrobial activity of CuNPs-G
For years, the antimicrobial mechanism of CuNPs has been anticipated. It involves their ability to enter cells through the cell membrane, induce damage to the membrane, interfere with biochemical processes, and disrupt metal cation homeostasis.60 Based on these considerations, an evaluation of the biological activity of the synthesized CuNPs-G was conducted in this study, encompassing both microorganisms and cancer cells. The results of these tests are presented in Fig. 8, wherein varying concentrations of CuNPs were employed to treat Gram-positive bacteria (B. subtilis, L. fermentum, and S. aureus), Gram-negative bacteria (E. coli, P. aeruginosa, and S. enterica), and cancer cells (A549, Hep-G2, KB, and MCF7) on CuNPs-G and CuNPs-wG. As depicted in Fig. 8a, Gram-positive bacteria displayed substantial inhibition when treated with CuNPs-G. However, the inhibitory response against Gram-negative bacteria was relatively modest, ranging from 38 to 53% (Fig. 8b). In Fig. 8c, the outcomes were particularly encouraging when cancer cells were tested, as CuNPs-G demonstrated significant effectiveness. Conversely, the produced CuNPs-wG sample exhibited inhibitory activity against bacterial and cancer cell growth, with a maximum inhibition percentage of around 30%. To overview these results, the half-maximal inhibitory concentration (IC50) and minimum inhibitory concentration (MIC) for each strain were also calculated, and the results are presented in Table 4. From the IC50 and MIC values, it can be observed that the control sample exhibited strong activity, followed by the CuNPs-G sample, whereas the CuNPs-wG sample displayed little to no inhibition of bacterial/cancer cell growth. Specifically, for the CuNPs-wG sample, the IC50 value could not be determined as the inhibition percentage remained below 50%, and the MIC exceeded the maximum studied CuNP concentration of 16.00 ppm. The control sample consistently showed IC50 and MIC values for bacteria/cancer cells, with better corresponding values than the CuNPs-G sample. The obtained results indicated that the presence of gelatin enhanced antibacterial and cell inhibition activity.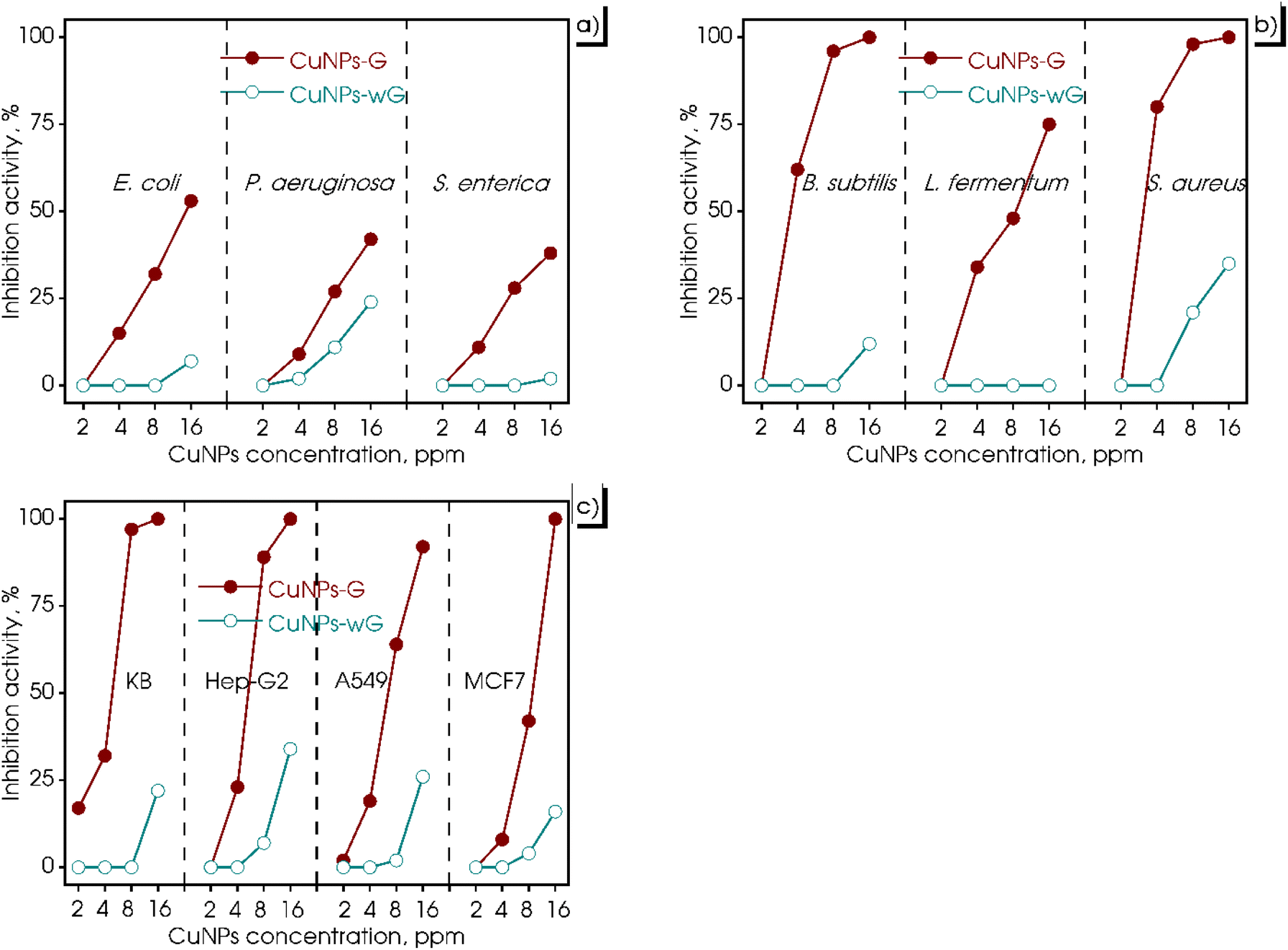 | ||
| Fig. 8 Antimicrobial activity of the produced CuNPs-G and CuNPs-wG with different CuNP concentrations against Gram-negative bacteria (a), Gram-positive bacteria (b), and cancer cells (c). | ||
| Bacteria/cancer cells | IC50, ppm | MIC, ppm | |||||
|---|---|---|---|---|---|---|---|
| CuNPs-G | CuNPs-wG | Controla | CuNPs-G | CuNPs-wG | Controla | ||
| a Ampicillin, cefotaxime, and ellipticine were used for testing on Gram-positive bacteria, Gram-negative bacteria, and cancer cells, respectively. | |||||||
| Gram-positive bacteria | S. aureus | 3.43 ± 0.24 | — | 0.05 ± 0.01 | 8.00 | >16.00 | 1.25 |
| B. subtilis | 7.16 ± 0.41 | — | 0.62 ± 0.07 | 8.00 | >16.00 | 2.00 | |
| L. fermentum | 2.86 ± 0.15 | — | 0.25 ± 0.06 | >16.00 | >16.00 | 1.75 | |
| Gram-negative bacteria | S. enterica | — | — | 2.74 ± 0.16 | >16.00 | >16.00 | 5.25 |
| E. coli | 10.74 ± 1.06 | — | 0.27 ± 0.05 | >16.00 | >16.00 | 1.75 | |
| P. aeruginosa | — | — | 1.86 ± 0.32 | >16.00 | >16.00 | 4.75 | |
| Cancer cells | KB | 4.28 ± 0.34 | — | 0.18 ± 0.02 | 8.00 | >16.00 | 1.50 |
| Hep-G2 | 5.06 ± 0.38 | — | 1.31 ± 0.15 | 12.00 | >16.00 | 4.25 | |
| A549 | 6.68 ± 0.44 | — | 0.06 ± 0.01 | >16.00 | >16.00 | 1.25 | |
| MCF7 | 9.11 ± 0.95 | — | 0.74 ± 0.04 | 16.00 | >16.00 | 2.25 | |
In the case of Gram-positive bacteria, CuNPs-G exhibited remarkable inhibitory effects, with significantly low IC50 values of 3.43, 7.16, and 2.86 ppm for S. aureus, B. subtilis, and L. fermentum, respectively. In contrast, for Gram-negative bacteria, the IC50 value of CuNPs for the CuNPs-G sample was only calculated for the most effective sample, E. coli, and was found to be a high value of 10.74 ppm. For the treatment of cancer cells, CuNPs-G demonstrated a more refined outcome, with IC50 values ranging from 4.28 to 9.11 ppm. The potent antibacterial properties of the synthesized CuNPs-G can be attributed to a common mechanism, where the released copper ions cause damage to microbial membranes.61 This mechanism is particularly effective against Gram-positive bacteria due to the more structural complexity of Gram-negative bacteria. The thinner cell wall of Gram-positive bacteria facilitates a more efficient exchange of compounds across the cell wall, leading to enhanced inhibition.62 In terms of anti-cancer activity, the synthesized CuNPs-G have demonstrated their ability to induce apoptosis and exhibit cytotoxic effects on various cancer cell lines. They achieve this by degrading isolated DNA molecules through the generation of reactive oxygen species, thereby contributing to their anti-cancer properties.63 These findings underscore the potential of CuNPs-G as a promising agent for targeting both cancer cells and microorganisms. The detailed antimicrobial mechanism of CuNPs is illustrated in Fig. 9. CuNPs damaged cellular components in several ways: (i) generating reactive oxygen species, (ii) directly disrupting the cell membrane, (iii) replacing/binding with the native cofactors in proteins, and (iv) damaging intracellular components.64 There was a similarity in the results of the current study with several recent studies on the antibacterial and cell inhibition activities of CuNPs. Wulandari et al. synthesized CuNPs by a chemical reduction method using NaBH4 as the reducing agent and cetyltrimethylammonium bromide as the stabilizing agent. The synthesized CuNPs exhibited good antibacterial activity against E. coli and S. aureus at a concentration of CuNPs reaching 10 and 15 ppm, respectively.65 Meanwhile, CuNPs synthesized from leaf extracts of Phragmanthera austroarabica and Foeniculum vulgare showed good inhibitory activity against cancer cells: MDA-MB-231 (IC50 = 67 ppm), NCI-H2126 (IC50 = 108 ppm), NCI-H1299 (IC50 = 168 ppm), and NCI-H1437 (IC50 = 122 ppm).66,67
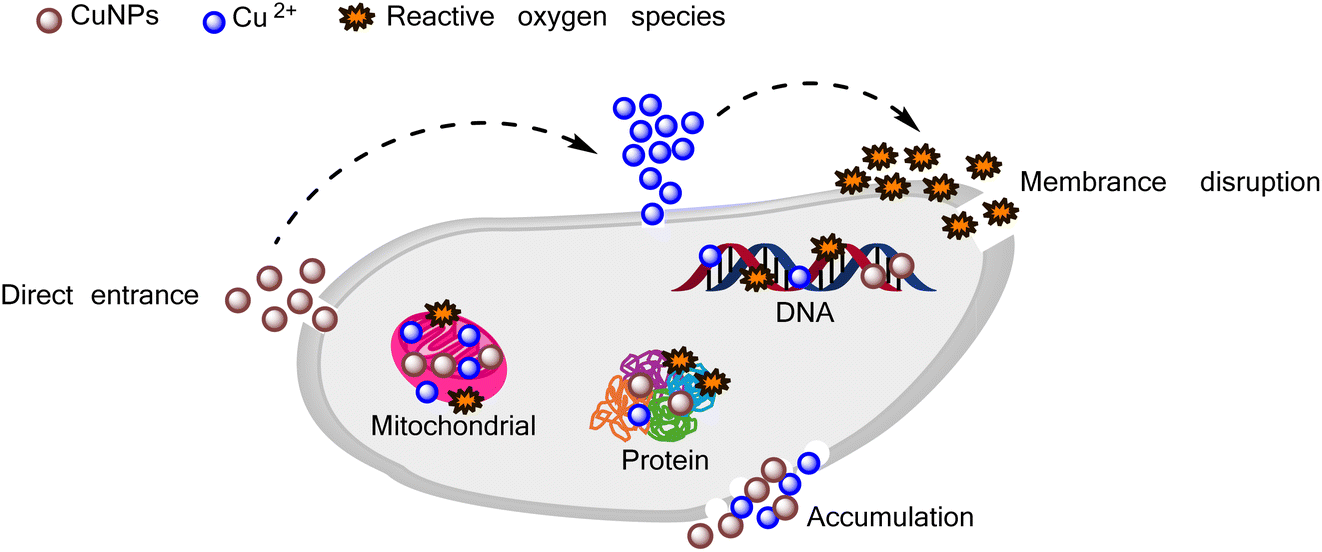 | ||
| Fig. 9 Antimicrobial mechanism of CuNPs. | ||
Fig. 10 illustrates the fungal growth, encompassing F. solani, C. gloeosporioides, and R. solani, across different concentrations of CuNPs. Generally, the growth diameter of the fungi diminished in the presence of CuNPs. The growth diameter of F. solani dropped from 74.8 mm to 20.4 mm with 450 ppm CuNPs. Temporarily, C. gloeosporioides was completely inhibited in the presence of 400 ppm CuNPs, and the growth diameter of R. solani was 13.6 mm in the presence of 1000 ppm CuNPs. The IC50 value for fungal growth was calculated from the graph in Fig. 11, showing the dependence of inhibitory capacity on CuNP concentration. Based on the obtained results, the order of antifungal activity of CuNPs-G was as follows: R. solani (IC50 = 760 ppm) < F. solani (IC50 = 386 ppm) < C. gloeosporioides (IC50 = 315 ppm). The value of CuNP concentration required to inhibit the growth of R. solani was approximately twice as high as that for F. solani and C. gloeosporioides.
 | ||
| Fig. 10 Antifungal activity of the prepared CuNPs-G: F. solani mixed with 0–450 mM CuNPs (a1–a6), C. gloeosporioides mixed with 0–400 mM CuNPs (b1–b6), and R. solani mixed with 0–1000 mM CuNPs (c1–c6). | ||
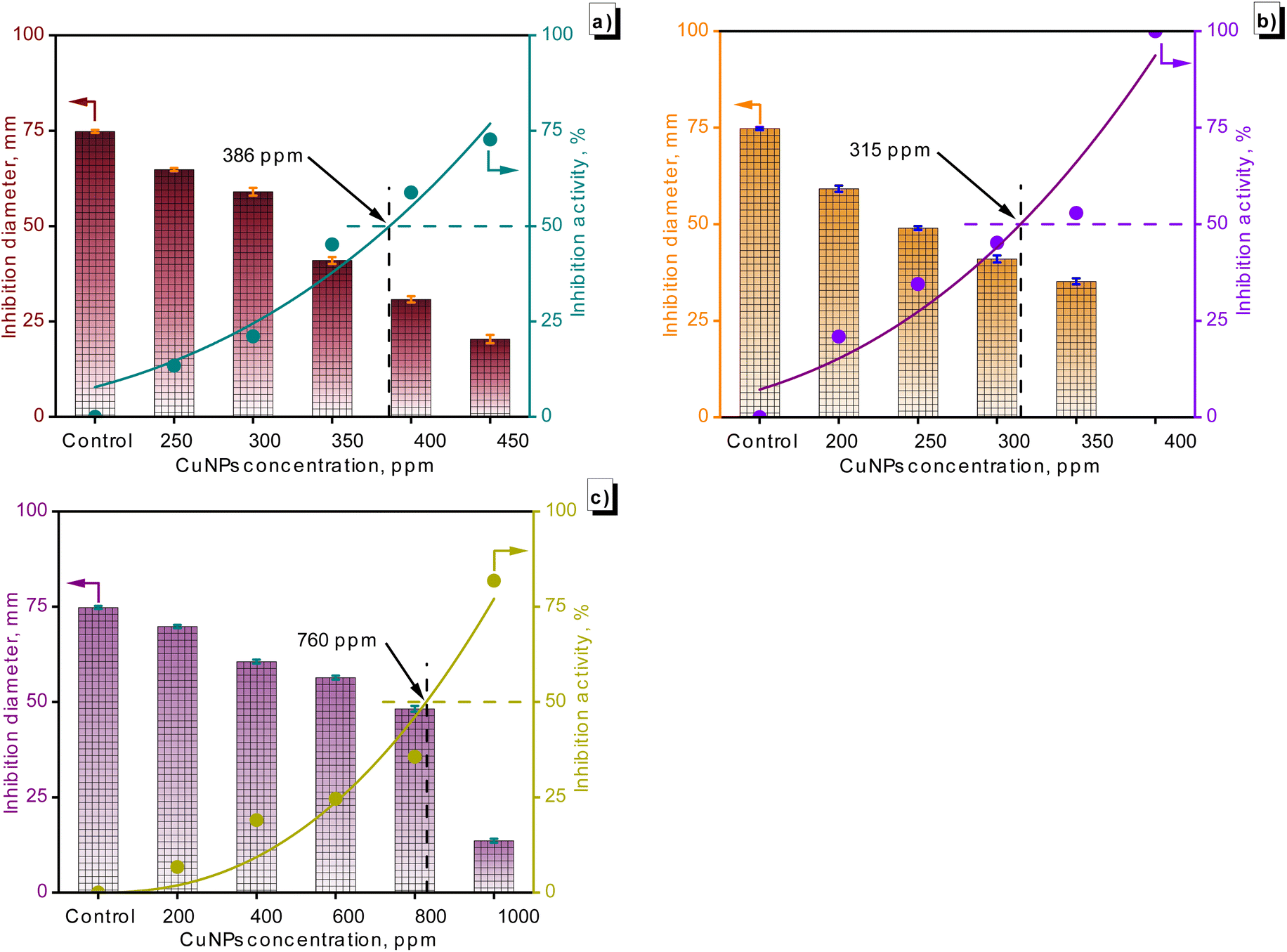 | ||
| Fig. 11 The suppression of F. solani (a), C. gloeosporioides (b), and R. solani (c) by the as-synthesized CuNPs was observed across various concentrations. | ||
3.4. Photocatalytic activity of CuNPs-G
Table 5 presents the values of PR conversion as a dependent variable obtained from experiments investigating independent variables, including the pH of PR solution, catalyst dosage, and NaBH4 concentration. PR conversion after 540 s was recorded in the range from 48.2 to 95.7%. The influence of the pH of the PR solution, catalyst dosage, and NaBH4 concentration on the PR removal and catalytic activity of the synthesized CuNPs-G is depicted in Fig. 12.| Run order | X 4 | X 5, ppm | X 6, mM | Y 3, % |
|---|---|---|---|---|
| 1 | 6 | 25 | 50 | 84.4 |
| 2 | 6 | 10 | 30 | 59.1 |
| 3 | 8 | 40 | 50 | 87.3 |
| 4 | 8 | 25 | 30 | 72.2 |
| 5 | 6 | 40 | 70 | 78.0 |
| 6 | 10 | 25 | 50 | 72.7 |
| 7 | 10 | 40 | 70 | 64.7 |
| 8 | 10 | 10 | 70 | 51.2 |
| 9 | 8 | 25 | 50 | 93.2 |
| 10 | 10 | 40 | 30 | 59.5 |
| 11 | 8 | 25 | 50 | 88.8 |
| 12 | 8 | 25 | 50 | 95.7 |
| 13 | 8 | 25 | 70 | 79.2 |
| 14 | 8 | 25 | 50 | 94.3 |
| 15 | 10 | 10 | 30 | 48.2 |
| 16 | 8 | 25 | 50 | 94.7 |
| 17 | 8 | 25 | 50 | 92.8 |
| 18 | 6 | 40 | 30 | 65.6 |
| 19 | 8 | 10 | 50 | 82.7 |
| 20 | 6 | 10 | 70 | 69.2 |
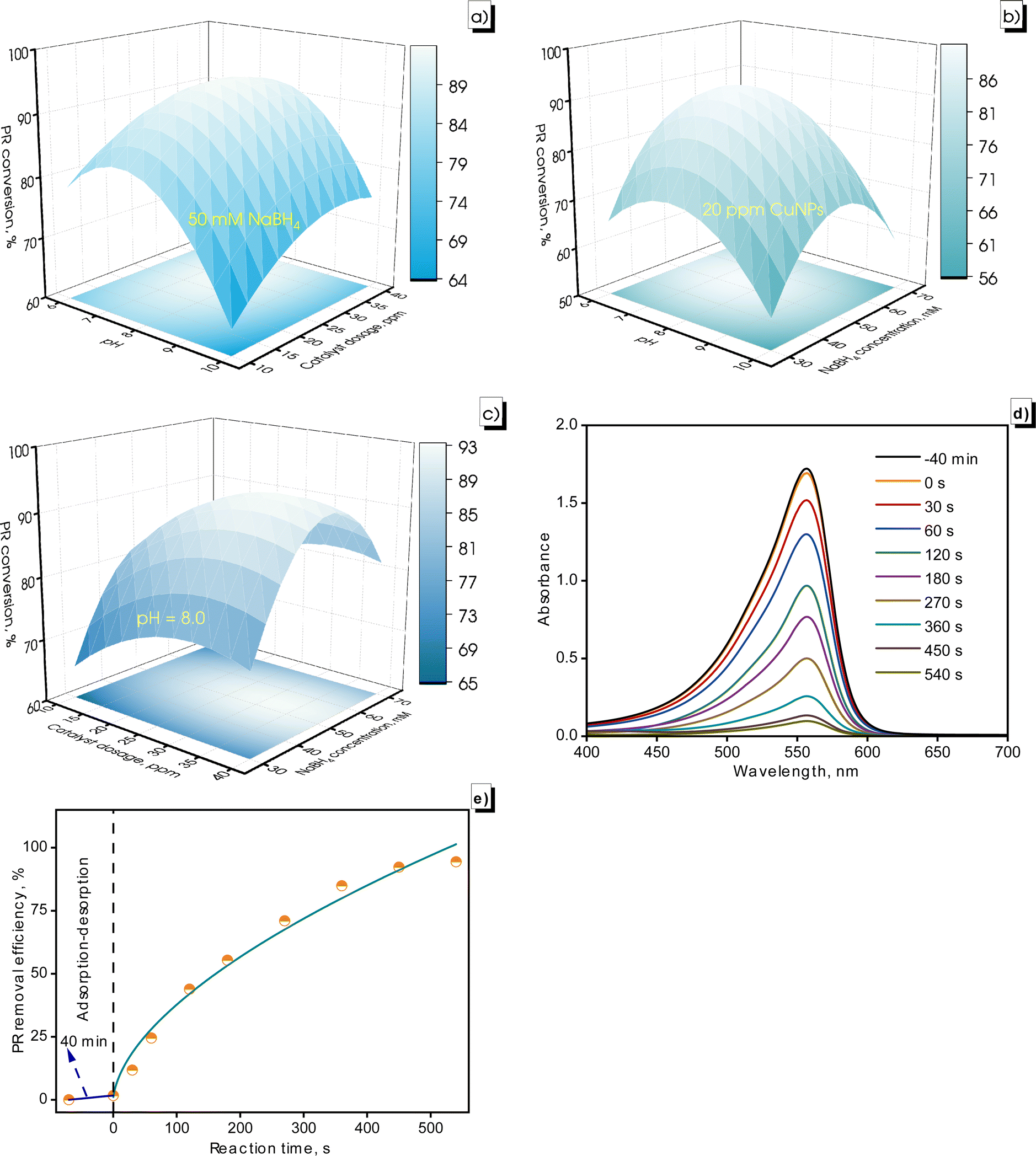 | ||
| Fig. 12 Surface plot with projection for the PR conversion at numerous CuNP dosages, NaBH4 concentrations, and pH solution of PR (a–c), UV-vis spectra of PR conversion on CuNPs-G (d), and PR conversion at different reaction times (e). | ||
The PR conversion was affected by the charge state of the CuNPs-G. In the pH range of 6.0 to 8.0, the positive charge of the produced CuNPs-G enhanced the adsorption of anionic substances, leading to increased PR removal. When the pH exceeds the PZC of 8.57, the CuNPs-G material converted negatively charged, reducing adsorption efficiency and lowering the PR degradation (Fig. 12a and b).
Regarding catalyst dosage, increasing the CuNPs-G dosage from 10 to 40 ppm led to a significant improvement in PR conversion (Fig. 12a and c). The primary reason was that the number of active sites increased, enhancing the catalytic activity for the degradation process.68 The active sites formed on the surface of CuNPs under light energy stimulation. Increasing the catalyst dosage added to the number of active sites on the catalyst surface.69 A slight decrease in the PR conversion rate was observed, and the catalyst dosage was further increased to 40 ppm. The increase in the catalyst dose lowered the dye removal efficiency due to increased turbidity, which caused a light scattering effect and the active sites on the surface were covered by the excessive catalyst.70
The PR conversion rate gradually increased as the NaBH4 concentration was increased from 30 to 50 mM, but then it gradually diminished as the NaBH4 concentration continued to increase to 70 mM (Fig. 12b and c). This result was due to the competitive adsorption between PR, surface hydrogens, and borohydride ions on the catalyst surface. As the amount of NaBH4 was increased, the surface hydrogens produced increased further and adsorbed more on the catalyst surface, leading to saturation of the active sites and reduction of the material's adsorption capacity.71
The regression analysis results of the model for PR conversion on the independent variables, including the pH of PR solution, catalyst dosage, and NaBH4 concentration, are presented in Table 6. The R2 value of 0.982 confirms that the proposed model accurately described the dependence of the PR removal on these independent variables. Additionally, the primary terms of independent variables showed a significant impact on the dependent variable with p-values <0.05, whereas the secondary terms have p-values >0.05, indicating a poor correlation among the independent variables. The optimal conditions for eliminating PR were determined at points where the PR conversion was the highest. According to computational results, the optimal conditions for PR removal included a catalyst dosage of 31.5 ppm, a NaBH4 concentration of 53.1 mM, and the pH of PR reaching 7.5.
| α 0 | α 4 | α 5 | α 6 | α 44 | α 55 | α 66 | α 45 | α 46 | α 56 | R 2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Y 3 | 91.9 | −6.0 | 4.5 | 3.8 | −11.4 | −4.9 | −14.2 | 1.2 | −1.8 | 0.982 | |
| p | <0.001 | <0.001 | 0.002 | <0.001 | <0.001 | <0.001 | 0.257 | 0.100 | 0.582 |
The previously mentioned conditions were applied to eliminate the 10 ppm PR solution. The absorbance of PR and PR conversion at different reaction times are recorded in Fig. 12d and e. The PR solution reached the adsorption–desorption equilibrium after 40 min, with the absorbance intensity of PR decreasing from 1.721 to 1.693, corresponding to a decrease in the PR concentration from 10 to 9.843 ppm. The absorbance intensity of PR gradually declined to 0.097 after a 540 s reaction time, corresponding to a PR concentration of 0.564 ppm in the solution, achieving a PR conversion of 94.4%.
In this study, a comprehensive analysis was conducted, comparing the obtained findings with recent investigations, as presented in Table 7, exploring various approaches to synthesizing CuNPs using diverse chemicals for reduction and stabilization. Some of these methods resulted in challenges such as a large particle size, irregular shape, and limited storage stability, which could impact their potential applications in biological and catalytic fields. While previous research primarily focused on the antimicrobial properties of CuNPs, investigations into their effective inhibition against Gram-positive bacteria and cancer cells were relatively scarce, especially for chemically synthesized CuNPs. On the contrary, CuNPs-G synthesis, utilizing environmentally friendly gelatin and ascorbic acid, demonstrated remarkable efficacy against Gram-positive bacteria, fungi, and cancer cells, suggesting promising applications in biomedicine.
| Reductant, stabilizer | Shape, size | Stability | Antimicrobial activity | Catalytic activity | References | |
|---|---|---|---|---|---|---|
| CuNPs-1 | Ascorbic acid | Spherical (50–60 nm) | 84 d | — | — | 72 |
| CuNPs-2 | Ascorbic acid, gelatin | Spherical (10–15 nm) | 2 d | Effective against Gram-negative bacteria | — | 73 |
| CuNPs-3 | Sodium borohydride | Spherical (50 nm) | — | — | Effective against Reactive Blue 4 degradation | 74 |
| CuNPs-4 | Ascorbic acid | Cubical (5–10 nm) | — | — | Effective against methylene blue, methyl red and Congo red degradation | 30 |
| CuNPs-5 | Ascorbic acid, sodium citrate | Spherical (200–500 nm) | — | Effective against fungi | — | 27 |
| CuNPs-G | Ascorbic acid, gelatin | Spherical (43.1 nm) | 28 d | Effective against Gram-positive bacteria, fungi, and cancer cells | Effective against phenol red degradation | This study |
| Cylindrical (105.2 nm) |
Additionally, an extra comparative analysis was conducted, as presented in Table 8, to comprehensively evaluate the effectiveness of nanoparticles in PR degradation. Most previous studies achieved almost complete PR conversion but required excessive catalyst quantities and prolonged treatment times. Some even employed an absorption method that did not effectively eliminate PR. In contrast, this study demonstrated that using a small quantity of CuNPs-G resulted in a satisfactory PR reduction within a short time. Analyzing samples with varying catalyst and reductant quantities over time provided valuable insights into the PR reduction process. These findings encourage further research to optimize PR reduction through alternative approaches.
| Material | Catalyst dosages, ppm | Degradation method | PR conversion | References |
|---|---|---|---|---|
| FeNPs | 100 | Adsorption | 98% for 180 min | 75 |
| AgNPs | 431 | Reduction by NaBH4 | 98% for 24 h | 31 |
| CuNPs | 6355 | Reduction by NaBH4 | 98% for 24 h | |
| TiO2 NPs | 40 | Irradiation | 74% for over 1 h | 76 |
| Nb(X)/TiO2 | 546 | Irradiation | 94% for 160 min | 77 |
| CuNPs-G | 31.5 | Reduction by NaBH4 | 94% for 540 s | This study |
4. Conclusions
To sum up, the use of ascorbic acid and gelatin played a pivotal role in the synthesis of CuNPs-G, and the material additionally showcased impressive biological effects against a range of microorganisms, along with its photocatalytic attributes during the PR degradation process. The stability of this substance was well maintained for 28 d, and the XRD examination verified its purity. The FTIR examination as well as the DLS test with both negative (−36.7 mV) and positive (+72.2 mV) charges for the zeta potential on their surface further confirmed the significant involvement of the stabilizer gelatin. The band gap energy of 2.14 eV and high specific surface area of 40.6 m2 g−1 further validated the notable characteristics of CuNPs-G. Taking advantage of these properties, CuNPs-G exhibited a unique inhibitory activity against cancer cells (KB, Hep-G2, A549, and MCF7) and Gram-positive bacteria (S. aureus, B. subtilis, and L. fermentum), while showing a minor inhibitory activity against Gram-negative bacteria (S. enterica, E. coli, and P. aeruginosa). The degradation of PR was achieved using the photocatalytic activity of CuNPs-G. An optimal CuNPs-G concentration of 10 mg L−1 was first identified when using excess NaBH4. This study highlights the captivating properties of ascorbic acid and gelatin-stabilized CuNPs, offering exciting prospects for novel applications in nanotechnology, biomedicine, and environmental remediation.Data availability
The raw/processed data required to reproduce these findings cannot be shared at this time as the data also form part of an ongoing study.Author contributions
Trung Dien Nguyen and Sang Thanh Ngo: investigation, data curation, software, formal analysis, writing – review and editing. Yen Hai Hoang and Sang Thanh Ngo: conceptualization, methodology, investigation, writing – original draft. Nhung Thi Tuyet Thai: data curation, investigation, methodology. Huong Thi Thu Nguyen: conceptualization, formal analysis, validation. Trung Dien Nguyen and Gia Thi Ngoc Trinh: supervision, writing – review and editing.Conflicts of interest
The authors have no competing interests to declare that are relevant to the content of this article.Acknowledgements
This study was funded in part by the Can Tho University, code: TSV2023-73.References
- Y. Zhang, T. P. Shareena Dasari, H. Deng and H. Yu, J. Environ. Sci. Health, Part C: Environ. Carcinog. Ecotoxicol. Rev., 2015, 33, 286–327 CrossRef CAS PubMed.
- A. Rozhin, S. Batasheva, M. Kruychkova, Y. Cherednichenko, E. Rozhina and R. Fakhrullin, Micromachines, 2021, 12, 1480 CrossRef PubMed.
- B. H. Shinde, S. N. Inamdar, S. A. Nalawade and S. B. Chaudhari, Mater. Today: Proc., 2023, 73, 412–417 CAS.
- T. Ishida, T. Murayama, A. Taketoshi and M. Haruta, Chem. Rev., 2020, 120, 464–525 CrossRef CAS.
- P. Priecel, H. Adekunle Salami, R. H. Padilla, Z. Zhong and J. A. Lopez-Sanchez, Chin. J. Catal., 2016, 37, 1619–1650 CrossRef CAS.
- A. B. Manohar and M. B. Bhalchandra, Curr. Org. Chem., 2015, 19, 708–727 CrossRef.
- L. Xu, Y. Y. Wang, J. Huang, C. Y. Chen, Z. X. Wang and H. Xie, Theranostics, 2020, 10, 8996–9031 CrossRef CAS PubMed.
- X. Hu, Y. Zhang, T. Ding, J. Liu and H. Zhao, Front. Bioeng. Biotechnol., 2020, 8, 990 CrossRef PubMed.
- G. Shobha, M. Vinutha and S. Ananda, Int. J. Pharm. Sci. Invent., 2014, 3, 28–38 Search PubMed.
- R. C. Kasana, N. R. Panwar, R. K. Kaul and P. Kumar, Environ. Chem. Lett., 2017, 15, 233–240 CrossRef CAS.
- M. B. Gawande, A. Goswami, F. X. Felpin, T. Asefa, X. Huang, R. Silva, X. Zou, R. Zboril and R. S. Varma, Chem. Rev., 2016, 116, 3722–3811 CrossRef CAS PubMed.
- N. K. Ojha, G. V. Zyryanov, A. Majee, V. N. Charushin, O. N. Chupakhin and S. Santra, Coord. Chem. Rev., 2017, 353, 1–57 CrossRef CAS.
- S. Taifa, A. Muhee, R. A. Bhat, S. U. I. Nabi, A. Roy, G. A. Rather, A. A. Khan, S. M. Bashir, M. Patwekar, S. Wahab, S. Alghamdi, A. S. Dablool, A. Kabrah, U. Hani, A. M. Bannunah, F. Patwekar and J. Mallick, J. Nanomater., 2022, 2022, 7124114 CrossRef.
- M. V. D. Prakash, S. Sampath, K. Amudha, A. Nadeem, B. S. Lopes, B. Durga and S. Muthupandian, Mater. Technol., 2023, 38, 2247908 CrossRef.
- V. K. Velisoju, J. L. Cerrillo, R. Ahmad, H. O. Mohamed, Y. Attada, Q. Cheng, X. Yao, L. Zheng, O. Shekhah, S. Telalovic, J. Narciso, L. Cavallo, Y. Han, M. Eddaoudi, E. V. Ramos-Fernández and P. Castaño, Nat. Commun., 2024, 15, 2045 CrossRef CAS PubMed.
- B. Khodashenas and H. R. Ghorbani, Korean J. Chem. Eng., 2014, 31, 1105–1109 CrossRef CAS.
- I. A. Tito, S. Uddin, S. Islam and S. Bhowmik, Orient. J. Chem., 2021, 37, 1030 CAS.
- W. Suttirak and S. Manurakchinakorn, Walailak J. Sci. Technol., 2011, 7, 5–14 Search PubMed.
- D. Njus, P. M. Kelley, Y. J. Tu and H. B. Schlegel, Free Radic. Biol. Med., 2020, 159, 37–43 CrossRef CAS PubMed.
- M. R. Khan and M. B. Sadiq, Polym. Bull., 2021, 78, 4047–4073 CrossRef CAS.
- A. Musa, M. B. Ahmad, M. Z. Hussein, M. I. Saiman and H. A. Sani, Nanoscale Res. Lett., 2016, 11, 438 CrossRef PubMed.
- M. S. Aguilar, R. Esparza and G. Rosas, Trans. Nonferrous Met. Soc. China, 2019, 29, 1510–1515 CrossRef CAS.
- P. H. Tseng, Y. Z. Wang, T. H. Hsieh, K. S. Ho, C. H. Tsai and K. T. Chen, Nanotechnology, 2018, 29, 085603 CrossRef PubMed.
- P. Cerchier, M. Dabalà and K. Brunelli, Green Process. Synth., 2017, 6, 311–316 CrossRef CAS.
- S. Chand Mali, A. Dhaka, S. Sharma and R. Trivedi, Inorg. Chem. Commun., 2023, 149, 110448 CrossRef CAS.
- M. I. Din, F. Arshad, Z. Hussain and M. Mukhtar, Nanoscale Res. Lett., 2017, 12, 638 CrossRef PubMed.
- N. Pariona, A. I. Mtz-Enriquez, D. Sanchez-Rangel, G. Carrion, F. Paraguay-Delgado and G. Rosas-Saito, RSC Adv., 2019, 9, 18835–18843 RSC.
- G. Naikoo, F. Al-Mashali, F. Arshad, N. Al-Maashani, I. U. Hassan, Z. Al-Baraami, L. H. Faruck, A. Qurashi, W. Ahmed, A. M. Asiri, A. A. Aljabali, H. A. Bakshi and M. M. Tambuwala, Curr. Pharm. Des., 2021, 27, 4416–4432 CrossRef CAS PubMed.
- E. Halevas and A. Pantazaki, J. Nanomed. Nanotechnol., 2018, 2, 119 Search PubMed.
- J. B. Fathima, A. Pugazhendhi, M. Oves and R. Venis, J. Mol. Liq., 2018, 260, 1–8 CrossRef CAS.
- S. Raina, A. Roy and N. Bharadvaja, Environ. Nanotechnol. Monit. Manag., 2020, 13, 100278 Search PubMed.
- M. Ismail, S. Gul, M. I. Khan, M. A. Khan, A. M. Asiri and S. B. Khan, Green Process. Synth., 2019, 8, 135–143 CAS.
- N. Masoudian, M. Rajabi and M. Ghaedi, Polyhedron, 2019, 173, 114105 CrossRef CAS.
- A. Kalaiselvi, S. M. Roopan, G. Madhumitha, C. Ramalingam and G. Elango, Spectrochim. Acta, Part A, 2015, 135, 116–119 CrossRef CAS PubMed.
- N. K. R. Bogireddy, H. A. Kiran Kumar and B. K. Mandal, J. Environ. Chem. Eng., 2016, 4, 56–64 CrossRef CAS.
- I. Kumar, M. Mondal, V. Meyappan and N. Sakthivel, Mater. Res. Bull., 2019, 117, 18–27 CrossRef CAS.
- A. Roy and N. Bharadvaja, Bioinspired, Biomimetic Nanobiomater., 2019, 8, 130–140 CrossRef.
- B. Moazzenchi and M. Montazer, Fibers Polym., 2020, 21, 522–531 CrossRef CAS.
- X. Wang and J. Gao, J. Nat. Fibers, 2019, 16, 307–318 CrossRef CAS.
- R. Seif El-Nasr, S. M. Abdelbasir, A. H. Kamel and S. S. M. Hassan, Sep. Purif. Technol., 2020, 230, 115860 CrossRef CAS.
- M. F. Al-Hakkani, SN Appl. Sci., 2020, 2, 505 CrossRef CAS.
- A. Salim, R. Vishnoi, H. Dixit, V. Chaudhary, J. Bhardwaj, D. Gupta, S. Aggarwal, U. K. Dwivedi, P. Kumar, S. Bishnoi, G. D. Sharma and R. Singhal, J. Mater. Sci.: Mater. Electron., 2022, 33, 15533–15545 CrossRef CAS.
- C. J. Wu, S. M. Chen, Y. J. Sheng and H. K. Tsao, J. Taiwan Inst. Chem. Eng., 2014, 45, 2719–2724 CrossRef CAS.
- I. Y. d. N. V. Brandão, E. F. de Macedo, P. H. B. de Souza Silva, A. F. Batista, S. L. G. Petroni, M. Gonçalves, K. Conceição, E. de Sousa Trichês, D. B. Tada and D. Maass, J. Environ. Manage., 2023, 338, 117804 CrossRef.
- G. Dinda, D. Halder, C. Vazquez-Vazquez, M. Arturo Lopez-Quintela and A. Mitra, J. Surf. Sci. Technol., 2015, 31, 117–122 CAS.
- H. S. Kim, Y. S. Seo, K. Kim, J. W. Han, Y. Park and S. Cho, Nanoscale Res. Lett., 2016, 11, 230 CrossRef PubMed.
- T. A. Gonçalves Martins, A. B. Botelho Junior, V. T. de Moraes and D. C. R. Espinosa, TMS 2020 149th Annu Meet Exhib Suppl Proc, 2020, pp. 1547–1557 Search PubMed.
- N. L. Pacioni, V. Filippenko, N. Presseau and J. C. Scaiano, Dalton Trans., 2013, 42, 5832–5838 RSC.
- S. Roy and J. W. Rhim, Appl. Sci., 2021, 11, 6307 CrossRef CAS.
- M. P. Das, P. Suguna, K. Prasad, J. Vijaylakshmi and M. Renuka, Int. J. Pharm. Pharmaceut. Sci., 2017, 9, 239–242 CAS.
- S. Roy, L. Van Hai, H. C. Kim, L. Zhai and J. Kim, Carbohydr. Polym., 2020, 231, 115729 CrossRef CAS PubMed.
- S. Shankar, X. Teng, G. Li and J. W. Rhim, Food Hydrocolloids, 2015, 45, 264–271 CrossRef CAS.
- A. Umer, S. Naveed, N. Ramzan, M. S. Rafique and M. Imran, Matéria, 2014, 19, 197–203 Search PubMed.
- V. Sreeja, K. N. Jayaprabha and P. A. Joy, Appl. Nanosci., 2014, 5, 435–441 CrossRef.
- J. Xiong, Y. Wang, Q. J. Xue and X. D. Wu, Green Chem., 2011, 13, 900–904 RSC.
- A. Sankhla, R. Sharma, R. S. Yadav, D. Kashyap, S. L. Kothari and S. Kachhwaha, Mater. Chem. Phys., 2016, 170, 44–51 CrossRef CAS.
- A. Pourjavadi and R. Soleyman, J. Nanoparticle Res., 2011, 13, 4647–4658 CrossRef CAS.
- E. N. Bakatula, D. Richard, C. M. Neculita and G. J. Zagury, Environ. Sci. Pollut. Res. Int., 2018, 25, 7823–7833 CrossRef CAS PubMed.
- M. A. Mekewi, A. S. Darwish, M. S. Amin, G. Eshaq and H. A. Bourazan, Egypt. J. Pet., 2016, 25, 269–279 CrossRef.
- A. P. Ingle, N. Duran and M. Rai, Appl. Microbiol. Biotechnol., 2014, 98, 1001–1009 CrossRef CAS PubMed.
- V. D. Cao, P. P. Nguyen, V. Q. Khuong, C. K. Nguyen, X. C. Nguyen, C. H. Dang and N. Q. Tran, Bull. Korean Chem. Soc., 2014, 35, 2645–2648 CrossRef.
- P. Sharma, S. Pant, V. Dave, K. Tak, V. Sadhu and K. R. Reddy, J. Microbiol. Methods, 2019, 160, 107–116 CrossRef CAS PubMed.
- M. C. Crisan, M. Teodora and M. Lucian, Appl. Sci., 2022, 12, 141 CrossRef CAS.
- X. Ma, S. Zhou, X. Xu and Q. Du, Front. Surg., 2022, 9, 905892 CrossRef PubMed.
- R. Wulandari, Y. Kusumastuti, A. Prasetya, Y. A. Purwestri, H. T. B. M. Petrus, A. Idrus and M. Tanaka, AIP Conf. Proc., 2024, 3073, 040001 CrossRef CAS.
- J. Gu, F. Chen, Z. Zheng, L. Bi, H. Morovvati and S. Goorani, Inorg. Chem. Commun., 2023, 150, 110442 CrossRef CAS.
- F. A. M. Alahdal, M. T. A. Qashqoosh, Y. K. Manea, R. K. A. Mohammed and S. Naqvi, Sustainable Mater. Technol., 2023, 35, 00540 Search PubMed.
- Y. G. Wang, D. Mei, V. A. Glezakou, J. Li and R. Rousseau, Nat. Commun., 2015, 6, 6511 CrossRef CAS PubMed.
- R. Phul, C. Kaur, U. Farooq and T. Ahmad, Mater. Sci. Eng. Int. J., 2018, 2, 90–94 Search PubMed.
- M. Jamil, Z. S. Khan, A. Ali and N. Iqbal, J. Alloys Compd., 2017, 694, 401–407 CrossRef CAS.
- S. H. Ammar, H. J. Khadim and A. Al-Farraji, Nanotechnol. Environ. Eng., 2020, 5, 16 CrossRef CAS.
- A. Umer, S. Naveed, N. Ramzan, M. S. Rafique and M. Imran, Materials, 2014, 19, 197–203 Search PubMed.
- B. Laban, M. Kosanin, G. Isic, U. Ralevic, M. Markovic, A. Jokic and V. Vasic, Univ. Thought, Publ. Nat. Sci., 2017, 7, 36–40 CrossRef.
- C. R. Marcelo, G. A. Puiatti, M. A. Nascimento, A. F. Oliveira and R. P. Lopes, J. Nanomater., 2018, 2018, 1–9 CrossRef.
- A. Gautam, S. Rawat, L. Verma, J. Singh, S. Sikarwar, B. C. Yadav and A. S. Kalamdhad, Environ. Nanotechnol. Monit. Manag., 2018, 10, 377–387 Search PubMed.
- S. Jamil and M. Fasehullah, Mater. Innovations, 2021, 1, 21–28 Search PubMed.
- N. Almulhem, C. Awada and N. M. Shaalan, Crystals, 2022, 12, 911 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |