
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDocetaxel-conjugated bile acid-derived nanomicelles can inhibit tumour progression with reduced toxicity†
Devashish
Mehta‡
 a,
Chhavi
Dua‡
b,
Ruchira
Chakraborty‡
b,
Poonam
Yadav
b,
Ujjaini
Dasgupta
a and
Avinash
Bajaj
*b
a,
Chhavi
Dua‡
b,
Ruchira
Chakraborty‡
b,
Poonam
Yadav
b,
Ujjaini
Dasgupta
a and
Avinash
Bajaj
*b
aAmity Institute of Biotechnology, Amity University Haryana, Panchgaon, Manesar, Gurgaon 122413, Haryana, India
bLaboratory of Nanotechnology and Chemical Biology, Regional Centre for Biotechnology, NCR Biotech Science Cluster, 3rd Milestone, Faridabad-Gurgaon Expressway, Faridabad 121001, Haryana, India. E-mail: bajaj@rcb.res.in
First published on 4th February 2025
Abstract
Docetaxel (DTX) is a highly effective chemotherapy drug commonly employed in the management of multiple cancers, such as breast, lung, and prostate cancer. However, its clinical usage is significantly hampered by its limited solubility, which limits its bioavailability, and its considerable toxic effects like neutropenia, neuropathy, and hypersensitive reactions. These limitations necessitate the development of innovative formulations to boost the therapeutic index of DTX. In this study, we aimed to enhance the tolerability and reduce the toxic effects of DTX by developing a novel hybrid scaffold (PIP-LCA-DTX), where we conjugated DTX to piperidine-derived lithocholic acid. This hybrid scaffold integrates the beneficial properties of bile acid-based drug conjugates and cationic amphiphiles to form stable and effective drug delivery systems. Our research demonstrates that PIP-LCA-DTX exhibits similar anticancer properties to DTX when tested against murine colon cancer (CT26) and melanoma (B16-F10) cell lines, indicating that the hybrid retains the therapeutic efficacy of the original drug. Our findings revealed that PIP-LCA-DTX forms stable nanomicelles (DTX-NMs) with an average hydrodynamic diameter of <150 nm and provides a promising delivery system by enhancing the solubility and stability of DTX. DTX-NMs showed significantly better tolerability and enhanced therapeutic efficacy (survival) compared to DTX alone. This improved tolerability, combined with the maintained therapeutic efficacy of DTX-NMs against murine cancer models, suggests that this hybrid scaffold could offer a more viable and safer option for cancer treatment.
1 Introduction
Cancer is a multifactorial complex disease characterised by abnormal cell proliferation and has emerged as one of the most formidable challenges in modern medicine.1 Emerging from genetic mutations and alterations, cancer cells defy the host regulatory mechanisms and lead to uncontrolled division with the potential to invade surrounding tissues.2,3 Docetaxel (DTX), part of the taxane class of chemotherapy agents, is a powerful anticancer drug commonly used to manage different solid tumours, including breast, lung and prostate cancers.4 Mechanistically, it disrupts microtubule dynamics, stabilises microtubules, and interferes with cell division, leading to apoptosis. DTX is commonly administered intravenously and has proven effective as a first- and second-line treatment for various cancers, either alone or in combination with other chemotherapy agents or targeted therapies.5 Despite its effectiveness, DTX can cause significant toxic effects, including neutropenia, neuropathy, fatigue, and alopecia, which impact patients' quality of life.6Drug delivery systems have tackled the challenges of chemotherapeutic drugs by protecting and transporting the active compounds to the target sites through controlled drug release.7–9 A notable breakthrough in this area is using nanoparticles to deliver chemotherapy drugs, which improves the pharmacological properties of drugs through the Enhanced Permeability and Retention (EPR) effect. The porous blood vessels and poor lymphatic drainage, characteristic of tumours, allow nanoparticles to accumulate preferentially in cancerous tissues, thereby reducing overall toxicity.10–12 This targeted accumulation boosts drug effectiveness and decreases off-target side effects typical of traditional chemotherapy. Nanoparticles also enhance the solubility and stability of drugs with poor solubility, such as DTX, making them more suitable for clinical use. Furthermore, they offer controlled and prolonged drug release, decrease the dosing frequency, and enhance patient adherence.13 The overexpression of esterases and acidic pH in the tumour microenvironment (TME) are suitable for developed ester-sensitive nanoformulations, allowing controlled drug release at the tumour site.14 Thus, nanoparticle-mediated chemotherapeutic delivery is a valuable asset in cancer therapy.
Hu et al. engineered core-crosslinked polymeric micelles (CCL-PMs) using poly(ethylene glycol)-b-poly[N-(2-hydroxypropyl) methacrylamide-lactate] (mPEG-b-p(HPMAm-Lacn)) copolymers and conjugated DTX to the micelles via ester bonds to improve tumour targeting.15 Etrych and colleagues developed a DTX-HPMA conjugate that exhibited stability under physiological conditions (pH 7.4) and in acidic environments (pH ∼5).16 Li's group developed Cellax, a polymer–drug conjugate composed of DTX, PEG, and carboxymethyl cellulose.17 Cellax therapy significantly reduced α-SMA + stroma and improved tumour perfusion, vascular permeability, and matrix depletion compared to DTX and nab-paclitaxel. Cellax significantly improved the pharmacokinetics and bio-distribution of DTX compared to free DTX, with reduced systemic toxicity and enhanced antitumour effectiveness.18 Demeule and colleagues developed a DTX-peptide conjugate, TH1902, targeting the sortilin (SORT1) receptor, which is associated with poor survival in triple-negative breast cancer (TNBC).19 Sanyal's group developed a micellar delivery system composed of amphiphilic polymer–dendron conjugates featuring a hydrophobic dendron core and a hydrophilic PEG shell.20 After loading DTX (10% wt), trastuzumab was conjugated to the micellar shell.20
Bile acids are the steroidal derivatives of cholesterol, which play an essential role in lipid homeostasis and possess anticancer properties at higher concentrations.21,22 Bile acids' amphiphilic nature and versatile chemistry encourage researchers to develop anticancer molecules and drug-delivery vehicles like nanomicelles and hydrogels.23,24 In our initial attempts, we designed and synthesised a series of lithocholic acid (LCA)-derived cationic amphiphiles to target tumour progression. Among the tested series, the piperidine-derivative of LCA (LCA-PIP) triggers apoptotic cell death in colon cancer cells.25 Furthermore, we witnessed that intratumoral injection of LCA-PIP inhibits tumour growth, highlighting that N-methyl piperidine at the C3 position of LCA is an essential anticancer pharmacophoric feature.25 The steroidal backbone, and hydroxyl and carboxylic acid groups make the bile acids amphiphilic. Numerous studies suggested that bile acid-based drug conjugates exhibit better therapeutic potency compared to the parent drugs.26 In a previous study, we synthesized two derivatives, a DTX-conjugated phosphatidylcholine derivative of LCA (PC-LCA-DTX) and a polyethyleneglycol derivative of LCA (LCA-PEG). Our findings revealed that combining PC-LCA-DTX with LCA-PEG resulted in nanomicelles (NMs) that showed enhanced anti-cancer efficacy and improved pharmacokinetic and biodistribution profiles compared to the standard polysorbate DTX formulation.27 Recently, our research has demonstrated that PC-LCA-DTX can also form NMs when combined with dexamethasone-tethered PEGylated LCA (PEG-LCA-DEX), and can inhibit tumour progression without inducing systemic toxicity.28 In addition, PC-LCA-DTX can also form chimeric NMs with combretastatin A4 (CA4) conjugated to PEGylated LCA (PEG-LCA-CA4), and exhibits promising anticancer properties.29
As DTX is classified as a Biopharmaceutics Classification System (BCS) class IV drug, it is characterized by low solubility and limited permeability.30 We propose linking DTX to LCA via an ester bond and incorporating a cationic piperidine charge at the C3 position, which would create a hybrid molecule (PIP-LCA-DTX, 1) (Fig. 1A) that can boost DTX's therapeutic effectiveness by addressing its solubility issues and minimising off-target effects. We show that PIP-LCA-DTX (1) can self-assemble with LCA-PEG (2) (Fig. 1A) and form stable NMs that are safer than the commercial formulations of DTX and inhibit tumour growth in murine colon cancer models.
 | ||
| Fig. 1 (A) Chemical structures of PIP-LCA-DTX and LCA-PEG. (B) Synthetic route for the synthesis of PIP-LCA-DTX. | ||
2 Materials and methods
2.1 Materials
Standard chromatography solvents, including hexane, ethyl acetate, dichloromethane (DCM), and methanol, were obtained from Merck and Rankem chemicals. Lithocholic acid (LCA), chloroacetic anhydride, anhydrous DCM, anhydrous methanol, and anhydrous N,N-dimethyl formamide (DMF) were procured from Sigma-Aldrich. p-Dimethyl aminopyridine (DMAP) was purchased from Spectrochemical Pvt. Ltd. Thin layer chromatography (TLC) was conducted on aluminium sheets coated with silica gel 60 GF 254 (Merck, Germany). TLC plates were examined under UV light and stained using a methanolic solution of phosphomolybdic acid. Bruker Avance 400 MHz spectrometer was used to record 1H NMR spectra in CDCl3 and DMSO-d6. Chemical shifts (δ) are reported in parts per million (ppm) relative to tetramethyl silane (TMS), which served as the internal standard. Peak descriptions include singlet (s), broad singlet (bs), doublet (d), triplet (t) or multiplet (m). An SCIEX 5600 was employed for high-resolution mass spectrometry (HRMS). Waters reverse-phase HPLC system with an Octyl-80Ts C18 column (S0005) measuring 4.6 × 250 mm (5 μm) and UV detection at 253 nm was used to assess the purity of PIP-LCA-DTX. The elution was performed using an isocratic program over 20 min. The solvent system includes MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O:THF (37.5:60:2.5 with 0.1% NH4OH) as solvent A and ACN as solvent B in a 5:5 ratio with the flow rate set at 1 mL min−1.
:7 mixture of ethyl acetate and hexane as the mobile phase. The purified compound was characterised through 1H NMR and HRMS. Compound 7: white solid; yield: 94.3%; 1H NMR (400 MHz, CDCl3) δ: 0.63 (s, 3H), 0.84–2.25 (m, steroid), 4.02 (s, 2H), 4.03–4.03 (d, 2H, J = 4 Hz), 4.70–4.80 (m, 1H), 5.59 (s, 1H). HRMS (ESI): calculated for C28H44ClNO3; observed (M + ACN + H) 531.2764.
:DCM (5 mL each). On the other hand, a CuSO4·5H2O (5 mg, 0.05 eq.) and sodium ascorbate (12 mg, 0.1 eq.) aqueous mixture (1 mL) was prepared and added to the reaction mixture. After 3 h, the completion of the reaction was confirmed using TLC. The compound was purified using CombiFlash chromatography using silica (230–400 mesh size) as a stationary phase and MeOH:DCM (0.5:9.5) as a mobile phase. The purified compound was characterized using 1H NMR and HRMS. Compound 8: white solid; yield: 90.2%; 1H NMR (400 MHz, CDCl3) δ: 0.63 (s, 3H), 0.88–2.47 (m, steroid), 3.97–3.98 (d, 2H, J = 4 Hz), 4.05 (s, 2H), 4.19–4.23 (m, 2H), 4.34–4.38 (m, 2H), 4.59–4.60 (d, 2H, J = 4 Hz), 4.75–4.81 (m, 1H), 4.98–5.00 (d, 1H, J = 8 Hz), 5.26 (s, 1H), 5.32 (s, 1H), 5.52–5.60 (m, 3H), 5.70–5.71 (d, 1H, J = 4 Hz), 6.23–6.35 (m, 2H), 7.27–7.29 (m, 1H), 7.39–7.43 (m, 4H), 7.51–7.55 (m, 2H), 7.60–7.64 (m, 1H), 7.85–7.87 (d, 2H, J = 8 Hz), 8.12–8.16 (m, 5H). HRMS (ESI): calculated for C79H100ClN5O8; observed (M + CH3OH + H): 1474.5781.
:H2O:THF (37.5:60:2.5 with 0.1% NH4OH) as solvent A and ACN as solvent B in a 5:5 ratio with the flow rate set at 1 mL min−1.
:7 mixture of ethyl acetate and hexane as the mobile phase. The purified compound was characterised through 1H NMR and HRMS. Compound 7: white solid; yield: 94.3%; 1H NMR (400 MHz, CDCl3) δ: 0.63 (s, 3H), 0.84–2.25 (m, steroid), 4.02 (s, 2H), 4.03–4.03 (d, 2H, J = 4 Hz), 4.70–4.80 (m, 1H), 5.59 (s, 1H). HRMS (ESI): calculated for C28H44ClNO3; observed (M + ACN + H) 531.2764.
:DCM (5 mL each). On the other hand, a CuSO4·5H2O (5 mg, 0.05 eq.) and sodium ascorbate (12 mg, 0.1 eq.) aqueous mixture (1 mL) was prepared and added to the reaction mixture. After 3 h, the completion of the reaction was confirmed using TLC. The compound was purified using CombiFlash chromatography using silica (230–400 mesh size) as a stationary phase and MeOH:DCM (0.5:9.5) as a mobile phase. The purified compound was characterized using 1H NMR and HRMS. Compound 8: white solid; yield: 90.2%; 1H NMR (400 MHz, CDCl3) δ: 0.63 (s, 3H), 0.88–2.47 (m, steroid), 3.97–3.98 (d, 2H, J = 4 Hz), 4.05 (s, 2H), 4.19–4.23 (m, 2H), 4.34–4.38 (m, 2H), 4.59–4.60 (d, 2H, J = 4 Hz), 4.75–4.81 (m, 1H), 4.98–5.00 (d, 1H, J = 8 Hz), 5.26 (s, 1H), 5.32 (s, 1H), 5.52–5.60 (m, 3H), 5.70–5.71 (d, 1H, J = 4 Hz), 6.23–6.35 (m, 2H), 7.27–7.29 (m, 1H), 7.39–7.43 (m, 4H), 7.51–7.55 (m, 2H), 7.60–7.64 (m, 1H), 7.85–7.87 (d, 2H, J = 8 Hz), 8.12–8.16 (m, 5H). HRMS (ESI): calculated for C79H100ClN5O8; observed (M + CH3OH + H): 1474.5781.
2.2 Maintenance of cell lines
CT26 and B16-F10 cells were maintained as monolayers. CT26 is an adherent murine colorectal carcinoma cell line with a fibroblast morphology and was cultured in the RPMI-1640 medium. B16-F10 is an adherent murine melanoma cell line with a spindle-shaped epithelial morphology and was cultured in the DMEM medium. The culture media were enriched with 10% (w/v) fetal bovine serum and antibiotics (100 I.U. mL−1 penicillin and 100 μg mL−1 streptomycin). Cells were incubated at 37 °C in a humidified environment containing 5% CO2. To maintain optimal cell density and promote continuous growth, the cultures underwent periodic trypsinization for passaging.2.3 Propidium iodide uptake analysis
In a 24-well plate, CT26 and B16-F10 cells were plated at 40000 cells per well. Following a 24 h adhesion period, the media were exchanged with fresh media. The cells were treated with 500 nM (DTX equivalent concentration) of DTX and PIP-LCA-DTX for 24 h. Subsequently, the cells were collected using trypsin, washed twice with DPBS, and the resulting pellet was resuspended in 200 μL DPBS. The suspension was stained with 5 μL of 50 μg mL−1 propidium iodide for 15 min. Flow cytometry analysis was performed using a BD FACSVerse to determine the percentage of dead cells.
2.4 Mitochondrial membrane potential analysis
In a 12-well plate, CT26 or B16-F10 cells were plated at 105 per well and left to adhere for 24 h. After this period, the media were exchanged with fresh media. The cells underwent treatment with 500 nM of DTX and PIP-LCA-DTX (DTX equivalent concentration) for 24 h. Following treatment, DiOC6 dye (20 nM) was added to each well, and the plate was incubated for 30–45 min at 37 °C. Subsequently, the cells were collected using trypsin, washed twice with DPBS, and the resulting pellet was resuspended in 200 μL of DPBS. Flow cytometry analysis was performed using a BD FACS Verse to quantify the percentage of DiOC6 negative cells.2.5 Cell cycle analysis
In a 12-well plate, cells were seeded at a density of 105 cells per well. After 24 h incubation, the culture medium was exchanged for a fresh medium. The cells were then treated with 500 nM of DTX and PIP-LCA-DTX (DTX equivalent concentration). Following 24 h incubation, cells were collected using trypsin and washed twice with DPBS. Cell fixation was achieved by slowly adding cold 70% ethanol with continuous vortexing, followed by overnight storage at 4 °C. Cells underwent two additional DPBS washes to remove residual ethanol. Subsequently, 50 μL of 100 μg mL−1 RNase was introduced, and cells were incubated at 37 °C for 1 h. Cell staining was performed at room temperature for 15 min using 5 μL of 50 μg mL−1 propidium iodide. Flow cytometric analysis was conducted on a BD FACSVerse to determine the proportion of cells in the G0/G1, S, and G2/M phases of the cell cycle.2.6 Preparation of PIP-LCA-DTX NMs
A round-bottom flask containing 2 mL of sodium chloride solution (normal saline) and a magnetic bead was stirred at 1500 rpm under ambient conditions. Tetrahydrofuran solution (300 μL) comprising a 1:1 (w/w) mixture of PIP-LCA-DTX (0.95 mg, 46% as mol%) and LCA-PEG (0.95 mg, 54% as mol%) was added dropwise to the saline in the flask over a period of 12–15 min using a Hamilton syringe. After complete addition, a cloudy turbid solution was formed. Rotary evaporation was carried out to remove the tetrahydrofuran solvent, which led to PIP-LCA-DTX nanomicelles (DTX-NMs).
2.7 Preparation of DiD-loaded PIP-LCA-DTX NMs
We prepared DTX-NMs as mentioned above, where 2% DiD ((1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine and 4-chlorobenzenesulfonate salt)) solution is added during the nanomicelle formulation process. After the NMs are formed, they are subjected to dialysis (1 kDa molecular weight cut-off) for 1 h to remove any excess, unincorporated dye. Following dialysis, the purified DiD-doped nanomicelles are collected for further experimental use.2.8 Characterization of DTX-NMs
The hydrodynamic diameter (Z-avg.), polydispersity index (PdI) and count rate of the NMs were measured by DLS using a Zetasizer NanoZS90. NMs (100 μL) were diluted to 1 mL using saline in a disposable cuvette, and three sets of 10 measurements were performed at a 90° scattering angle to obtain the Z-avg. and PdI for the NMs. The morphology of NMs was visualized through a JEM-1400Flash transmission electron microscope (Jeol, Japan). Prepared NMs were negatively stained with 1% PTA and subjected to TEM. For storage stability studies, NMs were kept at room temperature (25 °C), and 4 °C, and DLS measurements were performed at different time points for 30 days. For serum stability studies, NMs were kept under 10% and 50% FBS conditions at 37 °C and the Z-avg. and PdI were monitored at different time points.2.9 Cellular uptake of PIP-LCA-DTX NMs
In a 12-well plate, CT26 and B16-F10 cells were plated at a density of 105 cells per well and incubated to attach for 24 h. The cells were then exposed to DiD-loaded DTX-NMs at 500 nM, 1 μM, and 2 μM for 6 h. Following this treatment period, the cells were collected and analyzed using flow cytometry.2.10 Maximum tolerability dose (MTD) studies
C57BL/6 male mice (6–8 weeks) were employed to determine the MTD of DTX-NMs and DTX solution (DTX-Sol, a polysorbate solution of DTX). The treatment groups received either DTX-Sol (20 mg kg−1) or DTX-NMs (20 mg kg−1 of DTX equivalent). These formulations were administered via tail vein injection once daily for five consecutive days. Throughout the study, body weight was monitored daily for 15 days.2.11 Animal studies
Murine colorectal cancer cells (CT26) (1.5 × 106) in FBS (200 μL) were injected subcutaneously into male BALB/c mice (6–8 weeks). When the tumour volume reached ∼50 mm3, the mice were randomly assigned to three groups and subjected to different treatments: untreated, DTX-Sol (5 mg kg−1), and DTX-NMs (5 mg kg−1 DTX). All the doses were given by tail vein injection on alternate days. The mice's body weight and tumour volume were measured every alternate day. The length (L) and breadth (B) of the tumour were calculated using a digital vernier caliper, and the tumour volume was determined by using the formula L × B2/2. After the end of the experiment, mice were kept for survival study. For xenograft studies, HCT116 cells (3 × 106) were injected subcutaneously into 8-week-old male NOD/SCID mice. The mice underwent similar treatments to those described above.2.12 Statistical analysis
The results are presented as means with either the standard error of the mean (SEM) or standard deviation (SD), derived from a minimum of three independent samples. Statistical analysis was performed using either the unpaired Student's t-test or two-way analysis of variance (ANOVA) for comparisons. Statistical significance was determined by a p-value less than 0.05.3 Results and discussion
3.1 Synthesis of DTX-tethered bile acid cationic amphiphiles
We synthesised an azide derivative of DTX (4) from DTX (3) and used LCA (5) to synthesize an alkyne derivative of LCA (6) (Fig. 1B) as per previously published protocols.27 The alkyne derivative of LCA (6) was further reacted with chloroacetic anhydride using DMAP to yield compound 7. Next, we employed copper-mediated click chemistry to conjugate DTX-azide (4) with compound 7 to yield compound 8. Purified compound 8 was treated with N-methyl piperidine to obtain compound 1 (PIP-LCA-DTX). Intermediate molecules and PIP-LCA-DTX were characterised through 1H NMR and HRMS (Fig. S1–S3†). The purity of PIP-LCA-DTX was determined using reverse-phase HPLC (ESI†). LCA-PEG (2) was synthesized as per the earlier-mentioned procedure.283.2 PIP-LCA-DTX can target the mitochondrial membrane potential and arrest the cell cycle
We investigated the cytotoxic properties of DTX and PIP-LCA-DTX against murine colon cancer (CT26) and melanoma (B16-F10) cells through propidium iodide (PI) uptake assay. The intact plasma membrane of live cells halts the entry of PI. However, PI can permeate dead cells due to their loss of membrane integrity, and the quantification of PI fluorescence correlates directly with the percentage of dead cells within a population.31 We treated CT26 and B16-F10 cells with DTX and PIP-LCA-DTX (500 nM DTX equivalent) for 24 h, stained the cells with PI, and performed flow cytometry analysis. Our findings showed that DTX and PIP-LCA-DTX exhibit similar cytotoxic properties (Fig. 2A and B). The mitochondrial membrane potential (MMP) is a critical marker of cell vitality. The inner mitochondrial membrane typically prevents ion passage, while the matrix retains a highly anionic character due to the expulsion of protons. 3,3′-dihexyloxacarbocyanine iodide (DiOC6) is a lipophilic cationic dye that can accumulate in the metabolically active mitochondria. Loss of MMP leads to the leaking of DiOC6 dye from the mitochondria, which can be detected through flow cytometry.32 Therefore, we performed MMP analysis to determine whether DTX and PIP-LCA-DTX induce any change in the MMP of CT26 and B16-F10 cells. We treated CT26 and B16-F10 cells with DTX and PIP-LCA-DTX at 500 nM (DTX equivalent) for 24 h. Flow cytometric analysis showed that DTX and PIP-LCA-DTX treatment cause a shift of the DiOC6 fluorescence intensity towards the left compared to that of the untreated group, indicating leakage of the dye out of the mitochondria due to disruption of mitochondria and initiation of programmed cell death (Fig. S5†). Moreover, the proportion of DiOC6-negative cells was notably greater in all treatment groups compared to the untreated group (Fig. 2C and D).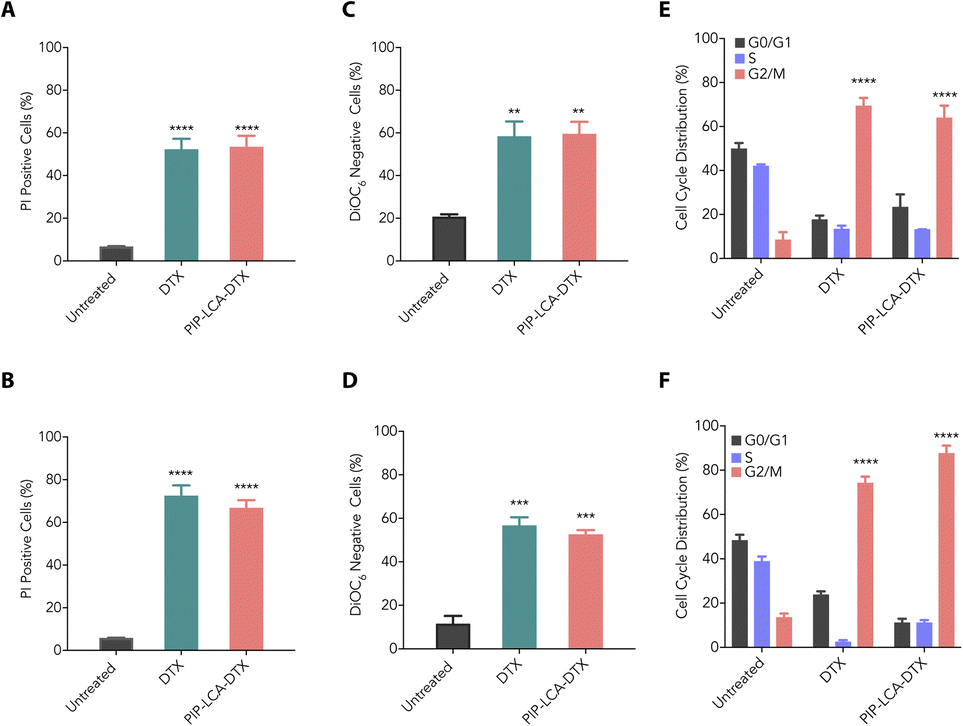 | ||
| Fig. 2 (A and B) Cytotoxic effects of DTX and PIP-LCA-DTX on CT26 (A) and B16-F10 (B) cells, shown as the percentage of PI-positive cells. (C and D) MMP disruption by DTX and PIP-LCA-DTX, shown as the percentage of DiOC6-negative CT26 (C) and B16-F10 (D) cells. (E and F) Cell cycle analysis of CT26 (E) and B16-F10 (F) cells following treatment with DTX and PIP-LCA-DTX. | ||
We assessed the impact of the treatment of DTX and PIP-LCA-DTX on cell cycle arrest using flow cytometry analysis. CT26 and B16-F10 cells were treated with DTX and PIP-LCA-DTX at 500 nM (DTX equivalent) for 24 h. A notable increase in the number of cells in the G2/M phase was observed in the treated groups relative to the untreated control (Fig. S6†). This suggests that DTX and PIP-LCA-DTX treatment can halt the cell cycle progression at the G2/M checkpoint in both CT26 (Fig. 2E) and B16-F10 (Fig. 2F) cells. These results suggest that the developed cationic drug conjugate demonstrates comparable pharmacological potential to that of DTX.
3.3 PIP-LCA-DTX can self-assemble and form stable NMs
Amphiphilic properties inherent in bile acids have paved the way for researchers to explore and create self-assembled formulations. These formulations capitalise on the unique ability of bile acids to spontaneously organise into structures such as micelles and liposomes in an aqueous environment due to their dual hydrophilic and hydrophobic characteristics. This versatility has facilitated the creation of diverse delivery systems for drugs and genes, presenting promising applications in healthcare biomaterials. We employed the nanoprecipitate method to develop DTX-NMs, where we dissolved PIP-LCA-DTX (1) and LCA-PEG (2) (helper lipid) in THF in different ratios and slowly added them to normal saline to find the optimized ratio. We concentrated the mixture to afford dispersed NMs (Fig. 3A). We characterised the NMs (prepared using a 1:1 (w/w) mixture of PIP-LCA-DTX and LCA-PEG) through DLS and transmission electron microscopy (TEM). DLS studies showed that these NMs have an average hydrodynamic diameter of ∼143 nm with a PDI of 0.201 (Fig. 3B). TEM studies revealed that these NMs are spherical and confirmed that their size is <150 nm (Fig. 3C).
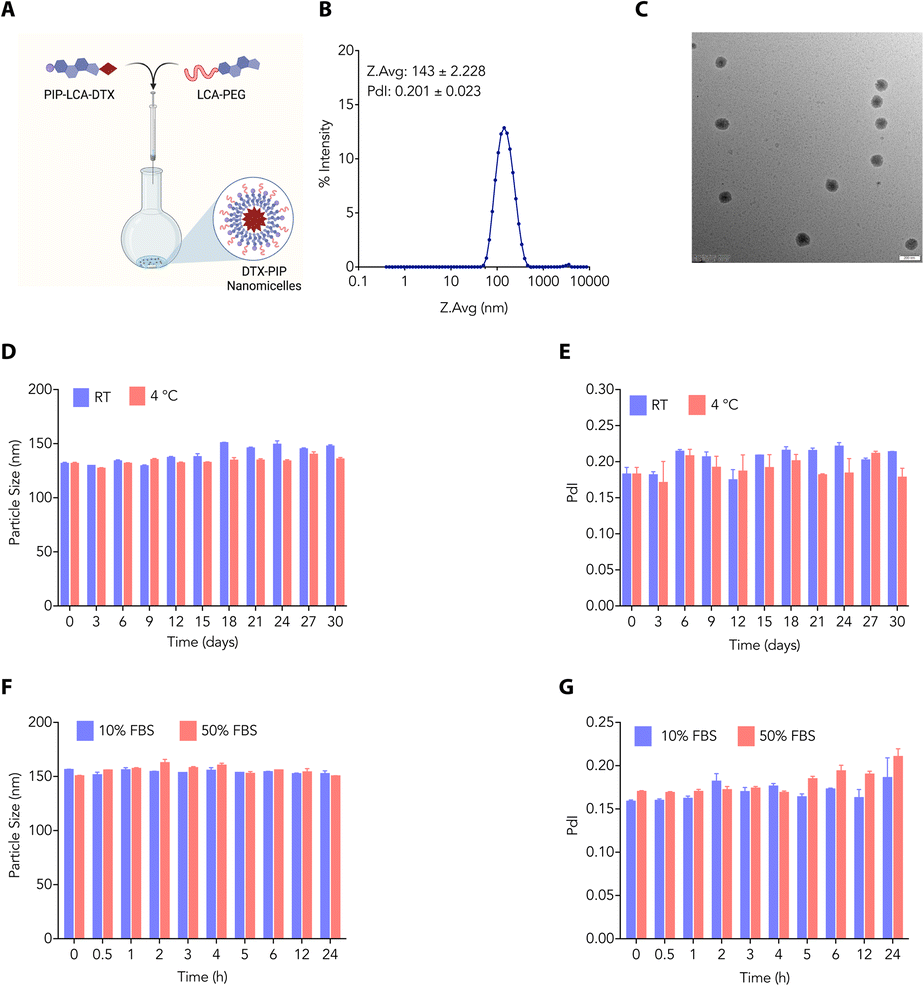 | ||
| Fig. 3 (A) A schematic illustrating the preparation of DTX-NMs. (B) Hydrodynamic diameter of DTX-NMs. (C) Transmission electron microscopy (TEM) images of DTX-NMs (scale bar: 200 nm). (D–G). Stability profile of DTX-NMs in terms of the hydrodynamic diameter (D and F) and PDI (E and G) under storage (D and E) and serum (F and G) conditions. | ||
As the stability of the nanoformulations plays an important role in clinical translation, we evaluated the stability profile of NMs under different conditions. We prepared and stored DTX-NMs at room temperature and 4 °C and checked the average size of these NMs at different time points. These NMs showed negligible changes in average size up to 30 days at room temperature and 4 °C (Fig. 3D and E). Furthermore, we prepared and incubated DTX-NMs under 10% and 50% FBS conditions and witnessed an enhancement in the average size of NMs due to protein corona formation (Fig. 3F and G). These findings suggest that DTX-NMs are <150 nm in size with spherical morphology and are stable under different conditions. Additionally, we examined the ability of cancer cells to take up these NMs. Therefore, we loaded the DTX-NMs with DiD dye to yield fluorescent NMs, which were then incubated with CT26 and B16-F10 cells. After 6 h of incubation, flow cytometry was employed to analyze their uptake. The fluorescence intensity of DiD, detected in the PI channel, indicated the percentage uptake of NMs. We observed that DTX-NMs exhibited a dose-dependent uptake by both CT26 and B16-F10 cells (Fig. 4A and B). Notably, we observed that the percentage uptake of DiD-loaded DTX-NMs was more pronounced in B16-F10 cells compared to CT26 cells.
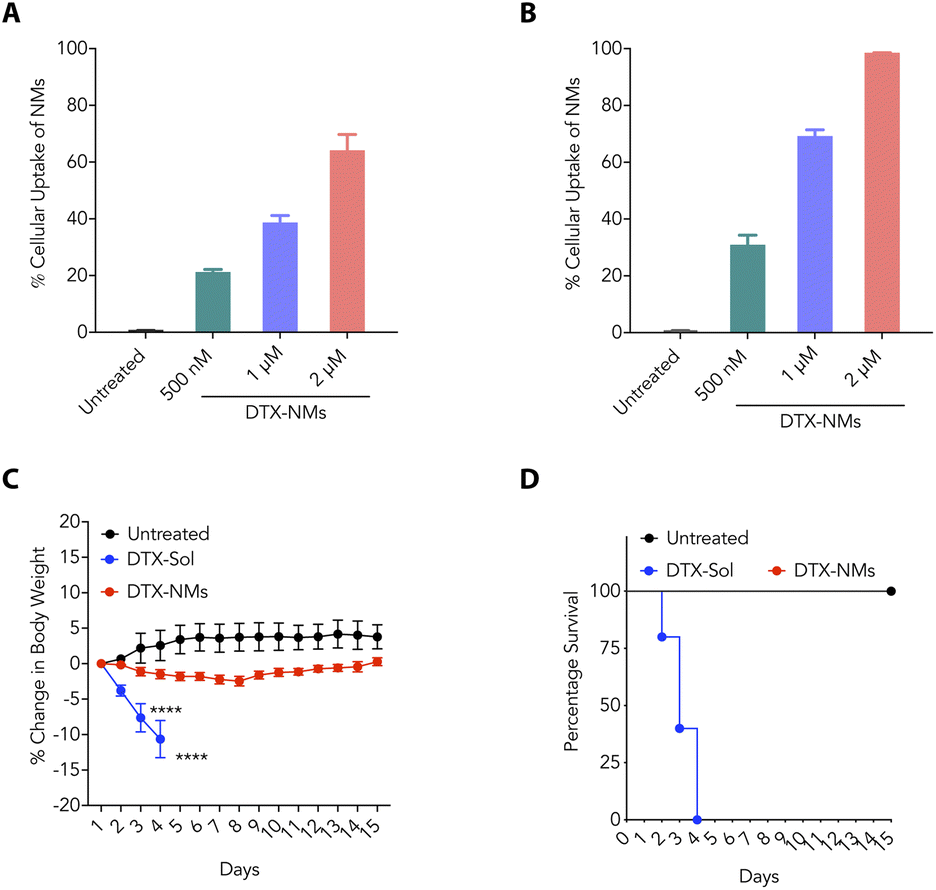 | ||
| Fig. 4 (A and B) Percent of DiD-positive CT26 (A) and B16-F10 (B) cells confirming the uptake of DTX-NMs. (C and D) Change in body weight of mice (C) and percentage survival (D) upon treatment of healthy mice (n = 5) with polysorbate solution of DTX (DTX-Sol) and DTX-NMs. | ||
3.4 DTX-NMs are safer than DTX-Sol
Since chemotherapeutic drugs are well-known for causing severe toxicity,33 it is important to investigate their safety profiles and maximum tolerability. To determine the maximum tolerable dose (MTD) of DTX-NMs, we employed C57BL/6 male mice (6–8 weeks old). We used DTX-Sol (20 mg kg−1) and DTX-NMs (20 mg kg−1 of DTX equivalent). These regimens were administered through the tail vein for five consecutive days. We observed that DTX-Sol-treated mice lost body weight and died after four doses (Fig. 4C and D). DTX-NMs treatment did not significantly impact the mice's body weight (Fig. 4C and D). These findings suggest that DTX-NMs are much safer than DTX-Sol.3.5 DTX-NMs effectively inhibit tumour progression
The compelling in vitro anticancer activity and favourable safety profile of DTX-NMs prompted us to evaluate the efficacy of these NMs in syngeneic and xenograft murine colon tumour models. In the syngeneic model, CT26 cells were implanted subcutaneously into the left flank of male BALB/c mice (6–8 weeks old, 20–25 g). When the tumours reached ∼50 mm3, the mice were randomly divided into three groups (n = 6 per group): untreated, DTX-Sol (polysorbate-based DTX suspension)-treated, and DTX-NMs-treated.We witnessed that DTX-NMs treatment resulted in a significant reduction in tumour growth compared to the untreated group and demonstrated effectiveness comparable to DTX-Sol (Fig. 5A). Notably, mice treated with DTX-Sol experienced a substantial body weight reduction (∼15%), indicating toxicity, whereas DTX-NMs showed no adverse effect (Fig. 5B). Median survival was 42 days for the DTX-NMs treated group, 33 days for the DTX-Sol group, and 28.5 days for the untreated group (Fig. 5C). This trend aligns with findings by Ernsting et al. where a polysorbate-based formulation of DTX at a dose of 40 mg kg−1 led to notable body weight loss in mice without improving survival compared to the untreated group. In contrast, a DTX-tethered CMC nanoformulation enhanced the survival outcomes.18 Similarly, Guo et al. observed that standard DTX treatment led to weight loss alongside tumour suppression, and their optimized DTX formulation minimized the weight loss, demonstrating reduced toxicity while maintaining efficacy.34
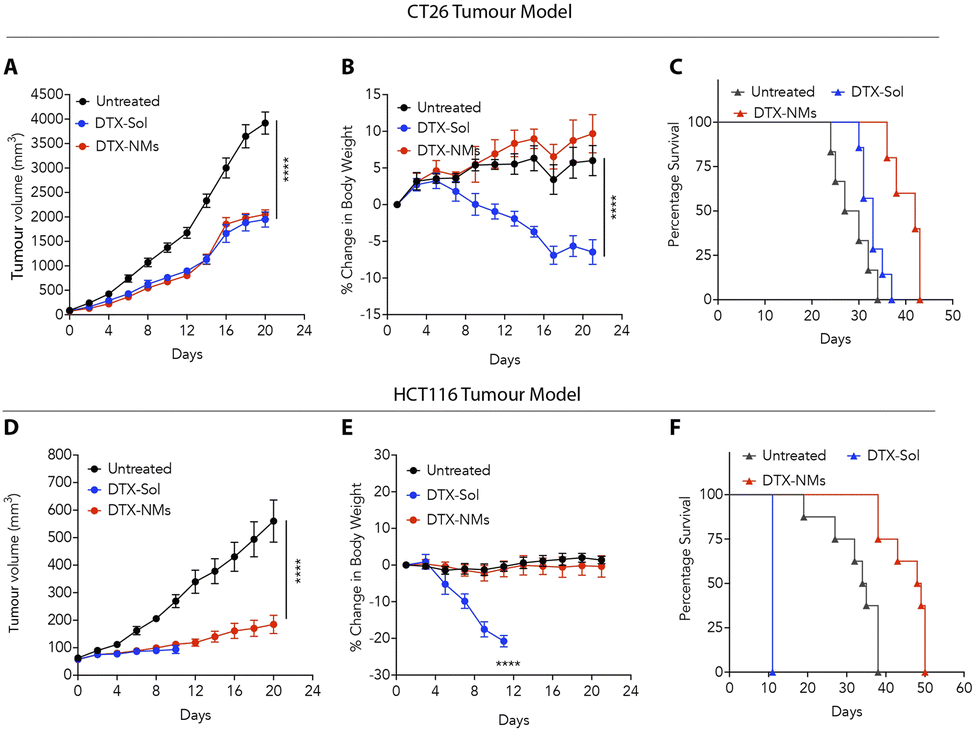 | ||
| Fig. 5 (A–C) Tumour growth kinetics (A), body weight change (B), and survival rate (C) in CT26 tumour-bearing BALB/c male mice (n = 5–6) under various treatments. (D–F) Tumour growth kinetics (D), body weight change (E), and survival rate (F) in HCT116 tumour-bearing NOD-SCID mice (n = 5–6) under various treatments. | ||
For xenograft studies, HCT116 cells (3 × 106) were subcutaneously injected into immunocompromised NOD-SCID mice. The same treatment protocol was followed with groups (n = 6 per group) receiving either DTX-Sol, DTX-NMs, or no treatment. DTX-NMs treated mice showed significant antitumour efficacy with no observable toxicity compared to DTX-Sol treatment (Fig. 5D and E). DTX-Sol treatment resulted in severe toxicity, requiring euthanasia of the mice on day 11 due to a drastic (∼20%) drop in body weight (Fig. 5E). As NOD-SCID mice lack functional B and T cells,35,36 they are more prone to drug toxicity due to their impaired immune response and altered metabolic pathways, which may lead to slower drug clearance. Furthermore, these mice exhibit heightened sensitivity to inflammation, which can further increase the risk of toxicity. These factors likely contribute to the observed adverse effects in the DTX-Sol group, making these mice more vulnerable to drug-induced toxicity compared to their immunocompetent counterparts. The median survival times for untreated, DTX-Sol and DTX-NMs treated mice were 34.5, 11, and 48.5 days, respectively (Fig. 5F), suggesting the advantages of DTX-NMs over DTX-Sol.
4 Conclusion
Our study demonstrates that the developed hybrid scaffold, PIP-LCA-DTX, forms stable NMs with an average size of ∼150 nm, offering a promising approach to enhance the therapeutic profile of DTX. DTX-NMs showed comparable anticancer efficacy to DTX against CT26 and HCT116 murine tumour models but with improved tolerability, as evidenced by the lack of weight loss in treated mice. These findings suggest that DTX-NMs can be optimized further as a viable alternative to standard DTX formulations, reducing adverse effects while maintaining therapeutic potency. Further investigation into the underlying mechanisms of these effects could enhance the clinical potential of these NMs in cancer therapy.Animal ethical statement
The Institutional Animal Ethics Committee of the Regional Centre for Biotechnology evaluated and sanctioned the animal experimental protocols (RCB/IAEC/2023/166). All studies were performed in compliance with the guidelines established by the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), under the Government of India.Data availability
The data supporting this article have been included in the ESI.†Author contributions
D. M. synthesised the compound, prepared the formulation, conducted the animal experiments, and wrote the manuscript. C. D. performed both the in vitro and animal experiments. R. C. carried out animal experiments. P. Y. assisted with formulation standardisation and in vitro experiments. U. D. supervised and edited the manuscript. A. B. conceived the idea and supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We sincerely thank RCB for providing intramural funding and acknowledge the support of the Department of Biotechnology (DBT), the Science and Engineering Research Board (SERB), and the Department of Science and Technology, Government of India, in facilitating the work conducted in the laboratories of UD and AB. The students acknowledge funding agencies, including DBT, UGC, SERB, and ICMR, for their research fellowships. The animal studies were carried out at the small animal facility of the Regional Centre for Biotechnology, with support from grant BT/PR5480/INF/22/158/2012 (DBT). We also thank the DBT e-Library Consortium (DeLCON) for granting availability to essential e-resources.References
- D. Hanahan, Cancer Discovery, 2022, 12, 31–46 CrossRef CAS PubMed.
- T. Armaghany, J. D. Wilson, Q. Chu and G. Mills, Gastrointest. Cancer Res., 2012, 5, 19 Search PubMed.
- C. Schiliro and B. L. Firestein, Cells, 2021, 10, 1056 CrossRef CAS PubMed.
- R. Gupta, M. M. Kadhim, A. T. Jalil, M. Q. Alasheqi, F. Alsaikhan, N. K. Mukhamedova, A. A. Ramírez-Coronel, Z. H. Jawhar, P. Ramaiah and M. Najafi, Int. Immunopharmacol., 2023, 119, 110214 CrossRef CAS PubMed.
- M. Imran, S. Saleem, A. Chaudhuri, J. Ali and S. Baboota, J. Drug Delivery Sci. Technol., 2020, 60, 101959 CrossRef CAS.
- J. Chen, Y. Zhang, X. Zhang, J. Zhao, Y. Ni, S. Zhu, B. He, J. Dai, Z. Wang, J. Liang, X. Zhu, P. Shen, H. Zeng and G. Sun, Front. Pharmacol, 2022, 12, 789319 CrossRef PubMed.
- X. Wang, C. Li, Y. Wang, H. Chen, X. Zhang, C. Luo, W. Zhou, L. Li, L. Teng, H. Yu and J. Wang, Acta Pharm. Sin. B, 2022, 12, 4098–4121 CrossRef CAS PubMed.
- S. Senapati, A. K. Mahanta, S. Kumar and P. Maiti, Signal Transduction Targeted Ther., 2018, 3, 7 CrossRef PubMed.
- L. Hong, W. Li, Y. Li and S. Yin, RSC Adv., 2023, 13, 21365–21382 RSC.
- T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. Yang and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 12320–12364 CrossRef CAS PubMed.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- H. Maeda, K. Tsukigawa and J. Fang, Microcirculation, 2016, 23, 173–182 CrossRef CAS PubMed.
- C. J. Prange, X. Hu and L. Tang, Biomatter, 2023, 303, 122353 CrossRef CAS PubMed.
- Q. Hu, C. J. Rijcken, R. Bansal, W. E. Hennink, G. Storm and J. Prakash, Biomater, 2015, 53, 370–378 CrossRef CAS PubMed.
- T. Etrych, M. Šírová, L. Starovoytova, B. Říhová and K. Ulbrich, Mol. Pharm., 2010, 7, 1015–1026 CrossRef CAS PubMed.
- M. Murakami, M. J. Ernsting, E. Undzys, N. Holwell, W. D. Foltz and S. D. Li, Cancer Res., 2013, 73, 4862–4871 CrossRef CAS PubMed.
- M. J. Ernsting, W. L. Tang, N. W. MacCallum and S. D. Li, Biomater, 2012, 33, 1445–1454 CrossRef CAS PubMed.
- M. Demeule, C. Charfi, J. C. Currie, A. Larocque, A. Zgheib, S. Kozelko, R. Béliveau, C. Marsolais and B. Annabi, Cancer Sci., 2021, 112, 4317–4334 CrossRef CAS PubMed.
- B. S. Bolu, B. Golba, A. Sanyal and R. Sanyal, Biomater. Sci., 2020, 8, 2600–2610 RSC.
- A. Molinaro, A. Wahlström and H. U. Marschall, Trends Endocrinol. Metab., 2018, 29, 31–41 CrossRef CAS PubMed.
- A. A. Goldberg, V. I. Titorenko, A. Beach and J. T. Sanderson, PeerJ, 2013, 1, e122 CrossRef PubMed.
- F. Meng, J. Wang, Y. He, G. M. Cresswell, N. A. Lanman, L. T. Lyle, T. L. Ratliff and Y. Yeo, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2122595119 CrossRef CAS PubMed.
- S. Pal, N. Medatwal, S. Kumar, A. Kar, V. Komalla, P. S. Yavvari, D. Mishra, Z. A. Rizvi, S. Nandan, D. Malakar, M. Pillai, A. Awasthi, P. Das, R. D. Sharma, A. Srivastava, S. Sengupta, U. Dasgupta and A. Bajaj, ACS Cent. Sci., 2019, 5, 1648–1662 CrossRef CAS PubMed.
- M. Singh, S. Bansal, S. Kundu, P. Bhargava, A. Singh, R. K. Motiani, R. Shyam, V. Sreekanth, S. Sengupta and A. Bajaj, MedChemComm, 2015, 6, 192–201 RSC.
- M. L. Navacchia, E. Marchesi and D. Perrone, Molecules, 2020, 26, 25 CrossRef PubMed.
- V. Sreekanth, A. Kar, S. Kumar, S. Pal, P. Yadav, Y. Sharma, V. Komalla, H. Sharma, R. Shyam, R. D. Sharma, A. Mukhopadhyay, S. Sengupta, U. Dasgupta and A. Bajaj, Angew. Chem., Int. Ed., 2021, 60, 5394–5399 CrossRef CAS PubMed.
- P. Yadav, K. Rana, V. Nardini, A. Khan, T. Pani, A. Kar, D. Jain, R. Chakraborty, R. Singh, S. K. Jha, D. Mehta, H. Sharma, R. D. Sharma, S. V. S. Deo, S. Sengupta, V. S. Patil, L. H. Faccioli, U. Dasgupta and A. Bajaj, J. Controlled Release, 2024, 368, 548–565 CrossRef CAS PubMed.
- P. Yadav, K. Rana, R. Chakraborty, A. Khan, D. Mehta, D. Jain, B. Aggarwal, S. K. Jha, U. Dasgupta and A. Bajaj, Nanoscale, 2024, 16, 10350–10365 RSC.
- K. Kreis, D. Horenkamp-Sonntag, U. Schneider, J. Zeidler, G. Glaeske and L. Weissbach, BJU Int., 2021, 129, 1–10 Search PubMed.
- L. C. Crowley, A. P. Scott, B. J. Marfell, J. A. Boughaba, G. Chojnowski and N. J. Waterhouse, Cold Spring Harb. Protoc., 2016, 2016, pdbprot087163 CrossRef PubMed.
- H. Rottenberg and S. Wu, Biochim. Biophys. Acta, Mol. Cell Res., 1998, 1404, 393–404 CrossRef CAS PubMed.
- U. Anand, A. Dey, A. K. S. Chandel, R. Sanyal, A. Mishra, D. K. Pandey, V. De Falco, A. Upadhyay, R. Kandimalla, A. Chaudhary, J. K. Dhanjal, S. Dewanjee, J. Vallamkondu and J. M. Pérez de la Lastra, Genes Dis., 2023, 10, 1367–1401 CrossRef CAS PubMed.
- L. Guo, X. Qin, L. Xue, J. Y. Yang, Y. Zhang, S. Zhu, G. Ye, R. Tang and W. Yang, Front. Pharmacol, 2022, 13, 964076 CrossRef CAS PubMed.
- M. Füchtenbusch, E. Larger, K. Thebault and C. Boitard, Horm. Metab. Res., 2005, 37, 63–67 CrossRef PubMed.
- K. Girdhar, Q. Huang, I. T. Chow, T. Vatanen, C. Brady, A. Raisingani, P. Autissier, M. A. Atkinson, W. W. Kwok, C. R. Kahn and E. Altindis, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2120028119 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00715h |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |