
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBinding affinity and transport studies of engineered photocrosslinkable affibody-enzyme-nanoparticle constructs†
Shane D. Currya,
Bryce M. Bowera,
Sven A. Saemundssona,
Andrew P. Goodwin *ab and
Jennifer N. Cha*abc
*ab and
Jennifer N. Cha*abc
aDepartment of Chemical and Biological Engineering, University of Colorado, Boulder, 596 UCB, Boulder, CO 80303, USA. E-mail: Jennifer.Cha@colorado.edu; Andrew.Goodwin@colorado.edu
bMaterials Science and Engineering Program, University of Colorado, Boulder, 596 UCB, Boulder, CO 80303, USA
cBiomedical Engineering Program, University of Colorado, Boulder, 596 UCB, Boulder, CO 80303, USA
First published on 24th February 2025
Abstract
Nanoparticle accumulation at tumor sites has been well reported in vivo, where targeting typically shows increased retention, but challenges remain for clinical translation. This work examines the effect of targeting ligand binding affinities and nanoparticle size on retention and transport through a solid tumor. We first show using cell affinity assays that modifying a wildtype (WT) anti-epidermal growth factor receptor (EGFR) affibody-enzyme fusion protein into a UV-photocrosslinkable (N23BP) version led to a significant decrease in affinity, whether as a free protein or as a conjugate to silica nanoparticles. Despite the reduced EGFR affinity, all protein conjugated nanoparticles showed binding and uptake to EGFR-overexpressing HTB9 bladder cancer cells as detected by confocal microscopy and flow cytometry. Next, transport studies of the protein conjugated nanoparticles using monoculture spheroids revealed that spheroid binding was higher for 17 nm particles bound with the WT proteins than N23BP, which was expected based on their respective KD values. However, the 17 nm particles conjugated with the photocrosslinkable N23BP affibody-enzymes showed an altered distribution profile that peaked further into the spheroid than the WT nanoparticle conjugates or in the absence of UV treatment. We correlate this finding with increased transport and retention of the photocrosslinked N23BP-nanoparticle conjugates in 3D spheroids to both the lower binding affinity of the affibodies for EGFR and the ability to introduce covalent linkages between the affibody and cell receptor. The larger 40 nm protein-conjugated nanoparticles showed limited penetration regardless of affinity or photocrosslinking on a 12 h timescale but did show overall increased transport after 24 h.
Introduction
Effective targeted therapy of solid tumors must balance specific binding to cancer cells while allowing for sufficient penetration to enable whole tumor treatment.1–7 For example, biomacromolecules with high binding affinities to cancer cell receptors often fail to disperse throughout the tumor itself, in large part due to the formation of a barrier caused by strong association with the most accessible ligands in the tumor, often referred to as a “binding site barrier”.1,4,7,8 Conjugation of these targeting agents to nanoparticles offers benefits for imaging and drug delivery, including sustained drug release, increased drug blood circulation time, greater selectivity, and improved targeting to tumor sites.9–13 Nanoparticles further exacerbate the challenges in tumor penetration of high affinity ligands due the multivalency of the targeting ligands and the overall sizes of the nanoparticle constructs.3,9,14–18 For example, Lee et al. showed inhibited distribution of block copolymer micelles upon addition of EGF as a targeting ligand.14 One potential way to reduce the binding site barrier effect is to inhibit binding of the targeting ligands to cellular receptors.1,3,19 For example, Bordeau et al. demonstrated that competitive inhibition of monoclonal antibody binding to HER2 allowed for enhanced tumor distribution in mouse model studies.19 Additionally, targeting proteins with low binding affinity or high KD to a specific cell receptor can be used to balance targeting and perfusion through the tumor microenvironment.1,20 Because high KD ligands are prone to dissociation and clearance, external triggers such as photoinduced crosslinking could be used to prolong their retention.21,22We previously demonstrated that photocrosslinking affibody-enzyme-nanoparticle complexes to CaCo2 colorectal cancer xenografts in mice led to inhibited tumor growth in the presence of prodrug administration.23 In said studies, ∼18 nm upconverting nanoparticles (UCNPs) were used to initiate crosslinking at clinically relevant wavelengths and the enzyme cytosine deaminase (CodA) was used for prodrug conversion. Despite the inhibited tumor growth, little information is known about the distribution of photocrosslinked, low affinity nanoparticle-affibody conjugates within the tumor. For the studies shown here, we first demonstrate methods to conjugate the EGFR binding affibody and affibody-enzyme fusions to ∼40 nm and ∼17 nm silica nanoparticles. Silica nanoparticles (SiNPs) were used in this study as a model for common silica-based functionalization of more complex nanoparticles like UCNPs. 17 nm SiNPs were chosen to have a size similar to the previous studies, while 40 nm particles were chosen as a larger group to analyze the impact of affinity alongside size. First, SiNPs were synthesized and bound with the wildtype (WT) and photocrosslinkable (N23BP) affibody-enzyme fusion proteins. Cell affinity assays were next run using the protein conjugated nanoparticles which showed highly disparate KD values ranging from the high nM for the WT-conjugated particles versus mid-to high μM for the N23BP affibody bound SiNPs. In comparing the protein conjugated 17 and 40 nm particles, a decrease in binding affinity was observed with the increase in particle size which may be due in part to the number of proteins bound to the differently sized SiNPs. The protein conjugated particles were next tested for 2D cell uptake against HTB9 bladder cancer cells by confocal microscopy, which showed significant binding and uptake regardless of affinity or particle size. Next, to measure transport in a tumor model, 3D HTB9 bladder cancer spheroids were formulated and reacted with the 17 and 40 nm WT and N23BP-affibody-enzyme modified SiNPs over 12–24 h. Analysis of confocal microscopy images was used to measure the binding and transport into the 3D spheroids. These conjugates demonstrated much higher binding for the 17 nm particles bound with the WT proteins as compared to N23BP, as expected based on their respective KD values. However, the 17 nm particles conjugated with the photocrosslinkable affibodies showed an altered diffusion profile upon UV irradiation, displaying greater distribution of retained particles within the spheroid. In studying the protein conjugated 40 nm SiNPs, the size barrier of the larger particles inhibited spheroid penetration regardless of affinity after 12 h of incubation, and there was minimal particle accumulation after 24 h compared to the 17 nm particles. All together, these results show the importance of optimizing particle size and affinity to enable maximum tumor penetration and accumulation in addition to the effects of photocrosslinking on nanoparticle diffusion profiles.
Results and discussion
First, wild-type (WT) and photocrosslinkable (N23BP) affibody proteins fused to CodA were produced using previously published procedures.24 The N23BP-CodA affibody enzyme contains a site-specific mutation at the 23rd amino acid position to introduce a benzophenone group, allowing for photocrosslinking upon UV excitation.20,21 This affibody enzyme has been previously demonstrated to crosslink to cellular EGFR.24 After purification, the proteins were characterized by SDS-PAGE and enzymatic analysis. As shown in Fig. 1, the as produced fusion proteins were pure and showed enzyme kinetics comparable to the native CodA, having an inhibited maximum rate but similar affinity for the substrate. Next, prior to particle conjugation, the WT-CodA and N23BP-CodA proteins were reacted with NHS-Alexa Fluor 488 using a 7.5-fold molar excess overnight at 4 °C for labeling and detection followed by dye removal. This procedure typically yielded a degree of labeling 0.57 ± 0.07 mole of dye per mole of protein (Fig. S1†).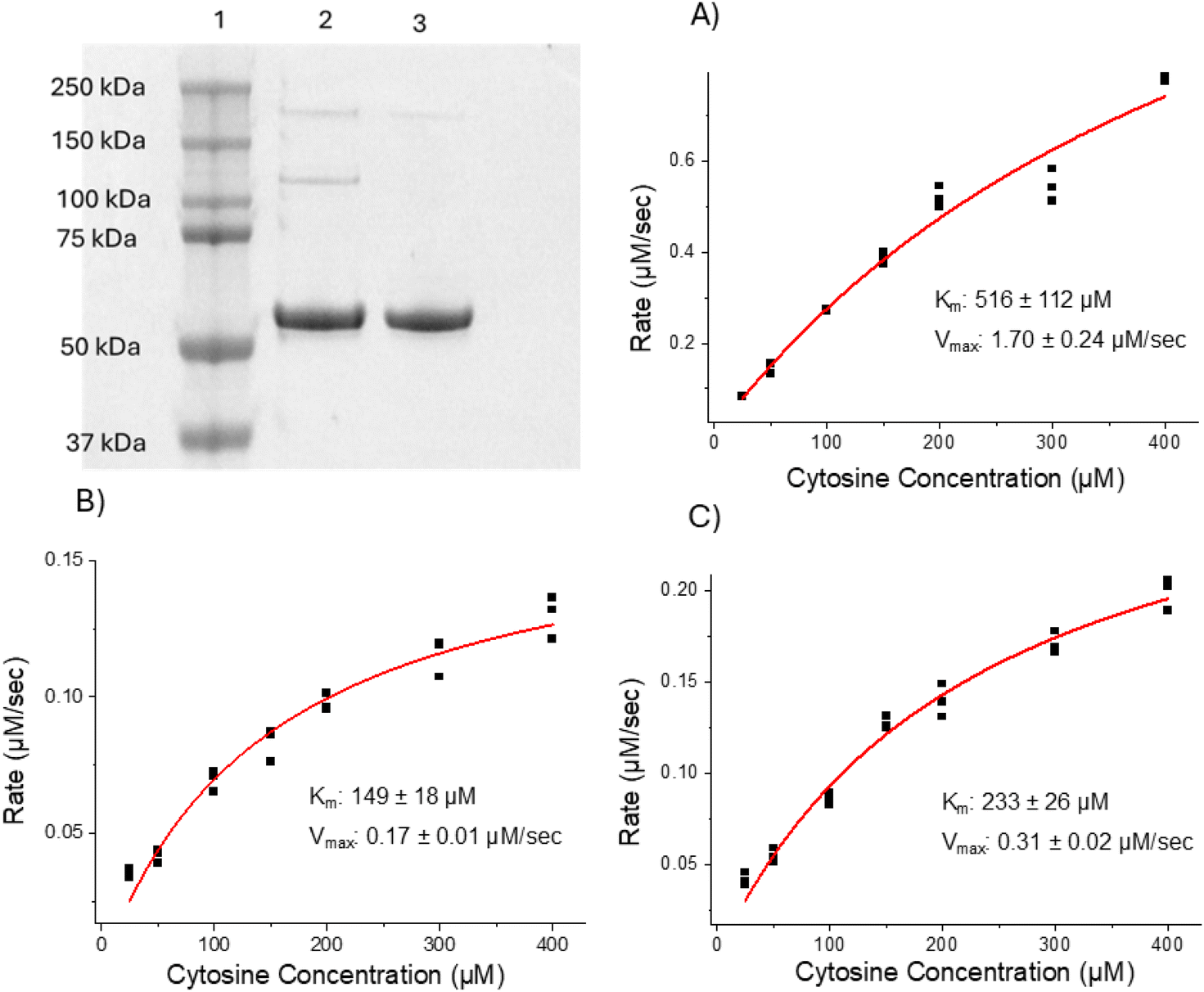 | ||
| Fig. 1 Purification and enzymatic activity of CodA and affibody-CodA. (Top Left) SDS-PAGE results displaying purity of fusion proteins (1: Ladder, 2: N23BP-CodA, 3: WT-CodA). Cytosine deaminase (CodA) activity at 50 nM concentration for (A) CodA, (B) WT-CodA, and (C) N23BP-CodA represented as the data points (black) and the best Michaelis–Menten equation fit (red). | ||
To study cellular interactions with nanoparticles conjugated with either the WT-CodA or N23BP-CodA, silica nanoparticles (SiNPs) were synthesized. As mentioned above, SiNPs were used due to the ease of large-scale synthesis and as a model for silica shell chemistry on nanoparticles such as UCNPs for use in vivo. For this, solid silica nanoparticles were synthesized using a sol gel process with an average particle diameter of ∼40 and ∼17 nm by TEM (Fig. 2a). The SiNPs were next modified with a functional polyethylene glycol (PEG) layer by reacting with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio of PEG-silane-2k to DBCO-PEG-silane-2k in anhydrous toluene followed by a slow centrifuge to remove aggregates. After reaction, zeta potential measurements of the particles showed a substantial increase in surface potential toward the positive as compared to silica nanoparticles alone (Table S1†). To orient the affibody-enzyme from the particle surface, the SiNPs were further reacted with nitrilotriacetic acid-PEG-azide (NTA-PEG-azide-3.4k) followed by the addition of 10 mM Ni2+ and the C-terminal His-tagged WT-CodA or N23BP-CodA (Fig. 2b). To remove any weakly bound protein, the particles were washed with 5–20 mM imidazole to yield protein–particle conjugates that showed minimal protein release at pH values 5–7 (Fig. S2†). While dynamic light scattering (DLS) measurements indicated particle aggregation, results showed particles remained on the nanoscale at each modification step (Table S1†). The final conjugation yield for the ∼40 nm SiNPs were ∼4–5 proteins per particle whereas ∼17 nm SiNP had ∼1–2 proteins per particle.
:5 ratio of PEG-silane-2k to DBCO-PEG-silane-2k in anhydrous toluene followed by a slow centrifuge to remove aggregates. After reaction, zeta potential measurements of the particles showed a substantial increase in surface potential toward the positive as compared to silica nanoparticles alone (Table S1†). To orient the affibody-enzyme from the particle surface, the SiNPs were further reacted with nitrilotriacetic acid-PEG-azide (NTA-PEG-azide-3.4k) followed by the addition of 10 mM Ni2+ and the C-terminal His-tagged WT-CodA or N23BP-CodA (Fig. 2b). To remove any weakly bound protein, the particles were washed with 5–20 mM imidazole to yield protein–particle conjugates that showed minimal protein release at pH values 5–7 (Fig. S2†). While dynamic light scattering (DLS) measurements indicated particle aggregation, results showed particles remained on the nanoscale at each modification step (Table S1†). The final conjugation yield for the ∼40 nm SiNPs were ∼4–5 proteins per particle whereas ∼17 nm SiNP had ∼1–2 proteins per particle.
 | ||
| Fig. 2 (Top) TEM images of as synthesized 40 nm (A) and 17 nm (B) SiNPs. (Bottom) SiNP modification and conjugation to affibody-CodA process. | ||
Next, cell affinity measurements were run to determine the binding of each affibody-CodA protein to EGFR-overexpressing HTB9 human bladder cancer cells to measure the affinity to cells.25,26 For this, 15000 HTB9 cells were treated with the affibody enzymes alone at various concentrations ranging from 0.5–250 nM for 1 h followed by centrifugation to collect the cells and remove unbound protein. Flow cytometry was then used to determine the median fluorescence intensity, which was analyzed and fit to a one site binding model to determine the apparent dissociation constant KD (Fig. S3†). This procedure yielded a KD of ∼6 nM for WT-CodA and in the millimolar range for N23BP-CodA against HTB9 cells, suggesting the actual KD was greater than what could be accurately measured by this method.
In order to determine the apparent affinity of the affibody-CodA-SiNPs after particle conjugation, a similar procedure was done with the protein conjugated nanoparticles. Fluorescence analysis was used to determine the affibody-CodA loading on each particle sample, which were then incubated with HTB9 cells at protein concentrations that ranged from 0.1 to 50 nM and 0.1 to 400 nM for the 17 and 40 nm particles, respectively. These protein concentrations corresponded to ∼0.67–333 μg mL−1 and 1 μg mL−1–3.8 mg mL−1 particle mass concentrations, respectively. 50 nM protein was chosen as a cutoff for the smaller particles as the presence of aggregates began to skew flow cytometry readings beyond this concentration. As shown in Fig. 3, for the 17 and 40 nm WT-CodA-SiNP, taking the median fluorescence intensity for each concentration yielded a KD of ∼112 and ∼517 nM, respectively. It should be noted though that, as with many particle systems, aggregation occurs which can skew the absolute values obtained. In the data obtained, both the 17 nm and 40 nm SiNPs modified with N23BP-CodA showed weak affinity to cells in comparison to the 17 and 40 nm WT-CodA-SiNPs, again yielding KD values in the millimolar and beyond range. Based on the obtained values, it appears that the photocrosslinkable proteins conjugated to nanoparticles do not show stronger binding to EGFR expressing cells even though multivalency often increases association.27 As a comparison, dye labeled, PEGylated, SiNPs were used to analyze the binding profile of particles with no affinity. As shown in Fig. S4,† the non-targeted particles demonstrate a sharp uptick after 125 μg mL−1, which would correlate with ∼19 and ∼13 nM protein for the 17 and 40 nm affibody-CodA-SiNPs, respectively. This uptick is not present with the targeted nanoparticles. Instead, the targeted particles resemble either the early part of a binding curve in which receptor saturation has not yet significantly affected the curve (N23BP-CodA-SiNPs) or after receptor saturation begins to show (WT-CodA-SiNPs). It is important to note that, due to the differences in dye loading, the median fluorescence intensity values cannot be compared directly.
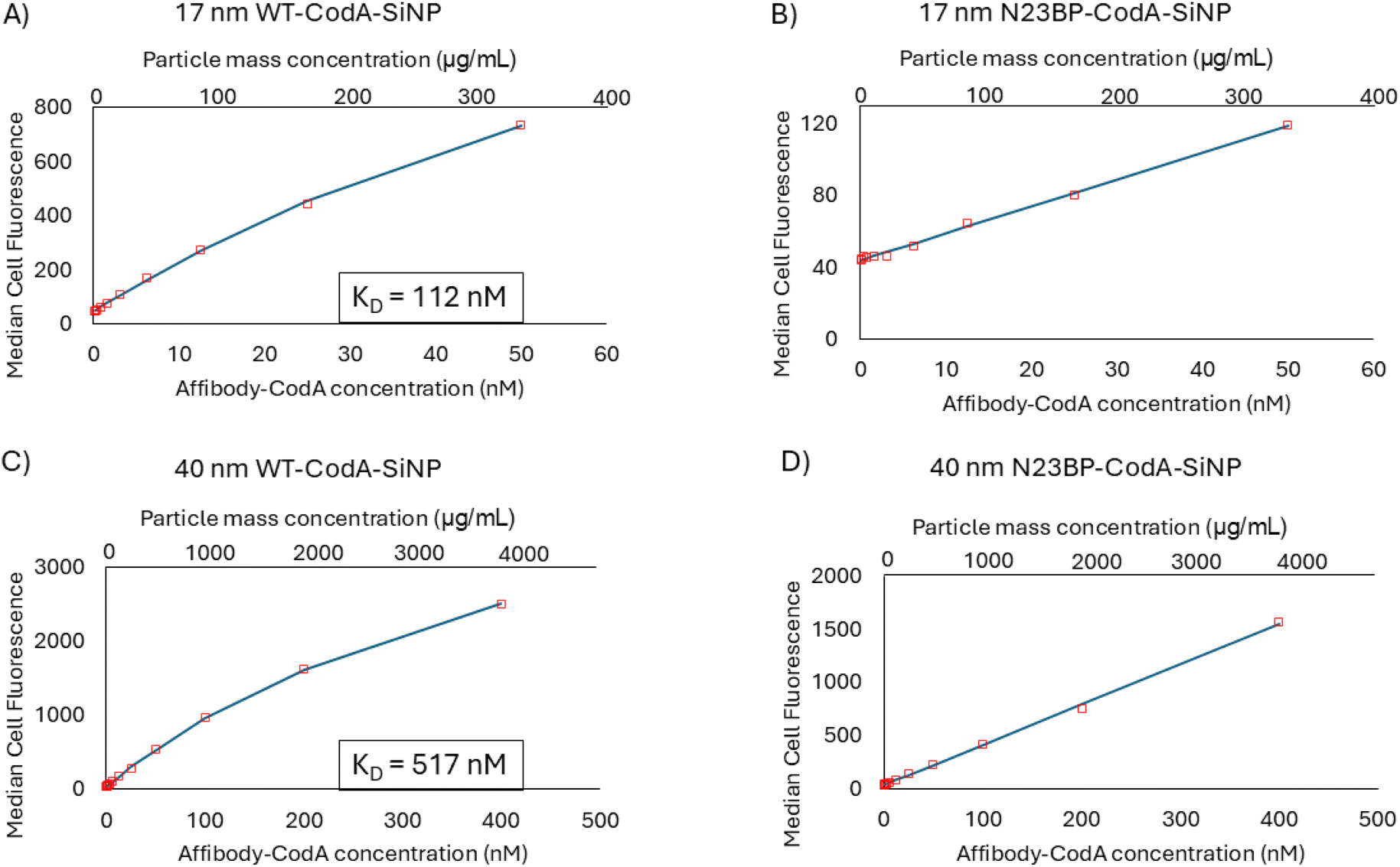 | ||
| Fig. 3 Affinity binding curve of (A) 17 nm WT-CodA-SiNP, (B) 17 nm N23BP-CodA-SiNP, (C) 40 nm WT-CodA-SiNP, and (D) 40 nm N23BP-CodA-SiNP. Particles were incubated with HTB9 cells at various concentrations and analyzed by flow cytometry. Median cell fluorescence vs. concentration was plotted and fitted to a one site binding model to determine apparent KD. | ||
Next, particle uptake by HTB9 cells was verified via fluorescence microscopy using 2D cell cultures. HTB9 cells were treated with each affibody-CodA-SiNP conjugate for 24 h at 100 nM protein equivalent concentration before washing, nuclear staining, and imaging. As shown in Fig. 4, each protein-NP conjugate type was shown to associate and be uptaken by the HTB9 cells. Not unexpectedly, the 17 nm WT-CodA particles at the same concentration added to cells as the 40 nm showed larger amounts of particles bound (Fig. S5†). Despite the high KD values of the N23BP affibodies for the EGFR expressing cells, a significant number of the particles bound to cells could be detected, as further confirmed by analyzing the proportion of particle fluorescent area in comparison to total nuclear area as a proxy for cell count (Fig. S5†). Additionally, when incorporating DiI membrane stain, the particles appear to be associated with the cells (either by internalization or on the cell membrane) (Fig. S6†). This was irrespective of the fact that only 100 nM protein were added to cells although the roughly measured KD values for the N23BP proteins were in the micromolar range.
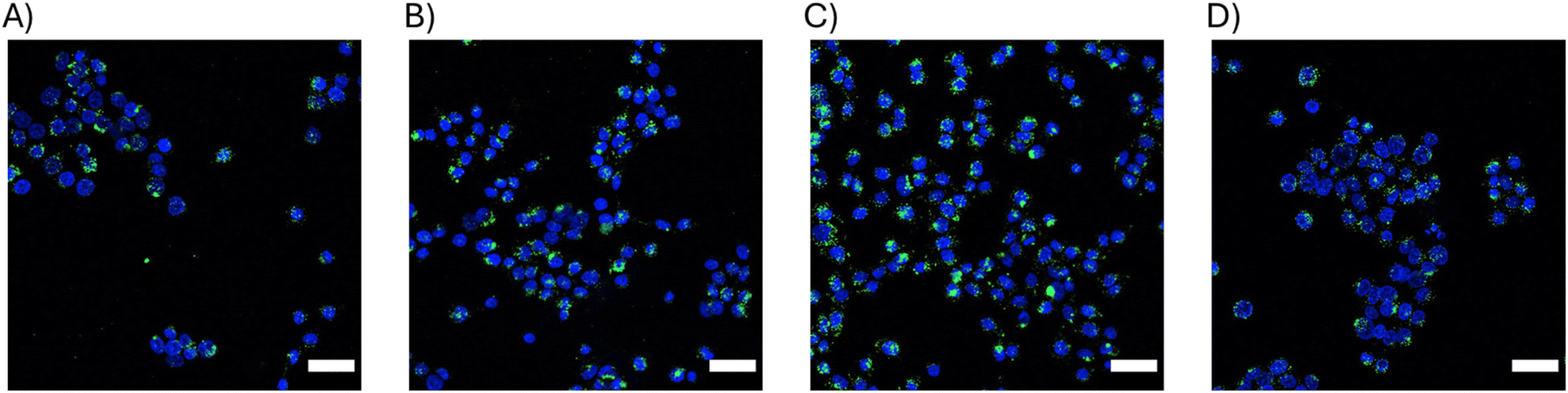 | ||
| Fig. 4 Confocal microscopy images showing HTB9 uptake of (A) 17 nm N23BP-CodA-SiNP, (B) 40 nm N23BP-CodA-SiNP (C) 17 nm WT-CodA-SiNP, and (D) 40 nm WT-CodA-SiNP. Affibody-enzyme-SiNP conjugates are labeled with Alexa Fluor 488 (green) and HTB9 nuclei are stained with Hoechst 33342 nuclear stain (blue). Scale bar 50 μm. | ||
The ability of the WT and N23BP conjugated SiNPs to accumulate and diffuse in tumor microenvironments was determined next. For this, monoculture 3D spheroids were formed by incubating 20000 HTB9 cells in polyHEMA coated 96 well plates for 72–96 h followed by characterization by widefield microscopy (Fig. S7†). Concurrently, the WT-CodA and N23BP-CodA proteins were modified with the Alexa Fluor 555 (AF555) dye prior to SiNP conjugation to track particle movement into the spheroids. AF555 was chosen to limit background autofluorescence often observed in cell imaging and to allow for greater spheroid light penetration. Spheroids were incubated with 250 nM of the AF555 labeled WT-CodA-SiNP and N23BP-CodA-SiNP for 12 h and 24 h, with UV irradiation occurring 4 h post particle treatment. They were then transferred to flat bottom imaging plates and fixed before nuclear staining and imaging. To analyze particle transport into spheroids, the Hoechst nuclear stain was used to create a mask around the entire spheroid. This mask was then eroded by 10 μm using the “imerode” MATLAB function to create a second mask which was subtracted from the first, creating a 10 μm slice on the outside of the spheroid. This process was continued to create a mask for every 10 μm slice until the center of the spheroid. For each slice, excessively bright aggregates were removed and the mean AF555 intensity was plotted against depth to yield a distribution profile.
As shown in Fig. 5, the 40 and 17 nm particles showed very different spheroid binding and penetration profiles as determined by tracking AF555 fluorescence. In each case, at 12 h post treatment, the WT-CodA-SiNPs showed overall higher binding throughout the spheroid in comparison to the photocrosslinkable affibody-nanoparticles, although this effect was only statistically significant for the 17 nm particles. Since the measured KD values of two different protein nanoparticle conjugates show multifold differences, an overall lower spheroid binding with the N23BP-CodA-SiNP conjugates is expected. Importantly, the 17 nm WT-CodA-SiNPs were able to accumulate past 70 μm from the edge of the spheroid, as shown by fluorescence detectable above background, suggesting that the high nM affinity was weak enough to still allow spheroid penetration at this particle size. Additionally, the WT-CodA-SiNPs showed greater accumulation for each depth up to 30 μm into the spheroid when compared to the crosslinked N23BP-CodA-SiNPs and up to 40 μm compared to the non-crosslinked N23BP-CodA-SiNPs (p < 0.05). For the N23BP-CodA-SiNPs, no statistical significance was obtained between the crosslinked and non-crosslinked groups. However, an altered penetration depth distribution profile was observed, as discussed further in Fig. 6 below. Regardless of affinity, the 40 nm protein conjugated nanoparticles showed little spheroid penetration, accumulating mostly on the periphery or having low overall binding, in comparison to the 17 nm particles (Fig. 5A and B). This result suggests that the size barrier of the 40 nm particles was enough to limit penetration on the 12 h timescale independent of affinity, while the 17 nm particles transported further after 12 h.
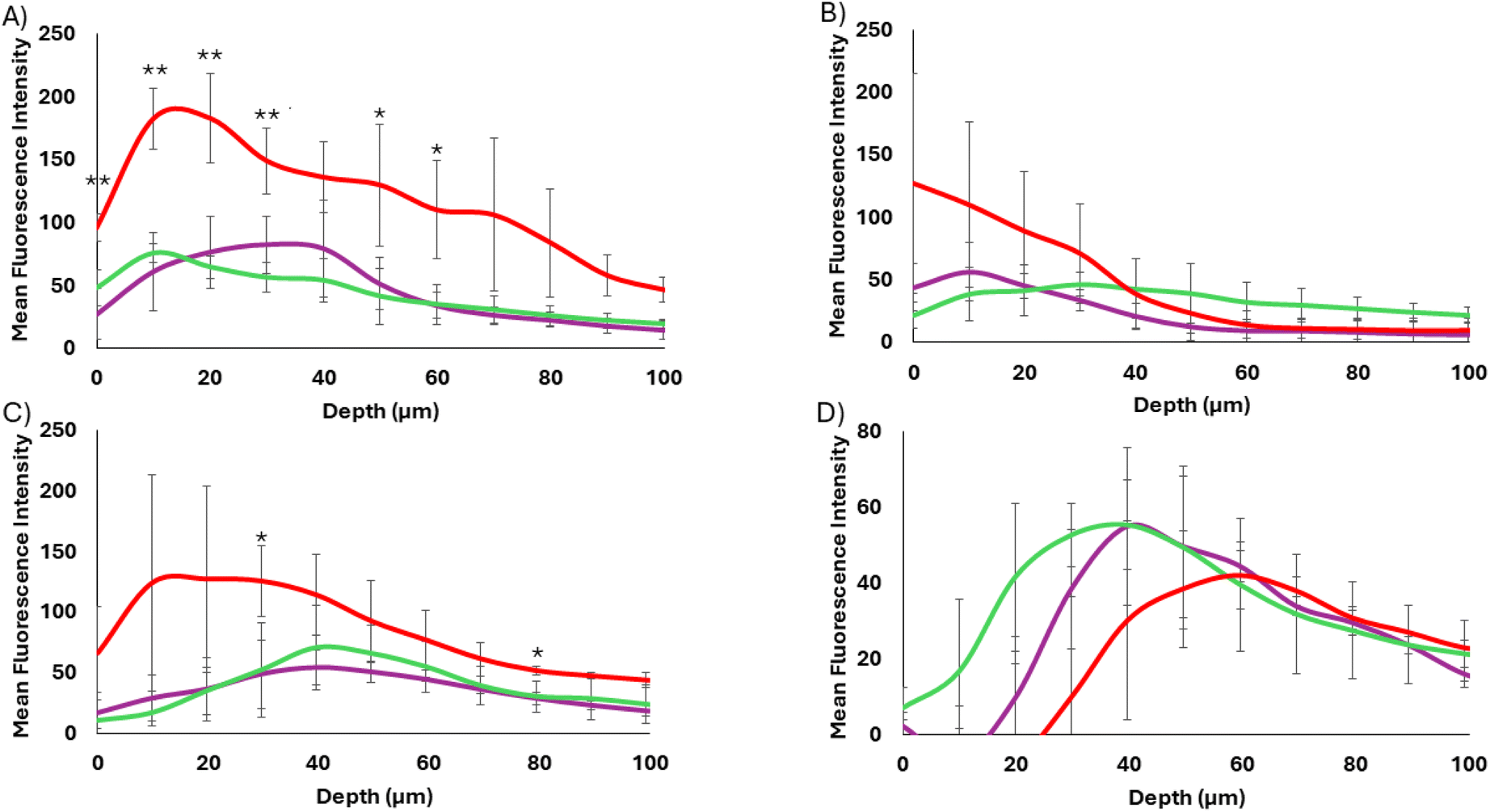 | ||
| Fig. 5 Affibody-enzyme-SiNP spheroid penetration of WT-CodA-SiNPs (red) and N23BP-CodA-SiNPs in the presence (purple) and absence (green) of photocrosslinking. (A) 17 nm particles, 12 h incubation. (B) 40 nm particles, 12 h incubation. (C) 17 nm particles, 24 h incubation. (D) 40 nm particles, 24 h incubation. *p < 0.1, **p < 0.05 for WT-CodA-SiNP vs. all other groups. | ||
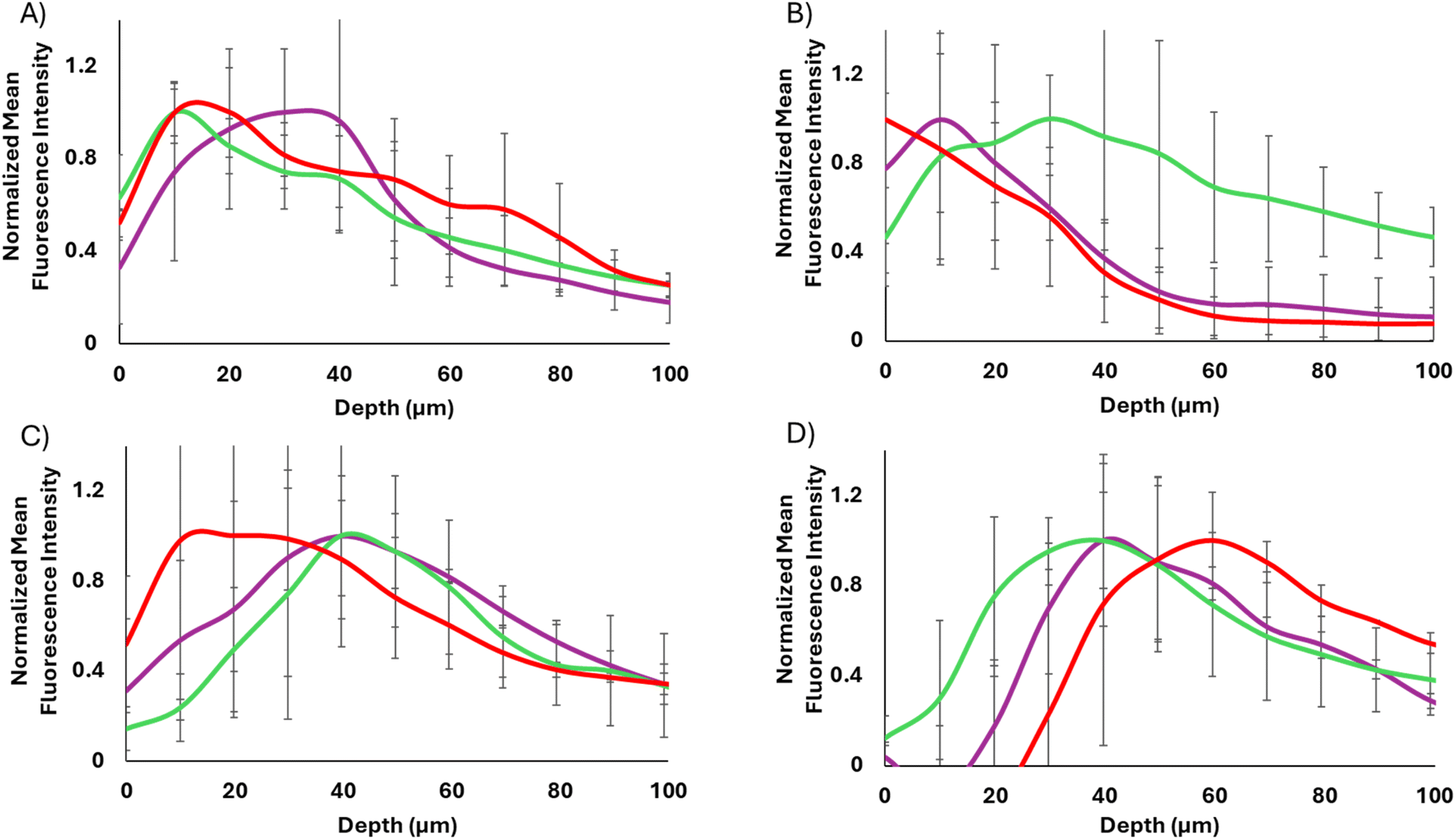 | ||
| Fig. 6 Normalized affibody-enzyme-SiNP spheroid penetration of WT-CodA-SiNPs (red) and N23BP-CodA-SiNPs in the presence (purple) and absence (green) of photocrosslinking. (A) 17 nm particles, 12 h incubation. (B) 40 nm particles, 12 h incubation. (C) 17 nm particles, 24 h incubation. (D) 40 nm particles, 24 h incubation. | ||
Incubating the spheroids with the affibody-enzyme-SiNP conjugates for 24 h revealed different trends (Fig. 5C and D). For the 40 nm particles, each particle set had traveled slightly deeper into the spheroids but showed low overall binding, even with the WT affibody. In the case of the 17 nm particles, the WT-CodA-SiNPs showed higher accumulation throughout the spheroid than the N23BP-CodA-SiNPs did (p < 0.1 at 30 and 80 μm depths). Similar to 12 h incubation, this result could be caused by the fact that the affinity is low enough where it serves to increase overall binding while still allowing some transportation into the spheroid.1 The 17 nm WT-CodA-SiNPs displayed their peak fluorescence closer to the edge of the spheroid as compared to the N23BP-CodA-SiNPs, which encouraged further observation of the distribution transport profiles.
Upon normalization of each group to its maximum fluorescence intensity, differences in transport became more evident for the 17 nm protein conjugated nanoparticles (WT-CodA-SiNP, N23BP-CodA-SiNP + UV, N23BP-CodA-SiNP − UV) (Fig. 6). First, at 12 h post treatment, the WT-CodA-SiNPs and the N23BP-CodA-SiNPs without UV irradiation showed similar trends. In both cases the mean fluorescence intensity peaked around 10 μm into the spheroid and decreased with further distance into the spheroid. However, with the photocrosslinked N23BP-CodA-SiNPs, the mean fluorescence intensity peaked around 20–40 μm into the spheroid, displaying a broader peak with a slower decline as depth increased. This unique profile was attributed to photocrosslinked particles being unable to exit the spheroid, leading to increased retention. As mentioned above, by 24 h post-treatment the low affinity N23BP-CodA-SiNPs had a distribution profile that peaked further into the spheroid than the WT-CodA-SiNPs did. This trend was evident with both the UV-treated and untreated particles and attributed to the lower affinity of the N23BP affibody reducing the binding site barrier impact. For the larger 40 nm protein-conjugated nanoparticles with 12 h of incubation, the WT-CodA-SiNPs peaked on the spheroid surface and the photocrosslinked particles peaked about 10 μm into the spheroid. In comparison, the non-crosslinked N23BP-CodA-SiNPs displayed a more uniform distribution profile, although this may be due to having a lower peak before normalization. Additionally, the 40 nm particles all showed peaks further into the spheroids after 24 h of incubation with limited surface bound particles. Only the particles with the strongest affinity (17 nm WT-CodA-SiNPs), had the fluorescence peak near the periphery of the spheroid.
Conclusion
In this work we demonstrated that both size and affinity are critical factors in determining penetration of targeted nanoparticles into spheroids. Although the protein conjugated nanoparticles were found to bind and be internalized in 2D HTB9 cell cultures, studies with 3D HTB9 spheroids revealed differences in uptake due to size, affinity, and crosslinking. The 17 nm WT-CodA-SiNPs were shown to have the greatest affinity to cellular EGFR, and showed detectable accumulation in the spheroid after both 12 and 24 h, suggesting that nanomolar affinities may strike a balance between cell binding and allowing some particle transport in 3D tissue.1 However, normalizing the results revealed this was the only group that peaked near the spheroid periphery after 24 h of incubation, with all lower affinity groups displaying deeper peaks in their distribution profiles. This was despite the fact that, with only 12 h of incubation, the 40 nm WT-CodA particles remained largely confined to the spheroid periphery, suggesting the barriers of size and affinity could not be overcome in only 12 h. Additionally, photocrosslinking the 17 nm N23BP-CodA-SiNPs led to deeper accumulation in the 3D tumor spheroids 12 h post treatment as compared to the non-crosslinked groups, however this difference was lost after 24 h. These results show the importance of optimizing affinity and particle size for nanoscale delivery to achieve greater tumor perfusion. Additionally, these studies also demonstrated the importance of 3D culture studies when analyzing nanoparticle uptake, as these results were different than those obtained in 2D studies. Future work will include analyzing retention of the affibody-CodA-SiNPs in the presence and absence of crosslinking and exploring the effects of valency.Experimental
Materials
HTB9 cells were grown in Corning T-25 TCPS flasks using RPMI-1640 (Gibco) supplemented with 1% penicillin–streptomycin antibiotic (HyClone), 10% HI Fetal Bovine Serum (FBS, Gibco), and 0.2% Plasmocin prophylactic (InvivoGen). DPBS and TryplE express for cell washing and detachment were obtained from Thermo Fisher. 15 mL conical tubes (Greiner Bio-One) were used for cell spin down and solutions. Spheroid formation was performed using polyHEMA (Sigma-Aldrich) coated round bottom well plates (Fisher Scientific) and spheroid imaging was done in Corning 96 well assay plates. Cell staining was done using Hoechst 33342 (Thermo Fisher) and DiI membrane stain (Thermo Fisher). SiNP synthesis and modification reagents included absolute ethanol (Decon Laboratories), 30 w/w% aqueous ammonia (LabChem), tetraethyl orthosilicate (Sigma-Aldrich), PEG-silane (MW 2000, Laysan Bio), DBCO-PEG-silane (MW 2000, Biopharma PEG), NTA-PEG-azide (MW 3400, Nanocs), and nickel chloride (Sigma-Aldrich). Particle characterization was done using an Anton Paar Litesizer 500 and a Tecnai ST20 200k V TEM. Protein and particle affinity measurements were done in the presence of BSA (Sigma-Aldrich) and sodium azide (Fisher Scientific). Affibody-CodA and CodA were produced using previously reported materials and methods with the additional use of HisTrap HP 1 mL columns and a HiTrap Desalting 5 mL column.24 Alexa Fluor dyes and Pierce Dye removal columns from Thermo Fisher were used for affibody-enzyme modification. Azide-fluor 545 from (Sigma-Aldrich) was used for direct modification of PEGylated SiNPs. UV-vis protein analysis was done using an Agilent Carry 100 UV-vis Spectrophotometer. Flow cytometry was performed using a BDFACSCelesta Flow Cytometer multicolor analyzer. Confocal and widefield microscopy were performed using a Nikon AXR Laser Scanning Confocal and Nikon Ti-E Widefield respectively.SiNP synthesis
Solid silica nanoparticle (SiNP) synthesis was carried out based on the Stöber method.28 46 mL of absolute ethanol, 1.55 mL of water, and 2.225 mL of 30 w/w% aqueous ammonia were stirred at 55 °C for 5 min before 3 mL of tetraethyl orthosilicate was added dropwise. The reaction was stirred at 400 rpm for 2 h at 55 °C. After this time, the particles were isolated by centrifugation at 12500 rcf for 15 min and washed twice in ethanol. For smaller particle production, the temperature was increased to 70 °C and centrifugation was done for 1 h. Isolated particles were characterized by transmission electron microscopy, dynamic light scattering spectroscopy, and zeta potential measurements.
SiNP functionalization
40 nm SiNP functionalization was done by first conferring azide reactivity onto the particles, followed by nickel reactivity and binding. 80 mg of SiNPs were reacted with 22 mg of PEG-silane (MW 2000) and 4.5 mg of DBCO-PEG-silane (MW 2000) at 50 °C for 24 h under constant stirring (400 rpm). Particles were isolated via centrifugation (12.5k rcf 15 min) and cleaned twice in ethanol followed by a slow centrifuge (0.6k rcf 3 min) to remove aggregates. 50 mg of these particles were reacted with 2 mg of NTA-PEG-azide (MW 3400) in ethanol for 8 h and isolated via centrifugation before activation in 10 mM nickel(II) Chloride in water for 1 h. The resulting Ni-SiNPs were centrifuged at 12.5k rcf for 20 min and washed twice with water. 17 nm SiNP functionalization was done using the same methods but with greater reagent concentrations due to a higher surface area to mass ratio. 65 mg of PEG-silane and 13 mg of DBCO-PEG-silane were used for the initial reaction followed by 4 mg of NTA-PEG-azide. Centrifugation was performed at 20k rcf for 1 h. After each modification, hydrodynamic radius and zeta potential measurements were taken to characterize particle aggregation and surface potential. For dye-modified SiNPs, 200 μl of 1 mM azide-fluor 545 was reacted with the DBCO-PEG-silane modified SiNPs for 4 h followed by centrifugation and two water washes.Protein synthesis and dye modification
The affibody-enzymes N23BP-CodA and WT-CodA were expressed synthesized, and modified with BP by methods reported previously.24 Purification was done using HisTrap HP 1 mL columns and a HiTrap Desalting 5 mL column.24 In the case of WT-CodA, no BP modification was performed. The affibody-CodA proteins were reacted with Alexa Fluor 488 NHS Ester in a 1:7.5 molar ratio overnight at 4 °C. Excess dye was removed with Pierce Dye Removal Columns. Degree of labeling was calculated following UV-vis spectroscopy.
CodA activity measurements
N23BP-CodA, WT-CodA, and the native CodA enzyme were incubated at 50 nM concentration with 25, 50, 100, 150, 200, 300, and 400 μM cytosine for 3 min while conversion to uracil was measured with UV-vis spectroscopy. Reaction rate as a function of substrate concentration was analyzed and fit to Michaelis–Menten kinetics to determine the Km and Kcat.Affibody-SiNP conjugation and characterization
Affibody-SiNP conjugates were formed by reacting the synthesized Ni2+ activated SiNPs with 300 μL of 40 μM dye labeled affibody-CodA for 12 h at 4 °C in PBS followed by centrifugation and redispersion in PBS. Loosely bound protein was removed via a low millimolar imidazole wash (7–15 mM) for 15 min. The particles were then centrifuged and washed twice in PBS before dispersion and storage in PBS at 4 °C. Fluorescence analysis was used to determine protein concentration and lyophilization followed by weighing was used to determine total particle concentration.Cell culture
HTB9 urinary bladder carcinoma cells were grown in 25 cm2 culture flasks at 37 °C under 5% CO2 in the presence of RPMI-1640 media supplemented with 10% FBS, 1% penicillin–streptomycin, and 0.2% Plasmocin Prophylactic.Cell affinity measurements
The affinity of the synthesized affibody-CodA-SiNPs was measured by incubating various concentrations (0.1 to 400 nM total protein) with 15000 HTB9 cells for 1 h at room temperature in PBS supplemented with 1% BSA and 0.1% sodium azide. The cells were then centrifuged at 150 rcf for 5 min and washed twice with ice cold BSA and sodium azide supplemented PBS. Cells were immediately analyzed via flow cytometry and characterized using median fluorescence intensity. The binding curve was used to calculate the apparent dissociation constant (KD) using a one site binding model using 0.1–400 nM for the 40 nm particles and 0.1–50 nM for the 17 nm particles. A similar process was used for the free affibody-enzymes using 0.5–250 nM protein. For the dye labeled SiNPs without affibody-enzymes, the same process was used with a 0.1–1000 μg mL−1 concentration range.
2D cell binding studies
HTB9 cells were seeded at 10000 cells per well in 96 well flat bottom imaging plates and grown for 48 h. Cells were then treated with 100 nM affibody-enzyme-SiNP for 24 h. Following this, cells were washed 2× with DPBS, stained with 5 μM Hoechst nuclear stain and 3 μM DiI membrane stain for 20 min, washed 2×, and imaged in DPBS. ImageJ was used for analysis by determining total fluorescent area of Hoechst and Alexa Fluor 488.
Spheroid penetration studies
HTB9 spheroids were made using polyHEMA coated 96 well round bottom plates. To do this, 200 μL of 30 mg per mL polyHEMA in 95% ethanol was added to each well and the ethanol was allowed to evaporate for 48 h followed by UV sterilization of the plate. HTB9 cells were then seeded in each well at 20000 cells per well and grown for 4 days. Following this, spheroids were treated with 250 nM of affibody-enzyme-SiNPs for 12 or 24 h. After 4 h of incubation, photocrosslinked samples were irradiated for 30 min with UV light to initiate crosslinking of the benzophenone group. After protein treatment, spheroids were transferred to 96 well flat bottom imaging plates, washed 1× with DPBS, and fixed with 4% paraformaldehyde for 30 min. They were then washed 1×, stained with 10 μM Hoechst nuclear stain for 30 min, washed 1×, and imaged in DPBS.
Image analysis was performed using a MATLAB script derived from Saemundsson et al.29 Briefly, TIF images were read and the nuclear channel was used to create a mask of the entire spheroid. This mask was then eroded by 10 μm using the “imerode” function to create a second mask which was subtracted from the first. This created a 10 μm slice on the outside of the spheroid. This process was continued to create a mask for every 10 μm slice until the center of the spheroid. For each slice, excessively bright aggregates were removed, background values were subtracted, and the mean Alexa Fluor 555 intensity was read and plotted against depth to yield a distribution profile. Student's t-test was used to determine statistical significance.
Data availability
The data for this article has also been included as part of the ESI.†Author contributions
SC was responsible for experimental design, data acquisition, analysis, and interpretation, and writing. AG and JC were responsible for conceptualization and funding acquisition and assisted with writing, experimental design, and data interpretation. BB and SS assisted with experimental design and analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for the research was provided by NSF CBET 2025547 and GAANN P200A210111. The imaging work was performed at the BioFrontiers Institute's Advanced Light Microscopy Core (RRID: SCR_018302). The Nikon AXR Laser Scanning Confocal is supported by NIH Grant 1S10OD034320. Nikon Ti-E Widefield is supported by NIH grant R01CA107098S1. We thank the Shared Instruments Pool (RRID: SCR_018986) of the Department of Biochemistry at the University of Colorado Boulder for use of the Typhoon 5. The Typhoon 5 is funded by NIH Shared Instrumentation Grant S10OD034218-01. Flow cytometric analysis was done with the help of the Flow Cytometry Shared Facility in JSCBB (RRID: SCR_019309). The authors acknowledge the Facility for Electron Microscopy of Materials at the University of Colorado at Boulder (CU FEMM, RRID: SCR_019306) for providing access to electron microscopy.Notes and references
- R. Tsumura, S. Manabe, H. Takashima, Y. Koga, M. Yasunaga and Y. Matsumura, J. Controlled Release, 2018, 284, 49–56 CrossRef CAS PubMed.
- C. Cilliers, H. Guo, J. Liao, N. Christodolu and G. M. Thurber, AAPS J., 2016, 18, 1117–1130 CrossRef CAS PubMed.
- K. T. Xenaki, S. Oliveira and P. M. P. van Bergen en Henegouwen, Front. Immunol., 2017, 8 DOI:10.3389/fimmu.2017.01287.
- C. Vasalou, G. Helmlinger and B. Gomes, PLoS One, 2015, 10, e0118977 CrossRef PubMed.
- G. M. Thurber, M. M. Schmidt and K. D. Wittrup, Adv. Drug Deliv. Rev., 2008, 60, 1421–1434 CrossRef CAS PubMed.
- Z.-F. Lim and P. C. Ma, J. Hematol. Oncol., 2019, 12, 134 CrossRef PubMed.
- S. I. Rudnick and G. P. Adams, Cancer Biother. Radiopharm., 2009, 24, 155 CrossRef CAS PubMed.
- J. W. G. Stroomer, J. C. Roos, M. Sproll, J. J. Quak, K.-H. Heider, B. J. Wilhelm, J. A. Castelijns, R. Meyer, M. O. Kwakkelstein, G. B. Snow, G. R. Adolf and G. A. M. S. van Dongen, Clin. Cancer Res., 2000, 6, 3046–3055 CAS.
- M. Kumari, A. Acharya and P. T. Krishnamurthy, Beilstein J. Nanotechnol., 2023, 14, 912–926 CrossRef CAS PubMed.
- E. Rostami, J. Drug Deliv. Sci. Technol., 2020, 58, 101813 CrossRef CAS.
- A. K. Bakshi, T. Haider, R. Tiwari and V. Soni, Drug Deliv. Transl. Res., 2022, 12, 2335–2358 CrossRef PubMed.
- J. Guo, X. Luan, Z. Cong, Y. Sun, L. Wang, S. L. McKenna, M. R. Cahill and C. M. O'Driscoll, J. Controlled Release, 2018, 286, 154–166 CrossRef CAS PubMed.
- H. Tian, T. Zhang, S. Qin, Z. Huang, L. Zhou, J. Shi, E. C. Nice, N. Xie, C. Huang and Z. Shen, J. Hematol. Oncol., 2022, 15, 132 CrossRef PubMed.
- H. Lee, H. Fonge, B. Hoang, R. M. Reilly and C. Allen, Mol. Pharm., 2010, 7, 1195–1208 CrossRef CAS PubMed.
- S. Huo, H. Ma, K. Huang, J. Liu, T. Wei, S. Jin, J. Zhang, S. He and X.-J. Liang, Cancer Res., 2013, 73, 319–330 CrossRef CAS PubMed.
- C. Wong, T. Stylianopoulos, J. Cui, J. Martin, V. P. Chauhan, W. Jiang, Z. Popović, R. K. Jain, M. G. Bawendi and D. Fukumura, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2426–2431 CrossRef CAS PubMed.
- S. Barua and S. Mitragotri, Nano Today, 2014, 9, 223–243 CrossRef CAS PubMed.
- W. C. Chen, A. X. Zhang and S.-D. Li, Eur. J. Nanomed., 2012, 4, 89–93 CrossRef.
- B. M. Bordeau, Y. Yang and J. P. Balthasar, Cancer Res., 2021, 81, 4145–4154 CrossRef CAS PubMed.
- M. Brasino, S. Roy, A. H. Erbse, L. He, C. Mao, W. Park, J. N. Cha and A. P. Goodwin, J. Am. Chem. Soc., 2018, 140, 11820–11828 CrossRef CAS PubMed.
- S. Roy, M. Brasino, J. M. Beirne, A. Harguindey, D. A. Chapnick, X. Liu, J. N. Cha and A. P. Goodwin, Bioconjug. Chem., 2020, 31, 104–112 CrossRef CAS PubMed.
- B. M. Bower, S. D. Curry, A. P. Goodwin and J. N. Cha, Biomacromolecules, 2024, 25, 7511–7517 CrossRef CAS PubMed.
- S. Roy, S. D. Curry, C. Corbella Bagot, E. N. Mueller, A. M. Mansouri, W. Park, J. N. Cha and A. P. Goodwin, ACS Nano, 2022, 16, 15873–15883 CrossRef CAS PubMed.
- S. Roy, S. D. Curry, M. G. Bibbey, D. A. Chapnick, X. Liu, A. P. Goodwin and J. N. Cha, Biotechnol. Bioeng., 2022, 119, 187–198 CrossRef CAS PubMed.
- M. Friedman, A. Orlova, E. Johansson, T. L. J. Eriksson, I. Höidén-Guthenberg, V. Tolmachev, F. Y. Nilsson and S. Ståhl, J. Mol. Biol., 2008, 376, 1388–1402 CrossRef CAS PubMed.
- X. Yang, E. Kessler, L.-J. Su, A. Thorburn, A. E. Frankel, Y. Li, F. G. La Rosa, J. Shen, C.-Y. Li, M. Varella-Garcia, L. M. Glodé and T. W. Flaig, Clin. Cancer Res., 2013, 19, 148–157 CrossRef PubMed.
- M.-H. Li, H. Zong, P. R. Leroueil, S. K. Choi and J. R. Jr. Baker, Bioconjug. Chem., 2017, 28, 1649–1657 CrossRef CAS PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- S. A. Saemundsson, S. D. Curry, B. M. Bower, E. J. DeBoo, A. P. Goodwin and J. N. Cha, Biomater. Sci., 2024, 12, 4759–4769 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00823e |
| This journal is © The Royal Society of Chemistry 2025 |