
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiological and mechanical properties of a self-curing acrylic resin enriched with AgNPs as a proposal for orthopedic aparatology
Christian Andrea
Lopez-Ayuso
 a,
Rene
Garcia-Contreras
a,
Ravichandran
Manisekaran
a,
Mario
Figueroa
b,
Manuel
Rangel-Grimaldo
c,
Mariano
Jacome
b,
Ruben Abraham
Dominguez-Perez
d,
Salvador
Lopez-Morales
e,
Sol
Cristians
f and
Laura Susana
Acosta-Torres
*a
a,
Rene
Garcia-Contreras
a,
Ravichandran
Manisekaran
a,
Mario
Figueroa
b,
Manuel
Rangel-Grimaldo
c,
Mariano
Jacome
b,
Ruben Abraham
Dominguez-Perez
d,
Salvador
Lopez-Morales
e,
Sol
Cristians
f and
Laura Susana
Acosta-Torres
*a
aLaboratorio de Investigación Interdisciplinaria, Área de Nanostructuras y Biomateriales, Escuela Nacional de Estudios Superiores Unidad León, Universidad Nacional Autónoma de México (UNAM), Predio el Saucillo y el Potrero, Comunidad de los Tepetates, León, 37684, Mexico. E-mail: lacosta@enes.unam.mx
bFacultad de Química, UNAM, Ciudad de México, 04510, Mexico
cInstituto de Química, UNAM, Ciudad de México, 04510, Mexico
dLaboratorio de Investigación Odontológica Multidisciplinaria, Universidad Autónoma de Querétaro, Querétaro, 76010, Mexico
eInstituto de Investigaciones en Materiales, Departamento de Reología y Mecánica de Materiales, UNAM, Ciudad de México, 04510, Mexico
fLaboratorio de Etnobotánica, Instituto de Biología, UNAM, Ciudad de México, 04510, Mexico
First published on 4th February 2025
Abstract
Polymethylmethacrylate (PMMA) is widely used in dentistry, but its inherent characteristics, such as roughness and porosity, can facilitate the formation of bacterial biofilms. However, the integration of silver nanoparticles (AgNPs) can provide antimicrobial properties. Ongoing research endeavors aim to preserve post-nanoaggregation biocompatibility without compromising the mechanical integrity of the material. In this study, we investigated the biological and mechanical attributes of a PMMA nanocomposite infused with AgNPs biosynthesized from Pelargonium × hortorum. A method has been described to incorporate nanoparticles into the polymer at minimum concentrations. In the results, LC-MS-MS revealed the presence of 56 biochemical compounds. UPLCHRESIMS-MS/MS was used to compare the phytochemical profiles of the leaf extract of Pelargonium × hortorum before and after the formation of AgNPs, which were identified with spherical morphology, an absorbance of 28.5 ± 8.16 nm and a particle size of 415 nm. The MIC of AgNPs was 10 μg mL−1. In bacterial MTT, a decrease to 18.2 ± 2.5% with PMMA-10 μg mL−1 was observed (p < 0.05). Decreased cell viability was found only in PMMA-0 μg mL−1 at 89.1 ± 6.7%, indicating no cytotoxicity. These findings suggest a promising bionano material that is suitable for orthodontic and orthopedic devices and warrants further research.
Introduction
Poly[1-(1-(methoxycarbonyl)-1-methylethylene])], is a polymeric material that is synthesized by the addition of free radicals to form polymethyl methacrylate [C5O2H8] (PMMA);1,2 its physical, mechanical, optical, low density and thermoplastic properties2,3 make it biocompatible with the cells of the human body, making it suitable for dental use;3 the applications in dentistry include dental prostheses,4 provisional prosthetic restorations5 and orthodontic appliances to treat malocclusions.6,7 According to the World Health Organization (WHO), dental malocclusions are considered the third most common oral health problem after caries and periodontal disease,8,9 with a prevalence of up to 80% in the primary dentition and 70% in the mixed dentition.10 To correct these alterations, orthopedic devices made of self-curing PMMA are used.11 However, due to the characteristics of its surface, such as porosity and roughness,12–14 bacterial colonization can quickly proliferate and cause an increase in volume and concentration of biofilms in this polymer, with an increase in species such as Streptococcus mutans and S. sobrinus, Actinomyces spp., Pseudomonas spp., Lactobacillus spp., and Candida albicans.15–18 This highlights the critical problem of PMMA's susceptibility to bacterial biofilm formation, necessitating improvements in its antimicrobial properties.Maintaining balance in the oral ecosystem is necessary to promote a healthy state.16 Still, when this balance is disrupted, dysbiosis occurs, allowing bacteria to manifest and cause diseases such as caries, gingivitis, and periodontitis.17,19 Due to these problems in clinical practice, in recent years, nanotechnology has shown advances in the development of biomaterials, giving rise to new bioproducts with antimicrobial, antifungal, and tissue regeneration properties, among others.3,20–22
To improve the properties of PMMA, it is necessary to incorporate different nanoparticles (NPs), such as zirconium dioxide (ZrO2),6 zinc oxide (ZnO),23 titanium dioxide (TiO2),24,25 silicon oxide (SiO2),26 aluminum and titanium (Al2O3/TiO2),27 resveratrol (RNPs),28 and silver nanoparticles (AgNPs).29–31 These new nanometric materials have contributed to advances in nanomedicine and biomedical sciences because of their excellent physical and chemical properties. Among these, AgNPs stand out for their antimicrobial effects, making them a promising addition to PMMA formulations.22,29–31 However, factors such as filler concentration, particle size, and the type of PMMA (heat-curing or self-curing) must be considered to maintain the biocompatibility and physical and mechanical properties of PMMA with NPs and consequently its biomedical characteristics. This emphasizes the gap in understanding the antimicrobial mechanisms and biocompatibility of PMMA-AgNPs, which this study aims to address.
Therefore, additional investigations are essential to elucidate the antimicrobial mechanisms and biocompatibility profiles of these materials, as well as their potential clinical utility. Consequently, this study aimed to evaluate the biological response encompassing antibiofilm activity against S. mutans, cytotoxicity, oxidative stress, genotoxicity, and proinflammatory responses in HGFs. It also assessed the chemical and mechanical properties, including flexural strength, flexural modulus, and residual monomer content, of self-curing PMMA and PMMA-AgNPs. This innovative formulation proposes a novel approach for orthodontic appliances, addressing the limitations of conventional PMMA materials, that warrants further exploration and validation. Therefore, additional investigations are imperative to elucidate the antimicrobial mechanisms and biocompatibility profiles of these materials, as well as their potential clinical utility (Fig. 1).
 | ||
| Fig. 1 Potential clinical applications for PMMA composites with AgNPs providing antimicrobial properties and biocompatibility (created in BioRender). | ||
Materials and methods
Pelargonium × hortorum extract
An aqueous extract of Pelargonium × hortorum was prepared; the specimen was taxonomically identified prior to the experiment, and a sample was deposited in the Herbarium of the Faculty of Sciences with voucher no. 182314. To obtain the extract, 12.15 g of fresh leaves were cut and added to 100 mL of deionized water (DI water) at boiling point; the extract was maintained at a constant temperature between 90 and 92 °C for 15 min, and then the solution was filtered using a 4–12 μm Whatman filter, to obtain a residue-free extract.Biosynthesis of AgNPs
The biosynthesis of AgNPs was carried out using 20 mL of ethylene glycol as a solvent, 10 mL of Pelargonium × hortorum extract as a reducing and stabilizing agent and 1.2 mL of 25 mM silver nitrate (AgNO3) (purity ≥ 99%) (Sigma-Aldrich® St Louis, Missouri, USA) as a precursor, added in 0.2 mL increments every 2 min. The solvent, reducing/stabilizing agent and precursor were added in the listed order, and the synthesis was maintained at a constant temperature of 110 °C using a recirculator for 30 min, as reported and detailed in previous experiments.32 The resulting AgNPs were washed twice with 100% acetone (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio) and once with absolute ethyl alcohol (1:1 ratio), sonicated for 15 min between each wash, followed by a 15 min centrifugation cycle at 4000 rpm and finally resuspended in deionized (DI) water. The final product was stored at 4 °C for further experiments.
:4 ratio) and once with absolute ethyl alcohol (1:1 ratio), sonicated for 15 min between each wash, followed by a 15 min centrifugation cycle at 4000 rpm and finally resuspended in deionized (DI) water. The final product was stored at 4 °C for further experiments.
Metabolomic analysis of Pelargonium × hortorum leaves extracts
The leaf extracts of Pelargonium × hortorum before and after AgNP formation were analyzed according to our previous work32 using an ultra-performance liquid chromatographic system (UPLC; Acquity, Waters Corp., Milford, MA, USA) coupled with a high-resolution mass spectrometer tandem MS/MS (HRESIMS-MS/MS; Q-Exactive Plus, Thermo Fisher Scientific, Waltham, MA, USA) equipped with an electrospray source (ESI) in positive and negative modes. The molecular networks were created using the GNPS (https://www.gnps.ucsd.edu) platform, using the same parameters as in our previous work.32 Finally, the MS data were subjected to principal component analysis (PCA) using the online platform MetaboAnalyst 5.0.33Preparation of self-curing acrylic resin compounds with AgNPs
Samples of self-curing PMMA NicTone (MDC Dental® Gardena, CA, USA) and Opti-Cryl (New Stetic, S.A. CDMX, Mexico), clear color, were prepared with 0, 5, 10, 15 and 20 μg mL−1 of AgNPs. The methodology was as follows: the concentration of AgNPs was calculated and they were placed in 1.5 mL Eppendorf tubes; the volume of all groups was adjusted to 0.5 mL with DI water and centrifuged at 13000 rpm for 10 min; the supernatant was removed and the pellet containing the AgNPs was retained; then 1 mL of monomer corresponding to each of the brands used was added and sonicated for 15 min to allow the dispersion of the NPs into the monomer; then a mixture of the monomer and polymer was made following the manufacturer's instructions in a 3:1 ratio (powder–liquid). The compounds were obtained by pressing this mixture in stainless-steel molds of 10 mm diameter and 2 mm thickness until polymerization was achieved (Fig. 1). Once the samples were obtained, they were polished to eliminate irregular edges. Finally, they were subjected to a sterilization cycle at 121 °C for 15 min and stored for biological experiments.
Characterization techniques
The optical properties of the AgNPs were analyzed with UV-visible spectroscopy (Multiskan Go, Thermo Fisher Scientific™, Finland) in the wavelength range of 300–600 nm with a resolution of 1 nm. The morphology was analyzed using transmission electron microscopy (TEM, JEOL, JEM-1010 Tokyo, Japan) at an accelerating voltage of 80 kV. The size distribution was estimated based on the TEM micrograph using ImageJ software (NIH, Maryland, USA). To confirm the crystalline nature of AgNPs, an X-ray diffraction (XRD) analysis (Bruker XDR, 2D Phaser, Billerica, Massachusetts, USA) with Cu-Kα radiation (1.54 Å), was carried out. To verify the presence of AgNPs in the monomer, an analysis was performed by inductively coupled plasma source mass spectrometry (ICP-MS, iCAP QR, Thermo Scientific, Waltham, Massachusetts, USA). Prior to sample analysis, the instrument was optimized using a certified aqueous solution of high-purity standards (Li, Co, In, Ba, Bi, Ce, and U at 1 μg L−1). For metal analysis, a 10-point calibration curve (0, 1, 1, 2.5, 5, 7.5, 10, 25, 50, 50, 75, and 100 μg L−1) was established from single-element solutions of Ag at 1000 μg mL−1. Instrumental drift was corrected using the internal standard rhodium (Rh at 10 μg L−1). The limits of detection were calculated using the following equation L. D. = 3(SD int BCO)(conc. STD)/(int STD – prom int BCO), where SD int BCO – standard deviation of the blank intensity; conc. STD – concentration of the standard solution; int STD – intensity of the standard solution and prom int BCO – average of the blank intensity.Biological response studies
 | ||
| Fig. 2 Schematic illustration of the processing of PMMA samples with and without AgNPs, biofilm formation and analysis using the bacterial MTT assay, scanning electron microscopy (SEM) and confocal microscopy for the LIVE/DEAD™ reagent (created in BioRender). | ||
:3 into 96-microwell plates and incubated for 24 h in fresh Eagle's Minimum Essential Medium (MEM) culture medium, supplemented with 10% sterile fetal bovine serum (FBS), 1% antibiotics (10000 IU mL−1 penicillin G and 10000 mg mL−1 streptomycin, Sigma-Aldrich, Mexico), and 1% glutamine (Glutamax, Gibco® Thermo Fisher Scientific™, Massachusetts, USA). Petri dishes were incubated at 37 °C with 5% CO2 and 95% relative humidity (Thermo Fisher Scientific™) to allow fixation for cell-based studies.
000 rpm for 10 min at 4 °C. A protease inhibitor cocktail (cOmplete Roche Diagnostics, Indianapolis, IN, USA) was added. The concentration of proteins in the lysates was determined by the Bradford method; then 30 μg mL−1 of proteins were loaded for 12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE), and the proteins were transferred to a membrane of polyvinylidene fluoride (PVDF, immobilon®-P Transfer Membranes, Sigma-Aldrich® St Louis, Missouri, USA). Blocking was performed with a 3% nonfat milk solution for 2 h at room temperature (RT) and anti-COX-1, anti-COX-2 and β-actin monoclonal antibodies were incubated (Santa Cruz Biotechnology, Dalla, TX, USA) at a dilution of 1:1000 in 3% blocking solution for 1 h 45 min, followed by four 5 min washes with 0.05% phosphate buffer saline with Tween detergent (PBST); then horseradish peroxidase-coupled polyclonal anti-mouse IgG antibodies were diluted (Sigma-Aldrich® St Louis, Missouri, USA) 1:10000 in 3% blocking solution and incubated for 1 h at room temperature. The formed complexes were visualized by chemiluminescence using the Cytiva Lifescience™ western Blot Detection Reagent (Amersham™ ECL™ Prime, Marlborough, USA) and the results were visualized using the C-Digit Blot Scanner (Li-Cor, Lincoln, NE, USA) and the density of the bands was analyzed using Software Image J (NIH, Maryland, USA).
Statistical analysis
Data obtained from all the experiments are expressed as mean and ± standard deviation of at least three independent experiments. Data analysis was performed using IBM® SPSS Statistics 29.0 and distribution was assessed using Shapiro-Wilk, ANOVA and Tukey's post hoc test, and p < 0.05 values were statistically significant.Results and discussion
Biosynthesis and characterization of AgNPs
The color change, from light green to amber, during the formation of AgNPs by green synthesis with Pelargonium × hortorum extract is shown, along with the surface plasmon resonance absorption at 412 nm in the UV-vis spectrum (Fig. 3A);this is typical of AgNP formation as reported in previous studies,32 and is consistent with findings from other researchers who suggest that the identification of high absorptions peaks in the spectrum between 400 and 450 nm is related to the formation of silver nanoclusters.38–40 Spherical spectra are related to a single band, while particles of different morphologies can give rise to two or more bands;41 so, our results from a single absorption band are consistent with this morphology, as shown in Fig. 3B, where the transmission electron microscope image reveals monodisperse nanoparticles of hemispherical morphology with an average size of 27.9 ± 9.2 nm. In Fig. 3C, the diffraction patterns of AgNPs are depicted, revealing diffraction peaks indicative of the presence of Ag0. Specifically, the peaks located at 2θ angles of 38.35°, 44.36°, 64.64°, and 77.65° correspond to the (111), (200), (220), and (311) crystal planes, respectively, according to the JCPDS: 04-0783 crystallographic chart. These peaks confirm the face-centered cubic (FCC) structure of the AgNPs obtained in this study, thus validating the presence of elemental silver, in line with the findings reported by other researchers.42,43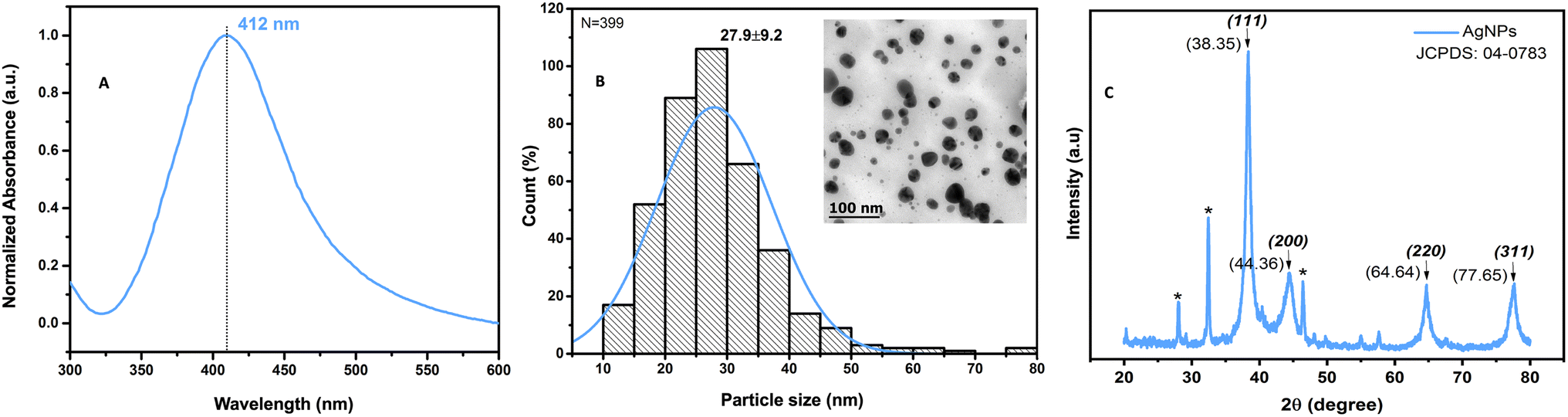 | ||
| Fig. 3 UV-vis spectrum, TEM micrograph, and XRD pattern of AgNPs. Absorption spectra of AgNPs obtained from Pelargonium × hortorum at 412 nm (A), the TEM micrograph taken at a magnification of 100k× and 80 kV showing monodisperse nanoparticles with a mean particle size of 27.9 ± 9.2 nm and hemispherical morphology (B), and the different diffraction peaks corresponding to the lattice plane values of silver crystals according to the standard (JCPDS) file no. 04-0783 (C). | ||
Chemical changes in the compositions of Pelargonium × hortorum leaf extracts after AgNP formation
Recently, we described the chemical composition of the Pelargonium × hortorum leaf extract using untargeted metabolomic analysis.32 The metabolite features present in the leaf extracts before and after AgNP formation were grouped into 1706 nodes arranged in 79 clusters with >3 nodes per cluster, 82 with two nodes, and 802 singletons (Fig. 4). From the total number of nodes, 412 were observed only in the extract after AgNP formation, while 691 were exclusive to the original extract before treatment. This indicates that the reaction during the formation of AgNPs affected the chemical composition of the extract. | ||
| Fig. 4 Molecular networking of Pelargonium × hortorum leaf extracts before and after AgNP formation. | ||
Principal component analysis (PCA) of the metabolite features observed by LCMS analysis clearly differentiated the Pelargonium × hortorum leaf extracts before and after treatment (PC1 = 97.8% versus PC2 = 1.1%; Fig. 5). The metabolites identified by GNPS for each treatment are shown in Table 1 and Fig. 6. Interestingly, the 24 metabolites identified in our previous work32 were not found in the leaf extract after AgNP formation. In addition, 24 metabolites were shared between the extracts from the 29 metabolites observed in the extract before AgNP formation. The major components of the plant extract were flavonoids and its glycosides, followed by O-glycosyl and phenylpropanoid compounds (tannins), and cyclitol derivatives (Fig. 6).
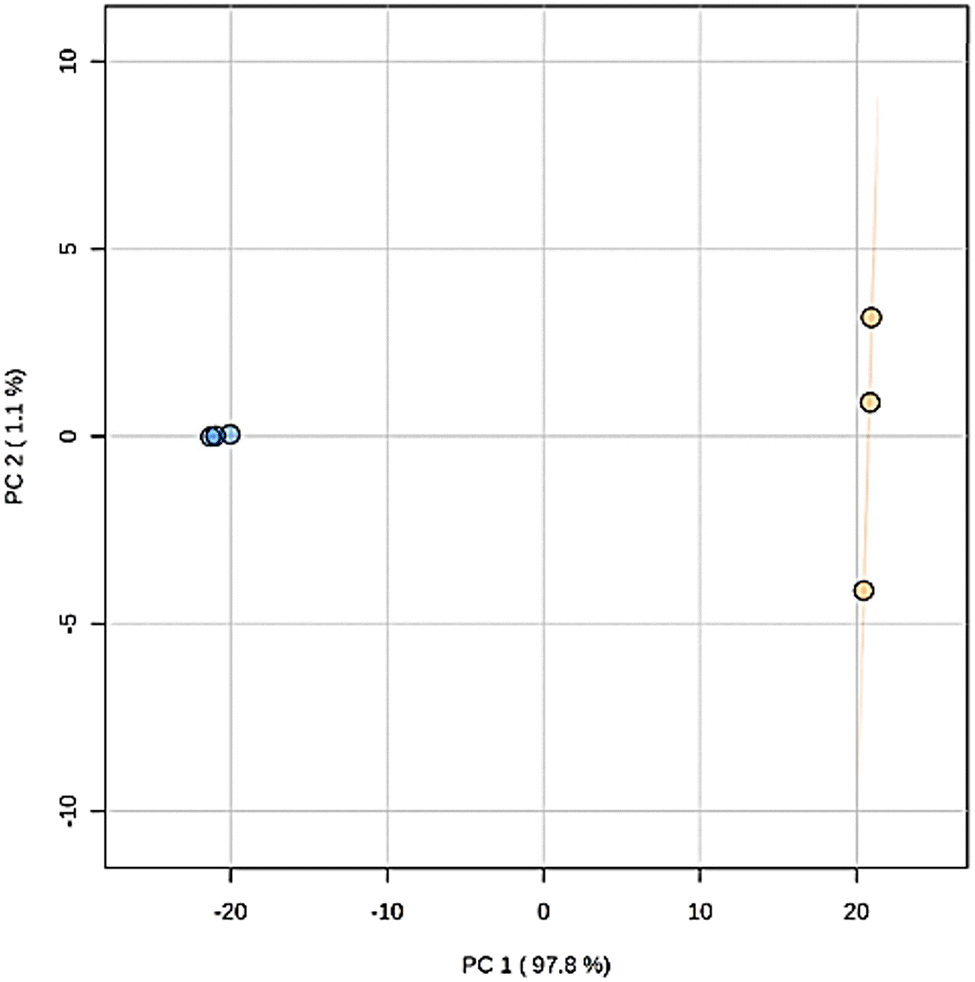 | ||
| Fig. 5 Nanoparticle treatment effect on the overall metabolomic profile determined by PCA of Pelargonium samples. Orange, original extract; blue, nanoparticle treatment. The PCA score plot derived from positive (ESI+) and negative (ESI−) modes in LC-MS metabolite profiles of Pelargonium × hortorum leaf extracts before and after AgNP formation. | ||
| Compounds | Observed iona (m/z) | Adduct | Molecular formula [adduct] | Exact mass (calculated) | Mass accuracy (ppm) |
|---|---|---|---|---|---|
| a Values taken from GNPS analysis. b Compounds observed in the extract before AgNP formation. c Compounds shared in the extracts. | |||||
| Galactaric acidc | 209.030 | [M − H]− | C6H9O8 | 209.0303 | −1.4 |
| Pantothenic acidb | 220.118 | [M + H]+ | C9H18NO5 | 220.1179 | +0.2 |
| Malic acidb | 289.018 | [2M + Na – 2H]− | C8H10O10Na | 289.0177 | +1.0 |
| 2-Phenylethyl β-D-glucopyranosidec | 302.161 | [M + NH4]+ | C14H24NO6 | 302.1598 | +3.9 |
| 2-Coumaroylquinic acidc | 339.109 | [M + H]+ | C16H19O8 | 339.1074 | +4.6 |
| Coumaric acid 4-O-glucosidec | 344.135 | [M + NH4]+ | C15H22NO8 | 344.1340 | +2.9 |
| Chlorogenic acidc | 353.088 | [M − H]− | C16H17O9 | 353.0878 | +0.6 |
| Coumaroyl + C6H9O8c | 355.068 | [M − H]− | C15H15O10 | 355.0671 | +2.6 |
| 3-O-Feruloylquinic acidc | 367.104 | [M − H]− | C17H19O9 | 367.1034 | +1.5 |
| NP-006680: 3-(benzoyloxy)-2-hydroxypropyl β-D-glucopyranosiduronic acidc | 371.099 | [M − H]− | C16H19O10 | 371.0984 | +1.7 |
| NP-016360: 3,5,5-trimethyl-4-[3-[(β-D-glucopyranosyl)oxy]butyl]-3-cyclohexen-1-olc | 375.239 | [M + H]+ | C19H35O7 | 375.2377 | +3.4 |
| NP-001173: [5-hydroxy-2-(3-hydroxybutyl)-3,3-dimethylcyclohexyl]methyl β-D-glucopyranosidec | 393.249 | [M + H]+ | C19H37O8 | 393.2483 | +1.8 |
| Kaempferol 3-α-L-arabinopyranosidec | 417.083 | [M − H]− | C20H17O10 | 417.0827 | +0.7 |
| Icariside F2c | 420.188 | [M + NH4]+ | C18H30NO10 | 420.1864 | +3.8 |
| Avicularinc | 435.093 | [M + H]+ | C20H19O11 | 435.0922 | +1.9 |
| Luteolin 4′-O-glucosideb | 447.094 | [M − H]− | C21H19O11 | 447.0933 | +1.6 |
| Astragalinc | 449.109 | [M + H]+ | C21H21O11 | 449.1078 | +2.6 |
| Isoquercetinc | 463.089 | [M − H]− | C21H19O12 | 463.0882 | +1.7 |
| 1,6-Digalloyl-β-D-glucopyranoseb | 502.121 | [M + NH4]+ | C20H24NO14 | 502.1191 | +3.7 |
| Procyanidin B2b | 577.136 | [M − H]− | C30H25O12 | 577.1352 | +1.5 |
| 6′′-O-(3-Hydroxy-3-methylglutaroyl)astragalinc | 591.137 | [M − H]− | C27H27O15 | 591.1355 | +2.5 |
| Nicotiflorinc | 593.152 | [M − H]− | C27H29O15 | 593.1512 | +1.4 |
| 2′′-O-Galloylhyperinc | 615.101 | [M + H]+ | C28H23O16 | 615.0992 | +3.0 |
| Mauritianinc | 739.211 | [M − H]− | C33H39O19 | 739.2091 | +2.6 |
| Kaempferol 3-O-(2,6-di-O-alpha-L-rhamnopyranosyl)-β-D-galactopyranosidec | 739.210 | [M − H]− | C33H39O19 | 739.2091 | +1.2 |
| Quercetin 3-(2R-apiosylrutinoside)c | 741.190 | [M − H]− | C32H37O20 | 741.1884 | +2.2 |
| Manghaslinc | 757.222 | [M + H]+ | C33H41O20 | 757.2186 | +4.5 |
| 1,2,3,6-Tetragalloylglucosec | 787.102 | [M − H]− | C34H27O22 | 787.0999 | +2.6 |
| Pentagalloylglucosec | 939.113 | [M − H]− | C41H31O26 | 939.1109 | +2.2 |
 | ||
| Fig. 6 Structures of the identified compounds in Pelargonium × hortorum leaf extracts. | ||
Self-curing acrylic resin compounds enriched with AgNPs
PMMA is a long polymer obtained through a chemical polymerization process in which the monomers react in steps between two molecular species (R and R′), which must contain at least two functional groups. When these compounds react, they join to form chains or networks of three-dimensional particles.44 The PMMA synthesis involves the addition of 0, 5, 10, 15, and 20 μL mL−1 of AgNPs to NicTone and Opti-Cryl monomers. Subsequently, the polymer–monomer mixture was prepared in a 3:1 ratio to initiate the polymerization reaction, following the methodology previously reported by Acosta-Torres et al.;22 the ICP-MS results indicated the presence of 15.623 ppm Ag in the monomer. Campos et al. reported the coating of AgNPs on Opti-Cryl using a sprayed solution with a concentration of 338 ppm; the authors analyzed the release of silver in an aqueous medium considering that the addition was by impregnation on the surface, identifying a release of 0.48 ppm after eight days of experimentation achieving antimicrobial activity.45 According to Flores-Arriaga et al., low concentrations of AgNPs at the filler level in the polymer interfere with the formation of bacterial biofilms through an anti-adherent effect.21
Biofilm formation
The adherence of S. mutans biofilms to PMMA and PMMA/AgNP composites was analyzed using the bacterial MTT assay.34,35Fig. 7 shows 91.6 ± 4.7% adherence of S. mutans biofilms on PMMA from the NicTone brand without the incorporation of AgNPs (0 μg mL−1), which decreased to 18.2 ± 2.8% with 10 μg mL−1 AgNPs (p < 0.05). The Opti-Cryl brand presented an adherence of 49.5 ± 3.3%, decreasing to 16.7 ± 2.4% with 15 μg mL−1 (p < 0.05). The AgNPs biosynthesized in this study showed antimicrobial activity against S. mutans at 10 μg mL−1,32 as described previously. These results are consistent with those of with Tavaf et al., who also reported a minimum inhibitory concentration at 10 μg mL−1, and are close to the ranges reported by Ma et al., at 8.5 μg mL−1.46,47 Campos et al.45 reported antimicrobial effects on Escherichia coli (6.74 μg mL−1) and Staphylococcus aureus (13.48 μg mL−1) in PMMA with AgNP compounds. These results, despite having been analyzed in other bacteria, are similar to our results, where a decrease in microbial adherence was found at 5, 10, and 15 μg mL−1 AgNPs. In the present study, the AgNPs were inside the material and were incorporated into the monomer, as described by other researchers.22,48 Several reports in the literature indicate that the antibacterial and antibiofilm efficacy of AgNP-incorporated materials depends on the release of silver ions and their direct contact with bacteria.38,48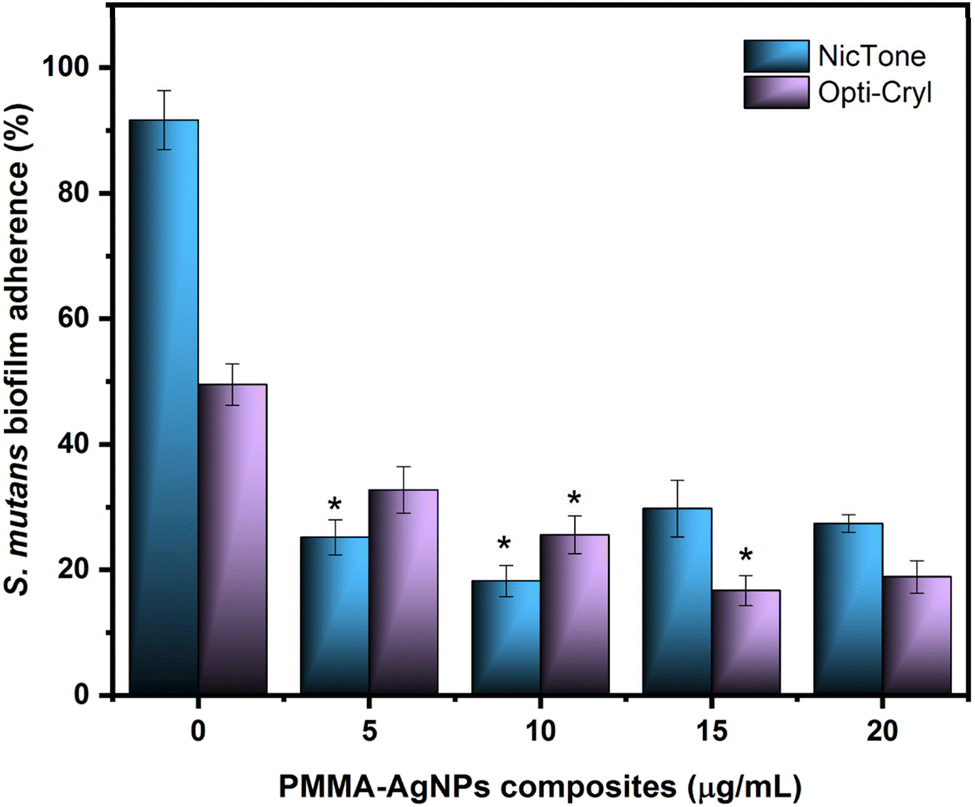 | ||
| Fig. 7 Adherence analysis of S. mutans biofilms to PMMA–AgNP composites on NicTone and Opti-Cryl with 0, 5, 10, 15, and 20 g mL−1 AgNPs was evaluated using bacterial MTT assay. Optical density 550 nm. Abs. range (0.062–0.709), where each value represents the percentage of the mean and S.D. One-way ANOVA, post hoc Tukey; (*) represents concentrations with significant difference; p < 0.05, n = 9 in independent experiments in triplicate. | ||
According to the SEM images (Fig. 8), notable differences were observed between the PMMA groups with and without AgNPs. A comparison of biofilm images showed changes in biofilm growth on self-curing PMMA, which was observed to be dispersed in the absence of sucrose. However, these biofilms developed homogeneously with areas of greater structural maturity and more homogeneous growth with a sucrose supplementation of 0.6% in the culture medium, suggesting that its presence favors the growth and structure of biofilms.49 This significantly higher accumulation of S. mutans is related to elevated levels of polysaccharides, such as glucans, which are the main constituents of the S. mutans biofilm matrix, providing a three-dimensional scaffold and attachment sites to support this bacterial coaggregation. The reduced biofilm formation on nanoparticle polymers may be due to the inability of bacteria to use these polyols as substrates for the synthesis of biofilm matrices.49,50 Since the SEM images only showed the biofilm volume, a Live/Dead™ BacLight bacterial viability kit was used to evaluate the effect of PMMA and PMMA/AgNP composites. Fig. 8 shows the presence of a higher biovolume of live-cell bacteria (green) in PMMA without the addition of AgNPs. This biofilm showed an increase in dead cells (red) in PMMA with 10 μg mL−1 AgNPs, mainly for the NicTone group with 10 μg mL−1 AgNPs and these results correlate with those identified in the MTT assay, showing the presence of a greater biovolume of live bacterial cells.
 | ||
| Fig. 8 Scanning electron microscopy and confocal fluorescence microscopy of S. mutans biofilms. Scanning electron microscopy at 6500× magnification shows the growth of the biofilm on the acrylic resin; a higher production of extracellular polysaccharides can be observed in the presence of 0.6% sucrose, which favors their formation; the incorporation of AgNPs allows the reduction of the density of the bacterial growth structure as observed in the NicTone group at 10 μg mL−1. Confocal microscopy images of biofilms stained with a Live/Dead™ BacLight bacterial viability kit show live cells in green and dead cells in red, indicating a greater presence of dead biofilms in the NicTone 10 μg mL−1 AgNPs group (magnification 40× and scale bar 20 μm). | ||
Cytotoxicity assay
The cytotoxic effect of biosynthesized AgNPs was identified in previous studies with a dose-dependent response, with moderate cytotoxicity, with the mean cytotoxic dose (CC50) being found at a concentration of 4.5 μg mL−1, as described in previous reports.32 For the HGF assays, from the cell viability test, 10 μg mL−1 AgNPs were incorporated into the PMMA, as this was the concentration at which statistically significant differences were found in both brands analyzed. A decrease in cell viability was observed for the NicTone groups with 0 and 10 μg mL−1 in direct contact, and for the Opti-Cryl group with 10 μg mL−1 in indirect contact (p < 0.05); however, the viability percentage in these groups was greater than 80%, which means that they did not present cytotoxicity.51 Furthermore, in all other groups, a hormesis effect was observed (Fig. 9A). These results are consistent with those of Sun et al., who confirmed the biocompatibility of PMMA with AgNPs at the cellular level using MTT and in vivo assays in experiments with rabbits and mice.52 In contrast, Pinheiro et al. found greater cytotoxicity in groups with AgNPs incorporated into or immersed in PMMA.53 For this reason, the nanoparticle incorporation technique plays an important role in the cellular response; in our study, the independent analysis of AgNPs showed a 50% decrease in cell viability at 4.5 μg mL−132 compared to incorporating 10 μg mL−1 in self-curing PMMA, which maintained it above 80%. When the polymers are immersed in PMMA, the decrease in cell viability is greater in the first 24 h, because AgNPs exhibit high diffusion in aqueous media;53 however, when incorporated during the polymerization process, the metallic silver is possibly encapsulated in the polymer, reducing the percentage of release into the medium and thus promoting cell viability.54 | ||
| Fig. 9 Biological assays of PMMA and PMMA–AgNP composites in direct and indirect contact with HGF. Groups: Opti-Cryl and NicTone, AgNPs concentrations of 0 and 10 μg mL−1. (A) Cytotoxicity determined by MTT assay. Optical density 570 nm. Abs. range (0.211–1.857). Each value in the graph represents the percentage of the mean and S.D. One-way ANOVA was performed, followed by post hoc Tukey's test; (*) represents concentrations with a significant difference p < 0.001, n = 9. (B) Oxidative stress induced by the intracellular ROS fluorometric assay. Negative control: sterile medium; positive control, H2O2 2 mM. Optical density λex = 490/λem = 520 nm. Abs. range (3.788–3.814). Each value in the graph represents the mean and S.D. One-way ANOVA was performed for p > 0.05, and n = 9. (C) 63× micrograph of nuclear morphology by DAPI staining using fluorescence microscopy. (1) Negative control: HGF in the culture medium; (B) positive control (H2O2 at 2 mM), (3) NicTone 0 μg mL; (4) NicTone 10 μg mL; (5) Opti-Cryl 0 μg mL; (6) Opti-Cryl 10 μg mL−1. The arrowheads indicate (I) normal morphology, (II) fragmented nuclei, (III) reniform nuclei, and (IV) shrunken nuclei (n = 3). (D) and (E) Western blotting showing the relative protein expression of anti-COX-1, anti-COX-2, and β-actin, and image analysis of western blot bands was performed by densitometry. Negative control: untreated cells (IL−); positive control: cells induced in a proinflammatory state (IL+) (n = 3). | ||
Oxidative stress
The reactive oxygen species (ROS) experiment showed a slight increase in the production of free radicals in the study groups without statistically significant results (p > 0.05) (Fig. 9B). While in the evaluation of nuclear morphology using DAPI and fluorescence microscopy, continuity of the cell nuclei can be observed for the negative control groups (HGF), in the PMMA and self-healing PMMA–AgNP groups, a change in morphology was observed, with more circular shapes and a decrease in size, as well as some reniform nuclei compared to the negative control group; in the positive control group (H2O2) a complete fragmentation of the nuclei was observed (Fig. 9C).Oxidative stress can cause chromosomal damage, nuclear abnormalities, and cytogenetic alterations. Some researchers have reported that reniform nuclei have a cytological cause, while others consider that genotoxic effects may induce nuclear morphological changes.55,56 Furthermore, excessive production of ROS within cellular mitochondria can potentially interfere with the proper mediation of cytochrome c release, which then triggers the activation of caspase-3, and consequently, can induce cell apoptosis.57
Tests for genotoxicity analysis are performed by different methods, such as the determination of DNA/protein crosslinking coefficients, mitochondrial enzymatic activity, cell proliferation, DNA break repair, mitotic index, comet assay, identification of damage, chromosomal aberrations and alterations in nuclear morphology.58 However, there are few studies reported in the literature to determine the genotoxicity of PMMA; Acosta-Torres et al. did not find alterations related to nuclear alterations when analyzing polymers with and without AgNPs,22 while others researchers identified genotoxic effects after 24 h of exposure to MMA vapour.59
Proinflammatory response and residual monomers
The toxic and inflammatory processes of PMMA reported in articles relate this response to the polymerization effect resulting from the exothermic reaction of the methacrylates in the material and the resulting residual monomers; this release can induce genetic mutations, causing cell cycle delay and cell death.60 Campaner et al. and Bacali et al. identified the presence of TNF-α as an inflammatory cellular response in PMMA in direct and indirect contact.60,61 It was also found that the addition of 2% AgNPs to PMMA had an anti-inflammatory effect.61 In relation to the release of pro-inflammatory cytokines in HGF, a significant release of cytokines has been reported in indirect contact with PMMA.61 On the other hand, Trubiani et al. reported that after 24 h of contact with PMMA, the release of interleukin-6 (IL-6) and interleukin-8 (IL-8) was identified, with lower levels observed when the samples were polished, favoring a reduction in the gingival inflammatory process.62To assess the inflammatory response in our study, HGFs were induced into a proinflammatory state with IL-1-β, which among its many functions, induces the production of IL-6. Cyclooxygenases (COX) 1 and 2, which are key enzymes involved in the synthesis of prostaglandins (PG), were detected. The COX-1 isoform maintains a normal physiological production of prostaglandins, while COX-2 is produced by cytokines and endotoxins responsible for inflammatory processes.63 Our results showed that PMMA with 10 μg mL−1 AgNPs both in direct and indirect contact did not show COX-1 related changes (Fig. 9D); however, in synergy with a pro-inflammatory state, an increase in COX-2 expression was identified in the case of direct contact with PMMA with 10 μg mL−1 AgNPs (p < 0.05) (Fig. 9E).
In relation to the residual monomer in Fig. 10A, the release amount does not change in the two brands analyzed with and without AgNPs, considering that both commercial self-curing PMMA comply with the provisions of the ISO-20975-2 STANDARD,36 in Section 5.2, because the maximum mass fraction of the residual monomer is less than 5.0%. In the specific case of Opti-Cryl, the standard deviation was large, although the addition of AgNPs did not change the system. This difference may be because the experimental group was not completely soluble in tetrahydrofuran (THF);37 however the solvent swells the sample sufficiently to extract any possible residual monomer, which does not affect the analysis of the residual monomer. Another possible explanation is that the sample contains a mixture of acrylates or different monomers that cross-link more strongly.
 | ||
| Fig. 10 (A) Residual monomers of Opti-Cryl and NicTone self-curing PMMA. AgNP concentrations of 0 and 10 g mL−1. Each value in the graph represents a percentage and S.D. One-way ANOVA was performed, along with post hoc Tukey; (*) represents concentrations with significant differences (p < 0.001, n = 3). (B) Flexural strength in the 3-point test for the PMMA and PMMA–AgNP composites. Standard (ISO 20795-2 values, minimum 50 Mpa). Each value in the graph represents the mean and S.D. One-way analysis of variance (ANOVA) was performed. p > 0.05, n = 10. (C) Flexural moduli of PMMA and PMMA–AgNP composites. Standard (ISO 20795-2, minimum 1500 Mpa). Each value in the graph represents the mean and S.D. One-way ANOVA was performed. p > 0.05, n = 10. | ||
Mechanical properties
In the results of the mechanical tests of strength and flexural modulus, values above the norm were observed for Opti-Cryl and NicTone with 0 μg mL−1 (control groups; Fig. 10). In Opti-Cryl with 10 μg mL−1 AgNPs, an increase was observed compared to its control group and in NicTone with 10 μg mL−1 of AgNPs a slight decrease was observed, although no statistically significant differences were found (p > 0.05) (Fig. 10B and C).Campos et al. reported a flexural strength of 67.89 ± 8.54 MPa for Opti-Cryl, which is above the ISO standard; when AgNPs were incorporated into the material, they reported improved mechanical properties (69.02 ± 6.14 MPa);45 our results show higher values (from 90 MPa) in the two brands of PMMA studied, even before the incorporation of AgNPs.
Research has shown that the mixing method can affect the dispersion of nanoparticles in a matrix. Furthermore, the type of NPs incorporated, their size, shape and concentration can determine the interaction with the polymer matrix.64,65 In our results, both mechanical tests for the NicTone group with 10 μg mL−1 AgNPs showed a slight decrease in comparison with the control group (0 μg mL−1). This can be explained by the dispersion of AgNPs in the polymer matrix, which reduced the reaction of the monomer and consequently the physical properties.65 However, these values were above the standard, so we can deduce that the proposed technique of adding AgNPs to the monomer avoids agglomeration sites that could negatively affect the physical properties of the nanocomposite.
It can be observed that the biological activity and mechanical characteristics of PMMA–AgNP composites are slightly affected, depending on the concentration and the incorporation technique of the AgNPs utilized. The cytotoxicity assays indicated that higher concentrations of AgNPs, up to 10 μg mL−1, can be integrated into PMMA composites without affecting the cell viability (below 80%). This suggests that the developed biocomposite can retain non-cytotoxic behavior, even under conditions that cause slight cell viability loss. This compatibility can be explained by the encapsulation of the AgNPs that occurred during the polymerization process, diminishing the chances of inward-cell exposure to the particles, and therefore, toxic interactions with the cells are minimized.64
Interestingly, the same method of encapsulation may also be responsible for the mechanical properties observed. For instance, addition of 10 μg mL−1 AgNPs resulted in a mild decrease in flexural strength in the NicTone group but an improvement in the Opti-Cryl group. These variations could be attributed to AgNP dispersions and interactions in the polymeric matrix.65 Inadequate dispersion can lead to microstructural inconsistencies, which reduces the monomer–polymer interaction and slightly compromises the mechanical properties, as observed in NicTone. In contrast, improved dispersion, as observed in Opti-Cryl, can enhance the reinforcing effect of AgNPs, leading to better mechanical performance.64,65
The encapsulation of AgNPs also seems to provide a potential reduction in trade-off between cytotoxicity and mechanical integrity. High concentrations of AgNPs have been linked with cytotoxicity in aqueous environments,53 but a polymeric matrix such as PMMA can hinder their release and enable diffusion over time. Our results show that not only we can achieve cell viability, but we also maintain mechanical properties above ISO standards.
Conclusions
A greater decrease in bacterial adhesion was observed with the incorporation of AgNPs into NicTone brand PMMA at 10 μg mL−1. At this concentration, cellular bioassays did not show cytotoxicity or ROS expression, but a genotoxicity response was observed with reniform nuclei, confirming that it is necessary to continue with more specific experiments to determine the mechanisms of inflammation or alteration in host cells. The present study demonstrates that the incorporation of AgNPs into PMMA matrices improves certain functional properties of the material, particularly its antimicrobial behaviour and mechanical performance; it was observed that the effect of AgNPs depends significantly on their concentration. This behaviour influences both the mechanical properties of the compound and its interaction with biological cells, highlighting the importance of optimizing the concentrations to avoid adverse effects such as cytotoxicity or embrittlement of the material. Overall, the cell bioassays analyzed in this research did not show any significant inflammatory effect with the PMMA–AgNP nanocomposite, suggesting that it could be a potential candidate for dental applications.Data availability
Raw data are available from the corresponding author on reasonable request.Author contributions
Conceptualization: C. A. L.-A., L. S. A.-T., and R. G.-C. Methodology and validation: L. S. A.-T., R. G.-C., and R. M. Result interpretation: C. A. L.-A., M. F., M. R.-G., M. J. A., and R. A. D.-P. HPLC analyses: M. F., M. R.-G., M. J. A. and S. C. Data curation: C. A. L.-A., M. F. R., G.-C. and R. M. Supervision: L. S. A.-T. Manuscript writing: C. A. L.-A. Manuscript review and editing: L. S. A.-T., R. G.-C., R. M., M. F., M. R.-G., M. J. A., R. A. D.-P. and S. C.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the financial support from UNAM-DGAPA-PAPIIT: IN211922, IT200922 & TA200123 and PAPIME: PE203622. They also acknowledge the technical assistance from Ing. Nydia Hernandez Rios (fluorescence microscopy), Ing. Ma. Lourdes Palma Tirado (TEM analysis), INB-Juriquilla, Mexico; Shigeru Amano (SEM images), Meikai University, Japan; M. C. Elizabeth Hernandez Alvarez for analytical support in the determination of trace elements (ICP-MS), Instituto de Geofisíca, UNAM, Mexico and Dr Dulce Guzman Rocha (XRD pattern), Centro de Investigaciones en Óptica, León, Guanajuato, Mexico.References
- A. M. Díez-Pascual, Int. J. Mol. Sci., 2022, 23, 10288, DOI:10.3390/ijms231810288.
- H. Kaur and A. Thakur, Mater. Today: Proc., 2022, 50, 1619–1625, DOI:10.1016/j.matpr.2021.09.125.
- R. de Souza Leão, S. L. D. de Moraes, J. M. de Luna Gomes, C. A. A. Lemos, B. G. da Silva Casado, B. C. do Egito Vasconcelos and E. P. Pellizzer, Mater. Sci. Eng., 2020, 106, 110292, DOI:10.1016/j.msec.2019.110292.
- M. S. Zafar, M. S. Polymers, 2020, 12, 10–2299. DOI:10.3390/polym12102299.
- S. M. Pituru, M. Greabu, A. Totan, M. Imre, M. Pantea, T. Spinu, A. M. C. Tancu, N. O. Popoviciu, I. Stanescu and E. Ionescu, Materials, 2020, 13, 2894, DOI:10.3390/ma13132894.
- K. Chęcińska, M. Chęciński, M. Sikora, Z. Nowak, S. Karwan and D. Chlubek, Polymers, 2022, 14, 1047, DOI:10.3390/polym14051047.
- I. Domagała, L. Gil, M. Firlej, D. Pieniak, J. Selech, D. Romek and B. Biedziak, Adv. Sci. Technol., 2020, 14, 4, DOI:10.12913/22998624/127437.
- G. Lombardo, F. Vena, P. Negri, S. Pagano, C. Barilotti, L. Paglia, S. Colombo, M. Orso and S. Cianetti, Eur. J. Paediatr. Dent., 2020, 21(2), 115–122, DOI:10.23804/ejpd.2020.21.02.05.
- M. S. Alhammadi, E. Halboub, M. S. Fayed, A. Labib and C. El-Saaidi, Dent. Press J. Orthod., 2018, 23(6), 40, DOI:10.1590/2177-6709.23.6.40.e1-10.onl.
- J. Murrieta-Pruneda, D. Varela-Ramírez, A. Rojano-Santillán, M. Adriano-Anaya and T. Caudillo-Joya, J. Oral. Res., 2020, 9(4), 293–299, DOI:10.17126/joralres.2020.070.
- J. O. Jorge, L. Corradi-Dias, C. Flores-Mir, I. A. Pordeus, S. M. Paiva and L. G. Abreu. The journal of evidence-based dental practice, 2020, 20, 3, 101423. doi: DOI:10.1016/j.jebdp.2020.101423.
- J. Bächle, C. Merle, S. Hahnel and M. Rosentritt, Materials, 2023, 16(6), 2373, DOI:10.3390/ma16062373.
- X. Wei, L. Gao, K. Wu, Y. Pan, L. Jiang, H. Lin and H. Cheng, Clin. Oral Investig., 2022, 26(12), 7287–7297, DOI:10.1007/s00784-022-04689-2.
- S. Keleştemur, Z. Çobandede and M. Çulha, Colloids Surf., B, 2020, 188, 110765, DOI:10.1016/j.colsurfb.2019.110765.
- A. Delgado, J. Carvalho, G. Borrecho, T. Nascimento, M. E. Silva, S. A. Félix and J. J. Mendes, Contemp. Clin. Dent., 2018, 9(3), 400–405, DOI:10.4103/ccd.ccd_141_18.
- T. Vanishree, G. S. Panchmal, R. Shenoy, P. Jodalli, L. Sonde and N. Kundapur, Oral Health Prev. Dent., 2017, 15(5), 453–459, DOI:10.3290/j.ohpd.a38776.
- C. Shukla, R. K. Maurya, V. Singh and M. Tijare, Dent. Res. J., 2016, 13(4), 309–314, DOI:10.4103/1735-3327.187876.
- N. Farhadian, R. Usefi Mashoof, S. Khanizadeh, E. Ghaderi, M. Farhadian and A. Miresmaeili, Am. J. Orthod. Dentofacial Orthop., 2016, 149(2), 155–160, DOI:10.1016/j.ajodo.2015.07.031.
- E. Chimenos-Küstner, M. L. Giovannoni and M. Schemel-Suárez, Med. Clínica, 2017, 149(7), 305–309, DOI:10.1016/j.medcli.2017.05.0.
- M. A. Huq, Int. J. Mol. Sci., 2020, 21(4), 1510, DOI:10.3390/ijms21041510.
- J. C. Flores-Arriaga, R. Garcia-Contreras, G. Villanueva-Sanchez and L. S. Acosta-Torres, Appl. Sci., 2020, 10(11), 4007, DOI:10.3390/app10114007.
- L. S. Acosta-Torres, I. Mendieta, R. E. Nuñez-Anita, M. Cajero-Juárez and V. M. Castaño, Int. J. Nanomed., 2012, 7, 4777–4786, DOI:10.2147/IJN.S32391.
- S. P. Singh, S. K. Sharma and D. Y. Kim, Solid State Sci., 2020, 99, 106046, DOI:10.1016/j.solidstatesciences.2019.106046.
- F. Yousefi, S. B. Mousavi, S. Z. Heris and S. Naghash-Hamed, Sci. Rep., 2023, 13(1), 7116, DOI:10.1038/s41598-023-34120-z.
- M. Cascione, V. De Matteis, P. Pellegrino, G. Albanese, M. L. De Giorgi, F. Paladini and R. Rinaldi, Nanomaterials, 2021, 11(8), 2027, DOI:10.3390/nano11082027.
- A. M. Alsaad, A. A. Ahmad, A. R. Al Dairy, A. S. Al-anbar and Q. M. Al-Bataineh, Results Phys., 2020, 19, 103463, DOI:10.1016/j.rinp.2020.103463.
- A. Nabhan, M. Taha and N. M. Ghazaly, Polym. Test., 2023, 117, 107848, DOI:10.1016/j.polymertesting.2022.107848.
- R. Bahrami, F. Gharibpour, M. Pourhajibagher and A. Bahador, Int. Orthod., 2024, 22(2), 100846, DOI:10.1016/j.ortho.2024.100846.
- H. A Widatalla, L. F. Yassin, A. A. Alrasheid, S. A. R. Ahmed, M. O. Widdatallah, S. H. Eltilib and A. A. Mohamed, Nanoscale Adv., 2022, 4(3), 911–915, 10.1039/d1na00509j.
- C. Takahashi and K. Moriguchi, Nanoscale Adv., 2024, 6(20), 5020–5030, 10.1039/d4na00249k.
- M. C. R. Pinheiro, J. A. O. Carneiro, M. M. Pithon and E. F. Martinez, Mater. Res., 2021, 24(2), e20200115, DOI:10.1590/1980-5373-MR-2020-0115.
- C. A. Lopez-Ayuso, R. Garcia-Contreras, R. Manisekaran, M. Figueroa, M. C. Arenas-Arrocena, G. Hernandez-Padron and L. S. Acosta-Torres, RSC Adv., 2023, 13(42), 29784–29800, 10.1039/d3ra00201b.
- Z. Pang, G. Zhou, J. Ewald, L. Chang, O. Hacariz, N. Basu and J. Xia, Nat. Protoc., 2023, 17(8), 1735–1761, DOI:10.1038/s41596-022-00710-w.
- E. Grela, J. Kozłowska and A. Grabowiecka, Acta Histochem., 2018, 120(4), 303–311, DOI:10.1016/j.acthis.2018.03.007.
- E. Brambilla, A. Ionescu, G. Cazzaniga, V. Edefonti and M. Gagliani, Am. J. Dent., 2014, 27(3), 160–166 Search PubMed.
- International Organization for Standardization. 20795-2:2013, Dentistry-Base Polymers, Part 2: Orthodontic Base Polymers.
- F. Solórzano Lemus, R. D. Venegas Lancón, V. Moreno Maldonado and S. López Morales, Rev. Odontol. Mex., 2010, 14(2), 91–98 Search PubMed.
- P. Serrano-Díaz, D. W. Williams, J. Vega-Arreguin, R. Manisekaran, J. Twigg, D. Morse and L. S. Acosta-Torres, Green Process. Synth., 2023, 12(1), 20228105, DOI:10.1515/gps-2022-8105.
- M. Marchioni, G. Veronesi, I. Worms, W. L. Ling, T. Gallon, D. Leonard and A. Deniaud, Nanoscale Horiz., 2020, 5(3), 507–513, 10.1039/c9nh00286c.
- R. D. Rivera-Rangel, M. P. González-Muñoz, M. Avila-Rodriguez, T. A. Razo-Lazcano and C. Solans, Colloids Surf., A, 2018, 536, 60–67, DOI:10.1016/j.colsurfa.2017.07.051.
- M. Mohammadlou, H. Jafarizadeh-Malmiri and H. Maghsoudi, Green Process. Synth., 2017, 6, 31–42, DOI:10.1515/gps-2016-0075.
- S. Jeong, S. Yang, Y. J. Lee and S. H. Lee, J. Mater. Chem. A, 2023, 11(25), 13409–13418, 10.1039/D3TA00691C.
- M. D. Ferreira, L. C. D. S. Neta, G. C. Brandão and W. N. L. Dos Santos, Biotechnol. Appl. Biochem., 2023, 70(3), 1001–1014, DOI:10.1002/bab.2415.
- A. Kaddouri, B. Serier, K. Kaddouri, M. Belhouari and M. Frattura, Integrità Strutturale, 2020, 14, 53, pp. 66–80. doi: DOI:10.3221/IGF-ESIS.53.06.
- V. Campos, I. DeAlba-Montero, F. Ruiz, C. Butrón-Téllez Girón, E. García-García and M. Loredo-Tovías, Superficies Vacio, 2017, 30(4), 51–55, DOI:10.47566/2017_syv30_1-040051.
- Z. Tavaf, M. Tabatabaei, A. Khalafi-Nezhad and F. Panahi, Eur. J. Integr. Med., 2017, 12, 163–171, DOI:10.1016/j.eujim.2017.05.011.
- X. Ma, J. Lang, P. Chen and R. Yang, AIChE J., 2021, 67, 12, DOI:10.1002/aic.17468.
- J. Méndez-Serrano, U. Velazquez-Enriquez, R. Contreras-Bulnes, I. De La Rosa-Gómez, T. Sawada and R. Yamaguchi, Interciencia, 2020, 45(1), 23–27 Search PubMed.
- A. Chan, K. Ellepola, T. Truong, P. Balan, H. Koo and C. J. Seneviratne, Arch. Oral Biol., 2020, 119, 104886, DOI:10.1016/j.archoralbio.2020.104886.
- J. N. Cai, H. M. Choi and J. G. Jeon, J. Oral Microbiol., 2021, 13(1), 1910443, DOI:10.1080/20002297.2021.1910443.
- International Organization for Standardization, 10993-5, Part 5: t-est for in vitro cytotoxicity.
- J. Sun, L. Wang, J. Wang, Y. Li, X. Zhou, X. Guo, T. Zhang and H. Guo, Dent. Mater. J., 2021, 40(5), 1100–1108, DOI:10.4012/dmj.2020-129.
- M. C. R. Pinheiro, J. A. O. Carneiro, M. M. Pithon and E. F. Martinez, Mater. Res., 2021, 24, e20200115, DOI:10.1590/1980-5373-MR-2020-0115.
- A. Shenava, Nanosci. Nanotechnol.--Asia, 2023, 13(3), 34–41, DOI:10.2174/2210681213666230413090403.
- I. Brandts, C. Barría, M. A. Martins, L. Franco-Martínez, A. Barreto, A. Tvarijonaviciute and M. Teles, J. Hazard. Mater., 2021, 403, 123590, DOI:10.1016/j.jhazmat.2020.123590.
- A. Barreto, A. Dias, B. Duarte, E. Pinto, A. Almeida, T. Trindade and M. Oliveira, Sci. Total Environ., 2020, 716, 137026, DOI:10.1016/j.scitotenv.2020.137026.
- A. F. Khafaga, M. A. Naiel, M. A. Dawood and H. M. Abdel-Latif, Aquat. Toxicol., 2020, 228, 105624, DOI:10.1016/j.aquatox.2020.105624.
- R. Landsiedel, N. Honarvar, S. B. Seiffert, B. Oesch and F. Oesch, Wiley Interdiscip. Rev.:Nanomed. Nanobiotechnol., 2022, 14(6), e1833, DOI:10.1002/wnan.1833.
- M. C. Arenas-Arrocena, L. Argueta-Figueroa, R. García-Contreras, O. Martínez-Arenas, B. Camacho-Flores, M. P. Rodriguez-Torres and L. S. Acosta-Torres, Acrylic Polym. Healthcare, 2017, 10, 43–74, DOI:10.5772/intechopen.69066.
- M. Campaner, A. S. Takamiya, S. B. Bitencourt, L. C. Mazza, S. H. P. de Oliveira, R. Shibayama and A. Pesqueira, Arch. Oral Biol., 2020, 111, 104643, DOI:10.1016/j.archoralbio.2019.104643.
- C. Bacali, I. Baldea, M. Moldovan, R. Carpa, D. E. Olteanu, G. A. Filip and F. Badea, Clin. Oral Invest., 2020, 24, 2713–2725, DOI:10.1007/s00784-019-03133-2.
- O. Trubiani, E. Toniato, D. Di Iorio, F. Diomede, I. Merciaro, C. D'Arcangelo and T. Oriana, Int. J. Immunopathol. Pharmacol., 2012, 25(3), 637–643, DOI:10.1177/039463201202500310.
- M. M. Taskan and F. Gevrek, Niger. J. Clin. Pract., 2020, 23(1), 46–53, DOI:10.4103/njcp.njcp_349_19.
- P. Soleymanijadidi, M. Moradi, F. Hamedirad, Z. Ghanavati, S. Maleki Dizaj and S. Salatin, Bioengineering, 2023, 10(5), 559, DOI:10.3390/bioengineering10050559.
- A. Mithran, J. Rakhra, S. K. Jain, M. Chikkanna, S. Gowrish, S. G. Pillai and A. S. Nayyar, J. Orthod. Sci., 2023, 12(1), 64, DOI:10.4103/jos.jos_43_23.
| This journal is © The Royal Society of Chemistry 2025 |