
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient catalytic hydrogenation of p-chloronitrobenzene: the synergistic effect and hybrid nano-structure†
Yanji Zhang *a and
Jicheng Zhoub
*a and
Jicheng Zhoub
aSchool of Chemistry and Materials Engineering, Quzhou University, Quzhou 324000, Zhejiang Province, China. E-mail: zhangyanji2017@126.com
bKey Laboratory of Green Catalysis and Chemical Reaction Engineering of Hunan Province, School of Chemical Engineering, Xiangtan University, Xiangtan 411105, Hunan Province, China
First published on 21st January 2025
Abstract
Noble metal catalysts have attracted worldwide attention for catalytic selective hydrogenation due to their high activity. However, constructing highly effective, selective, and stable noble metal catalysts remains challenging. Herein, zirconium/zeolite supported platinum catalysts have been prepared. These hybrid nano-structured Pt/ZrO2/ZSM-5 and Pt/ZrO2/MCM-22 catalysts (denoted as Pt/ZZ and Pt/ZM) exhibited superior catalytic performance for the selective hydrogenation of p-chloronitrobenzene (p-CNB), which was much higher than that of Pt/ZrO2, Pt/ZSM-5, and Pt/MCM-22, demonstrating the synergistic effect in these hybrid nano-structured catalysts. Especially, the turnover frequency (TOF) of Pt/ZM was as high as 8525 h−1, surpassing that of most reported noble metal and novel non-noble metal catalysts, and the catalyst can be recycled for six runs without any obvious decrease in activity and selectivity. The characterization indicated that the electron transfer from Pt to ZrO2 can greatly facilitate the performance of p-CNB hydrogenation through modulating the electronic structure and surface structure. This work provides an effective strategy for the design of noble metal catalysts with high activity and selectivity.
1 Introduction
Over the past few decades, noble metal catalysts have played an important role in selective hydrogenation due to their excellent performance.1–4 Notably, side reactions were unavoidable on these single-metal-based catalysts, such as complete hydrogenation, competition from other reducible functional groups, or group breaking. Furthermore, agglomeration or leaching of metal nanoparticles often occurred, leading to poor selectivity and catalytic activity.5–7 Therefore, the development of advanced catalysts that are stable, easy-to-handle, highly active, and selective has become a subject of urgent research, although this goal is challenging.As we all know, both noble metals and supports play a crucial role because H2 can be adsorbed and dissociated on a noble metal site, and the substrates hydrogenate on the support.8 Noble metals, especially Pt9–12 and Pd,13–15 have been intensively used in fabricating heterogeneous catalysts because of their remarkable activity for hydrogen activation. Meanwhile, the downside is that when C–X (X = F, Cl, Br, and I) coexists with –NO2, selective conversion of –NO2 to –NH2 will be accompanied by dehalogenation, affecting selectivity.16 Therefore, it is very important and urgent to design and prepare highly efficient noble metal catalysts.
In fact, the supports not only stabilize the dispersed metals but also modify their morphology and electronic structure through interaction with them, the so-called metal–support interaction.17 Taking advantage of this interaction to adjust the catalyst performance has been the most popular approach. Recently, some selective hydrogenation catalysts have been developed by choosing suitable supports, which enhance activity due to their architecture and interaction with noble metals. Various advanced materials have been reported because of significant improvements in synthetic methods and characterization techniques.
Zeolite, a unique inorganic crystalline aluminosilicate, with a high surface area, improves the dispersity of metals and provides reaction sites, contributing to the adsorption of substrate molecules. Thus, it has been widely used in selective hydrogenation.1,18,19 Chen et al.20 encapsulated Pt clusters into sodalite (SOD) zeolite, and the structure of SOD zeolite prevented the direct interaction between Pt and p-CNB, avoiding the simultaneous activation of the nitro group and C–Cl group on the encapsulated Pt surface. Instead, selective hydrogenation of nitro was achieved through the hydrogen overflow process. Zhang and Yin21 reported a synthesis strategy for encapsulating Pd nanoclusters into zeolite using a high-temperature soft gel system. The prepared Pd@S-1-SGP catalyst exhibited higher selectivity for p-chloroaniline (p-CAN) compared to Pd/SiO2 in the selective hydrogenation of p-CNB due to the steric structure of zeolite micropores favoring selective adsorption of the nitro group over the C–Cl group on the Pd surface. Yang22 fabricated a core–shell structured Pd@TS-1@meso-SiO2 catalyst, and the acid sites and protection provided by the mesoporous silica shell enhanced its activity. As can be seen, the most important design concept of the above-reported catalysts is the encapsulation of noble metals within zeolites. On the one hand, the mesoporous shell prevents noble metal leaching and improves stability. On the other hand, molecules are adsorbed in a sterically controlled manner and selectively transformed within the zeolite pores, improving product selectivity.
It is known that the catalytic performance is highly dependent on the establishment of a structure–performance relationship, which can generate additional interface effects, such as strong metal–support interaction (SMSI), oxygen vacancies, or electronic effect.23 Typically, noble metal nanoparticles are supported on metal oxide supports such as CeO2,24 TiO2,5,25 and Fe2O3.8 Since 2006, Corma and coworkers26–28 have reported the high chemoselectivity of Au/TiO2 and Pt/TiO2 catalysts, indicating that energetically and geometrically favored adsorption through the nitro group occurs on the TiO2 support and at the interface between the nanoparticle and the TiO2 support. In previous work,29–33 we combined metal oxides (TiO2, CeO2, La2O3, and ZrO2) and supports with a large specific surface area (activated carbon and SBA-15) to fabricate a series of hybrid nano-structured noble metal catalysts. Among these, the Pt/@-ZrO2/SBA-15 catalyst exhibited remarkable catalytic activity in the selective hydrogenation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, –NO2 groups, and CC bond, and the TOF reached 4822 h−1, 57
O bond, –NO2 groups, and CC bond, and the TOF reached 4822 h−1, 57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 588 h−1, and 66790 h−1 in the selective hydrogenation of benzaldehyde, p-CNB, and styrene, respectively. According to in-depth research, we found that the strong interface electronic effect and the unique properties of the special hybrid nano-structure contribute significantly.
588 h−1, and 66790 h−1 in the selective hydrogenation of benzaldehyde, p-CNB, and styrene, respectively. According to in-depth research, we found that the strong interface electronic effect and the unique properties of the special hybrid nano-structure contribute significantly.
Herein, we report simple and highly efficient Pt/ZrO2/ZSM-5 and Pt/ZrO2/MCM-22 catalysts (denoted as Pt/ZZ and Pt/ZM), obtained by a photochemical route, which exhibit excellent activity, selectivity, and stability for the selective hydrogenation of p-CNB under mild conditions. By combining metal oxides with zeolite, the prepared catalysts not only take advantage of the properties of ZrO2 but also retain the structures of ZSM-5 or MCM-22. It is not necessary to deliberately encapsulate the noble metal; instead, the catalysts can maintain stability and selectivity even at low noble metal loading (0.08% Pt). Furthermore, the special structure generates a synergistic effect, achieving a TOF of 5323 h−1 and 8525 h−1 for Pt/ZZ and Pt/ZM, respectively. This study provides a new approach for the design of supported metal catalysts that have great practical potential for selective hydrogenation of various nitroarenes.
2 Experimental
2.1 Materials
All chemicals were purchased from chemical companies and used without further purification: zirconium oxide chloride, ammonia monohydrate (Tianjin Ke Miou Chemical Reagent Co., Ltd), ZSM-5 or MCM-22 (the catalyst plant of Nankai University), H2PtCl6 (7.4 mg mL−1), p-chloroaniline (Shanghai Maclean Biochemical Technology Co., Ltd), and ethanol (Tianjin Ke Miou Chemical Reagent Co., Ltd).2.2 Preparation of catalysts
Typically, 0.291 g of ZrOCl2·8H2O and 10 mL of deionized water were added to a beaker and magnetically stirred until the solid was dissolved. Then, 1 g of MCM-22 or ZSM-5 was added to the above solution, and after thoroughly mixing, the pH was adjusted to 10 using 1.5 mol L−1 NH3·H2O, followed by aging for 24 h. Finally, the composite carrier ZrO2/MCM-22 or ZrO2/ZSM-5 was obtained by filtration and washing with deionized water several times, drying at 100 °C and calcining at 500 °C for 4 h. The preparation of ZrO2 was the same as above, with the addition of MCM-22 or ZSM-5. 0.497 g ZrO2/MCM-22 or ZrO2/ZSM-5, 100 mL deionized water, 5 mL methanol, and 0.68 mL H2PtCl6 were mixed and stirred for 10 h under ultraviolet light. Subsequently, the solution was filtered and washed with deionized water several times and then dried in a vacuum oven at 80 °C, yielding the Pt/ZZ and Pt/ZM catalysts.2.3 Catalytic hydrogenation reaction
0.05 g catalyst, 0.2 g p-CNB, and 20 mL ethanol were added into an autoclave (50 mL). The air was first replaced with 99.99% H2 five times, and then the autoclave was filled with H2 to 0.7 MPa. The temperature was raised to 40 °C, and the mixture was stirred at 800 rpm for 1.5 h. Finally, the reactor was moved to an ice-water bath to cool down. The reaction solution was obtained by filtration and analyzed using an Agilent 6890N GC with an HP-5 capillary column and FID detector.| Conversion (%) = (n0p-CNB − np-CNB)/n0p-CNB × 100% |
| Selectivity (%) = np-CAN/(n0p-CNB − np-CNB) × 100% |
| TOF = (n0p-CNB − np-CNB)/((load of Pt × m (cat.) × (Pt dispersion)/195.084) × reaction time) |
2.4 Catalyst characterization
X-ray diffraction (XRD) of samples was performed on a Rigaku D/max-II/2500 X-ray powder diffractometer with Cu Kα radiation and the working voltage and current were 40 kV and 30 mA, respectively. Scanning electron microscopy (SEM) images were collected on a ZEISS Sigma 300 to observe the morphology. Transmission electron microscope (TEM) images were obtained with a JEOL JEM-2100F at an acceleration voltage of 200 kV. Pt dispersions were measured by pulse adsorption of CO on an AutoChem II 2920 automated characterization system at 50 °C. The samples were pretreated in a 5 vol% H2/95 vol% Ar flow at 673 K for 2 h. The degree of dispersion and the mean particle size were estimated from the measured CO uptake, assuming a cross-sectional area for a surface platinum atom of 8.0 × 10−20 m2 and a stoichiometric factor of one, using the nominal platinum concentrations. The H2 temperature-programmed reduction (TPR) technique was performed using a Microtrac BELCat II, with the samples pretreated in a He flow at 400 °C for 1 h, cooled to 50 °C, and then heated from 50 to 800 °C at 10 °C min−1 in a 10% H2–Ar flow. The content of Pt in the catalysts was measured on an ICP-AES 7300DV instrument. X-ray photoelectron spectroscopy (XPS) was performed using an ESCALAB 250Xi (Thermo) with Al Kα radiation. The specific surface areas of the samples were calculated by the BET method by N2 adsorption–desorption using a NOVA-2200e volumetric adsorption analyzer.3 Results and discussion
3.1 Characterization
The crystal phase information of Pt and ZrO2 in catalysts was obtained by XRD and the patterns are depicted in Fig. 1. The ZSM-5, ZrO2/ZSM-5, and Pt/ZZ samples exhibit typical diffraction peaks corresponding to ZSM-5 (Fig. 1a). MCM-22, ZrO2/MCM-22, and Pt/ZM all exhibit typical diffraction peaks, corresponding to MCM-22 (Fig. 1b). Both ZrO2 and ZrO2/ZSM-5 (ZrO2/MCM-22) were prepared by the same method, and ZrO2 exhibited diffraction peaks of monoclinic and tetragonal ZrO2. But the characteristic ZrO2 peaks of the Pt/ZZ and Pt/ZM catalysts with 10% loading in the XRD patterns were not detected.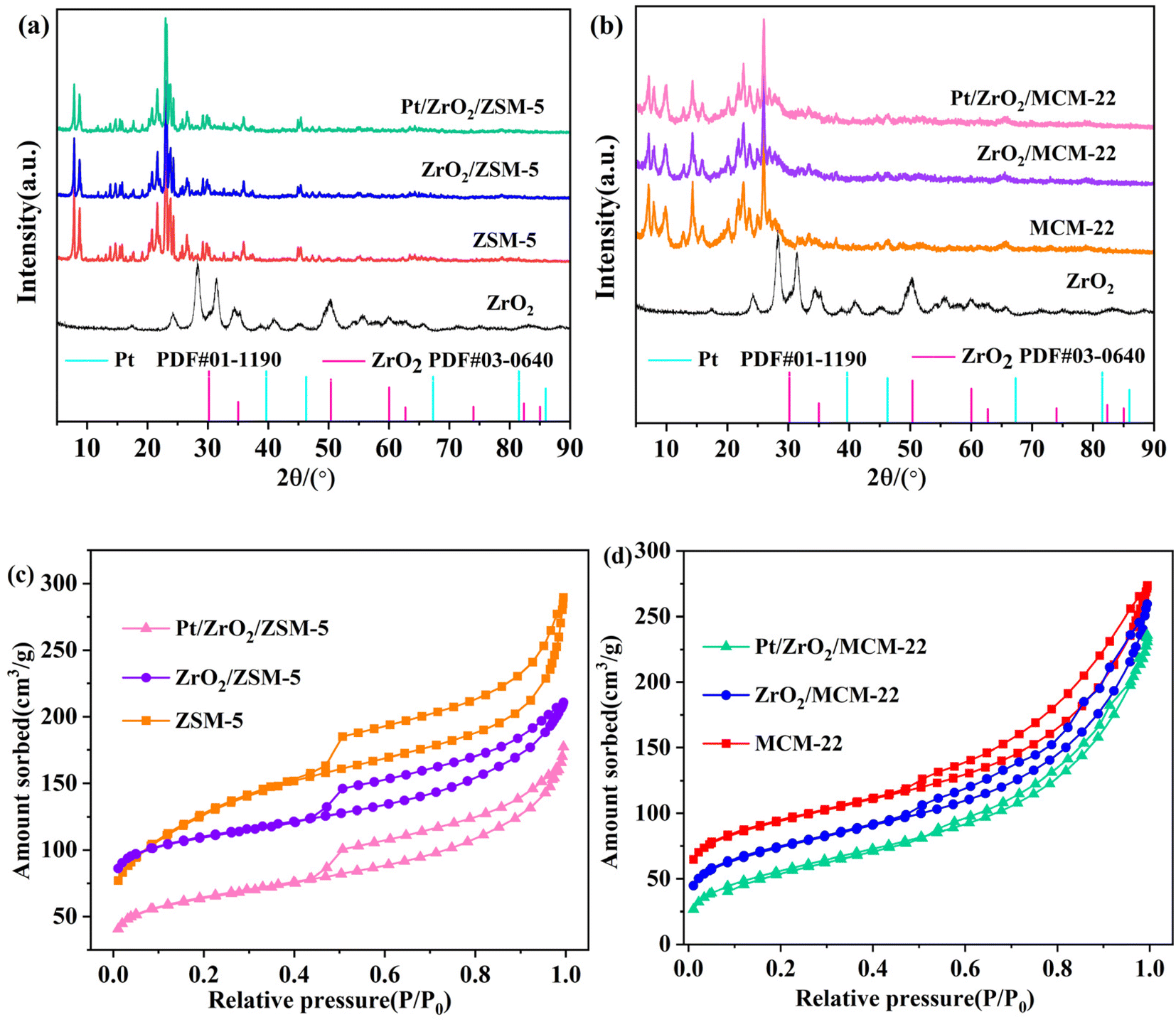 | ||
| Fig. 1 XRD patterns (a and b) and N2 adsorption–desorption isotherms (c and d) of Pt catalysts. | ||
Generally, the reasons why the component cannot be detected by XRD were as follows: (1) the content is too low; (2) it is in an amorphous glassy state; (3) it interacts to form a solid melt. In our previous work,34,35 ZrO2/AC with the same ZrO2 content was prepared using the same method. The XRD patterns of the ZrO2/AC samples showed distinct present ZrO2 peaks, indicating that ZrO2 was mainly coated on the surface of the AC (1419 m2 g−1). In this work, since ZSM-5 (442 m2 g−1) and MCM-22 (602 m2 g−1) have large specific surface areas, the lack of distinct ZrO2 diffraction peaks in their XRD patterns can be attributed to monolayer dispersion of ZrO2 within the channels of ZSM-5 or MCM-22, which inhibits crystal growth. Notably, no diffraction peaks of Pt were detected in related samples due to the relatively low loading (∼0.08%), small particle size, and high metal dispersion.
Furthermore, the physical structure of the catalysts was analyzed using N2 adsorption–desorption. The calculated BET specific surface areas of the samples are listed in Table 1, and the specific surface areas of Pt/ZZ (195 m2 g−1) and Pt/ZM (380 m2 g−1) were lower than those of ZSM-5 (442 m2 g−1) and MCM-22 (602 m2 g−1). Meanwhile, the pore volume of zeolite slightly decreased after the introduction of Pt and ZrO2, indicating that Pt nanoparticles and ZrO2 occupied some of the pores and surface, resulting in a decrease in the specific surface area and pore volume after loading of Pt species. Combined with the N2 adsorption–desorption isotherms (Fig. 1c and d), all ZSM-5 based samples exhibited a typical IV-type isotherm, indicating their microporous structure.35 Additionally, the isotherm curves of all MCM-22 based samples exhibit a Type IV pattern with an H4 hysteresis loop, suggesting the presence of mesoporous structures.36 These results indicate that Pt/ZZ and Pt/ZM successfully maintain the zeolite structure, making them suitable catalysts for the hydrogenation of p-CNB.
| Sample | Surface areaa (m2 g−1) | Pore volumea (cm3 g−1) | Pt loadingb (wt%) | Pt sizec (nm) | Pt dispersionc (%) |
|---|---|---|---|---|---|
| a Determined by N2 adsorption.b Determined by ICP.c Determined by CO chemisorption. | |||||
| ZSM-5 | 442 | 0.422 | — | — | — |
| ZrO2/ZSM-5 | 364 | 0.319 | — | — | — |
| Pt/ZZ | 195 | 0.131 | 0.087 | 1.55 | 63.1 |
| MCM-22 | 602 | 1.121 | — | — | — |
| ZrO2/MCM-22 | 429 | 0.436 | — | — | — |
| Pt/ZM | 380 | 0.411 | 0.082 | 2.25 | 43.5 |
A scanning electron microscope (SEM) and transmission electron microscope (TEM) were used to investigate the morphology and porous structure of the samples (Fig. 2). As can be seen from Fig. 2a–d, Pt/ZZ displayed a well-defined hexagonal or cuboid crystal morphology with some small particles on the outer surface, resulting in a rough surface. The EDS mapping showed that Pt/ZZ displayed uniform and high dispersion of Pt nanoparticles and ZrO2. CO chemisorption determined the distribution of Pt on the surface based on the amount of CO adsorbed and calculated the particle size of Pt nanoparticles, and the Pt dispersion and size were 63.1% and 1.55 nm, respectively (Table 1). For Pt/ZM (Fig. 2f–i), the morphology consisted of layered particles formed by a thin interpenetrating network. Similarly, uniform dispersion of Pt nanoparticles and ZrO2 was confirmed by EDS mapping (Fig. 2j). Accordingly, the CO chemisorption showed that the Pt dispersion and size of Pt/ZM were 43.5% and 2.25 nm, respectively.
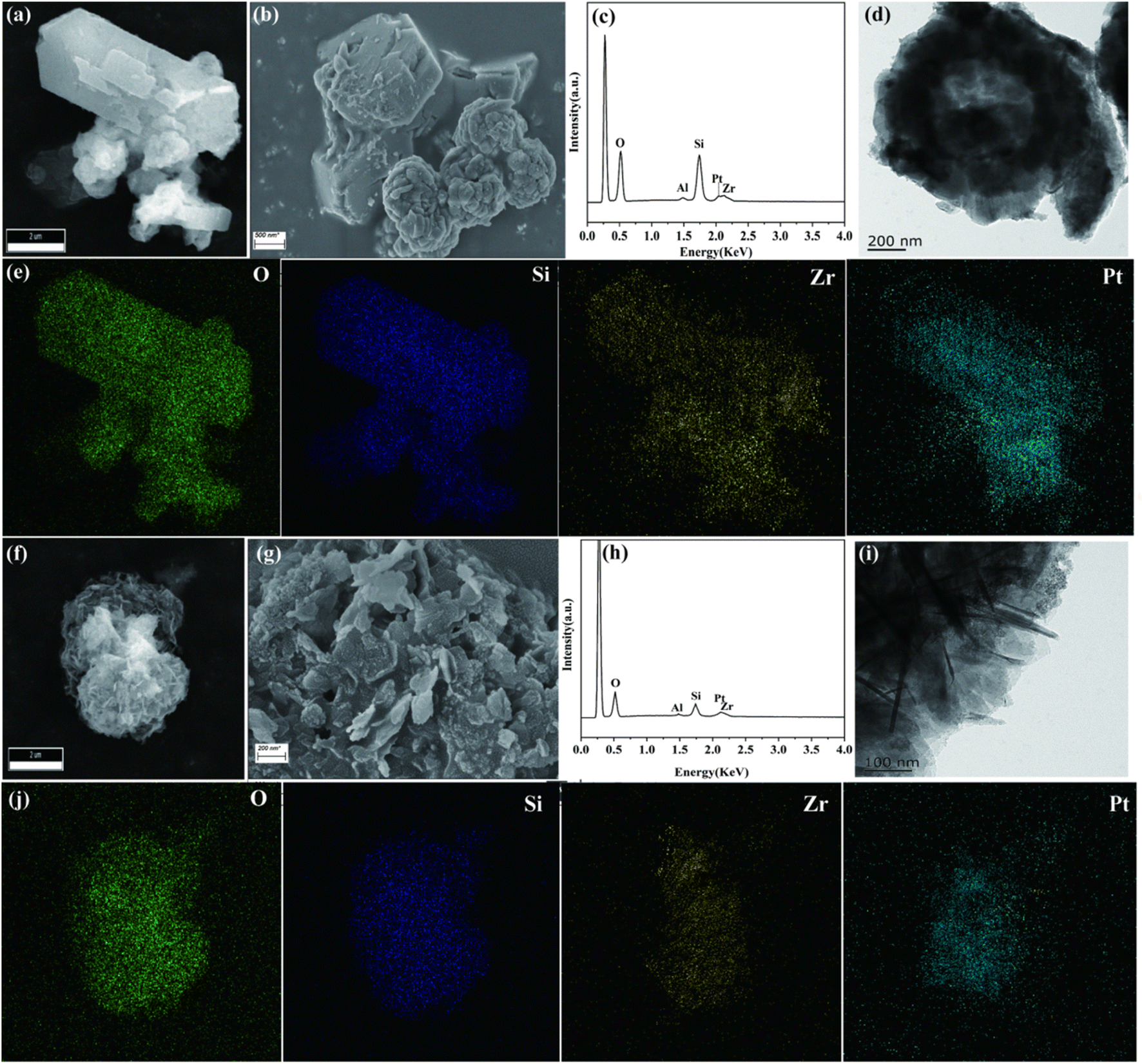 | ||
| Fig. 2 SEM, TEM images and EDS mapping of Pt/ZZ (a–e) and Pt/ZM (f–j). | ||
X-ray photoelectron spectroscopy (XPS) was further performed to investigate the electronic properties. The high-resolution spectra of Zr 3d peaks are shown in Fig. 3a. For Pt/ZrO2, two valence states of Zr are observed at the binding energies of around 182.1 and 184.6 eV for Zr3+(3d5/2) and Zr3+(3d3/2), and 182.7 and 185.1 eV for Zr4+(3d5/2) and Zr4+(3d3/2), respectively. The binding energies of Zr3+ 3d5/2/3d3/2 and Zr4+ 3d5/2/3d3/2 for Pt/ZZ are about 181.9/184.4 and 182.6/185.0 eV, respectively. The Zr3+ 3d5/2/3d3/2 and Zr4+ 3d5/2/3d3/2 peaks of Pt/ZM are centered at 182.4/184.7 and 183.2/185.6 eV, respectively. The presence of Zr3+ peaks can be attributed to the unsaturated coordination of Zr sites with defective O atoms in ZrO2,37 demonstrating a strong metal–support interaction between Pt nanoparticles and ZrO2 that facilitates electron transfer. The Pt 4f XPS spectrum (Fig. 3b) of Pt/ZrO2 shows four peaks at the binding energies of 75.6, 72.2, 76.6, and 73.4 eV. The first two binding energies can be assigned to the 4f5/2 and 4f7/2 peaks of Pt0 species, while the last two can be assigned to the 4f5/2 and 4f7/2 peaks of Ptδ+ species. For the Pt/ZZ catalyst, the binding energies at 75.0/71.6 and 76.3/72.9 eV can be assigned to the 4f5/2/4f7/2 of Pt0 and Ptδ+ species, respectively. For Pt/ZM, the Pt 4f5/2 and Pt 4f7/2 peaks were at 75.4/72.0 and 76.5/73.1 eV, respectively. Compared with Pt/ZrO2, the binding energy of Pt species in Pt/ZZ and Pt/ZM showed negative shifts, indicating partial electron transfer from Pt to ZrO2. In addition, according to the XPS results (Table S1†), the percentages of Pt0/Ptδ+ of Pt/ZrO2, Pt/ZZ, and Pt/ZM are calculated to be 48.9/51.1, 57.1/42.9, and 52.8/47.2, respectively, reflecting a significant electron transfer between Pt species and ZrO2. Specifically, the binding energy of Pt decreases more in Pt/ZM, further confirming the stronger interaction between Pt and ZrO2 in this sample. However, due to the overlap of the core energy levels of the Pt 4f and Al 2p spectra, analysis of the interaction based on the binding energy of Pt species is insufficient. Therefore, the Pt 4d spectrum of Pt/ZZ and Pt/ZM was recorded, and the corresponding results are shown in Fig. 3c. For Pt/ZZ, the Pt 4d5/2/Pt 4d3/2 regions located at 314.1/331.8 and 315.5/333.1 eV were attributed to Pt0 and Pt2+, respectively. The Pt 4d core level spectra of Pt/ZM were also deconvoluted into four peaks that were attributed to Pt0 and Pt2+.38,39 In addition, the Pt 4d peaks of Pt/ZZ and Pt/ZM were both shifted to higher binding energies compared with the typical Pt 4d5/2 (314.5 eV) and Pt 4d3/2 (331.2 eV), indicating that the electrons were transferred from Pt to ZrO2, and the percentages of Pt0/Ptδ+ of Pt/ZZ and Pt/ZM were calculated to be 60.5/39.5 and 57.9/42.1, respectively.
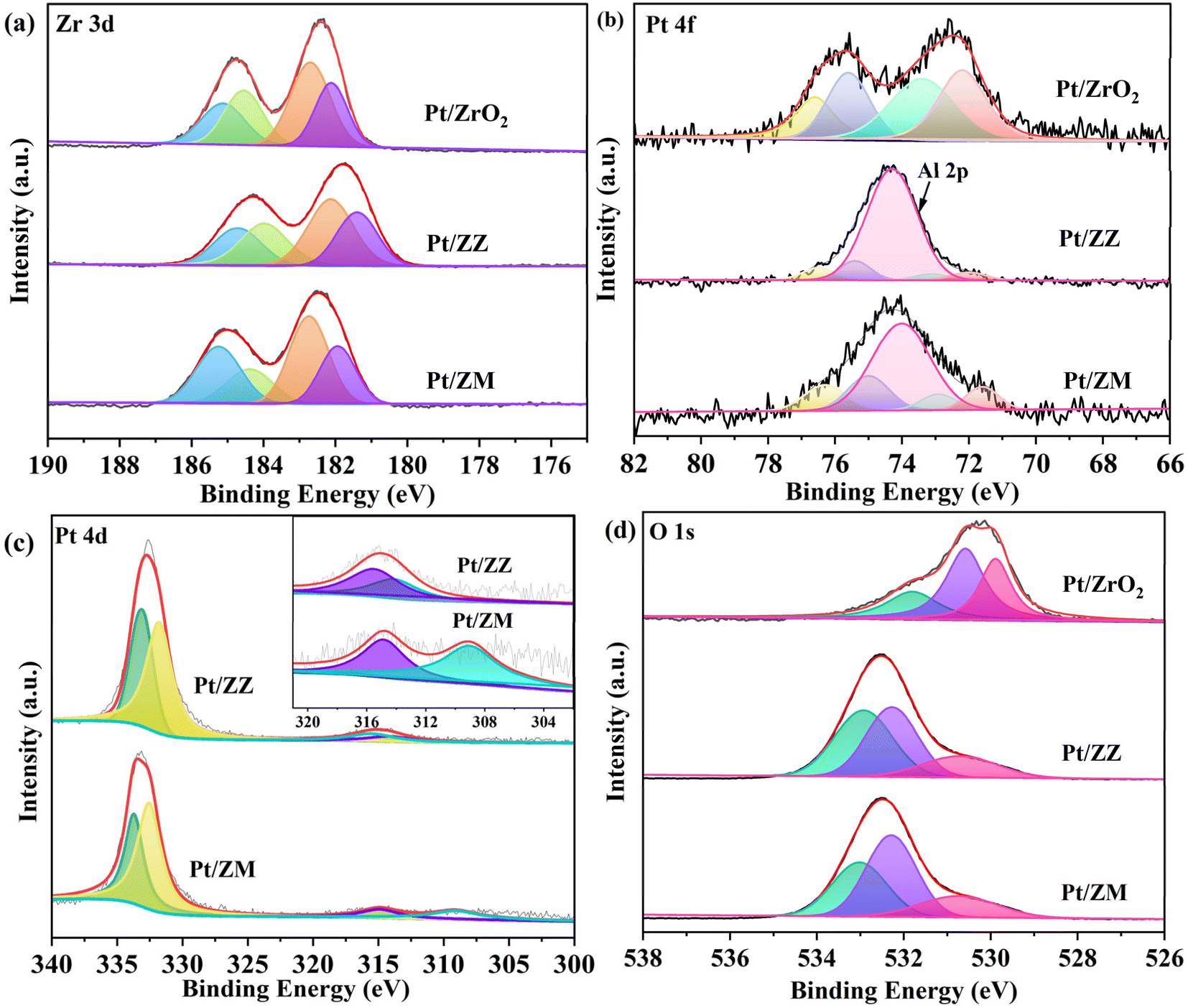 | ||
| Fig. 3 Zr 3d (a), Pt 4f (b), Pt 4d (c), and O 1s (d) XPS spectra of the samples. | ||
Because of the presence of Zr3+, oxygen vacancies form to achieve the charge balance. In the O XPS spectra shown in Fig. 3d, Oα, Oβ and Oγ represent the O element in the lattice, oxygen vacancy,40 and adsorbed oxygen, respectively.41 By calculating the proportion of the O species (Table S2†), the Oβ of Pt/ZrO2, Pt/ZZ, and Pt/ZM was 44.8, 39.8, and 47.2%, respectively. This result indicates that more oxygen vacancies were generated in Pt/ZM, which can be attributed to the electron transfer from Pt to ZrO2. Clearly, the characterization data validate the significant structural and electronic properties in these catalysts, which should significantly influence the catalytic reactivity.
The H2 consumption peaks at low and high reduction temperatures were assigned to the reduction of PtO with weak and strong interactions with the support.38,41,42 Fig. 4 shows the H2-TPR profiles of Pt catalysts. All samples showed two main signals centered at 70–90 °C and 550–650 °C. The first signal can be ascribed to the reduction of PtOx species, while the second can be assigned to the reduction of PtOx species having a stronger interaction with the support. For Pt/ZrO2, the reduction requires a slightly higher temperature (82.5 °C and 616 °C), indicating a relatively strong metal–support interaction. In addition, a negative peak at about 560 °C was detected, assigned to the bulk oxygen in ZrO2.43 Pt/ZZ exhibited two reduction peaks centered at 73.7 °C and 569.7 °C, and these peaks are slightly shifted to lower temperature compared to the corresponding peak of the Pt/ZrO2, suggesting that the support (ZSM-5 and MCM-22) influences the reduction of PtOx. In Pt/ZM, two peaks in the temperature range of 80–200 °C were clearly observed, one main peak (73.7 °C) assigned to PtOx and the other main peak at 173.2 °C related to the reduction of Pt2+ cations. The reduction region of strong interaction is restricted to a broader band centered around 400–600 °C, and the possible reason was the electron transfer and a strong interaction between Pt and ZrO2/MCM-22. All the results discussed so far suggest that a specific interaction exists between Pt and the support, and these metal–support interactions are strongly support dependent.
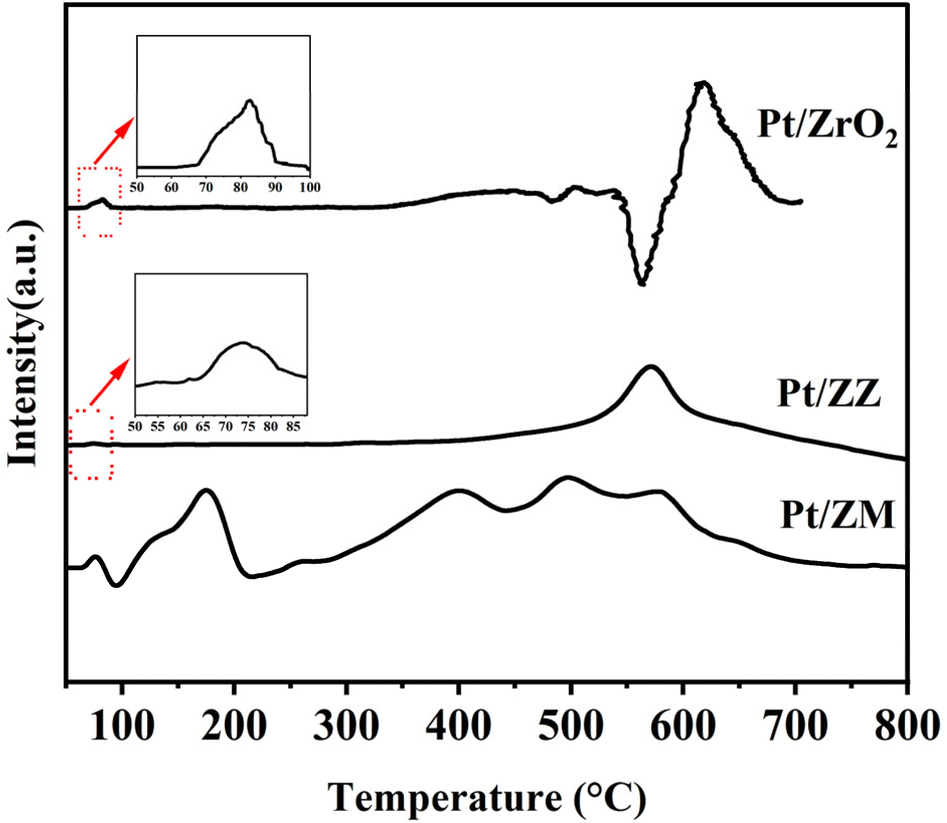 | ||
| Fig. 4 H2-TPR profiles of Pt catalysts. | ||
3.2 Hydrogenation of p-CNB
In this study, hydrogenation of p-CNB to aniline was chosen as a model reaction to demonstrate the catalytic advantage of Pt/ZZ and Pt/ZM (Fig. 5). For comparison, we also examined the performance of traditional Pt/ZrO2, Pt/ZSM-5, and Pt/MCM-22. Firstly, as shown in Fig. 5a, Pt/ZSM-5 and Pt/MCM-22 showed very poor activity under identical conditions, and 36.1% and 3.8% conversions for p-CAN were observed, respectively. In contrast, both Pt/ZZ and Pt/ZM exhibited exceptional catalytic activity, showing both outstanding conversion (>98%) and selectivity (>99.9%) and almost undetectable by-product aniline (AN). As can be seen, for the hydrogenation of p-CNB over the Pt/ZZ catalyst, the conversion of p-CNB was 2.76-fold higher than that of Pt/ZSM-5, and the Pt/ZM catalyst showed 25.8-fold higher conversion than Pt/MCM-22, indicating that the suitable amount of ZrO2 at the Pt/zeolite interface favors adjustment of the structure. Thus, the difference in conversion should be ascribed to the electronic modification of Pt from complex supports. These results clearly demonstrate the advantages of this particular unconventional structural catalyst in this hydrogenation system. Obviously, the Pt/ZZ and Pt/ZM catalysts exhibited a synergistic effect, which can greatly enhance the performance of p-CNB hydrogenation by modulating the electronic structure and surface structure.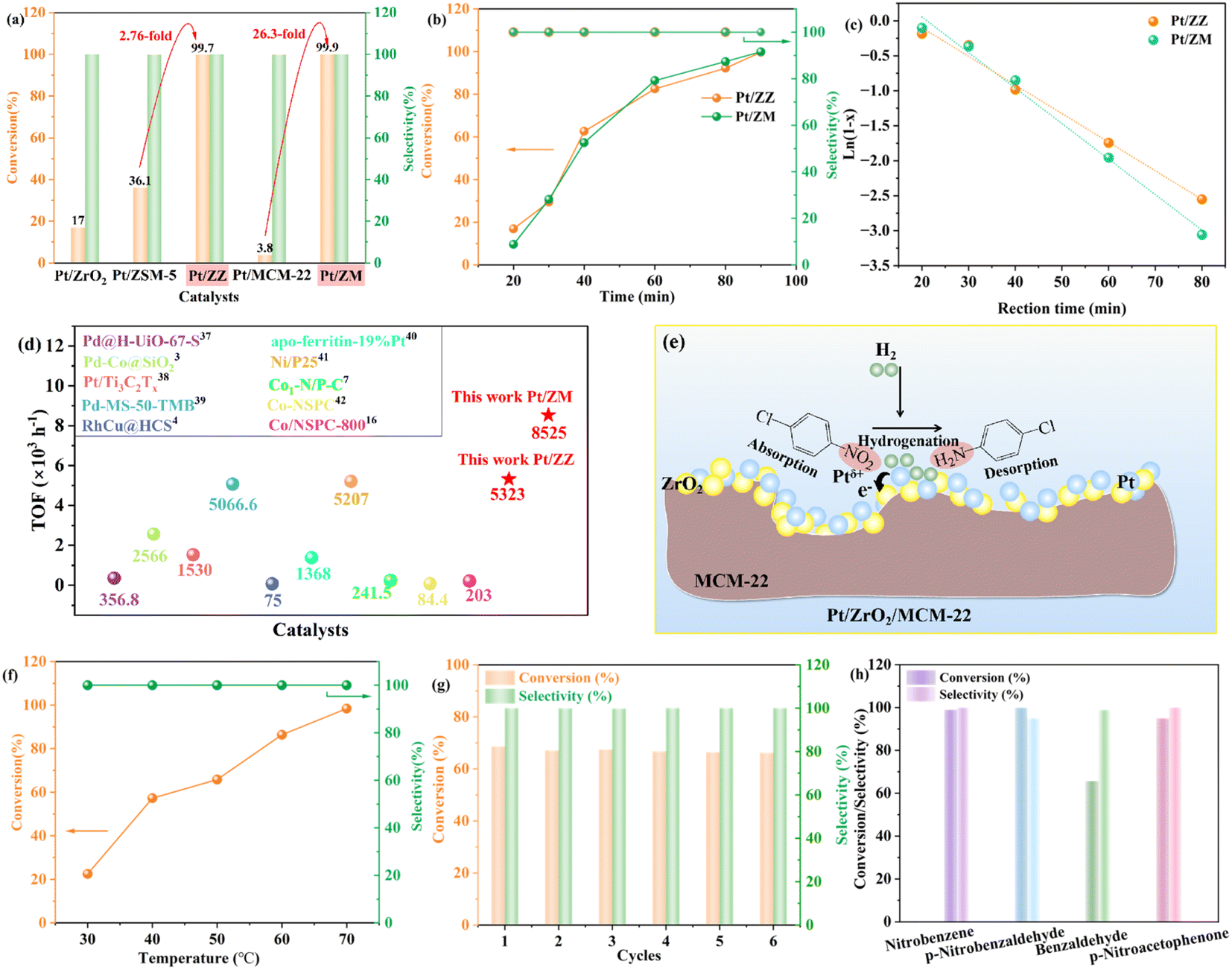 | ||
| Fig. 5 Catalytic performance of the catalysts in hydrogenation of p-CNB (a); effects of reaction time on p-CNB conversion and selectivity over Pt/ZZ and Pt/ZM (b); kinetic profiles (X represents the conversion) for the hydrogenation of p-CNB over Pt/ZZ and Pt/ZM (c); comparison of the TOF with reported catalysts3,4,7,16,45–50 (d); schematic diagram of p-CNB hydrogenation over the Pt/ZM catalyst (e); influence of temperature on the catalytic performance of Pt/ZM (f); stability test of the Pt/ZM catalyst (g); activity of Pt/ZM catalysts for hydrogenation of other substrates (h). Reaction conditions: aCat. (0.1 g), bp-CNB (0.4 g), cethanol (20 mL), d40 °C, eH2 (0.7 MPa), 90 min; f40 min; g50 min. | ||
In addition, Pt/ZrO2 afforded 17.0% conversion of p-CNB and 100% selectivity, and the catalytic activity was also lower than that of Pt/ZZ and Pt/ZM. On the one hand, the small specific surface area (52 m2 g−1), low Pt dispersion (8.5%), and relatively larger Pt size (5.65 nm) are not conducive to the hydrogenation reaction. On the other hand, although there is also an electronic effect caused by the electron transfer between Pt and ZrO2 in Pt/ZrO2, the negative impact of the size effect outweighs the positive influence of the electronic effect, resulting in unsatisfactory activity.
Then, the hydrogenation of p-CNB was performed using the Pt/ZZ and Pt/ZM catalysts at 40 °C, as shown in Fig. 5b. The p-CNB conversion increased from <20% at 20 min to >99.9% at 90 min, while the selectivity of p-CAN stayed at 99.9%. Significantly, although the Pt content was only 0.08%, the Pt/ZZ and Pt/ZM catalysts achieved >99% conversion, indicating that the strong synergistic effect within them fully developed the catalytic ability of Pt, thus reducing the Pt usage.
It seems that Pt/ZZ and Pt/ZM have similar catalytic activity. In Fig. 5c and Table S3,† the relative parameters and figures regarding the p-CNB conversion rate with reaction time show that both samples fit a pseudo-first-order kinetic model. However, the reaction rate of the Pt/ZM catalyst (1.068 min−1) is higher than that of Pt/ZZ (0.718 min−1). Moreover, the TOF of Pt/ZM is as high as 8525 h−1, much higher than that of Pt/ZZ (5323 h−1) (Fig. 5d). Generally, the smaller the nanoparticle size and the higher the dispersion, the higher the catalytic activity. Despite Pt/ZZ having a more favorable Pt size (1.55 nm) and dispersion (63.1%), its catalytic activity was behind that of Pt/ZM. This result indicates that the size effect is not dominant in this hybrid nano-structured catalyst, and the most possible reason may be the electronic effect. Indeed, the potential to enhance catalytic activity by modulating the electronic structure has been observed previously.12,44 Due to electron transfer from Pt to ZrO2 nanoparticles, the Ptδ+ percentage of Pt/ZM (42.1%) is higher than that of Pt/ZZ (39.5%). The electron-deficient Pt site is more favorable for p-CNB adsorption, and as the fraction of Ptδ+ increases, the catalytic activity increases.
Next, the effect of the Pt loading amount on the catalytic performance was investigated (Table S4†). The activity gradually increased with the increase of the Pt loading amount from 0.087% to 2.7%, and all these catalysts exhibited >99% conversion but required much shorter reaction times.
Finally, Pt/ZZ and Pt/ZM were also compared with the latest reported catalysts, and the results are displayed in Fig. 5d and Table S5.† Remarkably, the catalytic performance of Pt/ZZ and Pt/ZM surpasses that of most reported noble metal and novel non-noble metal catalysts. Notably, although these nano-structured Pt/ZZ and Pt/ZM have low Pt loading, they exhibited superior catalytic activity under mild reaction conditions, demonstrating that the developed strategy of supporting Pt on complex supports is effective in promoting the activity of supported Pt catalysts.
To gain deep insight into the hydrogenation process of the Pt/ZM catalyst, experiments were conducted at different temperatures under the same conditions (Fig. 5f). The conversion of p-CNB significantly increased from 22.5% to 98.4% as the reaction temperature was increased from 30 to 70 °C, while the selectivity to p-CAN was maintained at >99.9%, with almost no intermediate or by-product observed, indicating that Pt nanoparticles diffused in the pores of the support could effectively adsorb p-CNB and desorb p-CAN appropriately. As the temperature increases, the rapid consumption of p-CNB indicates that the hydrogenation of p-CNB to p-CAN is more favorable at elevated reaction temperature.
Then, the reusability of the Pt/ZM catalyst is shown in Fig. 5g. The catalyst was recycled through filtration, washing, and drying, and the conversions were 68.6, 67.1, 67.5, 66.8, 66.5, and 66.2% for the first to the sixth run, respectively. Moreover, the selectivity for p-CAN remained unchanged. The TEM image (Fig. S1†) of the Pt/ZM catalyst after several tests showed negligible differences compared with the fresh sample, implying the good retention of its structure. The unique structure of MCM-22 provided a limited spatial volume to anchor Pt nanoparticles and ZrO2, which contributed to the Pt/ZM catalyst maintaining its structure and activity. In conclusion, the Pt/ZM catalyst was efficient and reusable.
Subsequently, other substrates were also employed to investigate the properties of the Pt/ZM catalyst, and the results are shown in Fig. 5h. There is no doubt that the Pt/ZM catalyst shows good catalytic performance for nitrobenzene, affording the corresponding aniline with excellent conversion (>99%). In addition, p-nitrobenzaldehyde, an industrially important drug and pesticide intermediate, can be easily obtained by completely hydrogenating the corresponding nitroarene within 60 min with a 100% conversion. For the hydrogenation of benzaldehyde, the conversion can reach more than 65% with more than 98% selectivity. These results strongly indicate that the Pt/ZM catalyst preferentially adsorbs the nitro group and is a highly selective and effective catalyst for hydrogenating nitroarenes. In the case of p-nitroacetophenone, the conversion decreased slightly (95%), which may be due to the steric effect of the substituent group on the catalytic hydrogenation rates.
On the basis of the above results and previous literature reports,8,12,46 we propose a plausible catalytic mechanism for the hydrogenation of p-CNB over the Pt/ZM catalyst, as shown in Fig. 5e. The micropores of MCM-22 provide a limited spatial volume to anchor Pt nanoparticles and ZrO2, and the appropriate content of ZrO2 ensures the electron effect between Pt nanoparticles and ZrO2, forming the unique structure of Pt/ZM. On the one hand, the high specific surface area provides a perfect place for hydrogenation. On the other hand, MCM-22 micropores changed the spatial disposition of the adsorption of p-CNB on Pt sites in contact with these micropores, and the electron effect promoted the hydrogenation process. Hence, when p-CNB is adsorbed on Pt/ZM, it preferentially adsorbs onto Ptδ+ sites via the nitro group rather than the chloro group due to the strong interaction of the nitro group with the Pt surface, while H2 molecules were dissociated and activated on the Pt0 sites. Then, the nitro groups adsorbed on the catalyst undergo hydrogenation with hydrogen ions to form p-CAN. Simultaneously, the hydrogenolysis of the carbon–chlorine bond is suppressed, reducing byproduct formation and ensuring high selectivity for p-CAN.
4 Conclusion
In conclusion, we developed Pt catalysts with low loading but high performance by combining ZrO2 and zeolite as a complex support. The obtained Pt/ZZ and Pt/ZM catalysts exhibited a synergistic effect and demonstrated outstanding activity and excellent selectivity for selective hydrogenation of p-CNB to p-CAN, achieving TOFs of 5323 h−1 and 8525 h−1, respectively. Notably, the optimal Pt/ZM exhibited the best catalytic activity and good recyclability. Catalyst characterization indicated that the electron transfer from Pt to ZrO2 increased the fraction of electron-deficient Pt, which is more favorable for p-CNB adsorption, thereby enhancing catalytic activity. We believe that combining noble metals with metal oxides/zeolites to design high-efficiency, low-noble-metal-loading catalysts opens a new avenue for noble metal catalysts. This research also provides new ideas and insights for optimizing heterogeneous catalysts for chemoselective hydrogenation.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Natural Science Foundation of Zhejiang Province (LQ24B060005) and the Quzhou University PhD Research Start-up Project (BSYJ202103).References
- L. Gao, J. Liu, X. Zhong, Y. Zhang, J. Chen, M. Shakeri, X. Zhang and B. Zhang, ACS Appl. Nano Mater., 2023, 5685–5691 CrossRef CAS.
- A. Afzalinia and M. Mirzaee, J. Mol. Struct., 2023, 135322 CrossRef CAS.
- L. Xiao, C. Chuanqi, L. Zhixi, Z. Chaoyang, D. Jing, S. Meina, W. Yue, L. Shaopeng, G. Jun and M. Zhao, Nano Res., 2023, 16, 11334–11341 CrossRef CAS.
- F. Yang, H. Yu, C. Wu, S. Wang, T. Li and H. Yin, ACS Appl. Nano Mater., 2022, 11627–11635 CrossRef CAS.
- D. Liu, N. Ismail, F. Chaohe, H. Kuan, G. Binghui, C. Hailong and C. Zhuo, Appl. Catal., A, 2022, 118840 CrossRef CAS.
- L. Geng, F. Geng, X. Cheng, X. Wang, L. Wang, X. Pang, Y. Zhang, X. Zhang, D. Zhang, S. Zhuang, A. Abdukayum and G. Hu, Appl. Surf. Sci., 2024, 657, 159775 CrossRef CAS.
- F. Cao, W. Ni, Q. Zhao, L. Wang, S. Xue, Y. Li, D. Kong, M. Wu and L. Zhi, Appl. Catal., B, 2024, 123762 CrossRef CAS.
- G. Yue, Y. Yu, S. Li, H. Li, S. Gao, Y. Wang, W. Guo, N. Wang, X. Li, Z. Cui, C. Cao, L. Jiang and Y. Zhao, Small, 2023, 2207918 CrossRef CAS PubMed.
- K. P. Reddy, C. Swetha and A. Murugadoss, ACS Sustainable Chem. Eng., 2023, 1643–1654 CrossRef CAS.
- F. Zhang, Y. Jiang, S. Dai, X. Wei, Y. Ma, H. Liao, Y. Qin, Q. Peng, X. Zhao and Z. Hou, Ind. Eng. Chem. Res., 2023, 62, 5814–5825 CrossRef CAS.
- X. Zhang, Z. Li, W. Pei, G. Li, W. Liu, P. Du, Z. Wang, Z. Qin, H. Qi, X. Liu, S. Zhou, J. Zhao, B. Yang and W. Shen, ACS Catal., 2022, 12, 3634–3643 CrossRef CAS.
- Z. Wang, C. Wang, S. Mao, B. Lu, Y. Chen, X. Zhang, Z. Chen and Y. Wang, Nat. Commun., 2022, 13, 3561 CrossRef CAS PubMed.
- M. Cheng, P. Lv, X. Zhang, R. Xiong, Z. Guo, Z. Wang, Z. Zhou and M. Zhang, J. Catal., 2021, 399, 182–191 CrossRef CAS.
- N. Zengin, H. Goksu and F. Sen, Chemosphere, 2021, 130887 CrossRef CAS PubMed.
- B. Lu, X. Chen, C. Feng, J. Chang and F. Ye, ACS Appl. Nano Mater., 2021, 4, 2278–2284 CrossRef CAS.
- J. Xu, X. Chen, J. Bai, Z. Miao, Y. Tan, N. Zhan, H. Liu, M. Ma, M. Cai, Q. Cheng and S. Sun, Catal. Sci. Technol., 2024, 14, 1167–1180 RSC.
- B. Han, Y. Guo, Y. Huang, W. Xi, J. Xu, J. Luo, H. Qi, Y. Ren, X. Liu, B. Qiao and T. Zhang, Angew. Chem., Int. Ed., 2020, 59, 11824–11829 CrossRef CAS PubMed.
- G. Chen, J. Han, Z. Niu, P. She, L. Li, B. Guan and J. Yu, J. Am. Chem. Soc., 2023, 145, 9021–9028 CrossRef CAS PubMed.
- G. Tian, G. Chen, G. Yang, Z. Diao, R. Bai, J. Han, B. Guan and J. Yu, ACS Cent. Sci., 2024, 10, 1473–1480 CrossRef CAS PubMed.
- G. Yang, Y. He, C. Zhang, Z. Sun, M. Han, P. Peng, Y. Yue, H. Liu, L. Liu, J. Chen, L. Bai and Q. Chen, ChemCatChem, 2023, 15, e202300969 CrossRef CAS.
- T. Li, S. Wang, H. Yu, L. Yuan, D. Zhang and H. Yin, Ind. Eng. Chem. Res., 2022, 18762–18771 CrossRef CAS.
- Y. T. Wang, S. M. Wu, G. Q. Luo, S. T. Xiao, F. F. Pu, L. Y. Wang, G. G. Chang, G. Tian and X. Y. Yang, Chem.–Asian J., 2023, 18, e202300689 CrossRef CAS PubMed.
- J. Ma, X. Mao, C. Hu, X. Wang, W. Gong, D. Liu, R. Long, A. Du, H. Zhao and Y. Xiong, J. Am. Chem. Soc., 2024, 146, 970–978 CrossRef CAS PubMed.
- Q. Zhang, J. Bu, J. Wang, C. Sun, D. Zhao, G. Sheng, X. Xie, M. Sun and L. Yu, ACS Catal., 2020, 10350–10363 CrossRef CAS.
- D. Zhu, L. Long, J. Sun, H. Wan and S. Zheng, Appl. Surf. Sci., 2020, 504, 144329 CrossRef CAS.
- A. Corma and P. Serna, Science, 2006, 313, 332–334 CrossRef CAS PubMed.
- M. Boronat, P. Concepción, A. Corma, S. González, F. Illas and P. Serna, J. Am. Chem. Soc., 2007, 129, 16230–16237 CrossRef CAS PubMed.
- A. Corma, P. Serna, P. Concepción and J. J. Calvino, J. Am. Chem. Soc., 2008, 130, 8748–8753 CrossRef CAS PubMed.
- Y. Zhang, J. Zhou, F. Wang, M. Lv and K. Li, J. Catal., 2023, 12–19 CrossRef CAS.
- Y. Zhang, J. Zhou, F. Wang and X. Zhao, Langmuir, 2022, 38, 7699–7708 CrossRef CAS PubMed.
- Y. Zhang and J. Zhou, J. Catal., 2021, 395, 445–456 CAS.
- Y. Zhang and J. Zhou, Nanotechnology, 2020, 31, 295703 CAS.
- Y. Zhang, J. Zhou, K. Li and M. Lv, Catal. Today, 2020, 358, 129–137 CrossRef CAS.
- Y. Zhang, J. Zhou, K. Li and M. Lv, Mol. Catal., 2019, 478, 110567 CrossRef.
- T. Fu, Y. Wang and Z. Li, Langmuir, 2020, 36, 3737–3749 CrossRef CAS PubMed.
- D. Pithadia, A. Patel and V. Hatiya, Renewable Energy, 2022, 187, 933–943 Search PubMed.
- Z. Qin, L. Chen, Y. Li and K. Shen, ACS Catal., 2023, 8372–8383 CrossRef CAS.
- H. Pan, J. Li, J. Lu, G. Wang, W. Xie, P. Wu and X. Li, J. Catal., 2017, 354, 24–36 CrossRef CAS.
- W. Tan, S. Xie, X. Zhang, K. Ye, M. Almousawi, D. Kim, H. Yu, Y. Cai, H. Xi, L. Ma, S. N. Ehrlich, F. Gao, L. Dong and F. Liu, ACS Appl. Mater. Interfaces, 2024, 16, 454–466 CrossRef CAS PubMed.
- J. Wang, Y. Zhang, X. Xu and M. Bao, ACS Appl. Mater. Interfaces, 2023, 8149–8156 Search PubMed.
- Q. Wang, G. Wang, H. Xin, J. Liu, G. Xiong, P. Wu and X. Li, Catal. Sci. Technol., 2019, 9, 3226–3237 RSC.
- J. Lee, E. J. Jang, D. G. Oh, J. Szanyi and J. H. Kwak, J. Catal., 2020, 385, 204–212 CrossRef CAS.
- Z. Tang, P. Liu, H. Cao, S. Bals, H. J. Heeres and P. P. Pescarmona, ACS Catal., 2019, 9, 9953–9963 Search PubMed.
- Y. Zhang, K. Fulajtarova, M. Kubu, M. Mazur, M. Hronec and J. Cejka, Catal. Today, 2020, 345, 39–47 CAS.
- T. M. Bustamante, C. H. Campos, M. A. Fraga, J. L. G. Fierro and G. Pecchi, J. Catal., 2020, 385, 224–237 CrossRef CAS.
- X. Li, W. Gao, L. Li, B. Li, M. Chen, Y. Ge, S. Liu, L. Wang, L. Ma, L. Niu, Y. Li, Z. Liu and J. Chen, ACS Appl. Nano Mater., 2024, 14540–14548 CrossRef CAS.
- W. Yu, H. Lin and C. Tan, Chem. Eng. J., 2017, 325, 124–133 CrossRef CAS.
- Y. Zhou, Y. Zheng, C. Lu, B. Maity, Y. Chen, T. Ueno, Z. Liu and D. Lu, ACS Appl. Nano Mater., 2023, 5835–5843 CrossRef CAS.
- J. Bai, J. Xu, M. Ma, Z. Miao, J. Yu, H. Liu, Z. Qian, M. Cai, Q. Cheng, Y. Jiang and S. Sun, Langmuir, 2024, 12179–12190 CrossRef CAS PubMed.
- J. Xu, J. Bai, W. Si, Y. Zhang, J. Tan, M. Cai, Q. Cheng and S. Sun, Ind. Eng. Chem. Res., 2023, 3862–3872 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00870g |
| This journal is © The Royal Society of Chemistry 2025 |