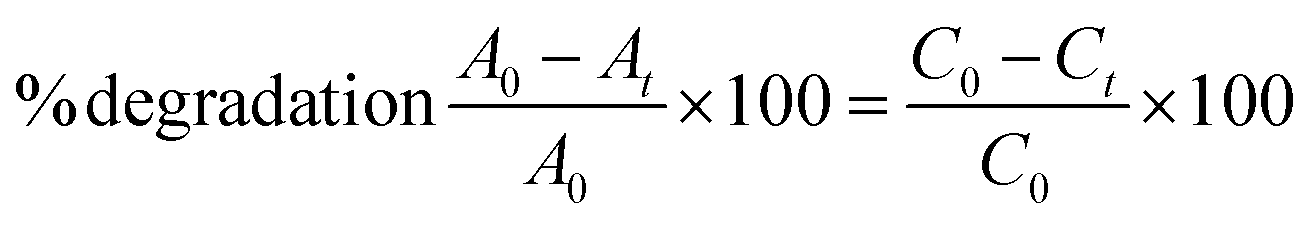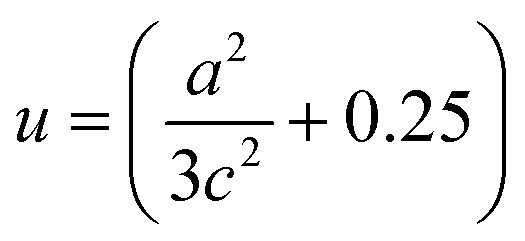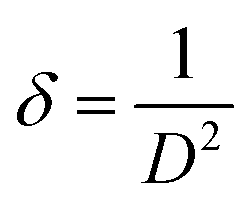DOI:
10.1039/D4NA00890A
(Paper)
Nanoscale Adv., 2025,
7, 1335-1352
Investigation of the photocatalytic potential of C/N-co-doped ZnO nanorods produced via a mechano-thermal process†
Received
28th October 2024
, Accepted 23rd December 2024
First published on 20th January 2025
Abstract
Doping in pure materials causes vital alterations in opto-electrical and physicochemical characteristics, which enable the produced doped material to be highly efficient and effective. The current work focused on the synthesis of C/N-co-doped-ZnO nanorods via a facile, eco-friendly, and solvent-free mechano-thermal approach. The synthesized C/N-co-doped ZnO nanorods were employed for the photocatalytic decay of methylene blue (MB) and brilliant cresyl blue (BCB) dyes, and their degradation capability was compared with that of pure ZnO nanoparticles prepared via a precipitation approach. The FESEM findings confirmed the formation of rod-shaped nanostructures of co-doped ZnO nanoparticles, and EDX and XPS results revealed the successful doping of C and N atoms in ZnO lattices. The XRD and XPS results substantiated that N-doping in the ZnO lattice followed substitutional and interstitial mechanisms, while C-doping followed a substitutional pathway. The co-doped ZnO nanorods exhibited highly enhanced degradation potential toward both MB (∼99%) and BCB (∼98%) dyes upon exposure to visible light for 60 min in a basic medium at pH = 10 owing to factors such as formation of new energy states within the band gap of ZnO, delayed recombination of photogenerated charge carriers, and formation of lattice defects in the ZnO lattice due to C and N doping. The MB and BCB dyes photodegraded at degradation rates of 637.23 × 10−4 and 775.25 × 10−4 min−1, respectively, and the photodegradation process showed good agreement with the pseudo-first-order kinetics in the presence of co-doped ZnO nanorods under visible light illumination. The ˙O2− radicals were the key reactive species involved in the decay of MB and BCB dyes over co-doped ZnO, as confirmed via scavenger studies, and the C/N-co-doped ZnO nanorods retained approximately 90% and 91% efficiencies for BCB and MB dyes, respectively, after three successive cycles of reuse, which confirmed their good stability and reusability under visible light.
1. Introduction
Semiconducting metal–oxide nanoparticle photocatalysts are promising candidates for environmental remediation,1 as they show excellent catalytic efficiency toward a wide range of water pollutants, such as organic dyes, nitro-compounds, and metal ions, when exposed to light. In the current scenario, various metal-based semiconducting materials, such as TiO2,2,3 CuO,4 ZnO,5 and Bi2O3,6,7 are extensively used in the photo-mineralization of organic dyes, such as crystal violet (CV),8 malachite green (MG),5 amido black-10B (AB-10B),6 alizarin red S (ARS),7 brilliant cresyl blue (BCB),9,10 Rhodamine B (RhB),8,11,12 and methylene blue (MB).13 Moreover, metal-based semiconducting materials have been used in the photocatalytic conversion of N2 into ammonia14–16 and the reduction of CO2.12
Zinc oxide (ZnO) is a diversified II–VI n-type semiconducting photocatalyst that possesses a broad band gap of 3.37 eV and an exciton binding energy of 60 meV;17,18 its space group is P63mc and lattice parameter a = b and c values are 3.2495 Å and 5.2069 Å, respectively.18 It is widely used in various fields including photocatalysis.19,20 Applying pure ZnO nanoparticles as photocatalysts under visible light irradiation exhibits poor activity owing to the deprived photo-energy conversion efficiency, wide band gap, and high e−/h+ pair recombining speed.21
Owing to the wide band gap of ZnO, it can only utilize 4% of solar radiation that covers the UV portion.22 In recent years, to broaden the photo-response of ZnO towards visible light and modify its band gap, substantial attempts and strategies have been developed,23 which include the intonation of band structure, doping with foreign elements as impurities, synthesis of hybrid materials by coupling with other semiconducting materials of narrow or wide band gap,8–11 and surface modification via which the band gap narrowing, creation of new energy states, slowdown of recombination rate of charge carriers, and increase in the delocalization speed of charge carriers are achieved.23–25
Doping is an attractive technique for modifying the optical and electrical characteristics of photocatalytic substances. Doping with foreign elements (metallic or nonmetallic) induces changes in the crystal structure, optical band gap, charge transporter concentration, and physicochemical features as well as it causes alteration in the energy levels inside the band gap of the supporting semiconducting material near to dopant the energy band.26 Electron-donor dopants generate new energy levels close to the CB of semiconducting materials, while electron-acceptor dopants create new levels near the VB. Thus, in comparison to the original band gap, the energy difference between the freshly generated levels is measured lower, which designates the decreasing band gap of the doped semiconductor than that of the pristine semiconductor.26 By doping of impurities, a wide range of alterations in the characteristics of ZnO occur, including transparency, ferromagnetism, band gap value, piezoelectrical and magneto-optical features.22,27–29 Currently, the generation of doped ZnO nanostructures with noteworthy superior characteristics and photocatalytic capacities by doping with nonmetal impurities employing various techniques is achieved,22 which results in electron-deficient and hole-richer ZnO, in which a significant delay in the recombination rate of photo-produced charge carriers is observed.30 In addition, the doping of nonmetal elements to the large band gap semiconductors causes a considerable decrease in band gap31 and absorption energy value by the generation of intermediate energy levels.22,32 Moreover, doping of nonmetallic impurities to ZnO does not create charge recombination centers under visible light.33–35
The n-type doping can be achieved by displacing Zn or O atoms with donor atoms, while p-type doping is by substitution with acceptor atoms. However, n-type doped ZnO can be easily obtained.27,36 The p-type doping with electron acceptor metal ions creates a fresh level below the original conduction band (CB), whereas n-type mixing of electron donor metal ions generates a new level above the valence band (VB).22 However, due to the generation of these new states, the absorption wavelength can shift toward a higher wavelength, and these states can work as charge recombination levels that may decline the photocatalytic capability.35 Besides, new levels formed by doping should hold adequate reduction potential of ˙O2− and oxidation potential of ˙OH radicals to create energetic ˙OH and ˙O2− radicals that participate in the photocatalytic process.37 The basic essentialities of non-metal doping are elements possessing similar atomic radii to O and a lower electronegativity than that of oxygen.37 Thus, carbon (C), nitrogen (N), and sulfur (S) are the appropriate nonmetal elements for lattice doping in ZnO.38–45 The doping of C, N, or S to ZnO can characteristically produce hole-richer and electron-poorer ZnO, which results in a decreased recombination rate of photoexcited electron–hole pairs.46,47
Nitrogen and carbon doping to ZnO can be achieved via two major ways, i.e. absorbing at interstitial sites or substitution of O by N or C;48 however, due to size dissimilarities of O2− (140 nm), N3− (132 nm), and C4− (260 nm) ions,49 the alterations caused in the ZnO lattice would be different on doping of C than that of N doping. The N-doping to ZnO can substitute O in the ZnO lattice without causing strain and changing the n-type of ZnO into p-type owing to the size and electronegativity similarity between O2− and N3− ions,50 and the mixing of N 2p orbital with O 2p generates new energy levels near the VB of ZnO that cause a decrease in the band gap of ZnO and enhance light absorption in the visible range.50,51 Moreover, the substitutional N-doping narrows the band gap due to the mixing of N 2p and O 2p states in the VB, while the interstitial N-doping creates deep energy states over the VB with no band gap contracting and increases the photocatalytic ability of ZnO.50
Conversely, C-doping to ZnO causes crystal strain owing to dissimilar ionic sizes,49 and new energy states under the CB of ZnO due to surface O vacancies.52 Hence, the doping of two different nonmetallic elements (N and C) to the ZnO lattice which holds size and electronegativity disparity with the O atom generates different optical, electrical, structural, and physicochemical modifications in the ZnO lattice. Co-doping is a process of doping two foreign metallic or nonmetallic elements rather than one kind of element.53 Thus, combined doping of C and N to the ZnO lattice structure would generate decidedly diverse and novel changes, which help in boosting the photocatalytic potential under light illumination than that of the individually doping of C and N to ZnO.
Although either N-doped or C-doped ZnO nanostructures have been fabricated by various researchers via different approaches and used for the photocatalytic degradation of numerous types of water pollutants, enough evidence of studies of synthesis, doping mechanism, and photocatalytic efficiency investigation of C and N co-doped ZnO is not available. For the synthesis of N-doped ZnO, mechanochemical,54,55 wet chemical,56 high energy milling,57 chemical vapor deposition (CVD),58,59 and calcination20 methods have been adopted. Carbon-doped ZnO was obtained via thermal decomposition,52 sonicated sol–gel technique,60 and hydrothermal methods.61 Few examples of the synthesis of C/N-co-doped ZnO have been reported in the literature as follows:41,45,62 Sun et al. have synthesized C- and N-doped ZnO through a vapor phase transport process using N2O as the N source,41 Liang et al. have prepared C, N-doped ZnO derived from ZIF-8 for the photocatalysis of the MB dye,45 and recently, Fu et al. and Shikuku et al. have fabricated C,N co-doped ZnO through pyrolysis of MOFs,62,63 and two-step sol–gel and physical milling63 processes, respectively and applied for the MO and MB degradation, respectively. Notably, conservative preparation procedures typically involve critical synthesis conditions, intricate formation processes, and high expenditures. The solid-phase synthesis processes of nanomaterials without using any solvent are potentially important options for the generation of nanomaterials due to their simplistic and environment-favoring nature and high yielding capacity because of no concentration constraints as possessed by solution-based methods.64 Thus, the generation of doped ZnO nanostructures using solid-based synthesis methods is highly efficient and simple, and gives high yields. Therefore, the main objective of the current study is the synthesis of C and N-co-doped ZnO nanostructures with higher photocatalytic ability and diversely different characteristics by simple, facile, and eco-friendly solid-phase synthesis via a mechanical grinding and thermal calcination approach under solvent-free conditions and the investigation of their photocatalytic potential against hazardous organic dye water toxicants under visible light illumination.
2. Experimental
2.1. Materials
For the synthesis of pure ZnO and C/N-co-doped-ZnO nanoparticles, urea and dihydrated zinc acetate were used as precursors and sodium hydroxide as the precipitating reagent. In scavenger experiments, disodium ethylenediamine tetraacetate (EDTA-2Na), tertiary butyl alcohol (t-BuOH), and p-benzoquinone (p-BQ) were used, which were procured from Loba chemicals. MB and BCB dyes were procured from TCI chemicals and elected as replica water pollutants.
2.2. Preparation of C/N-co-doped ZnO nanorods and pure ZnO nanoparticles
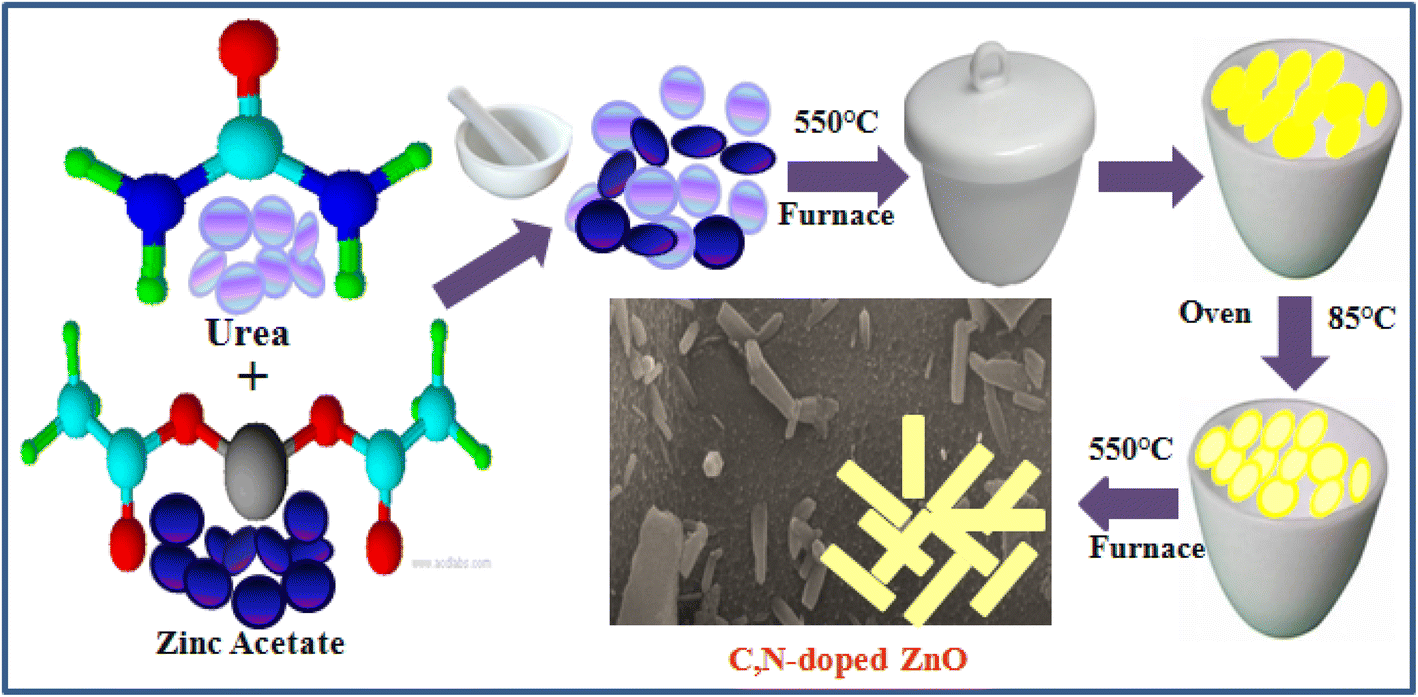
C/N-co-doped-ZnO nanorods were prepared via mechanical mince amalgamation of (NH2)2CO and Zn(CH3COO)2 and a thermal calcination approach without using any solvent. In a representative process, 8 g urea and 2 g zinc acetate (4/1 weight ratio) were mixed by mechanical grinding using a pestle and mortar, and the mixture was dehydrated by heating at 100 °C for 60 min on a pre-heated hot plate. The obtained solid mass was then calcined in a partially covered silica crucible at 550 °C temperature in a muffle furnace for 2 h, which resulted in a pale yellow-colored solid mass. The as-obtained solid material was mashed into a fine powder, sanitized using distilled water and ethyl alcohol, and dehydrated in an oven operating at 95 °C for 3 h, and at last, the dried material was again subjected to calcination at 550 °C for 2 h in a muffle furnace in an open crucible. The graphic illustration for the synthesis of C/N-co-doped-ZnO nanorods is shown in Fig. 1.
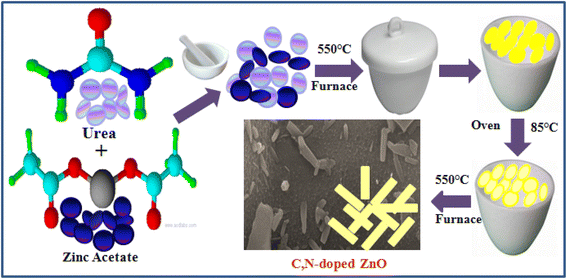 |
| | Fig. 1 Procedure for the synthesis of C/N-co-doped-ZnO nanorods. | |
In the preparation process of bare ZnO, 2 g zinc acetate is dispersed in 50 mL distilled water under magnetic stirring for 30 min. After that, the pH of the solution was adjusted to 9 by adding NaOH solution (1.5 M), and the mixture was kept under shaking at 75 °C for 80 min, cooled down to room temperature, filtered, washed with water and alcohol, and dried in an oven at 95 °C for 3 h, and at last the dried white mass was subjected to calcination at 550 °C for 2 h in a muffle furnace in an open crucible.
2.3. Characterization of the synthesized samples
A PANalytical's X'Pert Pro modeled X-ray diffractometer equipped with a Cu Kβ source (40 kV, and 50 mA, 0.01 step size and 10° min−1 of scan speed) at 2θ in the range of 10 to 80° was used for acquiring the XRD patterns of the developed samples. The surface texture, morphology, and particle size of the samples were analyzed using a field emission scanning electron microscope (FESEM, Nova Nano FESEM-450 (FEI)). The EDS spectrum and elemental mapping were recorded using a Nova Nano FESEM-450 (FEI) operating at an accelerating voltage of 5 kV to test the elemental composition and mapping. The existences of different functional groups in the reported nanomaterials were studied by Fourier transform infrared (FTIR) spectroscopy analysis (Perkin Elmer FT-IR). The UV-visible absorption spectra were recorded using a spectrophotometer (Shimadzu, UV-2600) for band gap calculation and optical feature investigation. For inferring composition and chemical states of constituent elements, X-ray photoelectron spectroscopy (XPS) and photoluminescence (PL) investigations were carried out with PL spectra.
2.4. Photocatalytic study
The photodegradation capabilities of the prepared bare ZnO NPs and C/N-co-doped ZnO nanorods were examined by conducting photodegradation experiments for MB and BCB dyes using an immersion-type photoreactor equipped with a sodium vapor lamp of 250 W. In the degradation experiments, 50 mL of dye (MB and BCB) solutions under the optimized reaction conditions were initially magnetically agitated in the darkness for 30 min to accomplish the adsorption–desorption equilibrium. Subsequently, the solutions were illuminated with the visible light, and the optical absorbance for the MB and BCB solutions at 664 nm and 624 nm was measured at various time intervals for finding the degradation abilities. The % photodegradation of the dyes was calculated using the following relation:| |  | (1) |
where C0 and Ct respectively are the concentrations at zero time and time ‘t’, and A0 and At respectively are the absorbance values of dye solutions at zero time and time ‘t’.
3. Results and discussion
3.1. Characterization of the prepared bare ZnO nanoparticles and C/N-co-doped ZnO nanorods
3.1.1. XRD analysis.
The XRD peaks for bare ZnO nanoparticles, as shown in Fig. 2(a), are diffracted at 31.80°, 34.46°, 36.29°, 47.50°, 56.63°, 62.88°, 66.39°, 67.98°, 69.05°, 72.57°, and 76.92° angles, which can be attributed to the {100}, {002}, (101), {102}, {110}, {103}, {200}, {112}, {201}, {004}, and {202} crystal planes of the hexagonal wurtzite structure of ZnO that possesses P63mc of space group and a = 3.23874 Å and c = 5.18707 Å lattice parameters. The peaks corresponding to these crystal planes of ZnO in C/N-co-doped ZnO are diffracted at 2 theta values of 31.81°, 34.41°, 36.33°, 47.54°, 56.60°, 62.87°, 66.43°, 67.96°, 69.09°, 72.65°, and 76.96° with the lattice parameter values of a = b = 3.24748 and c = 5.20114 Å, which are larger than that of the pure ZnO nanoparticles. Similar results are reported by Liang et al. for C/N-co-doped ZnO prepared from ZIF-8.45 The highly pointed and strong XRD peaks indicate the excellent crystallinity of the prepared materials. The doping of C and N induced slight alterations in diffraction peak positions in ZnO but caused a change in the crystal structure of ZnO. Moreover, no other XRD peaks except ZnO in C/N-co-doped-ZnO are noticed, which have ruled out the presence of residual C and N or any complex/compound of C and N in Co-doped ZnO nanorods.65,66
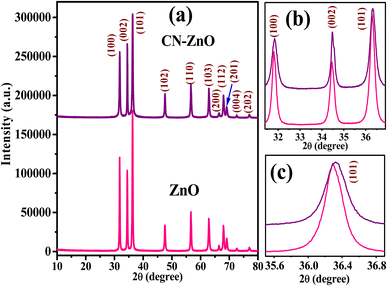 |
| | Fig. 2 (a) XRD patterns for pure ZnO and C/N-co-doped ZnO (b) enlarged view of peaks of (100), (002), (101), and for (c) (101) crystal planes of pure ZnO and C/N-co-doped ZnO. | |
It is well established that the XRD peak position shifting towards a lower or higher diffraction angle may be induced due to the lattice distortion caused by various types of lattice defects.67 Consequently, the variation in lattice parameters and particle size is observed due to the doping of impurities. In order to observe variation in lattice parameters and particle size of prepared co-doped ZnO, intense (100), (002), and (101) peaks and especially highly dominant (101) peaks were analyzed.66,68 The enlarged view of the (100), (002), and (101) peaks and the highly intense peak related to the crystal plane of (101) are shown in Fig. 2(b) and (c), respectively. The N-doping in the ZnO lattice causes a shifting of XRD peak position toward slightly lower diffraction angles, broadening of peaks, and a decrease in the lattice parameter (when O is replaced with N) values of the ZnO lattice structure,69,70 while shifting peaks toward higher angles with the increase in lattice parameters with the doping of C is observed. Because the ionic radius of the C4− ion is 2.60 Å, which is larger than the oxide ion (O2−) radius (1.40 Å),49 and the radius of the N3− ion (1.32 Å) is almost similar to O2− ion. Thus, the substitution of O with N in the ZnO lattice may cause a slight reduction in lattice parameter values, while substitution with C enlarges.71 In addition, the replacing of O-anions by C4− and N3− anions causes defects in the lattice because the radius of C4− is higher than that of O2−, while the N3− size is smaller than O2−.20 Moreover, the inequality of charges of C, N, and O also creates charge imbalance in doped ZnO, which requires O-loss.52
As illustrated in Fig. 2(b) and (c), which are the magnified XRD peaks between the diffraction angles of 31° to 37° of (100), (002), and (101), it can be observed that the shifting of XRD peaks of C/N-co-doped ZnO nanorods are towards the higher diffraction angles and along the broadening of peaks, especially for (100) crystal plane. Furthermore, the values of lattice parameters, c/a ratio, and cell volume (54.41 Å3 for ZnO and 54.85 Å3 for C/N-ZnO) for C/N-co-doped ZnO nanorods measured higher than pure ZnO. Moreover, due to the smaller size of N than O, the Zn–N bond length in N-doped-ZnO (O–Zn–N) is small in comparison to the Zn–O bond length and it causes shifting of XRD peaks and lattice parameters at lower values;70 however, the Zn–C bond length is higher than that of Zn–O in the C-doped-ZnO lattice (O–Zn–O) that shifts XRD peaks and lattice parameters toward higher values. Hence, the larger values of lattice parameters, higher lattice cell volume, and broadening of XRD peaks for prepared C/N-co-doped-ZnO nanorods than those of bare ZnO NPs confirm the substitution of O by C and N in ZnO.45,72
Furthermore, the grain size of pristine ZnO NPs and C/N-co-doped-ZnO nanorods was calculated employing Scherrer's formula as follows:
| |  | (2) |
where
D,
θ, and
β are the particle diameter (nm) of the prepared samples, XRD diffraction angle, and half-width at full maxima (FWHM) of the XRD peaks, respectively, and
λ = 0.15406 nm. The particle sizes for pure ZnO and C/N-co-doped-ZnO nanorods were estimated using the Scherrer formula as 30.18 and 37.33 nm, respectively. The Zn–O bond length values for pure and co-doped ZnO were estimated using
eqn (3):
| | 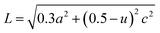 | (3) |
where
u denotes the internal factor (
Z-coordinate of O atoms), while ‘
a’ and ‘
c’ represent the lattice parameters of the prepared materials.
| |  | (4) |
The Zn–O bond lengths of bare and co-doped ZnO NPs were computed to be 1.880 and 1.885 Å, respectively, which reveals a slight increase in the Zn–O bond length that is linked with the increase in lattice parameter (a and c) values (Table 1).73,74 The increase in particle size is due to the expansion of bond length and lattice parameters owing to the substitution of O by C and N in the ZnO lattice.
Table 1 Lattice parameters for pristine ZnO nanoparticles and C/N-co-doped ZnO nanorods
| Sample |
a = b (Å) |
c (Å) |
Unit cell volume (Å3) |
Particle size (nm) |
c/a |
Zn–O bond length (Å) |
| ZnO |
3.23874 |
5.18707 |
54.4094 |
33.18 |
1.60157 |
1.8800 |
| C/N-ZnO |
3.24748 |
5.20114 |
54.8519 |
37.33 |
1.60159 |
1.8851 |
The particle size for pure and co-doped ZnO NPs was also evaluated by the Williamson–Hall (W–H) method using the following formula:75
| | 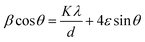 | (5) |
where
d is the particle size,
θ is the diffraction angle, and
A and
ε are constants. To estimate the W–H particle size, a graph was plotted between
β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
cos
θ and 4
sin
θ for samples and the intercept of the plot defined the particle size and slope to strain (
Fig. 3). The evaluated size values by the W–H method are 43.25 and 55.13 nm for pure ZnO and C/N-co-doped-ZnO NPs, respectively. Thus, it can be observed that the particle size values estimated using the Scherrer and W–H methods are slightly varying, which confirms that the micro-strain is inversely related to the grain size.
75
 |
| | Fig. 3 Williamson–Hall plots of (a) pure ZnO NPs (b) and C/N-co-doped ZnO nanorods. | |
Moreover, from the crystal parameter values, using eqn (6), the values of microstrain (ε) for bare and C/N-co-doped ZnO NPs were determined as follows:76
| |  | (6) |
where
β and
θ denote the FWHM and diffraction angles in radians, while
ε is the microstrain. The estimated values of microstrain for the pure and co-doped ZnO NPs are summarized in
Table 2, which show an increase in microstrain value for co-doped ZnO nanorods because of C and N atom doping and its effects on the crystallinity of ZnO.
Table 2 Physical parameters of pure ZnO and C/N-co-doped ZnO
| Samples |
Grain size (D) |
W–H size |
XRD (101) 2θ value |
Strain (ε) ×10−4 |
Dislocation (δ) ×10−5 |
| ZnO |
30.18 |
43.25 |
36.29 |
23.98 |
98.98 |
| C/N-ZnO |
37.33 |
55.12 |
36.33 |
24.86 |
106.00 |
The dislocation density (δ) designates the magnitude of defects in the crystal that can be determined using eqn (7):
| |  | (7) |
where
D is the particle diameter. The estimated mean values of dislocation density for the bare and co-doped ZnO NPs are illustrated in
Table 2, which reveal the increase in dislocation density for ZnO due to co-doping of C and N atoms in the ZnO lattice and reflect the reduction in crystalline character due to the doping of C and N atoms.
77
3.1.2. XPS analysis.
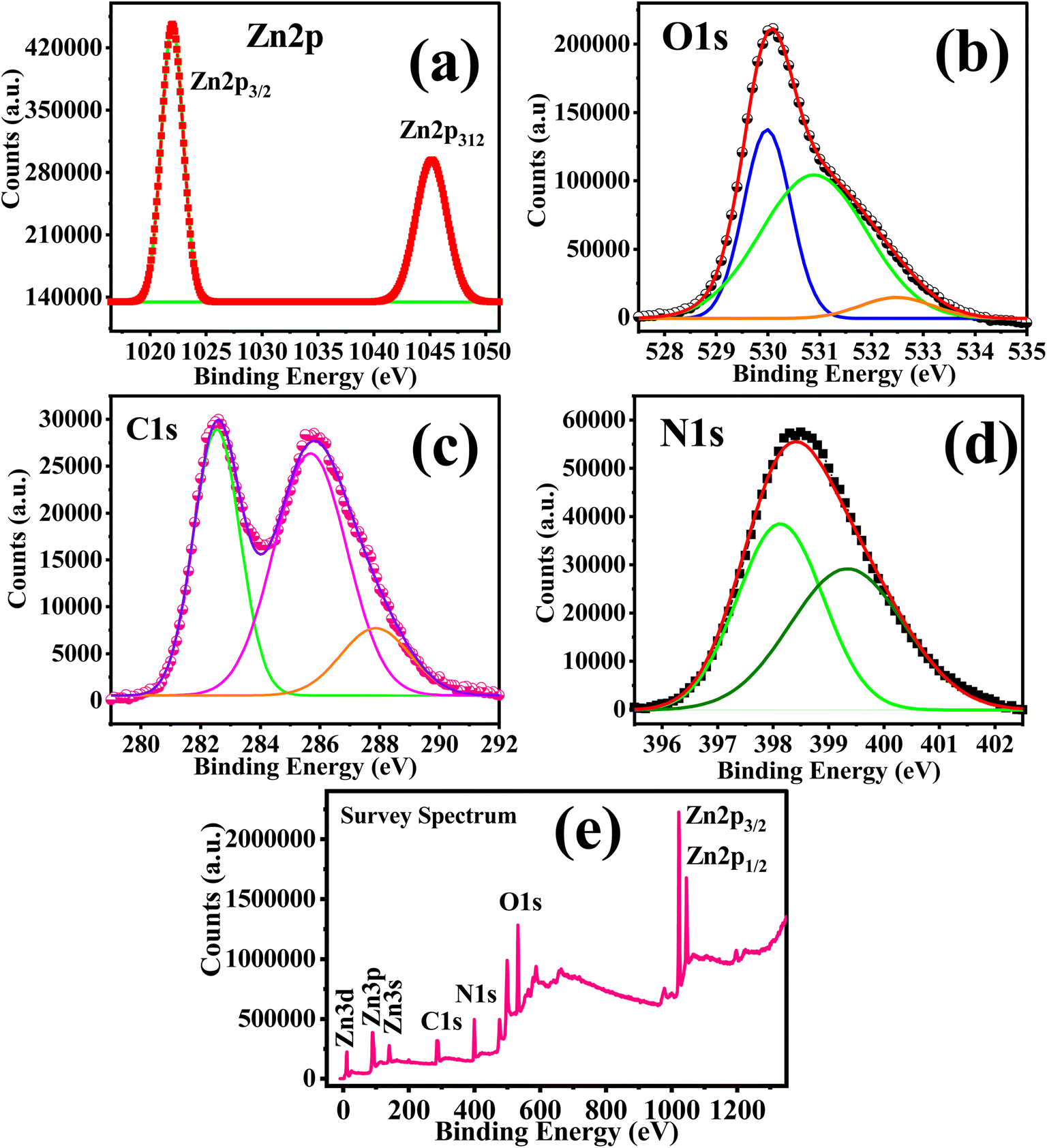
Fig. 4 shows the XPS spectra of C/N–ZnO; the Zn 2p spectra show two characteristic peaks at 1021.95 eV and 1045.07 eV, isolated by 23.1 eV, which can be indexed to Zn 2p3/2 and Zn 2p1/2, respectively,40,78 confirming the presence of zinc as Zn2+.40,78–80 The peak related to Zn 2p3/2 at 1021.95 eV can be attributed to the tetrahedrally coordinated Zn2+ ions with the O2− atoms in the wurtzite structure.81 However, the peaks corresponding to Zn 2p3/2 and Zn 2p1/2 for pure ZnO are observed at 1021.38 eV and 1044.40 eV, respectively (Fig. 2S; ESI†). The XPS spectrum of O 1s for C/N-co-doped ZnO is deconvoluted into three peaks at 529.95, 530.96, and 532.45 eV. The peak at 529.95 can be correlated to the O2− ions (lattice O) bound with Zn2+ (Zn–O bond) in the hexagonal wurtzite ZnO.82 The peak at 530.96 eV can be attributed to the oxygen vacancy (VO) or to the OH˙ radical. The peak observed at 532.45 eV is due to the presence of surface adsorbed oxygen as –OH groups owing to adsorption of water molecules.62,79,80,83 Conversely, in the XPS spectra of O 1s of pure ZnO, two peaks appear after deconvolution at binding energy values of 531.48 eV and 533.06 eV corresponding to O2− ions bound with Zn2+ (Zn–O bond) and –OH group from water molecules adsorbed on the surface of ZnO79 (Fig. 2S; ESI†).
 |
| | Fig. 4 (a) N 1s, (b) Zn 2p, (c) O 1s, (d) C 1s XPS spectra, and (e) survey spectrum of C/N-co-doped ZnO. | |
The N 1s XPS spectrum is de-convoluted into two peaks at 398.13 eV and 399.80 eV binding energy values. Various studies reported that the existence of the peak in the 397–398 eV range entails substitutional N-doping O–Zn–N (Nsubs) and the peak at 399–400 eV reveals the presence of oxidized N species such as NOx.42,84,85 The N 1s peak at 499.90 eV established the interstitial N-doping N–O–Zn (Ninters).20 N 1s XPS peak at 398.13 eV can be attributed to the O–Zn–N bond due to substitutional doping86 and the peak at 399.80 eV is due to the anionic N in Zn–O–N bonds in ZnO. The interstitial N forms interstitial Zn–O–N bonds.
The C 1s spectrum of C/N-co-doped ZnO nanorods is de-convoluted into three peaks at 282.50, 285.76 and 287.95 eV. The peaks at 285.76 and 287.95 eV are corresponding to the C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, while the peak at a binding energy value of 282.50 eV shows the presence of carbon as carbide (C4−).18,87 These peaks establish the successful doping of C into the ZnO lattice. The occurrence of a peak around 282 eV indicates the presence of C4− ions in the ZnO lattice and also shows the substitution of oxygen atom by C4−,18,88 and the formation of Zn–C bonds in the C/N-co-doped ZnO nanorods.18 Due to the bigger size of C4− (0.26 nm) compared to O2− (0.14 nm), the doping of carbon causes an increase in the bond length and lattice parameters in C/N-co-doped ZnO,45 as confirmed by XRD analysis.
O bonds, while the peak at a binding energy value of 282.50 eV shows the presence of carbon as carbide (C4−).18,87 These peaks establish the successful doping of C into the ZnO lattice. The occurrence of a peak around 282 eV indicates the presence of C4− ions in the ZnO lattice and also shows the substitution of oxygen atom by C4−,18,88 and the formation of Zn–C bonds in the C/N-co-doped ZnO nanorods.18 Due to the bigger size of C4− (0.26 nm) compared to O2− (0.14 nm), the doping of carbon causes an increase in the bond length and lattice parameters in C/N-co-doped ZnO,45 as confirmed by XRD analysis.
The widening of XRD peaks, increases in diffraction angles, increase of lattice parameters (a, b, and c), a/c ratio, and cell volume for C/N-co-doped ZnO compared to that of pure ZnO which confirmed the substitution of lattice O by C atoms in the ZnO lattice. Furthermore, the presence of the C 1s XPS peak around 282 eV indicated the presence of carbon in the C4− form that generates Zn–C bonding in the ZnO lattice, agreeing the substitution of oxygen atom by carbon. Thus, the N-doping in the ZnO lattice occurred through substitutional as well as interstitial mechanisms, and the C-doping through substitutional doping in the ZnO lattice.
3.1.3. FTIR analysis.
The FTIR spectra for the pure ZnO nanoparticles and carbon and nitrogen-doped ZnO NPs are shown in Fig. 5. The pure ZnO nanoparticles had IR bands at wavenumber values of 525, 703, 881, 1520, 3625, and 3863 cm−1. The bands at 3600–3870 cm−1 show the presence of O–H stretching vibrations, an intense peak at 525 cm−1 indicates Zn–O bond vibration,89–91 a band at 881 cm−1 signifies the weak vibration of the Zn–O bond,91,92 and peaks at a wavenumber value of 1520 cm−1 can be ascribed to the CO bond vibration.91,92 The FTIR bands for C/N-co-doped-ZnO are observed at 526, 881, 985, 1520, 1697, 2312, 3620, 3739, and 3866 cm−1. The extra peaks noticed in doped ZnO nanorods at 1122, 1743, and 2315 cm−1 can be indexed to the C–O, CC, and CO bond vibrations, respectively.89 The band observed at 985 cm−1 may be assigned to the strain vibrations of Zn–O45 and the sharp and intense peak observed at 526 cm−1 indicates the Zn–O bond vibration.89–91 The peak at 1520 cm−1 corresponding to Zn–N bond vibrations57 and also N–Zn–O bond vibrations93 confirm the doping of N in the ZnO lattice.
 |
| | Fig. 5 (a) FTIR spectra and (b) UV-vis spectra and Tauc plots for pure ZnO NPs and C/N-co-doped-ZnO nanorods. | |
3.1.4. Optical property investigation.
The optical properties of the pure ZnO nanoparticles and C/N-co-doped-ZnO nanorods were investigated by UV-vis spectroscopy for the analysis of optical absorbance characteristics and band gap assessment. The pure ZnO NPs show an absorbance shift near 400 nm and a characteristic absorbance peak at 305 nm (Fig. 5(b)) in the UV region due to the electronic transition from the VB to the CB.20,94 The indirect bandgap energy values for the fabricated samples were calculated using Tauc's relation as follows:where α is the absorption coefficient, hv is the light photon energy, Eg is the bandgap energy, and A is a constant. The Tauc plots for the estimation of direct bandgap energy for bare and co-doped ZnO NPs are sketched between (αhν)2 and hν (inset in Fig. 5(b)), and the reported values are 3.18 and 3.03 eV for bare ZnO and C/N-co-doped ZnO, respectively. Ray et al. in their study reported a similar band gap value (3.02 eV) for the C/N-doped ZnO.79 The lessening of the band gap is due to the doping of C and N in the ZnO lattice, which created localized occupied levels,95 and defect (ionic) states owing to the exchange of lattice O by C and N, which are situated over the VB of ZnO.56 The creation of defects (Ovac) in the ZnO lattice due to heteroatom doping directs the absorption band to shift toward a higher wavelength in the visible light range and narrow the band gap of ZnO,96 and sufficiently suppresses the recombination rate of charge carriers under the illumination of visible light photons that led to boost up the photocatalytic performance of C/N-co-doped-ZnO nanorods in comparison to pristine ZnO under similar reaction conditions. Furthermore, the interstitial N-doping creates localized states possessing the π character of NO, while the substitutional N-doping produces some occupied N 2p localized levels somewhat above the VB of ZnO. Thus, substitutional doping causes a little decrease in the band gap, while interstitial doping causes a significant reduction in the band gap.97–99
3.1.5. FESEM and EDX analysis.
The surface texture and morphological analysis of pure ZnO NPs and C/N-co-doped ZnO nanorods were carried out by the analysis of FESEM images. The FESEM images illustrate (Fig. 6(a and b)) that the pure ZnO NPs are made up of granular and sheet-shaped nanostructures having regular edges and smooth surface texture, whereas the C/N-co-doped-ZnO particles are composed of rod-shaped nanostructures possessing smooth surfaces and regular edges (Fig. 6(c and d)). The elemental compositions in the prepared nanosamples were investigated from the EDX analysis. The constituent elements in C/N-co-doped ZnO nanorods are displayed by EDX elemental maps, as depicted in Fig. 6(e–h), which suggest the homogenous distribution of elements in C/N-co-doped-ZnO nanorods.
 |
| | Fig. 6 FESEM images of (a and b) pure ZnO NPs and (c and d) C/N-co-doped-ZnO nanorods. Elemental mapping of C/N-co-doped-ZnO nanorods: (e) Zn, (f) O, (g) N and (h) C. EDX spectra of (i) pure ZnO and (j) C/N-co-doped-ZnO nanorods. | |
As depicted in the EDX spectra in Fig. 6(i) for pure ZnO and Fig. 6(j) for C/N-co-doped ZnO, the dominant peaks for pure ZnO are observed at X-ray energy values of 0.52 keV, 0.98 keV, 8.43 keV and 9.41 keV (Fig. 6(i)) while for C/N-co-doped-ZnO nanorods peaks at 0.2 keV, 0.36 keV, 0.61 keV, 1.08 keV, 8.6 keV and 9.5 keV (Fig. 6(j)). The peaks observed at the X-ray energy values of 0.52 keV and 0.95 keV are due to the emissions from the K-shell of O and L-shell of Zn, respectively.67,100 The peak at 0.95 keV due to L-shell emission is the convolution of photoelectron energies of Zn 2p3/2 and Zn 2p1/2.67
The peaks at 8.43 keV and 9.41 keV X-ray energy values are due to the additional emissions from the core levels of Zn.67,100 Thus, the appearance of basic emission peaks of O and Zn approved the subsistence of Zn and O atoms in the prepared pure ZnO nanoparticles. The K-shell of O and L-shell of Zn emission peaks in the EDS spectrum of C/N-co-doped-ZnO nanorods are observed at X-ray energy values of 0.61 keV and 0.1.08 keV, respectively.67 The N 1s core level emission observed an X-ray energy value near 0.36 keV,101 and the C 1s core–shell emission appears at 0.24 keV (Fig. 6(j)), The co-existence of C and N core–shell emission peaks at 0.0.24 keV and 0.36 keV values of X-ray energy confirmed the presence of C and N atoms with Zn and O atoms in C/N-co-doped-ZnO nanorods.
The atomic composition (at%) of the constituent atoms in the prepared nanoparticles Zn and O for pure ZnO (inset in Fig. 6(i)) and C, N, Zn, and O for doped ZnO (inset in Fig. 6(j)) can be analyzed using EDX spectra. Though the ratio of atoms is not in exact proportion, the higher percentage of nitrogen might be due to the extra doping owing to the presence of the excess amount of urea as a precursor in the synthesis mixture. Additionally, it is found that the pure ZnO sample shows almost unequal percent of O (42) and Zn (58) (O/Zn ratio = 1/38), which indicates an excess of Zn or O vacancies in pure ZnO NPs, while in C/N-co-doped-ZnO nanorods, the at% values for O, C, N, and Zn are 12.8, 15, 23 and 52, respectively (at% ratio of O + C + N to Zn = 0.94), revealing slight excess of Zn.
3.1.6. Photoluminescence study.
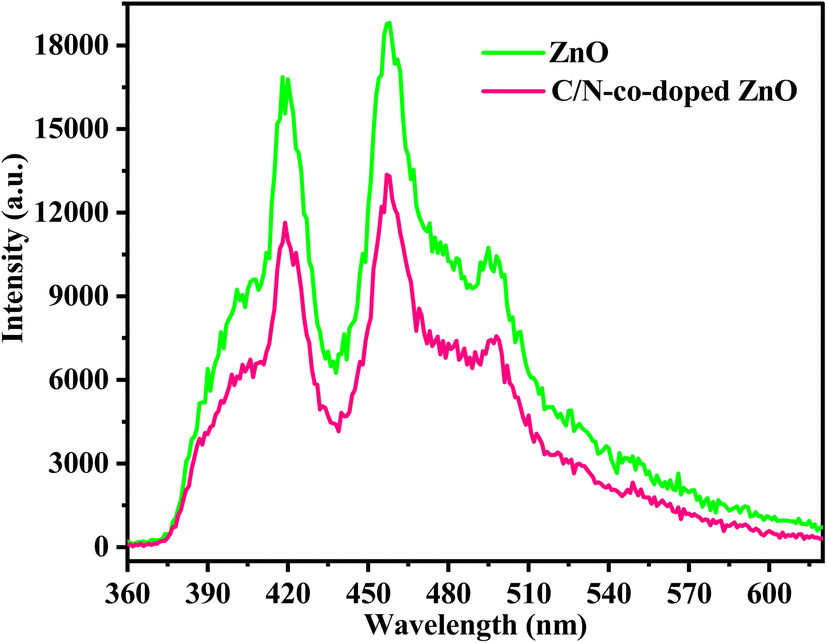
The PL spectra of C/N-co-doped ZnO and pure ZnO recorded at 340 nm wavelength are shown in Fig. 7. Usually, ZnO exhibits emission peaks in the near-ultraviolet and visible emission regions, with an emission peak at 390–400 nm representing the inherent luminescence process via the transfer of electrons from the CB to the VB in the excited state. The emissions in the visible region at 430–500 nm occur due to zinc vacancy (Vzn) defects or oxygen vacancy (VO), commonly due to the oxygen vacancies, which can produce the localized levels of free excitons and show blue-light emission at room temperature, and the emission peaks at 500–550 nm can be indexed to the occurrence of Zn interstitials.62 The intense narrow emission peaks in the range of 415–420 and 455–460 nm and a low-intensity peak in the range of 480–485 nm for pure ZnO nanoparticles and C/N-co-doped ZnO nanorods are noticed in the PL spectra. The lower intensity of the excitation peaks in C/N-co-doped ZnO nanorods reveals the lower rate of recombination of photo-produced charge carriers, which is accredited to the C and N doping and the synergistic role of the oxygen vacancies (VO) and changes in size and morphology. Thus, the synergistic effect due to C and N, oxygen vacancies (VO), alteration in size and morphology, and reduced rate of electron–hole recombination are accountable for enhancement in the photocatalytic potential of C/N-co-doped ZnO.62,102
 |
| | Fig. 7 PL spectra of pure ZnO and C/N-co-doped ZnO nanorods. | |
3.2. Photocatalytic applications of bare ZnO nanoparticles and C/N-co-doped ZnO nanorods
3.2.1. Photocatalytic degradation of organic dyes.
The photocatalytic competencies of pure ZnO NPs and C/N-co-doped ZnO nanorods were tested for the photo-mineralization of organic dyes BCB and MB under the illumination of visible light. The results of the photo-decay of dye solutions over pure ZnO and C/N-co-doped-ZnO as percent degradation are displayed in Fig. 8. To investigate the stability of dyes, standard experiments were also carried out in the absence of a catalyst. The adsorption abilities of the catalysts were tested before illumination to light photons by stirring the catalyst with dye solutions in the darkness for 30 min. The non-degraded quantities of BCB and MB dyes were evaluated from the UV-vis absorbance data of UV-vis. Spectra of dye solutions recorded at different time intervals at 625 nm and 664 nm wavelength values of radiations for BCB and MB dyes, respectively. Additionally, the degradation potential of dyes under dark conditions was also studied via stirring for 90 min in the presence of C/N-co-doped ZnO, which exhibited a slight removal due to adsorption and not via degradation of dyes (Fig. 1S; ESI†); this confirms that in the absence of light, C/N-co-doped ZnO is inactive. However, as demonstrated in the results of standard experiments, auto-mineralization of dyes in the absence of a catalyst is negligible, which is indicative of the photostability of dyes and confirms that the auto-degradation of BCB and MB dyes is very low and dyes are stable under visible light exposure (Fig. 8(a) for BCB and Fig. 8(b) for MB). However, in the presence of a C/N-co-doped ZnO nanorod catalyst, the decay potential of BCB and MB dyes is observed to be exceptionally very high. With more photodegradation experiments, the absorbance values of dye solutions demonstrated regular declining trends and after 60 min of photo-illumination, the dye solutions were almost completely turned into colorless forms and the absorption intensities for dye solutions approached zero, as depicted in the UV-vis absorption spectra in Fig. 8(c) for BCB and Fig. 8(d) for MB dye. Nevertheless, in the presence of pure ZnO NPs, sufficient intensities of blue colors in dye solutions were reported with higher absorbance values of UV-vis absorbance spectra for both BCB and MB dyes after 60 min of light exposure.
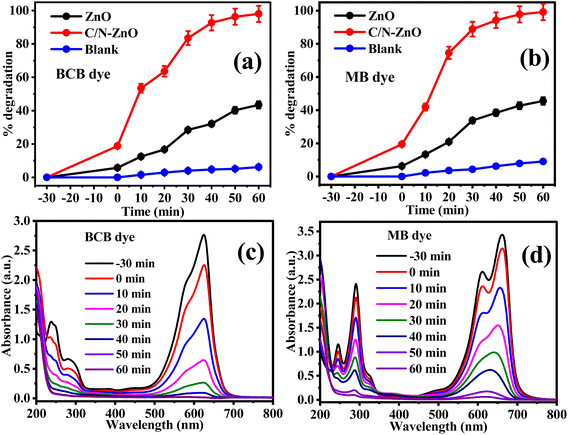 |
| | Fig. 8 Photodegradation performance of pure ZnO and C/N-co-doped-ZnO under visible light for (a) BCB (b) MB dyes, and UV-vis absorbance spectra for (c) BCB and (d) MB dyes in the presence of C/N-co-doped-ZnO under visible light. | |
In 60 min of visible light exposure, ∼98% of BCB (Fig. 8(a)) and ∼99% of MB (Fig. 8(b)) dyes were disintegrated at pH = 10 with 20 mg of C/N-co-doped ZnO nanorod catalyst at optimized dye concentrations. However, in the presence of undoped ZnO under similar reaction conditions, the degradation was achieved at only 43 and 45% for BCB and MB, respectively. Thus, the degradation aptitude of BCB and MB dyes followed the bandgap energy values of pure and doped ZnO. Due to the higher band gap of pure ZnO NPs, it has exhibited poor excitation under visible light photons, and the fast rate of recombining of photo-produced charge carrier species suppressed the photocatalytic potential of pure ZnO. On the contrary, the estimated band gap for C/N-co-doped-ZnO nanorods is less than undoped ZnO as of this photo-excitation of doped ZnO is enhanced and the recombination of electron–holes is significantly suppressed due to the creation of new energy states above the VB, due to the nonmetallic doping in the ZnO lattice, which leads to an increase in the photodegradation potential of the C/N-co-doped ZnO nanorods for BCB and MB dyes.
3.2.2. Optimization of parameters the for photocatalytic degradation of organic dyes.
The photo-decolorization potential of the prepared C/N-co-doped-ZnO nanorods was further tested with BCB and MB dyes by alterations of their initial concentration at pH = 10 using 20 mg of catalyst to optimize the concentrations of the dyes for photocatalytic degradation experiments. The initial dye concentrations were varied from 20 to 35 mg L−1 using 50 mL of dye solutions. The percent photo-decay of BCB and MB dyes at different initial concentrations is depicted in Fig. 9(a and b). As shown in Fig. 9(a)) for BCB and Fig. 9(b) for MB, the degradation indicates increasing trends with the increase for both BCB and MB dye concentrations from 20 mg L−1 to 25 mg L−1; however, upon further increase to 30 mg L−1 and 35 mg L−1, it indicates decreasing trends. Hence, the utmost degradations for BCB (98%) and MB (99.16%) dyes were obtained for 25 mg L−1 of dye solutions. At 20 mg L−1 of initial concentrations, 82.07% and 83.58% of BCB and MB dye were degraded, while at 30 mg L−1, the degradation was 92.26% and 93.96%, respectively, and at 35 mg L−1 concentration, 80.12% and 86.48% respectively. At a lower concentrations of dye solutions, it might be possible that all the active sites present at the surface of the catalyst are not occupied by dye molecules which results in poor degradation.103 Conversely, beyond the optimized limit, excess dye concentrations hinder the approach of light photons to the catalyst, due to which the catalyst is not activated appropriately to generate a sufficient amount of charge carriers. Moreover, a higher quantity of dye molecules also suppress the path length of photons.104 Consequently, the BCB and MB dyes showed decreased photo-decay capacity above the optimized levels of concentrations.
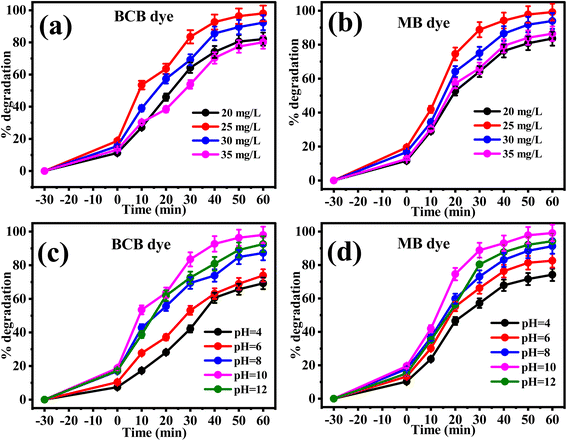 |
| | Fig. 9 Photodegradation performance of C/N-co-doped-ZnO at different initial concentrations of (a) BCB and (b) MB dyes and at different solution pH values for (c) BCB and (d) MB dyes under visible light. | |
The surface charges on the catalyst depend on the solution pH value, and surface charges directly affect the adsorption of molecules at the catalyst surface which finely determines the photocatalytic performance of the catalyst. Hence, the solution pH is an important parameter on which the photocatalytic power of a catalyst for any pollutant or chemical species being degraded under exposure to light photons is dependent. To examine the influence of solution pH on the photo-mineralization potential of C/N-co-doped-ZnO nanorods for the photo-decay of BCB and MB dyes with visible light photons, the solution pH values of dye solutions were considered in the range of 4–12 and the obtained results are illustrated by the percent degradation plots, as illustrated in Fig. 9(c and d). When the photodegradation experiments were performed in acidic solutions, the degradation activities of BCB and MB dyes were calculated less, at pH = 4, 69.13% and 74.18% for BCB and MB, respectively, and 73.89% and 82.54%, respectively at pH = 6. However, with the increase in solution pH, the degradation potential of both BCB and MB dyes over doped ZnO was reported to be enhanced, and determined at 87.31% and 91.30%, respectively for BCB and MB dyes at pH = 8, and 98.03% and 99.16%, respectively at pH = 10. Furthermore, the increase in solution pH values and a slight decrease in degradation capacities for both BCB and MB dyes were observed and determined to be 92.56% and 94.23%, respectively. Thus, in the alkaline phase, the photo-decay ability of the prepared C/N-co-doped-ZnO nanorods was enhanced but in the highly basic medium at higher pH, it was found to be decreased for both dyes. However, the utmost degradation percentage for BCB and MB dyes with doped ZnO nanorods was calculated at a basic pH of 10.
The contact of the ZnO surface with water molecules in the aqueous phase produces a hydroxide layer at the surface (ZnO–OH or C/N-co-doped ZnO) (eqn (9)), which can acquire charge in an acidic or basic medium by protonation and deprotonation processes.100 The point of zero charge (pHpzc) as demonstrated in Fig. 10 is 9.02 for the pure ZnO nanoparticles, whereas 7.31 for C/N-co-doped ZnO. Therefore, at pH below 7.31, the hydroxylated surface of ZnO turns positively charges via accepting protons (protonation), as illustrated in eqn (10), while above pH 7.31, the deprotonation of the –OH group of hydroxylated surface generates a negative charge on the surface (eqn (11)). Hence, the functionalities at the surface of C/N-co-doped ZnO at pHpzc, pHpzc > pH, and pHpzc < pH conditions are C/N–Zn–OH, C/N–Zn–OH2+, and C/N–Zn–O−, respectively.100 Conversely, the MB and BCB dyes are cationic dyes. Thus, dye molecules experience strong electrostatic repulsion in acidic pH below 7.31, and competitive adsorption with H+ ions on the photocatalyst surface, which reduces the adsorption of dye molecules, and consequently, results in lower photodegradation efficiencies. With the increase in pH value, the concentration of hydrogen ions in the solution decreases, resulting in a reduction in the intensity of positive charge on the catalyst surface and favoring the attachments of BCB and MB dye molecules, and the degradation efficacy enhanced accordingly. Moreover, at a basic pH value above 7.31, the deprotonation of the hydroxylated surface of C/N-co-doped ZnO changes negatively charged, which favors the adsorption of dye molecules on the surface. This might be accredited to the enhanced photodegradation of MB and BCB dyes. In addition, the basic pH favors the conversion of OH− ions into hydroxyl radicals, which are responsible for degradation dyes that also favor the degradation activities of BCB and MB dyes in alkaline pH.
| | | C/N-ZnO + H2O → C/N-ZnO⋯OH | (9) |
| | | C/N-ZnO⋯OH + H+ → C/N-ZnO⋯OH2+ (acidic pH) | (10) |
| | | C/N-ZnO⋯OH + OH− → C/N-ZnO⋯O− + H2O (basic pH) | (11) |
 |
| | Fig. 10 Point of zero charge (pHpzc) for pure ZnO and C/N-co-doped ZnO. | |
Conversely, in a very strong alkaline medium, the Zn2+ ions from the catalyst may also turn into complex species such as Zn(OH)2 or [Zn(OH)4]2−, which affects the photodegradation potential of Zn-based nanomaterials negatively.
The photocatalyst dose quantity for the decay of BCB and MB dyes varied from 10 to 25 mg using optimal dye concentrations and solution pH values. The uppermost degradation activities for the BCB and MB dyes were determined for 20 mg amount of C/N-co-doped-ZnO nanorod catalyst in 60 min of visible light exposure at optimized dye concentrations and solution pH. At a lower dose amount of catalyst, the poor degradation activity for dye is determined because of the lower surface area available for the binding of dye molecules. Beyond the optimized dose of catalysts, the decreased degradation efficiencies are determined for BCB and MB dyes because the effective surface area of the used catalyst becomes lower owing to the generation of aggregates and agglomerates and deprived infiltration of light photons in the solutions. Therefore, 20 mg of catalyst was elected as the optimized dose amount for the photocatalytic decay processes of BCB and MB dyes.
3.2.3. Kinetics of photodegradation processes.
The get a better understanding of the kinetic behaviors of the photodegradation reactions of BCB and MB dyes catalyzed by prepared catalysts, pristine ZnO NPs and C/N-co-doped-ZnO nanorods, the degradation experimental data were treated with the pseudo-first-order kinetic model.| |  | (12) |
where C0 and Ct respectively represent the dye concentrations at initial and time t, while A0 and At respectively represent the absorbance values for dye solutions at initial and time t, and k is the kinetic rate constant (min−1).
The values of rate constants were estimated from the slopes of the fitting lines of kinetics plots sketched between ln(Ct/C0) and reaction time (t) (Fig. 11). When the catalyst was not used in dye solutions during the photodegradation processes, the organic dye pollutant amounts show a little decrease, which indicates very slow auto-decays of BCB and MB dyes with visible light photons (Fig. 11(a) for BCB and Fig. 11(b) for MB). The undoped ZnO catalyst exhibited poor activity for the degradation of both dyes, and the C/N-co-doped-ZnO nanorod catalyst demonstrated very high photo-decay performances towards both BCB and MB dye pollutants under visible light illumination. The k values for pure ZnO were evaluated at 89.23 × 10−4 and 96.76 × 10−4 min−1 for BCB and MB dyes, respectively; however, for doped ZnO, it is 637.23 × 10−4 and 775.25 × 10−4 min−1, respectively, which reveals that the degradation of BCB dyes by doped ZnO nanorods is almost seven times higher than that of bare ZnO NPs, while for MB, it is approx. eight times.
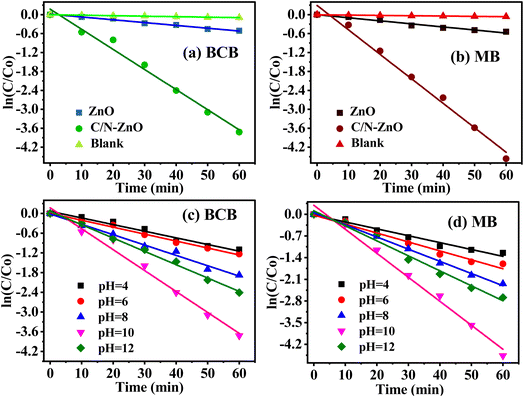 |
| | Fig. 11 Kinetic plots for the photodegradation of (a) BCB and (b) MB dyes over C/N-co-doped-ZnO at optimized dye concentrations, catalyst dose amount, and solution pH values under visible light. Kinetic plots for the photodegradation of (c) BCB and (d) MB dyes over C/N-co-doped-ZnO at different pH values of dye solutions at optimized dye concentrations and catalyst dose amounts. | |
Furthermore, the effect of pH on the kinetic behavior of photo-decays of BCB and MB dyes was investigated in the presence of C/N-co-doped-ZnO nanorod catalysts, and the results are shown in Fig. 11(c and d) and the calculated values of kinetics rate constants at different pH values are portrayed by a bar diagram, as shown in Fig. 12, which indicates that the uppermost degradation rates for BCB (637.23 × 10−4 min−1) and MB (775.25 × 10−4 min−1) dyes are observed at pH = 10. The experimental data exhibiting good agreement with the pseudo-first-order kinetics model established that the photodegradation of BCB and MB dyes over the prepared catalyst followed the pseudo-first-order kinetics.
 |
| | Fig. 12 Pseudo-first-order kinetics rate constants for the photodegradation of (a) BCB and (b) MB dyes over C/N-co-doped-ZnO nanorods at different pH values of dye solutions at optimized dye concentrations and catalyst dose amounts. | |
3.3. Scavenger and reusability studies
The role of active species in the photodegradation of BCB and MB dyes in the presence of C/N-co-doped ZnO nanorods under visible light illumination is confirmed by performing scavenger experiments using p-BQ, EDTA-2Na, and t-BuOH for scavenging of ˙O2−, ˙OH, and h+, respectively. The results of the trapper study observed that upon mixing of p-BQ in BCB and MB dye solutions during photocatalytic reactions (Fig. 13(a)), a drastic reduction in the degradation of both dyes is reported, while with EDTA-2Na and t-BuOH, a slight decrease is measured for both dyes. Thus, ˙O2− played a key role in the photodegradation of BCB and MB dyes in the presence of C/N-co-doped ZnO nanorods under visible light illumination.
 |
| | Fig. 13 (a) Scavenger experiment and (b) recyclability results for BCB and MB dyes over C/N-co-doped-ZnO nanorods under visible light photons. | |
The photo-durability and reusability of the as-synthesized C/N-co-doped-ZnO nanorod catalyst were investigated to examine their potential uses. The recycling experiments were performed for the BCB and MB dye photo-mineralization separately in four recycling runs in the presence of used C/N-co-doped-ZnO nanorod catalysts collected at the end of every run of reuse and utilized in the next cycle with proper sanitation and drying following similar reaction conditions, as described in the experimental section, and the results are shown in Fig. 13(b).
The photodegradation capabilities of C/N-co-doped-ZnO nanorod catalysts for BCB and MB dyes in every recycling test were established to be approximately comparable, and at the end of the 4th recycling test, the degradation efficacies for both dyes were reported excellent with limited decreasing from 98% to 90% for BCB and 99% to 91% MB dyes. Thus, the doped ZnO nanorods exhibited excellent stability and reusability under visible light irradiation and showed good potential for realistic uses.
In addition, the photocatalytic performance of the C/N-co-doped ZnO nanorod is compared with some previously prepared doped metal oxide (TiO2 and ZnO) photocatalysts, as demonstrated in Table 3, which reveals excellent photocatalytic performance of C/N-co-doped ZnO under visible light against the BCB and MB dyes.
Table 3 Comparison of the photocatalytic ability of C/N-co-doped ZnO with previously prepared photocatalysts
| Photocatalyst |
Pollutant |
Light source |
Synthesis method |
Time (min) |
Degradation (%) |
References |
| C/N-co-doped ZnO |
BCB |
Visible |
Calcination |
70 |
∼98 |
Current work |
| MB |
∼99 |
| C/N-doped ZnO |
MB |
Solar simulated light |
Two-step pyrolysis |
90 |
100 |
45
|
| N-doped ZnO/CNT |
MB |
Visible |
Cocrystallization |
25 |
96.58 |
59
|
| C-doped ZnO |
PHE |
Sun light |
Sonicated sol–gel |
30 |
100 |
60
|
| C-, N-co-doped ZnO |
MO |
Sun light |
MOFs pyrolysis |
30 |
50 |
62
|
| C–N-co-doped ZnO |
MB |
Visible |
Sol–gel & physical grinding |
140 |
91 |
63
|
| C-ZnO |
MB |
Visible |
Thermal decomposition |
180 |
98.2 |
71
|
| C-ZnO |
RhB |
Visible |
MOF pyrolysis |
210 |
>90 |
105
|
| C-doped g-C3N4 grafted C, N co-doped ZnO |
BPA |
Simulated solar |
Bio-template hydrothermal |
180 |
92.5 |
106
|
| C/N-Au/TiO2 |
RhB |
Visible |
Template |
150 |
90 |
107
|
| N-ZnO/C-dots |
MG |
Visible |
Hydrothermal |
160 |
85 |
108
|
| C-ZnO |
MB |
Visible |
Pyrolysis |
120 |
— |
109
|
3.4. Photodegradation mechanism
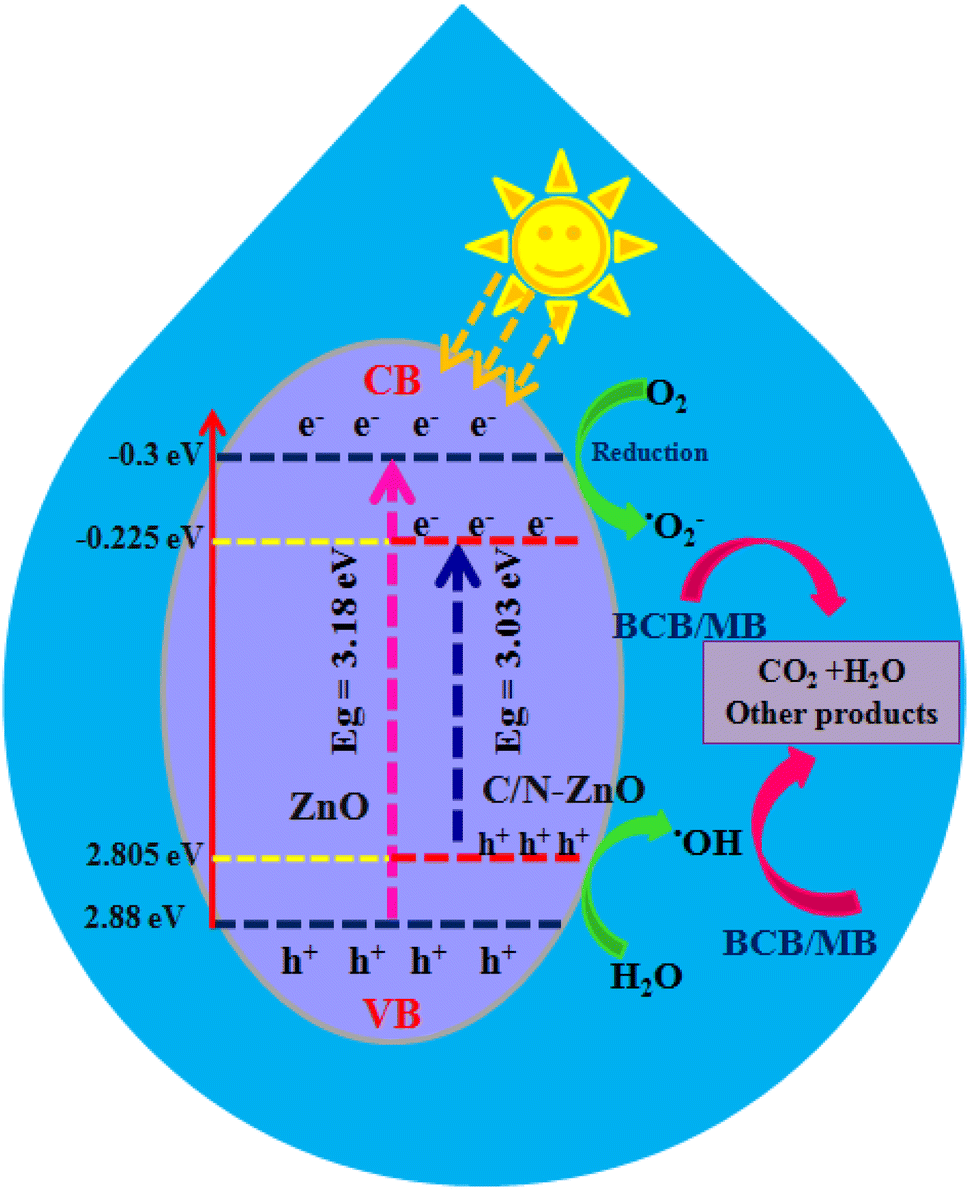
The photodegradation plausible mechanisms for both BCB and MB dyes in the presence of the prepared C/N-co-doped-ZnO nanorod photocatalyst are intended based on the results of active species capture experiments and band edge potential values, which are displayed in Fig. 14. The VB and CB edge potential values for C/N-co-doped-ZnO nanorods were estimated considering Evb = X − Ee + 0.5Eg and ECB = EVB − Eg relations, where X represents the absolute electronegativity (X = 5.79 for ZnO), Ee = ∼4.5 eV indicates free electron energy value on the hydrogen scale and Eg = bandgap (eV) energy. The bandgap energy values for pure ZnO and C/N-co-doped-ZnO nanorods are 3.18 and 3.03 eV; therefore, the calculated EVB values are 2.88 eV and 2.805 eV, respectively while the ECB values are −0.30 eV and −0.225 eV, respectively. Upon illumination of C/N-co-doped-ZnO nanorods with visible light photons, photogenerated electron–hole pairs are generated in the VB. The photo-produced electrons move to the CB from the VB, while the produced holes stay at VB. Usually, the O vacancy acts as e− trapper center, and the atoms close to O vacancies act as reaction locations.70 Hence, photo-produced e− at the CB and energy states of O vacancies could be entrapped by surface-adsorbed oxygen molecules and generate superoxide radical anions.71 The holes that stay at the VB diffuse to the surface and interact with H2O or –OH to generate hydroxyl radicals. The role of produced active species is further established from the findings of scavenging studies, which confirmed the active role of ˙OH in the degradation of BCB and MB dyes under photo-illumination conditions (Fig. 13(a)).
 |
| | Fig. 14 Photodegradation pathway for the BCB and MB dyes over C/N-co-doped-ZnO nanorod catalysts under visible light irradiation. | |
Alternatively, the occurrence of O vacancies also helped in reducing the recombination of photo-produced electron–hole pairs, due to which sufficient time of survival is availed by these charge carriers for the generation of reactive species (oxidative and reactive), which can be used in the disintegration of BCB and MB dyes. Ansari et al. and Sawant et al. reported similar results from carbon-doped-ZnO nanostructures and found that extra energy levels in band gap were generated due to doping of C, and the enhanced visible light response of C-ZnO nanostructures could be due to the generation of extra energy states, O vacancies and interactions of C with ZnO.99,110
On the whole, the doping of heteroatoms (C and N) to the ZnO enhanced visible light absorption and photodegradation of BCB and MB dyes. First, due to the formation of new energy states in the ZnO lattice, through the interaction of the O 2p orbital with the C 2p and N 2p orbital1,111 that leads to a reduction in band gap and an increase in light absorption in the visible range. In addition, the generation of superoxide radicals occurs via the interaction of electrons of the CB of ZnO and oxygen vacancy (Ovac) energy states with the oxygen molecules, which turns into hydroxyl radicals that actively participate in the degradation of dyes.71,111
Moreover, owing to the higher positive value of the VB of ZnO, the photo-produced holes present in the VB of ZnO and the dopant (C and N) energy levels interact with water molecules to generate ˙OH that are involved in the decay of BCB and MB dyes.111 Hence, C/N-co-doped ZnO adsorbs higher visible light, generates more photoexcited e−–h+, and hampers the charge carrier recombination, and by this means, increases the photodegradation of BCB and MB dyes. The key reactions which may occur in the photo-decay process of BCB/MB dyes in the presence of C/N-co-doped-ZnO nanorods are demonstrated using eqn (13)–(19).
| | | C/N-ZnO + hv → e− + h+ | (13) |
| | | C/N-ZnO(VBe−) → C/N-ZnO(CBe−) | (14) |
| | | C/N-ZnO(VBh+) + OH− → OH˙ | (15) |
| | | C/N-ZnO(CBe−) + O2 → O2˙− | (16) |
| | | O2˙− + H2O → OOH˙ + OH− | (17) |
| |  | (18) |
| | | OḢ/O2˙− + BCB/MB → degradation products | (19) |
4. Conclusion
The synthesis of C/N-co-doped ZnO nanorods by a facile and eco-friendly mechano-thermal method via a solvent-free approach was reported. The synthesized C/N-co-doped-ZnO nanorods were employed for the photocatalytic decay of water pollutants, namely, MB and BCB dyes, which demonstrated highly enhanced degradation capacity compared to the undoped ZnO NPs under visible light illumination. The structural and textural features of the fabricated nanospheres were elucidated by XRD, FT-IR spectroscopy, UV-vis spectroscopy, FESEM, EDX, XPS, and PL spectroscopy. The XRD patterns confirmed the hexagonal wurtzite crystal structure of C/N-co-doped-ZnO nanorods with the P63mc space group having higher purity and crystallinity. In addition, the FESEM findings confirmed the formation of rod-shaped nanostructures of co-doped ZnO nanoparticles, and EDX and XPS established the doping of C and N atoms in the ZnO lattice, and N-doping in the ZnO lattice followed substitutional and interstitial mechanisms, while C-doping occurred via a substitutional pathway as substantiated by XRD and XPS results. Furthermore, the doping of heteroatoms in the ZnO lattice increased the lattice parameter values, unit cell volume, Zn–O bond length, particle diameter, strain, and dislocation density while observing the lower band gap energy value, and shifted absorption in the visible range. The co-doped ZnO nanorods showed highly enhanced degradation potential toward both MB (99.16%) and BCB (98%) dyes under visible light exposure for 60 min in the basic medium at pH = 10 due to increased light absorption in the visible region, decreased band gap, generation of extra energy states within the VB and CB levels, and formation of defects in the lattice. MB and BCB degraded with rates of 637.23 × 10−4 and 775.25 × 10−4 min−1, respectively, and showed good agreement with the pseudo-first-order kinetics over C/N-co-doped-ZnO nanorods. The superoxide radicals were the key reactive species engaged in the decay of MB and BCB dyes over co-doped ZnO as confirmed by scavenger studies. The reusability studies carried out for the successive reuse in four cycles established good reusability and photo-stability of co-doped ZnO nanorods under visible light, which retained efficiencies of approx. 90% and 91% after four cycles of reuse. Therefore, the doping of ZnO with heteroatoms such as C and N causes significant alterations in structural, physicochemical, and optoelectrical properties and increases the photocatalytic capacity of ZnO nanoparticles under visible light, making it a superior photocatalyst for the degradation of water pollutants.
Data availability
The datasets generated and analysed during the current study are confidential and are available from the authors on reasonable request.
Conflicts of interest
Authors proclaim no conflicts of interest.
Acknowledgements
Authors are thankful to the Department of Science and Technology (DST), Govt. of Rajasthan for providing necessary assistance for this work. The authors also show their gratitude to the Department of Chemistry, University of Rajasthan, Jaipur (India) for the necessary assistance.
References
- S. T. Kochuveedu, Y. H. Jang, Y. J. Jang and D. H. Kim, J. Mater. Chem. A, 2013, 1, 898–905 RSC.
- R. Jain, M. Upadhyaya, A. Singh and R. P. Jaiswal, J. Environ. Chem. Eng., 2024, 12(5), 113881, DOI:10.1016/j.jece.2024.113881.
- C. Mao, H. Lei, Z. Guo, X. Jia, X. Cui, J. Huang, L. Fei, Y. Jia and W. Chen, Ceram. Int., 2024, 50(3), 4737–4745, DOI:10.1016/j.ceramint.2023.11.218.
- P. L. Meena, A. K. Surela and L. K. Chhachhia, J. Mol. Struct., 2024, 1303, 137575, DOI:10.1016/j.molstruc.2024.137575.
- P. L. Meena, K. Poswal and A. K. Surela, Water Environ. J., 2022, 36(3), 513–524, DOI:10.1111/wej.12783.
- P. L. Meena, A. K. Surela, K. Poswal, J. K. Saini and L. K. Chhachhia, Environ. Sci. Pollut. Res., 2022, 29, 79253–79271, DOI:10.1007/s11356-022-21435-z.
- P. L. Meena, A. K. Surela, K. Poswal, J. K. Saini and L. K. Chhachhia, Biomass Convers. Biorefin., 2024, 14, 3793–3809, DOI:10.1007/s13399-022-02605-y.
- P. L. Meena, A. K. Surela, K. Poswal and J. K. Saini, Water Sci. Technol., 2021, 84(9), 2615, DOI:10.2166/wst.2021.431.
- P. L. Meena, K. Poswal, A. K. Surela and J. K. Saini, J. Water Environ. Nanotechnol., 2022, 6(3), 196–211, DOI:10.22090/jwent.2021.03.001.
- P. L. Meena, K. Poswal, J. K. Saini and A. K. Surela, Adv. Compos. Hybrid Mater., 2023, 6, 16, DOI:10.1007/s42114-022-00577-1.
- P. L. Meena, K. Poswal, A. K. Surela, B. Mordhiya and K. S. Meena, Environ. Sci. Pollut. Res., 2023, 30, 68770–68791, DOI:10.1007/s11356-023-27215-7.
- Y. Chu, C. Zhao, Y. Zheng, X. Ren, S. Yuan, L. Zhao, Y. Wu and Y. He, J. Environ. Sci., 2025, 154, 444–456, DOI:10.1016/j.jes.2024.08.033.
- P. L. Meena, A. K. Surela and L. K. Chhachhia, Opt. Mater., 2024, 148, 114916, DOI:10.1016/j.optmat.2024.114916.
- J. Wang, L. Guan, S. Yuan, J. Zhang, C. Zhao, X. Hu, B. Teng, Y. Wu and Y. He, Sep. Purif. Technol., 2023, 314, 123554, DOI:10.1016/j.seppur.2023.123554.
- C. Zhao, X. Li, L. Yue, X. Ren, S. Yuan, Z. Zeng and Y. He, Mater. Res. Bull., 2023, 167, 112377, DOI:10.1016/j.materresbull.2023.112377.
- C. Zhao, C. Wang, X. Ren, S. Yuan, L. Zhao, L. Zhuang, B. Teng, Y. Wu and Y. He, Chem. Eng. J., 2024, 498, 155202, DOI:10.1016/j.cej.2024.155202.
-
C. Jagadish and S. J. Pearton, Zinc Oxide Bulk, Thin Films and Nanostructures: Processing, Properties, and Applications, Elsevier, Amsterdam, 2011 Search PubMed.
- S. Cho, J.-W. Jang, J. S. Lee and K.-H. Lee, CrystEngComm, 2010, 12, 3929–3935 RSC.
- A. F. Borzyszkowska, A. Sulowska, P. Czaja, A. Bielicka-Giełdon, I. Zekkere and A. Zielinska-Jurek, RSC Adv., 2023, 13, 25529–25551 RSC.
- D. G. Ayu, S. Gea, Andriayani, D. J. Telaumbanua, A. F. R. Piliang, M. Hrahap, Z. Yen, R. Goei and A. I. Y. Tok, ACS Omega, 2023, 8, 14965–14984, DOI:10.1021/acsomega.2c07546.
- Y. P. Zhu, M. Li, Y. L. Liu, T. Z. Ren and Z. Y. Yuan, J. Phys. Chem. C, 2014, 118, 10963–10971 CrossRef CAS.
- M. Samadi, M. Zirak, A. Naseri, E. Khorashadizade and A. Z. Moshfegh, Thin Solid Films, 2016, 605, 2–19, DOI:10.1016/j.tsf.2015.12.064.
- Z. Wang, Y. Liu, B. Huang, Y. Dai, Z. Lou, G. Wang, X. Zhang and X. Qin, Phys. Chem. Chem. Phys., 2014, 16, 2758–2774 RSC.
- S. Dong, J. Feng, M. Fan, Y. Pi, L. Hu, X. Han, M. Liu, J. Sun and J. Sun, RSC Adv., 2015, 5, 14610–14630 RSC.
- Y. Wang, Q. Wang, X. Zhan, F. Wang, M. Safdar and J. He, Nanoscale, 2013, 5, 8326–8339 RSC.
-
P. Yadav, P. K. Dwivedi, S. Tonda, R. Boukherroub and M. V. Shelke, Environmental Chemistry for a Sustainable World, Springer, Cham, 2020, vol. 34, DOI:10.1007/978-3-030-15608-4_4.
- C. Klingshirn, J. Fallert, H. Zhou, J. Sartor, C. Thiele, F. Maier-Flaig, D. Schneider and H. Kalt, Phys. Status Solidi B, 2010, 247, 1424–1447 CrossRef CAS.
-
H. Morkoç and Ü. Özgür, Zinc Oxide: Fundamentals, Materials and Device Technology, John Wiley & Sons, New York, 2008 Search PubMed.
- J. C. Fan, K. M. Sreekanth, Z. Xie, S. L. Chang and K. V. Rao, Prog. Mater. Sci., 2013, 58, 874–985 CrossRef CAS.
- W. Yu, J. Zhang and T. Peng, Appl. Catal., B, 2016, 181, 220–227 CrossRef CAS.
- K. S. Ahn, Y. Yan, S. Shet, T. Deutsch, J. Turner and M. Al-Jassim, Appl. Phys. Lett., 2007, 91, 1–3 Search PubMed.
- X. Y. Yang, A. Wolcott, G. Wang, A. Sobo, R. C. Fitzmorris, F. Qian, J. Z. Zhang and Y. Li, Nano Lett., 2009, 9, 2331–2336 CrossRef CAS PubMed.
- S. M. Lam, J. C. Sin, A. Z. Abdullah and A. R. Mohamed, Desalin. Water Treat., 2012, 41, 131–169 CrossRef CAS.
- J. Li and N. Wu, Catal. Sci. Technol., 2015, 5, 1360–1384 RSC.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- H. von Wenckstern, H. Schmidt, M. Brandt, A. Lajn, R. Pickenhain, M. Lorenz, M. Grundmann, D. M. Hofmann, A. Polity, B. K. Meyer, H. Saal, M. Binnewies, A. Börger, K. D. Becker, V. A. Tikhomirov and K. Jug, Prog. Solid State Chem., 2009, 37, 153–172 CrossRef CAS.
-
J. M. Coronado, F. Fresno, M. D. Hernández-Alonso and R. Portela, Design of Advanced Photocatalytic Materials for Energy and Environmental Applications, Springer, New York, 2013 Search PubMed.
- S. R. Kadam, V. R. Mate, R. P. Panmand, L. K. Nikam, M. V. Kulkarni, R. S. Sonawane and B. B. Kale, RSC Adv., 2014, 4, 60626–60635 RSC.
- L. C. Chen, Y. J. Tu, Y. S. Wang, R. S. Kan and C. M. Huang, J. Photochem. Photobiol., A, 2008, 199, 170–178 CrossRef CAS.
- K. Dib, M. Trari and Y. Bessekhouad, Appl. Surf. Sci., 2020, 505, 144541, DOI:10.1016/j.apsusc.2019.144541.
- L. Sun, H. He, L. Hu and Z. Ye, Phys. Chem. Chem. Phys., 2013, 15, 1369–1373 RSC.
- X. Zong, C. Sun, H. Yu, Z. G. Chen, Z. Xing, D. Ye, G. Q. Lu, X. Li and L. Wang, J. Phys. Chem. C, 2013, 117, 4937–4942 CrossRef CAS.
- M. Zhou, X. Gao, Y. Hu, J. Chen and X. Hu, Appl. Catal., B, 2013, 138–139, 1–8 CrossRef CAS.
- S. Lian, H. Huang, J. Zhang, Z. Kang and Y. Liu, Solid State Commun., 2013, 155, 53–56 CrossRef CAS.
- P. Liang, C. Zhang, H. Sun, S. Liu, M. O. Tade and S. Wang, RSC Adv., 2016, 6, 95903–95909, 10.1039/C6RA20667K.
- T. Tang, C. Li, W. He, W. Hong, H. Zhu, G. Liu, Y. Yu and C. Lei, J. Alloys Compd., 2022, 894, 162559, DOI:10.1016/j.jallcom.2021.162559.
- L. M. Huong, C. Q. Cong, N. M. Dat, N. D. Hai, N. T. H. Nam, H. An, L. T. Tai, T. D. Dat, N. T. Dat, M. T. Phong and N. H. Hieu, J. Cleaner Prod., 2023, 392, 136269, DOI:10.1016/j.jclepro.2023.136269.
- C. Di Valentin and G. Pacchioni, Acc. Chem. Res., 2014, 47, 3233–3241 CrossRef CAS PubMed.
- S. Zhou, Q. Xu, K. Potzger, G. Talut, R. Grötzschel, J. Fassbender, M. Vinnichenko, J. Grenzer, M. Helm and H. Hochmuth,
et al.
, Appl. Phys. Lett., 2008, 93, 232507 CrossRef.
- V. Kumari, A. Mittal, J. Jindal, S. Yadav and N. Kumar, Front. Mater. Sci., 2019, 13(1), 1–22, DOI:10.1007/s11706-019-0453-4.
- R. Swapna and M. C. K. Santhosh, Mater. Sci. Eng., B, 2013, 178(16), 1032–1039 CrossRef CAS.
-
M. S. Jyothi, V. Nayak, K. R. Reddy, S. Naveen and A. V. Raghu, Environmental Chemistry for a Sustainable World, Springer, Cham, 2019, vol. 29, DOI:10.1007/978-3-030-10609-6_3.
- N. M. Hosny, I. Gomaa and M. A. Elmahgary, Sci. Rep., 2023, 13, 14173, DOI:10.1038/s41598-023-41106-4.
- R. Kabir, Md. A. K. Saifullah, A. Z. Ahmed, S. Md. Masum and M. A. I. Molla, J. Compos. Sci., 2020, 4, 49, DOI:10.3390/jcs4020049.
- M. A. I. Molla, M. Furukawa, I. Tateishi, H. Katsumata and S. Kaneco, J. Environ. Sci. Health, Part A, 2019, 54, 914–923 CrossRef CAS PubMed.
- A. P. Bhirud, S. D. Sathaye, R. P. Waichal, L. K. Nikam and B. B. Kale, Green Chem., 2012, 14, 2790–2798 RSC.
- J. Lu, Q. Zhang, J. Wang, F. Saito and M. Uchida, Powder Technol., 2006, 162, 33–37 CrossRef CAS.
- C. L. Perkins, S.-H. Lee, X. Li, S. E. Asher and T. J. Coutts, J. Appl. Phys., 2005, 97, 034907 CrossRef.
- Md. A. Hanif, Y.-S. Kim, J. Akter, H. G. Kim and L. K. Kwac, ACS Omega, 2023, 8, 16174–16185 CrossRef CAS PubMed.
- Y. Shaban and N. A. Alharbi, Environ. Sci. Pollut. Res., 2022, 29, 47818–47831, DOI:10.1007/s11356-022-19214-x.
- S. N. Backer, S. E. Oussadou, I. W. Almanassra, M. K. Mousa, M. A. Atieh and A. Shanableh, Results Eng., 2023, 20, 101432 CrossRef CAS.
- S. Fu, W. Xi, J. Ren, H. Wei and W. Sun, Materials, 2024, 17, 855, DOI:10.3390/ma17040855.
- R. Shikuku and M. Abdulla-Al-Mamun, MRS Adv., 2024, 9, 1015–1022, DOI:10.1557/s43580-024-00857-6.
- C. Tian, Q. Zhang, A. Wu, M. Jiang, Z. Liang, B. Jiang and H. Fu, Chem. Commun., 2012, 48, 2858–2860 RSC.
- R. Lamba, A. Umar, S. K. Mehta and S. K. Kansal, J. Alloys Compd., 2015, 620, 67–73 CrossRef CAS.
- R. Kumari, A. Sahai and N. Goswami, Prog. Nat. Sci.: Mater. Int., 2015, 25(4), 300–309 CrossRef CAS.
- A. Sahai and N. Goswami, Ceram. Int., 2014, 40, 14569–14578 CrossRef CAS.
- S. Dutta, S. Chattopadhyay, A. Sarkar, M. Chakrabarti, D. Sanyal and D. Jana, Prog. Mater. Sci., 2009, 54, 89–136 CrossRef CAS.
- B. M. Rajbongshi, A. Ramchiary and S. Samdarshi, Mater. Lett., 2014, 134, 111–114 CrossRef CAS.
- Z. Li, S. Sun, X. Xu, B. Zheng and A. Meng, Catal. Commun., 2011, 12, 890–894 CrossRef CAS.
- X. Zhang, J. Qin, R. Hao, L. Wang, X. Shen, R. Yu, S. Limpanart, M. Ma and R. Liu, J. Phys. Chem. C, 2015, 119, 20544–20554, DOI:10.1021/acs.jpcc.5b07116.
- M. Samadi, H. A. Shivaee, A. Pourjavadi and A. Z. Moshfegh, Appl. Catal., A, 2013, 466, 153–160 CrossRef CAS.
- T. Hurma and M. Caglar, Mater. Sci. Semicond. Process., 2020, 110, 104949 CrossRef CAS.
- E. Gunasekaran, M. Ezhilan, G. K. Mani, P. Shankar, A. J. Kulandaisamy, J. B. B. Rayappan and K. J. Babu, Semicond. Sci. Technol., 2018, 33, 095005 CrossRef.
- M. F. Sanakousar, C. C. Vidyasagar, V. M. Jiménez-Pérez, B. K. Jayanna, Mounesh, A. H. Shridhar and K. Prakash, J. Hazard. Mater. Adv., 2021, 2, 100004, DOI:10.1016/j.hazadv.2021.100004.
- P. Bindu and S. Thomas, J. Theor. Appl. Phys., 2014, 8, 123–134, DOI:10.1007/s40094-014-0141-9.
- K. I. John, A. A. Adenle, A. T. Adeleye, I. P. Onyia, C. Amune-Matthews and M. O. Omorogie, Chem. Phys. Lett., 2021, 776, 138725, DOI:10.1016/j.cplett.2021.138725.
- J. Xue, S. Ma, Y. Zhou, Z. Zhang and P. Jiang, RSC Adv., 2015, 5, 18832–18840, 10.1039/C5RA00217F.
- A. Ray, S. Subudhi, S. P. Tripathy, L. Acharya and K. Parida, Adv. Mater. Interfaces, 2022, 9, 2201440, DOI:10.1002/admi.202201440.
- P. Yan, Y. Ye and W. Mingwen, Environ. Sci. Pollut. Res., 2022, 29, 88172–88181, DOI:10.1007/s11356-022-21696-8.
- J. J. Beltran, C. A. Barrero and A. Punnoose, Phys. Chem. Chem. Phys., 2019, 21, 8808–8819, 10.1039/C9CP01277J.
- J. P. Wang, Z. Y. Wang, B. B. Huang, Y. D. Ma, Y. Y. Liu, X. Y. Qin, X. Y. Zhang and Y. Dai, ACS Appl. Mater. Interfaces, 2012, 4, 4024–4030 CrossRef CAS PubMed.
- C. Shuai, X. Yuan, Y. Shuai, G. Qian, J. Yao, W. Xu, S. Peng and W. Yang, Mater. Today Nano, 2022, 18, 100210 CrossRef CAS.
- I. M. P. Silva, G. Byzynski, C. Ribeiro and E. Longo, J. Mol. Catal. A: Chem., 2016, 417, 89–100 CrossRef CAS.
- Y. L. Pang and A. Z. Abdullah, Chem. Eng. J., 2013, 214, 129–138 CrossRef CAS.
- S. Cho, J.-W. Jang, K.-j. Kong, E. S. Kim, K.-H. Lee and J. S. Lee, Adv. Funct. Mater., 2013, 23, 2348–2356 CrossRef CAS.
- L. Ramqvist, K. Hamrin, G. Johansson, A. Fahlman and C. Nordling, J. Phys. Chem. Solids, 1969, 30, 1835 CrossRef CAS.
- V. Kiran and S. Sampath, ACS Appl. Mater. Interfaces, 2012, 4, 3818–3828 CrossRef CAS PubMed.
- S. Muthulingam, K. Bin Bae, R. Khan, I.-H. Lee and P. Uthirakumar, RSC Adv., 2015, 5, 46247, 10.1039/c5ra07811c.
- Y. Li, B. P. Zhang, J. X. Zhao, Z. H. Ge, X. K. Zhao and L. Zou, Appl. Surf. Sci., 2013, 279, 367–373 CrossRef CAS.
- M. Kaur, V. Kumar, P. Kaur and R. Sharma, Mater. Technol., 2022, 37(9), 1156–1167, DOI:10.1080/10667857.2021.1926777.
- R. Patino-Herrera, R. Catarino-Centeno and M. M. Robles-Martinez,
et al.
, Powder Technol., 2018, 327, 381–391 CrossRef CAS.
- E. Prabakaran and K. Pillay, RSC Adv., 2019, 9, 7509–7535 RSC.
- Y. Li and C. Liao, Int. J. Mol. Sci., 2020, 21, 8836 CrossRef CAS PubMed.
- Z. Yu, L. C. Yin, Y. Xie, G. Liu, X. Ma and H. M. Cheng, J. Colloid Interface Sci., 2013, 400, 18–23 CrossRef CAS PubMed.
- R. Gupta, N. Krishnarao Eswar, J. M. Modak and G. Madras, RSC Adv., 2016, 6, 85675–85687 RSC.
- C. Thambiliyagodage and L. Usgoda, Curr. Res. Green Sustainable Chem., 2021, 4, 100186 CrossRef CAS.
- C. Di Valentin, E. Finazzi and G. Pacchioni,
et al.
, Chem. Phys., 2007, 339(1–3), 44–56, DOI:10.1016/J.CHEMPHYS.2007.07.020.
- S. A. Ansari, S. G. Ansari, H. Foaud and M. H. Cho, New J. Chem., 2017, 41, 9314, 10.1039/c6nj04070e.
- A. Yushau and U. I. Gaya, J. Phys. Chem. Funct. Mater., 2023, 6(1), 1–17 Search PubMed.
- T. D. Veal, I. Mahboob, L. Piper, C. F. McConville and M. Hopkinson, Appl. Phys. Lett., 2004, 85, 1550–1552 CrossRef CAS.
- L. Motelica, B.-S. Vasile, A. Ficai, A.-V. Surdu, D. Ficai, Q.-C. Oprea, E. Andronescu, D. C. Jinga and A. M. Holban, Pharmaceutics, 2022, 14, 2842 CrossRef CAS PubMed.
- H. Osman, Z. Su, X. Ma, S. Liu, X. Liu and D. Abduwayit, Ceram. Int., 2016, 42, 10237–10241 CrossRef CAS.
- R. C. Pawar, Y. Son, Y. J. Kim, S. H. Ahn and C. S. Lee, Curr. Appl Phys., 2018, 16, 101–108 CrossRef.
- Y. Zhang, J. Zhou, X. Chen, Q. Feng and W. Cai, J. Alloys Compd., 2018, 777, 109–118, DOI:10.1016/j.jallcom.2018.10.383.
- M. A. Mohamed, M. F. M. Zain, L. J. Minggu, M. B. Kassim, J. Jaafar, N. A. S. Amin, M. S. Mastuli, H. Wu, R. J. Wong and Y. H. Ng, J. Ind. Eng. Chem., 2019, 77, 393–407, DOI:10.1016/j.jiec.2019.05.003.
- Y. Li, S. Cao, A. Zhang, C. Zhang, T. Qu, Y. Zhao and A. Chen, Appl. Surf. Sci., 2018, 445, 350–358, DOI:10.1016/j.apsusc.2018.03.187.
- S. Sharma, S. K. Mehta and S. K. Kansal, J. Alloys Compd., 2017, 699, 323–333, DOI:10.1016/j.jallcom.2016.12.408.
- A. S. Alshammari, L. Chi, X. Chen, A. Bagabas, N. Rashidi-Alavijeh, D. Kramer, A. Alromaeh and Z. Jiang, RSC Adv., 2015, 5, 27690, 10.1039/b000000x.
- S. Y. Sawant and M. H. Cho, RSC Adv., 2016, 6, 70644–70652 RSC.
- J.-J. Xu, Y.-N. Lu, F.-F. Tao, P.-F. Liang and P.-A. Zhang, ACS Omega, 2023, 8, 7845–7857 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Ajay Kumar
Surela
,
Lata Kumari
Chhachhia
,
Jugmohan
Meena
and
Rohitash
Meena
*,
Ajay Kumar
Surela
,
Lata Kumari
Chhachhia
,
Jugmohan
Meena
and
Rohitash
Meena