
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTetrahedral DNA framework-directed hybridization chain reaction controlled self-assembly†
Dongdong
He
ac,
Pengyao
Wei
ac,
Lin
Li
a,
Pan
Fu
a,
Jianping
Zheng
*ab and
Kaizhe
Wang
*ab
aNingbo Key Laboratory of Biomedical Imaging Probe Materials and Technology, Ningbo Cixi Institute of Biomedical Engineering, Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, Ningbo, 315300, P. R. China. E-mail: zhengjianping@nimte.ac.cn; wangkaizhe@nimte.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049, P. R. China
cCixi Biomedical Research Institute, Wenzhou Medical University, Ningbo, 315300, P. R. China
First published on 21st January 2025
Abstract
Nonenzymatic isothermal nucleic acid self-assembly techniques (e.g., the hybridization chain reaction, HCR) hold potential in building materials and biological sensing. However, a traditional HCR is triggered by the random diffusion and disordered conformations of ssDNA initiators, resulting in asynchronous initiation and inherently highly heterogeneous products that do not meet the standards of well-defined nanomaterials. Herein, we developed a nanomechanical restricted strategy directed by tetrahedral DNA frameworks (TDFs) to control HCR self-assembly. We found that the restricted initiator at TDF vertices could induce DNA hairpin assembly to form homogeneous products in solution. Mechanistically, we found that TDFs accelerated the strand displacement rate of the starting H1 and synchronized the assembly process of the HCR. Furthermore, the TDF exhibited strict vertex specificity for HCR controllable assembly, and side extension of the initiator could not result in homogeneous products. This work presents a straightforward and efficient approach for controlling the living self-assembly of macromolecular DNA, thus providing a novel tool for HCR-based nanomanufacturing and quantitative sensing applications.
In nature, organisms guide molecular building blocks through specific physicochemical microenvironments to undergo ordered assembly, leading to the construction of functional higher-order structures to execute their intricate functionality.1–6 Inspired by biological systems, controlled self-assembly in vitro can provide a route to understanding the mechanism of biological processes while offering promising prospects for the development of precise nanomanufacturing with sophisticated functions.7,8 For example, Nils and coworkers recently achieved control of supramolecular polymerization processes by simulating crowding effects in biological systems.9 Unlike traditional assembly control, there is no need to change external parameters such as solvent or temperature; moreover, more product morphologies can be obtained by restricting the molecular state. In the field of supramolecular assembly, DNA self-assembly technology is a prominent example. Owing to their unique predictable and programmable properties, DNA molecules have been used for bottom-up assembly of various complex functional materials.10–16 In particular, the hybridization chain reaction (HCR), a type of toehold-mediated strand displacement reaction, involves the addition of DNA initiator strands under mild conditions, leading to the alternating hybridization of two metastable complementary DNA hairpins.17 With its enzyme-free nature, isothermal conditions, and simple protocols, the HCR has been a popular toolbox in biosensing,18,19 bioimaging,20,21 DNA computing,22 and nanodevice construction.23,24 However, in traditional HCR systems, the uncontrolled free state of initiator strands in solution leads to asynchronous initiation and extension reactions, ultimately causing self-assembled products to exhibit unpredictable high dispersity, thereby preventing formation of a well-defined nanomaterial (Scheme 1A).
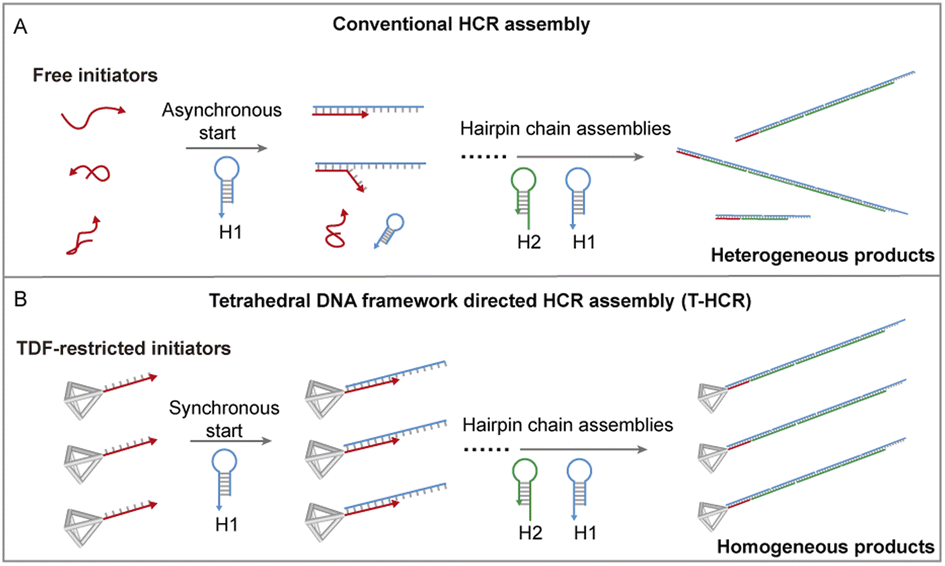 | ||
| Scheme 1 Schematic of TDF-directed HCR controlled self-assembly. Traditionally, HCR initiators in solution vary in their degree of curling, affecting the rate at which the initiator H1 opens and leads to asynchronous assembly processes and resulting in heterogeneous products. In TDF-directed HCR systems, the TDF vertex restricts initiators and maintains these initiators in an extended state, synchronously accelerating the opening of H1. This synchronization extends to subsequent propagation reactions, producing homogeneous products. | ||
In recent years, numerous strategies have been employed to control HCRs. Some cationic polymers, such as poly(L-lysine)-graft-dextran (PLL-g-Dex), have been introduced to accelerate both the initiation and subsequent amplification rates of the HCR.25 Furthermore, Li designed a metastable domain region on the initiator strand H2, adjusting the number of spacer bases between H1 and the metastable domain region to accelerate both initiation and propagation rates of the HCR.26 In addition, the initiator or hairpin strands have been fixed on DNA origami or magnetic columns, increasing their local concentration and thereby promoting reaction rates.27,28 However, although these strategies enhance the reaction rates of the HCR, they do not effectively reduce the production of heterogeneous products. To address this issue, Mirkin et al. introduced mismatched sequences on the initiator and hairpin strands to regulate the initiation and propagation rate, thereby achieving homogeneous self-assembled products.29 However, this method is only suitable for high-concentration reactions (micromolar level) and requires a long reaction time (∼16 h). More importantly, the introduction of mismatched bases limits the availability of HCR sequences. Therefore, controlling HCR polymerization through a simple and efficient method remains a significant challenge.
The rapid development of DNA nanotechnology offers a precise method for creating addressable DNA nanostructures, enabling a thorough understanding and manipulation of DNA hybridization from a nanomechanical perspective.30,31 In particular, tetrahedral DNA frameworks (TDFs) have been reported to improve the recognition hybridization of their vertex extension ssDNA with their complementary strand.32–34 Herein, we constructed a TDF nanotrigger to control HCR oligomerization (Scheme 1B). We found that the TDF restricts via vertex-specific initiators and synchronously initiates and accelerates HCR polymerization, resulting in uniform and homogeneous self-assembled products.
We hypothesize that the TDF-triggered HCR can produce uniform-length self-assembling products (Fig. 1A). Initially, we designed a tetrahedral DNA vertex-modified HCR initiator strand, termed TDF-In (Fig. S1†). TDF-In was self-assembled by four single-stranded DNA components (A−C, and D-In in Table S1†) as detected using polyacrylamide gel electrophoresis. The morphology of TDF-In was then confirmed through AFM imaging, which demonstrated a characteristic tetrahedral framework structure. Zeta potential analysis indicated that the synthetic TDF-In exhibits a strong negative charge, further confirming its successful assembly (Fig. S1†). To investigate the characteristics of HCR products induced by TDF-In, we incubated TDF-In/In with the hairpin chains H1 and H2 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio (200 mM:1000 mM) in PBS/Na+ (100 mM Na+) at 22 °C for 1 hour. Gel electrophoresis analysis using 2% agarose gel showed dispersed bands in the conventional HCR. Interestingly, T-HCR products exhibited relatively aggregated bands and clustered around 250 bp, which was close to the ideal expected product size (275 bp) (Fig. 1B). AFM imaging demonstrated that HCR products comprise dot-like short chains and variably sized linear chains, with lengths approximately averaging at 103.7 ± 78.85 nm, while T-HCR products primarily comprised uniform-sized linear chains, with lengths approximately averaging at 115.2 ± 22.52 nm (Fig. 1C and D). To summarize, these results indicate that the TDF can control HCR oligomerization to produce homogeneous self-assembled products.
:5 ratio (200 mM:1000 mM) in PBS/Na+ (100 mM Na+) at 22 °C for 1 hour. Gel electrophoresis analysis using 2% agarose gel showed dispersed bands in the conventional HCR. Interestingly, T-HCR products exhibited relatively aggregated bands and clustered around 250 bp, which was close to the ideal expected product size (275 bp) (Fig. 1B). AFM imaging demonstrated that HCR products comprise dot-like short chains and variably sized linear chains, with lengths approximately averaging at 103.7 ± 78.85 nm, while T-HCR products primarily comprised uniform-sized linear chains, with lengths approximately averaging at 115.2 ± 22.52 nm (Fig. 1C and D). To summarize, these results indicate that the TDF can control HCR oligomerization to produce homogeneous self-assembled products.
 | ||
| Fig. 1 Homogeneity analysis of HCR/T-HCR products. (A) Schematic of the T-HCR. (B) Agarose gel (2.5%) electrophoresis of HCR and T-HCR products. (C) AFM images of HCR and T-HCR products. Scale bar = 200 nm. (D and E) The length frequency distribution of HCR and T-HCR products. | ||
Previous research has shown that TDFs can facilitate the hybridization of ssDNA with complementary strands owing to the framework effect and electrostatic repulsion.32,33 To investigate whether TDF vertices can accelerate the H1 strand displacement reaction initiating the formation of DNA duplex structures, we conducted fluorescence quenching experiments. The stem-loop region of the H1 hairpin was labeled with a Cy3 fluorescent group (H1-Cy3), and the 5′ end of the initiator chain was modified with a quenching group black hole quencher (In-BHQ/TDF-In-BHQ) (Fig. 2A). We observed that the fluorescence quenching half-life of the TDF-restricted group was approximately 56 seconds, which was about five times faster than that (212 s) of the In-free group (Fig. 2B and C). These results indicate that the TDFs successfully accelerate the HCR initial strand displacement reaction rate.
 | ||
| Fig. 2 The hybrid kinetic of the T-HCR. (A) Schematic of kinetic quenching experiments. (B) Patterns of fluorescence intensity decay over time in the HCR and T-HCR. (C) The half-life of fluorescence decay in the HCR and T-HCR. Data are represented as mean ± SD. | ||
To investigate the influence of temperature on the TDF-directed HCR, we conducted T-HCR experiments at various temperatures. T-HCRs (TDF-In: H1:H2 was 1:5) were performed at 4 °C, 22 °C, 37 °C, and 45 °C for 1 hour, followed by characterization of assembly products using 2% agarose gel electrophoresis. As depicted in Fig. 3A, at 4 °C, 22 °C, and 37 °C, T-HCR assembly products showed aggregated states, with increasing product sizes observed as the temperature rose. However, at 45 °C, the electrophoretic bands of T-HCR products exhibited pronounced dispersion. Further grayscale analysis revealed that the reaction efficiencies of the T-HCR were 59% and 72% at 4 °C and 45 °C, respectively. In contrast, the reaction efficiencies of the T-HCR were 83% and 84% at 22 °C and 37 °C, indicating that these temperatures promoted the assembly of the HCR (Fig. S2†). The traditional HCR exhibited the same trends (Fig. S2†). These findings suggest that within a certain temperature range, increasing temperature accelerates T-HCRs, whereas excessively high or low temperatures reduce assembly efficiency. The T-HCR demonstrates optimal controllable assembly efficiency at 22 °C.
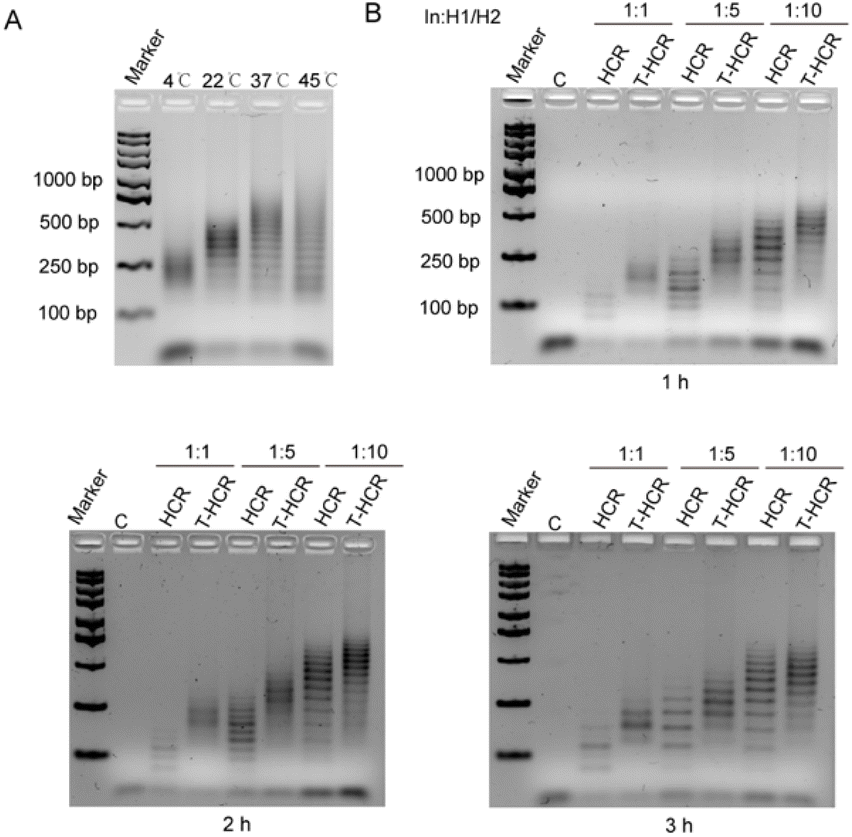 | ||
| Fig. 3 T-HCR under different conditions. (A) Agarose gel (2%) for T-HCR products in the characterization of different temperatures. The initiator and hairpin strands (TDF-In: H1:H2 was 1:5) were incubated in PBS/Na+ (Na+ 100 mM) buffer for 1 h. (B) Agarose gel (2%) characterization of the reaction products of the HCR and T-HCR with different initiator chain ratios and reaction times. Lane C served as a control, which only had hairpin H1 and H2 strands and no initiator strands. Lanes 1, 3, and 5 were HCRs corresponding to initiator to hairpin chain ratios of 1:1, 1:2.5, and 1:5, respectively. Lanes 2, 4, and 6 are T-HCRs corresponding to initiator to hairpin chain ratios of 1:1, 1:2.5, and 1:5, respectively. The corresponding reaction times were 1 h, 2 h, and 3 h, respectively. | ||
For the traditional HCR, increasing the initiator-to-monomer ratio enhances the average molecular weight of self-assembled products and is accompanied with an increase in oligomer dispersion. To investigate whether the introduction of the TDF can generate homogeneous products under varying initiator-to-monomer ratios, we targeted an HCR product with DPn of 2, 5, and 10. We conducted reactions at 22 °C in PBS/Na+ (100 mM Na+) solution with an initiator chain concentration of 20 nM for different durations. As shown in Fig. 3B, as the initiator-to-monomer ratio increases, traditional HCR and T-HCR products exhibit noticeable upward shifts in gel electrophoresis bands, indicating a positive correlation between the product size and initiator-to-monomer ratio. Interestingly, under different initiator-to-monomer ratios, T-HCR products consistently display fewer bands on electrophoresis gels than traditional HCR products, suggesting that the T-HCR can generate homogeneous products even at high initiator-to-monomer ratios. With an increase in the reaction time, the number of bands in T-HCR groups slightly increased, but the products remained homogeneous. In addition, we observed that high sodium ion concentrations promote the heterogeneous of T-HCR products (Fig. S3†), possibly owing to the strong screen electrostatic forces between the TDF and initiators.32
To explore the effect of the positioning of the initiator strand in TDFs on mediating the HCR response, based on the precise addressing capability inherent in TDFs, we designed TDF nanostructures with initiator chains extended from the vertices or edges of the tetrahedron (position 13, TDF13-In) (Fig. 4A). Gel electrophoresis analysis indicated that TDF13-In migrated slower compared to TDF-In, confirming the successful synthesis of its structure (Fig. 4B). Subsequently, agarose gel characterization demonstrated that assemblies induced by TDF13-In and TDF-In. TDF13-HCR products exhibited heterogeneity (Fig. 4C). Furthermore, the reaction efficiency of the T13-HCR was significantly lower (70%) than that of the T-HCR (87%) (Fig. S4†). The distinct differences in the HCRs may be attributed to spatial hindrance introduced by edge-modified initiator chains on the tetrahedron, which can lead to reduced efficiency in interacting with hairpin strands. These findings highlight the vertex-specific of controlled HCRs induced by TDFs.
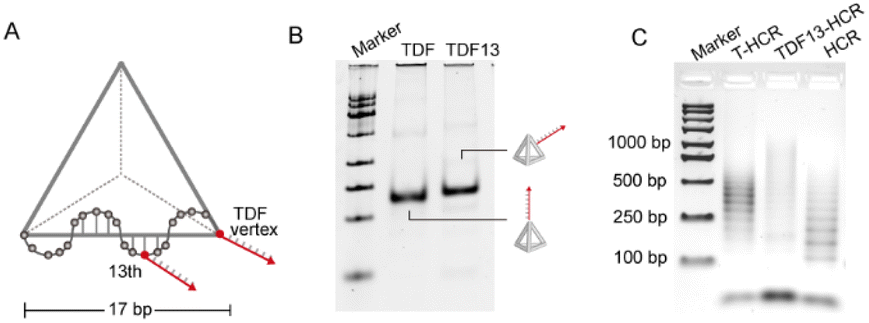 | ||
| Fig. 4 Modified initiator in different positions on the TDF. (A) Schematic of the modifiable sites of the initiator on the TDF edge. (B) Polyacrylamide gel (6%) electrophoresis (PAGE) image of TDFs and TDFs-13. (C) Agarose gel (2%) characterization of reaction products triggered by modification of initiators at different sites on TDFs. | ||
In this study, we employed tetrahedral DNA frameworks as restricted nanotriggers to control HCR self-assembled living polymerization, achieving homogeneous product control. This strategy is easily implemented and avoids complex design. This nanomechanical-restricted DNA polymerization provides insights into understanding molecular assembly mechanisms and lays the groundwork for creating well-defined materials that can be used in quantitative biosensing, drug delivery, and other applications.
Data availability
The data supporting this article have been included as part of the ESI†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Ningbo Science and Technological Innovation Yongjiang 2035 Major Project (2024Z183), the National Natural Science Foundation of China (No. U24A20377), and the Ningbo Yongjiang Talent Introduction Programme (No. 2023A-114-G).References
- J. A. Marsh, H. Hernandez, Z. Hall, S. E. Ahnert, T. Perica, C. V. Robinson and S. A. Teichmann, Cell, 2013, 153, 461–470 CrossRef CAS PubMed.
- S. Bhatia and J. B. Udgaonkar, Chem. Rev., 2022, 122, 8911–8935 CrossRef CAS PubMed.
- J. Janowski, V. A. B. Pham, S. Vecchioni, K. Woloszyn, B. Lu, Y. Zou, B. Erkalo, L. Perren, J. Rueb, J. Madnick, C. Mao, M. Saito, Y. P. Ohayon, N. Jonoska and R. Sha, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2321992121 CrossRef CAS PubMed.
- C. Jiang, B. Lu, W. Zhang, Y. P. Ohayon, F. Feng, S. Li, N. C. Seeman and S. J. Xiao, J. Am. Chem. Soc., 2022, 144, 6759–6769 CrossRef CAS PubMed.
- B. Lu, Y. P. Ohayon, K. Woloszyn, C. F. Yang, J. B. Yoder, L. J. Rothschild, S. J. Wind, W. A. Hendrickson, C. Mao, N. C. Seeman, J. W. Canary, R. Sha and S. Vecchioni, J. Am. Chem. Soc., 2023, 145, 17945–17953 CrossRef CAS PubMed.
- B. Lu, K. Woloszyn, Y. P. Ohayon, B. Yang, C. Zhang, C. Mao, N. C. Seeman, S. Vecchioni and R. Sha, Angew Chem. Int. Ed. Engl., 2023, 62, e202213451 CrossRef CAS PubMed.
- H. Zhang, Y. Wang, H. Zhang, X. Liu, A. Lee, Q. Huang, F. Wang, J. Chao, H. Liu, J. Li, J. Shi, X. Zuo, L. Wang, L. Wang, X. Cao, C. Bustamante, Z. Tian and C. Fan, Nat. Commun., 2019, 10, 1006 CrossRef CAS PubMed.
- O. Kimchi, C. P. Goodrich, A. Courbet, A. I. Curatolo, N. B. Woodall, D. Baker and M. P. Brenner, Sci. Adv., 2020, 6(51), eabc1939 CrossRef CAS PubMed.
- N. Baumer, E. Castellanos, B. Soberats and G. Fernandez, Nat. Commun., 2023, 14, 1084 CrossRef PubMed.
- P. W. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- N. Akter, B. S. Alladin-Mustan, Y. Liu, J. An and J. M. Gibbs, J. Am. Chem. Soc., 2024, 146, 18205–18209 CrossRef CAS PubMed.
- B. Lu, S. Vecchioni, Y. P. Ohayon, J. W. Canary and R. Sha, Biophys. J., 2022, 121, 4759–4765 CrossRef CAS PubMed.
- S. Vecchioni, R. Lo, Q. Huang, K. Wang, Y. P. Ohayon, R. Sha, L. J. Rothschild and S. J. Wind, Small, 2024, e2407604 Search PubMed.
- S. Vecchioni, B. Lu, W. Livernois, Y. P. Ohayon, J. B. Yoder, C. F. Yang, K. Woloszyn, W. Bernfeld, M. P. Anantram, J. W. Canary, W. A. Hendrickson, L. J. Rothschild, C. Mao, S. J. Wind, N. C. Seeman and R. Sha, Adv. Mater., 2023, 35, e2210938 CrossRef PubMed.
- T. Wang, T. Bai, Z. Tan, Y. P. Ohayon, R. Sha, S. Vecchioni, N. C. Seeman and B. Wei, J. Am. Chem. Soc., 2023, 145, 2455–2460 CrossRef CAS PubMed.
- K. Woloszyn, S. Vecchioni, Y. P. Ohayon, B. Lu, Y. Ma, Q. Huang, E. Zhu, D. Chernovolenko, T. Markus, N. Jonoska, C. Mao, N. C. Seeman and R. Sha, Adv. Mater., 2022, 34, e2206876 CrossRef PubMed.
- R. M. Dirks and N. A. Pierce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15275–15278 CrossRef CAS PubMed.
- J. Huang, Y. Wu, Y. Chen, Z. Zhu, X. Yang, C. J. Yang, K. Wang and W. Tan, Angew Chem. Int. Ed. Engl., 2011, 50, 401–404 CrossRef CAS PubMed.
- X. Qiu, P. Wang and Z. Cao, Biosens. Bioelectron., 2014, 60, 351–357 CrossRef CAS PubMed.
- X. Liu, D. Mao, Y. Song, L. Zhu, A. N. Isak, C. Lu, G. Deng, F. Chen, F. Sun, Y. Yang, X. Zhu and W. Tan, Sci. Adv., 2022, 8, eabk0133 CrossRef CAS PubMed.
- D. Cao, X. Qin, W. Wang, Y. Zhang, S. Peng, H. Gong, Q. Luo and J. Yang, ACS Nano, 2024, 18, 618–629 CrossRef CAS PubMed.
- D. Y. Zhang and G. Seelig, Nat. Chem., 2011, 3, 103–113 CrossRef CAS PubMed.
- S. Venkataraman, R. M. Dirks, P. W. Rothemund, E. Winfree and N. A. Pierce, Nat. Nanotechnol., 2007, 2, 490–494 Search PubMed.
- K. Chen, M. Mao, L. Huo, G. Wang, Z. Pu and Y. Zhang, ACS Appl. Mater. Interfaces, 2024, 16, 29760–29769 Search PubMed.
- J. Wang, N. Shimada and A. Maruyama, ACS Appl. Mater. Interfaces, 2022, 14, 39396–39403 Search PubMed.
- L. Zhang, Y. Fu, Y. Tong, G. Xie and S. Deng, Nano Lett., 2024, 24, 2603–2610 CrossRef CAS PubMed.
- R. Wang, Z. Yin, J. Yang, X. Yang, Z. Tang and G. Mustafa, J. Math., 2021, 2021, 1–6 Search PubMed.
- H. Bui, S. Shah, R. Mokhtar, T. Song, S. Garg and J. Reif, ACS Nano, 2018, 12, 1146–1155 CrossRef CAS PubMed.
- C. A. Figg, P. H. Winegar, O. G. Hayes and C. A. Mirkin, J. Am. Chem. Soc., 2020, 142, 8596–8601 Search PubMed.
- T. Zhai, Q. Li, J. Shen, J. Li and C. Fan, Aggregate, 2020, 1, 107–116 Search PubMed.
- Y. Yang, H. Lu, D. Fang, Y. Zhang, Y. Tang, S. Zhao, J. Yan, X. Qin, J. Shen and F. Yang, Aggregate, 2024, e636 CrossRef CAS.
- Z. Qu, Y. Zhang, Z. Dai, Y. Hao, Y. Zhang, J. Shen, F. Wang, Q. Li, C. Fan and X. Liu, Angew Chem. Int. Ed. Engl., 2021, 60, 16693–16699 CrossRef CAS PubMed.
- F. Li, X. Mao, F. Li, M. Li, J. Shen, Z. Ge, C. Fan and X. Zuo, J. Am. Chem. Soc., 2020, 142, 9975–9981 CrossRef CAS PubMed.
- M. Lin, J. Wang, G. Zhou, J. Wang, N. Wu, J. Lu, J. Gao, X. Chen, J. Shi, X. Zuo and C. Fan, Angew Chem. Int. Ed. Engl., 2015, 54, 2151–2155 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00912f |
| This journal is © The Royal Society of Chemistry 2025 |