
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrothermal carbonization synthesis of amorphous carbon nanoparticles (15–150 nm) with fine-tuning of the size, bulk order, and the consequent impact on antioxidant and photothermal properties†
Francesco Barbero *a,
Elena Destroa,
Aurora Belloneb,
Ludovica Di Lorenzoa,
Valentina Brunellaa,
Guido Perroneb,
Alessandro Damina and
Ivana Fenoglioa
*a,
Elena Destroa,
Aurora Belloneb,
Ludovica Di Lorenzoa,
Valentina Brunellaa,
Guido Perroneb,
Alessandro Damina and
Ivana Fenoglioa
aDepartment of Chemistry, University of Torino, Torino, Italy. E-mail: francesco.barbero@unito.it
bDepartment of Electronics and Telecommunications, Politecnico di Torino, Torino, Italy
First published on 8th January 2025
Abstract
Hydrothermal carbonization (HTC) of carbohydrates has been reported as a sustainable and green technique to produce carbonaceous micro- and nano-materials. These materials have been developed for several applications, including catalysis, separation science, metal ion adsorption and nanomedicine. Carbon nanoparticles (CNPs) obtained through HTC are particularly interesting for the latter application since they exhibit photothermal properties when irradiated with near-infrared (NIR) light, act as an antioxidant by scavenging reactive oxygen species (ROS), and present good colloidal stability and biocompatibility. However, due to the highly disordered structure, there is still a poor understanding of the mechanism of synthesis of CNPs. Consequently, the modulation of the CNP properties by controlling the synthetic parameters is still a challenge. In this work, a novel and simplified HTC synthetic strategy to obtain non-aggregated glucose derived CNPs in the 15–150 nm size range with precise control of the diameter is presented, together with an advance in the understanding of the reaction mechanism behind the synthesis. Modifications of the synthetic parameters and a post-synthesis hydrothermal process were applied to increase the bulk order of CNPs, resulting in an increase of the photothermal and ROS scavenging activities, without affecting the morphological and colloidal properties of the nanomaterial.
1. Introduction
Carbon nanomaterials are a large family of different materials, including carbon nanotubes,1 graphene,2 carbon quantum dots,3 nanodiamonds,4 carbon black5 and fullerenes.6 They have attracted continuous interest due to their wide current and potential applications. In the last twenty years, increasingly greater attention has also been paid to the use of carbohydrates or carbohydrate-rich biomass to produce carbonaceous microparticles and, more recently, nanoparticles (CNPs), due to their broad potential applications and derived economic, environmental and societal issues.7 To synthesise CNPs, among different techniques, the hydrothermal carbonization (HTC) of biomass, a traditional but recently revived method, is the most effective, simple, and intriguing one and is a promising route with broad potential applications.7–9 HTC of polysaccharides is considered a low cost, sustainable and green technique to produce nanomaterials (NMs) because carbohydrates and carbohydrate-rich biomass are renewable and highly abundant precursors, the use of water as solvent reduces pollution and the hydrothermal process employed proceeds under relatively low reaction temperatures (normally less than 200 °C).10 CNPs derived from HTC of carbohydrates, like other carbonaceous particles (soot, carbon black, etc.), are predominantly composed of amorphous carbon. However, they may exhibit graphitic patches with variable abundance. On the other hand, in contrast to combustion derived carbonaceous particles like soot or carbon black that are produced as powder, CNPs are synthesized as colloids, and can be produced in high yield as spherical monodisperse particles in water.11These NMs have several potential applications such as sorbents for heavy metals,12 composites for energy storage,13 catalysis or catalyst supports,14,15 photocatalysis,16 electrode materials17 and sacrificial templates.18 Several studies suggest a possible use also in medicine as nanoplatforms for different diagnostic and/or therapeutic applications, including photoacoustic imaging,19 photothermal therapy,20,21 photodynamic therapy,11 antioxidants,11 drug delivery,20,22 and antimicrobial applications.23
A primary target when synthesising CNPs is to ensure that they are monodisperse and that their size can be accurately controlled.9 Strict control of these parameters and the production of particles with diameters less than 150 nm are prerequisites for many applications.24
Controlling CNP properties is also of primary importance in medical applications since it is well known that there is a strong correlation between the physicochemical properties of NMs, in particular size, aggregation state, shape, surface and bulk chemistry, and their interactions with biological systems.25,26 In particular, the particle size strongly influences their biodistribution, since it modulates cellular internalization, barrier crossing, and excretion from the body.27 In the case of CNPs, the possibility of controlling their bulk structure would open the way to the design of CNPs with optimized photothermal and redox properties.
Sevilla et al.28 work and several following studies9,11,29 clearly showed that the parameters affecting the HTC synthesis of CNPs using carbohydrates as carbon sources are mainly five: (i) precursor molecule/s; (ii) additives; (iii) synthesis temperature; (iv) precursor concentration; and (v) synthesis time. These parameters were shown to affect the shape, mean size, size distribution, aggregation state, bulk structure and reaction yield of the synthesized CNPs. The most common morphology of CNPs is micro/nano-spheres, while the other cited characteristics can vary significantly depending on the synthesis parameters.9 Spherical morphology is typical of polymeric nanoparticles,30 since this shape corresponds to the minimum state of energy. This is in line with the previously reported formation mechanism of CNPs, claiming that the steps preceding the formation of CNPs is the polycondensation of polyfuranic nanostructures.9,31
Many studies focused on the control of CNP size and size distribution and good control and fine-tuning were achieved for diameters greater than 400 nm thanks to the addition of sodium polyacrylate as the dispersing agent.29 With the use of poly(sodium-p-styrenesulfonate) as the additive, CNPs with a mean diameter of 70 nm were obtained by the HTC of glucose and fructose but without the possibility of tuning the size and forming particle aggregates in the dispersion.32 The addition of poly(ionic liquid)s allowed the production of particles with a size tuneable from 28 to 110 nm; however, these CNPs were strongly interconnected.33 To the best of our knowledge, the smallest mean hydrodynamic diameter reported for non-interconnected and non-aggregated CNPs is around 130 nm.11
Few studies have reported on the effect of synthetic parameters on the resulting bulk structure. CNPs with different bulk structures were obtained by modifying the precursor molecule.28 However, the reason for this effect was not clarified and the correlation between CNP bulk chemistry and their applicative properties is not studied yet. Moreover, the possibility of tuning the bulk structure without affecting the particle aggregation state has never been reported.
In the present study, a simplified one-pot hydrothermal synthesis was developed to produce stable colloidal suspensions of monodisperse CNPs in the hydrodynamic diameter range of 15–150 nm. The synthetic parameters were modified to tune the CNP bulk structure and, in turn, their photothermal and antioxidant properties.
2. Materials and methods
2.1 Chemicals
D-(+)-Glucose, 30% (w/w) hydrogen peroxide, iron(II) sulfate heptahydrate, and phosphate buffered saline powder were obtained from Sigma-Aldrich (Steinheim, Germany). 5,5-Dimethyl-1-pyrroline-N-oxide (DMPO) from Enzo Life Science (Farmingdale, NY, USA). Ultrapure water was obtained from a Millipore Milli-Q system.2.2 CNP synthesis
All the CNPs were synthesised heating aqueous solutions of glucose (50 mL) in a Teflon-lined stainless-steel autoclave (100 mL, Büchi AG) at 190 °C, in a pre-heated oven. The initial glucose concentrations and synthesis times are reported in Table 1. After the synthesis, CNP dispersions were purified with ultrapure water by tangential flow filtration (Sartorius Vivaflow® 50R, MW 30 kDa).| Precursor | Time of reaction | |||||||
|---|---|---|---|---|---|---|---|---|
| Glucose (M) | 1 h | 3 h | 4 h | 4.5 h | 5 h | 6 h | 9 h | 15 h |
| 0.022 | x | x | x | x | ||||
| 0.22 | x | x | x | x | x | x | ||
| 2.2 | x | |||||||
2.3 Gravimetric analysis
After the purification step, the CNP concentration was measured by gravimetric analysis. 3 mL of CNP suspension were transferred into a pre-weighed vial, weighed and dried in a lab oven at 70 °C, and then the CNP mass was measured. All weighings have been carried out using an analytical balance.2.4 Electron microscopy
A droplet of the suspension was added to a TEM holey carbon grid (copper mesh support). Once the solvent evaporated, the material remained on the grid for analysis. TEM was carried out using a Tecnai F30 microscope (FEI Company) at a working voltage of 300 kV. TEM images were obtained with a coupled CCD camera (Gatan). Field emission scanning electron microscopy was carried out on a FESEM TESCAN S9000G equipped with a Schottky-type field emission gun (FED) operating at 5 keV. Size distribution analyses were performed using Fiji-ImageJ software. At least 150 particles from different regions of the grid were counted.2.5 Light scattering techniques
The hydrodynamic diameter (DH) of CNPs was determined by dynamic light scattering (DLS) and by nanoparticle tracking analysis (NTA). DLS measurements were performed using a Malvern Zetasizer Nano ZS instrument (Malvern Panalytical Ltd, UK), equipped with a light source wavelength of 632.8 nm with the following settings: measurement angle = 173 °C, temperature = 25 °C and equilibration time = 120 s. The parameters used for the carbon materials were as follows: refractive index = 2.420 and absorption = 1.00. The parameters used for the water dispersant were as follows: refractive index = 1.330; viscosity (25 °C) = 0.8872 cP; and dielectric constant = 78.5. Measurements were performed diluting pristine CNPs in ultrapure water at concentrations that led the instrument to automatically select an attenuator between 6 and 9. The results were expressed as mean values of 3 independent measurements and the diameters were reported as distribution by intensity calculated by non-negative least squares analysis. NTA was performed using a ZetaView® PMX-120 (Particle Metrix GmbH, Germany) instrument, equipped with a light source wavelength of 488 nm and a 90° laser scattering video microscope with 10× magnification. Before the measurements, the pristine samples were diluted in double filtered ultrapure water to have around 150 NPs measured per frame. After the optimization of the instrumental parameters, the sensitivity and the shutter were set at 80 and 100, respectively; 3 × 33 videos of 1 second for each sample were recorded analysing a total of ∼2500 NPs per measurement.ζ-Potentials were measured using an electrophoretic light scattering analyser (Zetasizer Nano ZS, Malvern). The samples were diluted to have an optimal scattering intensity.
2.6 Vibrational spectroscopy
Raman spectra of CNP dry powders were collected in backscattering configuration by using a Renishaw Raman Microscope spectrometer. An Ar+ laser emitting at 514 nm was used as the exciting source, limiting the final power on the sample to 0.029 mW in order to avoid sample damage; note that at higher power, variations in I(G)/I(D) were observed over time. A laser beam has been focused on the sample through a 50× objective (NA = 0.75) and an 1800 L mm−1 dispersive grating has been adopted to analyse the backscattered light, previously cleaned with Rayleigh light through the adoption of an edge filter. Each registered Raman spectrum consisted of 20 acquisitions, each with a duration of 10 s. Data are reported as the average of three measurements performed at different points. The baseline was corrected using Origin software with a rigid baseline set between 1950 cm−1 and 800 cm−1.FT-IR spectra were recorded using a PerkinElmer Spectrum Two FT-IR spectrometer equipped with a universal attenuated total reflection (UATR) module performing 16 scans per sample.
2.7 Evaluation of the photo-thermal properties
The evaluation of the photothermal properties was conducted by exposing the CNP dispersions to a collimated laser beam at 915 nm. The setup consisted of a cuvette holder from Thorlabs (CVH100), to which a beam expander was attached to ensure unchanged distances, thus beam size, and thus reproducibility of measurement conditions. The cuvette contained 2 mL of dispersion at 0.85 mg mL−1.The laser beam was generated by a laser diode at a wavelength of 915 nm and then expanded with a combination of cylindrical lenses to a diameter of 23 mm. The power density delivered to the sample in the cuvette was 2.4 W cm−2. The tests were conducted three times under the same conditions for each sample; the duration of the laser exposure was 10 minutes. The photothermal effect involved an increase of temperature of the sample, monitored with an optical sensor based on the fibre Bragg grating technology. Optical fibre sensors offer many advantages in the application described in this work, in particular the capability to not interact with the measured system, altering the results. The fabrication and the calibration are described in ref. 34 and 35.
2.8 Scavenging activity toward hydroxyl radicals
The scavenging activity of CNPs towards hydroxyl radicals was evaluated using the electronic paramagnetic resonance (EPR)/spin trapping technique. To 0.2 mL of CNP suspension, the following were successively added under stirring in the dark: (i) 0.1 mL phosphate buffer saline (PBS), (ii) 0.25 mL of a solution of 5,5-dimethyl-1-pyrroline N-oxide (DMPO) in water, (iii) 0.08 mL of a solution of FeSO4 and (iv) the reaction was started by adding 0.1 mL of a solution of H2O2. The final concentrations were as follows: CNPs, 6.5 × 1016 nm2 mL−1; PBS, 1.4 mM; DMPO, 62 mM; FeSO4, 0.5 mM; H2O2, 10.5 mM. EPR spectra were collected using a Miniscope MS100, Magnettech (Berlin, Germany). The gain was set at 500. EPR spectra were recorded for the CNP suspensions and in the absence of NPs (negative control) after 15, 30, 45 and 60 minutes. The experiments have been repeated almost three times. The data in Fig. 8 represent the average of different experiments after 60 minutes of incubation, while ESI Fig. S9† reports the trend over time of the average values and the standard deviation of the 3305 G peak intensities of the different experiments.3. Results and discussion
3.1 Effect of precursor concentration and synthesis time on the size of CNPs
First, the role of precursor concentration and synthesis time was systematically investigated. As one of the most common and abundant monosaccharides, glucose was chosen as the carbohydrate precursor.9 Biomass is a precursor cheaper than pure saccharides; however, the use of pure glucose increases the reproducibility of the synthesis and facilitates the understanding of the mechanism of carbonization, simplifying the HTC process since the initial hydrolytic steps occurring with complex carbohydrates are skipped. Moreover, the synthesis was performed without any addition of additives with the aim of obtaining a simple, one-pot synthesis. The synthesis temperature was fixed at 190 °C (typical for HTC), while the precursor concentration and reaction time have been varied one by one with a systematic approach, to understand their effect on the resulting nanomaterial. In Table 1, the different parameters used are illustrated.Morphological analysis of the resulting CNPs was performed by Transmission Electron Microscopy (TEM) and Field Emission Scanning Electron Microscopy (FESEM) (Fig. 1). Under all synthetic conditions, nanospheres were obtained, in line with previous studies.9 Interestingly, decreasing the precursor concentration seems to bring about a rougher surface and less perfect spherical morphology (ESI Fig. S1†). Electron microscopy (EM) analysis evidenced a strong dependency of size on the synthetic conditions. The results suggest that increasing the synthesis time or the precursor concentration increases the mean CNP size (Fig. 1), similarly to what was previously reported for the synthesis in the presence of polyacrylate with larger diameters.11 Light scattering techniques evidenced a slightly larger mean diameter and diameter range (Fig. 1; Table 2; ESI Table S1†). This is expected since TEM measures the geometrical diameter on the dry sample, while DLS and NTA measure the hydrodynamic diameters (DH) in colloidal suspension. Moreover, DLS is known to overestimate large particles due to their higher intensity of the scattered light.36 The size distributions evaluated by EM were close enough to those obtained by DLS and NTA to prove that the particles in water dispersion were mainly formed from single spherical CNPs and not aggregates or agglomerates (Fig. 1; Table 2; ESI Table S1†), with the exception of CNPs prepared at the higher synthesis times tested (2.2 M, 3 h; 0.22 M, 6 h; 0.022 M, 15 h), as inferred by the discrepancy between EM and light scattering techniques. The reproducibility of the synthesis and the stability of the colloidal suspension were also monitored by DLS, for the latter case by repeating the measurement on a sample stored for 1 year at room temperature. The results demonstrate very good stability and reproducibility (ESI Fig. S2A–C†).
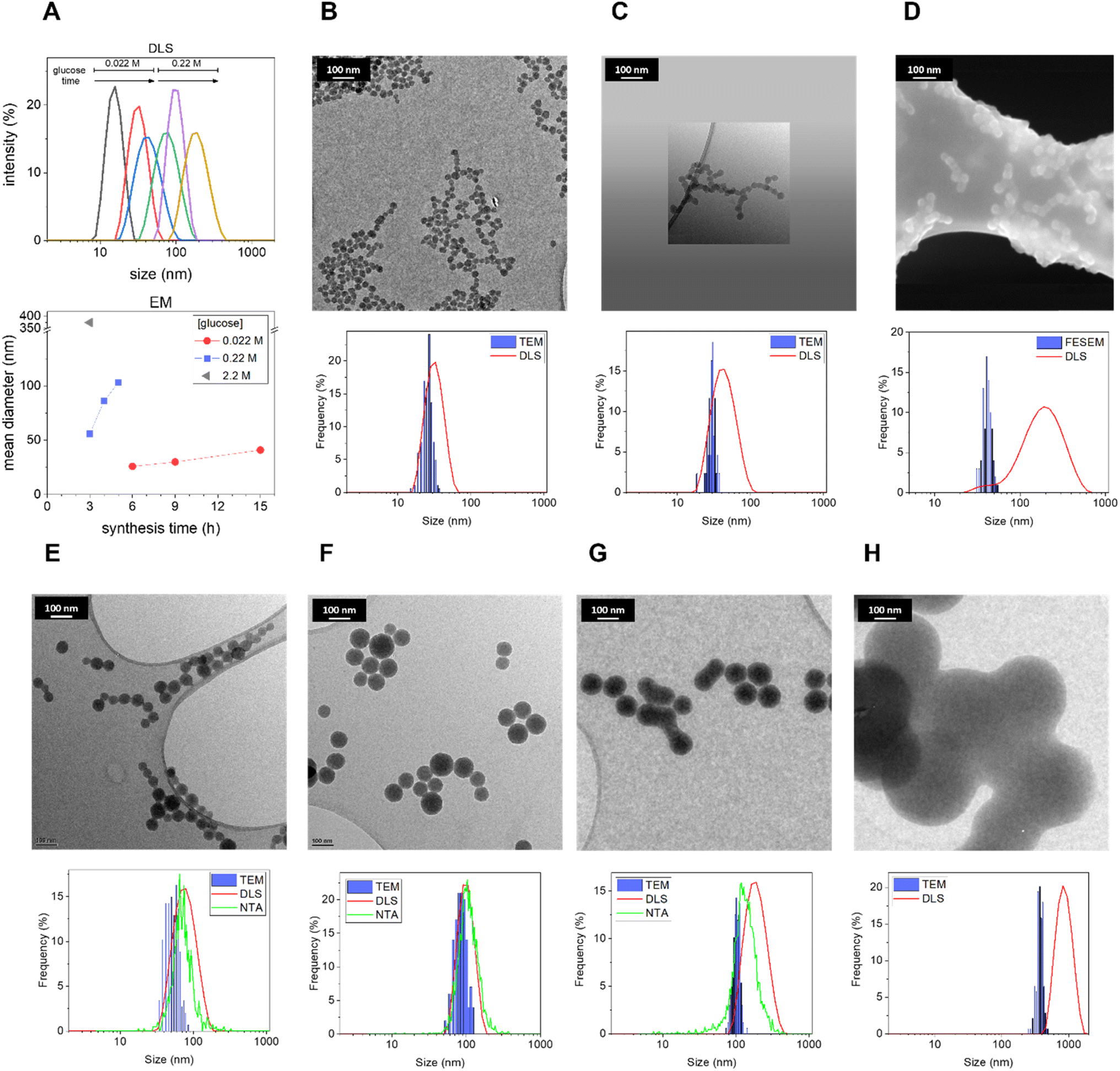 | ||
| Fig. 1 Effect of the synthetic parameters on the CNP size distribution. (A) – upper graph – DLS size distribution (by intensity) of CNP dispersions obtained at different concentrations and synthesis times (from the smallest diameter: glucose, 0.022 M – synthesis time, 3 h (black); 0.022 M – 6 h (red); 0.022 M – 9 h (blue); 0.22 M – 3 h (green); 0.22 M – 4 h (violet); 0.22 M – 5 h (mustard)); (A) – bottom graph – CNP mean diameters determined by EM as a function of initial glucose concentration (0.022 M red circles, 0.22 M blue squares, and 2.2 M grey triangle) and synthesis time (temperature fixed at 190 °C). (B–H) TEM and FESEM images, DLS and NTA measurements of CNPs synthesised at different precursor concentrations and synthesis times. (B–D) glucose, 0.022 M: (B) synthesis time, 6 h, (C) 9 h, (D) and 15 h; (E–G) glucose, 0.22 M: (E) 3 h; (F) 4 h; (G) 5 h; (H) glucose 2.2 M – 3 h. The hydrodynamic diameter evaluated by DLS is expressed as the relative percentage in intensity. DLS and NTA were performed on suspensions of CNPs in water. | ||
| Size (nm) | 0.022 M glucose | 0.22 M | 2.2 M | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 3 h | 6 h | 9 h | 15 h | 1 h | 3 h | 4 h | 5 h | 6 h | 3 h | |
| a The values refer to the mean size of the distribution and the standard deviation refers to the width of the distribution.b After 1 h of synthesis no evidence of particle formation was observed in any of the above used techniques. | ||||||||||
| EM | nd | 26 ± 4a | 30 ± 3 | 41 ± 5 | b | 53 ± 11 | 86 ± 16 | 103 ± 11 | nd | 374 ± 40 |
| DLS | 16 ± 4 | 33 ± 9 | 45 ± 16 | 164 ± 62 | b | 80 ± 27 | 101 ± 24 | 195 ± 64 | 672 ± 195 | 830 ± 233 |
| NTA | <LOD | <LOD | <LOD | <LOD | b | 80 ± 33 | 92 ± 30 | 142 ± 54 | 175 ± 76 | nd |
These results show that small variations in the synthetic conditions allowed the fine-tuning of the size of non-aggregated CNPs with a resolution in the order of a few nanometers in a mean DH range between ∼15 and 150 nm (Fig. 1; Table 2), in the absence of any stabilizer. Note that after 1 h of synthesis no evidence of CNP formation with any of the used techniques using glucose 0.22 M was observed (Table 2), and just a decrease of pH from 6.6 to 4 was observed (ESI Fig. 3A†), an indication of the formation of organic acids, as previously reported.28 The yield of the reaction increases with the increase of the synthesis time. A minimum yield of 2.3% up to 21.2% (with respect to the initial precursor concentration) was measured by gravimetric analysis (ESI Fig. 3B–D†). The concentration of particles increases with the concentration of the precursor, with values from 0.1 mg mL−1 up to 28.1 mg mL−1 (ESI Fig. 3B–D†).
The kinetics of formation of particles appears to be relatively slow, on the time scale of hours, and it is modulated by the precursor concentration. For example, particles obtained after 5 h with 0.22 M glucose showed a DH of around 140 nm, while with 0.022 M glucose in 9 h just a DH of 45 nm was observed (Table 2).
Overall, the results presented herein demonstrate the possibility of finely tuning the size of CNPs without the use of any dispersant. The precursor was always in excess under all conditions tested allowing the growth to be interrupted at different time points by decreasing the temperature in order to choose the particle size of interest. The relationship between time and particle size strongly suggests that further varying the synthesis parameters with values in between the ones tested is possible to further fine-tune the size of CNPs.
3.2 Surface properties of CNPs
ζ-Potential analysis of the obtained colloidal dispersions was performed to get information on the CNP surface chemistry, since they are strongly correlated,37 and on the colloidal stabilization mechanism (Fig. 2).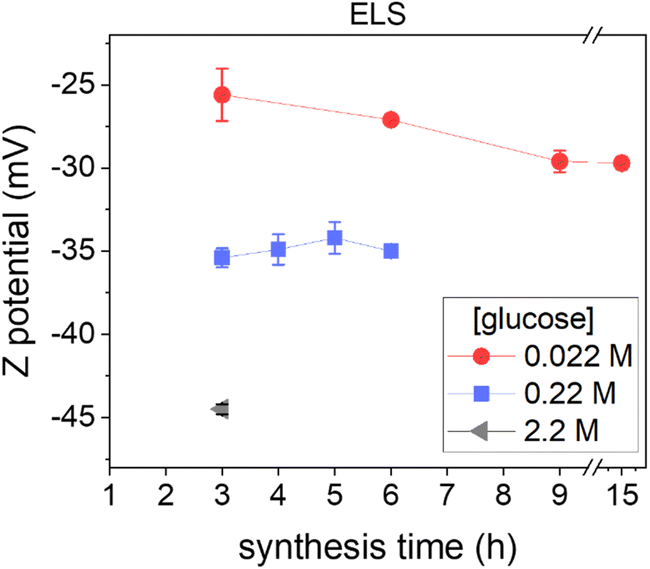 | ||
| Fig. 2 Effect of the synthetic parameters on CNP ζ-potential. ζ-potential of CNPs synthesized starting from 0.022 M (red circles), 0.22 M (blue squares) and 2.2 M glucose solutions (grey triangle) at the different synthesis times tested, all measured at pH ∼4. | ||
ζ-Potential measurements in all cases showed negative values in a range between −25 and −45 mV (measured at pH ∼4), in line with the presence of hydrophilic negatively charged groups on the NP surface, likely acidic carboxylic or phenolic groups, as previously reported for this type of NP.11 Another piece of evidence of acidic moieties on the CNP surface was the acidic pH values (∼4) of the dispersion obtained, which remain constant no matter how many purification steps were performed (ESI Fig. S3A†), discarding an effect due to soluble secondary products. Note that, as reported above, pH analysis indicates the formation of organic acids during the synthesis process, even before particle formation (ESI Fig. S3A†). The ζ-potential (measured at the same pH and conductivity) becomes more negative by increasing the precursor concentration (Fig. 2), suggesting a higher acidity of the surface groups or a higher surface density of charged moieties.38 In contrast, the ζ-potential did not change significantly as the synthesis time increased (Fig. 2).
The highly negative ζ-potential well explains the hydrophilicity and the colloidal stability of CNPs: according to the DLVO, a high ζ-potential provides electrostatic stabilization to the NPs.39,40 However, in the present case when the conductivity of the dispersant was increased by NaCl (0.137 M corresponding to ∼15 mS cm−1), the CNPs did not lose their colloidal stability (ESI Fig. S4†) even if the ζ-potential, as expected, dropped to −13.0 ± 0.9 mV, due to the increase of ion concentration, suggesting the presence of a steric stabilizing component that prevented the aggregation of the NPs. In fact, a ζ-potential larger than approximately ±30 mV is needed to sustain colloidal stability in the case of electrostatic stabilization,39,40 while steric stabilization is independent of the ionic concentration.40 It could be speculated that, in addition to the moieties responsible for the negative charge, the CNP surface possesses a roughness due, for example, to hydrophilic polymeric-like organic chains responsible for the observed steric component of stabilization.
To reveal the degree of hydrophilicity, CNPs were dried and suspended in solvents with different polarity. CNPs formed stable colloids in acetone or dichloromethane, while in n-hexane the particles tended to aggregate as evidenced by visual observation (ESI Fig. S5†). These results indicate that these particles possess a peculiar amphiphilic nature that makes them different from other carbon nanomaterials synthesized by HTC like for example carbon quantum dots3 that can be precipitated just by adding ethanol.
The amphiphilic nature and the electro-steric stabilization in water are properties exploitable for all applications that need media change or for the manufacture of composites in which CNPs need to be embedded in matrices of different polarity.
3.3 Tuning the bulk structure of CNPs
The bulk structure of CNPs was first investigated by Raman spectroscopy. Raman spectroscopy is widely used to characterize crystalline, nanocrystalline, and amorphous carbons and provide information on the degree of order and crystallinity of carbonaceous materials.41,42 Disordered graphite and also amorphous carbons exhibit two main Raman modes, the G (from “graphene”) peak between 1520 and 1600 cm−1 and the D (from “defect”) peak around 1350 cm−1.41Fig. 3A reports the normalized (with respect to the G band) and baseline-subtracted Raman spectra of the CNP obtained after 3 h of synthesis at the three different precursor concentrations tested. As expected, the profiles are those of carbonaceous materials containing sp2 sites, in all cases presenting G and D modes, albeit in different proportions. The G peaks of all three samples were found centred at around 1580 cm−1. Regarding the D bands, decreasing the initial concentration of glucose increases the D band intensity and consequently the I(D)/I(G) ratio.
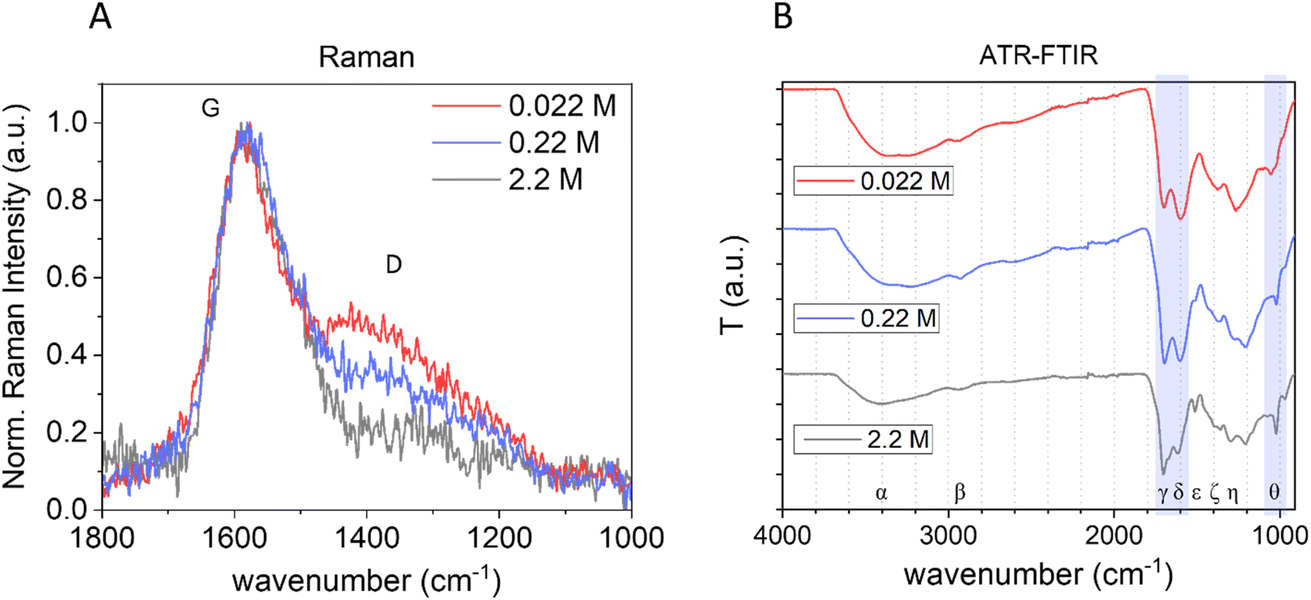 | ||
| Fig. 3 Effect of the precursor concentration on the bulk structure of CNPs. (A) Normalized baseline-subtracted Raman spectra of CNPs synthesized starting from 0.022 M (red), 0.22 M (blue) and 2.2 M (grey) glucose aqueous solutions with a synthesis time of 3 h at 190 °C. (B) ATR-FTIR of the same samples (Greek letters refer to band assignation, see Table 3). | ||
The G mode occurs at all sp2 sites, even in olefinic molecules, not only in the presence of sixfold aromatic rings.41 Oppositely, the D mode intensity depends exclusively on the presence of sixfold aromatic rings and at the same time is forbidden in perfect graphite, becoming permitted only if disorder is present.41 The intensity ratio between D and G peaks is known to vary inversely with the graphitic cluster diameter, i.e., I(D)/I(G) will grow with increasing disorder (Tuinstra and Koenig relationship).43 However, as the disorder further increases, clusters become more distorted, smaller and fewer in number. As the G peak is just associated with C sp2 atoms, I(D) will now decrease with respect to I(G) and the Tuinstra and Koenig relationship will not be valid anymore. It has been reported that for graphitic clusters with diameters smaller than ∼20 Å, the D band intensity is proportional to the probability of finding a sixfold ring in the cluster, which in turn is proportional to the area of the cluster. Consequently, in amorphous carbons the growth of a D peak indicates ordering, exactly opposite of what occurs in the case of graphite.41 The D band can be deconvoluted in several other modes;44 however, the well-established relationship between G and D bands for disordered and amorphous carbon described by Ferrari et al.41 gives, for the purpose of this work, the needed information. It is well defined that CNP bulk is formed mainly by amorphous carbon with the presence of graphitic patches,11 consequently in terms of Raman behaviours it follows the second relationship, described above, and therefore the observed increase of the I(D)/I(G) ratio with decreasing precursor concentration (Fig. 3A) is an indication of an increase of order. This is further supported by the decrease of the photoluminescence (PL) background along with the decrease of the precursor concentration in raw Raman spectra of the same samples (ESI Fig. S6†).45 In fact, a decrease of PL was observed for carbonaceous nanoparticles with increasing graphitization grade obtained by annealing.11 Moreover, for partially hydrogenated amorphous carbon materials, the increasing Raman PL background was found to be a typical signature of the increasing saturation, due to recombination of electron–hole pairs within sp2 bonded clusters in a sp3 bonded amorphous matrix.46,47
The chemical composition of the three samples produced at different precursor concentrations was further investigated by Attenuated Total Reflection Fourier Transform Infrared Spectroscopy (ATR-FTIR) (Fig. 3B). In all samples, several functional groups containing C, H and O were present, as reported in other studies. The main bands were assigned based on the literature16,28,48–53 and are summarized in Table 3.
| α | Broad band around 3400 cm−1 | Stretching (ν) of O–H (primary, secondary, phenol, and carboxyl) |
| β | 2964 cm−1 | Aliphatic C–H ν |
| γ | Around 1710 cm−1 | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ν (carboxyl, carbonyl, quinone, and ester) O ν (carboxyl, carbonyl, quinone, and ester) |
| δ | Around 1605 cm−1 | CC (six-fold aromatic ring, furanic and olefinic) νa |
| ε | 1513 cm−1 | CC ν |
| ζ | 1380 cm−1 | C–H ν in the carbonyl |
| η | Between 1280 and 1208 cm−1 | C–O–C and C–O (carboxyl) ν |
| θ | 1023 cm−1 | Vibration characteristic of furan |
However, a different intensity ratio of the bands was observed depending upon the synthesis conditions, an indication of chemical structures composed of similar moieties but with different abundances. In particular, a different relative intensity ratio between CO and CC stretching bands (at approx. 1703 and 1605 cm−1, respectively) is observed. As the glucose concentration increases, the CO/CC intensity ratio increases, suggesting that high precursor concentrations promote the formation of carboxylic moieties in line with more negative ζ-potential and/or result in fewer CC moieties ascribable to a less ordered bulk structure observed by Raman. Moreover, this CC band shifts from 1618 to 1604 to 1600 cm−1 decreasing the glucose concentration, ascribable to a more sp2 aromatic carbon contribution.52,53 It is also notable that there is a strong difference in the peak at 1513 cm−1 (CC ν) that disappears with decreasing precursor concentration and a difference in the zone 1280–1208 cm−1, attributed to C–O–C, further indicating different chemical composition. Furthermore, the signal at 1023 cm−1, widely claimed in the literature as typical for the presence of furan, is more predominant in the CNP obtained from higher glucose concentration, likely due to a more polyfuranic structure. Polycondensation of polyfuranic nanostructures was shown to be the step preceding the formation of CNPs and several studies indicate this polymer as the main component of NPs obtained from HTC of carbohydrates.9,31 Therefore, tuning from NPs having mainly a polyfuranic skeleton to amorphous carbon particles with increasing graphitization is possible by modifying the initial precursor concentration.
The effect of the synthesis time on the chemical composition of CNPs was also studied by FTIR and Raman spectroscopy (Fig. 4).
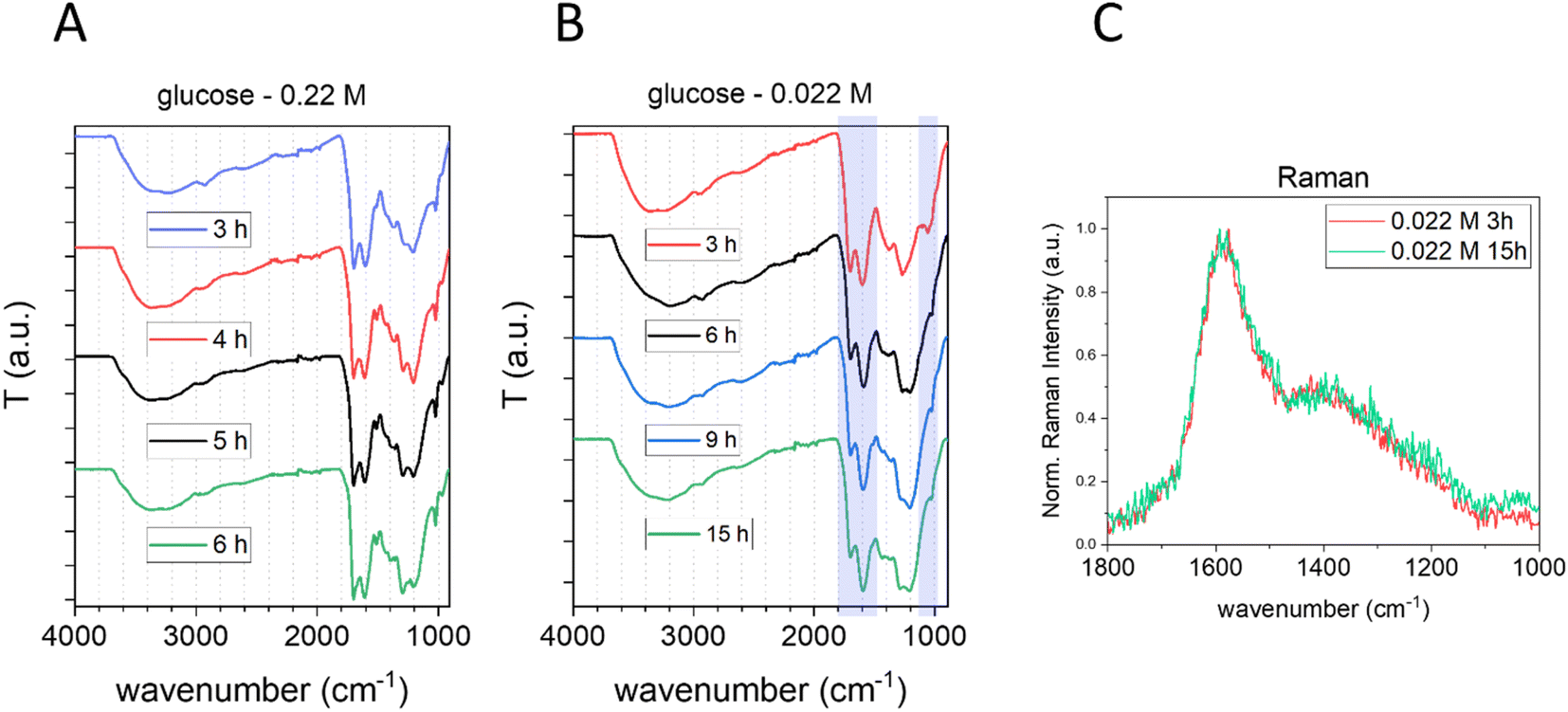 | ||
| Fig. 4 Effect of the time of synthesis on the bulk structure of CNPs. (A and B) ATR-FTIR spectra of CNPs synthesized starting from 0.22 M (A) and 0.022 M (B) glucose at different synthesis times. (C) Normalized baseline-subtracted Raman spectra of CNPs synthesized in 3 h (red) and 15 h (green) starting from 0.022 M glucose. | ||
No significant differences were observed in the sample prepared starting from 0.22 M glucose at different synthesis times (Fig. 4A). In contrast, a slightly more intense CC vibration peak, along with a small decrease of the peak at 1023 cm−1 (typical of furan), was observed increasing the synthesis time for CNPs prepared starting from 0.022 M glucose between 3 h and 6 h, a possible indication of an increase in the carbonization grade (Fig. 4B). However, after 6 h of synthesis no significant differences were observed in the FTIR spectra (Fig. 4B) and Raman analysis of CNPs synthesized in 3 h or 15 h starting from 0.022 M glucose shows no significant differences between the two spectra (Fig. 4C and ESI Fig. S6B†).
Overall, the results obtained from vibrational spectroscopy techniques indicate that the graphitization grade and the chemical composition of CNPs are poorly affected by the synthesis time, especially if compared with the effect of the precursor concentration. However, it has to be taken into account that these are bulk techniques that give an average measurement of the samples; consequently, possible variation of the particle core, which experiences longer residence time, could be not detected. In contrast, vibrational spectroscopy data suggest that the precursor concentration modulates not only the particle size and the surface chemistry, but also the bulk structure of the particles, in particular their carbonization grade.
The results presented so far evidenced that, with this approach, it is not possible to modulate the carbonization degree independently from the glucose concentration and partially from their size. To obtain particles with a higher carbonization degree, the precursor concentration needs to be decreased, which leads to a decrease in the concentration of the synthesized CNPs.
To further modulate the bulk structure, independently from the initial glucose concentration and keeping constant the size and concentration, CNPs synthesized in 3 h with 0.22 M glucose were purified from the remaining precursor/byproducts (by tangential dialysis) and subjected again to a hydrothermal treatment at 190 °C for 24 h. First, the size distribution of the obtained sample was measured by DLS and NTA. Interestingly, the treated CNPs maintained their colloidal stability and presented a very similar diameter and narrow size distribution; just a slight decrease in the size (∼15 nm) was observed by both light scattering techniques and TEM (Fig. 5A and B and ESI Fig. S7B†). Concurrently, the ELS analysis indicated a decrease of ∼10 mV in the re-heated CNP ζ-potential value. Interestingly, a clear increase of I(D)/I(G) was observed in the Raman spectra of the re-heated CNPs (Fig. 5C). Moreover, a shift to a higher wavenumber of the G band (from 1576 cm−1 to 1597 cm−1) was observed, a well-known indication of graphitization.41 Furthermore, the raw Raman spectra of the same samples (ESI Fig. S6C†) clearly show a decrease of the PL background, additional evidence of increased graphitization. The FTIR spectrum (Fig. 5D) clearly indicates a transformation of the chemical structure, with an increase of the contribution of CC (around 1600 cm−1) and with a shift of this band from 1604 cm−1 to 1582 cm−1, an indication of a more sp2 aromatic carbon contribution.52,53 Both results are in line with the result obtained by Raman spectroscopy. Furthermore, a substantial reduction of the 1023 cm−1 vibration (typical of furan), the disappearance of the 1513 cm−1 band (CC ν) and the appearance of a pronounced band peaking at 1225 cm−1 (C–O–C) are observed. As observed above, the 1023 cm−1 band is more prominent in less graphitic CNPs, hypothesized to be an indication of a more polyfuranic structure, and consequently its strong reduction is another piece of evidence of more advanced carbonization. Moreover, the C–O–C zone undergoes an evolution with an increase of ether moieties (as inferred by the higher intensity) and ether species (as inferred by the presence of a broad single band). Note that the FTIR of the same CNP re-heated for a shorter time (6 h) showed similar results, but with less pronounced variation (ESI Fig. S7A†).
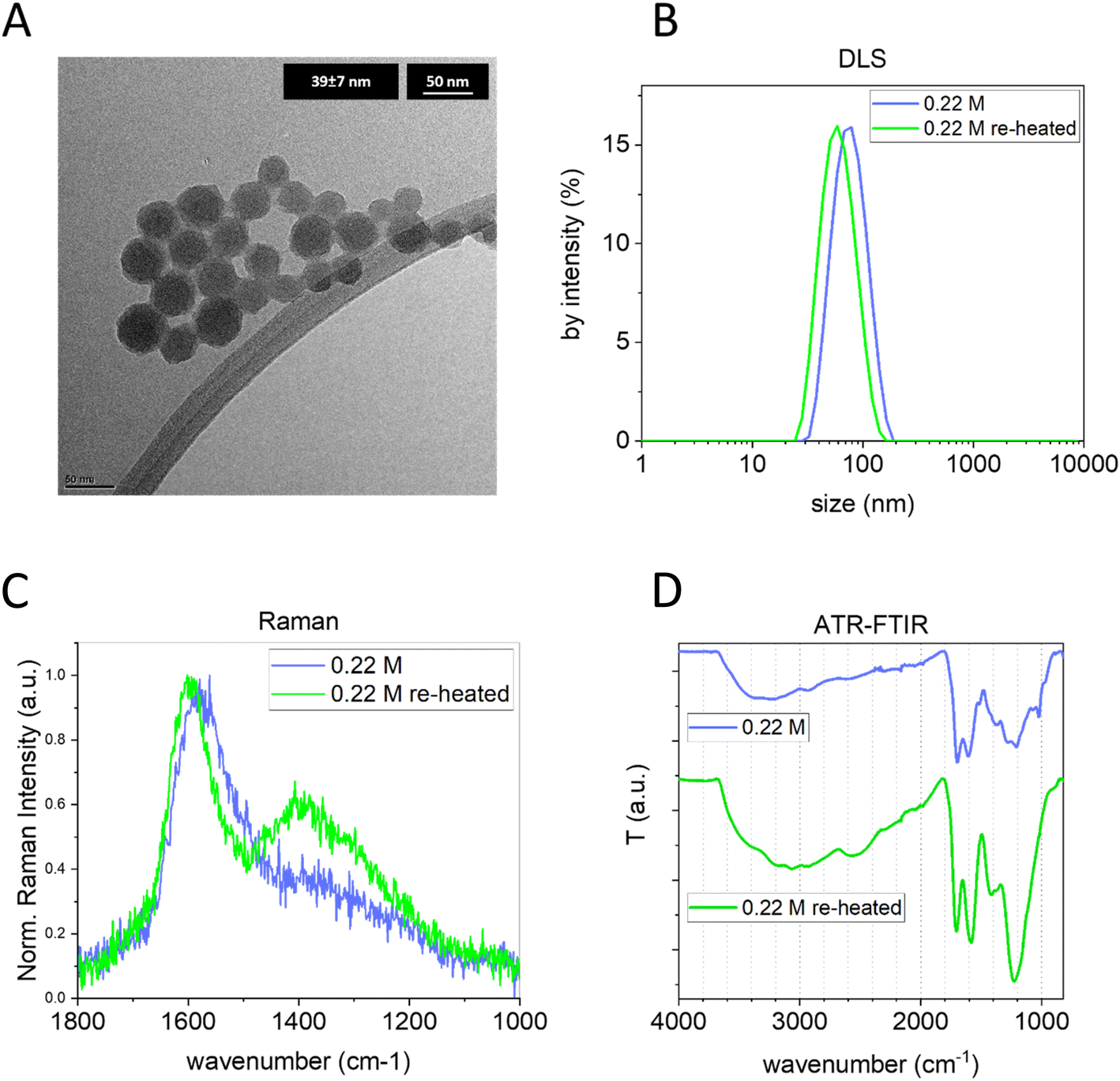 | ||
| Fig. 5 (A) TEM images of CNPs (0.22 M – 3h) purified and after hydrothermal treatment at 190 °C for 24 h. (B) DLS, (C) Raman, and (D) ATR-FTIR of CNPs before and after hydrothermal treatment at 190 °C for 24 h (both samples were purified by 30 kDa tangential dialysis). | ||
These results highlight the possibility of modulating the bulk structure of pre-synthesized CNPs, increasing their graphitization and, unlike the commonly used annealing process, performing this procedure in aqueous dispersion at relatively low temperature without having particle aggregation and retaining their narrow size distribution.
3.4 Mechanism of CNP formation
There is not a general consensus on the mechanism of formation of hydrothermal CNPs. The most accepted formation process for CNPs is the “LaMer model”,9,54 namely, the concepts of “instantaneous” nucleation and “diffusion-controlled” growth of the nucleus until all the monomers are consumed. Conversely, Jung et al.55 and Qi et al.48 claimed that, after the particle nucleation, the CNP growth results from the primary particle coalescence/aggregation.To shed light on these discrepancies, the particle concentration of the dispersions obtained at the different time points starting from 0.22 M glucose was quantified by NTA (Fig. 6A, blue squares).
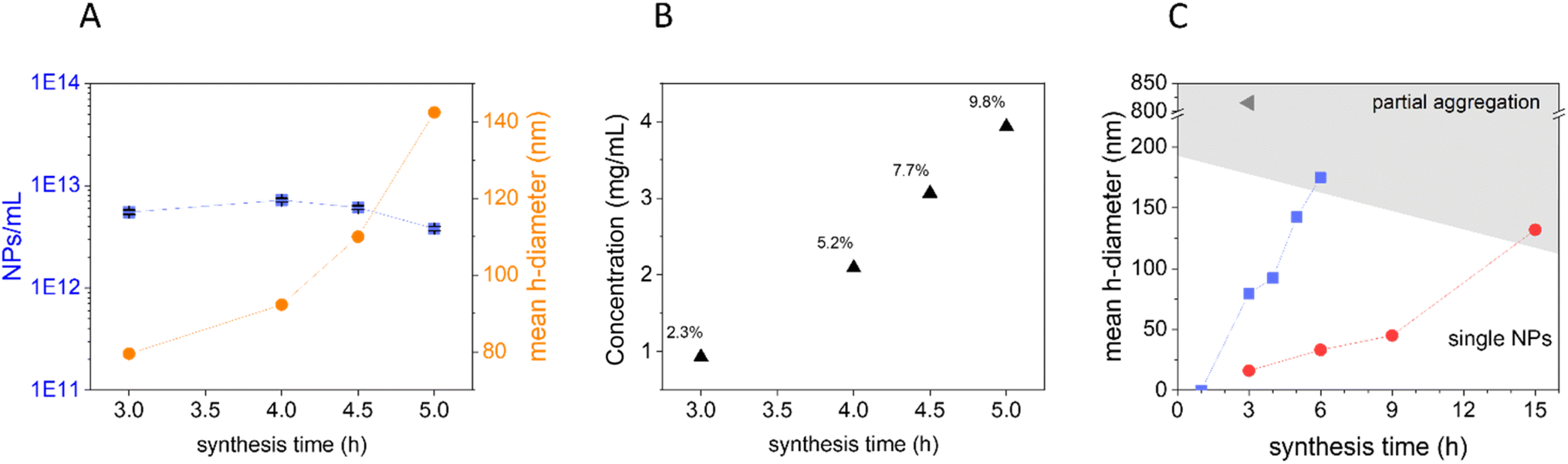 | ||
| Fig. 6 CNP formation process. (A) Concentration of particles in NPs per mL (blue squares) and mean DH (orange circles) of CNPs synthesised starting from 0.22 M glucose solution at different synthesis times, measured by NTA, and concentration values reported are the average of 3 measurements. (B) Yield of reaction of CNPs prepared starting with 0.22 M glucose solution. (C) CNP mean DH (DLS < 50 nm, NTA > 50 nm) as a function of initial glucose concentration (0.022 M red circles, 0.22 M blue squares, and 2.2 M grey triangle) and synthesis time. | ||
Interestingly, as the synthesis time and in turn the particle size increased, the concentration of CNPs remained almost constant, a strong indication of a seeded growth mechanism where at the beginning of the synthetic process small particles are formed and subsequently on these nuclei new material is deposited increasing the initial size. This observation is in line with the measured yield of the reaction that increases with the increase of the synthesis time and without reaching 100% (Fig. 6B). These results suggest that under these conditions, the growth process is not driven by the coalescence/aggregation of the previously formed particles. Instead, it resembles the LaMer model, which conceptualizes two distinct stages, the particle nucleation and their subsequent growth. According to the LaMer mechanism, a rapid nucleation occurs, which significantly reduces the concentration of free monomers in the solution, thus inhibiting further nucleation. Following this, particle growth proceeds as a result of the diffusion of the remaining monomers through the solution.56 However, although the mechanism is similar, in our case the reaction yields are low (ranging from 2.3% to 21.2%). Therefore, the mechanism involving nucleation inhibition due to a significant reduction in precursor concentration, which in turn favours nucleus growth, in our case, is likely to be excluded.
The process appears to be quite slow with kinetics depending upon the precursor concentrations. In fact, at equal synthesis time, lower glucose concentration leads to smaller CNPs (Fig. 6C). As shown above, the variation of the glucose concentration also has a remarkable effect on the graphitization degree and on the ζ-potential of the particles: low precursor concentration led to a more ordered bulk structure (Fig. 3) and less negative ζ-potential (Fig. 2). It could be speculated that the slower particle growth kinetics observed upon decreasing the precursor concentration allows the formation of a more ordered carbon framework and a less defective surface.
In the previous paragraph, it was shown that the increase of the residence time during the synthesis did not lead to a strong modification of the average bulk structure, while re-heating at the same temperature pre-synthesized CNPs (purified from the precursor/byproducts) had a notable effect on the bulk structure, basically increasing the carbonization degree. This suggests that the CNP bulk structure evolves with increasing the residence time at the synthesis temperature. It could be hypothesised that, in the growing process, which follows a seeded growth mechanism, the new layers deposited on already formed CNPs are composed of amorphous carbon with chemical characteristics that depend upon the precursor concentration. In the meantime, the CNP core experiences a longer residence time and it likely evolves to a more ordered structure. However, the increase of the particle volume is probably faster than the graphitization process; for instance, the volume of CNPs synthesized from 0.22 M glucose between 3 h and 4 h increases by ∼80%, while differences in the average bulk chemistry in purified pre-synthesized CNPs were observed after several hours. Ultimately, the CNPs synthesized at different times (at an equal glucose concentration) present an overall similar carbon framework order, indicating that essentially there is no impact of the residence time on the average bulk chemistry while particles are growing due to the deposition of new layers.
3.5 Photothermal properties
In previous studies, the ability of CNPs to generate heat once irradiated with NIR light has been demonstrated.11 Being also biocompatible,21,57 CNPs appear to be promising agents for photothermal therapy (PTT).21 PT conversion arises as a result of vibrational relaxation processes (photon to phonon conversion).58 To improve the PT conversion, high light absorption and good thermal conductivity are required so that the photon energy can be effectively converted into heat,59 along with inhibition of light emission (PL), intersystem crossing and a reduction of scattered light.60,61 It is reported that in amorphous carbon, the thermal conductivity is greatly affected by phonon scattering on grain boundaries or by disorder62 and that carbon black particles with higher crystallinity possess a more efficient PT conversion once illuminated with NIR light in water.11Since the bulk structure of the CNPs can be tuned using the synthetic parameters, tuning of the PT properties is also expected. The correlation between graphitic orders in CNPs and their PT activity in water dispersion under NIR laser beam irradiation (915 nm diode laser) was investigated. CNPs presenting different carbonization grades, prepared starting from different glucose concentrations, were used in PT experiments. Specifically, CNPs obtained from: (i) 2.2 M glucose in 3 h (mean DH 830 nm); (ii) 0.22 M glucose in 3 h (mean DH 80 nm); (iii) 0.022 M glucose in 15 h (mean DH 164 nm) were first tested. For the PT experiments, all the dispersions were normalized for mass concentration (0.85 mg mL−1). As shown in Fig. 7A, all the samples produced heat release. However, the capability of the samples to generate heat was different and was found to increase directly with the carbonization grade of the CNPs (from a ΔT, measured after 10 minutes of irradiation, of 11.4 ± 0.5 °C to 16.7 ± 0.6 °C). A lower ability to generate heat could be due to a lower efficiency in converting the absorbed energy into heat or a lower adsorption of the light. In particular, light scattering (which increases with the particle size) decreases the adsorption of light, consequently having an effect on the PT activity.61 In the present case, no correlation between size and PT activity was found, although this effect cannot be totally discarded. To distinguish between these two effects, the PT efficiency of CNPs obtained from 0.22 M glucose in 3 h was compared with that of the same sample re-heated for 24 h at 190 °C. In this case, the two samples have a similar size but different graphitization grades (Fig. 5). Note that the re-heated sample had a higher absorption at the laser wavelength used (915 nm) when measured at the same mass concentration (ESI Fig. S8†). Fig. 7B clearly shows that the re-heating significantly increases the ability to generate heat compared to the non-re-heated one (ΔT from 13.3 ± 0.5 °C to 17.3 ± 0.6 °C). Consequently, these results further indicate that the PT ability of CNPs increases with the graphitic degree of the material, which can be correlated with an increase in the NIR light absorption and a higher thermal conductivity, thus confirming what was found previously on other types of carbon nanomaterials.11,63 It is then possible to tune the PT efficiency by modifying the CNP precursor concentration or re-heating pre-synthesised CNPs.
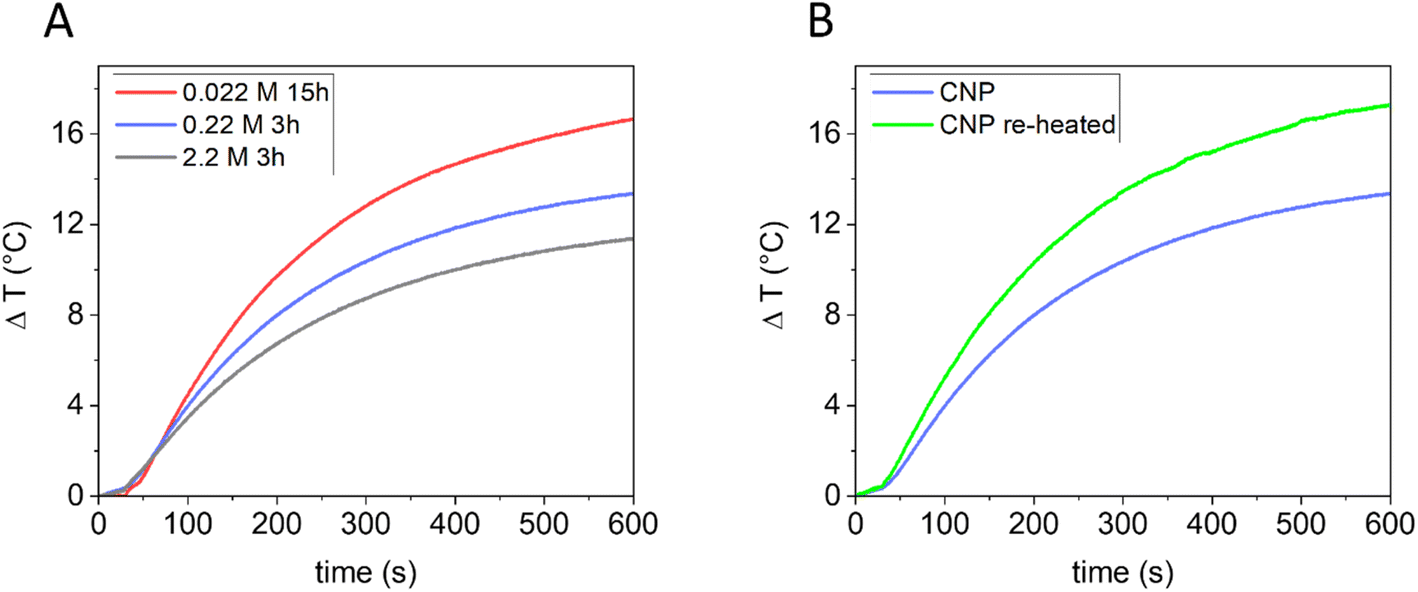 | ||
| Fig. 7 Photothermal activities of CNPs irradiated with NIR light: (A) CNPs obtained as follows: 2.2 M glucose in 3 h (grey); (i) 0.22 M glucose in 3 h (blue); (ii) 0.022 M glucose in 15 h (red). (B) CNPs obtained as follows: from 0.22 M glucose in 3 h (blue) and the same sample subsequently re-heated for 24 h at 190 °C (green). | ||
3.6 Antioxidant properties
Carbon nanomaterials exhibit antioxidant properties due to their ability to scavenge free radicals. This ability has been observed for graphene-based materials,64 fullerenes,65 carbon nanotubes,66 carbon soot and carbon black,67,68 and CNPs.11 These properties are a promising feature in all applications where radical reactions need to be controlled. In the medical field, the ability to scavenge oxygenated radical species like hydroxyl or superoxide radicals makes these materials useful in several oxidative stress-associated disorders, including cancer.58Particularly relevant is the ability of carbon nanomaterials to scavenge hydroxyl radicals, the most aggressive among the reactive oxygen species (ROS). This activity is related to the structural characteristics of carbon nanomaterials.64,69 For disordered carbon nanomaterials like soot and carbon black11,67 and for graphene-based materials,64 the scavenging ability has been shown to increase with the degree of graphitization, suggesting that the radical scavenging is associated with the presence of sp2 carbon domains. We therefore hypothesized that the modulation of the crystallinity through re-heating treatment would also affect the antioxidant activity of this material. To investigate this, CNPs obtained from 0.22 M glucose by 3 h of heating and the same sample re-heated for 24 h were compared for their scavenging ability toward hydroxyl radicals (HO˙) by electron paramagnetic resonance (EPR) spectroscopy coupled with spin trapping using DMPO as the spin-trap molecule. Hydroxyl radicals were firstly generated by the Fenton reaction. The generation of HO˙ radicals was confirmed by the intense (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1) four-line EPR signal recorded (Fig. 8, red line) corresponding to the DMPO/HO˙ adduct (aN = aH = 14.6 G).11 When the reaction was performed in the presence of the CNPs (Fig. 8, blue line and ESI Fig. S9†), the EPR signal was slightly reduced, indicating a negligible scavenging ability. When the reaction was repeated in the presence of the re-heated CNPs (dosed at an equal surface area, Fig. 8, green line and ESI Fig. S9†), the EPR signal was almost suppressed. These results highlight that a higher bulk order in the CNPs leads to an increase in the ROS scavenging ability, thus confirming what was found previously on other types of carbon nanomaterials.11,64,67 The results indicate that it is possible to tune the antioxidant properties of these NMs by adjusting the CNP synthesis, specifically the precursor concentration or introducing a second hydrothermal treatment on pre-synthesised particles.
:2:2:1) four-line EPR signal recorded (Fig. 8, red line) corresponding to the DMPO/HO˙ adduct (aN = aH = 14.6 G).11 When the reaction was performed in the presence of the CNPs (Fig. 8, blue line and ESI Fig. S9†), the EPR signal was slightly reduced, indicating a negligible scavenging ability. When the reaction was repeated in the presence of the re-heated CNPs (dosed at an equal surface area, Fig. 8, green line and ESI Fig. S9†), the EPR signal was almost suppressed. These results highlight that a higher bulk order in the CNPs leads to an increase in the ROS scavenging ability, thus confirming what was found previously on other types of carbon nanomaterials.11,64,67 The results indicate that it is possible to tune the antioxidant properties of these NMs by adjusting the CNP synthesis, specifically the precursor concentration or introducing a second hydrothermal treatment on pre-synthesised particles.
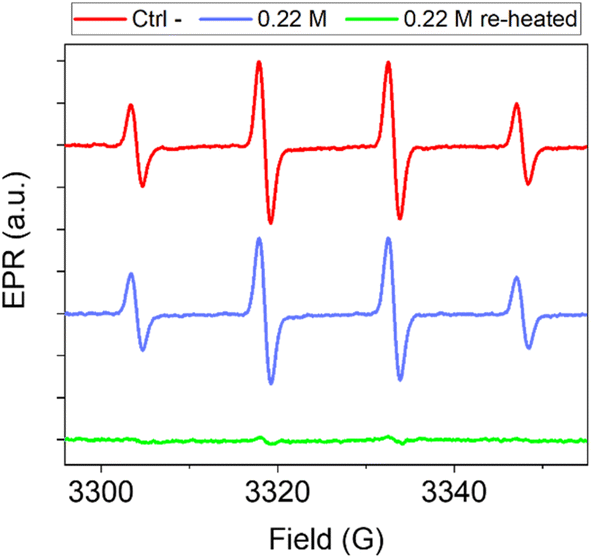 | ||
| Fig. 8 Scavenging activity of CNPs toward hydroxyl radicals generated by a Fenton reaction measured by the EPR spin-trapping technique. DMPO was used as the spin-trap molecule. EPR spectra recorded after 60 minutes of incubation in the absence of CNPs (red, Ctrl −); EPR spectra recorded in the presence of CNPs obtained from 0.22 M glucose in 3 h (blue) and the same sample subsequently re-heated for 24 h at 190 °C (green). The number of radicals produced is proportional to the intensity of the EPR signal. The reported signals are the average of at least 3 independent experiments. | ||
4. Conclusions
This study represents a significant step forward towards the understanding of the HTC process, enabling the synthesis of CNPs for medical applications with definite size, bulk order, PT and antioxidant properties.By varying the HTC synthetic parameters, CNP sizes between 15 and 150 nm forming highly stable colloids in water have been obtained, a range that allows for fine-tuning the biodistribution of CNPs. A novel post-synthesis treatment has been applied to increase the graphitic degree of the particles without affecting the colloidal stability. Both PT and antioxidant properties of CNPs were enhanced with the increase of sp2 carbon domains.
Data availability
The authors state that all data supporting the results of this study can be found within the paper and its ESI.† If raw data in different formats are required, they can be provided by the corresponding author upon reasonable request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We acknowledge financial support from the ReUNITA project (Grant Agreement No. 101035810). The authors acknowledge the use of instrumentation as well as the technical advice provided by the National Facility ELECMI ICTS, node “Laboratorio de Microscopias Avanzadas (LMA)” at “Universidad de Zaragoza”.References
- R. H. Baughman, A. A. Zakhidov and W. A. De Heer, Carbon nanotubes – the route toward applications, Science, 2002, 297, 787–792, DOI:10.1126/SCIENCE.1060928.
- A. K. Geim, Graphene: status and prospects, Science, 2009, 324, 1530–1534, DOI:10.1126/SCIENCE.1158877.
- Z. Tian, X. Zhang, D. Li, D. Zhou, P. Jing, D. Shen, S. Qu, R. Zboril, A. L. Rogach and Z. Tian, et al., Full-color inorganic carbon dot phosphors for white-light-emitting diodes, Adv. Opt. Mater., 2017, 5(19), 1700416, DOI:10.1002/adom.201700416.
- F. Picollo, L. Mino, A. Battiato, S. Ditalia Tchernij, J. Forneris, K. Martina, M. Sacco, S. Tagliapietra, E. Vittone and P. Olivero, et al., Synthesis and characterization of porphyrin functionalized nanodiamonds, Diamond Relat. Mater., 2019, 91, 22–28, DOI:10.1016/J.DIAMOND.2018.11.001.
- M.-J. Wang, C. A. Gray, S. A. Reznek, K. Mahmud, Y. Kutsovsky, Carbon black, in Kirk-Othmer Encyclopedia of Chemical Technology, 2003, DOI:10.1002/0471238961.0301180204011414.A01.PUB2.
- F. Wudl, Fullerene materials, J. Mater. Chem., 2002, 12, 1959–1963, 10.1039/B201196D.
- B. Hu, S. Yu, K. Wang, L. Liu and X. X.-D. Transactions, functional carbonaceous materials from hydrothermal carbonization of biomass: an effective chemical process, Dalton Trans., 2008, 5414–5423 RSC.
- F. Bergius, Production of hydrogen from water and coal from cellulose at high temperatures and pressures, J. Soc. Chem. Ind., 1913, 32(9), 462–467 CrossRef.
- Y. Gong, L. Xie, C. Chen, J. Liu, M. Antonietti and Y. Wang, Bottom-up hydrothermal carbonization for the precise engineering of carbon materials, Prog. Mater. Sci., 2023, 132, 101048, DOI:10.1016/J.PMATSCI.2022.101048.
- Y. Gong, H. Wang, Z. Wei, L. Xie and Y. Wang, An efficient way to introduce hierarchical structure into biomass-based hydrothermal carbonaceous materials, ACS Sustainable Chem. Eng., 2014, 2, 2435–2441 CrossRef CAS.
- I. Kokalari, R. Gassino, A. M. Giovannozzi, L. Croin, E. Gazzano, E. Bergamaschi, A. M. Rossi, G. Perrone, C. Riganti and J. Ponti, et al., Pro- and anti-oxidant properties of near-infrared (NIR) light responsive carbon nanoparticles, Free Radic Biol. Med., 2019, 134, 165–176, DOI:10.1016/j.freeradbiomed.2019.01.013.
- R. Demir-Cakan, N. Baccile, M. Antonietti and M. M. Titirici, Carboxylate-rich carbonaceous materials via one-step hydrothermal carbonization of glucose in the presence of acrylic acid, Chem. Mater., 2009, 21, 484–490 CrossRef CAS.
- Z. Yi, Y. Liang, X. Lei, C. Wang and J. Sun, Low-temperature synthesis of nanosized disordered carbon spheres as an anode material for lithium ion batteries, Mater. Lett., 2007, 61, 4199–4203, DOI:10.1016/J.MATLET.2007.01.054.
- L. Xie, X. Li, J. Deng, Y. Gong, H. Wang, S. Mao and Y. Wang, Sustainable and scalable synthesis of monodisperse carbon nanospheres and their derived superstructures, Green Chem., 2018, 20, 4596–4601, 10.1039/C8GC02196A.
- T. Sun, W. Li, M. Yang, H. Chen, Y. Liu and H. Li, Nitrogen and iron codoped porous carbon spheres derived from tetrazine-based polyindole as efficient catalyst for oxygen reduction reaction in acidic, J. Power Sources, 2019, 434, 226738 CrossRef CAS.
- W. Miao, D. Yao, C. Chu, Y. Liu, Q. Huang, S. Mao and K. Ostrikov, (Ken) Highly-efficient photocatalytic H2O2 evolution using hydrothermal carbons with donor-acceptor furan couples, Appl. Catal., B, 2023, 332, 122770, DOI:10.1016/J.APCATB.2023.122770.
- M. Inada, N. Enomoto, J. Hojo and K. H.-A. P. Technology, Structural analysis and capacitive properties of carbon spheres prepared by hydrothermal carbonization, Adv. Powder Technol., 2017, 28, 884–889 CrossRef CAS.
- B. Réti, G. I. Kiss, T. Gyulavári, K. Baan, K. Magyari and K. Hernadi, Carbon sphere templates for TiO2 hollow structures: preparation, characterization and photocatalytic activity, Catal. Today, 2017, 284, 160–168, DOI:10.1016/J.CATTOD.2016.11.038.
- Z. H. Miao, H. Wang, H. Yang, Z. Li, L. Zhen and C. Y. Xu, Glucose-derived carbonaceous nanospheres for photoacoustic imaging and photothermal therapy, ACS Appl. Mater. Interfaces, 2016, 8, 15904–15910, DOI:10.1021/ACSAMI.6B03652.
- N. Li, T. Li, C. Hu, X. Lei, Y. Zuo and H. Han, Targeted near-infrared fluorescent turn-on nanoprobe for activatable imaging and effective phototherapy of cancer cells, ACS Appl. Mater. Interfaces, 2016, 8, 15013–15023, DOI:10.1021/ACSAMI.5B02037.
- I. Kokalari, S. Keshavan, M. Rahman, E. Gazzano, G. Barzan, L. Mandrile, A. Giovannozzi, J. Ponti, G. Antonello and M. Monopoli, et al., Efficacy, biocompatibility and degradability of carbon nanoparticles for photothermal therapy of lung cancer, Nanomedicine, 2021, 16, 689–707, DOI:10.2217/NNM-2021-0009.
- M. Ajmal, U. Yunus, A. Matin and N. U. Haq, Synthesis, characterization and in vitro evaluation of methotrexate conjugated fluorescent carbon nanoparticles as drug delivery system for human lung cancer targeting, J. Photochem. Photobiol. B, 2015, 153, 111–120, DOI:10.1016/J.JPHOTOBIOL.2015.09.006.
- A. Scattareggia Marchese, E. Destro, C. Boselli, F. Barbero, M. Malandrino, G. Cardeti, I. Fenoglio and L. Lanni, Inhibitory effect against listeria monocytogenes of carbon nanoparticles loaded with copper as precursors of food active packaging, Foods, 2022, 11, 2941, DOI:10.3390/FOODS11192941/S1.
- L. Zhang, B. Peng, L. Wang, C. Guo and Q. Wang, Sustainable and high-quality synthesis of carbon nanospheres with excellent dispersibility via synergistic external pressure- and PSSMA-assisted hydrothermal carbonization, Adv. Powder Technol., 2021, 32, 2449–2456, DOI:10.1016/J.APT.2021.05.016.
- S. Soares, J. Sousa, A. Pais and C. Vitorino, Nanomedicine: principles, properties, and regulatory issues, Front. Chem., 2018, 6, 356901, DOI:10.3389/FCHEM.2018.00360/BIBTEX.
- F. Barbero, S. Michelini, O. H. Moriones, J. Patarroyo, J. Rosell, M. F. Gusta, M. Vitali, L. Martín, F. Canals and A. Duschl, et al., Role of common cell culture media supplements on citrate-stabilized gold nanoparticle protein corona formation, aggregation state, and the consequent impact on cellular uptake, Bioconjugate Chem., 2022, 33, 1505–1514, DOI:10.1021/ACS.BIOCONJCHEM.2C00232.
- L. Tang, X. Yang, Q. Yin, K. Cai, H. Wang, I. Chaudhury, C. Yao, Q. Zhou, M. Kwon and J. A. Hartman, et al., Investigating the optimal size of anticancer nanomedicine, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15344–15349, DOI:10.1073/PNAS.1411499111.
- M. Sevilla and A. B. Fuertes, Chemical and structural properties of carbonaceous products obtained by hydrothermal carbonization of saccharides, Chem.–Eur. J., 2009, 15, 4195–4203, DOI:10.1002/CHEM.200802097.
- Y. Gong, L. Xie, H. Li and Y. Wang, Sustainable and scalable production of monodisperse and highly uniform colloidal carbonaceous spheres using sodium polyacrylate as the dispersant, Chem. Commun., 2014, 50(84), 12633–12636 RSC.
- L. Peltonen, M. Singhal and J. Hirvonen, Principles of nanosized drug delivery systems, Nanoeng. Biomater. Adv. Drug Deliv., 2020, 3–25, DOI:10.1016/B978-0-08-102985-5.00001-2.
- Q. Wang, H. Li, L. Chen and X. Huang, Monodispersed hard carbon spherules with uniform nanopores, Carbon N. Y., 2001, 39, 2211–2214, DOI:10.1016/S0008-6223(01)00040-9.
- Y. Gong, Z. Wei, J. Wang, P. Zhang, H. Li and Y. Wang, Design and fabrication of hierarchically porous carbon with a template-free method, Sci. Rep., 2014, 4(1), 6349 CrossRef CAS.
- P. Zhang, J. Yuan, T.-P. Fellinger, M. Antonietti, H. Li, Y. Wang, P. F. Zhang, H. R. Li, Y. Wang and J. Yuan, et al., Improving hydrothermal carbonization by using poly (ionic liquid)s, Angew. Chem., Int. Ed., 2013, 52, 6028–6032, DOI:10.1002/anie.201301069.
- A. Beccaria, A. Bellone, A. Mirigaldi, V. Serafini, M. Olivero, A. Vallan and G. Perrone, Temperature monitoring of tumor hyperthermal treatments with optical fibers: comparison of distributed and quasi-distributed techniques, Opt. Fiber Technol., 2020, 60, 102340 CrossRef CAS.
- A. Bellone, E. Ullo, M. Olivero, G. Coppa, A. Vallan, and G. Perrone, Preliminary analysis of the estimation of tissue thermal parameters for tumor laser ablation with minimally invasive techniques, in Conference Record – IEEE Instrumentation and Measurement Technology Conference, 2024, DOI:10.1109/I2MTC60896.2024.10561198.
- S. Bhattacharjee, DLS and zeta potential – what they are and what they are not?, J. Controlled Release, 2016, 235, 337–351, DOI:10.1016/J.JCONREL.2016.06.017.
- T. L. Doane, C. H. Chuang, R. J. Hill and C. Burda, Nanoparticle ζ-potentials, Acc. Chem. Res., 2012, 45, 317–326, DOI:10.1021/AR200113C.
- F. Barbero, O. H. Moriones, N. G. Bastús and V. Puntes, Dynamic equilibrium in the cetyltrimethylammonium bromide-au nanoparticle bilayer, and the consequent impact on the formation of the nanoparticle protein corona, Bioconjugate Chem., 2019, 2917–2930, DOI:10.1021/ACS.BIOCONJCHEM.9B00624.
- T. Cosgrove, Colloid Science: Principles, Methods and Applications, 2010 Search PubMed.
- S. Abbott and N. Holmes, Nanocoatings: Principles and Practice: from Research to Production, 2013 Search PubMed.
- A. Ferrari and J. Robertson, Interpretation of raman spectra of disordered and amorphous carbon, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095, DOI:10.1103/PhysRevB.61.14095.
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Pöschl, Raman microspectroscopy of soot and related carbonaceous materials: spectral analysis and structural information, Carbon N. Y., 2005, 43, 1731–1742, DOI:10.1016/J.CARBON.2005.02.018.
- F. Tuinstra and J. L. Koenig, Raman spectrum of graphite, J. Chem., 1970, 53, 1126, DOI:10.1063/1.1674108.
- S. I. Moseenkov, V. L. Kuznetsov, N. A. Zolotarev, B. A. Kolesov, I. P. Prosvirin, A. V. Ishchenko and A. V. Zavorin, Investigation of amorphous carbon in nanostructured carbon materials (a comparative study by TEM, XPS, Raman spectroscopy and XRD), Materials, 2023, 16, 1112, DOI:10.3390/MA16031112.
- J. Robertson, Recombination and photoluminescence mechanism in hydrogenated amorphous carbon, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 16302, DOI:10.1103/PhysRevB.53.16302.
- C. Casiraghi, A. C. Ferrari and J. Robertson, Raman spectroscopy of hydrogenated amorphous carbons, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 085401, DOI:10.1103/PHYSREVB.72.085401.
- B. Marchon, Photoluminescence and Raman spectroscopy in hydrogenated carbon films, IEEE Trans. Magn., 1997, 33, 3148–3150, DOI:10.1109/20.617873.
- Y. Qi, M. Zhang, L. Qi and Y. Q.-R. Advances, Mechanism for the formation and growth of carbonaceous spheres from sucrose by hydrothermal carbonization, RSC Adv., 2016, 6(25), 20814–20823, 10.1039/c5ra26725k.
- F. Alakhras and R. Holze, In situ UV–vis- and FT-IR-spectroscopy of electrochemically synthesized furan–thiophene copolymers, Synth. Met., 2007, 157, 109–119, DOI:10.1016/J.SYNTHMET.2006.12.011.
- J. Ryu, Y.-W. Suh, D. J. Suh and D. J. Ahn, Hydrothermal preparation of carbon microspheres from mono-saccharides and phenolic compounds, Carbon, 2010, 48(7), 1990–1998, DOI:10.1016/j.carbon.2010.02.006.
- D. Wu, R. Fu and Z. Yu, Organic and carbon aerogels from the NaOH-catalyzed polycondensation of resorcinol–furfural and supercritical drying in ethanol, J. Appl. Polym. Sci., 2005, 96, 1429–1435, DOI:10.1002/app.21582.
- J. Robertson, Diamond-like amorphous carbon, Mater. Sci. Eng., 2002, 37, 129–281, DOI:10.1016/S0927-796X(02)00005-0.
- V. Ţucureanu, A. Matei and A. M. Avram, FTIR spectroscopy for carbon family study, Crit. Rev. Anal. Chem., 2016, 46, 502–520, DOI:10.1080/10408347.2016.1157013.
- V. K. Lamer and R. H. Dinegar, Theory, production and mechanism of formation of monodispersed hydrosols, J. Am. Chem. Soc., 1950, 72, 4847–4854, DOI:10.1021/JA01167A001.
- D. Jung, M. Zimmermann and A. Kruse, Hydrothermal carbonization of fructose: growth mechanism and kinetic model, ACS Sustainable Chem. Eng., 2018, 6, 13877–13887, DOI:10.1021/ACSSUSCHEMENG.8B02118.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Mechanisms of nucleation and growth of nanoparticles in solution, Chem. Rev., 2014, 114, 7610–7630, DOI:10.1021/CR400544S.
- G. Antonello, A. Marucco, E. Gazzano, P. Kainourgios, C. Ravagli, A. Gonzalez-Paredes, S. Sprio, E. Padín-González, M. G. Soliman and D. Beal, et al., Changes of physico-chemical properties of nano-biomaterials by digestion fluids affect the physiological properties of epithelial intestinal cells and barrier models, Part Fibre Toxicol., 2022, 19(1), 49, DOI:10.1186/S12989-022-00491-W.
- F. Barbero, S. Gul, G. Perrone and I. Fenoglio, Photoresponsive inorganic nanomaterials in oncology, Technol. Cancer Res. Treat., 2023, 22 DOI:10.1177/15330338231192850.
- B. Han, Y.-L. Zhang, Q.-D. Chen, H.-B. Sun, A. B. Han, Y.-L. Q. Zhang, D. Chen and B. H. Sun, Carbon-based photothermal actuators, Adv. Funct. Mater., 2018, 28, 1802235, DOI:10.1002/adfm.201802235.
- L. Zhao, Y. Liu, R. Chang, R. Xing and X. Yan, Supramolecular photothermal nanomaterials as an emerging paradigm toward precision cancer therapy, Adv. Funct. Mater., 2019, 29(4), 1806877, DOI:10.1002/adfm.201806877.
- M. A. MacKey, M. R. K. Ali, L. A. Austin, R. D. Near and M. A. El-Sayed, The most effective gold nanorod size for plasmonic photothermal therapy: theory and in vitro experiments, J. Phys. Chem. B, 2014, 118, 1319–1326, DOI:10.1021/JP409298F.
- A. A. Balandin, Thermal properties of graphene and nanostructured carbon materials, Nat. Mater., 2011, 10(8), 569–581, DOI:10.1038/nmat3064.
- R. Singh and S. V. Torti, Carbon nanotubes in hyperthermia therapy, Adv. Drug Deliv. Rev., 2013, 65, 2045–2060, DOI:10.1016/J.ADDR.2013.08.001.
- Y. Qiu, Z. Wang, A. C. E. Owens, I. Kulaots, Y. Chen, A. B. Kane and R. H. Hurt, Antioxidant chemistry of graphene-based materials and its role in oxidation protection technology, Nanoscale, 2014, 6, 11744–11755, 10.1039/C4NR03275F.
- L. L. Dugan, D. M. Turetsky, C. Du, D. Lobner, M. Wheeler, C. R. Almli, C. K. F. Shen, T. Y. Luh, D. W. Choi and T. S. Lin, Carboxyfullerenes as neuroprotective agents, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 9434–9439 CrossRef CAS.
- I. Fenoglio, M. Tomatis, D. Lison, J. Muller, A. Fonseca, J. B. Nagy and B. Fubini, Reactivity of carbon nanotubes: free radical generation or scavenging activity?, Free Radical Biol. Med., 2006, 40, 1227–1233, DOI:10.1016/J.FREERADBIOMED.2005.11.010.
- E. Carella, M. Ghiazza, M. Alfè, E. Gazzano, D. Ghigo, V. Gargiulo, A. Ciajolo, B. Fubini and I. Fenoglio, Graphenic nanoparticles from combustion sources scavenge hydroxyl radicals depending upon their structure, Bionanoscience, 2013, 3, 112–122, DOI:10.1007/S12668-013-0077-6.
- J. Mwila, M. Miraftab and A. R. Horrocks, Effect of carbon black on the oxidation of polyolefins—an overview, Polym. Degrad. Stab., 1994, 44, 351–356, DOI:10.1016/0141-3910(94)90094-9.
- I. Fenoglio, G. Greco, M. Tomatis, J. Muller, E. Raymundo-Piñero, F. Béguin, A. Fonseca, J. B. Nagy, D. Lison and B. Fubini, Structural defects play a major role in the acute lung toxicity of multiwall carbon nanotubes: physicochemical aspects, Chem. Res. Toxicol., 2008, 21, 1690–1697, DOI:10.1021/TX800100S.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00923a |
| This journal is © The Royal Society of Chemistry 2025 |