
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Engineering Au single-atom sites embedded in TiO2 nanostructures for boosting photocatalytic methane oxidation†
Qui Thanh Hoai Ta a and
Ly Tan Nhiem*b
a and
Ly Tan Nhiem*b
aInstitute of Chemical Technology, Vietnam Academy of Science and Technology, 1A TL29 Street, Thanh Loc Ward, District 12, Ho Chi Minh City 700000, Vietnam. E-mail: tathanhhoaiqui2292@gmail.com
bFaculty of Chemical and Food Technology, Ho Chi Minh City University of Technology and Education, 01 Vo Van Ngan Street, Linh Chieu Ward, Thu Duc City, Ho Chi Minh City 700000, Vietnam. E-mail: nhiemlt@hcmute.edu.vn
First published on 13th January 2025
Abstract
Photocatalytic methane oxidation under mild conditions using single-atom catalysts remains an advanced technology. In this work, gold single atoms (Au SAs) were introduced onto TiO2 nanostructures using a simple method. The resulting performance demonstrated effective conversion of methane into H2 and C2 products at room temperature. The as-synthesized Au SA/TiO2 exhibited a high hydrogen production rate of 2190 μmol g−1, with selectivity reaching up to 58% under optimized conditions. The methane oxidation mechanism was investigated, revealing a methyl radical pathway for generating value-added chemicals. This research provides a strategy for photocatalytic methane conversion over single-atom-supported photocatalysts.
1. Introduction
Methane (CH4) is a crucial fossil fuel and an important component of natural resources, serving as a significant chemical raw material. It constitutes approximately 80% of natural gas, making it a massive contributor to the greenhouse effect and global warming, particularly during the combustion process compared to carbon dioxide (CO2).1,2 Therefore, the sustainable harvesting of CH4 and its conversion into valuable products is essential for the sustainable utilization of natural resources and environmental protection. However, conventional methods for CH4 conversion typically require high operating conditions (>100 °C and 100 MPa), which can result in excessive oxidation and low product selectivity. This is due to the bond dissociation energy of the initial C–H bonds being higher than that of the subsequent C–H bonds, which prevents the activation of the first C–H bond when relying solely on thermal energy to overcome the activation barrier.3–5 Thus, it remains a challenge to enhance the selectivity of CH4 conversion to high-value-added products under mild conditions.6,7 Photocatalysis has been recognized as a green technique and a unique method for harnessing light to generate electron–hole pairs in semiconducting materials.8–11 The application of single-atom catalysts in methane oxidation shows significant advancement over traditional catalysts, as they exhibit enhanced catalytic performance by uniformly decorating noble metal atoms. This arrangement facilitates active exposure and increases the surface area for the oxidation process. Additionally, single-atom catalysts reduce the amount of catalyst required, addressing concerns about high costs and the scarcity of precious metals, making them economically viable even in small quantities.12–14 It is crucial to understand the overall reaction rate of photocatalysts with varying active atom loadings due to their low coverage, which helps prevent the formation of nanoparticles. Single-atom catalysts also integrate seamlessly with advanced nanomaterials, such as reduced graphene oxide and carbon nanotubes, further improving catalytic activity and stability. These catalysts have demonstrated the ability to reduce dependence on noble metals while enhancing selectivity, owing to the abundance of active sites. Several studies have focused on engineering noble metal atoms (e.g., Pt and Ag) on titania catalysts, exploring the role of titanium cations and oxygen vacancies in the localization of single atoms on oxo ligands, thereby facilitating superior photocatalytic performance.15–20Titanium dioxide (TiO2) has garnered significant attention within the scientific community and has become a prominent commercial product due to its unique properties and diverse applications. Its excellent chemical stability, strong catalytic ability, and wide band gap energy (Eg) under light illumination contribute to its utility.21–23 The application of TiO2 as a catalyst in photocatalytic methane oxidation and water splitting presents a promising solution in response to the urgent demand for net-zero emissions and sustainable energy applications. However, one of the main disadvantages of TiO2 is its wide band gap and the rapid recombination rate of electron–hole pairs, which poses challenges for researchers. Numerous strategies have been explored to enhance the photocatalytic performance of TiO2, including metal doping, non-metal loading, and surface modification with other semiconductors or quantum dots.3,24,25 These combinatorial techniques aim to reduce the electron–hole recombination rate, extend light harvesting capabilities, and improve surface reaction kinetics.
Building on this foundation, the highly selective methane oxidation using an Fe/TiO2 catalyst was investigated in 2018, which demonstrated a 15% conversion rate after 3 hours of reaction, with methanol selectivity exceeding 90%. The presence of FeOOH and Fe2O3 plays a crucial role in enhancing charge transfer and separation in TiO2, thereby reducing the overall overpotential of the reaction. Photocatalytic methane oxidation over the TiO2 nanostructure is commonly considered owing to its low cost, stable chemical properties, and non-toxic nature.26
In the same context, photocatalytic CH4 oxidation to C1 oxygenates over Pd/TiO2 composites was investigated using O2 and H2O as oxidants. Palladium (Pd) serves as a hole acceptor, while oxygen vacancies function as electron acceptors, enhancing charge separation efficiency. Under optimized conditions, C1 products were obtained with approximately 99% selectivity and a high yield of 54.7 mmol g−1 h−1.27 Shuang et al. utilized Au–ZnO/TiO2 composites for the direct oxidation of CH4 to valuable C2H6 in a flow reactor under mild conditions. A C2H6 production rate of over 5 mmol g−1 h−1 was achieved after 50 minutes of reaction, with a high selectivity of 90% attributed to the weak overoxidation ability of ZnO. In this system, Au facilitates the desorption of ˙CH3 radicals in the gas phase, thereby preventing overoxidation to CO2.28
In this work, Au single-atom (Au SA) supported TiO2 nanostructures were investigated for driving CH4 oxidation under mild conditions. Due to energetic active sites of Au SAs, the optimized production yield of H2 reached 2190 μmol g−1, with a selectivity of 58%. The investigation into the plausible mechanism indicated that Au inhibits the charge recombination rate and activates the reactant gases. These results suggest that this approach can effectively enhance H2 production through improved photocatalytic performance at room temperature.
2. Experimental
2.1. Materials
Titania powder (TiO2, P25, 99%), gold(III) chloride tetrahydrate (HAuCl4·4H2O), sodium borohydride powder (NaBH4, 98%), and sodium hydroxide (NaOH) were purchased from Sigma-Aldrich. Deionized (DI) water and ethanol (C2H5OH) were used as a solvent for material cleaning steps. All chemicals were used without further purification.2.2. Preparation of catalysts
The Au SA-supported photocatalysts were synthesized using a wet precipitation process, as presented in Fig. 1. Specifically, 0.3 g of TiO2 was added to an aqueous solution of HAuCl4 (1.5 mg mL−1), with the pH pre-adjusted to approximately 9 using 0.1 M NaOH solution and NaBH4 solution, followed by stirring at 80 °C for 1 hour. After the reaction, the precipitates were collected, thoroughly washed with deionized water, and dried at 60 °C overnight. The loading content of Au SAs was estimated at 2 wt% as the standard dose. The final product was calcined at 400 °C for 3 hours, ground, and denoted as Au SA/TiO2, which was then stored for subsequent photocatalytic experiments. For comparison, Au NPs/TiO2 and Pt NPs/TiO2 were also prepared via the traditional impregnation technique without pH control, as in previous publications.29,30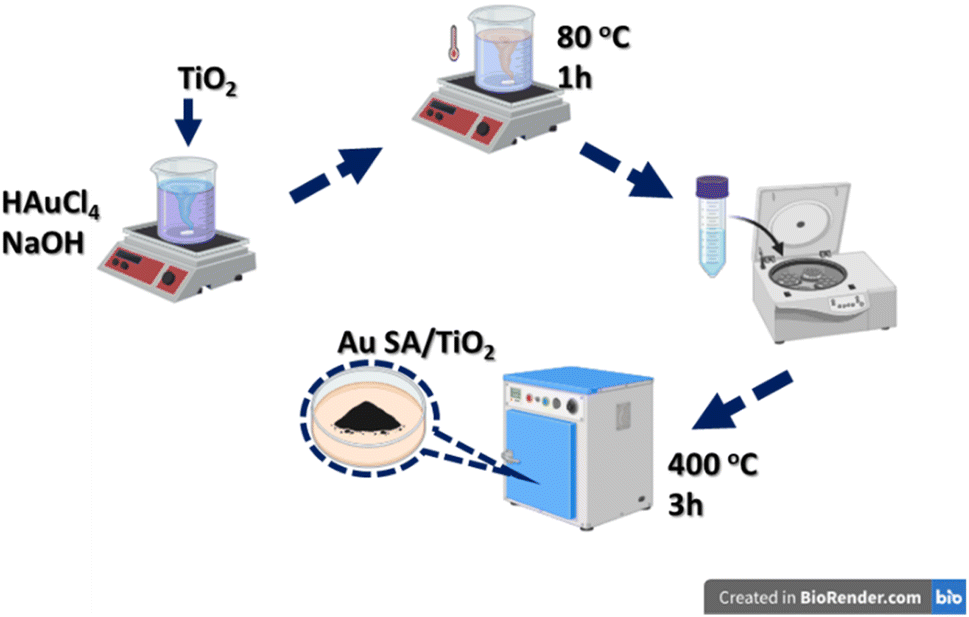 | ||
| Fig. 1 The schematic illustration of Au single-atom (Au SA) – supported catalysts. | ||
2.3. Characterization
The crystal structure of the prepared photocatalysts and pure TiO2 was examined by using an X-ray diffractometer (XRD), equipped with a Cu-Kα radiation source working at 40 kV and 30 mA. The presence of single atoms was observed by using a scanning transmission electron microscope (S/TEM) equipped with an HAADF detector (electron energy of 300 keV). The optical absorption and radiative recombination of photocatalysts were studied by ultraviolet-visible diffuse reflectance spectroscopy (UV-vis DRS) and photoluminescence (PL) using a spectrometer (PerkinElmer Lambda 650S) and HR Labram, Horiba, respectively. The optical properties of selected samples were confirmed by using Fourier transform infrared spectroscopy (FTIR/JASCO-4600), Raman spectroscopy (Olympus-BX5), a semiconductor device analyzer (Keysight B1500A), and a CH instruments electrochemical workstation.The analysis of paramagnetic species was performed by continuous-wave electron paramagnetic resonance (EPR). These experiments were performed on a Bruker ELEXSYS E500 spectrometer operating in the X-band (9.5 GHz). The EPR spectra were recorded at 94 K to avoid electron–hole recombination. The detection of radical species was conducted by spin-trapping tests using 5-tert-butoxycarbonyl-5-methyl-1-pyrroline-N-oxide (BMPO) as the scavenger. The atomic-scale and electronic structure of the sample were investigated by scanning tunnelling microscopy (STM) measurement. For this, the Au SA/TiO2 solution was drop-cast on a gold plate, that was quickly loaded in the load lock chamber of a UHV system (base pressure < 1 × 10−10 Torr) to be heated for twelve hours at 85 °C. The STM experiments were performed at room temperature with a tungsten tip and under tunneling conditions: Vsample = 1.8 V, It = 200 pA.
2.4. Photocatalytic methane oxidation
The photocatalytic oxidation of CH4 by CO2 molecules was carried out in a batch stainless-steel reactor (100 mL), with a circular quartz window attached on top. In brief, 50 mg of the sample was placed in the reactor, which was closed and evacuated for 10 min prior to the provision of 10 bars of the CH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO2 gas mixture (3:1 pressure ratio) and kept under dark conditions for 30 min. Afterwards, a 300 W xenon lamp (200–1000 nm) was switched on as the light source. The wavelength of light irradiation could be varied by specific filters. The chamber was kept at room temperature by using a chiller. The gaseous products were analyzed and quantified using a gas chromatography system (HP-6890, Agilent).
:CO2 gas mixture (3:1 pressure ratio) and kept under dark conditions for 30 min. Afterwards, a 300 W xenon lamp (200–1000 nm) was switched on as the light source. The wavelength of light irradiation could be varied by specific filters. The chamber was kept at room temperature by using a chiller. The gaseous products were analyzed and quantified using a gas chromatography system (HP-6890, Agilent).
3. Results and discussion
3.1. Structural and morphological properties
The crystal structures of the samples were characterized by X-ray diffraction (XRD). As shown in Fig. 2a, the TiO2 nanostructures exhibit mixed crystal phases of rutile and anatase, corresponding to the standard JCPDS data (no. 21-1272 for rutile and no. 21-1276 for anatase).31–33 Upon calcination at 400 °C, the well-defined diffraction peaks at 25.8°, 28.1°, 37.1°, 48.3°, and 55.3° were assigned to the (101), (110), (004), (220), and (105) planes of TiO2, respectively. Additionally, the broadened peaks at 44.1° and 64.1° were observed, indicating the presence of Au SAs on the TiO2 nanostructures. The relatively small size of the Au diffraction peaks suggests high dispersion of the Au SA. Fig. 2b–d illustrate the Ti 2p, Au 4f, and O 1s region spectra of the selected Au SA/TiO2 composite. The main photoelectron peaks at binding energies of 463.8 eV and 458.3 eV are assigned to Ti4+ 2p1/2 and 2p3/2, respectively (Fig. 2b).34 The Au 4f spectra were deconvoluted into two major peaks at 86.5 eV and 82.9 eV, corresponding to the characteristic peaks of Au 4f5/2 and Au 4f7/2, respectively (Fig. 2c). These findings suggest that Au is reduced from Au3+ to Au1+ upon the formation of single atoms.35,36 As observed in Fig. 2d, the O 1s peak at 529.4 eV can be deconvoluted into two peaks, with primary peaks at 529.1 eV and 529.6 eV corresponding to lattice oxygen (OL) and surface-adsorbed oxygenated species (OHs/Os), respectively.34 This confirms the creation of oxygen vacancies. These characterization studies confirm the phase and surface chemical states of the Au SA/TiO2 catalyst.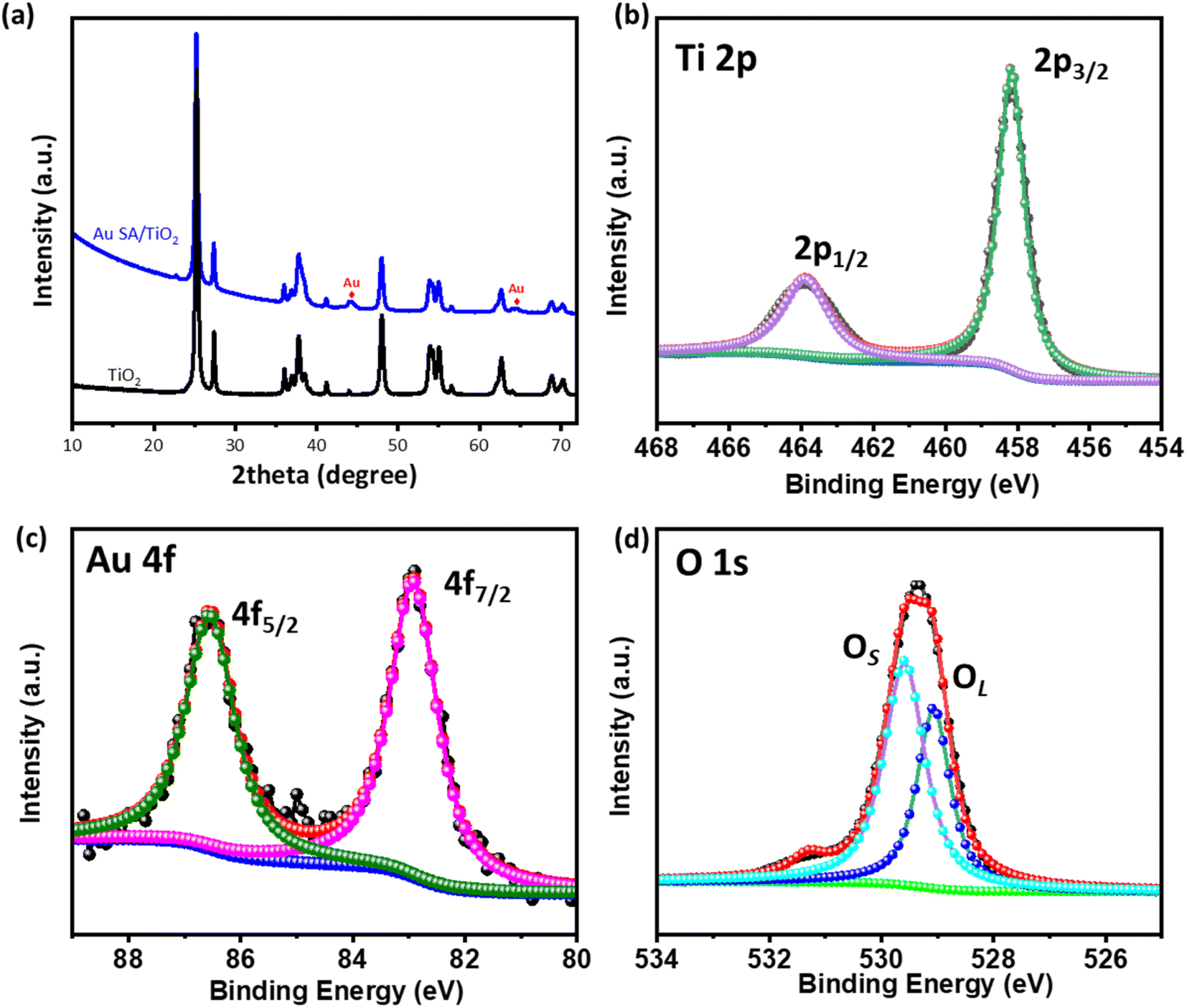 | ||
| Fig. 2 (a) XRD patterns of pristine TiO2 and the Au SA/TiO2 composite and (b)–(d) XPS spectra of Ti 2p, Au 4f, and O 1s of the selected Au SA/TiO2 composite. | ||
Due to the inherent resolution limitations of the SEM system, these images cannot definitively confirm the formation of single atoms or nanoparticle assemblies. Therefore, high-resolution TEM (HR-TEM) analysis provides clear evidence of the distribution of Au SAs on the surface of TiO2 nanostructures. As illustrated in Fig. 3, the Au SAs are atomically dispersed across the TiO2 surface, visible through their brighter contrast (highlighted by the blue circle). This confirms the enhanced distribution of Au SAs as a result of the calcination process, with no noticeable agglomeration compared to the Au NPs/TiO2 synthesized via traditional impregnation methods. In other words, TiO2, as a reducible oxide, is well-suited for the atomic dispersion of metal catalysts due to its inherent surface defects, which help stabilize metal atoms by forming metal–oxygen-support bonds.37,38
 | ||
| Fig. 3 The HR-TEM image and SAED images of Au SA/TiO2 powder, respectively (a) and (b). The bright spots in (a) indicate Au single atoms (SA) over the TiO2 matrix. | ||
To further investigate the Au SA/TiO2 heterostructure at the atomic scale, the STM analysis has been carried out (schematized in Fig. 4a). The topography STM image indicated the atomic arrangement of TiO2 powder, although the signals of Au SAs were unable to be detected (Fig. 4b). By selecting a specific area on the catalyst surface, STM measurement was conducted and revealed the electronic structure of Au/TiO2 (Fig. 4c). By measuring the distance between peaks of the valence band (EV) and conduction band (EC), the bandgap energy of Au SA/TiO2 was estimated to be 2.71 eV, which is smaller than 3.2 eV of pristine TiO2 P25, leading to favorable benefits for photocatalytic reactions.39 Besides, the bright defects (Ebd) near the valence band were also observed, indicating shallow trapping states of TiO2 crystals.
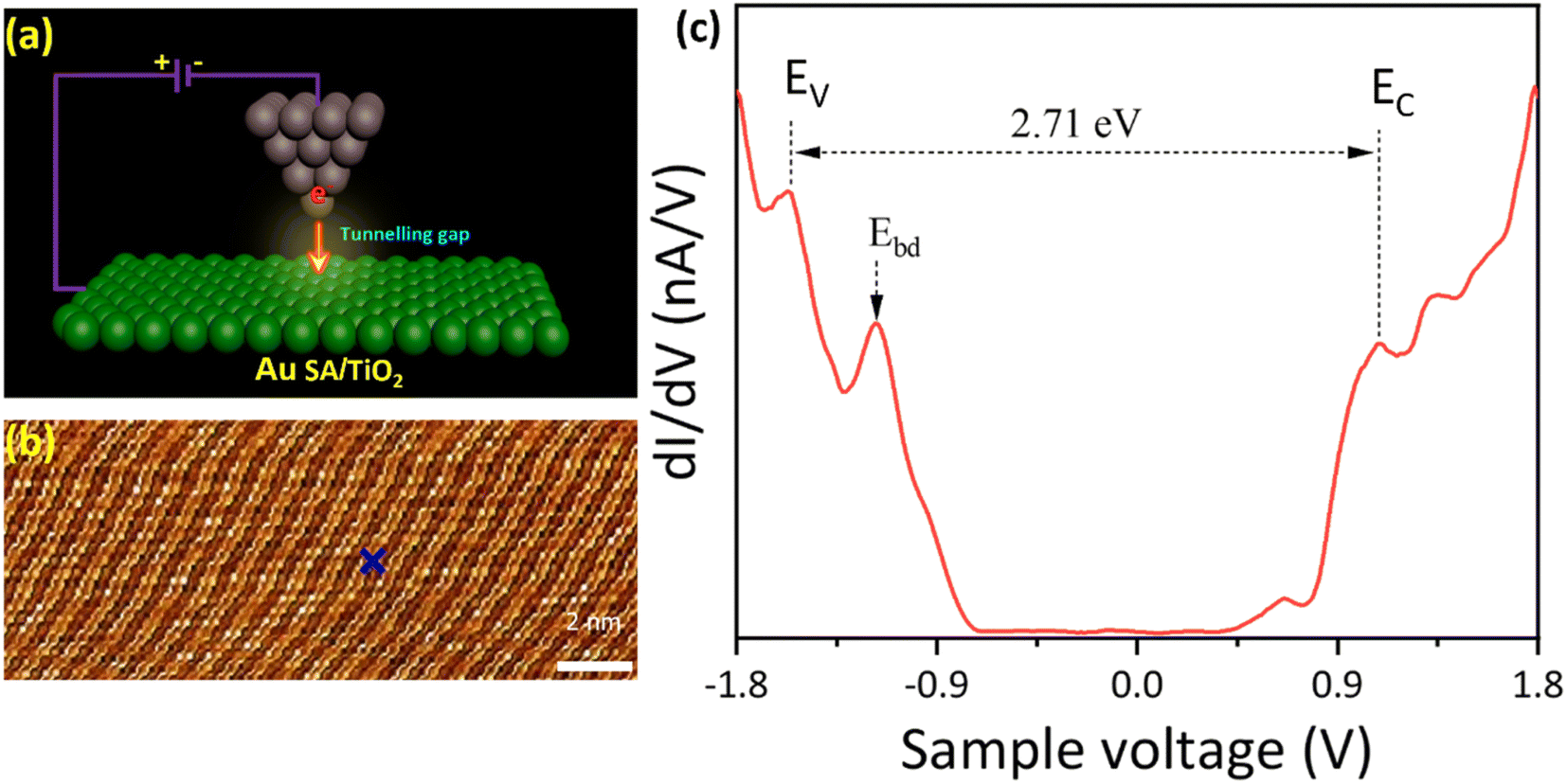 | ||
| Fig. 4 (a) The schematic illustration of scanning tunneling microscopy (STM) analysis, (b) and (c) topography STM image and tunneling spectroscopy of the Au SA/TiO2 sample recorded from the green cross in (b). Tunneling conditions: Vsample = 1.8 V, It = 200 pA. The valence and conduction band edges of Au SA/TiO2 are indicated by vertical segments, labeled as EV and EC. The Ebd stands for bright defects in the TiO2 structure. | ||
3.2. Optical properties
To understand the effect of Au loading on the light absorption properties of TiO2, UV-vis spectra were analyzed. As shown in Fig. 5a, apart from strong absorption of pristine TiO2 below 400 nm, the Au SA/TiO2 powder exhibited another absorption peak at 550 nm, indicating the plasmonic effects, which were absent in the case of the pristine TiO2 spectrum. The plasmon resonance of Au SAs could significantly contribute to photocatalytic performance later. Moreover, the steady-state photoluminescence (PL) spectra were obtained to confirm the separation properties of photogenerated carriers (Fig. 5b). The pure TiO2 samples exhibited stronger photoluminescence emission signals compared to Au SA/TiO2, indicating a higher recombination rate of electrons and holes. Notably, Au SA/TiO2 displayed the lowest fluorescence intensity, suggesting efficient separation of photocarriers. This finding indicates that the transfer of excited electrons occurred rapidly between Au and TiO2, effectively reducing the recombination rate in the photocatalysts.40,41 The photocatalysts were analyzed using the photocurrent response and electrochemical impedance spectroscopy. Fig. S1a† displays the current–voltage (I–V) curve for the Au SA/TiO2 photocatalyst under both dark and light conditions, with the applied voltage ranging from −1 V to +1 V. Under light illumination, the slope of the I–V curve begins to increase, indicating that the generated charge carriers significantly influence the electrical behavior of the optimized sample. Furthermore, the Nyquist plot in Fig. S1b† shows that the electron transport capability of the electrode surface has been slightly enhanced in the Au SA/TiO2 sample compared to pure TiO2.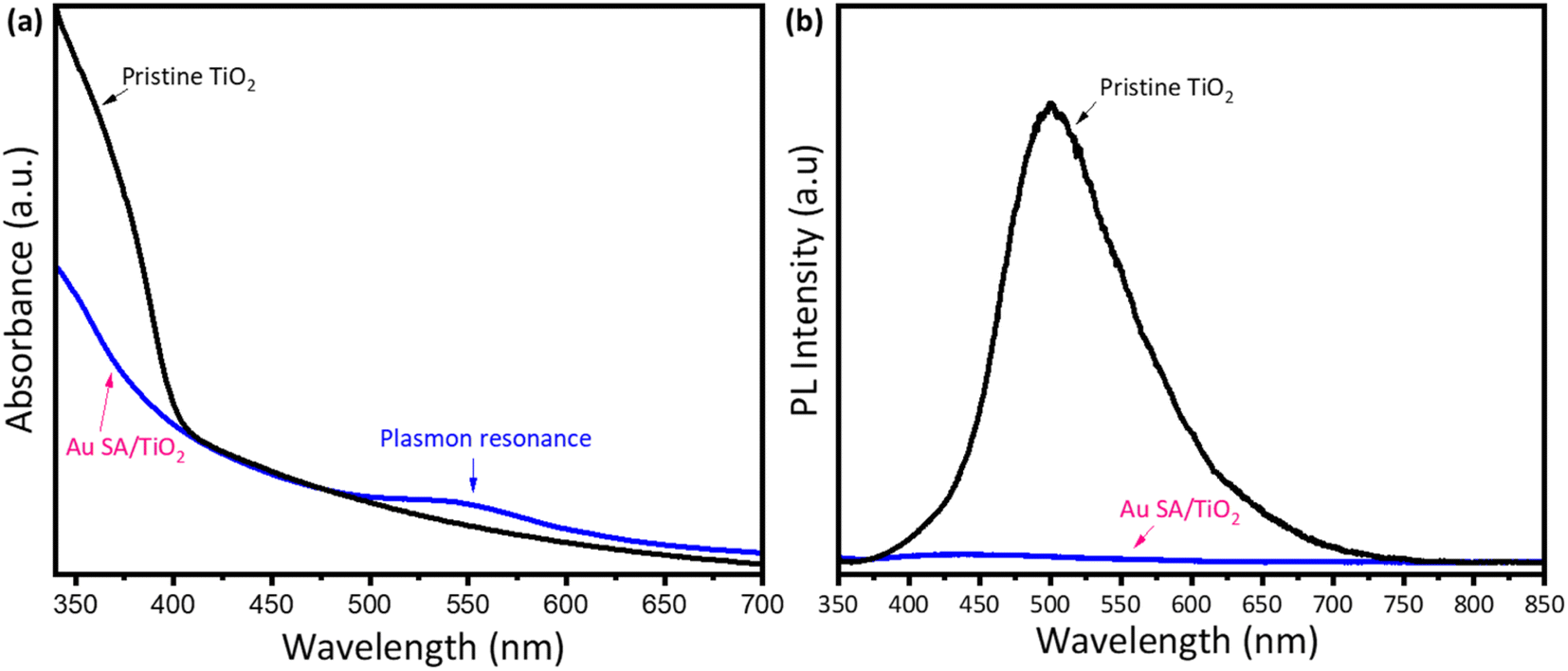 | ||
| Fig. 5 The UV-vis absorption spectra of pristine TiO2 and Au SA/TiO2, respectively (a) and (b). | ||
The interaction between TiO2 and Au SAs was analyzed using Raman spectroscopy and FTIR. As shown in Fig. 6a and S2a,† the Raman spectrum of the selected samples displays five characteristic modes located at 128, 182, 390, 514, and 643 cm−1, which correspond to the Eg, Eg, Bg, Ag, and Eg modes, respectively. The main peak at 128 cm−1, characteristic of the Ti–O stretching mode in pristine TiO2, positively shifts to 133 cm−1, confirming that Au has interacted with the Ti–O bond. This shift indicates the strengthening of the Ti–O bond, as well as phonon confinement effects associated with the creation of oxygen vacancies.42,43 The FTIR spectra of all samples show bands corresponding to TiO2, with a broad band from 400 to 900 cm−1 assigned to Ti–O–Ti vibrations, as well as bands at around 1621 cm−1 and 3416 cm−1, which are attributed to O–H bending and O–H stretching vibrations, respectively (Fig. 6b and S2b†).44,45 The FTIR spectrum of TiO2 after modification with Au does not exhibit significant changes compared to pristine TiO2, likely due to the formation of Au single atoms on the overall TiO2 structure.
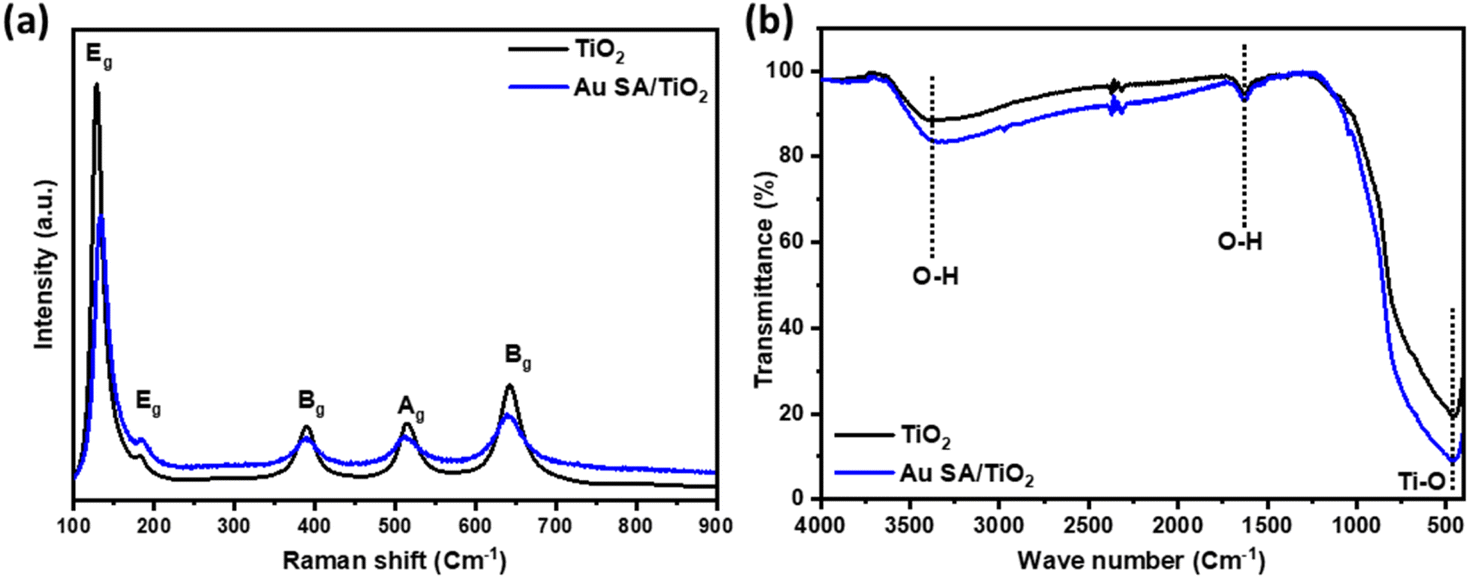 | ||
| Fig. 6 (a) The Raman and (b) FTIR spectra of pristine TiO2 and Au SA/TiO2, respectively. | ||
3.3. Photocatalytic performance
According to unique properties, the Au SA/TiO2 sample was employed for photocatalytic CH4 oxidation by CO2 molecules, as depicted in Fig. 7. Note that there were no products generated in the absence of catalysts and light irradiation (Fig. S3†). By contrast, under light conditions, the Au SA/TiO2 catalyst was able to convert CH4 and CO2 into several gaseous products as a function of reaction time (Fig. 7a). Obviously, Au SA/TiO2 could produce a significant number of products after 8 hours (around 3800 μmol g−1 in total) including H2, C2H6, C3H8 and CO, with a H2 selectivity of about 58%. This performance surpassed that of pure TiO2 powder (Fig. S4†) and other reported composites (Table 1). Although the Au SA/TiO2 photocatalyst did not exhibit the highest performance in comparison with other composites listed in Table 1, it remains a promising candidate for CH4 oxidation reactions.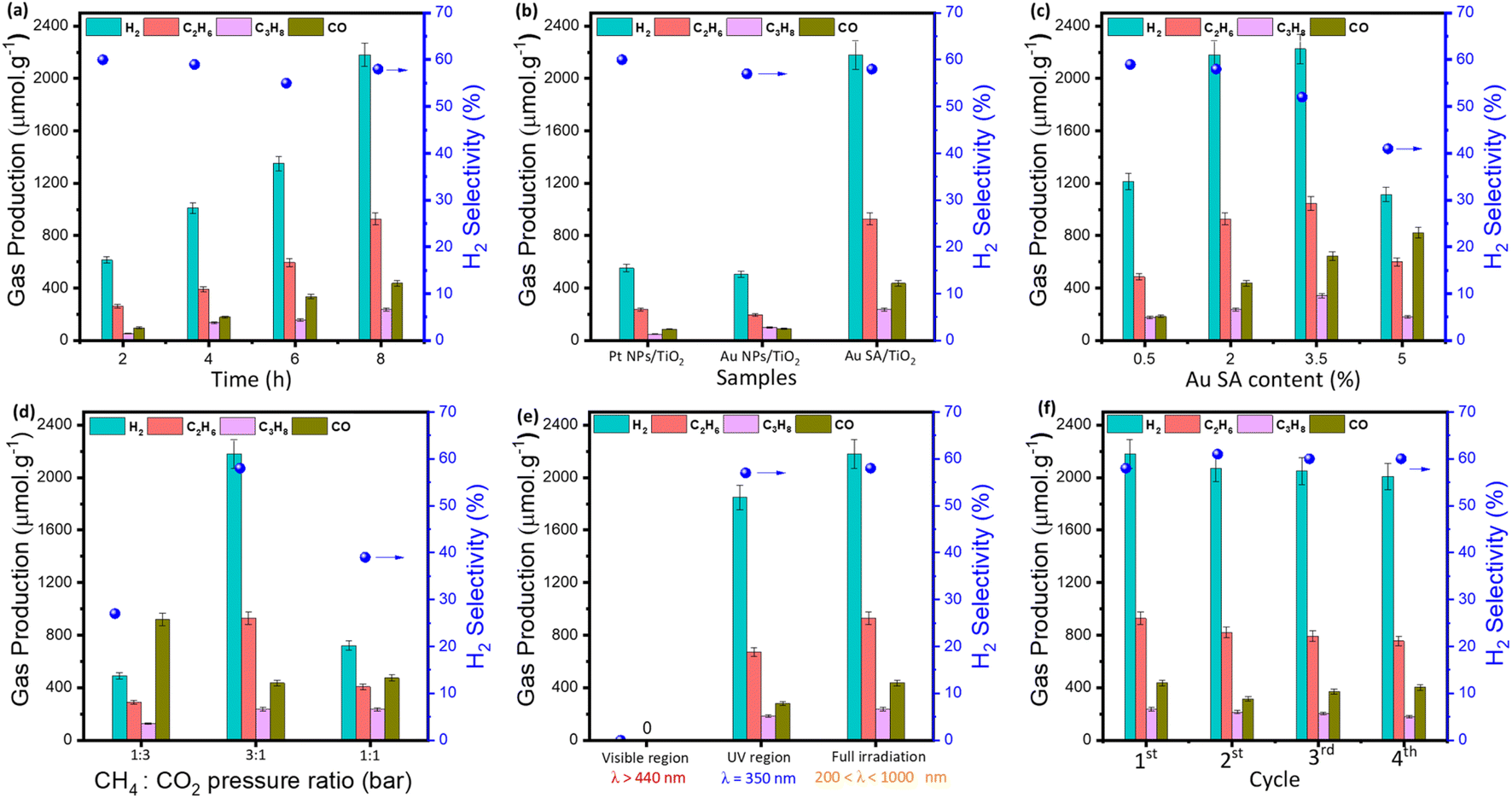 | ||
| Fig. 7 Photocatalytic methane oxidation performances and the H2 selectivity as a function of reaction time over the standard Au SA/TiO2 sample (2 wt% Au) (a), comparable photocatalytic experiments of different catalysts (b), comparison of photocatalytic activity with different Au loading contents, reactant ratios, and irradiation regions, respectively (c)–(e), and stability tests over four cycles (f). Reaction conditions: 50 mg of the standard Au SA/TiO2 catalyst with 2% Au loading, CH4:CO2 = 3:1 (stand pressure ratio), xenon lamp 300 W. In figures (a), (d) and (f), standard Au SA/TiO2 was used. | ||
| Photocatalysts | Amount of catalyst | Reaction conditions | Performance | Ref. |
|---|---|---|---|---|
| Cu@TiO2 | 10 mg | 3 MPa CH4 | CH3OH: 20000 μmol g−1 h−1 |
46 |
| H2O2 1 M 10 mL | ||||
| UV-vis light | ||||
| Ru/TiO2–H2 | 5.0 mg | 8 vol% CO2 | CO: 708.4 mmol g−1 h−1, H2: 645.5 mmol g−1 h−1 | 47 |
| 8 vol% CH4 | ||||
| 84 vol% Ar | ||||
| 50 mL min−1 | ||||
| 300 W Xe lamp | ||||
| Pt@TiO2 | 2 mg | 3 MPa CH4 | CH3OH: 300 μmol g−1 h−1 | 48 |
| 300 W Xe lamp | ||||
| Rh/TiO2 | 8.6 mg | 1% CH4 | H2: 21.5 mmol g−1 h−1, CO: 21.2 mmol g−1 h−1 | 49 |
| 1% CO2 | ||||
| 98% Ar | ||||
| 150 W Hg–Xe lamp | ||||
| Cr@TiO2 | 10 mg | 3 MPa CH4 | CH3OH: 3.45 μmol g−1 h−1 | 50 |
| 9.5 mL H2O2 (30%) | ||||
| Mild conditions | ||||
| 2% Rh/TiO2 | 20 mg | 10% CH4 | H2: 117 μmol g−1 min−1 | 51 |
| 3% H2O vapor | ||||
| 10 mL min−1 | ||||
| LA-251 Xe lamp, 260 °C | ||||
| Au SA/TiO2 | 50 mg | CH4:CO2 ∼ 3:1 |
H2: 2190 μmol g−1 | This work |
| H2O 10 mL | ||||
| 300 W Xe lamp |
To determine the role of Au single atoms, the photocatalytic activities of Au SA/TiO2 catalysts were compared with those of Au nanoparticle (NP)-based samples. The morphology of typical Au NP-supported TiO2 is depicted in Fig. S5.† Under the same reaction conditions, Au SA/TiO2 exhibited the highest product yield, outperforming Pt NPs/TiO2 and Au NPs/TiO2 (Fig. 7b). These results confirm that photocatalytic activities of the single-atom catalyst are more efficient for driving CH4/CO2 conversion compared to that of nanoparticle-based samples, which could be attributed to its abundant active sites and highly energetic features. Moreover, the effect of Au SA loading on product formation was then investigated. As shown in Fig. 7c, the catalysts with Au SA loading contents in the range of 2–3.5% showed outstanding performances, which were remarkably higher than that of the 0.5% and 5% Au SA loadings. This enhanced performance can be attributed to the improved charge separation rate in the photocatalyst powders.
On top of that, the reactant composition is also a key element and essential to study (Fig. 7d). By varying the pressure ratios of CH4 to CO2, the photocatalytic yields of the catalyst were changed drastically. Particularly, by feeding CH4 and CO2 with ratios of 1:3 and 1:1, the yields of H2 and hydrocarbon products were relatively low, while the amount of O-based products increased to around 900 μmol g−1 for CO formation, compared to the ratio of 3:1, which contains more CH4 composition. It is evident that the main products were preferentially generated from CH4 rather than CO2.
Next, the wavelength-dependent yields were investigated in the visible region and UV region, and under full irradiation for the standard Au SA/TiO2 photocatalyst. The catalyst exhibited impressive activities under full illumination and in the UV range, while the visible region was unable to activate CH4 and CO2 molecules (Fig. 7e). The nonappearance of photocatalytic activities in the visible range could be explained by its deficient energy to excite charge carriers in deep 3d orbitals of the TiO2 crystal for CH4 activation. As another important factor, the cycling catalytic performance tests were conducted to study the lifetime of the catalyst. As shown in Fig. S6,† there are virtually no changes in the surface morphology of the sample before and after four recycling cycles. The activity of Au SA/TiO2 during photocatalytic conversion remained almost stable, with a high yield of 2000 μmol g−1 after four cycles (Fig. 7f). A slight decrease in product yield was observed, which was attributed to the reduction of catalyst efficiency during collection and washing processes between cycles.
To elucidate the reaction pathway and mechanism, the electron paramagnetic resonance analysis was carried out. As shown in Fig. 8, unlike under dark conditions, obvious signals of radical oxygen species (ROS) were detected under light irradiation, which are responsible for reaction activation. Importantly, by conducting the spin-trapping test with the BMPO scavenger, the presence of the hydroxyl (˙OH) radical was definitely confirmed, which played an important role in the CH4 oxidation process.
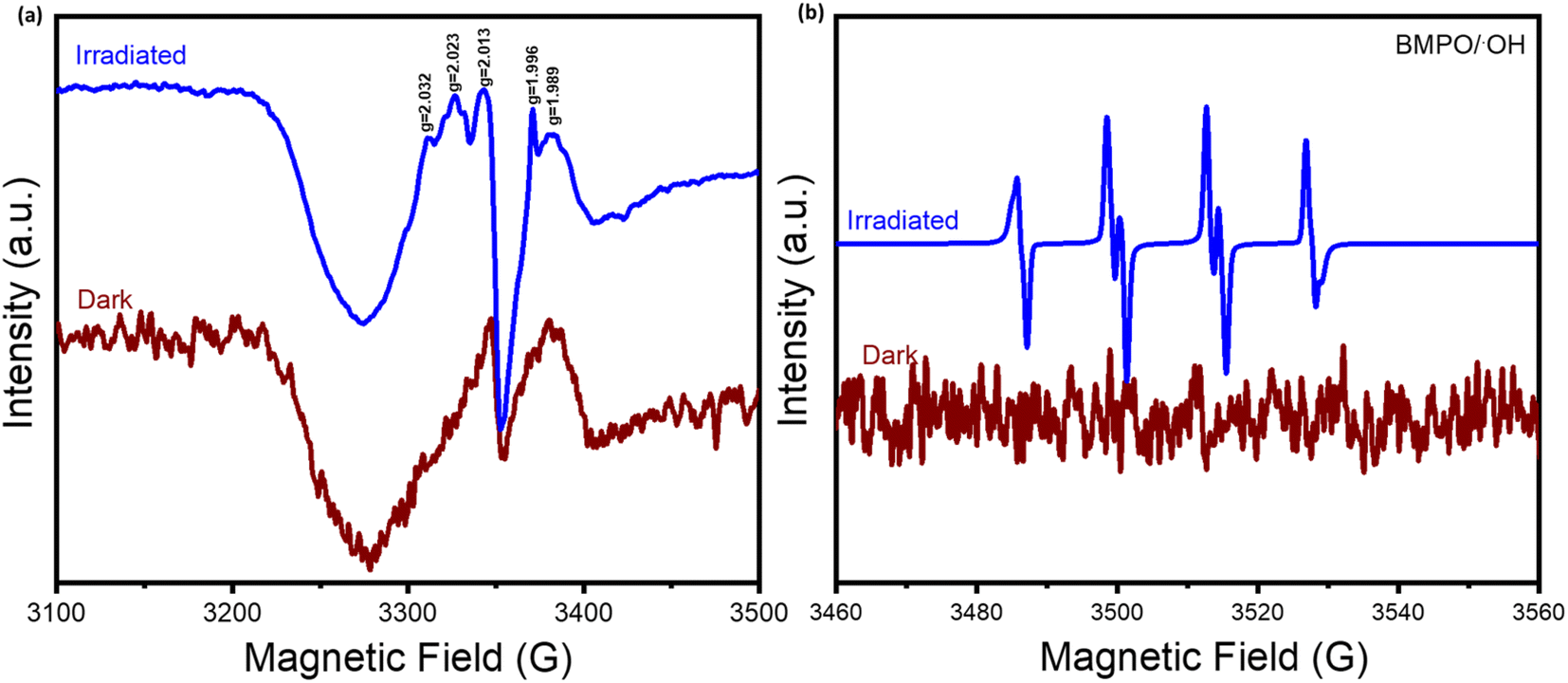 | ||
| Fig. 8 (a) EPR spectra of the Au SA/TiO2 sample under subsequential dark and light conditions at 90 K. (b) The spin trapping tests of Au SA/TiO2 for detecting hydroxyl (˙OH) radical formation by using 5-tert-butoxycarbonyl-5-methyl-1-pyrroline-N-oxide (BMPO) as a spin trapping agent. | ||
Based on the energy band structure and the aforementioned photocatalytic methane oxidation performance, we propose a plausible reaction pathway for CH4 on Au SA/TiO2. Under dark conditions, CH4 adsorbs onto the catalyst surface. Upon illumination, electrons are excited and jump from the valence band to the conduction band, leaving holes behind. As a result, dehydrogenation of CH4 occurs on the catalyst surface, forming methyl radicals (˙CH3). Furthermore, during the photogenerated charge transfer process, Au plays a crucial role in preventing electron–hole pair recombination, which facilitates the oxidation to OH˙ and H+. In subsequent steps, the generated radicals combine with other species to produce H2, C2H6, and C3H8.52–54
4. Conclusion
In summary, we have prepared an atomically dispersed Au catalyst supported on TiO2 nanostructures with a high loading capacity. Photocatalytic CH4 oxidation measurements confirmed that the as-synthesized composite exhibited superior catalytic activity and long-term durability compared to pure TiO2 under mild conditions. The main products, including H2 and C2H6, reached a high yield of 2190 μmol g−1 with approximately 58% selectivity after 8 hours of operation. We anticipate that this simple approach holds great promise for enhancing single-atom engineering without the formation of nanoclusters.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.References
- M. Filonchyk, M. P. Peterson, L. Zhang, V. Hurynovich and Y. He, Sci. Total Environ., 2024, 935, 173359 Search PubMed.
- P. Martin, I. B. Ocko, S. Esquivel-Elizondo, R. Kupers, D. Cebon, T. Baxter and S. P. Hamburg, Energy Sci. Eng., 2024, 12, 3995–4009 CrossRef.
- Y. Jiang, S. Li, Y. Fan and Z. Tang, Angew. Chem., 2024, 136, e202404658 CrossRef.
- M. C. Alvarez-Galvan, N. Mota, M. Ojeda, S. Rojas, R. M. Navarro and J. L. G. Fierro, Catal. Today, 2011, 171, 15–23 CrossRef CAS.
- M. M. Zain and A. R. Mohamed, Renewable Sustainable Energy Rev., 2018, 98, 56–63 CrossRef CAS.
- P. Kumar, T. A. Al-Attas, J. Hu and M. G. Kibria, ACS Nano, 2022, 16, 8557–8618 CrossRef CAS PubMed.
- Q. T. H. Ta, L. T. Nhiem, D. T. Y. Oanh, N. H. Hieu and P. K. T. Nguyen, Vietnam J. Chem., 2024 DOI:10.1002/VJCH.202400155.
- N. Sun, X. Si, L. He, J. Zhang and Y. Sun, Int. J. Hydrogen Energy, 2024, 58, 1249–1265 CrossRef CAS.
- M. J. Molaei, Fuel, 2024, 365, 131159 CrossRef CAS.
- F. Mohamadpour and A. M. Amani, RSC Adv., 2024, 14, 20609–20645 RSC.
- V. H. Dang, T. A. Nguyen, M. V. Le, D. Q. Nguyen, Y. H. Wang and J. C. S. Wu, Chem. Eng. J., 2024, 484, 149213 CrossRef CAS.
- A. Dong, Q. Jiang and Y. Zhou, Int. J. Hydrogen Energy, 2023, 48, 29542–29551 CrossRef CAS.
- T. H. Nguyen, D. T. Tran, N. H. Kim and J. H. Lee, Int. J. Hydrogen Energy, 2023, 48, 32294–32303 CrossRef CAS.
- H. Xu, Y. Zhao, G. He and H. Chen, Int. J. Hydrogen Energy, 2022, 47, 14257–14279 CrossRef CAS.
- Y. Wang, F. Chu, J. Zeng, Q. Wang, T. Naren, Y. Li, Y. Cheng, Y. Lei and F. Wu, ACS Nano, 2021, 15, 210–239 CrossRef CAS PubMed.
- S. S. A. Shah, T. Najam, M. S. Bashir, L. Peng, M. A. Nazir and M. S. Javed, Energy Storage Mater., 2022, 45, 301–322 CrossRef.
- A. Kumar, V. Q. Bui, J. Lee, L. Wang, A. R. Jadhav, X. Liu, X. Shao, Y. Liu, J. Yu, Y. Hwang, H. T. D. Bui, S. Ajmal, M. G. Kim, S. G. Kim, G. S. Park, Y. Kawazoe and H. Lee, Nat. Commun., 2021, 12(1), 1–10 CrossRef PubMed.
- S. Weon, M. J. Suh, C. Chu, D. Huang, E. Stavitski and J. H. Kim, ACS ES&T Eng., 2021, 1, 512–522 Search PubMed.
- D. Wang, B. Zhang, H. Ding, D. Liu, J. Xiang, X. J. Gao, X. Chen, Z. Li, L. Yang, H. Duan, J. Zheng, Z. Liu, B. Jiang, Y. Liu, N. Xie, H. Zhang, X. Yan, K. Fan and G. Nie, Nano Today, 2021, 40, 101243 CrossRef CAS PubMed.
- S. Hejazi, H. Mehdi-pour, C. O. Otieno, J. Müller, S. Pour-Ali, M. Shahsanaei, S. Sarabadani Tafreshi, B. Butz, M. S. Killian and S. Mohajernia, Int. J. Hydrogen Energy, 2024, 51, 222–233 CrossRef CAS.
- R. Rashid, I. Shafiq, M. R. H. S. Gilani, M. Maaz, P. Akhter, M. Hussain, K. E. Jeong, E. E. Kwon, S. Bae and Y. K. Park, Chemosphere, 2024, 349, 140703 CrossRef CAS PubMed.
- Y. Zhao, Y. Kondo, Y. Kuwahara, K. Mori and H. Yamashita, Appl. Catal., B, 2024, 351, 123945 CrossRef CAS.
- M. Chandra Sekhar, B. Purusottam Reddy, C. Kuchi, C. Kamal Basha, F. A. M. Al-Zahrani and R. Mangiri, Ceram. Int., 2024, 50, 38679–38687 CrossRef CAS.
- R. Kaveh, H. Alijani, E. Falletta, C. L. Bianchi, M. Mokhtarifar and D. C. Boffito, Prog. Org. Coat., 2024, 190, 108347 CrossRef CAS.
- N. Nair, V. Gandhi, A. Shukla, S. Ghotekar, V. H. Nguyen and K. Varma, J. Phys.: Condens. Matter, 2024, 36, 413003 CrossRef CAS PubMed.
- J. Xie, R. Jin, A. Li, Y. Bi, Q. Ruan, Y. Deng, Y. Zhang, S. Yao, G. Sankar, D. Ma and J. Tang, Nat. Catal., 2018, 1(11), 889–896 CrossRef CAS.
- Z. Gong, L. Luo, C. Wang and J. Tang, Sol. RRL, 2022, 6 DOI:10.1002/SOLR.202200335.
- S. Song, H. Song, L. Li, S. Wang, W. Chu, K. Peng, X. Meng, Q. Wang, B. Deng, Q. Liu, Z. Wang, Y. Weng, H. Hu, H. Lin, T. Kako and J. Ye, Nat. Catal., 2021, 4(12), 1032–1042 CrossRef CAS.
- A. Das, P. Dagar, S. Kumar and A. K. Ganguli, J. Solid State Chem., 2020, 281, 121051 CrossRef CAS.
- S. Wu, X. Tan, J. Lei, H. Chen, L. Wang and J. Zhang, J. Am. Chem. Soc., 2019, 141, 6592–6600 Search PubMed.
- T. Wang, J. Zhou, D. Li and Z. Ao, Environmental Surfaces and Interfaces, 2024, 2, 19–25 Search PubMed.
- F. Ariaeinezhad, G. Mohammadnezhad, M. Zare, O. Akintola and W. Plass, J. Mater. Chem. A, 2024, 12, 6488–6506 RSC.
- T. D. N. Thi, X. H. Nguyen, H. V. Phung, M. T. Le and L. H. Nguyen, Vietnam Journal of Science, Technology and Engineering, 2023, 65, 81–88 CrossRef.
- N. Roy, K. Bhunia, C. Terashima, A. Fujishima and D. Pradhan, ACS Omega, 2017, 2, 1215–1221 CrossRef CAS PubMed.
- Y. Du, M. Guo, Y. Chen, X. Mo, J. Cao and F. Hu, Anal. Chim. Acta, 2024, 1303, 342462 CrossRef CAS PubMed.
- L. Zeng, C. Dai, B. Liu and C. Xue, J. Mater. Chem. A, 2019, 7, 24217–24221 RSC.
- S. Hejazi, H. Mehdi-pour, C. O. Otieno, J. Müller, S. Pour-Ali, M. Shahsanaei, S. Sarabadani Tafreshi, B. Butz, M. S. Killian and S. Mohajernia, Int. J. Hydrogen Energy, 2024, 51, 222–233 CrossRef CAS.
- J. Chen, Y. Wanyan, J. Zeng, H. Fang, Z. Li, Y. Dong, R. Qin, C. Wu, D. Liu, M. Wang, Q. Kuang, Z. Xie and L. Zheng, ACS Sustainable Chem. Eng., 2018, 6, 14054–14062 CrossRef CAS.
- C. Zhang, T. Uchikoshi and T. Akashi, Mater. Lett., 2024, 367, 136604 CrossRef CAS.
- L. A. Al-Hajji, A. A. Ismail, A. Bumajdad, M. Alsaidi, S. A. Ahmed, F. Almutawa and A. Al-Hazza, J. Mater. Sci.: Mater. Electron., 2021, 32, 19764–19777 CrossRef CAS.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35–54 CrossRef CAS.
- N. Lopez and J. K. Norskov, Surf. Sci., 2002, 515, 175–186 CrossRef CAS.
- L. Torrente-Murciano, B. Solsona, S. Agouram, R. Sanchis, J. M. López, T. García and R. Zanella, Catal. Sci. Technol., 2017, 7, 2886–2896 RSC.
- N. González-Ballesteros, P. M. Martins, C. J. Tavares and S. Lanceros-Méndez, J. Ind. Eng. Chem., 2025, 143, 526–537 CrossRef.
- F. Boccuzzi, A. Chiorino, M. Manzoli, D. Andreeva and T. Tabakova, J. Catal., 1999, 188, 176–185 CrossRef CAS.
- W. Li, Y. Ren, Z. Xie, Y. Wang, H. Zhang, D. Peng, H. Shen, H. Shi, J. Cai, P. Wang, T. Zhang and Z. Zhao, Nano Res., 2024, 17, 3844–3852 CrossRef CAS.
- Q. Li, H. Wang, M. Zhang, G. Li, J. Chen and H. Jia, Angew. Chem., Int. Ed., 2023, 62, e202300129 CrossRef CAS PubMed.
- Y. Sun, C. Bo, Z. Cheng, X. Zhang, J. Liu and L. Piao, Nano Res., 2023, 16, 12942–12948 CrossRef CAS.
- M. Kushida, A. Yamaguchi and M. Miyauchi, J. Energy Chem., 2022, 71, 562–571 CrossRef CAS.
- Q. Shen, C. Cao, R. Huang, L. Zhu, X. Zhou, Q. Zhang, L. Gu and W. Song, Angew. Chem., Int. Ed., 2020, 59, 1216–1219 CrossRef CAS PubMed.
- H. Song, X. Meng, Z. J. Wang, Z. Wang, H. Chen, Y. Weng, F. Ichihara, M. Oshikiri, T. Kako and J. Ye, ACS Catal., 2018, 8, 7556–7565 CrossRef CAS.
- X. Zhang, Y. Wang, K. Chang, S. Yang, H. Liu, Q. Chen, Z. Xie and Q. Kuang, Appl. Catal., B, 2023, 320, 121961 CrossRef CAS.
- X. Lu, H. Luo, B. Xu, Z. Liu, Y. Cao, K. Li, X. Yang, L. Xie, T. Guan, W. Zhu and Y. Zhou, Chem. Commun., 2024, 60, 14276–14279 RSC.
- T. M. Vo, T. M. H. Nguyen and C. W. Bark, ACS Appl. Electron. Mater., 2024, 6, 4391–4405 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00947a |
| This journal is © The Royal Society of Chemistry 2025 |