
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
pH-Sensitive oligopeptide magnetic mesoporous silica beads for deoxyribonucleic acid extraction†
Sihua
Qian‡
 b,
Yiting
Wang‡
ab,
Junjie
Fan
b,
Tong
Kong
b,
Yuhui
Wang
b,
Kaizhe
Wang
b,
Yufeng
Liao
*c,
Li
Wang
*a and
Jianping
Zheng
*b
b,
Yiting
Wang‡
ab,
Junjie
Fan
b,
Tong
Kong
b,
Yuhui
Wang
b,
Kaizhe
Wang
b,
Yufeng
Liao
*c,
Li
Wang
*a and
Jianping
Zheng
*b
aCollege of Chemistry, Jilin Normal University, Siping 136000, P. R. China. E-mail: liwang@jlnu.edu.cn
bNingbo Cixi Institute of Biomedical Engineering, Laboratory of Advanced Theranostic Materials and Technology, Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, Ningbo 315300, P. R. China. E-mail: zhengjianping@nimte.ac.cn
cDepartment of Clinical Laboratory, Ningbo No. 2 Hospital, Ningbo 315010, P.R. China. E-mail: liaoyufengs@163.com
First published on 21st January 2025
Abstract
Exploring novel synthesis strategies for magnetic beads to extract nucleic acids is of great significance in the field of in vitro diagnostics. In the present research, monodisperse magnetic mesoporous silica beads were synthesized via the thermolysis reaction of Fe(acac)3 by using large-pore dendritic silica colloids as templates, and were further functionalized with a highly pH-sensitive histidine-glutamate co-oligopeptide for deoxyribonucleic acid extraction. The large-pore dendritic silica colloid scaffolds were utilized for high-density incorporation of superparamagnetic iron oxide nanoparticles within the vertical channels. The morphology and properties of the as-prepared pH-sensitive oligopeptide magnetic mesoporous silica beads were evaluated by transmission electron microscopy, scanning electron microscopy, vibrating sample magnetometry, X-ray photoelectron spectroscopy, X-ray diffraction testing and so on. The average size of the obtained magnetic beads was 370 nm in diameter with a narrow size distribution. The saturation magnetization and magnetic content of the resultant magnetic beads were 25 emu g−1 and 59%, respectively. Moreover, the magnetic mesoporous silica beads exhibited an obvious pH-responsive behavior. Due to these remarkable features, successful deoxyribonucleic acid capture using the as-prepared pH-sensitive oligopeptide magnetic mesoporous silica beads was achieved.
Introduction
Deoxyribonucleic acid (DNA) extraction is an essential process in molecular biology and a fundamental step for subsequent sequencing, amplification, and biodetection.1–8 The traditional method for isolation of DNA based on phenol/chloroform extraction suffers from several drawbacks, including using highly toxic solvents, being labor- and time-consuming, and being unsuitable for processing of trace samples.9–11 Nowadays, employing various solid-phase supports for DNA extraction has become more and more attractive,12–16 among which magnetic beads are preferred due to their easy manipulation and low cost.17–22 Magnetic DNA purification is a clear improvement upon centrifuge-dependent isolation techniques when semi-automatic or fully automatic systems are considered.23–26 With the assistance of an external magnetic field, magnetic beads as solid-phase adsorbents can be removed readily.Various methods have been developed for the preparation of magnetic beads,27–31 among which Ugelstad's activated swelling method is the most successful route and has formed a series of commercial products, Dynabeads.32–36 The preparation process of the activated swelling method generally involves the fabrication of monodisperse macroporous polymeric beads using seed polymerization technology. Subsequently, the internal and external surfaces of the macroporous beads are modified with –SO3 or –NO2 to impart hydrophilic properties to them. The beads are then immersed in an aqueous solution of iron salt, leading to the generation of superparamagnetic Fe3O4 or γ-Fe3O4 within the pores under suitable reaction conditions. Finally, a monomer containing active functional groups is chosen for swelling, polymerization, and coating of the beads to seal off pores and functionalize their surfaces.37 To meet the requirements of rapid isolation, magnetic beads produced by swelling methods have a micron-level size with a diameter typically ranging from 1 to 100 μm to contain enough magnetic components.33,38 However, micron-sized magnetic beads exhibit poor suspension and quick sedimentation, which are unfavorable for DNA extraction.
Compared with organic polymeric materials, inorganic ones possess the advantages of low toxicity, special structures, stable physicochemical properties and so on.39,40 Among inorganic substrates, mesoporous silica materials have been a hot topic due to their larger surface area and specific mesoporous channels, which can dramatically increase the binding capacity of target molecules onto the surface and pores.41–45 Furthermore, the integration of magnetic nanoparticles with mesoporous silica materials has attracted considerable attention in recent years owing to their excellent properties.46–51 Magnetic mesoporous silica nanocomposites are generally core–shell-type, which would reduce the magnetic response of the pure magnetic nanoparticles. Therefore, an alternative magnetic nanoparticles-mesoporous silica material integration type needs to be urgently developed.
Herein, to increase the suspension and magnetic response of magnetic beads, magnetic mesoporous silica beads with several hundreds of nanometers were prepared, and were further functionalized with a highly pH-sensitive histidine-glutamate (HE) co-oligopeptide for DNA extraction, as illustrated in Fig. 1. We first prepared dendritic silica colloids (dSiO2) with uniform size and expected monodispersity through an anion-assisted approach. Then, the Fe3O4@dSiO2 composite was obtained via the thermolysis reaction of Fe(acac)3 by using large-pore dSiO2 as templates, and the ultra-large pore channels of dSiO2 allowed the complete confinement of superparamagnetic Fe3O4 nanoparticles in the interior of the supports. Finally, the pH-responsive Fe3O4@dSiO2-(HE)10 composite was acquired via conjugate addition reactions between thiol functional groups and maleimide moieties. Oligopeptides have been particularly widely used to provide physical and biological stabilization to magnetic nanoparticles.52 The structures, properties, and surface morphologies of dSiO2, Fe3O4@dSiO2 and Fe3O4@dSiO2-(HE)10 were investigated by transmission electron microscopy, scanning electron microscopy, dynamic light scattering, inductively coupled plasma (ICP, magnetic content 59%) spectrometry, and vibrating sample magnetometry, respectively. Meanwhile, DNA separation experiments were carried out with these particles to evaluate their adsorption ability.
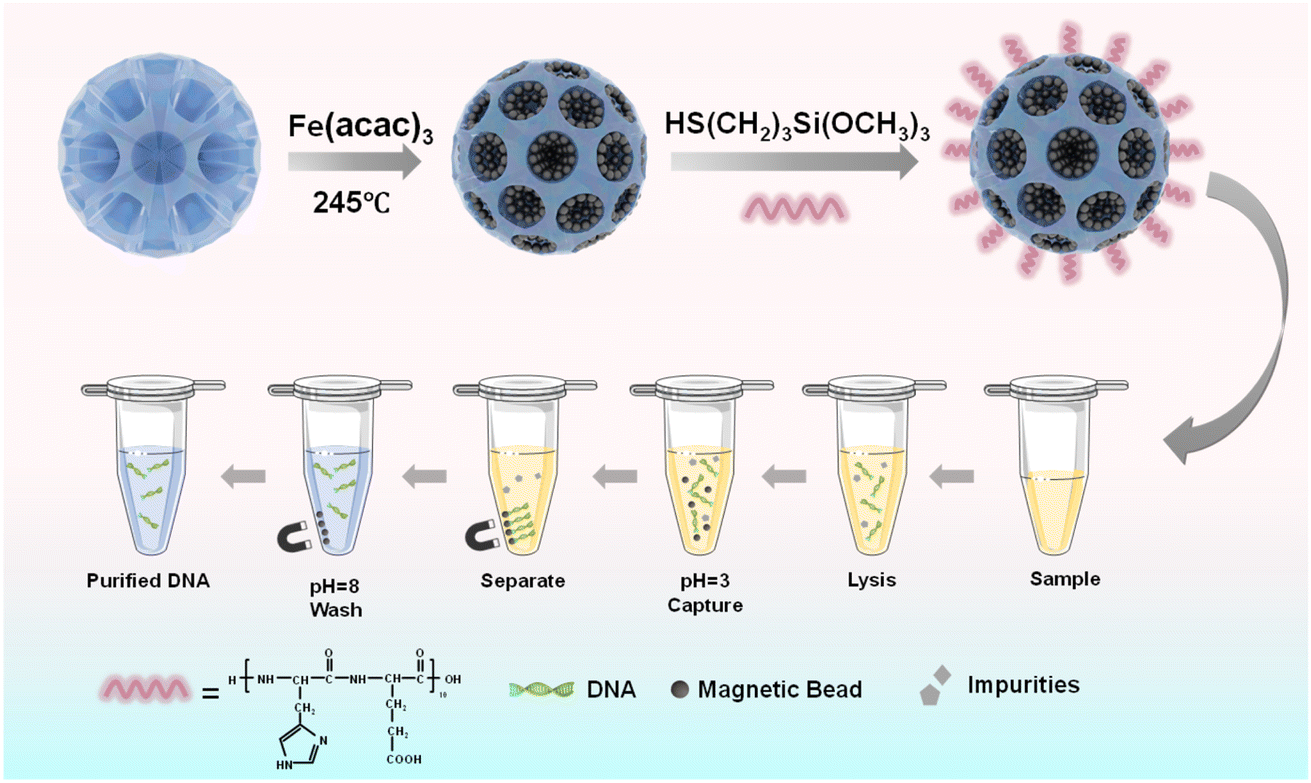 | ||
| Fig. 1 Schematic illustrations of the preparation process of pH-sensitive oligopeptide magnetic mesoporous silica beads and their application in deoxyribonucleic acid extraction. | ||
Results and discussion
Characterization of Fe3O4@dSiO2-(HE)10
The dSiO2 templates were synthesized using an anion-assisted approach, using cationic surfactant CTAB and NaSal as structure directing agents, TEA as a catalyst and TEOS as a silica source. In the TEM image of dSiO2, shown in Fig. 2a, the central-radial pores can be clearly seen. The SEM image of dSiO2 revealed an average pore-width of 20 nm at the external surface (Fig. 3a), and the large pore channels presented in the SEM image indicated the high accessibility of dSiO2's inner surface for Fe3O4 immobilization. Additionally, the dSiO2 particles were monodisperse and highly uniform in size. The Fe3O4 nanoparticles were synthesized by the thermolysis reaction of Fe(acac)3 in 2-pyrrolidone at 245 °C. As shown in Fig. S1,† the diameter of Fe3O4 nanoparticles was 5–10 nm, and 2-pyrrolidone was chosen as both a stabilizer and solvent. To obtain a strong magnetic response and uniform magnetic nanoparticles, the dSiO2 that can serve as substrates for Fe3O4 deposition was added before the thermal decomposition of the iron precursor. As presented in Fig. 2b and 3b, all the radial channels of dSiO2 were occupied by a dense Fe3O4 layer, and the high coverage of small sized Fe3O4 confirmed a strong magnetic response and maintained the superparamagnetism of the Fe3O4@dSiO2 composites. After modification with the pH-sensitive histidine-glutamate co-oligopeptide via conjugate addition reactions between thiol functional groups and maleimide moieties, the morphology of Fe3O4@dSiO2-(HE)10 was similar to that of Fe3O4@dSiO2 (Fig. 2c). The distribution of hydrated particle sizes of dSiO2, Fe3O4@dSiO2 and Fe3O4@dSiO2-(HE)10 in an aqueous medium was also characterized, as presented in Fig. S2.† It can be found in Fig. S2† that the particle sizes of dSiO2, Fe3O4@dSiO2 and Fe3O4@dSiO2-(HE)10 in the aqueous medium satisfied the normal distribution and that the particle size distribution was narrow and symmetric, indicating there was little presence of either very small particles or very large ones. Most of the particles of dSiO2, Fe3O4@dSiO2 and Fe3O4@dSiO2-(HE)10 were distributed around the average hydrated particle sizes of 340 nm, 370 nm and 370 nm, respectively. The scanning transmission electron microscopy (STEM) image of Fe3O4@dSiO2-(HE)10 illustrated the even and compact distribution of Fe3O4 confined in the silica matrix (Fig. 2d). The energy-dispersive X-ray spectroscopy (EDS) mapping of Fe3O4@dSiO2-(HE)10 (Fig. 2e–i) confirmed the constituent elements of Fe3O4 (Fe), dSiO2 (Si and O), and histidine-glutamate co-oligopeptide modification (N and S). | ||
| Fig. 2 TEM images of dSiO2 templates (a), Fe3O4@dSiO2 (b), and Fe3O4@dSiO2-(HE)10 (c). STEM image (d) and EDS elemental (Fe, N, O, Si, and S) mapping images (e–i) of a single Fe3O4@dSiO2-(HE)10 nanosphere. | ||
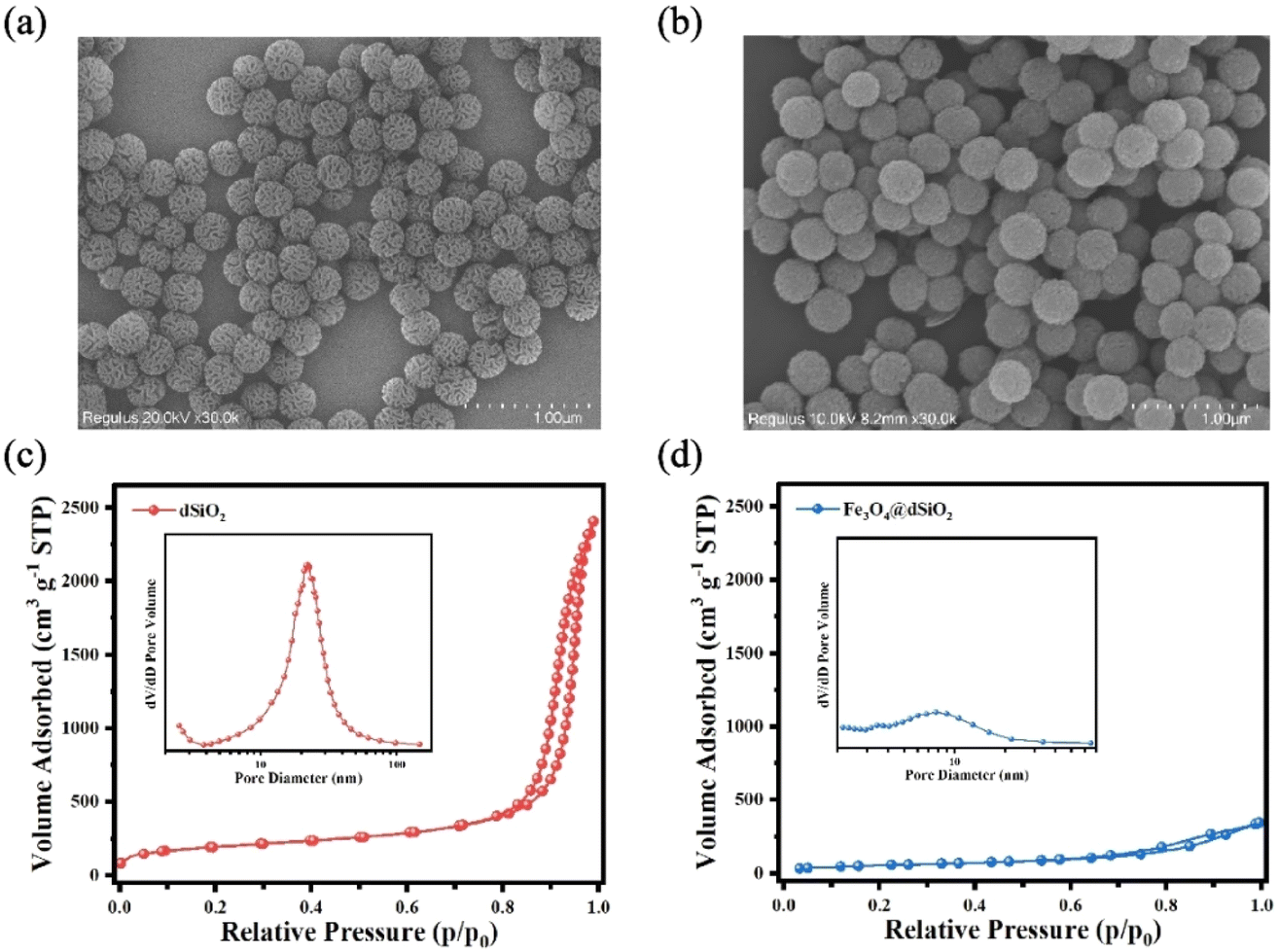 | ||
| Fig. 3 SEM images of dSiO2 templates (a) and Fe3O4@dSiO2 (b); nitrogen adsorption–desorption isotherms and the BJH pore size distributions (inset) of dSiO2 (c) and Fe3O4@dSiO2 (d). | ||
The nitrogen adsorption–desorption isotherms reflected the evolution of the pore structure during the synthesis of Fe3O4@dSiO2 composites, as shown in Fig. 3c and d. They all exhibited typical IV-type curves with a hysteresis loop of the H2-type, which proved that the mesoporous structure of dSiO2 was not altered after loading Fe3O4. The dSiO2 templates exhibited large Brunauer–Emmett–Teller (BET) surface area (678.68 m2 g−1) and total pore volume (13.74 cm3 g−1). After loading Fe3O4, the BET surface area and total pore volume of dSiO2 decreased remarkably to 206.24 m2 g−1 and 0.53 cm3 g−1 due to the blockage of loaded Fe3O4 in the pore channels of dSiO2, which agreed well with the smooth spherical morphology. The Barrett–Joyner–Halenda (BJH) pore size distributions of the microspheres also indicated a shrinking of the pore size from 23.15 nm (inset in Fig. 3c) to 9.25 nm (inset in Fig. 3d), which was consistent with the TEM observations.
X-ray photoelectron spectroscopy (XPS) was utilized to analyse the elemental composition and chemical valence changes of Fe3O4@dSiO2-(HE)10. As shown in Fig. 4a, peaks at 711, 532.3, 400.4, 285.5 and 100.35 eV of the prepared Fe3O4@dSiO2-(HE)10 were assigned to binding energies of Fe 2p, O 1s, N 1s, C 1s and Si 2p, respectively, which indicated that the sample was composed of the elements Fe, O, N, C, and Si. The spectra for Fe 2p in the Fe3O4@dSiO2-(HE)10 nanoparticles exhibited peaks at 711 eV and 724.3 eV for Fe 2p3/2 and Fe 2p1/2, respectively (Fig. 4b), which revealed that Fe3O4 was successfully prepared. The spectra for Si 2p exhibited a Si peak at 100.35 eV, which corresponded to Si–O, as described in Fig. 4c. Since the oligopeptide contained numerous –NH2 groups, the peak for the amino group confirmed the successful modification of the oligopeptide. The spectra for N 1s had a peak at 400.4 eV (Fig. 4d), which corresponded to the –NH2 group, demonstrating the presence of amino groups in nanoparticles. The O 1s binding energies at 531.3 eV and 533.5 eV were attributed to the lattice oxygen in Fe3O4 and Si–O bonds in SiO2, respectively, as presented in Fig. 4e. As shown in Fig. 4f, the C 1s peak can be deconvoluted into three peaks at 285.5, 256.4 and 289.2 eV, corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–N and CO, respectively. The XPS results also confirmed the successful formation of Fe3O4@dSiO2-(HE)10.
C, C–N and CO, respectively. The XPS results also confirmed the successful formation of Fe3O4@dSiO2-(HE)10.
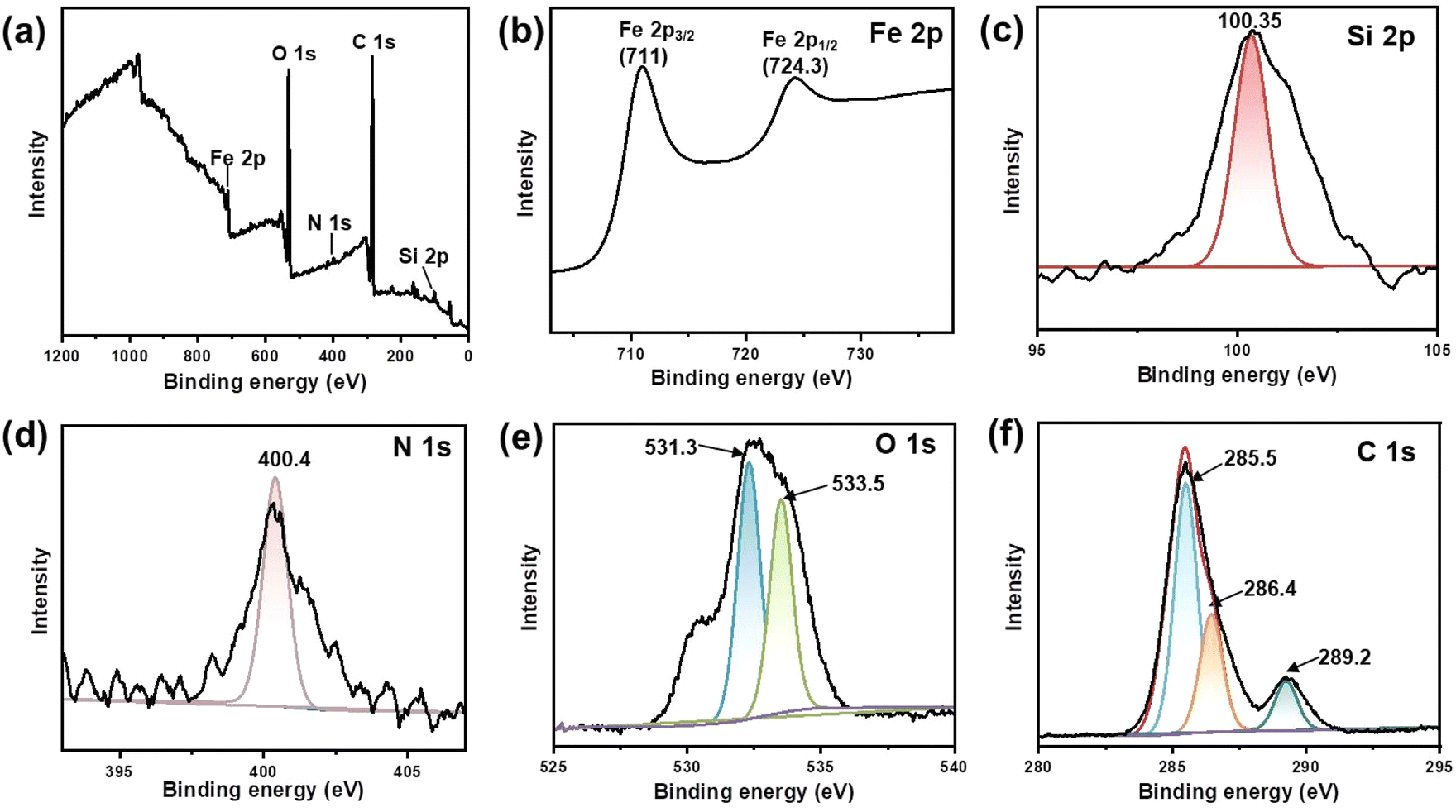 | ||
| Fig. 4 XPS spectra of Fe3O4@dSiO2-(HE)10. (a) Survey spectrum, (b) Fe 2p spectrum, (c) Si 2p spectrum, (d) N 1s spectrum, (e) O 1s spectrum, and (f) C 1s spectrum. | ||
The zeta potentials of Fe3O4@dSiO2, Fe3O4@dSiO2-SH and Fe3O4@dSiO2-(HE)10 were measured in binding solution (10 mM BR, pH 3.0), and their zeta potentials were +5.9, −0.5 and +17.5, respectively, as shown in Fig. 5a. The zeta potentials of Fe3O4@dSiO2-(HE)10 in different pH solutions indicated that the isoelectric point of Fe3O4@dSiO2-(HE)10 was 3.75 (Fig. 5b). The XRD pattern showed characteristic diffractograms of dSiO2, Fe3O4@dSiO2 and Fe3O4@dSiO2-(HE)10 (Fig. 5c). The 30.3°, 35.6°, 43.2°, 53.5°, 57.4° and 62.7° diffraction peaks of Fe3O4@dSiO2 at 2θ values were assigned to the crystal planes (220), (311), (400), (422), (511), and (440), respectively, which confirmed the successful synthesis of Fe3O4 with a cubic spinel structure (JCPDS# 19-0629). Fe3O4@dSiO2-(HE)10 showed a similar diffraction peak to Fe3O4@dSiO2, suggesting that Fe3O4@dSiO2-(HE)10 also had a typical magnetite structure. Moreover, the (HE)10 modification revealed no effect on the crystal structure of the functional unit. The broad diffraction peak near 22.7° was indexed to amorphous mesoporous SiO2. These diffraction peaks further confirmed that Fe3O4@dSiO2-(HE)10 was synthesized. Magnetic characteristics of Fe3O4, Fe3O4@dSiO2 and Fe3O4@dSiO2-(HE)10 at 300 K were investigated, and the magnetic hysteresis curves revealed their superparamagnetic features, as shown in Fig. 5d. The saturation magnetizations of Fe3O4, Fe3O4@dSiO2 and Fe3O4@dSiO2-(HE)10 were 39.47, 24.87 and 28.21 emu g−1, respectively, indicating a complete and quick magnetic separation of them from solution. The inset in Fig. 5d shows a very strong magnetic response of the prepared magnetic mesoporous silica beads. Typically, 15 s were long enough to completely collect the beads, suspended in pure water in a 1 × 1 cm cuvette. According to Fig. 5d, we have drawn normalized magnetic hysteresis curves of Fe3O4, Fe3O4@dSiO2 and Fe3O4@dSiO2-(HE)10 in Fig. S3.† As shown in Fig. S3,† the saturation magnetization of Fe3O4@dSiO2 was about 60% of that of Fe3O4, which indicated a decreased saturation magnetization of Fe3O4 after conjugation with dSiO2. Additionally, the (HE)10 modification had no effect on the saturation magnetization of Fe3O4@dSiO2.
 | ||
| Fig. 5 (a) Zeta potentials of Fe3O4@dSiO2, Fe3O4@dSiO2-SH and Fe3O4@dSiO2-(HE)10 at pH = 3.0; (b) zeta potentials of Fe3O4@dSiO2-(HE)10 in different pH solutions, and pH 3.75 turned out to be the isoelectric point of Fe3O4@dSiO2-(HE)10; (c) XRD patterns of Fe3O4, Fe3O4@dSiO2 and Fe3O4@dSiO2-(HE)10 nanospheres; (d) the magnetic hysteresis curves of Fe3O4, Fe3O4@dSiO2 and Fe3O4@dSiO2-(HE)10 at 300 K, and the inset shows pictures of the aqueous suspension of Fe3O4@dSiO2-(HE)10 (left) and the same suspension magnetically separated after 15 s (right). | ||
Application of Fe3O4@dSiO2-(HE)10
To investigate the potential of Fe3O4@dSiO2-(HE)10 for DNA extraction, a 100 bp DNA ladder marker was selected as a model to determine the recovery yields of DNA. The zeta potentials of Fe3O4@dSiO2-(HE)10 in different pH solutions (Fig. 5b) indicated that the isoelectric point of Fe3O4@dSiO2-(HE)10 was 3.75, and a certain amount of DNA can be captured by positively charged Fe3O4@dSiO2-(HE)10 in the pH range of 2.0–3.5. When the pH was above 3.75, the Fe3O4@dSiO2-(HE)10 and DNA were both negatively charged, leading to almost no DNA capture by Fe3O4@dSiO2-(HE)10 in the pH range of 4.0–10.0. Therefore, the solutions with pH = 3.0 and pH = 8.0 were selected as the binding solution for DNA capture and the elution solution for DNA release, respectively. As shown in PAGE analysis (Fig. 6a) and agarose gel electrophoresis (Fig. 6b), the lane of Fe3O4@dSiO2-(HE)10 was similar to that of the 100 bp DNA marker, while the lane of sulfhydrated Fe3O4@dSiO2 without modification of (HE)10 was different from that of the 100 bp DNA marker. The above results revealed that the obtained Fe3O4@dSiO2-(HE)10 magnetic beads could be used for DNA extraction based on the electrostatic interactions between Fe3O4@dSiO2-(HE)10 and DNA.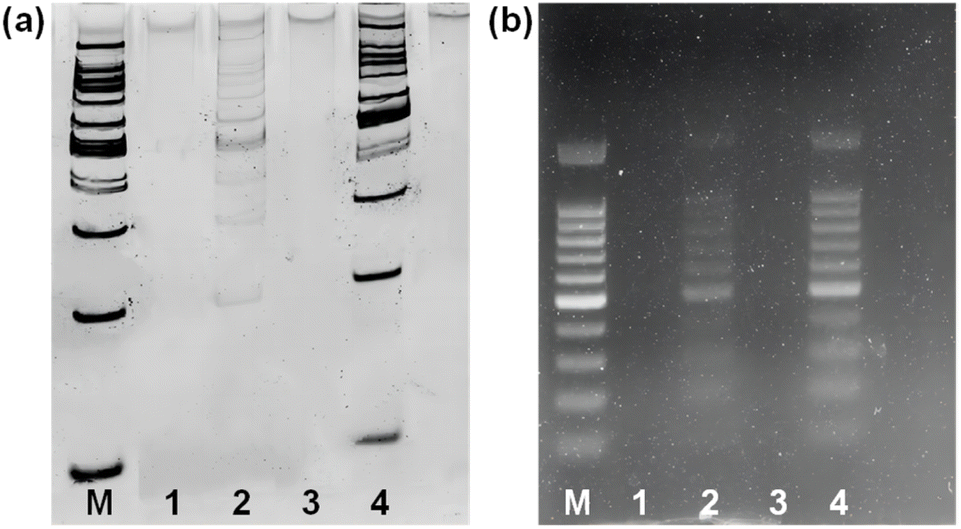 | ||
| Fig. 6 (a) 6% PAGE analysis; (b) 1% agarose gel electrophoresis of the DNA marker. Lane M: 100 bp DNA marker; lane 1: supernatant buffer (pH = 3.0) of sulfhydrated Fe3O4@dSiO2; lane 2: elution buffer of sulfhydrated Fe3O4@dSiO2; lane 3: supernatant buffer (pH = 8.0) of Fe3O4@dSiO2-(HE)10; lane 4: elution buffer of Fe3O4@dSiO2-(HE)10. | ||
The DNA capture efficiency of Fe3O4@dSiO2-(HE)10 compared to that of Dynabeads MyOne Silane was investigated. As presented in agarose gel electrophoresis (Fig. S4†), the lane of Dynabeads MyOne Silane was similar to that of the 100 bp DNA marker, and the results were nearly the same those for Fe3O4@dSiO2-(HE)10, indicating the similar DNA capture efficiency of Fe3O4@dSiO2-(HE)10 and Dynabeads MyOne Silane. Finally, the potential applications of the obtained magnetic beads in DNA purifying for NGS library preparation were demonstrated, as described in Fig. S5.†
Conclusions
In summary, monodisperse pH-sensitive oligopeptide magnetic mesoporous silica beads were successfully prepared via the thermolysis reaction of Fe(acac)3 by using large-pore dendritic silica colloids as templates. The ultra-large pore channels of dendritic silica colloids allowed the complete confinement of superparamagnetic iron oxide nanoparticles in the interior of the supports, enabling controlled dimension and monodispersity of the obtained magnetic beads. Due to the effective loading of superparamagnetic iron oxide nanoparticles, the as-prepared magnetic beads possessed an extremely high magnetic content of 59% and showed a strong magnetic response in magnetic fields. Furthermore, the magnetic mesoporous silica beads exhibited a pH-responsive behavior after modification with a highly pH-sensitive histidine-glutamate co-oligopeptide. All these remarkable features together with the relatively narrow dispersity of the resultant magnetic beads made them highly desirable for bioapplications, as demonstrated by deoxyribonucleic acid capture experiments.Experimental
Reagents and instruments
Iron(III) acetylacetonate (Fe(acac)3), cetyltrimethylammonium bromide (CTAB), sodium salicylate (NaSal), triethanolamine (TEA), and tetraethyl orthosilicate (TEOS) were purchased from Aladdin Chemical Reagent Co., Ltd. Hydrochloric acid (HCl), methanol, ethanol, (3-mercaptopropyl)trimethoxysilane (MPTMS), 2-pyrrolidone, and ammonia aqueous solution (25–28%) were received from Sinopharm Chemical Reagent Co., Ltd. Maleimide modified (HE)10 was obtained from Shanghai HongTide Biotechnology Co., Ltd. All chemicals were used as received without purification.Zeta potentials and hydrodynamic diameters were measured using a Zetasizer Nano ZS dynamic light scattering particle size analyzer (Malvern, U.K.). Transmission electron microscopy (TEM) images were recorded using an FEI-Talos-S electron microscope operated at an accelerating voltage of 200 kV. The morphology of the magnetic beads was characterized using scanning electron microscopy (SEM, Regulus 8230). Magnetization curves of Fe3O4@dSiO2 and Fe3O4@dSiO2-(HE)10 were measured on a vibrating sample magnetometer (PPMS-1, Quantum Design, San Diego, USA) at 300 K. The magnetic content was determined using an inductively coupled plasma atomic emission spectrometer (Spectro, Arcos, Germany). Small-angle powder X-ray diffraction (XRD) testing was performed using a Rigaku D/max-2000 X-ray powder diffractometer (Rigaku) with Cu/Kα radiation of 1.5405 Å. Surface states of the magnetic mesoporous silica beads were determined by X-ray photoelectron spectroscopy (XPS, SUPRA).
Preparation of Fe3O4
In a typical preparation of Fe3O4, a solution of 0.7063 g of Fe(acac)3 in 20 mL of 2-pyrrolidone was first purged with nitrogen to remove oxygen, and then heated to 245 °C and refluxed for 30 min. After cooling to room temperature, the product was collected by magnetic separation and washed several times with ethanol. The final black product was re-dispersed in 10 mL of ethanol.Synthesis of Fe3O4@dSiO2
The dendritic silica colloids (dSiO2) were synthesized according to a reported method with slight modifications. Briefly, 0.068 g of TEA was added to 25 mL of deionized water and stirred gently at 80 °C for 15 min. Then, 0.38 g of CTAB and 0.218 g of NaSal were added to the above solution, followed by stirring for another 15 min. Finally, 4 mL of TEOS was added into the water–TEA–CTAB–NaSal solution, which was stirred at 80 °C for another 3 h. The white product was collected by high-speed centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm) and washed several times with ethanol. The residual template was removed by HCl/methanol extraction at 60 °C for 12 h, and the purified product was re-dispersed in 30 mL of ethanol.
000 rpm) and washed several times with ethanol. The residual template was removed by HCl/methanol extraction at 60 °C for 12 h, and the purified product was re-dispersed in 30 mL of ethanol.
The typical synthesis of Fe3O4@dSiO2 was as follows: 20 mL of a 2-pyrrolidone solution containing 0.7063 g of Fe(acac)3 and 0.05 g of dSiO2 was purged with nitrogen for 30 min to remove oxygen. Then the reaction system was heated to 245 °C and refluxed for 30 min. After cooling to room temperature, the product was collected by magnetic separation and washed several times with ethanol. The final black product was re-dispersed in 10 mL of ethanol.
Preparation of Fe3O4@dSiO2-(HE)10
10 mL of ethanol solution containing Fe3O4@dSiO2 was mixed with 56 μL of MPTMS and 250 μL of ammonia, and the mixture was stirred (600 rpm) at room temperature for 12 h. The obtained product was collected by magnetic separation and washed several times with ethanol. The obtained thiolated Fe3O4@dSiO2 (Fe3O4@dSiO2-SH) was re-dispersed in 10 mL of ethanol.Conjugate addition reactions between thiolated Fe3O4@dSiO2 (0.05 g) and maleimide modified (HE)10 (0.001 g) were carried out in HEPES buffer (10 mM, pH 7.4). After being stirred for 30 min, the product was collected by magnetic separation and washed several times with ethanol/deionized water. The final product was re-dispersed in 1.0 mL of deionized water.
DNA capture and elution assays with Fe3O4@dSiO2-(HE)10
To investigate the potential for DNA extraction from real samples, a 100 bp DNA ladder marker was selected as a model to determine the recovery yields of DNA using Fe3O4@dSiO2-(HE)10. The DNA ladder marker (1 μL) was dissolved in 1× TE buffer (9 μL, pH = 7.4) to prepare a DNA standard solution. The DNA standard solution, 25 μL of binding solution (10 nM BR buffer at pH = 3.0), and 2 μL of Fe3O4@dSiO2-(HE)10 suspension (30 mg mL−1) were mixed fully. Then the mixture was incubated at room temperature for 5 min. Afterwards, a magnetic stand was employed for magnetic separation and the supernatant was carefully removed. The DNA–Fe3O4@dSiO2-(HE)10 conjugates were washed twice with 100 μL of 80% (v/v) ethanol and allowed to dry at room temperature. Then the adsorbed DNA molecules were eluted from the conjugates by addition of 15 μL of elution solution (10 mM BR buffer at pH = 8.0) under vigorous shaking for 5 min. Finally, an appropriate amount of the supernatant was carefully taken for gel electrophoresis.Data availability
The data supporting this study are available within the article and its ESI.†Author contributions
Sihua Qian: formal analysis, writing – original draft, designed the study, performed experiments, analyzed the results, and wrote the manuscript. Yiting Wang: performed DNA capture and elution assays, analyzed the results, and wrote part of the manuscript. Junjie Fan: drew Fig. 3c, d, 4 and 5. Tong Kong: designed and drew Fig. 1. Yuhui Wang: provided valuable suggestions and discussed the results. Kaizhe Wang: provided valuable suggestions and discussed the results. Yufeng Liao: analyzed the results, conceived the idea and designed the study. Li Wang: conceived the idea and designed the study, writing – review & editing. Jianping Zheng: conceived the idea and designed the study, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the Ningbo Science and Technology Bureau (No. 2023S179, No. 2024Z044 and No. 2024Z183).References
- M. Volaric, D. Veseljak, B. Mravinac, N. Mestrovic and E. Despot-Slade, Genes, 2021, 12, 1114–1125 CrossRef CAS PubMed.
- S. Jaudou, M. L. Tran, F. Vorimore, P. Fach and S. Delannoy, PLoS One, 2022, 17, e0270751 CrossRef CAS PubMed.
- T. B. McAtee, L. J. Pinnell, S. A. Powledge, C. A. Wolfe, P. S. Morley and J. T. Richeson, Front. Microbiol., 2023, 14, 1203498–1203513 CrossRef PubMed.
- R. Sivakumar and N. Y. Lee, Analyst, 2023, 148, 3153–3168 RSC.
- L. H. Thang, W. Han, J. Shin and J. H. Shin, Sens. Actuators, B, 2023, 375, 132948–132956 CrossRef CAS.
- Z. J. Fei, C. Cheng, R. B. Wei, G. L. Tan and P. F. Xiao, Chem. Eng. J., 2022, 431, 133953–133964 CrossRef CAS.
- Y. H. Cheng, C. H. Wang, K. F. Hsu and G. B. Lee, Anal. Chem., 2022, 94, 4311–4318 CrossRef CAS PubMed.
- Y. A. Katayama, S. Hayase, R. Iwamoto, T. Kuroita, T. Okuda, M. Kitajima and Y. Masago, Sci. Total Environ., 2024, 907, 167866–167875 CrossRef CAS PubMed.
- A. Ayoib, U. Hashim, S. C. B. Gopinath and M. K. M. Arshad, Appl. Microbiol. Biotechnol., 2017, 101, 8077–8088 CrossRef CAS PubMed.
- R. Dieki, E. N. Emvo and J. P. Akue, PLoS One, 2022, 17, e0265582 CrossRef CAS PubMed.
- I. Lutz, J. Miranda, P. Santana, T. Martins, C. Ferreira, I. Sampaio, M. Vallinoto and G. E. Gomes, PLoS One, 2023, 18, e0282369 CrossRef CAS PubMed.
- C. Teixeira, A. Ipatov, J. Carvalho, A. Purwidyantri, N. Fontes and M. Prado, ACS Omega, 2023, 35, 31738–31746 CrossRef PubMed.
- X. H. Du, M. Lu, H. Z. Lan, Z. D. Cai, D. D. Pan and Y. C. Wu, J. Food Compos. Anal., 2024, 127, 105977–105985 CrossRef CAS.
- S. De Silva, I. Ocaña-Rios, C. Cagliero, M. R. Gostel, G. Johnson and J. L. Anderson, Anal. Chim. Acta, 2023, 1245, 340858–340867 CrossRef CAS PubMed.
- H. Seong, J. Park, M. Bae and S. Shin, Biomedicines, 2022, 10, 1883–1892 CrossRef CAS PubMed.
- S. M. Lee, Y. H. Nai, E. H. Doeven, H. K. Balakrishnan, D. Yuan and R. M. Guijt, Talanta, 2024, 266, 125006–125013 CrossRef CAS PubMed.
- A. Z. Hu, L. Chen, X. L. Geng, L. Zhu, Y. Liu, K. Yang, H. Q. Zhu and C. C. Zhu, Forensic Sci. Int.:Genet., 2023, 67, 102932–102941 CrossRef CAS PubMed.
- A. Szymczyk, M. Drozd, A. Kaminska, M. Matczuk, M. Trzaskowski, M. Mazurkiewicz-Pawlicka, R. Ziólkowski and E. Malinowska, Int. J. Mol. Sci., 2022, 23, 8860–8881 CrossRef CAS PubMed.
- M. Fukuchi, M. Hanamura, K. Otake, M. Nakamori, N. Toyoda and K. Matsuda, J. Non-Cryst. Solids, 2023, 609, 122284–122289 CrossRef CAS.
- Q. B. Yuan, Z. Q. Liang, S. J. Wang, P. X. Zuo, Y. Wang and Y. Luo, Colloids Surf., A, 2022, 644, 128831–128840 CrossRef CAS.
- N. V. T. Nguyen, C. Smadja, M. Taverna, L. T. H. Nguyen, S. Descroix and T. D. Mai, Anal. Chim. Acta, 2023, 1255, 341141–341149 CrossRef CAS PubMed.
- Z. M. Tian, H. Yan and Y. Zeng, ACS Appl. Mater. Interfaces, 2024, 16, 14445–14456 CrossRef CAS PubMed.
- G. Kibar, B. Sariarslan, S. Doganay, G. Yildiz, O. B. Usta and B. Çetin, Anal. Chem., 2024, 96, 1985–1992 CrossRef CAS PubMed.
- A. Hatami, M. Saadatmand and M. Garshasbi, Talanta, 2023, 267, 125245–125252 CrossRef PubMed.
- E. Polatoglou, Z. Mayer, V. Ungerer, A. J. Bronkhorst and S. Holdenrieder, Diagnostics, 2022, 12, 2550–2557 CrossRef CAS PubMed.
- S. Y. Wang, W. Z. Qi, S. Y. Wu, J. Yuan, H. Duan, Y. B. Li and J. H. Lin, Lab Chip, 2022, 22, 3780–3789 RSC.
- H. Z Tang, L. He, Z. B. Zhou, W. B. Cheng, Z. Z. Liu, X. X. Yan and P. L. Bai, Sep. Purif. Technol., 2025, 353, 128488–128498 CrossRef.
- Z. J. Fei, P. Liu, C. Cheng, R. B. Wei, P. F. Xiao and Y. F. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 4924–4934 CrossRef CAS PubMed.
- M. Niu, M. H. Du, Z. Y. Gao, C. H. Yang, X. Y. Lu, R. R. Qiao and M. Y. Gao, Macromol. Rapid Commun., 2010, 31, 1805–1810 CrossRef CAS PubMed.
- Y. Wang, X. D. Ma, C. Ding and L. Jia, Anal. Chim. Acta, 2015, 862, 33–40 CrossRef CAS PubMed.
- X. H. Pham, A. Baek, T. H. Kim, S. H. Lee, W. Y. Rho, W. J. Chung, D. E. Kim and B. H. Jun, Chem.–Asian J., 2017, 12, 1883–1888 CrossRef CAS PubMed.
- O. Olsvik, T. Popovic, E. Skjerve, K. S. Cudjoe, E. Hornes, J. Ugelstad and M. Uhlen, Clin. Microbiol. Rev., 1994, 7, 43–54 CrossRef CAS PubMed.
- J. Ugelstad, P. Stenstad, L. Kilaas, W. S. Prestvik, R. Herje, A. Berge and E. Hornes, Blood Purif., 1993, 11, 349–369 CrossRef CAS PubMed.
- T. Ellingsen, O. Aune, A. Berge, L. Kilaas, R. Schmid, P. Stenstad, J. Ugelstad, S. Hagen, E. Weng and L. Johansen, Makromol. Chem., Macromol. Symp., 1993, 70, 315–326 CrossRef.
- J. Ugelstad, P. Stenstad, L. Kilaas, W. S. Prestvik, A. Rian, K. Nustad, R. Herje and A. Berge, Macromol. Symp., 1996, 101, 491–500 CrossRef CAS.
- J. Ugelstad, P. C. Mork, R. Schmid, T. Ellingsen and A. Berge, Polym. Int., 1993, 30, 157–168 CrossRef CAS.
- J. Ugelstad, A. Berge, T. Ellingsen, R. Schmid, T. N. Nilsen, P. C. Mork, P. Stenstad, E. Hornes and O. Olsvik, Prog. Polym. Sci., 1992, 17, 87–161 CrossRef CAS.
- C. Zhang, W. J. Liu, T. Y. Wang, T. T. Qian, N. U. Ain, L. Q. Wu, X. N. Wu, X. P. Cai, Q. Zeng and H. P. Xie, J. Pharm. Biomed. Anal., 2020, 189, 113439–113447 CrossRef CAS PubMed.
- F. Q. Tang, L. L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS PubMed.
- Y. Chen, H. R. Chen and J. L. Shi, Adv. Mater., 2013, 25, 3144–3176 CrossRef CAS PubMed.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- Q. Cai, Z. S. Luo, W. Q. Pang, Y. W. Fan, X. H. Chen and F. Z. Cui, Chem. Mater., 2001, 13(2), 258–263 CrossRef CAS.
- M. Vallet-Regí, F. Schüth, D. Lozano, M. Colilla and M. Manzano, Chem. Soc. Rev., 2022, 51, 5365–5451 RSC.
- Q. Q. Xu, Y. Q. Yang, J. Y. Lu, Y. Z. Lin, S. P. Feng, X. Y. Luo, D. H. Di, S. L. Wang and Q. F. Zhao, Coord. Chem. Rev., 2022, 469, 214687–214711 CrossRef CAS.
- L. Huang, Y. X. Zhang, T. Liao, K. Xu, C. X. Jiang, D. L. Zhuo, Y. Wang, H. M. Wen, J. Wang, L. J. Ao and J. Hu, Small, 2021, 17, 2100862–2100873 CrossRef CAS PubMed.
- D. Q. Li, L. J. Ao, R. Hu, X. Q. Zhang, L. Huang, C. X. Jiang, G. S. Gao, Z. L. Shen, J. Hu and J. Wang, Small, 2024, 2402676–2402686 CrossRef CAS PubMed.
- Y. Wang and H. C. Gu, Adv. Mater., 2015, 27, 576–585 CrossRef CAS PubMed.
- Z. M. Chang, Z. Wang, D. Shao, J. Yue, H. Xing, L. Li, M. F. Ge, M. Q. Li, H. Z. Yan, H. Z. Hu, Q. B. Xu and W. F. Dong, ACS Appl. Mater. Interfaces, 2018, 10, 10656–10663 CrossRef CAS PubMed.
- J. Kim, J. E. Lee, J. Lee, J. H. Yu, B. C. Kim, K. An, Y. Hwang, C. H. Shin, J. G. Park, J. Kim and T. Hyeon, J. Am. Chem. Soc., 2006, 128(3), 688–689 CrossRef CAS PubMed.
- Y. H. Deng, D. W. Qi, C. H. Deng, X. M. Zhang and D. Y. Zhao, J. Am. Chem. Soc., 2008, 130, 28–29 CrossRef CAS PubMed.
- C. D. Spicer, C. Jumeaux, B. Gupta and M. M. Stevens, Chem. Soc. Rev., 2018, 47, 3574–3620 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00987h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |