DOI:
10.1039/D5NA00342C
(Paper)
Nanoscale Adv., 2025,
7, 4161-4168
Sensitive, selective, and pretreatment-free detection of ferric ions in different solvents based on organic-soluble carbon dots†
Received
11th April 2025
, Accepted 16th May 2025
First published on 3rd June 2025
Abstract
The presence of ferric ion (Fe3+) impurities can reduce the stability and efficiency of organic solvents, which can significantly impact the production of chemicals, food, medical supplies, and daily necessities. Herein we describe the synthesis of organic-soluble carbon dots (CDs) and their first application for Fe3+ detection in various organic solvents without requiring pretreatment procedures. The CA-CDs were prepared via a solvothermal method using caffeic acid (CA) as a precursor and anhydrous ethanol as the solvent. The presence of Fe3+ triggered changes in the fluorescence intensity of CA-CDs and exhibited good linearity in five protonic and non-protonic solvents with different polarities, including ethanol (3.0–50 μM, R2 = 0.9915), methanol (5.0–50 μM, R2 = 0.9903), ethyl acetate (5.0–50 μM, R2 = 0.9945), acetonitrile (6.0–50 μM, R2 = 0.9998), and dichloromethane (4.0–40 μM, R2 = 0.9940). The corresponding detection limits were 0.96, 1.66, 1.54, 1.73, and 1.19 μM, respectively. Owing to the formation of an iron hydroxyl complex, CA-CDs demonstrated high selectivity towards Fe3+ over other potentially interfering metal ions in both pure solvents and solvents containing 1% (v/v) water. The accuracy of CA-CDs was validated by comparison with results from the Inductively Coupled Plasma-Optical Emission Spectrometer method. With the above outstanding properties, the proposed CA-CDs were successfully employed for Fe3+ quantification in automotive ethanol gasoline with a detection limit of 2.82 μM. Compared to the contamination and errors associated with sample pretreatment in most conventional assays, the CA-CD-based platform offers low-cost, high sensitivity, selectivity, operational simplicity, and contamination-free detection.
Introduction
Ferric ions (Fe3+) play an important role in the geochemical cycle by driving biogeochemical transformations via redox reactions.1 Furthermore, Fe3+ serves as a pivotal component in human industrial applications, with its utilization spanning diverse fields such as chemical production,2,3 biomedical engineering,4 food safety management,5,6 and environmental remediation.7 Monitoring of Fe3+ impurities is of critical significance in product quality control, especially in the organic phase matrix. For instance, trace Fe3+ impurities in high-purity organic solvents can significantly degrade their chemical stability, compromising their efficiency and safe utilization.8 In chemical reactions that employ iron salts as catalysts, monitoring the residual Fe3+ in organic solutions is essential to prevent the production of iron contamination-induced side products.9,10 Similarly, detection of the presence of Fe3+ in oil products (e.g., petroleum and lubricants) can provide quantifiable indicators of mechanical component wear during transportation and storage.11 Therefore, it can be seen that the development of convenient and accurate methods for detecting Fe3+ in organic systems has important practical application significance. Currently, contemporary advancements in Fe3+ detection have been predominantly confined to aqueous environments, with the critical domain of organophilic-phase Fe3+ quantification remaining substantially underexplored.
Conventional approaches to Fe3+ detection include phenanthroline spectrophotometry,12,13 flame atomic absorption spectroscopy,14 inductively coupled plasma optical emission spectrometry,15etc. These methods are typically involved with relatively complex pre-treatment procedures, such as digestion and roasting, as well as the use of potentially hazardous reagents, which may raise environmental concerns.16
Although direct organic solvent injection analysis is gaining increasing popularity, and various analytical methods such as ICP-based techniques and their coupled systems are available, they still face limitations. These include specific technical requirements that elevate assay costs, such as the need for hardware materials compatible with organic solvents in the sample introduction system, careful selection of an internal standard system, and integration of oxygen-assisted combustion.17,18 In contrast, fluorescence analysis emerges as an analytical method characterized by low-cost instrumentation, simple operation, high sensitivity, and accurate detection. However, fluorescence-based determination of Fe3+ in organic systems remains underdeveloped, constrained by limited adaptability to diverse organic matrices and the complex synthesis and purification of fluorescent materials.19
Significantly, fluorescence analysis utilizing carbon dots (CDs) has gained widespread attention due to its low cost, simple operation, and environmental friendliness.20 Nowadays, CDs have been extensively studied for Fe3+ detection taking advantages of diverse synthetic precursors, easy preparation, low toxicity, and tunable fluorescence emissions.21–23 However, as noted earlier, existing methods for applying CDs to Fe3+ detection still face limitations, particularly the insufficient emphasis on organic phase applications. Therefore, given the merits of CD-based fluorescence analysis, there is a high demand to develop organic-soluble CDs to monitor Fe3+, especially in diverse solvent systems.24–26 So far, many strategies have been reported to fabricate organic-soluble CDs, demonstrating that their optical properties and solubility can be manipulated by adjusting several factors including precursors, solvents, and preparation methods.27–30 For instance, Zhang et al. reported CDs prepared with o-phenylenediamine as the precursor by hydrothermal treatment, which exhibited constant optical properties in six alcohols but solvent-dependent fluorescence emission in other solvents.27 Gyulai et al. prepared CDs with identical precursors (citric acid and urea) in different solvents and found that the dispersing solvents significantly influenced emission intensities rather than peak positions.28 It is worth noting that, on the one hand, the fluorescence emission peaks of certain CDs typically shift and change in intensity with changes in solvent, which affects the sensitivity of the detection method to some extent, making it difficult to develop a universal detection platform. On the other hand, the fluorescence emission intensity of CDs is usually susceptible to interference by various metal cations such as Cu2+, Hg2+, Pb2+, etc.31–34 Therefore, it is still a challenge to develop CD-based analysis methods with sensitive and selective Fe3+ detection ability in diverse solvents.
In this work, we synthesized organic-soluble CDs for highly sensitive, selective, simple operation, and contamination-free detection of Fe3+ in a wide range of solvents with simplified operational procedures. The obtained CDs were prepared with caffeic acid (CA) as the carbon source (CA-CDs) in a one-step anhydrous ethanol-based solvothermal method (Scheme 1). The CA-CDs presented relatively stable fluorescence emission and good solubility in a diversity of organic solvents. Furthermore, CA-CDs exhibited Fe3+-induced fluorescence quenching, which could be further employed for Fe3+ quantification in various solvents. In the end, Fe3+ in automotive ethanol gasoline with complex solvent composition was quantitatively detected using CA-CDs, confirming their practical utility as Fe3+ sensors.
 |
| | Scheme 1 Preparation of CA-CDs and the working mechanisms of Fe3+ detection in organic solvents. | |
Experimental
Chemicals and reagents
Caffeic acid (CA) and metal cations (as chloride or phosphate salts) were purchased from Sinopharm Chemical Reagent Co., Ltd (China, Shanghai). Anhydrous ethanol, dichloromethane, methanol, acetonitrile, and ethyl acetate were purchased from Macklin Biochemical Technology Co., Ltd (China, Shanghai). The automotive ethanol gasoline (E10, 92#) was obtained from a gas station of China Petroleum & Chemical Corporation.
Measurements
High-resolution transmission electron microscopy (HRTEM) images were captured using an FEI Tecnai F20 microscope (Thermo Fisher, USA). The particle size distribution of CDs was characterized by a dynamic light scattering (DLS) study with a Zetasizer Nano ZS90 (Malvern, U.K.). A D8 X-ray diffractometer (XRD) was used for X-ray photoelectron spectroscopy analysis (Bruker AXS GmbH, USA). X-ray photoelectron spectra (XPS) were obtained on an ESCALAB 250XI (Thermo Fisher, USA). Fourier transform infrared spectroscopy (FTIR) was conducted on an iS50 FTIR spectrometer (Nicolet, Japan). UV absorption and fluorescence emission spectra were recorded on a UV-2700 spectrophotometer (Shimadzu, Japan) and a RF-6000 spectrophotometer (Shimadzu, Japan), respectively. The fluorescence lifetime test was performed on an FLS 1000 spectrometer (Edinburgh, UK).
Synthesis of CA-CDs
The CA-CDs were synthesized by heating caffeic acid in anhydrous ethanol. Briefly, 1.8 g of caffeic acid was dissolved in 20.0 mL of anhydrous ethanol solvent. The resulting suspension was transferred to a Teflon-sealed autoclave reactor and heated at 200 °C for 6 h. Subsequently, the reactor was naturally cooled to room temperature. The obtained yellowish solution was purified through a 0.22 μm filter membrane and then dialyzed against anhydrous ethanol for 48 h. After evaporating the solvent, the CA-CD powder was obtained and stored in a desiccator for further use.
Determination of quantum yield (QY)
The relative fluorescence quantum yield (Φ) of CA-CDs in anhydrous ethanol was determined with that of L-tryptophan in water as the reference according to the following equation:35| | | Φs = Φr × (Fs/Fr) × (ηs2/ηr2) × (Ar/As) | (1) |
where the subscripts “r” and “s” refer to the reference (L-tryptophan) and sample (CA-CDs), respectively. “F”, “A”, and “η”, respectively, refer to the integrated fluorescence intensity, absorbance at 280 nm, and refractive index (1.3330 and 1.3618 for water and ethanol, respectively).
Quantification of Fe3+ in various organic solvents
The quantification of Fe3+ in ethanol was performed as follows. Briefly, CA-CD solution (85 μg mL−1) and standard Fe3+ solutions with different concentrations were prepared by dissolving CA-CDs and FeCl3 in anhydrous ethanol, respectively. Subsequently, the CA-CD solution was mixed with different concentrations of standard Fe3+ solutions according to desired amounts in the volume ratio of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. After incubation at room temperature for 1 h, fluorescence intensity spectra of CA-CDs before and after Fe3+ addition were measured at 340 nm with an excitation wavelength of 290 nm. Each measurement was conducted in triplicate. The calibration curve for Fe3+ quantification in ethanol was obtained by plotting the quenching efficiency (F0/F) against the concentration of Fe3+. The quantification of Fe3+ in other organic solvents was conducted similarly but with different solvents (methanol, ethyl acetate, acetonitrile, and dichloromethane) replacing ethanol.
:1. After incubation at room temperature for 1 h, fluorescence intensity spectra of CA-CDs before and after Fe3+ addition were measured at 340 nm with an excitation wavelength of 290 nm. Each measurement was conducted in triplicate. The calibration curve for Fe3+ quantification in ethanol was obtained by plotting the quenching efficiency (F0/F) against the concentration of Fe3+. The quantification of Fe3+ in other organic solvents was conducted similarly but with different solvents (methanol, ethyl acetate, acetonitrile, and dichloromethane) replacing ethanol.
Determination of anti-interference ability
Various cationic solutions (1.0 mM) were prepared by dissolving chloride or nitrate salts in organic solvents. The obtained solutions were then incubated with CA-CDs at the volume ratio of 1:9 (v/v) at room temperature for 1 h. Fluorescence intensity spectra of the mixtures were recorded before and after the addition of interfering ions. Each measurement was conducted in triplicate.
Results and discussion
Characterization of CA-CDs
CA-CDs were prepared as described in the Experimental section. The influences of experimental parameters including CA mass (1.4, 1.6, 1.8, 2.0, 2.2, and 2.4 g), reaction temperature (140, 160, 180, 200, and 220 °C), and reaction duration (2, 4, 6, 8, and 10 h) on the QY performance of CA-CDs were comprehensively investigated. Table S1† shows that as the CA amount increases from 1.4 g to 2.4 g, the QY value of CA-CDs initially increased and then declined, which results in a maximum QY value at 1.8 g. In addition, the QY value of CA-CDs was enhanced as the reaction temperature increased and relatively stabilized at 200 °C. The QY value of CA-CDs exhibited a positive correlation with reaction time, peaking at 6 h. Accordingly, CA-CDs achieved a maximum QY of 13.8% under the optimized conditions (1.8 g CA, 200 °C, 6 h).
The morphological feature of CA-CDs was characterized with transmission electron microscopy (TEM), dynamic light scattering (DLS), and X-ray diffraction (XRD) study. The individual CA-CDs exhibit a particle size of less than 5 nm with a monodisperse morphology and no evident aggregation (Fig. 1A). Distinct lattice structures were observed, characterized by a lattice spacing of 0.220 nm, corresponding to the lattice plane (100) of graphitic carbon (Fig. 1A, inset). Additionally, the CA-CDs displayed a weak and broad diffraction peak at 20.7°, indicative of an amorphous carbon matrix (Fig. 1B). Combined HRTEM and XRD analyses confirm the presence of localized graphitized domains within the CA-CDs, despite their overall amorphous nature. Dynamic light scattering (DLS) evaluation revealed a particle size distribution ranging from 3.62 to 6.50 nm, with a predominant size of 4.78 ± 0.46 nm (Fig. S1†). Moreover, the chemical structure of CA-CDs was explored by FTIR and XPS analyses. As illustrated in Fig. 1C, typical peaks could be found in the FTIR spectra of both CA and CA-CDs. The peak at 3390 cm−1 was attributed to O–H in the carboxyl group, peaks at 1605, 1516, and 1441 cm−1 suggested the tensile vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO, and the peak at 1280 cm−1 is derived from the C–O stretching of phenolic hydroxyl groups. The XPS analysis revealed the presence of two elements, carbon (75.37%) and oxygen (24.63%), on the surface of CA-CDs (Fig. 1D). Deconvolution of C 1s high-resolution spectra showed three contributions including C–C/C–H at 240.80 eV, C–O at 286.24 eV, and O–CO at 289.02 eV (Fig. 1E). The binding energies of O 1s high-resolution spectra at 532.37 and 533.54 eV were attributed to the formation of CO and C–O, respectively (Fig. 1F).
C and CO, and the peak at 1280 cm−1 is derived from the C–O stretching of phenolic hydroxyl groups. The XPS analysis revealed the presence of two elements, carbon (75.37%) and oxygen (24.63%), on the surface of CA-CDs (Fig. 1D). Deconvolution of C 1s high-resolution spectra showed three contributions including C–C/C–H at 240.80 eV, C–O at 286.24 eV, and O–CO at 289.02 eV (Fig. 1E). The binding energies of O 1s high-resolution spectra at 532.37 and 533.54 eV were attributed to the formation of CO and C–O, respectively (Fig. 1F).
 |
| | Fig. 1 Characterization of CA-CDs. (A) A representative HRTEM image of CA-CDs. The inset shows a crystalline structure image. (B) The XRD pattern. (C) FTIR spectra. (D) XPS survey scan. (E and F) The high-resolution scan of C 1s peaks (E) and O 1s peaks (F). | |
Fundamental optical properties of CA-CDs
The optical properties of CA-CDs were explored by recording their UV-vis absorption and fluorescence emission spectra. The ethanol solution of CA-CDs is transparent yellowish and shows blue emission under UV light (Fig. 2A). It displays a maximum absorption peak at 286 nm, which is attributed to the n–π* electronic transition of CO bonds. The fluorescence emission intensity and emission wavelength of CA-CDs exhibited distinct excitation wavelength dependencies, as systematically characterized in Fig. 2B. The maximum fluorescence emission peak appeared at 340 nm with an excitation wavelength of 290 nm. Normalized spectral analysis revealed a pronounced 18 nm redshift that occurred across the 280–320 nm excitation range, whereas a negligible shift (Δλ ≤ 5 nm) occurred within the 240–270 nm excitation window (Fig. S2†). Integrated with the preceding characterization data, the observed fluorescence behaviour of CA-CDs can be attributed to a synergistic interplay of the surface domain effect (diverse states of multiple C- and O-containing surface functional groups) and the quantum effects modulated by the graphitization of the carbon core.36–39
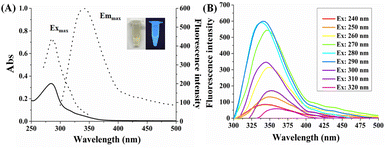 |
| | Fig. 2 (A) The absorption (solid black line) spectrum, the fluorescence excitation and emission (dashed black line) spectra of CA-CDs in ethanol solution. (B) Fluorescence emission spectra of CA-CDs at different excitation wavelengths. | |
Responsiveness study of Fe3+
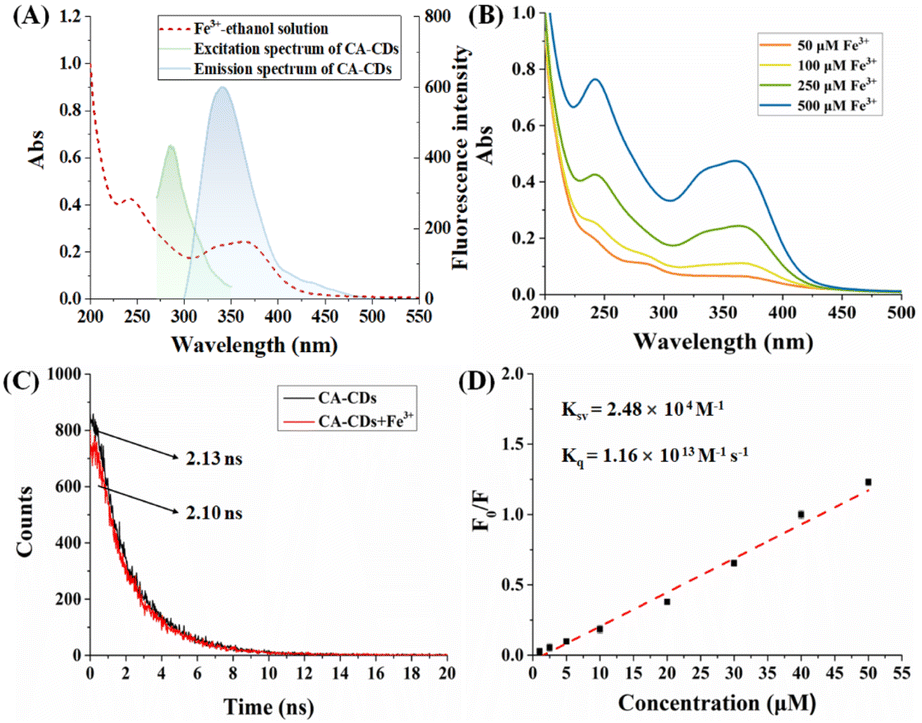
The responsiveness of CA-CDs to various cations (Ag, Al, Ca, Cu, Fe, Hg, K, Mg, Na, Pb, and Zn) in ethanol was preliminarily investigated. It turns out that the fluorescence emission of CA-CDs could be significantly quenched by Fe3+ with prolonged incubation time, which implies the potential use of CA-CDs as a fluorescent Fe3+ sensor. However, it was also observed that Cu2+ interfered with the detection selectivity, as it partially quenched the fluorescence emission of CA-CDs. Therefore, we optimized the measurement conditions by varying the incubation time to achieve satisfactory selectivity. As shown in Fig. S3A,† the quenching efficiency of Fe3+ toward CA-CDs was 42.4% immediately after addition of 100 μM and gradually increased to 93.8% after 8 h of incubation. In contrast, the addition of 100 μM Cu2+ resulted in only 16.6% quenching efficiency within 1 h, ultimately reaching 44.0% after an incubation period of 8 h. Thus, selective detection of Fe3+ over Cu2+ can be realized by setting the incubation time to be 1 h (Fig. S3B†). Based on this optimization, a detailed investigation of Fe3+ detection using CA-CDs in ethanol was performed. The fluorescence intensity of CA-CDs gradually decreased as the concentration of Fe3+ increased from 0 to 100 μM (Fig. 3A). In particular, a good linear relationship (R2 = 0.9915) in the range of 3.0–50 μM was obtained by plotting the relative quenching efficiency (F0/F) at 340 nm against the concentration of Fe3+ (Fig. 3B). The limit of detection (LOD) was calculated to be 0.96 μM by the 3δ/S method, where δ represents the standard deviation of 20 blank samples, and S is the slope of the standard curve (Table S2†). The LOD obtained in this work is lower than the iron limit specified in some quality standards, such as the quality standard for the iron limit in ethanol (chemical reagent) in China (GB/T 678-2002), indicating its potential for sensitive monitoring of trace iron impurities in ethanol-based chemical reagents.
 |
| | Fig. 3 (A) Fluorescence emission spectra of CA-CDs in ethanol with increasing concentrations of Fe3+ from 0 to 100 μM. (B) The corresponding plot of the relative quenching efficiency (F0/F) at 340 nm against the concentration of Fe3+ in the range of 3.0–50 μM. | |
Fe3+ detection in various organic solvents
The sensing ability of CA-CDs for Fe3+ in other organic solvents was also investigated. Five protonic and non-protonic solvents with different polarities including methanol, ethyl acetate, acetonitrile, dichloromethane, and acetone were utilized. It was found that the addition of 1.25% (v/v) acetone to the CA-CD ethanol solution immediately resulted in a significant drop by 50% in the fluorescence intensity. When the acetone concentration increased to 10% (v/v), the fluorescence intensity was quenched over 80%. It indicates that it is difficult to sense Fe3+ in acetone due to the strong solvent quenching effect (data not shown). On the contrary, as shown in Fig. S4,† the fluorescence intensity of CA-CDs remained above 90% with 10% (v/v) of other solvents, except for dichloromethane (54.2%). It is also worth noting that the maximum emission wavelength of CA-CDs was unaffected by the addition of those solvents. Therefore, the detection linear relationship could be further established in methanol, ethyl acetate, acetonitrile, and dichloromethane, demonstrating the potential of CA-CDs for detecting Fe3+ in diverse solvents.
Fig. 4 shows the plots of relative quenching efficiency (F0/F) of CA-CDs against the concentration of Fe3+ in methanol (Fig. 4A), ethyl acetate (Fig. 4B), acetonitrile (Fig. 4C), and dichloromethane (Fig. 4D), and they all display linear relationship with good correlation coefficients. The linear detection ranges and the corresponding LOD values in these solvents are as follows: methanol (5.0–50 μM, 1.66 μM), ethyl acetate (5.0–50 μM, 1.54 μM), acetonitrile (6.0–50 μM, 1.73 μM), and dichloromethane (4.0–40 μM, 1.19 μM). Additionally, the reliability of LOD values was confirmed by calculating the signal-to-noise ratio (SNR), with a requirement of SNR ≥ 3. As shown in Table S3,† the SNR values at LOD concentrations all exceeded 3.0, thus collectively validating the accuracy of our LOD thresholds.
 |
| | Fig. 4 The linear relationship of relative quenching efficiency (F0/F) versus different concentrations of Fe3+ in (A) methanol, (B) ethyl acetate, (C) acetonitrile, and (D) dichloromethane. | |
Investigation of interfering ions
The evaluation of anti-interference capability serves as a critical performance metric for the practical application of CD-based fluorescence analysis. Due to the limited solubility of interfering ionic impurities in organic solvents, we systematically evaluated the anti-interference performance of CA-CDs in the presence of interfering ions at a final concentration of 100 μM (equivalent to twice the upper detection limit concentration of the linear range). As demonstrated in Fig. S5A–C,† the common interfering ions found in ethanol, methanol, and acetonitrile exhibited slight impact on Fe3+ detection after 1 h of incubation. In addition, we investigated the response of CA-CDs to Fe3+ in organic solvents containing 1% (v/v) water, as the presence of trace amounts of water can enhance the solubility of metal ions. Fig. S5D–F† demonstrate that interfering ions with increased solubility can induce partial fluorescence quenching of CA-CDs. However, the overall impact of these ions remains negligible at this concentration. In response to higher concentrations of interfering ions, it was found that Cu2+ can stably coexist in these solvents within 100–500 μM while inducing pronounced fluorescence quenching of CA-CDs (data not shown). Notably, the presence of elevated concentrations of Cu2+ did not compromise the analytical validity. For instance, in anhydrous ethanol solution, the reaction between CA-CDs and high concentrations of Fe3+ is rapid (less than 1 min) and results in a substantial colour change to grey-black. Conversely, although high concentrations of Cu2+ can expeditiously quench the fluorescence of CA-CD, there is no propensity for the solution colour to blacken (Fig. S6†). These findings collectively demonstrate that the CA-CDs developed in this study exhibit effective anti-interference properties for Fe3+ detection.
Photostability evaluation of CA-CDs for practical applications
Photostability is a critical indicator of the practical applicability of carbon dots for detection purposes. To this end, a comprehensive analysis of the photostability of CA-CDs was performed and detailed experimental procedures are described in Text S1.† Initially, the effects of storage and ambient temperatures on the practical utility of CA-CDs were investigated. CA-CD solutions were mixed with various solvents (ethanol, methanol, ethyl acetate, acetonitrile, and dichloromethane) at a 9:1 (v/v) ratio and incubated at 4 °C, 25 °C, and 35 °C prior to fluorescence measurement. As shown in Fig. 5A, the fluorescence intensities of these CA-CD solutions decreased with increasing temperature, with a decrease of approximately 20% across the tested temperature range. Subsequently, the long-term photostability of the CA-CD solutions under ambient light was assessed at room temperature (Fig. 5B). The relative fluorescence intensities of CA-CDs exposed to indoor light for 30 days remained stable compared to light-protected control solutions, demonstrating the potential for practical applications. Furthermore, their resistance to photobleaching was evaluated by monitoring fluorescence intensity changes during continuous scanning for 120 min at an excitation wavelength of 290 nm. The fluorescence intensity of CA-CDs showed negligible variation, with all solvent systems retaining over 95% after 120 min of irradiation (Fig. 5C). Combining these results, the carbon dots synthesized in this work exhibit significant advantages for practical applications, particularly in Fe3+ detection across various solvents.
 |
| | Fig. 5 (A) Fluorescence intensity of CA-CD solutions under different temperature conditions. Abbreviations: EtOH (ethanol), MeOH (methanol), EA (ethyl acetate), ACN (acetonitrile), and DCM (dichloromethane), representing CA-CD solutions mixed with each solvent at a 9:1 (v/v) ratio. (B) The relative fluorescence intensity of CA-CDs in different solvents (9:1, v/v) under ambient light exposure for 30 days. (C) Photobleaching resistance of CA-CDs in different solvents (9:1, v/v) during continuous irradiation for 120 min. Excitation and emission wavelengths were 290 nm and 340 nm, respectively. | |
Accuracy evaluation
The quantitative accuracy of Fe3+ in dichloromethane using CA-CDs was verified by comparing the recovery results of spiked samples with those obtained by the Inductively Coupled Plasma-Optical Emission Spectrometer (ICP-OES) method (Table 1). The recovery rate achieved by the CA-CD-based method ranged from 100.4% to 105.1%, with a relative standard deviation (RSD) of 1.03% to 8.38%, confirming that the accuracy of the proposed method is comparable to that of ICP-OES. The International Organization for Standardization (ISO number: 6353-3:1987) specifies that the iron content of the analytical reagent dichloromethane should not exceed 0.001% (w/%). Therefore, the CA-CD-based fluorescence analysis method for Fe3+ detection can achieve relatively high accuracy, sensitivity, precision, operational simplicity and low-cost.
Table 1 Recovery results of spiked samples in dichloromethane using CA-CD-based fluorescence analysis and ICP-OES methodsa
| Spiked samples |
Recovery rate (this method) |
Recovery rate (ICP-OES method) |
|
Sample 1, 2, and 3 are spiked samples with 3.0 μM, 7.0 μM, and 15.0 μM of Fe3+, respectively.
|
| Sample 1 |
101.4% ± 1.03% |
107.7% ± 0.07% |
| Sample 2 |
105.1% ± 4.70% |
108.2% ± 0.09% |
| Sample 3 |
100.4% ± 8.38% |
109.6% ± 0.05% |
CA-CDs for Fe3+ detection in actual samples
CA-CDs were further practically applied for Fe3+ detection in real samples. An automotive ethanol gasoline (E10, 92#), a mixture of multiple organic solvents, was used as the analyte of interest. Fig. 6A depicts the fluorescence emission spectra of CA-CDs in ethanol after mixing with automotive ethanol gasoline with various Fe3+ concentrations in the volume ratio of 9:1. It clearly shows that the fluorescence intensity decreased as the Fe3+ concentration increased, and a good linear relationship between F0/F and Fe3+ concentration was observed in the range of 10–50 μM (R2 = 0.9822), which showed that CA-CDs are able to effectively monitor Fe3+ in complex solvents (Fig. 6B). The detection limit of the proposed method was calculated to be 2.82 μM, which complies with the requirement for iron detection specified in the National Standard of the People's Republic of China (GB 18351: Ethanol gasoline for motor vehicles (E10); ≤ 0.010 g L−1, equivalent to 179.05 μM). Moreover, a comparative table has been included in terms of sensitivity, convenience, cost and time consumption of the present method with other existing approaches. As demonstrated in Table S4,† the CA-CD-based fluorescence analysis method developed in this study is well-suited for the detection of trace iron in organic solvents, exhibiting advantages including reduced cost, simple operation, high accuracy and precision, as well as broad applicability to various organic matrices.
 |
| | Fig. 6 (A) Fluorescence spectra of CA-CDs with different concentrations of Fe3+ in automotive ethanol gasoline and (B) the corresponding plot of the relative quenching efficiency (F0/F) against the concentration of Fe3+. | |
Exploration of the possible sensing mechanism
Encouraged by the remarkable fluorescence responsiveness of CA-CDs to ferric ions across various solvent systems, a series of experiments were conducted to elucidate the underlying detection mechanism. Typically, the fluorescence quenching mechanism may involve one or more processes, such as fluorescence internal filtration effect (IFE), dynamic quenching and static quenching. The IFE is usually presented as a decrease in fluorescence intensity due to the absorption of the excitation/emission of the fluorophore in the solution. Therefore, the overlapping of absorption and excitation/emission spectra of CA-CDs in the presence of Fe3+ in ethanol was studied (Fig. 7A). In the Fe3+–ethanol solution, it was clearly observed that typical peaks appeared at 240 nm and a broad peak appeared between 330 nm and 370 nm. With the increase in concentration of Fe3+, the absorption intensity increased gradually, indicating the formation of ligand complexes between Fe3+ and the ethanol molecule (Fig. 7B). Therefore, competitive absorption of UV light may occur between the complexes and the CA-CDs at the excitation wavelength of 290 nm. Meanwhile, the broad absorption between 330 nm and 370 nm precisely overlaps the emission spectrum of CA-CDs, resulting in fluorescence quenching of the IFE mechanism. In addition, the fluorescence lifetime of CA-CDs with or without the presence of Fe3+ was determined to confirm their interaction (Fig. 7C). The average fluorescence lifetime of CA-CDs remains almost unchanged with the presence of Fe3+ (τ0 = 2.13 ns) compared to that without Fe3+ (τ = 2.10 ns), which indicates the formation of non-fluorescent complexes due to the static interaction between CA-CDs and the Fe3+ quencher. Subsequently, the quenching constant (Ksv) and bimodular quenching constant (Kq) were determined to provide further evidence of this conclusion as they are also common indices to distinguish static quenching from dynamic quenching (Fig. 7D).40–42 The quenching constant (Ksv) is measured by using the standard Stern–Volmer equation (2).where F0 and F refer to fluorescence intensities of CA-CDs in the absence and presence of Fe3+, respectively. Ksv is the Stern–Volmer quenching constant and [Q] is the concentration of the Fe3+ quencher. The Kq value can be obtained by calculating the ratio of the Stern–Volmer quenching constant (Ksv) and unquenched fluorescence lifetime of CA-CDs (τ0), and it is reported that the Kq value is usually several orders of magnitude larger than 1010 M−1 s−1 in static quenching while it is in the order of 1010 M−1 s−1 in dynamic quenching. Fig. 7D shows that the estimated Ksv was 2.48 × 104 M−1 and the corresponding Kq value was 1.16 × 1013 M−1 s−1, which provides convincing evidence of static quenching. Collectively, these results indicate that the fluorescence quenching phenomenon in this study is caused by the synergistic effect of IEF and static quenching.
 |
| | Fig. 7 (A) UV-vis absorption spectrum of Fe3+–ethanol solution (red line) and the excitation/emission spectra of CA-CDs (green/black line). (B). UV-vis absorption spectra of different concentrations of Fe3+ in ethanol. (C) Fluorescence lifetime investigation of CA-CDs in the absence (black line) and presence (red line) of Fe3+. (D) The quenching constant (Ksv) and bimodular quenching constant (Kq) calculated using the Stern–Volmer equation. | |
Conclusions
To summarize, we present organic-soluble CA-CDs for selective, sensitive, and pretreatment-free detection of Fe3+ in the organic phase. CA-CDs were fabricated via a one-pot solvothermal method using caffeic acid and anhydrous ethanol. The resulting CA-CDs exhibit high fluorescence stability, good solvent compatibility, and a quantum yield of 13.8%. The relative quenching efficiency (F0/F) of CA-CDs against the concentrations of Fe3+ presents good linearity in a diversity of organic solvents (ethanol, methanol, ethyl acetate, acetonitrile, and dichloromethane) with detection limits of 0.96, 1.66, 1.54, 1.72, and 1.19 μM, respectively. Furthermore, CA-CDs display high specificity towards Fe3+via Fe–O bond formation in both pure solvents and water-containing solvents 1% (v/v) under the optimized conditions. The precision and accuracy of CA-CD-based fluorescence analysis were validated by comparison with the ICP-OES method. Their detection capability in real samples was also confirmed using automotive ethanol gasoline as a complex matrix. In comparison with other analytical methods requiring complex pretreatment steps, the proposed CA-CD fluorescence sensor provides a green and facile approach for Fe3+ detection across a broad range of organic solvents. It serves as a cost-effective tool for monitoring trace Fe3+ impurities, offering low-cost, high accuracy and precision detection capabilities.
Data availability
The data supporting the findings of this study are available within the article and/or its ESI.†
Author contributions
JS contributed to experimental design, funding acquisition, and manuscript composition. CQ contributed to methodology and investigation. YZ and NL contributed to research execution. TW and ZC contributed to data analysis. YY contributed to funding acquisition. QL and LL contributed to writing – review & editing. WL contributed to project administration and writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was jointly supported by Henan Provincial Science and Technology Research Project (grant number 232102231029), National Natural Science Foundation of China (grant number 21804116), Doctoral Starting Fund of Xinxiang Medical University (grant number XYBSKYZZ201807), and the Key Scientific Research Projects of Colleges and Universities in Henan Province (grant number 24A350014).
References
- A. Kappler, C. Bryce, M. Mansor, U. Lueder, J. M. Byrne and E. D. Swanner, Nat. Rev. Microbiol., 2021, 19, 360–374, DOI:10.1038/s41579-020-00502-7.
- K. Zhang, Z. Pei and D. Wang, Bioresour. Technol., 2016, 199, 21–33 CrossRef CAS PubMed.
- R. Ferreira, J. Chaar, M. Baldan and N. Braga, Fuel, 2021, 291, 120104, DOI:10.1016/j.fuel.2020.120104.
- T. Schikarski, H. Trzenschiok, M. Avila and W. Peukert, Powder Technol., 2023, 415, 118032, DOI:10.1016/j.powtec.2022.118032.
- Y. Deng, W. Wang, S. Zhao, X. Yang, W. Xu, M. Guo, E. Xu, T. Ding, X. Ye and D. Liu, Trends Food Sci. Technol., 2022, 122, 83–96, DOI:10.1016/j.tifs.2022.01.034.
- R. Manjusha, R. Shekhar and S. J. Kumar, Food Chem., 2019, 294, 384–389, DOI:10.1016/j.foodchem.2019.04.104.
-
ASTM, D5863-22: Standard Test Methods for Determination of Nickel, Vanadium, Iron, and Sodium in Crude Oils and Residual Fuels by Flame Atomic Absorption Spectrometry, ASTM International, 2022 Search PubMed.
-
Standardization Administration of China, Chemical Reagent—Dichloromethane (GB/T 16983-2021), Standardization Administration of the People's Republic of China, Beijing, China, 2021 Search PubMed.
-
N. E. Leadbeater, Impurities in Organometallic Catalysis, Comprehensive Organometallic Chemistry, 4th edn, 2021, DOI:10.1016/b978-0-12-820206-7.00036-6.
- Y. Huang, M. Wang, W. Liu, Q. Wu and P. Hu, J. Org. Chem., 2024, 89, 4156–4164, DOI:10.1021/acs.joc.4c00155.
- M. D. O. Souza, M. A. Ribeiro, M. T. W. D. Carneiro, G. P. B. Athayde, E. V. R. D. Castro, F. L. F. D. Silva, W. O. Matos and R. D. Q. Ferreira, Fuel, 2015, 154, 181–187, DOI:10.1016/j.fuel.2015.03.079.
- B. Shao, J. Deng, H. Dong, S. Wang, E. Li and X. Guan, Environ. Sci. Technol., 2023, 57, 17144–17153, DOI:10.1021/acs.est.3c04589.
- Z. Wu, Y. Zhang, L. Zhang, B. Zhou, Z. Wei, D. Wang, W. Lu, J. Jia, L. Tao, T. Wang and S. Wang, Adv. Funct. Mater., 2023, 33, 2212479, DOI:10.1002/adfm.202212479.
- G. A. Antunes, H. S. Santos, Y. P. Silva, M. M. Silva, C. M. S. Piatnicki and D. Samios, Energy Fuels, 2017, 31, 2944–2950, DOI:10.1021/acs.energyfuels.6b03360.
- S. V. Smirnova, D. V. Ilin and I. V. Pletnev, Talanta, 2021, 221, 121485, DOI:10.1016/j.talanta.2020.121485.
-
R. M. Barnes Jr, D. Dário and F. J. Krug, Introduction to sample preparation for trace element determination, Microwave-Assisted Sample Preparation for Trace Element Determination, 2014, pp. 1–58, DOI:10.1016/B978-0-444-59420-4.00001-5.
- R. Sánchez, J. L. Todolí, C. P. Lienemann and J. M. Mermet, Spectrochim. Acta, Part B, 2013, 88, 104–126, DOI:10.1016/j.sab.2013.06.005.
- P. A. Mello, J. S. F. Pereira, M. F. Mesko, J. S. Barin and E. M. M. Flores, Anal. Chim. Acta, 2012, 746, 15–36, DOI:10.1016/j.aca.2012.08.009.
- L. He, C. Liu and J. H. Xin, Sens. Actuators, B, 2015, 213, 181–187, DOI:10.1016/j.snb.2015.02.060.
- D. N. Bloch, M. Sandre, S. B. Zichri, A. Masato, S. Kolusheva, L. Bubacco and R. J. Li, Nanoscale Adv., 2023, 5, 1356, 10.1039/d2na00804a.
- X. Li, T. Zhu, Y. Du, H. Yan, R. Yan, W. F. Dong and L. Li, Analyst, 2023, 148, 2564–2572, 10.1039/D3AN00319A.
- H. Liu, X. Zhong, Q. Pan, Y. Zhang, W. Deng, G. Zou, H. Hou and X. Ji, Coord. Chem. Rev., 2024, 498, 215468, DOI:10.1016/j.ccr.2023.215468.
- R. Atchudan, T. N. J. I. Edison, K. R. Aseer, S. Perumal, N. Karthik and Y. R. Lee, Biosens. Bioelectron., 2018, 99, 303–311, DOI:10.1016/j.bios.2017.07.076.
- Z. He, Y. Sun, C. Zhang, J. Zhang, S. Liu, K. Zhang and M. Lan, Carbon, 2023, 204, 76–93, DOI:10.1016/j.carbon.2022.12.052.
- Y. Zhang, S. Zhang, B. Tan, L. Guo and H. Li, J. Colloid Interface Sci., 2021, 604, 1–14, DOI:10.1016/j.jcis.2021.07.034.
- S. Pandit, S. Mondala and M. De, J. Mater. Chem. B, 2021, 9, 1432–1440, 10.1039/D0TB02007A.
- D. Chao, W. Lyu, Y. Liu, L. Zhou, Q. Zhang, R. Deng and H. Zhang, J. Mater. Chem. C, 2018, 6, 7527–7532, 10.1039/c8tc02184h.
- G. Gyulai, F. Ouanzi, I. Bertóti, M. Mohai, T. Kolonits, K. Horváti and S. Bősze, J. Colloid Interface Sci., 2019, 549, 150–161, DOI:10.1016/j.jcis.2019.04.058.
- H. J. Lee, J. Jana, T. Y. L. Ngo, L. L. Wang, J. S. Chung and S. H. Hur, Mater. Res. Bull., 2019, 119, 110564, DOI:10.1016/j.materresbull.2019.110564.
- V. K. Jayswal, A. M. Ritcey and J. F. Morin, Nanoscale Adv., 2023, 5, 337, 10.1039/d2na00619g.
- P. Singh, S. A. Kumar, P. Kumar, N. Kataria, V. Bhankar, K. Kumar, R. Kumar, C. T. Hsieh and K. S. Khoo, Nanoscale, 2023, 15, 16241–16267, 10.1039/d3nr01966g.
- S. Pang and S. Liu, Anal. Chim. Acta, 2020, 1105, 155e161, DOI:10.1016/j.aca.2020.01.033.
- J. Tan, Y. Song, X. Dai, G. Wang and L. Zhou, Nanoscale Adv., 2022, 4, 4035, 10.1039/D2AN01674E.
- Y. F. Vadia, S. Ghosh, N. V. Mehta, S. Jha, I. N. Malek, T. J. Park and K. S. Kailasa, Food Chem., 2023, 428, 136796, 10.1039/d2na00498d.
- F. R. Chen, Anal. Lett., 1967, 1(1), 35–42, DOI:10.1080/00032716708051097.
- A. B. Siddique, S. M. Hossain, A. K. Pramanick and M. Ray, Nanoscale, 2021, 13, 16662, 10.1039/d1nr04301c.
- N. Dhenadhayalan, K.-C. Lin, R. Suresh and P. Ramamurthy, J. Phys. Chem. C, 2016, 120, 1252–1261, DOI:10.1021/acs.jpcc.5b08516.
- Y. Hu and A. Bianco, Carbon, 2025, 235, 120073, DOI:10.1016/j.carbon.2025.120073.
- R. Atchudan, T. N. J. I. Edison, S. Perumal, R. Vinodh and Y. R. Lee, J. Mol. Liq., 2019, 296, 111817, DOI:10.1016/j.molliq.2019.111817.
- R. Atchudan, T. N. J. I. Edison, S. Perumal, N. Muthuchamy and Y. R. Lee, Fuel, 2020, 275, 117821, DOI:10.1016/j.fuel.2020.117821.
- X. Sun, Y. Wang and Y. Lei, Chem. Soc. Rev., 2015, 44, 8019, 10.1039/c5cs00496a.
- F. Zu, F. Yan, Z. Bai, J. Xu, Y. Wang, Y. Huang and X. Zhou, Microchim. Acta, 2017, 184, 1899–1914, DOI:10.1007/s00604-017-2318-9.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a